 Sylvia Kerschbaum-Gruber1,2,3*
Sylvia Kerschbaum-Gruber1,2,3* Ava Kleinwächter1,2,4,5,6Katerina Popova1,2,3Alexandra Kneringer1,2,3
Ava Kleinwächter1,2,4,5,6Katerina Popova1,2,3Alexandra Kneringer1,2,3 Lisa-Marie Appel1,2,4,5Katharina Stasny3Anna Röhrer1,2,3Ana Beatriz Dias1,2,3,6
Lisa-Marie Appel1,2,4,5Katharina Stasny3Anna Röhrer1,2,3Ana Beatriz Dias1,2,3,6 Johannes Benedum1,2,4,5,6Lena Walch4,5
Johannes Benedum1,2,4,5,6Lena Walch4,5 Andreas Postl1,2,3
Andreas Postl1,2,3 Sandra Barna1,2,3
Sandra Barna1,2,3 Bernhard Kratzer7
Bernhard Kratzer7 Winfried F. Pickl7,8Altuna Akalin9Filip Horvat4,5Vedran Franke9Joachim Widder1,2
Winfried F. Pickl7,8Altuna Akalin9Filip Horvat4,5Vedran Franke9Joachim Widder1,2 Dietmar Georg1,2,3
Dietmar Georg1,2,3 Dea Slade1,2,3,4,5*
Dea Slade1,2,3,4,5*- 1Department of Radiation Oncology, Medical University of Vienna, Vienna, Austria
- 2Comprehensive Cancer Center, Medical University of Vienna, Vienna, Austria
- 3MedAustron Ion Therapy Center, Wiener Neustadt, Austria
- 4Max Perutz Labs, Vienna Biocenter Campus (VBC), Vienna, Austria
- 5Center for Medical Biochemistry, Medical University of Vienna, Vienna, Austria
- 6Vienna Biocenter PhD Program, a Doctoral School of the University of Vienna and the Medical University of Vienna, Vienna, Austria
- 7Center for Pathophysiology, Infectiology and Immunology, Institute of Immunology, Medical University of Vienna, Vienna, Austria
- 8Karl Landsteiner University of Health Sciences, Krems, Austria
- 9Max Delbrück Center, The Berlin Institute for Medical Systems Biology, Berlin, Germany
Introduction: Pancreatic ductal adenocarcinoma (PDAC) remains a leading cause of cancer-related deaths worldwide with limited treatment options due to extensive radiation and chemotherapy resistance. Monotherapy with immune checkpoint blockade showed no survival benefit. A combination of immunomodulation and radiotherapy may offer new treatment strategies, as demonstrated for non-small cell lung cancer. Radiation-induced anti-tumour immunity is mediated through cytosolic nucleic acid sensing pathways that drive the expression of interferon beta-1 (IFNB1) and proinflammatory cytokines.
Methods: Human PDAC cell lines (PANC-1, MIA PaCa-2, BxPC-3) were treated with X-rays and protons. Immunogenic cell death was measured based on HMGB1 release. Cytosolic dsDNA and dsRNA were analysed by immunofluorescence microscopy. Cell cycle progression, MHC-I and PD-L1 expression were determined by flow cytometry. Galectin-1 and IFNB1 were measured by ELISA. The expression levels and the phosphorylation status of the cGAS/STING and RIG-I/MAVS signalling pathways were analysed by western blotting, the expression of IFNB1 and proinflammatory cytokines was determined by RT-qPCR and genome-wide by RNA-seq. CRISPR-Cas9 knock-outs and inhibitors were used to elucidate the relevance of STING, MAVS and NF-κB for radiation-induced IFNB1 activation.
Results: We demonstrate that a clinically relevant X-ray hypofractionation regimen (3x8 Gy) induces immunogenic cell death and activates IFNB1 and proinflammatory cytokines. Fractionated radiation induces G2/M arrest and accumulation of cytosolic DNA in PDAC cells, which partly originates from mitochondria. RNA-seq analysis shows a global upregulation of type I interferon response and NF-κB signalling in PDAC cells following 3x8 Gy. Radiation-induced immunogenic response is regulated by STING, MAVS and NF-κB. In addition to immunostimulation, radiation also induces immunosuppressive galectin-1. No significant changes in MHC-I or PD-L1 expression were observed. Moreover, PDAC cell lines show similar radiation-induced immune effects when exposed to single-dose protons or photons.
Conclusion: Our findings provide a rationale for combinatorial radiation-immunomodulatory treatment approaches in PDAC using conventional photon-based or proton beam radiotherapy.
