
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 March 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1302587
This article is part of the Research Topic Reviews in Breast Cancer: 2023 View all 28 articles
 Tosin Akinsipe1
Tosin Akinsipe1 Rania Mohamedelhassan2
Rania Mohamedelhassan2 Ayuba Akinpelu3
Ayuba Akinpelu3 Satyanarayana R. Pondugula2
Satyanarayana R. Pondugula2 Panagiotis Mistriotis3
Panagiotis Mistriotis3 L. Adriana Avila1
L. Adriana Avila1 Amol Suryawanshi4*
Amol Suryawanshi4*The breast cancer tumor microenvironment (TME) is dynamic, with various immune and non-immune cells interacting to regulate tumor progression and anti-tumor immunity. It is now evident that the cells within the TME significantly contribute to breast cancer progression and resistance to various conventional and newly developed anti-tumor therapies. Both immune and non-immune cells in the TME play critical roles in tumor onset, uncontrolled proliferation, metastasis, immune evasion, and resistance to anti-tumor therapies. Consequently, molecular and cellular components of breast TME have emerged as promising therapeutic targets for developing novel treatments. The breast TME primarily comprises cancer cells, stromal cells, vasculature, and infiltrating immune cells. Currently, numerous clinical trials targeting specific TME components of breast cancer are underway. However, the complexity of the TME and its impact on the evasion of anti-tumor immunity necessitate further research to develop novel and improved breast cancer therapies. The multifaceted nature of breast TME cells arises from their phenotypic and functional plasticity, which endows them with both pro and anti-tumor roles during tumor progression. In this review, we discuss current understanding and recent advances in the pro and anti-tumoral functions of TME cells and their implications for developing safe and effective therapies to control breast cancer progress.
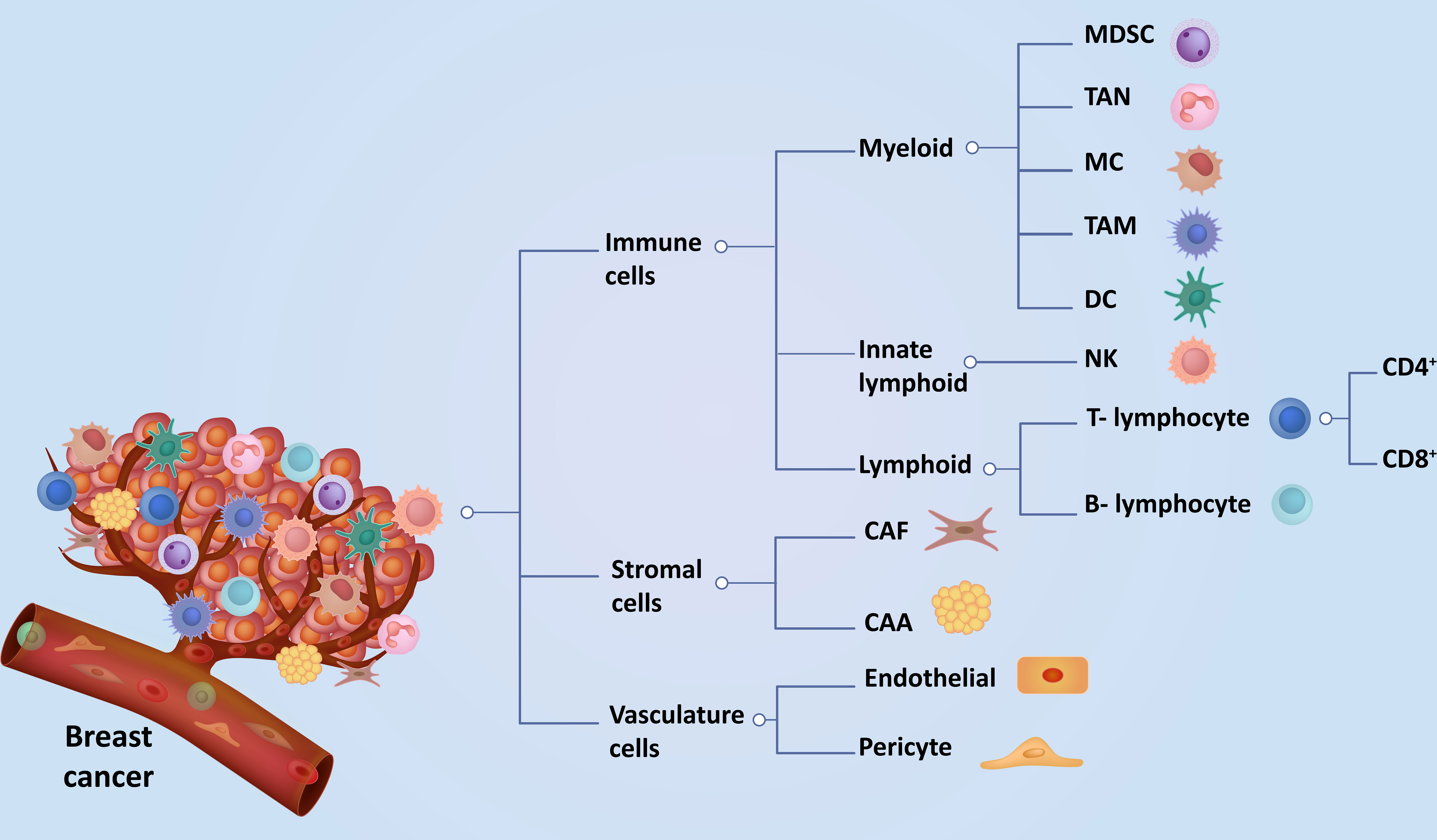
Graphical Abstract
Breast cancer is the most common and frequently diagnosed cancer in women worldwide, with more than 2 million new breast cancer cases reported annually (1). Globally, breast cancer is the leading cause of cancer deaths in women (2, 3). Based on gene profiling, breast cancer can be classified into five molecular subtypes - Luminal A, Luminal B, Human epidermal growth factor receptor 2 (HER2), Basal-like, and Triple-negative breast cancer (TNBC) (4). Although these classifications are not static, ongoing research may lead to refinements or the discovery of new subtypes. Molecular classification plays a crucial role in tailoring treatment strategies for breast cancer patients, helping oncologists choose the most effective therapies based on the specific molecular characteristics of the tumor (5). Additionally, the TNM staging system proficiently evaluates patients by effectively assessing the extent of the tumor (T), involvement of lymph nodes (N), and presence of metastasis (M). Due to differences in molecular characteristics of breast cancer sub-types, the use of biomarkers, histologic grade, HER2 expression, hormone receptor, and multigene panels have now been incorporated into the conventional TNM staging (6). The tumor microenvironment (TME) of breast cancer plays a central role in tumor progression, immune evasion, and resistance to conventional anti-cancer therapy (7). Breast TME mainly comprises cancer, immune, and stroma cells. Apart from cancer cells, the cellular components of breast TME can be broadly classified as immune cells (myeloid, innate lymphoid, and lymphocytes), stromal cells (fibroblasts and adipocytes), and vasculature cells (endothelial cells and pericytes) (Graphical Abstract). The various cellular components of breast TME exhibit intricate and dynamic interactions that significantly impact cancer progression, metastasis, immunosuppression, and resistance to both conventional and emerging immunotherapies (8, 9). The complex molecular and cellular interplay among the TME constituents provides essential nutrients, oxygen, and growth factors that facilitate efficient tumor cell proliferation and progression (10, 11). The surrounding stroma’s cellular, genetic, structural, functional, and epigenetic alterations profoundly impact the plasticity and morphogenesis of epithelial cells, thereby contributing to tumorigenesis (12). Recent breakthroughs and extensive studies from preclinical studies (Table 1) and clinical trials (Table 2) have indicated that alterations in breast TME signatures can serve as valuable prognostic indicators and aid in the development of innovative anti-cancer therapies (27). Consequently, there has been a notable shift towards targeting the key components of the TME in the development of novel treatments (27, 28). In this review, we discuss the current understanding of cancer, stromal, vasculature, and immune cell interactions within the breast TME and their implications for developing novel, safe, and effective breast cancer treatments.
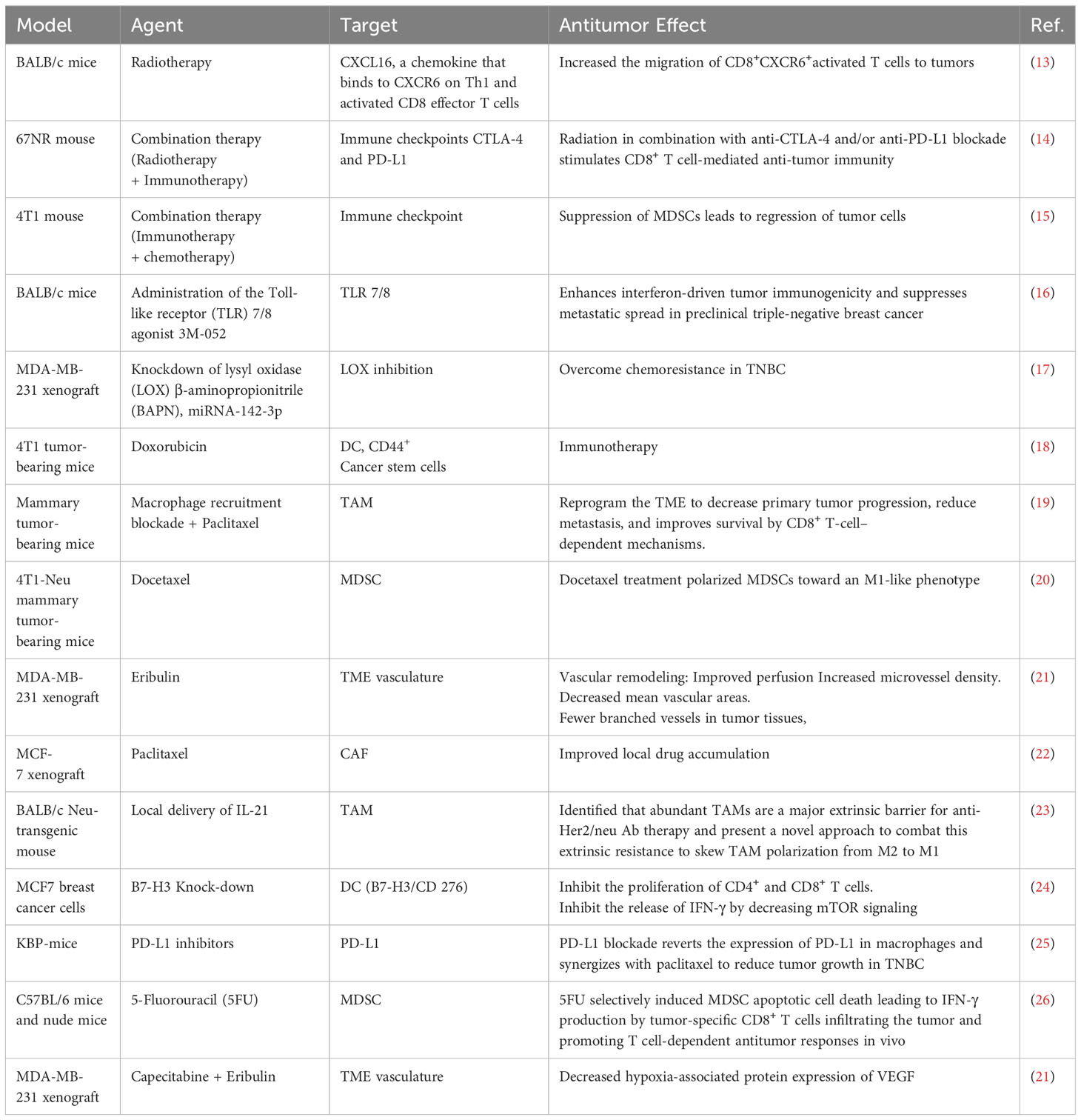
Table 1 Selected pre-clinical studies showing the suppression of tumor progression by targeting TME-associated cells and effector molecules.
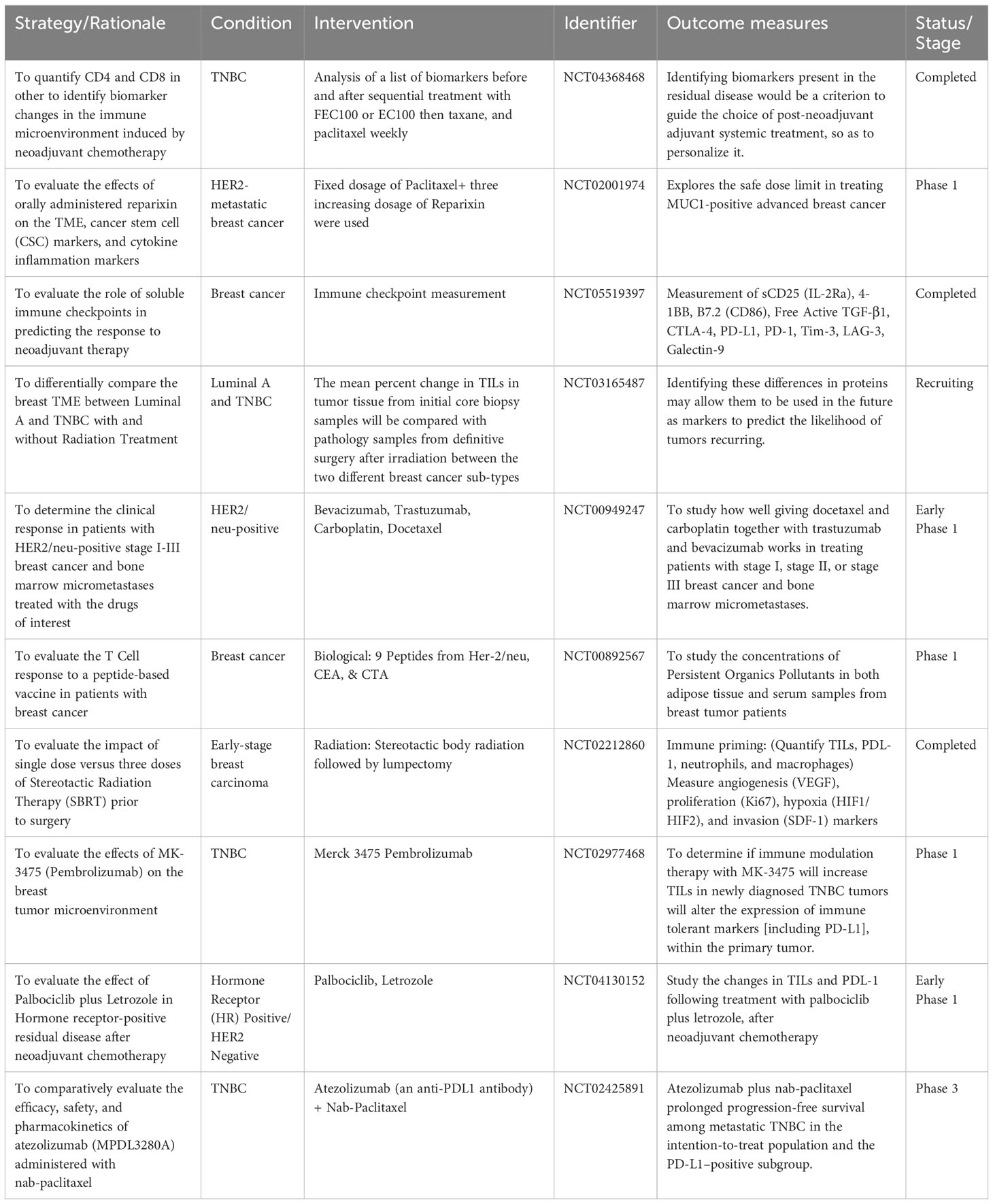
Table 2 Selected clinical trials on breast TME-related targeting modalities (https://www.clinicaltrials.gov/).
Breast TME is highly plastic and undergoes constant changes and stage-specific adaptations depending on numerous cancer cell-intrinsic and extrinsic factors. These alterations in the TME are characterized by networks of cytokines and growth factors, disrupted signaling pathways, and modified molecular signatures in the stroma (29). Extensive research on TME characterization has highlighted the crucial role of communication between tumor cells and stroma in driving breast cancer oncogenesis, progression, and metastasis (Figure 1) (30, 31). The breast tumor stroma comprises various components, including fibroblasts, immune cells, endothelial cells, adipocytes, and pericytes (32). Throughout the progression of breast cancer, the stroma undergoes significant changes, including the formation of cancer-associated fibroblasts (CAFs), infiltration of immune cells, inflammation, angiogenesis, and remodeling of the extracellular matrix (ECM) (33, 34). These alterations disrupt the integrity of the basement membrane, facilitating the spread of tumor epithelial cells into the stroma (35). Since these various molecular and cellular components have a direct influence on breast cancer progression, they represent attractive targets for therapeutic development. The immune cells within the breast TME play a critical and dynamic role in cancer progression and anti-tumor immunity (36). Most immune cells are plastic in their functional phenotype and can adapt in response to local TME factors, allowing them to play dual pro or anti-tumor roles (37). Effector immune cells infiltrating the TME can directly eliminate neoplastic cells expressing neo-antigens on their surface and suppress tumor progression (Figure 2) (38). However, tumors employ numerous immune evasion strategies to impede immune cell infiltration and hinder their effector functions within the TME (37). The immune cell repertoire within the breast TME can be broadly classified as myeloid, innate lymphoid, and lymphoid cells. Myeloid cells include myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs), dendritic cells (DCs), mast cells (MCs), etc. Natural killer (NK) are innate lymphoid cells with cytotoxic effector functions and play a crucial role in anti-tumor immunity. Lymphoid cells include B lymphocytes and numerous subsets of T-lymphocytes that play a central role in tumor-antigens-specific anti-tumor immunity (39).
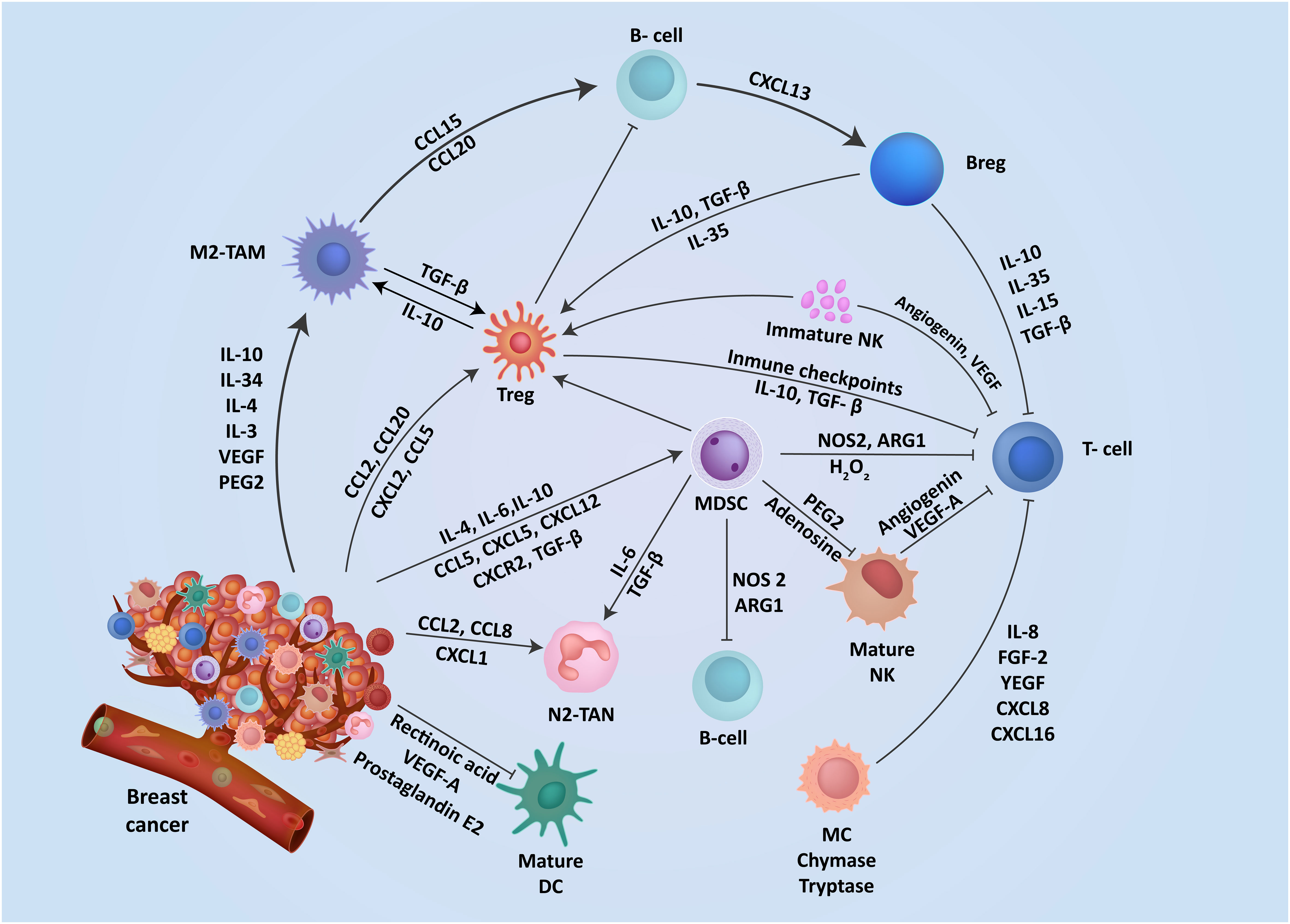
Figure 1 The interplay of mediators aid immunosuppression in breast TME. The TME contains a range of resident cells playing a key role in the progression and metastasis of breast cancer cells. These resident cells and their associated secretory elements and receptors including cytokines, chemokines, and stimulatory growth factors are shown. Cells in the TME exhibit a diverse network of mediators that actively engage in promoting an immunosuppressive TME.
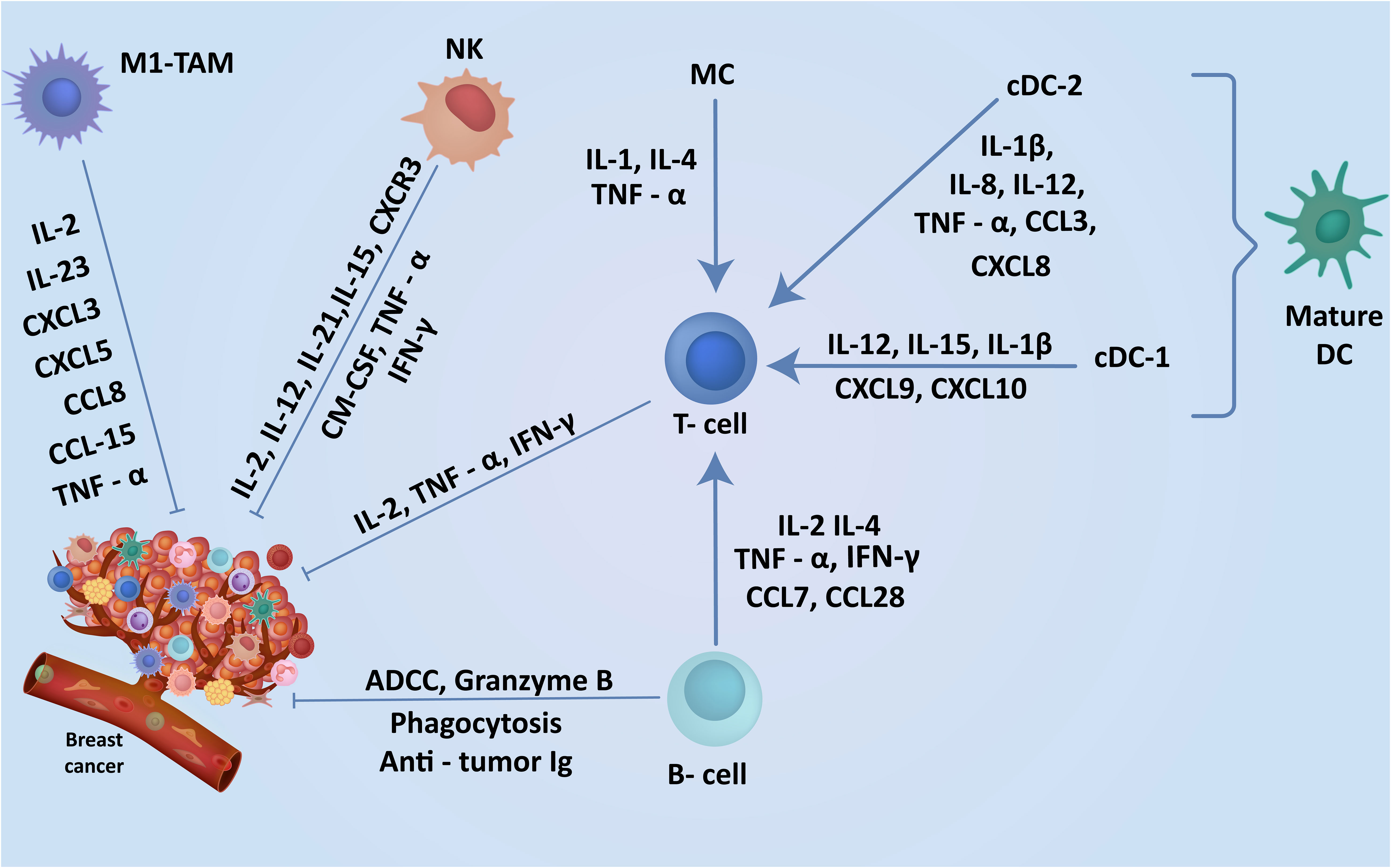
Figure 2 Cells in breast TME regulate the induction of robust anti-tumor immunity. The TME contains a range of anti-tumor cells including TILs, DCs and macrophages in the breast playing a key role in the breast cancer suppression. The expression of these cells within the breast cancer TME and understanding their anti-tumor function may enhance the discovery of new markers associated with specific subtypes leading to earlier diagnosis and better clinical outcomes.
In subsequent sections, we will briefly discuss the interplay of immune and non-immune (stromal and vasculature) cells and how these complex interactions can be strategically targeted to develop novel, safe, and highly effective therapies for breast cancer patients.
Myeloid cells, derived from hematopoietic stem cells in the bone marrow, play a crucial role in initiating innate and adaptive immune responses (40). However, these cells undergo impaired differentiation during cancer progression, resulting in immature phenotypes with reduced phagocytic capacity and immunosuppressive function (41). MDSCs are prominent cell types in the breast TME that rapidly proliferate and promote tumor progression, angiogenesis, and metastases (42, 43). When activated, MDSCs contribute to immunosuppression and cancer invasiveness through increased production of reactive nitrogen species (RNS), reactive oxygen species (ROS), and arginase 1 (ARG1) expression (44, 45). Human MDSCs in the bloodstream can be classified into two types: granulocytic MDSCs (G-MDSCs) and monocytic MDSCs (M-MDSCs) (46). G-MDSCs are further categorized based on cell surface marker expression as CD11b+CD14-CD66+ and CD11b+CD14-CD15+. Similarly, M-MDSCs are characterized by the cell surface markers CD11b+CD14+CD15- (47). MDSCs exhibit low expression of Human Leukocyte Antigen–DR isotype (HLA-DR) and CD14, the cell surface receptors essential for proper immune responses to antigens, resulting in an immune response defect (43, 48). The activation and recruitment of MDSCs in the TME is mediated through increased production of specific chemokines, cytokines, and factors, including IL-4, IL-6, IL-10, IL-12, IL-13, CCL5, CCL2, CXCL2, CXCL5, CXCL12, vascular endothelial growth factor (VEGF)-A, transforming growth factor (TGF)-β, and granulocytic-colony stimulating factor (G-CSF) in TME (49–52). These molecules are critical in shaping the tumor microenvironment and promoting MDSC-mediated immune suppression. MDSCs have been found to play a crucial role in the immunosuppressive microenvironment by facilitating the development of CD4+Foxp3+ regulatory T (Treg) cells and promoting immunosuppressive phenotype in macrophages (53, 54). Additionally, MDSCs express CD40, increasing Treg-mediated tumor immune tolerance (55). CD40, a member of the tumor necrotic factor (TNF) receptor superfamily, is expressed on antigen-presenting cells (APCs), while its ligand (CD40L) is primarily expressed on activated T and B cells (56). The interaction between CD40 and CD40L promotes the development of adaptive immunity (57). When exposed to increased stimulation by IFN-γ, G-MDSCs upregulate the expression of CD40 and MHC II, leading to the induction of Tregs and the suppression of T cell proliferation (55). MDSCs also contribute to angiogenesis, maintain cancer stem cells (CSCs), and inhibit CD8+ T cell activation through the expression of nitric oxide synthase 2 (NOS2) and ARG1 (58, 59). Using microarray analysis, Hix et al. compared the low-aggressive TM40D and highly aggressive TM40D-MD mouse mammary carcinoma cells and discovered a positive correlation between tumor-recruited CD33+ myeloid cells and the progression of human breast cancer from DCIS to IDC (60). Additionally, they found a significant association between CD33+ MDSCs and poor prognosis and worsened overall survival (OS) in the ER- subtype (61). Furthermore, the transcriptional factor deltaNp63 enhanced the recruitment of MDSCs and correlated with poor prognosis and metastasis in TNBC (62). A pre-clinical study revealed the major role of CXCR2+ MDSCs, a subtype of MDSCs, in breast cancer metastases (63). Moreover, MSDCs indirectly regulate immune response and hinder cancer immunotherapy by interacting with other components of the TME (64). The critical role of TME MDSCs in causing immunosuppression and resistance to cancer immunotherapies during breast cancer progression underscores the need for further comprehensive studies to successfully develop innovative immunotherapies.
Neutrophils, comprising 50-70% of circulating leukocytes, represent the body’s primary defense against infections (65). Additionally, they play a crucial role in tumor progression by infiltrating the TME. The TME regulates the recruitment and polarization of neutrophils, allowing them to develop either an anti-tumor (N1) or pro-tumor (N2) phenotype in response to cytokines present in the TME (66, 67). N1 polarized neutrophils exhibit a robust immune profile characterized by elevated levels of TNF-α, CCL3, ICAM-1, and reduced arginase expression. On the other hand, N2 TANs overexpress several chemokines, including CCL2, CCL8, CXCL1, CXCL2, etc (68). The increased NADPH oxidase activity of N1-like neutrophils leads to the generation of cytotoxic ROS, which can effectively target tumor cells (69). Despite the critical role of neutrophils in the TME (70, 71), further studies are warranted to investigate molecular and cellular networks that drive immunosuppressive phenotype in neutrophils in TME. Studies have demonstrated the preferential migration of neutrophils into specific breast tumor subtypes, such as hormonal negative ductal adenocarcinoma and TNBC (72). Additionally, TGF-β has been shown to promote the N2 phenotype in neutrophils infiltrating the TME (66). TANs in the TNBC TME are a source of proangiogenic factors and matrix metalloproteinase (MMP)-9, a protease crucial in ECM remodeling (73). MMPs and gelatinase B/MMP-9 actively degrade the extracellular matrix, promoting tumor invasiveness and metastasis (74, 75). MMP-9 also contributes to angiogenesis and tumor progression by releasing VEGF-A and inhibiting anti-angiogenic molecules (70). TANs’ inability to express tissue inhibitors of metalloproteinase-1 (TIMP-1) enhances the angiogenic potential of neutrophil-derived MMP-9 in the TME, unlike cells expressing the MMP-9/TIMP-1 complexes (76). Recent findings have shown that in the presence of CD90, TIMP-1 expressed by TANs induces epithelial-mesenchymal transition (EMT) in breast cancer, facilitating metastasis (77). As a result, a significant reduction in the spread of cancer has been observed through CD90 blockade (77). Several strategies can be employed to target TANs, including preventing neutrophil migration to tumors, hindering their polarization into N2-type, and targeting neutrophil-associated mediators (71, 78). However, further studies are warranted to characterize TANs’ pro-tumoral role during breast cancer progression properly.
MCs demonstrate a vital role in both innate and adaptive immunity. Positioned within epithelial and mucosal tissues throughout the body, MCs effectively regulate various immune and non-immune cell types, including T and B lymphocytes, endothelial cells, fibroblasts, macrophages, and DCs (79). Notably, MCs exhibit a dual function in breast cancer progression (80). Their ability to produce anti-tumoral cytokines, such as IL-1, IL-4, IL-6, and TNF-α facilitates CD8+ priming and maturation (81). Conversely, MCs can assume pro-tumor roles by increasing the production of immunoregulatory molecules, including IL-8, fibroblast growth factor (FGF)-2, TGF-β, VEGF-A, CXCL8, and CXCL16 (82). Such effector molecules released by MCs hinder immunity, degrade the ECM, and enhance tumor vascularization, thus modifying the TME (82, 83).
In the context of breast TME, MCs actively promote cell proliferation, invasiveness, and metastases, ultimately correlating with a poor prognosis (84). Additionally, MCs are crucial in promoting angiogenesis through secretion of angiogenic cytokines (85). MC stabilizer, disodium cromolyn, has demonstrated its ability to induce an anti-tumor effect by effectively inhibiting the production of VEGF and platelet derived growth factor (PDGF) (86). The infiltration of human MC subpopulations within the TME can be classified based on their expression of the proteases chymase, tryptase, or tryptase-chymase (87). The involvement of chymase and tryptase in ECM remodeling and the production of angiogenic factors highlights their significant role in promoting invasiveness (88). The functions of tryptase and chymase MCs in breast cancer are specific to subtypes. Research by Glajcar et al. revealed a significantly higher presence of the MC tryptase-chymase subset in luminal A and B tumors compared to HER2+ and TNBC, indicating relevance in these subtypes (89). Various studies have also shown the contribution of tryptase+ MCs to tumor progression in TNBC and luminal A breast cancer (89, 90). Although MC stabilizers and protease inhibitors have been successfully used in other cancers, their clinical effectiveness in breast cancer remains uncertain (91). Conversely, a recent report suggested the increased infiltration of MCs is associated with lower tumor grade, reduced tumor proliferation, and decreased HER2 overexpression (92). Further studies are needed to fully comprehend MC function and explore their potential as therapeutic targets in breast cancer.
TAMs are abundant immune cells within the TME (93). Human blood monocytes undergo differentiation into naïve macrophages (M0) and subsequent polarization into M1 and M2 phenotypes mediated by IFN-γ and IL-4, respectively (94, 95). M1 macrophages are highly phagocytic and are associated with CD4+ polarization towards IFN-γ producing Th1 cells (95). M1-like macrophages possess the capability to induce acute inflammatory responses through the production of inflammatory cytokines and chemokines such as IL-2, IL-12, IL-23, TNF-α, CXCL3, CXCL 5, CCL8, CCL15, as well as reactive nitrogen and oxygen intermediates, which exert antitumor effects (96, 97). Resident macrophages play a critical role in host defense (98). However, macrophage populations in TME adapt to an anti-inflammatory, M2-like phenotype (99). The recruitment of TAMs to the TME is promoted by stromal and tumor cells’ production of chemokines and growth factors (100). Peripheral blood monocytes derived from the bone marrow are recruited to the tumor site and undergo differentiation into TAMs (101). The CSF is an integral factor in regulating the recruitment of macrophage populations (102). The recruitment of peripheral blood monocytes to the tumor site is facilitated through chemokine receptors expressed on monocytes and chemokine gradient in TME. One such example is the binding of CCL2 to CCR2 and CCR5 receptors on monocytes, leading to monocyte recruitment to the TME (103). Another example is the binding of CCL20 to CCR6 receptors (104).
In breast cancer, the polarization of monocytes to TAMs is influenced by various factors, including tumor-derived factors produced by breast cancer cells and other cells in the TME (105). Monocyte differentiation into TAMs is mediated by VEGF-A and IL-4 (106). M2 TAM differentiation can occur through IL-4 secreted from Th2 cells and IL-10 derived from Tregs (107). IL-10 inhibits the production of pro-inflammatory chemokines by macrophages and promotes the self-polarization of TAMs (108). Furthermore, alternative M2 activation of TAMs is elicited by IL-34 and IL-13 derived from Th2 cells, eosinophils, or basophils (102). These macrophages serve as a significant source of proteolytic enzymes that facilitate the destruction of the ECM and promote neoplastic cell invasion (74).
TAMs contribute to immune evasion by producing IL-10, EGF, and TGFβ (99, 109). The EGF produced by TAMs actively stimulates the proliferation of breast carcinoma cells (110), whereas TAM-produced IL-10 promotes the accumulation of tumor cells at distant sites (111). Furthermore, TGFβ originating from TAMs enables monocyte efflux (112). Also, TAMs facilitate tumor cell growth, angiogenesis, metastasis, and immune evasion by recruiting Tregs (113). TAMs can also establish cancer stem-cell niches, leading to tumor chemotherapy resistance (114). In TNBC, TAMs consistently activate hepatic leukemia factor (HLF) through the IL-6-TGF-β1 axis. HLF transactivates gamma-glutamyltransferase 1 (GGT1), which promotes ferroptosis and cisplatin resistance, ultimately driving malignancy in tumor cells (115). High infiltration of TAMs is associated with a worsened prognosis in breast cancer patients (116). TAMs and DCs play a pivotal role in inducing and regulating effector T cell and Treg responses within the TME, thereby influencing resistance to recently developed immune-checkpoint blockade (ICB) therapies (93). The Wnt/β-catenin pathway is critical for several biological processes (117). However, its dysregulation has been associated with the development of cancer and other diseases. TAMs and DCs activate the Wnt/β-catenin pathway to induce immune tolerance, inhibiting effector T-cell responses and promoting regulatory T-cell responses (118). Consequently, targeting the Wnt/β-catenin pathway holds promise for effective therapeutic interventions in breast cancer (119). Research on TAMs has led to the development of macrophage-focused treatment approaches, which are currently undergoing clinical trials for breast cancer (120). These strategies involve suppressing macrophage recruitment, reprogramming TAMs towards an anti-tumor phenotype, and enhancing macrophage-mediated phagocytosis or tumor cell killing (100, 116).