1 Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a disease with dismal prognosis, especially when locally advanced. PDAC has the lowest 5-year survival of any solid tumour of 12.8% (1). Treatment is exceedingly difficult; the majority of patients present with advanced, unresectable disease to whom the standard of care with combinational radio-chemotherapy confers limited benefit (2). Immunotherapy is an emerging treatment strategy considered the fourth pillar of cancer treatment; its success, however, is restricted to a few malignancies, excluding PDAC (3). Immune checkpoint blockade (ICB) failed to show clinical efficacy as monotherapy in PDAC (4), which can be at least partially attributed to its highly desmoplastic and immunosuppressive tumour microenvironment (TME). PDAC are largely immunologically cold, phenotypically devoid of effector CD4+ and CD8+ T cells and dominated by immunosuppressive immune cell populations (5, 6). Radiotherapy is being increasingly recognized for its ability to induce anti-tumour effects beyond cell killing. Multiple preclinical studies including preclinical PDAC models have demonstrated increased efficacy when radiotherapy and ICB are combined (7–12). Moreover, early clinical data demonstrated encouraging results when combining ICB with radiotherapy for PDAC treatment (13–16).
Radiation may drive an anti-tumour response through multiple mechanisms, including a pro-immunogenic cellular state and immunogenic cell death (ICD). This type of regulated cell death initiates cytotoxic T-cell mediated adaptive immunity through the release or cell surface presentation of damage-associated-molecular pattern (DAMP) (17). Mechanistically, radiation-induced anti-tumour immunity can be induced by the accumulation of DNA in the cytosol of irradiated cells, which activates the cGAMP synthase (cGAS)/stimulator of interferon genes (STING) pathway and leads to the production of type 1 interferons (IFN-1), specifically interferon-beta 1 (IFNB1) and interferon-stimulated genes (ISGs) such as CXCL10 (18, 19). Radiation was also shown to induce the mitochondrial antiviral-signalling (MAVS) pathway through the RNA sensor retinoic acid-inducible gene I (RIG-I), which also results in IFN-1 production (20–22). Both pathways rely on transcription factors interferon regulatory factors 3 and 7 (IRF3/IRF7) or nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NF-κB can induce CXCL10 in an IFN-1-independent manner and promotes the expression of pro-inflammatory cytokines such as interleukin 6 (IL-6) and tumour necrosis factor alpha (TNF-α) as part of its canonical function (23, 24). Together, IFNB1, ISGs and NF-κB signalling orchestrate the recruitment and activation of immune cells, thus creating an in situ vaccine (25). Accretive evidence for the cGAS/STING-mediated anti-tumour immunity has emerged for multiple malignancies including PDAC (26). PDAC clinical samples expressing both cGAS and STING show infiltration of cytotoxic T-cells and higher overall survival (27).
Different radiotherapy parameters may influence the induction of an anti-tumour immune response, including type of radiation, radiation dose, fraction size and time post-radiation (28, 29). Photon-based radiotherapy of PDAC is limited due to the proximity of several radiation-sensitive organs at risk to the tumour, which can be effectively spared with protons due to the inverted depth-dose profile and reduced lateral scatter (30). In addition, protons may also lead to a different biological radiation response that could be therapeutically beneficial. Only a limited number of studies have investigated differential immune responses to photons versus protons and none have investigated proton-mediated immune effects in PDAC (31).
Here we compared the effects of photon and proton therapy on the activation of anti-tumour immunogenicity markers including the nucleic acid sensing/IFNB1 axis and canonical NF-κB using multiple dose levels and a clinically relevant fractionation regimen. In addition, we examined markers of immunosuppressive TME such as galectin-1 (Gal-1), which was recently implicated in impaired immune surveillance during PDAC progression (32), programmed death-ligand 1 (PD-L1) as the most prominent immune checkpoint protein expressed by tumour cells, and major histocompatibility complex class I (MHC-I), which is necessary for antigen presentation and T-cell activation.
We show that hypofractionated radiation induces the release of an immunogenic cell death signal in human PDAC cell lines. Compared to single dose treatment, hypofractionation is superior in activating IFNB1 through cytosolic nucleic acid sensing via STING and MAVS pathways. We found that radiation induces the secretion of Gal-1 but no change in PD-L1 or MHC-I expression, and that protons yield similar effects compared to photons with respect to cell survival and immunogenicity. Our findings demonstrate that radiation can stimulate the key signalling axis of anti-tumour immunity in human PDAC cells, supporting the potential therapeutic benefit of combinatorial neoadjuvant radiotherapy followed by immunotherapy using conventional photon-based radiotherapy or proton beam therapy.
2 Results
2.1 Radiation elicits immunogenic cell death in human PDAC cell lines
Clonogenic survival was used to assess the effect of X-rays and protons on the viability of human PDAC cell lines PANC-1, MIA PaCa-2 and BxPC-3. All cell lines showed a dose-dependent decline of survival. RBE values were calculated at 10% cell survival (RBE10) from the applied linear-quadratic fit. For PANC-1, MIA PaCa-2 and BxPC-3, RBE10 values of 1.08 ± 0.02 Gy, 1.13 +/- 0.07 Gy and 1.11 ± 0.28 Gy were calculated, respectively (Figure 1A). To determine whether radiation-induced cancer cell death can elicit an anti-tumour immune response, we examined the release of HMGB1. HMGB1 acts as DAMP and is recognized by dendritic cells to elicit a potent tumour-specific T-cell response (33). Both irradiation modalities induced the ICD marker HMGB1 in a dose-dependent manner, while 3x8 Gy showed the strongest effect in all three cell lines. HMGB1 release kinetics were similar following X-rays or protons at equal physical doses (Figure 1B).