DCs are critical in maintaining immune surveillance and achieving a delicate equilibrium between protective immunity and immune tolerance (121). However, tumors exploit these mechanisms to regulate anti-tumor immunity (122). DCs can be categorized into various subsets based on their location, phenotype, and antigen presentation abilities (123). As professional APCs, DCs are pivotal in initiating and activating anti-tumor T cell responses in tumor-draining lymph nodes and the TME (124). During breast cancer progression, DCs engage in phagocytosis of apoptotic tumor cells, process and present tumor antigens on MHC-I and MHC-II molecules, migrate to local lymph nodes, and present antigens to naive CD4+ and CD8+ T cells to elicit an anti-tumor immune response (125, 126). Additionally, DCs in the TME secrete chemokines and cytokines that play a crucial role in recruiting and activating effector CD4+ and cytotoxic CD8+ T cells for effective anti-tumor immune responses (127).
Transcriptional profiling has identified specific subsets of DCs in both normal breast tissue and breast TME (128). While the nomenclature and classification of DCs in the TME can be complex, DCs in TME can be broadly classified into three subsets, including plasmacytoid DCs (pDCs), monocytic DCs (moDCs), and conventional DCs (cDCs), which are further classified as cDC-1 and cDC-2 (129, 130). pDCs play a significant role in cross-presenting tumor antigens on MHC-I molecules to initiate cytotoxic CD8+ T Lymphocytes (CTL)-mediated anti-tumor responses (131). pDCs also secrete large amounts of type I interferons (IFN-α/β) (131). Recent studies have shown that pDCs in breast TME promote Treg responses, negatively impacting prognosis and survival rates (132, 133). Additionally, gene expression analysis has revealed that pDC-related genes are among the top genes associated with an increased risk of breast cancer metastasis (134). However, other studies have contradicted these findings, highlighting a better prognosis and increased survival linked to pDCs (135–137). Circulating monocytes can differentiate into moDCs within the TME and are primarily responsible for inducing CD4+ T cell-mediated responses (138). However, the immunosuppressive TME often leads to a tolerogenic phenotype in moDCs, increasing pro-tumor Treg responses (139). cDC-1 and cDC-2 in the TME play a critical role in capturing tumor antigens to activate CD8+ and CD4+ effector T-cell responses (134). cDC-1 can be identified by the expression of markers such as IRF8, BDCA3, BATF3, CLEC9A, and CD103 (140). They produce cytokines (IL-12, IL-15, IFN-β) and chemokines (CXCL9 and CXCL10) to mount a robust immune response (128). In the luminal and TNBC subtypes, cDC-1 has been associated with improved disease-free survival (DFS) and positive patient outcomes through its activation and expansion of CD103, enhancing the tumor response to therapeutic programmed death-ligand 1 (PD-L1) and BRAF inhibition (141, 142). cDC-2 express various markers such as IRF4, CD11b, SIRPα, CLEC10A, and CD1C, and produce IL-1β, IL-6, IL-8, IL-12, TFN-α, CCL3, and CXCL8 to activate anti-tumor T cell responses (143, 144).
DCs have been found to exhibit a dual pro-tumoral and anti-tumoral role depending on the cytokine milieu in the TME and their maturation state (145, 146). For instance, immature DCs support angiogenesis in rapidly growing angiogenic tumors, while mature DCs suppress angiogenic characteristics (147). Additionally, infiltration of mature DCs in primary tumors is associated with reduced metastasis and improved clinical outcomes (148). The increased expression of CD83, a marker for mature DCs, is strongly linked to improved survival in node-positive tumor patients, particularly in TNBC patients with mature CD11c+ (149, 150). Moreover, the presence of CD83+ in the peri-tumoral region of IDC lesions suggests a potential role for mature DCs, while immature CD1a+ DCs are found at tumor edges (151). Previous studies indicate that the immature DC phenotype promotes primary tumor progression to IDC (148, 152). Furthermore, elevated levels of pro-tumor molecules like VEGF-A and prostaglandin E2 in the TME hinder DC maturation, thereby inhibiting T-cell proliferation (153, 154). In response, therapeutic strategies targeting VEGF-A, such as anti-VEGF-A antibodies like bevacizumab, have shown promise in promoting T cell and DC infiltration in TNBC (155). Extensive research has unveiled the pivotal role played by Wnt/β-catenin signaling in the advancement of breast cancer, spanning both tumor cells and immune cells (156). The upregulation of Wnt ligands triggers the activation of canonical β-catenin signaling in DCs, thereby facilitating the generation of IL-10, TGF-β, and retinoic acid (RA) synthesizing enzymes (122, 157). This augmented production of immunoregulatory molecules by DCs within the TME fosters the development of Treg responses, overshadowing Th1 and CTLs (156, 158). These findings underscore the potential of targeting DCs in breast cancer progression as a viable therapeutic strategy, capable of stimulating robust anti-tumor immunity and suppressing regulatory T-cell responses.
NKs are innate lymphoid cells that can directly eliminate tumor cells by releasing anti-tumor cytokines and cytolytic granules (159). Their development primarily occurs from hematopoietic stem cells (HSCs) in the bone marrow, although other origin sites, such as the thymus and liver, have been proposed (160). Essential transcriptional factors for NK cell precursors include Nfil3, Id2, and Tcf1, while maturation relies heavily on Smad4, Tox, Eomes, Gata3, T-bet, and Runx3 (161–163). Cytokines also play a crucial role in NK cell development and maturation (164–166). For example, IL-7 is responsible for generating CD122+ NK progenitors from HSCs, while IL-15 is critical for the development of NKs from CD122+ NK progenitors into mature NKs (mNKs) (167, 168). Additionally, IL-17 modulates the activities of IL-15 (166). Functionally, mNKs can be differentiated into two major subtypes: CD56brightCD16dim NK subtypes, which make up approximately 90% and are involved in cytotoxicity, and CD56dimCD16bright NK subtypes, which make up the remaining 10% and are responsible for antibody-dependent cell-mediated cytotoxicity (ADCC) (169). Unlike T-lymphocytes, NK cells recognize target cells expressing aberrant cell surface proteins, such as virus-infected or tumor cells, through their Fc receptors (170). The binding of NK cell Fc receptors to antibody-coated target cells leads to targeted killing through ADCC. Moreover, NK cells can eradicate cells that lack or display diminished MHC class I molecules on their cell surface, a common strategy that cancer cells employ to avoid CTL responses (171).
Tumor-infiltrating NK cells engage in immunosurveillance using a combination of activating and inhibitory receptors, effectively identifying and eliminating target cells while sparing healthy ones (172, 173). This process is facilitated by the production of various cytokines and chemokines, including TNF-α, IFN-γ, IL-2, IL-12, IL-21, IL-15, IL-18, CXCR3, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which actively promote anti-tumor immunity (174, 175). Additionally, the receptors on NK cells can selectively target tumor cells by recognizing growth factors like PDGF, thereby triggering the release of IFN-γ and TNF-α to inhibit tumor growth (176). Although NK cells exhibit anti-tumor capabilities, they can also produce immunosuppressive cytokines that hinder anti-tumor immunity. NK cells secrete angiogenic factors like VEGF-A and angiogenin, which contribute to the progression of breast cancer (177). A recent study uncovered a new mechanism of cancer immune evasion, which involves inhibiting NK cells’ cytotoxic granule machinery by chitinase-3-like protein 1 (CHI3L1) (178). This protein, synthesized by tumor cells, plays a significant role in inflammation, tissue injury, and remodeling responses (179). Analysis conducted in vitro revealed elevated levels of CHI3L1 in the sera of trastuzumab-resistant patients compared to responders (178). CHI3L1 inhibits NK cell cytotoxicity and ADCC by disrupting the cytotoxic machinery, preventing lytic granule polarization to the immune synapse, and hindering downstream JNK signaling, a crucial process for cancer cell apoptosis (180). Furthermore, administering CHI3L1 in vivo weakens the control of NK cell-sensitive tumors while blocking CHI3L1 in conjunction with ADCC effectively treats HER2+ xenografts in mice (178).
NK cell exhaustion has been observed in an immunosuppressive TME and characterized by reduced activating receptors, decreased production of effector cytokines (181), impaired signaling/transcriptional pathways, hypoxia (182), low pH (183), upregulation of inhibitory receptors like NKG2A, TIM-3, PD-1, TIGIT, LAG-3, KIR (184, 185), and the presence of Tregs (186), Bregs (187), and MDSCs (188). This NK cell exhaustion phenotype presents a significant obstacle to developing NK cell-targeting immunotherapies. However, new strategies are being developed to combat NK cell exhaustion and enhance their anti-tumor function. For example, IL-21 treatment increases IFN-γ and granzyme B levels through Tim-3+PD-1+NK cells, reversing NK cell exhaustion (189). This highlights the potential therapeutic approach of using IL-21 to restore NK cell immunity function (190). In addition, IL-15 plays a crucial role in NK cell proliferation and survival (191). However, repetitive exposure to IL-15 during cancer treatment can diminish viable cell cycle signaling, decreased tumor control, and reduced fatty acid oxidation, resulting in NK cell exhaustion (192–194). Alternatively, an immunotherapy with membrane-bound IL-15 (mbIL15) is proposed (193, 195). By linking the human IL-15 gene to the CD8α transmembrane domain gene, mbIL15 can be created. NK cells expressing mbIL15 have been shown to activate cell cycle signaling and exhibit higher cytotoxicity against leukemia, lymphoma, and sarcoma in vitro and in vivo mouse xenograft tumor models (193). Expression profiling of NK cells can help identify dysfunction and exhaustion markers relevant to each breast cancer subtype. However, further studies on NK cell exhaustion in breast cancer are necessary.
Moreover, TME has demonstrated the capacity to modify the functionality and phenotype of NK cells (196). In a recent study by Mamessier et al., the dysfunctional tendencies of tumor-infiltrating NK cells in invasive and non-invasive breast cancer were characterized (197). Their findings unveiled a gradual reduction in the expression of NK cell activating receptors, such as NKp30, NKG2D, DNAM-1, CD16, CD226, and 2B4, as breast cancer progressed. Conversely, there was an upregulation of the inhibitory receptor NKG2A, which diminishes NK cell cytotoxic function and evasion of NK cell-mediated anti-tumor immunity (197, 198). Another study revealed a decline in the levels of NKp46, a lysis receptor responsible for direct tumor cell elimination, within the TME compared to normal cells (199). Immunotherapies targeting NK cells encompass various strategies to improve their activity, including promoting ADCC with mABs (200), blocking inhibitory signals (201), utilizing cytokines to augment NK cell proliferation and cytotoxicity through CAR NKs (202), IL-15 (203), and adoptive transfer of NK cells (204). In recent years, adoptive cell therapy strategies have emerged as a promising approach for utilizing NK cells (205). These immunotherapies entail the isolation, activation, and expansion of immune cells, which are then reintroduced into patients to combat tumor cells. A noteworthy application of this technique involves equipping NK cells with cancer-targeting CARs (206). However, the potential of engineered NK cells is hindered by immunometabolism limitations caused by factors such as hypoxia and cytokine stimulation in the TME (194, 207). Further studies are needed to understand how NK cell immunometabolism in TME regulates their anti-tumor properties.
Tumor-infiltrating lymphocytes (TILs) in TME regulate the induction of robust anti-tumor immunity, immunosuppression, efficacy of ICB therapy, cancer metastasis, and resistance to novel combinational ICB therapies (208). The TILs found in the TME primarily consist of CTLs, B cells, NK T cells, and CD4+ T helper cell subsets, including IFN-γ-producing CD4+ (Th1) cells, IL-4-producing CD4+ (Th2) cells, Foxp3+CD4+ regulatory T cells (Tregs) (209). Recent advancements in sub-type classification of TILs, using techniques such as flow cytometry, genomic approaches (single-cell RNA-seq, 10X genomic sequencing), and ICB therapies targeting T cells, have resulted in an increased emphasis on identifying TILs and potential immunological prognostic biomarkers specific to different subtypes of breast cancer (210, 211). Despite the ability of Th1 and CTLs to stimulate strong anti-tumor immunity, the TME employs various immune evasion strategies to suppress the infiltration, activation, and effector functions of CTLs and Th1 cells, inhibiting host anti-tumor effector responses. One extensively studied mechanism involved in this process is the upregulation of inhibitory receptors on T cells and higher expression of inhibitory ligands by tumor cells and APCs within the TME (212). APCs define the T cell differentiation and activation through tumor antigen presentation on MHC molecules to T cell receptors (TCR), expression of CD80 and CD86 ligands, which bind to co-receptors (such as CD28, ICOS, PD-1, CTLA4), and secretion of specific cytokines that define the fate of T cell differentiation (213). These co-signaling receptors can stimulate or inhibit T cell activation and effector functions. Examples of inhibitory receptors include PD-1, CTLA-4, LAG3, and TIM-3 (214). These receptors are crucial in maintaining immune balance and preventing excessive T cell activation during infections. However, tumors highly promote the expression of co-inhibitory receptors on T cells in TME to promote immune evasion. PD-1, for instance, binds to PD-L1 or PD-L2 ligands expressed by various immune cells or cancer cells to facilitate immune evasion (214). PD-1 possesses an inhibitory immunoreceptor tyrosine-based inhibition motif (ITIM) and immunoreceptor tyrosine-based switch (ITSM) motif in its cytoplasmic tail (215). When T cells engage with tumor cells and APCs, PD-L1 phosphorylates ITIM/ITSM, resulting in the recruitment of TCR-phosphorylating kinase, cytosolic tyrosine phosphatases (SHP-1 and SHP-2), and the inhibitory tyrosine kinase (216). As a result, the PI3K/Akt and Ras/MEK/Erk pathways necessary for initiating T cell activation are weakened. Recent research has shown the potential of blocking the PD-1/SHP-2 interaction as a novel approach to PD-1 inhibition (217). Accordingly, several monoclonal antibodies (mAbs) targeting PD-1 (pembrolizumab and nivolumab) and PD-L1 (atezolimumab) interaction have received FDA approval for the treatment of various lethal cancers including metastatic melanoma, Hodgkin’s lymphoma, head and neck squamous cell carcinoma, and breast cancer, among others (218).
In breast TME, infiltrating T cells demonstrate an upregulation of PD-1, while APCs (DCs and macrophages) and tumor cells exhibit higher expression of PD-L1 (219). The expression of PD-1 on CD4+ TILs is correlated with the invasiveness of breast cancer (220). Moreover, recent studies have shown decreased CD4+ and CD8+ T lymphocyte infiltration in DCIS and IDC breast cancer subtypes (221). These findings suggest that the reduced number of T lymphocytes in TME contributes to the transition of TNBC and HER2+ cancer subtypes from DCIS to IDC, resulting in a poor prognosis and worsened overall survival (OS) (222). Another recent study revealed the efficacy of CD3-HAC, a bifunctional fusion protein engineered to target EA1-mesenchymal stromal cells against metastatic breast cancer (223). CD3-HAC specifically binds to PD-L1-positive tumor cells to attenuate the impact of PD-1/PD-L1 on T cells exposed to MDA-MB-231, leading to enhanced T cell activation and stimulated lymphocyte-mediated lysis both in vitro and in vivo (223). In addition to immune evasion, the heightened expression of PD-1 on T cells indicates T cell exhaustion. CD8+ T cell exhaustion was initially identified in mice infected with chronic lymphocytic choriomeningitis virus (LCMV) infection (224). In this condition, the chronic presence of viral antigens constantly activates and stimulates CD8+ T cells, resulting in a decline in their effector functions (224, 225). In the TME, immune cells experience continuous stimulation from tumor antigens (226). Consequently, their metabolism and transcription profile change, ultimately leading to functional exhaustion (227). Immune cell exhaustion in TME is characterized by persistent tumor antigens stimulation, reduced proliferation capacity, enhanced inhibitory receptor expression, and decreased production of effector cytokines such as IL-2, TNFα, or IFN-γ (228).
In a comprehensive cohort study of breast cancer patients, it was discovered that despite the prevalence of T lymphocytes in IDCs, a significant portion of T cells exhibited reduced activity or were inactive due to exhaustion. These exhausted T cells displayed heightened expression of co-inhibitory receptors, PD-1 and CTLA-4, and diminished levels of active anti-tumor T cell subsets, CD62-L and CD127 (229). Phenotyping and functional analysis studies unveiled a distinctive T cell differentiation subset associated with exhaustion (230). It was observed that the underlying transcriptional mechanisms differed between effector T cells and exhausted T cells (231). This distinction was reflected in the expression of phenotypic markers, with effector CD8+ T cells exhibiting high levels of CD44 and killer cell lectin-like receptor subfamily G member 1 (KLRG1), while exhausted T cells displayed low or intermediate levels of these markers (232). Conversely, inhibitory receptor markers were highly expressed on exhausted T cells compared to effector CD8+ T cells (231). Additionally, exhausted T cells exhibited disparate expression of the transcription factors EOMES and T-bet, whereas effector CD8+ T cells expressed both simultaneously (233). The TME plays a critical role in inducing functional exhaustion in CD8+ T cells by promoting the cell surface expression of CD39, an immunosuppressive molecule (234). CD39+CD8+ T lymphocytes displayed an exhausted phenotype characterized by reduced production of IFNγ, TNF-α, and IL-2 and increased expression of co-inhibitory receptors such as PD-1 and CTLA-4. Targeting CD39+ appears promising in restoring T cell function and as a potential therapeutic intervention (234, 235). Revitalization of exhausted CD8+ T lymphocytes can be achieved through the inhibition of PD-1:PD-L1 interaction (236), CTLA-4 (237), and LAG-3 (238). Clinical studies that block the PD-1/PDL-1 inhibitory pathway to restore CD8+ T cell ability to proliferate and carry out its cytotoxic functions have been reported in other cancers. For instance, while pembrolizumab and atezolizumab are effective PD-1/PDL-1 inhibitors in second-line advanced non-small cell lung cancer (NSCLC), avelumab and durvalumab were effective in late-phase clinical testing (239). Another clinical study proposed donor lymphocyte infusion (DLI) targeting T cell exhaustion in hematology malignancy (240). In a clinical study, their findings reveal that patients who received DLI had a significant increase in CD8 cell counts, while the levels of CD4 T cells and B cells remained unaffected, indicating the potential of DLI to reverse CD8+ T cell exhaustion (241). However, the use of DLI alongside other T cell exhaustion revitalization methods has been suggested (242). While research on T cell exhaustion in breast cancer subtypes remains limited, future investigations aimed at revitalizing exhausted T cells and enhancing active T lymphocyte proliferation hold immense potential for the development of safe and effective immunotherapies against breast cancer.
The presence of regulatory T lymphocytes (Tregs), specifically the Foxp3 expressing subtype, is associated with a negative prognosis in breast cancer patients (243). Tregs express co-inhibitory receptors such as PDL-1, CTLA-4, and PD-1, which promote local immunosuppression and contribute to the spread of breast cancer (244, 245). Targeting Tregs can lead to a breakthrough in immunotherapy. Current strategies developed to inhibit Tregs’ harmful impact in the TME include inhibiting their recruitment, favoring their transformation into effector CD4+ T-cell subsets, blocking their expansion, depleting Tregs, and impeding their suppressive function (246). Further research and clinical trials are needed to fully understand the dynamics of T cell exhaustion and explore the use of combination therapies that can enhance T cells’ effector and cytotoxic functions.
B lymphocytes are primary mediators of humoral immunity. In the induction of adaptive immunity, B cells stimulated by antigens, along with the assistance of helper T cells, undergo differentiation into antibody-secreting plasma cells, initiating adaptive immune responses (247). In tumors, B lymphocytes are commonly found in the lymph nodes and invasive margins (248). Their impact on tumor onset and progression can be positive, negative, or passive (249). Upon activation by antigens, B cells undergo differentiation into antibody-secreting plasma cells. There are five subtypes of human immunoglobulins (Ig): IgG, IgA, IgM, IgD, and IgE (250). Among these five Ig types, IgG accounts for approximately 75% of the antibodies found in human serum (251). Despite being highly preserved, IgG is classified into four, namely IgG1 – IgG4, which exhibit varying effector functions based on their interaction with Fcγ receptors (FcγR) (252). Activation of FcγR-expressing cells triggers ADCC and phagocytosis of tumor cells (253). Conversely, when expressed by tumors, IgG-FcγR interaction can promote tumor progression (254, 255). A study conducted by Ma et al. revealed an abundance of IgG-expressing cancer cells in 68 breast cancer cases, encompassing 40 primary cancers and 28 metastatic cancers (256). Their findings demonstrated that IgG-expressing breast cancer cells exhibit more aggressive biological behavior, indicating the progression and metastasis of breast cancer. Moreover, the formation of circulating immune complexes (CICs) from the Ag-Ab complex can activate FcγR on myeloid cells, leading to the generation of MDSCs (257). These MDSCs effectively suppress the anti-tumor function of CD4+ and CD8+ T cells (42). B cells actively induce tumor cell apoptosis by producing granzyme B, a potent cytolytic molecule (258). These granzyme B-producing B cells can perform vital effector and regulatory functions during immune responses (258). Notably, granzyme B derived from carcinoma sources has been observed to effectively eliminate tumor cells in vitro (259). However, it is worth noting that the presence of granzyme B in breast tumor tissue can degrade the TCR-zeta subunit in the TCR, thereby impeding TCR assembly, expression, and anti-tumor signaling. This phenomenon occurs particularly in continuous antigen exposure and chronic inflammation (260). Moreover, the production of cytokines such as IL-2, IL-4, IL-6, IL-7, IFN-γ, IFN-α, TNF-α, CCL7, and CCL28 can stimulate an anti-tumor response (261, 262). These vital molecules are crucial in B cell maturation, differentiation, and survival (261). Notably, CCL28 and CCL27 direct the migration of plasma cells to mucosal sites during breast cancer anti-tumor response, correlating with improved prognosis (263). Conversely, other chemokines produced by B cells like CCL5, CCL20, and CCL1 are known to attract TAMs, Tregs, and MDSCs and induce EMT in breast cancer cell (264).
The immunosuppressive B cell subtype, Bregs, produce IL-10, IL-35, IL-15, and TGF-β cytokines that suppress CD8+ T-cell cytotoxicity, Treg recruitment, and M2/Th2 polarization (265–268). A study with a mouse 4T1 model of breast cancer demonstrated that the secretion of IL-10 by B lymphocytes acts in a TGF-β-dependent manner to promote the conversion of naive CD4+ T cells to Foxp3+ Tregs (269). Also, a chemokine, CXCL13, functions to recruit B cells to TME, where they differentiate into Bregs and stimulate EMT in tumor cells (270). A study showed that nano-trapping CXCL13 reduces Bregs differentiation, leading to prolonged cancer-free survival (271). Bregs-specific phenotype PD-1-PD-L1+CD19+ has been reported to exert the greatest suppressive effects on T effector cells (272). Two separate groups, Campbell et al. and Miligy et al. in 2017, revealed that B cells with phenotypes CD19+, CD24+, and CD38+ were correlated with increased tumor proliferation and risk of recurrence in breast cancer subtypes ER-, PR- and HER2+ (273, 274). The findings from their studies also suggested that CD20+ is a prognostic marker for better patient outcomes. Conversely, numerous studies have shown that Foxp3+ Tregs also express CD20+ and can be indicative of poor prognosis in breast cancer (273–277). Hence, conducting in-depth research to accurately define and differentiate the CD20+ anti-tumor role in B cells and the pro-tumor role in T cells is necessary.
CAFs are heterogenous cells that demonstrate their significance in various aspects of breast cancer, including growth, metastasis, response to treatment, and resistance to anti-cancer therapies (278). These cells derive from a range of sources, including normal fibroblasts, myofibroblasts, mesenchymal cells, stellate cells, fibrocytes, pericytes, smooth muscle cells, preadipocytes, or bone marrow-derived cells (279). Additionally, recent research by Flores et al. has identified CD34+ stromal cells/telocytes as another origin of CAFs, particularly in the invasive lobular carcinoma (ILC) subtype (280). Throughout tumor progression, CAFs contribute to the production of crucial structural proteins like elastin and collagen type I-V, which are involved in basement membrane formation (281), inflammation (282), epithelial differentiation (283), and angiogenesis (284). Moreover, CAFs produce MMPs, which are responsible for the degradation of the ECM and play a role in ECM homeostasis (285). Increased proliferation and secretion of growth factors, immunomodulatory factors, and ECM proteins have also been observed in CAFs and linked to their role in breast cancer (286). These CAF-specific markers can be used to identify breast cancer biomarkers and hold significant importance in diagnosis, prognosis, and the development of novel therapeutic approaches against breast cancer (287, 288). CAF biomarkers are not exclusive to CAFs, thereby requiring a comprehensive characterization to accurately define CAFs. Notably, biomarkers such as α-SMA, vimentin, desmin, cadherin-11, integrin α1β1, and MMPs are utilized for identifying CAFs originating from myofibroblast (286). However, there remains a lack of clear understanding regarding the detailed characterization of pro-tumor phenotypes of CAFs and their associated biomarkers. Initial studies employing SNP array analyses, multi-gene sequencing, and whole exome sequencing have reported the absence of somatic mutations in CAF phenotypes (289, 290). Subsequent findings have suggested somatic mutations and loss of heterozygosity as indicative of CAFs in the tumor stroma (291, 292). Furthermore, additional reports have demonstrated that epigenetic modifications, such as DNA methylation, may be responsible for maintaining the CAF phenotype and contributing to cancer cell growth and progression (293, 294). Hence, further studies are required to precisely characterize the pro-tumor properties of CAFs.
CAFs secrete growth factors such as TGF-β, EGF, FGF-2, TNF-α, platelet-derived growth factor (PGDF), and VEGF-A, and express cell surface and extracellular matrix proteins (295, 296). Extensive research links CAFs to breast cancer progression, with studies showing that CAFs secrete SDF-1/CXCL12 and HGF, both of which promote breast cancer growth and metastasis (297). HGF activates c-Met on tumor cells, leading to enhanced metastasis, while SDF-1 facilitates tumor growth and angiogenesis through the CXCR4 receptor on breast carcinoma cells. These functions promote the transition of breast carcinoma from ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) (297). Recent targeting of HGF/c-Met interaction has emerged as a significant breakthrough in breast cancer therapy (298). Additionally, SDF-1 secretion by breast CAFs contributes to the proliferation of breast cancer stem cells (CD44+CD24-) and the induction of drug resistance (299, 300). Therefore, targeting SDF-1 holds great promise for breast cancer therapeutics. Furthermore, CAFs play a crucial role in immune evasion by regulating the miR-92/PD-L1 pathway during breast cancer progression (301, 302). Molecular profiling of CAFs in breast tissue and carcinoma has identified differentially expressed genes (DEGs) that can serve as diagnostic and prognostic biomarkers and be targeted for developing new therapies (303, 304). Notably, high PDGF expression by CAFs indicates a shorter median survival for breast cancer patients (305).
Numerous oncogenic and immune cell signaling pathways within the TME cross-regulate CAFs and immune cells (Figure 3), promoting tumor progression, immunosuppression, and drug resistance (306). These pathways encompass TGF-β/Smad, Wnt/β-catenin, EGFR, TGF-β, PI3k/AKT/mTOR, JAK/STAT3, etc (307). Shangguan et al. previously demonstrated that inhibiting the TGF-β/Smad signaling pathway in human bone marrow mesenchymal cells hinders their differentiation into CAFs (308). Additionally, suppressing the EGFR signaling pathway, a crucial factor in EbbB/HER subtype metastasis has shown potential to inhibit CAF-associated cancer stemness (309). Therapies targeting CAFs have proved effective in overcoming treatment resistance in HER2+ breast cancer, with increased expression of NK-IL2RS, NK, and NKT cell signatures before treatment correlating with improved response to anti-HER2 mAbs-based therapy (310). Therapeutic targeting of CAF signaling pathways within the TME presents a promising approach for achieving breast cancer remission. Considering the significant role of CAFs in breast cancer metastasis and the complexity of cancer cell molecular signatures (311), further research and clinical trials are imperative to establish their potential utility in breast cancer prognosis and therapeutic intervention.
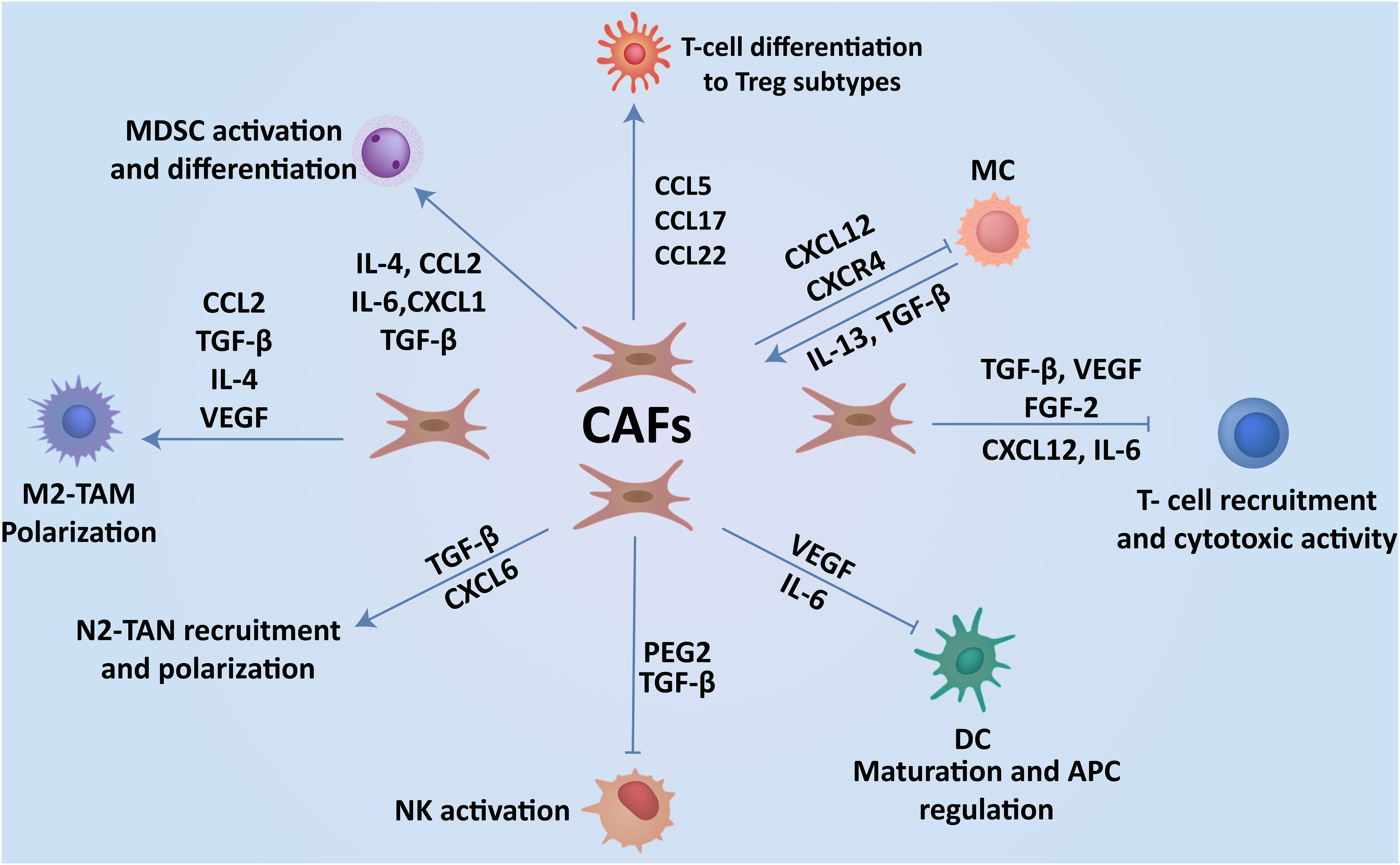
Figure 3 CAFs-immune cell interplay contributes to breast cancer progression. Interaction of CAFs with immune cells via the production of cytokines and soluble factors create an immunosuppressive TME, which enhances the progression of cancer to metastasis.
CAAs are adipocytes that actively reside near cancer cells, promoting crucial communication by releasing factors that can induce localized and systemic effects (312). Adipocytes in the TME can change in response to signals from cancer cells, leading to the formation of CAAs. These CAAs may release fatty acids into the surrounding tissue which can be taken up by breast tumor cells (313). The increased demand for energy and building blocks for rapidly dividing cancer cells makes fatty acids a valuable substrate for their metabolic needs. Within the TME, fatty acids undergo β-oxidation and serve as the principal source of ATP which promotes tumor survival and proliferation (314). Breast cancer cells can utilize fatty acid oxidation (FAO) as a metabolic pathway to oxidize fatty acids and generate energy. This process becomes particularly relevant in situations where other energy sources, such as glucose, are limited. Enhanced fatty acid metabolism, including FAO, has been associated with increased tumor aggressiveness in breast cancer (315). Fatty acids not only serve as an energy source but also play a role in various signaling pathways that can influence cell survival, proliferation, and invasiveness. Fatty acids can also activate specific lipid signaling pathways within tumor cells leading to changes in gene expression and metabolic pathways (316, 317). For instance, fatty acids can activate peroxisome proliferator-activated receptors (PPARs) beside other nuclear receptors, which can regulate genes involved in lipid metabolism, inflammation, and cell growth (318). PPARs are a group of nuclear receptors that play a crucial role in the regulation of fatty acid metabolism and energy homeostasis (319). Activation of PPARs occurs when ligands, such as fatty acids or their derivatives, bind to the receptors. Once activated, PPARs form heterodimers with retinoid X receptors (RXRs) and bind to specific DNA sequences called PPAR response elements (PPREs) in the promoters of target genes (320). This binding regulates the transcription of genes involved in lipid metabolism, energy homeostasis, and inflammation (321). In hormone receptor-positive breast cancer, estrogen receptor-positive (ER+) tumors can be influenced by adipose tissue-derived factors. Fatty acids and other adipokines may affect the growth and behavior of ER+ breast cancer cells (322). Although all adipose depots can secrete inflammatory factors, such as TNF, IL-6, IL-1β, and TGF-β (323) obese visceral adipose primarily releases excessive fatty acids, cholesterol, triglycerides, hormones, and adipokines, closely associated with metabolic dysfunction and unfavorable cancer outcomes (324). Additionally, adipocytes can contribute to chemotherapeutic drug resistance, as their co-culture with fibroblasts can deactivate the effectiveness of anti-cancer drugs by metabolizing them into less potent secondary metabolites (325). Understanding the metabolic interactions between CAAs and breast cancer cells, specifically involving fatty acids and FAO, has implications for developing targeted therapies. Researchers are exploring ways to disrupt these metabolic pathways as potential strategies to inhibit tumor growth and improve treatment outcomes.
Furthermore, TME’s plasticity allows for transdifferentiation, a process whereby cells undergo a significant shift in their identity, thereby acquiring new transcriptional or morphological characteristics typical of a different cell lineage. Microenvironmental cues including neighboring cells, extracellular matrix, blood vessels, and immune cells can induce shifts in cancer cell phenotypes (326). Emerging research has shown that cancer cells, can exhibit plasticity and undergo transdifferentiation, which can contribute to tumor heterogeneity and complicate treatment strategies (327). Despite these challenges, researchers are exploring ways to harness lineage plasticity for therapeutic purposes. In a research conducted by Ronen et al, to capitalize on the plasticity of cancer cells, breast cancer cell differentiation was redirected towards a non-malignant and non-proliferative adipocyte fate (328). In this study, the utilization of Rosiglitazone and an MEK inhibitor as part of the therapy appears to be particularly effective against aggressive characteristics of breast cancer cells, consequently inhibiting metastasis. The transdifferentiated adipogenesis-induced cancer cells, MTDECad and 3T3-L1 cells formed become functional post-mitotic adipocytes which have comparable characteristics with functional adipocytes. For instance, both differentiated cell types express the adipocyte-specific markers C/EPBa, PPARg2, and fatty acid binding protein 4 (FABP4), and they secrete the adipocyte-specific adipokine adiponectin (328). Therapeutic strategies need to consider the evolving nature of cancer cells and the potential for phenotypic changes under different microenvironmental conditions. Generally, CAA significantly influences various aspects of breast cancer, including risk, progression, migration, metastasis, and resistance to existing treatments (329). Therefore, targeting the interaction between adipose tissue and breast cancer may be a promising approach to overcoming immune tolerance and drug resistance.
ECs are a constitutive part of the cardiovascular system and are critical to homeostasis, angiogenesis, and immune response (330). They regulate the passage of substances through tight cell junctions and line the basement membrane of capillaries (331). ECs, along with a basal lamina and strategically positioned pericytes, form the structure of blood vessel walls (332). ECs facilitate intravasation, allowing cancer cells to migrate into the blood vessel lumen, a critical step in cancer metastasis (333). Tumor growth relies on a blood supply, and during rapid growth, tumors stimulate neovascularization by weakening the basement membrane of existing blood vessels (334). Upon the secretion of angiogenic factors like VEGF-A, PDGF, hypoxia-inducing factors (HIF-1), and MMPs, the basement membrane degrades. This basement membrane degradation triggers the migration of endothelial cells and pericytes to the tumor region, contributing to TME angiogenesis (334). Additionally, tumor-associated hypoxia, mediated by HIF-1α and HIF-2α, plays a role in malignant conversion and metastasis, as well as influencing immune cell functions within the TME (335). Schneider & Miller’s study revealed that angiogenesis precedes the progression of mammary hyperplasia to malignancy in breast cancer (336). They demonstrated that transfection of tumor cells with angiogenic stimulatory peptides promoted tumor growth, invasiveness, and metastasis (337). Clinical outcomes have substantiated the efficacy of anti-angiogenic therapy as a viable treatment approach. However, the use of antiangiogenic drugs in conjunction with conventional chemotherapy in metastatic breast cancer has shown limited clinical impact on overall survival (337). It is essential to conduct further studies by addressing potential obstacles, such as toxicity, drug resistance, and alternative angiogenesis mechanisms, in order to optimize the effectiveness of anti-angiogenic therapies in breast cancer progression.
Recently, correlation between neurogenesis and angiogenesis in breast TME has been linked to aggressive breast cancer breast cancer (338). Tumors release neurotrophic factors that can initiate innervation, a process that imitates angiogenesis (339). Hence, tumor neurogenesis is intricately linked to metastasis, as the presence of ingrown nerve endings can release neurotransmitters that significantly enhance the development of metastatic cells (339). In an immunohistochemistry analysis of carcinoma breast tissues, it was observed that protein gene product (PGP) 9.5 protein was present in 61% of IDC tissues compared to fibroadenoma and DCIS, particularly in ER-negative and node-negative subtypes (340). PGP 9.5, a ubiquitin-carboxyl hydrolase, is an enzyme expressed throughout the stages of differentiation in nerve tissue of mice brains and is a useful marker for detecting central nervous system damage (341). Likewise, in both ER-negative and node-negative subgroups of breast IDC, a significant association was observed between PGP 9.5 expression and higher microvessel density (MVD), compared to less expression of PGP 9.5 and MVD identified in DCIS. The analysis reveals a clear correlation between neurogenesis and angiogenesis, particularly in ER-negative and node-negative subtypes of breast cancer (340). In a human breast cancer cohort study, a significant association between neurogenesis, consolidated neuro-angiogenic signature, and high-grade breast cancer features was observed (342). Single cell-based spatial mapping with imaging mass cytometry was used to identify the colocalization of neural and vascular structures, indicating the presence of neurovascular niches within tumor tissue. Cancer cells can release various signaling molecules, including growth factors and cytokines, that play a role in recruiting both sprouting axons (microaxons) and endothelial cells (microvessels) to the TME (343). This phenomenon is referred to as neurotropism, and it has been observed in several types of cancers, including breast cancer (343, 344). The exact mechanisms by which cancer cells influence axon recruitment are still an area of active research. The coexistence and potential coregulation of microaxons and microvessels suggest a complex interplay between neural and vascular elements within the tumor stroma.
Pericytes are mural cells that envelop blood vessels and reside adjacent to the endothelial cells lining the capillaries. Pericytes play a crucial role in the development and stabilization of the vasculature through TGF-β signaling activation (345). Also, pericytes actively enhance the physical stability and support of endothelial tubule function during the initial phase of angiogenesis by co-occupying endothelial tubules (346). Within the TME, the tumor vasculature serves multiple functions, such as supporting tumor growth and facilitating metastasis to distant organ sites (347). Notably, breast cancer is a highly vascularized tumor with extensive pericyte coverage (347). Targeting angiogenesis during breast cancer progression can be approached by inhibiting the vessel-stabilizing properties of vascular pericytes (348, 349). Depleting pericytes has been shown to increase intra-tumoral hypoxia and lung metastasis in advanced-stage hypoxic tumors with pre-established vasculature (348). The presence of perfusion defects in breast cancer blood vessels is associated with vessel dilation, tortuosity, and inadequate perivascular coverage (347, 350). This abnormal vascular system is partly attributed to morphological and molecular alterations in pericytes and significant population heterogeneity (347). The presence of pericytes in the primary TME impedes cancer progression and metastasis (350). Distinguishing pericytes can be achieved through morphological characteristics and molecular markers, including α-SMA, desmin, PDGFR-β, CD248, NG2, and angiopoietin-2 (349, 351). Many of the pericyte markers are used in several studies to calculate the mean microvascular pericyte coverage index (MPI). For instance, α-SMA expression in breast cancer yielded an estimated MPI range of 32%-80%. Other markers such as NG2, PDGFRβ, desmin, and CD248 have also been employed for MPI measurement (351). Other markers such as NG2, PDGFRβ, desmin, and CD248 have also been applied in MPI measurement (350). Many anti-angiogenic treatments involve targeting endothelial cells or proangiogenic factors to suppress neovascularization cause tumor cell death. In the context of anti-angiogenic treatments, simultaneously targeting both endothelial cells and pericytes has been suggested (352). According to some studies, non-selective elimination of pericytes may not provide benefits but may instead promote tumor aggressiveness and metastasis. Therefore, gaining a comprehensive understanding of pericyte heterogeneity in response to changes in the TME can inform effective pericyte targeting strategies (351, 353).
The current research findings on the interaction between the TME and cancer cells have significantly advanced our understanding of their crucial roles in cancer progression and treatment response. Traditionally, treatment strategies for breast cancer predominantly focused on promoting tumor cell death. However, the emergence of immunotherapy has revolutionized cancer treatment by incorporating anti-tumor immune responses and targeting TME cells. Successful research of some clinical trials targeting breast TME has provided a promising outlook for utilizing these cells in cancer therapies (Table 2). The cells within the breast TME can either act against or promote tumor cells; in certain conditions, they may exhibit dual roles. The ability of TME cells to switch from anti-tumor to pro-tumor functions poses a significant challenge for immunotherapy. The anti-tumor and pro-tumor functions of these cells primarily depend on specific mediators such as cytokines, chemokines, and growth factors in TME. The interplay between these mediators generated by the cellular components modulates the TME towards either an anti-tumor or an immunosuppressive environment. Despite the initial findings on breast TME, future studies should focus on understanding the evasion of anti-tumor immunity and exploiting TME cell mediators to target cancer cells. Immunotherapy has emerged as a critical component in the treatment of various types of cancer, including breast cancer.
ICB therapies have shown remarkable efficacy when used alone or in combination with other treatment modalities. Reprogramming CTLs through ICB immunotherapies has been successful, but the resistance caused by TAMs to recently developed ICB therapies remains challenging. Therefore, there is a need for targeted inhibition of TAMs to enhance tumor cell-killing capacity, as well as further investigation into repolarizing TAMs towards an anti-tumor phenotype. Moreover, the emerging field of immunometabolism and understanding how TME regulates metabolism in immune cells to suppress anti-tumor immunity is crucial to developing novel immunotherapies and overcoming resistance against conventional and ICB therapies. The potential of adoptive immunotherapy, specifically equipping NK cells with cancer-targeting CARs, is hindered by immunometabolism. Extensive research is required to understand how regulating the metabolism of NK and other immune cells in TME can promote their anti-tumor activities. Furthermore, there is a pressing need for further investigation into the recovery of exhausted T-cells and NK cells to promote their effector functions. The complexity and heterogeneity of TME cells, such as the CAFs and pericytes, present challenges in their proper characterization. The recent advances in next-generation sequencing, metabolomics, and bioinformatics, which study cancer progression at both tissue and single-cell levels, can be employed to identify novel breast cancer stage-specific biomarkers, functional phenotype of immune and non-immune cells in TME, resistance to cancer therapies, and development of novel targeted immunotherapies. Such investigations will lead to improved stage-specific breast cancer diagnosis, the development of innovative TME cell-specific targeted immunotherapies with fewer side effects, and overall improved quality of life and survival for women with highly metastatic breast cancer.
TA: Conceptualization, Writing – original draft, Writing – review & editing. RM: Writing – original draft. AA: Writing – original draft. SP: Writing – review & editing. PM: Writing – review & editing. LA: Writing – review & editing. AS: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AA and PM were supported by a grant from the Breast Cancer Research Foundation of Alabama (project number: 0142-22P).
We thank Ferrin Antony, Auburn University for giving feedback during manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
4. Mohammed EA, Solyman MTM, Omar NN, Hasan NMA. Imaging features of breast cancer molecular subtypes: An Updated Review of the Literature. SVU-International J Med Sci. (2022) 5:92–103. doi: 10.21608/svuijm.2021.104214.1238
5. Jones RL, Constantinidou A, Reis-Filho JS. Molecular classification of breast cancer. Surg Pathol clinics. (2012) 5:701–17. doi: 10.1016/j.path.2012.06.008
6. Amin MB, Edge SB, Greene FL, Byrd DR, Brookland RK, Washington MK, et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin. (2017) 67(2):93–99. doi: 10.3322/caac.21388
7. Ren G, Liu Y, Zhao X, Zhang J, Zheng B, Yuan Z, et al. Tumor resident mesenchymal stromal cells endow naive stromal cells with tumor-promoting properties. Oncogene. (2014) 33:4016–20. doi: 10.1038/onc.2013.387
8. Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. (2020) 39:e104063. doi: 10.15252/embj.2019104063
9. Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L. The crosstalk between tumor cells and the immune microenvironment in breast cancer: implications for immunotherapy. Front Oncol. (2021) 11:610303. doi: 10.3389/fonc.2021.610303
10. Hieken TJ, Chen J, Chen B, Johnson S, Hoskin TL, Degnim AC, et al. The breast tissue microbiome, stroma, immune cells and breast cancer. Neoplasia. (2022) 27:100786. doi: 10.1016/j.neo.2022.100786
11. Huang Z, Yu P, Tang J. Characterization of triple-negative breast cancer MDA-MB-231 cell spheroid model. OncoTargets Ther. (2020) 13:5395. doi: 10.2147/OTT.S249756
12. Pruitt K. Molecular and cellular changes during cancer progression resulting from genetic and epigenetic alterations. Prog Mol Biol Trans Science. (2016) 144:3–47. doi: 10.1016/bs.pmbts.2016.09.001
13. Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. (2008) 181:3099–107. doi: 10.4049/jimmunol.181.5.3099
14. Derer A, Frey B, Fietkau R, Gaipl US. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunology Immunother. (2016) 65:779–86. doi: 10.1007/s00262-015-1771-8
15. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci. (2014) 111:11774–9. doi: 10.1073/pnas.1410626111
16. Zanker DJ, Spurling AJ, Brockwell NK, Owen KL, Zakhour JM, Robinson T, et al. Intratumoral administration of the Toll-like receptor 7/8 agonist 3M-052 enhances interferon-driven tumor immunogenicity and suppresses metastatic spread in preclinical triple-negative breast cancer. Clin Trans Immunol. (2020) 9:e1177. doi: 10.1002/cti2.1177
17. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun. (2020) 11:2416. doi: 10.1038/s41467-020-16199-4
18. Liu Y, Qiao L, Zhang S, Wan G, Chen B, Zhou P, et al. Dual pH-responsive multifunctional nanoparticles for targeted treatment of breast cancer by combining immunotherapy and chemotherapy. Acta Biomaterialia. (2018) 66:310–24. doi: 10.1016/j.actbio.2017.11.010
19. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
20. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. (2010) 16:4583–94. doi: 10.1158/1078-0432.CCR-10-0733
21. Funahashi Y, Okamoto K, Adachi Y, Semba T, Uesugi M, Ozawa Y, et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer science. (2014) 105:1334–42. doi: 10.1111/cas.12488
22. Ji T, Zhao Y, Ding Y, Wang J, Zhao R, Lang J, et al. Transformable peptide nanocarriers for expeditious drug release and effective cancer therapy via cancer-associated fibroblast activation. Angewandte Chemie Int Edition. (2016) 55:1050–5. doi: 10.1002/anie.201506262
23. Xu M, Liu M, Du X, Li S, Li H, Li X, et al. Intratumoral delivery of IL-21 overcomes anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. J Immunol. (2015) 194:4997–5006. doi: 10.4049/jimmunol.1402603
24. Shao L, Yu Q, Xia R, Zhang J, Gu S, Yu D, et al. B7-H3 on breast cancer cell MCF7 inhibits IFN-γ release from tumour-infiltrating T cells. Pathology-Research Practice. (2021) 224:153461. doi: 10.1016/j.prp.2021.153461
25. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci. (2019) 116:4326–35. doi: 10.1073/pnas.1819473116
26. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell–dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
27. Danenberg E, Bardwell H, Zanotelli VR, Provenzano E, Chin S-F, Rueda OM, et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat Genet. (2022) 54:660–9. doi: 10.1038/s41588-022-01041-y
28. Singh S, Lamichhane A, Rafsanjani Nejad P, Heiss J, Baumann H, Gudneppanavar R, et al. Therapeutic targeting of stromal-tumor HGF-MET signaling in an organotypic triple-negative breast tumor model. Mol Cancer Res. (2022) 20:1166–77. doi: 10.1158/1541-7786.MCR-21-0317
29. Andersson P, Yang Y, Hosaka K, Zhang Y, Fischer C, Braun H, et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight. (2018) 3(20):e122375. doi: 10.1172/jci.insight.122375
30. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
31. Yu P, Huang Y, Han Y, Lin L, Sun W, Rabson A, et al. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene. (2017) 36:482–90. doi: 10.1038/onc.2016.217
32. Ramamonjisoa N, Ackerstaff E. Characterization of the tumor microenvironment and tumor–stroma interaction by non-invasive preclinical imaging. Front Oncol. (2017) 7:3. doi: 10.3389/fonc.2017.00003
33. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol. (2019) 7:60. doi: 10.3389/fcell.2019.00060
34. Chen L, Li J, Wang F, Dai C, Wu F, Liu X, et al. Tie2 Expression on Macrophages Is Required for Blood Vessel Reconstruction and Tumor Relapse after ChemotherapyTie2+ Macrophages Promote Tumor Relapse after Chemotherapy. Cancer Res. (2016) 76:6828–38. doi: 10.1158/0008-5472.CAN-16-1114
35. Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell. (2022) 185:299–310. e18. doi: 10.1016/j.cell.2021.12.023
36. Mehraj U, Dar AH, Wani NA, Mir MA. Tumor microenvironment promotes breast cancer chemoresistance. Cancer chemotherapy Pharmacol. (2021) 87:147–58. doi: 10.1007/s00280-020-04222-w
37. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
38. Burugu S, Asleh-Aburaya K, Nielsen TO. Immune infiltrates in the breast cancer microenvironment: detection, characterization and clinical implication. Breast cancer. (2017) 24:3–15. doi: 10.1007/s12282-016-0698-z
39. Issa-Nummer Y, Darb-Esfahani S, Loibl S, Kunz G, Nekljudova V, Schrader I, et al. Prospective validation of immunological infiltrate for prediction of response to neoadjuvant chemotherapy in HER2-negative breast cancer–a substudy of the neoadjuvant GeparQuinto trial. PloS One. (2013) 8:e79775. doi: 10.1371/journal.pone.0079775
40. Kawamoto H, Minato N. Myeloid cells. Int J Biochem Cell Biol. (2004) 36:1374–9. doi: 10.1016/j.biocel.2004.01.020
41. Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, Osiecka O, Hu X, Rundgren IM, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J immunotherapy Cancer. (2020) 8(2):e001223. doi: 10.1136/jitc-2020-001223
42. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduction Targeted Ther. (2021) 6:362. doi: 10.1038/s41392-021-00670-9
43. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:1–10. doi: 10.1038/ncomms12150
44. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. (2017) 32:869–83. e5. doi: 10.1016/j.ccell.2017.11.004
45. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol. (2020) 11:938. doi: 10.3389/fimmu.2020.00938
46. Guha P, Gardell J, Rabinowitz B, Lopes M, DaSilva NA, Rowley D, et al. Monocytic and granulocytic myeloid-derived suppressor cell plasticity and differentiation are organ-specific. Oncogene. (2021) 40:693–704. doi: 10.1038/s41388-020-01559-7
47. Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. (2020) 5:eaay6017. doi: 10.1126/sciimmunol.aay6017
48. Toor SM, Syed Khaja AS, El Salhat H, Faour I, Kanbar J, Quadri AA, et al. Myeloid cells in circulation and tumor microenvironment of breast cancer patients. Cancer Immunology Immunother. (2017) 66:753–64. doi: 10.1007/s00262-017-1977-z
49. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. (2017) 8:3649. doi: 10.18632/oncotarget.v8i2
50. Li R, Mukherjee MB, Lin J. Coordinated regulation of myeloid-derived suppressor cells by cytokines and chemokines. Cancers. (2022) 14:1236. doi: 10.3390/cancers14051236
51. Oh K, Lee O-Y, Shon SY, Nam O, Ryu PM, Seo MW, et al. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. (2013) 15:1–16. doi: 10.1186/bcr3473
52. Li Y, He H, Jihu R, Zhou J, Zeng R, Yan H. Novel characterization of myeloid-derived suppressor cells in tumor microenvironment. Front Cell Dev Biol. (2021) 9:698532. doi: 10.3389/fcell.2021.698532
53. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
54. Pyzer AR, Cole L, Rosenblatt J, Avigan DE. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int J cancer. (2016) 139:1915–26. doi: 10.1002/ijc.30232
55. Pan P-Y, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
56. van Kooten C, Banchereau J. Functions of CD40 on B cells, dendritic cells and other cells. Curr Opin Immunol. (1997) 9:330–7. doi: 10.1016/S0952-7915(97)80078-7
57. Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. (1998) 16:111–35. doi: 10.1146/annurev.immunol.16.1.111
58. Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signalingMDSC and cancer stem cell. Cancer Res. (2016) 76:3156–65. doi: 10.1158/0008-5472.CAN-15-2528
59. Monu NR, Frey AB. Myeloid-derived suppressor cells and anti-tumor T cells: a complex relationship. Immunol investigations. (2012) 41:595–613. doi: 10.3109/08820139.2012.673191
60. Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem. (2013) 288:11676–88. doi: 10.1074/jbc.M112.441402
61. Ali HR, Chlon L, Pharoah PD, Markowetz F, Caldas C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PloS Med. (2016) 13:e1002194. doi: 10.1371/journal.pmed.1002194
62. Kumar S, Wilkes DW, Samuel N, Blanco MA, Nayak A, Alicea-Torres K, et al. ΔNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J Clin Invest. (2018) 128:5095–109. doi: 10.1172/JCI99673
63. Bergenfelz C, Roxå A, Mehmeti M, Leandersson K, Larsson A-M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunology Immunother. (2020) 69:435–48. doi: 10.1007/s00262-019-02472-z
64. Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, et al. Targeting MDSC for immune-checkpoint blockade in cancer immunotherapy: current progress and new prospects. Clin Med Insights: Oncol. (2021) 15:11795549211035540. doi: 10.1177/11795549211035540
65. Trump LR, Nayak RC, Singh AK, Emberesh S, Wellendorf AM, Lutzko CM, et al. Neutrophils derived from genetically modified human induced pluripotent stem cells circulate and phagocytose bacteria in vivo. Stem Cells Trans Med. (2019) 8:557–67. doi: 10.1002/sctm.18-0255
66. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β:”N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
67. Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro-vs. antitumor TANs. Oncoimmunology. (2016) 5:e1232221. doi: 10.1080/2162402X.2016.1232221
68. Eruslanov EB. Phenotype and function of tumor-associated neutrophils and their subsets in early-stage human lung cancer. Cancer Immunology Immunother. (2017) 66:997–1006. doi: 10.1007/s00262-017-1976-0
69. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. (2011) 20:300–14. doi: 10.1016/j.ccr.2011.08.012
70. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. (2012) 33:949–55. doi: 10.1093/carcin/bgs123
71. Wu L, Saxena S, Singh RK. Neutrophils in the tumor microenvironment. Tumor Microenvironment. (2020), 1–20. doi: 10.1007/978-3-030-35723-81
72. Soto-Perez-de-Celis E, Chavarri-Guerra Y, Leon-Rodriguez E, Gamboa-Dominguez A. Tumor-associated neutrophils in breast cancer subtypes. Asian Pacific J Cancer prevention: APJCP. (2017) 18:2689. doi: 10.22034/APJCP.2017.18.10.2689
73. Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, Quigley JP. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia. (2014) 16:771–88. doi: 10.1016/j.neo.2014.08.013
74. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
75. Das S, Amin SA, Jha T. Inhibitors of gelatinases (MMP-2 and MMP-9) for the management of hematological Malignancies. Eur J Medicinal Chem. (2021) 223:113623. doi: 10.1016/j.ejmech.2021.113623
76. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci. (2007) 104:20262–7. doi: 10.1073/pnas.0706438104
77. Wang Y, Chen J, Yang L, Li J, Wu W, Huang M, et al. Tumor-contacted neutrophils promote metastasis by a CD90-TIMP-1 juxtacrine–paracrine LoopNeutrophils induce EMT by a CD90-TIMP-1 loop. Clin Cancer Res. (2019) 25:1957–69. doi: 10.1158/1078-0432.CCR-18-2544
78. Hajizadeh F, Maleki LA, Alexander M, Mikhailova MV, Masjedi A, Ahmadpour M, et al. Tumor-associated neutrophils as new players in immunosuppressive process of the tumor microenvironment in breast cancer. Life Sci. (2021) 264:118699. doi: 10.1016/j.lfs.2020.118699
79. Hanes MR, Giacomantonio CA, Marshall JS. Mast cells and skin and breast cancers: a complicated and microenvironment-dependent role. Cells. (2021) 10:986. doi: 10.3390/cells10050986
80. Majorini MT, Cancila V, Rigoni A, Botti L, Dugo M, Triulzi T, et al. Infiltrating mast cell–mediated stimulation of estrogen receptor activity in breast cancer cells promotes the luminal phenotype. Cancer Res. (2020) 80:2311–24. doi: 10.1158/0008-5472.CAN-19-3596
81. Dudeck J, Ghouse SM, Lehmann CH, Hoppe A, Schubert N, Nedospasov SA, et al. Mast-cell-derived TNF amplifies CD8+ dendritic cell functionality and CD8+ T cell priming. Cell Rep. (2015) 13:399–411. doi: 10.1016/j.celrep.2015.08.078
82. Majorini MT, Colombo MP, Lecis D. Few, but efficient: the role of mast cells in breast cancer and other solid tumors. Cancer Res. (2022) 82:1439–47. doi: 10.1158/0008-5472.CAN-21-3424
83. Ribatti D, Annese T, Tamma R. Controversial role of mast cells in breast cancer tumor progression and angiogenesis. Clin Breast Cancer. (2021) 21:486–91. doi: 10.1016/j.clbc.2021.08.010
84. Aponte-López A, Muñoz-Cruz S. Mast cells in the tumor microenvironment. Tumor Microenvironment: Hematopoietic Cells–Part B. (2020), 159–73. doi: 10.1007/978-3-030-49270-0_9
85. Cimpean AM, Tamma R, Ruggieri S, Nico B, Toma A, Ribatti D. Mast cells in breast cancer angiogenesis. Crit Rev Oncology/Hematology. (2017) 115:23–6. doi: 10.1016/j.critrevonc.2017.04.009
86. Cimpean AM, Raica M. The hidden side of disodium cromolyn: from mast cell stabilizer to an angiogenic factor and antitumor agent. Archivum Immunologiae Therapiae Experimentalis. (2016) 64:515–22. doi: 10.1007/s00005-016-0408-8
87. de Souza Junior DA, Santana AC, da Silva EZM, Oliver C, Jamur MC. The role of mast cell specific chymases and tryptases in tumor angiogenesis. BioMed Res Int. (2015) 2015:142359. doi: 10.1155/2015/142359
88. Ammendola M, Leporini C, Marech I, Gadaleta CD, Scognamillo G, Sacco R, et al. Targeting mast cells tryptase in tumor microenvironment: a potential antiangiogenetic strategy. BioMed Res Int. (2014) 2014:154702. doi: 10.1155/2014/154702
89. Glajcar A, Szpor J, Pacek A, Tyrak KE, Chan F, Streb J, et al. The relationship between breast cancer molecular subtypes and mast cell populations in tumor microenvironment. Virchows Archiv. (2017) 470:505–15. doi: 10.1007/s00428-017-2103-5
90. Carpenco E, Ceauşu RA, Cimpean AM, Gaje PN, Șaptefraţi L, Fulga V, et al. Mast cells as an indicator and prognostic marker in molecular subtypes of breast cancer. vivo. (2019) 33:743–8. doi: 10.21873/invivo.11534
91. Rigoni A, Colombo M, Pucillo C. The role of mast cells in molding the tumor microenvironment. Cancer microenvironment. (2015) 8:167–76. doi: 10.1007/s12307-014-0152-8
92. Raica M, Cimpean AM, Ceauşu R, Ribatti D, Gaje P. Interplay between mast cells and lymphatic vessels in different molecular types of breast cancer. Anticancer Res. (2013) 33:957–63.
93. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal transduction targeted Ther. (2021) 6:75. doi: 10.1038/s41392-021-00484-9
94. Vogel DY, Glim JE, Stavenuiter AW, Breur M, Heijnen P, Amor S, et al. Human macrophage polarization in vitro: maturation and activation methods compared. Immunobiology. (2014) 219:695–703. doi: 10.1016/j.imbio.2014.05.002
95. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. (2015) 53:676–88. doi: 10.1165/rcmb.2015-0012OC
96. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J leukocyte Biol. (2009) 86:1065–73. doi: 10.1189/jlb.0609385
97. Anfray C, Ummarino A, Torres Andon F, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. (2019) 9:46. doi: 10.3390/cells9010046
98. Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, et al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci. (2013) 110:15919–24. doi: 10.1073/pnas.1312197110
99. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
100. Hourani T, Holden JA, Li W, Lenzo JC, Hadjigol S, O’Brien-Simpson NM. Tumor associated macrophages: Origin, recruitment, phenotypic diversity, and targeting. Front Oncol. (2021) 11:788365. doi: 10.3389/fonc.2021.788365
101. Lee H-W, Choi H-J, Ha S-J, Lee K-T, Kwon Y-G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim Biophys Acta (BBA)-Reviews Cancer. (2013) 1835:170–9. doi: 10.1016/j.bbcan.2012.12.007
102. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
103. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
104. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. (2017) 10:1–12. doi: 10.1186/s13045-017-0430-2
105. Amer HT, Stein U, El Tayebi HM. The monocyte, a maestro in the tumor microenvironment (TME) of breast cancer. Cancers. (2022) 14:5460. doi: 10.3390/cancers14215460
106. Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation. (2016) 23:95–121. doi: 10.1111/micc.12259
107. Gocheva V, Wang H-W, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. (2010) 24:241–55. doi: 10.1101/gad.1874010
108. Orekhov AN, Orekhova VA, Nikiforov NG, Myasoedova VA, Grechko AV, Romanenko EB, et al. Monocyte differentiation and macrophage polarization. Vessel Plus. (2019) 3:10. doi: 10.20517/2574-1209.2019.04
109. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. (2016) 126:4157–73. doi: 10.1172/JCI87252
110. Jung K-H, Lee EJ, Park JW, Lee JH, Moon SH, Cho YS, et al. EGF receptor stimulation shifts breast cancer cell glucose metabolism toward glycolytic flux through PI3 kinase signaling. PloS One. (2019) 14:e0221294. doi: 10.1371/journal.pone.0221294
111. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. (2006) 25:315–22. doi: 10.1007/s10555-006-9001-7
112. Yang Q, Guo N, Zhou Y, Chen J, Wei Q, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B. (2020) 10:2156–70. doi: 10.1016/j.apsb.2020.04.004
113. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. (2007) 67:5064–6. doi: 10.1158/0008-5472.CAN-07-0912
114. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. doi: 10.1101/gad.180331.111
115. Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y, et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol. (2022) 15:1–6. doi: 10.1186/s13045-021-01223-x
116. Qiu S-Q, Waaijer SJ, Zwager MC, de Vries EG, van der Vegt B, Schröder CP. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat Rev. (2018) 70:178–89. doi: 10.1016/j.ctrv.2018.08.010
117. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal transduction targeted Ther. (2022) 7:3. doi: 10.1038/s41392-021-00762-6
118. Suryawanshi A, Tadagavadi RK, Swafford D, Manicassamy S. Modulation of inflammatory responses by Wnt/β-catenin signaling in dendritic cells: a novel immunotherapy target for autoimmunity and cancer. Front Immunol. (2016) 7:460. doi: 10.3389/fimmu.2016.00460
119. Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J pathology. (2010) 176:2911–20. doi: 10.2353/ajpath.2010.091125
120. Wang S, Yang Y, Ma P, Huang H, Tang Q, Miao H, et al. Landscape and perspectives of macrophage targeted cancer therapy in clinical trials. Mol Therapy-Oncolytics. (2022), 1–15. doi: 10.1016/j.omto.2022.02.019
121. Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, et al. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev. (2010) 234:177–98. doi: 10.1111/j.0105-2896.2009.00868.x
122. Suryawanshi A, Hussein MS, Prasad PD, Manicassamy S. Wnt signaling cascade in dendritic cells and regulation of anti-tumor immunity. Front Immunol. (2020) 11:122. doi: 10.3389/fimmu.2020.00122
123. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PloS One. (2009) 4:e4204. doi: 10.1371/journal.pone.0004204
124. Wang Y, Xiang Y, Xin VW, Wang X-W, Peng X-C, Liu X-Q, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. (2020) 13:1–18. doi: 10.1186/s13045-020-00939-6
125. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ–resident dendritic cells. J Exp Med. (2013) 210:1035–47. doi: 10.1084/jem.20121103
126. Belz GT, Behrens GM, Smith CM, Miller JF, Jones C, Lejon K, et al. The CD8α+ dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J Exp Med. (2002) 196:1099–104. doi: 10.1084/jem.20020861
127. Mailliard RB, Son Y-I, Redlinger R, Coates PT, Giermasz A, Morel PA, et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol. (2003) 171:2366–73. doi: 10.4049/jimmunol.171.5.2366
128. Michea P, Noël F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. (2018) 19:885–97. doi: 10.1038/s41590-018-0145-8
129. Carenza C, Calcaterra F, Oriolo F, Di Vito C, Ubezio M, Della Porta MG, et al. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front Immunol. (2019) 10:1325. doi: 10.3389/fimmu.2019.01325
130. Robinson SP, Patterson S, English N, Davies D, Knight SC, Reid CDL. Human peripheral blood contains two distinct lineages of dendritic cells. Eur J Immunol. (1999) 29:2769–78. doi: 10.1002/(ISSN)1521-4141
131. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
132. Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cellsICOS in pDC and treg immunosuppression in breast cancer. Cancer Res. (2012) 72:6130–41. doi: 10.1158/0008-5472.CAN-12-2409
133. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, et al. Impaired IFN-α Production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progressionpDC in breast cancer pathophysiology. Cancer Res. (2012) 72:5188–97. doi: 10.1158/0008-5472.CAN-11-3468
134. Holsbø E, Olsen KS. Metastatic breast cancer and pre-diagnostic blood gene expression profiles—the Norwegian Women and Cancer (NOWAC) post-genome cohort. Front Oncol. (2020) 10:575461. doi: 10.3389/fonc.2020.575461
135. Kini Bailur J, Gueckel B, Pawelec G. Prognostic impact of high levels of circulating plasmacytoid dendritic cells in breast cancer. J Trans Med. (2016) 14:1–10. doi: 10.1186/s12967-016-0905-x
136. Oshi M, Newman S, Tokumaru Y, Yan L, Matsuyama R, Kalinski P, et al. Plasmacytoid dendritic cell (pDC) infiltration correlate with tumor infiltrating lymphocytes, cancer immunity, and better survival in triple negative breast cancer (TNBC) more strongly than conventional dendritic cell (cDC). Cancers. (2020) 12:3342. doi: 10.3390/cancers12113342
137. Tian S, Yan L, Fu L, Zhang Z, Zhang J, Meng G, et al. A comprehensive investigation to reveal the relationship between plasmacytoid dendritic cells and breast cancer by multiomics data analysis. Front Cell Dev Biol. (2021) 9:640476. doi: 10.3389/fcell.2021.640476
138. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. (2013) 38:336–48. doi: 10.1016/j.immuni.2012.10.018
139. Ramos RN, Chin LS, Dos Santos APS, Bergami-Santos PC, Laginha F, Barbuto JAM. Monocyte-derived dendritic cells from breast cancer patients are biased to induce CD4+ CD25+ Foxp3+ regulatory T cells. J leukocyte Biol. (2012) 92:673–82. doi: 10.1189/jlb.0112048
140. Shin J-Y, Wang C-Y, Lin C-C, Chu C-L. A recently described type 2 conventional dendritic cell (cDC2) subset mediates inflammation. Cell Mol Immunol. (2020) 17:1215–7. doi: 10.1038/s41423-020-0511-y
141. Hubert M, Gobbini E, Couillault C, Manh T-PV, Doffin A-C, Berthet J, et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci Immunol. (2020) 5:eaav3942. doi: 10.1126/sciimmunol.aav3942
142. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
143. Del Prete A, Sozio F, Barbazza I, Salvi V, Tiberio L, Laffranchi M, et al. Functional role of dendritic cell subsets in cancer progression and clinical implications. Int J Mol Sci. (2020) 21:3930. doi: 10.3390/ijms21113930
144. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell. (2019) 177:556–71. e16. doi: 10.1016/j.cell.2019.02.005
145. Verneau J, Sautés-Fridman C, Sun C-M. Dendritic cells in the tumor microenvironment: prognostic and theranostic impact. Semin Immunol. (2020) 48:1–11. doi: 10.1016/j.smim.2020.101410
146. Azeem W, Bakke RM, Appel S, Øyan AM, Kalland K-H. Dual pro-and anti-inflammatory features of monocyte-derived dendritic cells. Front Immunol. (2020) 11:438. doi: 10.3389/fimmu.2020.00438
147. Fainaru O, Almog N, Yung CW, Nakai K, Montoya-Zavala M, Abdollahi A, et al. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J. (2010) 24:1411. doi: 10.1096/fj.09-147025
148. Szpor J, Streb J, Glajcar A, Frączek P, Winiarska A, Tyrak KE, et al. Dendritic cells are associated with prognosis and survival in breast cancer. Diagnostics. (2021) 11:702. doi: 10.3390/diagnostics11040702
149. Lee H, Lee HJ, Song IH, Bang WS, Heo S-H, Gong G, et al. CD11c-positive dendritic cells in triple-negative breast cancer. vivo. (2018) 32:1561–9. doi: 10.21873/invivo.11415
150. Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J immunotherapy cancer. (2018) 6:1–13. doi: 10.1186/s40425-018-0446-3
151. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. (1999) 190:1417–26. doi: 10.1084/jem.190.10.1417
152. Giorello MB, Matas A, Marenco P, Davies KM, Borzone FR, MdL C, et al. CD1a-and CD83-positive dendritic cells as prognostic markers of metastasis development in early breast cancer patients. Breast Cancer. (2021) 28:1328–39. doi: 10.1007/s12282-021-01270-9
153. Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology. (2020) 9:434. doi: 10.3390/biology9120434
154. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. (2011) 121:3804–9. doi: 10.1172/JCI57099
155. Boucher Y, Kumar AS, Posada JM, Gjini E, Pfaff K, Lipschitz M, et al. Bevacizumab improves tumor infiltration of mature dendritic cells and effector T-cells in triple-negative breast cancer patients. NPJ Precis Oncol. (2021) 5:62. doi: 10.1038/s41698-021-00197-w
156. Xu X, Zhang M, Xu F, Jiang S. Wnt signaling in breast cancer: biological mechanisms, challenges and opportunities. Mol cancer. (2020) 19:1–35. doi: 10.1186/s12943-020-01276-5
157. Suryawanshi A, Manicassamy S. Tumors induce immune tolerance through activation of β-catenin/TCF4 signaling in dendritic cells: A novel therapeutic target for cancer immunotherapy. Oncoimmunology. (2015) 4:e1052932. doi: 10.1080/2162402X.2015.1052932
158. Zhu S, Yang N, Wu J, Wang X, Wang W, Liu Y-J, et al. Tumor microenvironment-related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharmacol Res. (2020) 159:104980. doi: 10.1016/j.phrs.2020.104980
159. Cózar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-infiltrating natural killer cellsTumor-infiltrating natural killer cells. Cancer discovery. (2021) 11:34–44. doi: 10.1158/2159-8290.CD-20-0655
160. Tang Y, Peitzsch C, Charoudeh HN, Cheng M, Chaves P, Jacobsen SEW, et al. Emergence of NK-cell progenitors and functionally competent NK-cell lineage subsets in the early mouse embryo. Blood J Am Soc Hematology. (2012) 120:63–75. doi: 10.1182/blood-2011-02-337980
161. Ramirez K, Chandler KJ, Spaulding C, Zandi S, Sigvardsson M, Graves BJ, et al. Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity. (2012) 36:921–32. doi: 10.1016/j.immuni.2012.04.006
162. Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med. (2014) 211:635–42. doi: 10.1084/jem.20132398
163. Ali AK, Oh JS, Vivier E, Busslinger M, Lee S-H. NK cell–specific gata3 ablation identifies the maturation program required for bone marrow exit and control of proliferation. J Immunol. (2016) 196:1753–67. doi: 10.4049/jimmunol.1501593
164. Matsuda M, Ono R, Iyoda T, Endo T, Iwasaki M, Tomizawa-Murasawa M, et al. Human NK cell development in hIL-7 and hIL-15 knockin NOD/SCID/IL2rgKO mice. . Life Sci alliance. (2019) 2(2):e201800195. doi: 10.26508/lsa.201800195
165. Vosshenrich CA, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, et al. Roles for common cytokine receptor γ-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol. (2005) 174:1213–21. doi: 10.4049/jimmunol.174.3.1213
166. Wang X, Sun R, Hao X, Lian Z-X, Wei H, Tian Z. IL-17 constrains natural killer cell activity by restraining IL-15–driven cell maturation via SOCS3. Proc Natl Acad Sci. (2019) 116:17409–18. doi: 10.1073/pnas.1904125116
167. Moore TA, von Freeden-Jeffry U, Murray R, Zlotnik A. Inhibition of gamma delta T cell development and early thymocyte maturation in IL-7-/-mice. J Immunol (Baltimore Md: 1950). (1996) 157:2366–73. doi: 10.4049/jimmunol.157.6.2366
168. Lee GA, Liou Y-H, Wang S-W, Ko K-L, Jiang S-T, Liao N-S. Different NK cell developmental events require different levels of IL-15 trans-presentation. J Immunol. (2011) 187:1212–21. doi: 10.4049/jimmunol.1100331
169. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. (2020) 10:3038. doi: 10.3389/fimmu.2019.03038
170. Ran GH, Lin YQ, Tian L, Zhang T, Yan DM, Yu JH, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal transduction targeted Ther. (2022) 7:205. doi: 10.1038/s41392-022-01058-z
171. Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC cancer. (2018) 18:1–10. doi: 10.1186/s12885-018-4389-3
172. Paul S, Kulkarni N, Shilpi, Lal G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. Oncoimmunology. (2016) 5:e1235106. doi: 10.1080/2162402X.2016.1235106
173. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
174. Susek KH, Karvouni M, Alici E, Lundqvist A. The role of CXC chemokine receptors 1–4 on immune cells in the tumor microenvironment. Front Immunol. (2018) 9:2159. doi: 10.3389/fimmu.2018.02159
175. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug discovery. (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
176. Lapeyre-Prost A, Terme M, Pernot S, Pointet A-L, Voron T, Tartour E, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. (2017) 330:295–342. doi: 10.1016/bs.ircmb.2016.09.007
177. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. (2015) 6:13835. doi: 10.18632/oncotarget.v6i15
178. Darwich A, Silvestri A, Benmebarek M-R, Mouriès J, Cadilha B, Melacarne A, et al. Paralysis of the cytotoxic granule machinery is a new cancer immune evasion mechanism mediated by chitinase 3-like-1. J Immunotherapy Cancer. (2021) 9(11):e003224. doi: 10.1136/jitc-2021-003224
179. Zhao T, Su Z, Li Y, Zhang X, You Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal transduction targeted Ther. (2020) 5:201. doi: 10.1038/s41392-020-00303-7
180. Ji S, Yu H, Zhou D, Fan X, Duan Y, Tan Y, et al. Cancer stem cell-derived CHI3L1 activates the MAF/CTLA4 signaling pathway to promote immune escape in triple-negative breast cancer. J Trans Med. (2023) 21:721. doi: 10.1186/s12967-023-04532-6
181. Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood J Am Soc Hematology. (2012) 119:5758–68. doi: 10.1182/blood-2012-03-415364
182. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
183. Zhang W, Zhao Z, Li F. Natural killer cell dysfunction in cancer and new strategies to utilize NK cell potential for cancer immunotherapy. Mol Immunol. (2022) 144:58–70. doi: 10.1016/j.molimm.2022.02.015
184. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
185. Fisher JG, Doyle AD, Graham LV, Khakoo SI, Blunt MD. Disruption of the NKG2A: HLA-E immune checkpoint axis to enhance NK cell activation against cancer. Vaccines. (2022) 10:1993. doi: 10.3390/vaccines10121993
186. Geng X, Li M, Cui B, Lu C, Liu X, Zhang P, et al. CD4+ CD25+ Foxp3+ regulatory T cells suppress NKG2D-mediated NK cell cytotoxicity in peripheral blood. Medicine. (2019) 98(22):e15722. doi: 10.1097/MD.0000000000015722
187. Zhang Y, Husnain M, Luo Z, Bayati MA, Rosenblatt JD. EMT-6 tumor conditioned Breg cells inhibit NK cell proliferation and cytotoxic activity. Cancer Res. (2020) 80:3333. doi: 10.1158/1538-7445.AM2020-3333
188. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing fc receptor–mediated natural killer cell functionMDSC inhibit antibody therapy via nitric oxide production. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
189. Jin Y, Sun Z, Geng J, Yang L, Song Z, Song H, et al. IL-21 reinvigorates exhausted natural killer cells in patients with HBV-associated hepatocellular carcinoma in STAT1-depedent pathway. Int Immunopharmacology. (2019) 70:1–8. doi: 10.1016/j.intimp.2019.02.007
190. Seo H, Jeon I, Kim B-S, Park M, Bae E-A, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. doi: 10.1038/ncomms15776
191. Wang Y, Zhang Y, Yi P, Dong W, Nalin AP, Zhang J, et al. The IL-15–AKT–XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat Immunol. (2019) 20:10–7. doi: 10.1038/s41590-018-0265-1
192. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res. (2019) 25:7126–38. doi: 10.1158/1078-0432.CCR-19-0421
193. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood J Am Soc Hematology. (2014) 124:1081–8. doi: 10.1182/blood-2014-02-556837
194. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. (2018) 3(3):e96219. doi: 10.1172/jci.insight.96219
195. Liu K, Cui J-J, Zhan Y, Ouyang Q-Y, Lu Q-S, Yang D-H, et al. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Mol Cancer. (2022) 21:1–23. doi: 10.1186/s12943-022-01561-5
196. Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol. (2016) 13:628–39. doi: 10.1038/cmi.2015.42
197. Mamessier E, Sylvain A, Thibult M-L, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
198. Donatelli SS, Zhou J-M, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-β–inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci. (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
199. Glasner A, Levi A, Enk J, Isaacson B, Viukov S, Orlanski S, et al. NKp46 receptor-mediated interferon-γ production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity. (2018) 48:107–19. e4. doi: 10.1016/j.immuni.2017.12.007
200. Li F, Liu S. Focusing on NK cells and ADCC: A promising immunotherapy approach in targeted therapy for HER2-positive breast cancer. Front Immunol. (2022) 13:7709. doi: 10.3389/fimmu.2022.1083462
201. Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, de Cerio AL-D, Cabo M, et al. Targeting NK-cell checkpoints for cancer immunotherapy. Curr Opin Immunol. (2017) 45:73–81. doi: 10.1016/j.coi.2017.01.003
202. Zhang J, Zheng H, Diao Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int J Mol Sci. (2019) 20:317. doi: 10.3390/ijms20020317
203. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastomaCAR-NKTs mediate long-term control of neuroblastoma in mice. Clin Cancer Res. (2019) 25:7126–38. doi: 10.1158/1078-0432.CCR-19-0421
204. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
205. Ruella M, Kalos M. Adoptive immunotherapy for cancer. Immunol Rev. (2014) 257:14–38. doi: 10.1111/imr.12136
206. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. (2019) 10:1205. doi: 10.3389/fimmu.2019.01205
207. Velásquez SY, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J Biol Chem. (2016) 291:12960–77. doi: 10.1074/jbc.M116.721753
208. Brummel K, Eerkens AL, de Bruyn M, Nijman HW. Tumour-infiltrating lymphocytes: From prognosis to treatment selection. Br J Cancer. (2023) 128:451–8. doi: 10.1038/s41416-022-02119-4
209. Pruneri G, Vingiani A, Denkert C. Tumor infiltrating lymphocytes in early breast cancer. Breast. (2018) 37:207–14. doi: 10.1016/j.breast.2017.03.010
210. Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol. (2015) 26:259–71. doi: 10.1093/annonc/mdu450
211. Dieci MV, Radosevic-Robin N, Fineberg S, Van den Eynden G, Ternes N, Penault-Llorca F, et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: A report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol. (2018) 25:16–25. doi: 10.1016/j.semcancer.2017.10.003
212. Shergold AL, Millar R, Nibbs RJ. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res. (2019) 145:104258. doi: 10.1016/j.phrs.2019.104258
213. Hwang J-R, Byeon Y, Kim D, Park S-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. (2020) 52:750–61. doi: 10.1038/s12276-020-0435-8
214. Shi X, Li C-W, Tan L-C, Wen S-S, Liao T, Zhang Y, et al. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: a large cohort study. J Clin Endocrinol Metab. (2021) 106:120–32. doi: 10.1210/clinem/dgaa701
215. Liu J, Chen Z, Li Y, Zhao W, Wu JB, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Frontiers in pharmacology (2021) 12:731798. doi: 10.3389/fphar.2021.731798
216. Zhao Y, Harrison DL, Song Y, Ji J, Huang J, Hui E. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. (2018) 24:379–90. e6. doi: 10.1016/j.celrep.2018.06.054
217. Fan Z, Tian Y, Chen Z, Liu L, Zhou Q, He J, et al. Blocking interaction between SHP2 and PD-1 denotes a novel opportunity for developing PD-1 inhibitors. EMBO Mol Med. (2020) 12:e11571. doi: 10.15252/emmm.201911571
218. Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed science. (2020) 27:1–30. doi: 10.1186/s12929-019-0592-z
219. Bertucci F, Gonçalves A. Immunotherapy in breast cancer: the emerging role of PD-1 and PD-L1. Curr Oncol Rep. (2017) 19:1–11. doi: 10.1007/s11912-017-0627-0
220. Zhao Y-J, Zhang J, Shi F, Hu Z-P, Wu J-P, Wu G-J, et al. Expression of PD-1 on CD4+ tumor-infiltrating lymphocytes in tumor microenvironment associated with pathological characteristics of breast cancer. J Immunol Res. (2018) 2018:1278. doi: 10.1155/2018/5690258
221. Gil Del Alcazar C, Huh S, Ekram M, Trinh A, Liu L, Beca F, et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discovery. (2017) 7:1098–115. doi: 10.1158/2159-8290.CD-17-0222
222. Posabella A, Schaerli A, Muenst S, Gao F, Daster S, Trella E, et al. The expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Eur J Surg Oncol. (2015) 41:S2. doi: 10.1016/j.ejso.2014.10.007
223. Yang Y, Zhang X, Lin F, Xiong M, Fan D, Yuan X, et al. Bispecific CD3-HAC carried by E1A-engineered mesenchymal stromal cells against metastatic breast cancer by blocking PD-L1 and activating T cells. J Hematol Oncol. (2019) 12:1–16. doi: 10.1186/s13045-019-0723-8
224. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
225. Gallimore A, Glithero A, Godkin A, Tissot AC, Plückthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus–specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I–peptide complexes. J Exp Med. (1998) 187:1383–93. doi: 10.1084/jem.187.9.1383
226. Lei X, Lei Y, Li J-K, Du W-X, Li R-G, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer letters. (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
227. Tang L, Zhang Y, Hu Y, Mei H. T cell exhaustion and CAR-T immunotherapy in hematological Malignancies. BioMed Res Int. (2021) 2021:6616391. doi: 10.1155/2021/6616391
228. Lopez de Rodas M, Schalper KA. Tumour antigen-induced T cell exhaustion—The archenemy of immune-hot Malignancies. Nat Rev Clin Oncol. (2021) 18:749–50. doi: 10.1038/s41571-021-00562-5
229. Shi F, Chang H, Zhou Q, Zhao Y-J, Wu G-J, Song Q-K. Distribution of CD4+ and CD8+ exhausted tumor-infiltrating lymphocytes in molecular subtypes of Chinese breast cancer patients. OncoTargets Ther. (2018), 6139–45. doi: 10.2147/OTT
230. Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. (2012) 37:1130–44. doi: 10.1016/j.immuni.2012.08.021
231. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
233. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. (2005) 6:1236–44. doi: 10.1038/ni1268
234. Canale FP, Ramello MC, Montes CL. CD39 as a marker of pathogenic CD8+ T cells in cancer and other chronic inflammatory diseases. Oncoscience. (2018) 5:65. doi: 10.18632/oncoscience.v5i3-4
235. Canale FP, Ramello MC, Núñez N, Furlan CLA, Bossio SN, Serrán MG, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cellsCD39 defines exhaustion in CD8+ TILs. Cancer Res. (2018) 78:115–28. doi: 10.1158/0008-5472.CAN-16-2684
236. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
237. Jiang W, He Y, He W, Wu G, Zhou X, Sheng Q, et al. Exhausted CD8+ T cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol. (2021) 11:622509. doi: 10.3389/fimmu.2020.622509
238. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol Rev. (2017) 276:80–96. doi: 10.1111/imr.12519
239. Kumar R, Collins D, Dolly S, McDonald F, O’Brien ME, Yap TA. Targeting the PD-1/PD-L1 axis in non–small cell lung cancer. Curr problems cancer. (2017) 41:111–24. doi: 10.1016/j.currproblcancer.2016.12.002
240. Bachireddy P, Hainz U, Rooney M, Pozdnyakova O, Aldridge J, Zhang W, et al. Reversal of in situ T-cell exhaustion during effective human antileukemia responses to donor lymphocyte infusion. Blood J Am Soc Hematology. (2014) 123:1412–21. doi: 10.1182/blood-2013-08-523001
241. Martin PJ. Reversing CD8+ T-cell exhaustion with DLI. Blood J Am Soc Hematology. (2014) 123:1289–90. doi: 10.1182/blood-2014-01-547000
242. Oostvogels R, Kneppers E, Minnema M, Doorn R, Franssen L, Aarts T, et al. Efficacy of host-dendritic cell vaccinations with or without minor histocompatibility antigen loading, combined with donor lymphocyte infusion in multiple myeloma patients. Bone Marrow Transplantation. (2017) 52:228–37. doi: 10.1038/bmt.2016.250
243. Kim ST, Jeong H, Woo OH, Seo JH, Kim A, Lee ES, et al. Tumor-infiltrating lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am J Clin Oncol. (2013) 36:224–31. doi: 10.1097/COC.0b013e3182467d90
244. Zhao X, Li Y, Wang X, Wu J, Yuan Y, Lv S, et al. Synergistic association of FOXP3+ tumor infiltrating lymphocytes with CCL20 expressions with poor prognosis of primary breast cancer: a retrospective cohort study. Medicine. (2019) 98(50):e18403. doi: 10.1097/MD.0000000000018403
245. Khedr R, Ghannam A, El-Rashidy M, El-Deen A. The prognostic role of tumor-infiltrating lymphocytes CD8 and Foxp3 and their impact on recurrence in breast cancer patients. J Cancer Sci Ther. (2016) 8:206–12. doi: 10.4172/1948-5956
246. Cinier J, Hubert M, Besson L, Di Roio A, Rodriguez C, Lombardi V, et al. Recruitment and expansion of tregs cells in the tumor environment—How to target them? Cancers. (2021) 13:1850. doi: 10.3390/cancers13081850
247. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev. (2016) 270:8–19. doi: 10.1111/imr.12396
248. Tanaka M, Iwakiri Y. The hepatic lymphatic vascular system: structure, function, markers, and lymphangiogenesis. Cell Mol Gastroenterol hepatology. (2016) 2:733–49. doi: 10.1016/j.jcmgh.2016.09.002
249. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
250. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. B cells and antibodies. Mol. Biol. Educ. Garland Science. (2002) 36:317–318. doi: 10.1002/bmb.20192
251. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
252. Bruggeman CW, Dekkers G, Bentlage AE, Treffers LW, Nagelkerke SQ, Lissenberg-Thunnissen S, et al. Enhanced effector functions due to antibody defucosylation depend on the effector cell Fcγ receptor profile. J Immunol. (2017) 199:204–11. doi: 10.4049/jimmunol.1700116
253. Petricevic B, Laengle J, Singer J, Sachet M, Fazekas J, Steger G, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Trans Med. (2013) 11:1–11. doi: 10.1186/1479-5876-11-307
254. Niu N, Zhang J, Huang T, Sun Y, Chen Z, Yi W, et al. IgG expression in human colorectal cancer and its relationship to cancer cell behaviors. PloS One. (2012) 7:e47362. doi: 10.1371/journal.pone.0047362
255. Kdimati S, Mullins CS, Linnebacher M. Cancer-cell-derived IgG and its potential role in tumor development. Int J Mol Sci. (2021) 22:11597. doi: 10.3390/ijms222111597
256. Ma C, Wang Y, Zhang G, Chen Z, Qiu Y, Li J, et al. Immunoglobulin G expression and its potential role in primary and metastatic breast cancers. Curr Mol Med. (2013) 13:429–37. doi: 10.2174/1566524011313030012
257. Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol. (2020) 20:294–307. doi: 10.1038/s41577-019-0257-x
258. Arabpour M, Rasolmali R, Talei A-R, Mehdipour F, Ghaderi A. Granzyme B production by activated B cells derived from breast cancer-draining lymph nodes. Mol Immunol. (2019) 114:172–8. doi: 10.1016/j.molimm.2019.07.019
259. Pimentel VO, Yaromina A, Marcus D, Dubois LJ, Lambin P. A novel co-culture assay to assess anti-tumor CD8+ T cell cytotoxicity via luminescence and multicolor flow cytometry. J Immunol Methods. (2020) 487:112899. doi: 10.1016/j.jim.2020.112899
260. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21–induced granzyme B–expressing B cells infiltrate tumors and regulate T cellsGranzyme B+ Regulatory B cells infiltrate tumors. Cancer Res. (2013) 73:2468–79. doi: 10.1158/0008-5472.CAN-12-3450
261. Vazquez MI, Catalan-Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine. (2015) 74:318–26. doi: 10.1016/j.cyto.2015.02.007
262. Xia Y, Tao H, Hu Y, Zhou L, Bao Y, Lundy SK, et al. Antitumor effector B cells directly kill tumor cells involving the CXCL12/CXCR4 pathway and their therapeutic efficacy is enhanced by IL-2. Cancer Res. (2015) 75:1340. doi: 10.1158/1538-7445.AM2015-1340
263. Gong D-H, Chen Y-Y, Ma D, Chen H-Y, Ding K-F, Yu K-D. Complicated prognostic values of CCL28 in breast cancer by subtype. J Thorac disease. (2019) 11:777. doi: 10.21037/jtd
264. Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska-Bosiacka I. CC chemokines in a tumor: a review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 ligands. Int J Mol Sci. (2020) 21:7619. doi: 10.3390/ijms21207619
265. Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. (2017) 14:662–74. doi: 10.1038/cmi.2017.35
266. Tao H, Lu L, Xia Y, Dai F, Wang Y, Bao Y, et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur J Immunol. (2015) 45:999–1009. doi: 10.1002/eji.201444625
267. Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina T. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front Cell Dev Biol. (2021) 1282. doi: 10.3389/fcell.2021.678127
268. Zhang Y, Morgan R, Podack ER, Rosenblatt J. B cell regulation of anti-tumor immune response. Immunologic Res. (2013) 57:115–24. doi: 10.1007/s12026-013-8472-1
269. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-evoked regulatory b cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cellscancer-promoting role of B cellscancer-promoting role of B cells. Cancer Res. (2011) 71:3505–15. doi: 10.1158/0008-5472.CAN-10-4316
270. Gao S-H, Liu S-Z, Wang G-Z, Zhou G-B. CXCL13 in cancer and other diseases: biological functions, clinical significance, and therapeutic opportunities. Life. (2021) 11:1282. doi: 10.3390/life11121282
271. Shen L, Li J, Liu Q, Das M, Song W, Zhang X, et al. Nano-trapping CXCL13 reduces regulatory B cells in tumor microenvironment and inhibits tumor growth. J Controlled Release. (2022) 343:303–13. doi: 10.1016/j.jconrel.2022.01.039
272. Shen M, Wang J, Yu W, Zhang C, Liu M, Wang K, et al. A novel MDSC-induced PD-1– PD-L1+ B-cell subset in breast tumor microenvironment possesses immuno-suppressive properties. Oncoimmunology. (2018) 7:e1413520. doi: 10.1080/2162402X.2017.1413520
273. Miligy I, Mohan P, Gaber A, Aleskandarany MA, Nolan CC, Diez-Rodriguez M, et al. Prognostic significance of tumour infiltrating B lymphocytes in breast ductal carcinoma in situ. Histopathology. (2017) 71:258–68. doi: 10.1111/his.13217
274. Campbell MJ, Baehner F, O’Meara T, Ojukwu E, Han B, Mukhtar R, et al. Characterizing the immune microenvironment in high-risk ductal carcinoma in situ of the breast. Breast Cancer Res Treat. (2017) 161:17–28. doi: 10.1007/s10549-016-4036-0
275. Arias-Pulido H, Cimino-Mathews A, Chaher N, Qualls C, Joste N, Colpaert C, et al. The combined presence of CD20+ B cells and PD-L1+ tumor-infiltrating lymphocytes in inflammatory breast cancer is prognostic of improved patient outcome. Breast Cancer Res Treat. (2018) 171:273–82. doi: 10.1007/s10549-018-4834-7
276. Kuroda H, Jamiyan T, Yamaguchi R, Kakumoto A, Abe A, Harada O, et al. Tumor-infiltrating B cells and T cells correlate with postoperative prognosis in triple-negative carcinoma of the breast. BMC cancer. (2021) 21:1–10. doi: 10.1186/s12885-021-08009-x
277. Guan H, Lan Y, Wan Y, Wang Q, Wang C, Xu L, et al. PD-L1 mediated the differentiation of tumor-infiltrating CD19+ B lymphocytes and T cells in Invasive breast cancer. Oncoimmunology. (2016) 5:e1075112. doi: 10.1080/216240X.2015.1075112
278. Sharma U, Medina-Saenz K, Miller PC, Troness B, Spartz A, Sandoval-Leon A, et al. Heterotypic clustering of circulating tumor cells and circulating cancer-associated fibroblasts facilitates breast cancer metastasis. Breast Cancer Res Treat. (2021) 189:63–80. doi: 10.1007/s10549-021-06299-0
279. Chen P-Y, Wei W-F, Wu H-Z, Fan L-S, Wang W. Cancer-associated fibroblast heterogeneity: A factor that cannot be ignored in immune microenvironment remodeling. Front Immunol. (2021) 12:2760. doi: 10.3389/fimmu.2021.671595
280. Díaz-Flores L, Gutiérrez R, García MP, González-Gómez M, Rodríguez-Rodriguez R, Hernández-León N, et al. CD34+ stromal cells/telocytes in normal and pathological skin. Int J Mol Sci. (2021) 22:7342. doi: 10.3390/ijms22147342
281. Hanley CJ, Noble F, Ward M, Bullock M, Drifka C, Mellone M, et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget. (2016) 7:6159. doi: 10.18632/oncotarget.v7i5
282. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. (2019) 570:246–51. doi: 10.1038/s41586-019-1263-7
283. Tang X, Hou Y, Yang G, Wang X, Tang S, Du Y, et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differentiation. (2016) 23:132–45. doi: 10.1038/cdd.2015.78
284. Wang FT, Sun W, Zhang JT, Fan YZ. Cancer−associated fibroblast regulation of tumor neo−angiogenesis as a therapeutic target in cancer. Oncol letters. (2019) 17:3055–65. doi: 10.3892/ol
285. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. (2015) 33:230–6. doi: 10.1016/j.tibtech.2015.01.004
286. Younesi FS, Son DO, Firmino J, Hinz B. Myofibroblast markers and microscopy detection methods in cell culture and histology. Myofibroblasts: Springer;. (2021), 17–47. doi: 10.1007/978-1-0716-1382-5_3
287. Gao Y, Li X, Zeng C, Liu C, Hao Q, Li W, et al. CD63+ cancer-associated fibroblasts confer tamoxifen resistance to breast cancer cells through exosomal miR-22. Advanced Science. (2020) 7:2002518. doi: 10.1002/advs.202002518
288. Schoppmann SF, Berghoff A, Dinhof C, Jakesz R, Gnant M, Dubsky P, et al. Podoplanin-expressing cancer-associated fibroblasts are associated with poor prognosis in invasive breast cancer. Breast Cancer Res Treat. (2012) 134:237–44. doi: 10.1007/s10549-012-1984-x
289. Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. (2008) 40:650–5. doi: 10.1038/ng.117
290. Rohan TE, Miller CA, Li T, Wang Y, Loudig O, Ginsberg M, et al. Somatic mutations in benign breast disease tissue and risk of subsequent invasive breast cancer. Br J Cancer. (2018) 118:1662–4. doi: 10.1038/s41416-018-0089-7
291. Soysal SD, Ng CK, Costa L, Weber WP, Paradiso V, Piscuoglio S, et al. Genetic alterations in benign breast biopsies of subsequent breast cancer patients. Front Med. (2019) 6:166. doi: 10.3389/fmed.2019.00166
292. Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol. (2010) 21:19–25. doi: 10.1016/j.semcdb.2009.10.002
293. Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. (2020) 31:107701. doi: 10.1016/j.celrep.2020.107701
294. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. (2015) 6:1–15. doi: 10.1038/ncomms10204
295. Wan X, Guan S, Hou Y, Qin Y, Zeng H, Yang L, et al. FOSL2 promotes VEGF-independent angiogenesis by transcriptionnally activating Wnt5a in breast cancer-associated fibroblasts. Theranostics. (2021) 11:4975. doi: 10.7150/thno.55074
296. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Advanced Drug delivery Rev. (2016) 99:186–96. doi: 10.1016/j.addr.2015.07.007
297. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
298. Fu J, Su X, Li Z, Deng L, Liu X, Feng X, et al. HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene. (2021) 40:4625–51. doi: 10.1038/s41388-021-01863-w
299. Kong L, Guo S, Liu C, Zhao Y, Feng C, Liu Y, et al. Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int J Oncol. (2016) 48:1085–94. doi: 10.3892/ijo.2016.3343
300. Kinugasa Y, Matsui T, Takakura N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of Malignant cancer cells in the tumor microenvironment. Stem Cells. (2014) 32:145–56. doi: 10.1002/stem.1556
301. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79. e10. doi: 10.1016/j.ccell.2018.01.011
302. Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer-associated fibroblasts-derived exosomes suppress immune cell function in breast cancer via the miR-92/PD-L1 pathway. Front Immunol. (2020) 11:2026. doi: 10.3389/fimmu.2020.02026
303. Jiang G, Tu J, Zhou L, Dong M, Fan J, Chang Z, et al. Single-cell transcriptomics reveal the heterogeneity and dynamic of cancer stem-like cells during breast tumor progression. Cell Death Disease. (2021) 12:979. doi: 10.1038/s41419-021-04261-y
304. Park SY, Kim HM, Koo JS. Differential expression of cancer-associated fibroblast-related proteins according to molecular subtype and stromal histology in breast cancer. Breast Cancer Res Treat. (2015) 149:727–41. doi: 10.1007/s10549-015-3291-9
305. Paulsson J, Rydén L, Strell C, Frings O, Tobin NP, Fornander T, et al. High expression of stromal PDGFRβ is associated with reduced benefit of tamoxifen in breast cancer. J Pathology: Clin Res. (2017) 3:38–43. doi: 10.1002/cjp2.56
306. Conciatori F, Ciuffreda L, Bazzichetto C, Falcone I, Pilotto S, Bria E, et al. mTOR cross-talk in cancer and potential for combination therapy. Cancers. (2018) 10:23. doi: 10.3390/cancers10010023
307. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduction Targeted Ther. (2021) 6:218. doi: 10.1038/s41392-021-00641-0
308. Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, et al. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. (2012) 30:2810–9. doi: 10.1002/stem.1251
309. Grasset EM, Bertero T, Bozec A, Friard J, Bourget I, Pisano S, et al. Matrix stiffening and EGFR cooperate to promote the collective invasion of cancer cellsMechanical regulation of EGFR drives collective invasion. Cancer Res. (2018) 78:5229–42. doi: 10.1158/0008-5472.CAN-18-0601
310. Rivas EI, Linares J, Zwick M, Gómez-Llonin A, Guiu M, Labernadie A, et al. Targeted immunotherapy against distinct cancer-associated fibroblasts overcomes treatment resistance in refractory HER2+ breast tumors. Nat Commun. (2022) 13:1–16. doi: 10.1038/s41467-022-32782-3
311. Sun X-x, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacologica Sinica. (2015) 36:1219–27. doi: 10.1038/aps.2015.92
312. Wei X, Li S, He J, Du H, Liu Y, Yu W, et al. Tumor-secreted PAI-1 promotes breast cancer metastasis via the induction of adipocyte-derived collagen remodeling. Cell communication Signaling. (2019) 17:1–18. doi: 10.1186/s12964-019-0373-z
313. Lyu X, Zhang Q, Fares HM, Wang Y, Han Y, Sun L. Contribution of adipocytes in the tumor microenvironment to breast cancer metabolism. Cancer Letters. (2022) 534:215616. doi: 10.1016/j.canlet.2022.215616
314. Chu D-T, Nguyen Thi Phuong T, Tien NLB, Tran D-K, Nguyen T-T, Thanh VV, et al. The effects of adipocytes on the regulation of breast cancer in the tumor microenvironment: an update. Cells. (2019) 8:857. doi: 10.3390/cells8080857
315. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang X-Y, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer letters. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
316. Ferreri C, Sansone A, Ferreri R, Amézaga J, Tueros I. Fatty acids and membrane lipidomics in oncology: a cross-road of nutritional, signaling and metabolic pathways. Metabolites. (2020) 10:345. doi: 10.3390/metabo10090345
317. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J cancer. (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
318. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta (BBA)-Molecular Basis Disease. (2011) 1812:1007–22. doi: 10.1016/j.bbadis.2011.02.014
319. Muzio G, Barrera G, Pizzimenti S. Peroxisome proliferator-activated receptors (PPARs) and oxidative stress in physiological conditions and in cancer. Antioxidants. (2021) 10:1734. doi: 10.3390/antiox10111734
320. Bai P, Houten SM, Huber A, Schreiber V, Watanabe M, Kiss B, et al. Peroxisome proliferator-activated receptor (PPAR)-2 controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid X receptor/PPARγ heterodimer. J Biol Chem. (2007) 282:37738–46. doi: 10.1074/jbc.M701021200
321. Zhao B, Xin Z, Ren P, Wu H. The role of PPARs in breast cancer. Cells. (2022) 12:130. doi: 10.3390/cells12010130
322. Wu Y, Li X, Li Q, Cheng C, Zheng L. Adipose tissue-to-breast cancer crosstalk: Comprehensive insights. Biochim Biophys Acta (BBA)-Reviews Cancer. (2022), 188800. doi: 10.1016/j.bbcan.2022.188800
323. Kern L, Mittenbühler MJ, Vesting AJ, Ostermann AL, Wunderlich CM, Wunderlich FT. Obesity-induced TNFα and IL-6 signaling: the missing link between obesity and inflammation—driven liver and colorectal cancers. Cancers. (2018) 11:24. doi: 10.3390/cancers11010024
324. Ackerman SE, Blackburn OA, Marchildon F, Cohen P. Insights into the link between obesity and cancer. Curr Obes Rep. (2017) 6:195–203. doi: 10.1007/s13679-017-0263-x
325. Sheng X, Parmentier J-H, Tucci J, Pei H, Cortez-Toledo O, Dieli-Conwright CM, et al. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res. (2017) 15:1704–13. doi: 10.1158/1541-7786.MCR-17-0338
326. Hass R, von der Ohe J, Ungefroren H. Impact of the tumor microenvironment on tumor heterogeneity and consequences for cancer cell plasticity and stemness. Cancers. (2020) 12:3716. doi: 10.3390/cancers12123716
327. Quintanal-Villalonga Á, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. (2020) 17:360–71. doi: 10.1038/s41571-020-0340-z
328. Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain fat—lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell. (2019) 35:17–32. e6. doi: 10.1016/j.ccell.2018.12.002
329. Kothari C, Diorio C, Durocher F. The importance of breast adipose tissue in breast cancer. Int J Mol Sci. (2020) 21:5760. doi: 10.3390/ijms21165760
330. Sturtzel C. Endothelial cells. Adv Exp Med Biol. (2017) 1003:71–91. doi: 10.1007/978-3-319-57613-8_4
331. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. (2004) 84:869–901. doi: 10.1152/physrev.00035.2003
332. Alberts B, Johnson A, Walter P, Lewis J, Raff M, Roberts K. Molecular cell biology. New York: Garland Science (2008).
333. Sobierajska K, Ciszewski WM, Sacewicz-Hofman I, Niewiarowska J. Endothelial cells in the tumor microenvironment. Adv Exp Med Biol. (2020) 1234:71–86. doi: 10.1007/978-3-030-37184-5_6
334. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes cancer. (2011) 2:1117–33. doi: 10.1177/1947601911423654
335. Lee S-H, Golinska M, Griffiths JR. HIF-1-independent mechanisms regulating metabolic adaptation in hypoxic cancer cells. Cells. (2021) 10:2371. doi: 10.3390/cells10092371
336. Schneider BP. Angiogenesis of breast cancer. J Clin Oncol. (2005) 23:1782–90. doi: 10.1200/JCO.2005.12.017
337. Madu CO, Wang S, Madu CO, Lu Y. Angiogenesis in breast cancer progression, diagnosis, and treatment. J Cancer. (2020) 11:4474–94. doi: 10.7150/jca.44313
338. Tomita T. Localization of nerve fibers in colonic polyps, adenomas, and adenocarcinomas by immunocytochemical staining for PGP 9. 5. Digestive Dis Sci. (2012) 57:364–70. doi: 10.1007/s10620-011-1876-7
339. Entschladen F, Palm D, Lang K, Drell TL IV, Zaenker KS. Neoneurogenesis: tumors may initiate their own innervation by the release of neurotrophic factors in analogy to lymphangiogenesis and neoangiogenesis. Med hypotheses. (2006) 67:33–5. doi: 10.1016/j.mehy.2006.01.015
340. Zhao Q, Yang Y, Liang X, Du G, Liu L, Lu L, et al. The clinicopathological significance of neurogenesis in breast cancer. BMC cancer. (2014) 14:1–6. doi: 10.1186/1471-2407-14-484
341. Pirim İ, Eyerci N. Assesment of protein gene product (Pgp9. 5) enzyme activity against potential peptide substrate. Eurasian J Med. (2009) 41:102.
342. Wik E, Aziz S, Vethe H, Askeland C, Finne K, Knutsvik G, et al. Neurogenesis and angiogenesis are associated features of aggressive breast cancer. Medicine, Biology (2022) 20:13. doi: 10.1101/2022.01.28.477898
343. Hu J, Chen W, Shen L, Chen Z, Huang J. Crosstalk between the peripheral nervous system and breast cancer influences tumor progression. Biochim Biophys Acta (BBA)-Reviews Cancer. (2022), 188828. doi: 10.1016/j.bbcan.2022.188828
344. Tan X, Sivakumar S, Bednarsch J, Wiltberger G, Kather JN, Niehues J, et al. Nerve fibers in the tumor microenvironment in neurotropic cancer—pancreatic cancer and cholangiocarcinoma. Oncogene. (2021) 40:899–908. doi: 10.1038/s41388-020-01578-4
345. Payne LB, Hoque M, Houk C, Darden J, Chappell JC. Pericytes in vascular development. Curr Tissue Microenviron Rep. (2020) 1:143–54. doi: 10.1007/s43152-020-00014-9
346. Davis GE, Stratman AN, Sacharidou A, Koh W. Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol. (2011) 288:101–65. doi: 10.1016/B978-0-12-386041-5.00003-0
347. Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. (2000) 60:1388–93.
348. Keskin D, Kim J, Cooke VG, Wu C-C, Sugimoto H, Gu C, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep. (2015) 10:1066–81. doi: 10.1016/j.celrep.2015.01.035
349. Kim J, de Sampaio PC, Lundy DM, Peng Q, Evans KW, Sugimoto H, et al. Heterogeneous perivascular cell coverage affects breast cancer metastasis and response to chemotherapy. JCI Insight. (2016) 1(21):e90733. doi: 10.1172/jci.insight.90733
350. Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. (2012) 21:66–81. doi: 10.1016/j.ccr.2011.11.024
351. Kim J. Pericytes in breast cancer. In: Pericyte biology in disease, vol. p. Springer (2019) 1147:93–107. doi: 10.1007/978-3-030-16908-4_3
352. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro-oncology. (2005) 7:452–64. doi: 10.1215/S1152851705000232
Keywords: tumor microenvironment, stroma, immune cells, breast cancer, metastasis
Citation: Akinsipe T, Mohamedelhassan R, Akinpelu A, Pondugula SR, Mistriotis P, Avila LA and Suryawanshi A (2024) Cellular interactions in tumor microenvironment during breast cancer progression: new frontiers and implications for novel therapeutics. Front. Immunol. 15:1302587. doi: 10.3389/fimmu.2024.1302587
Received: 26 September 2023; Accepted: 16 February 2024;
Published: 12 March 2024.
Edited by:
Sulma Mohammed, Purdue University, United StatesReviewed by:
Catarina Roma Rodrigues, New University of Lisboa, PortugalCopyright © 2024 Akinsipe, Mohamedelhassan, Akinpelu, Pondugula, Mistriotis, Avila and Suryawanshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amol Suryawanshi, YW1vbC5zdXJ5YXdhbnNoaUBhdWJ1cm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.