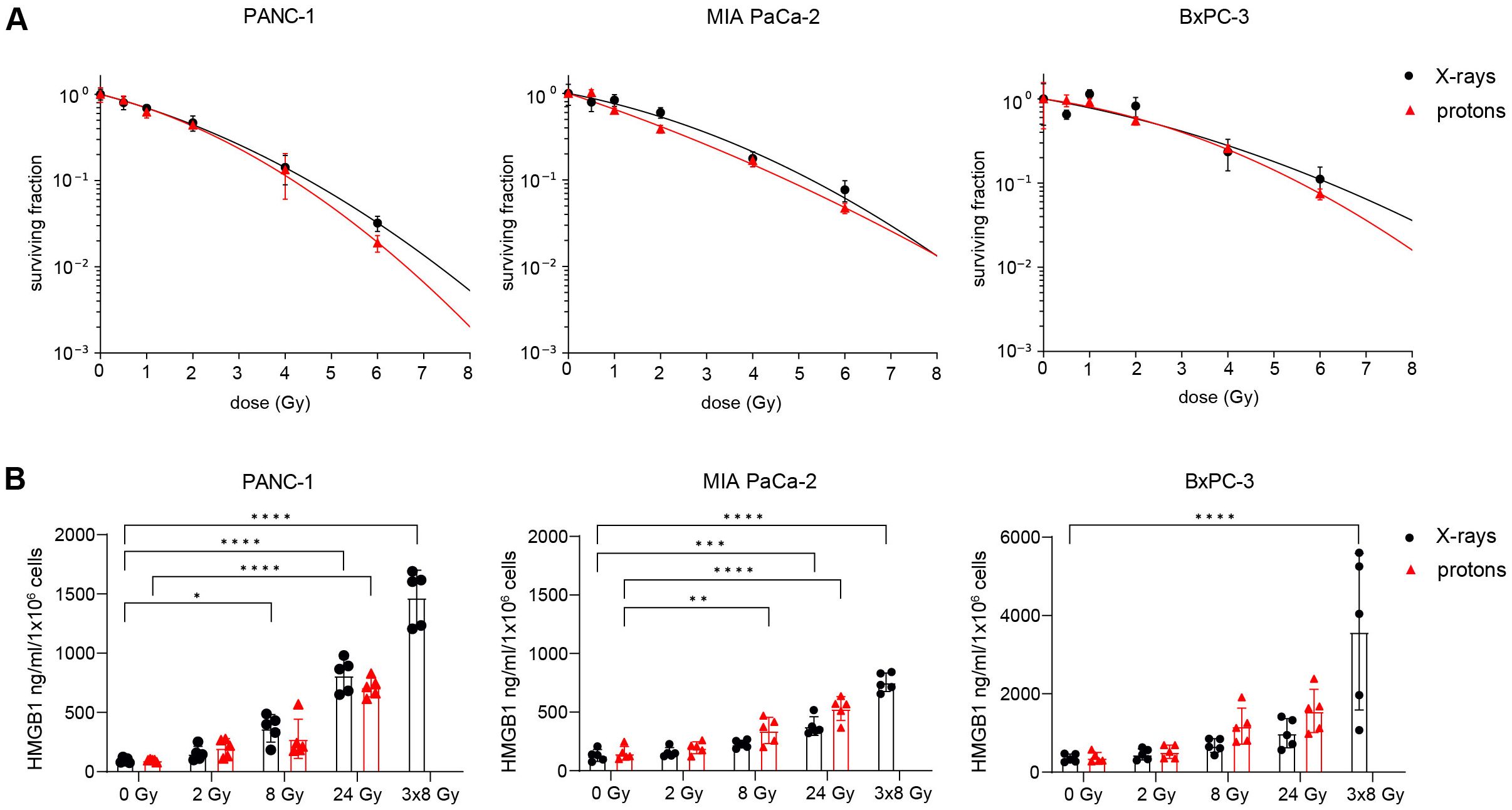
Figure 1. Radiation induces immunogenic cell death in human PDAC cell lines. (A) Cell survival curves of PANC-1, MIA PaCa-2 and BxPC-3 after reference X-ray (black circles) or proton irradiation (red triangles) (N=4-5). (B) Soluble HMGB1 was measured by ELISA in the supernatant 72 h post radiation (N=5). Data points represent mean values ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001
2.2 PDAC cells accumulate cytosolic dsDNA following irradiation
Radiation-induced activation of IFNB1, which is crucial for radiation-induced anti-tumour immunogenicity, can be a consequence of cytosolic dsDNA or dsRNA accumulation. Thus, we tested whether radiation can cause cytosolic dsDNA and dsRNA accumulation in human PDAC cells using immunofluorescence microscopy. We observed a significant accumulation of cytosolic dsDNA after 3x8 Gy X-rays in all three cell lines (Figures 2A, B; Supplementary Figure 1). Only BxPC-3 showed an increase in cytosolic dsRNA after 8 Gy X-rays (Supplementary Figure 2).
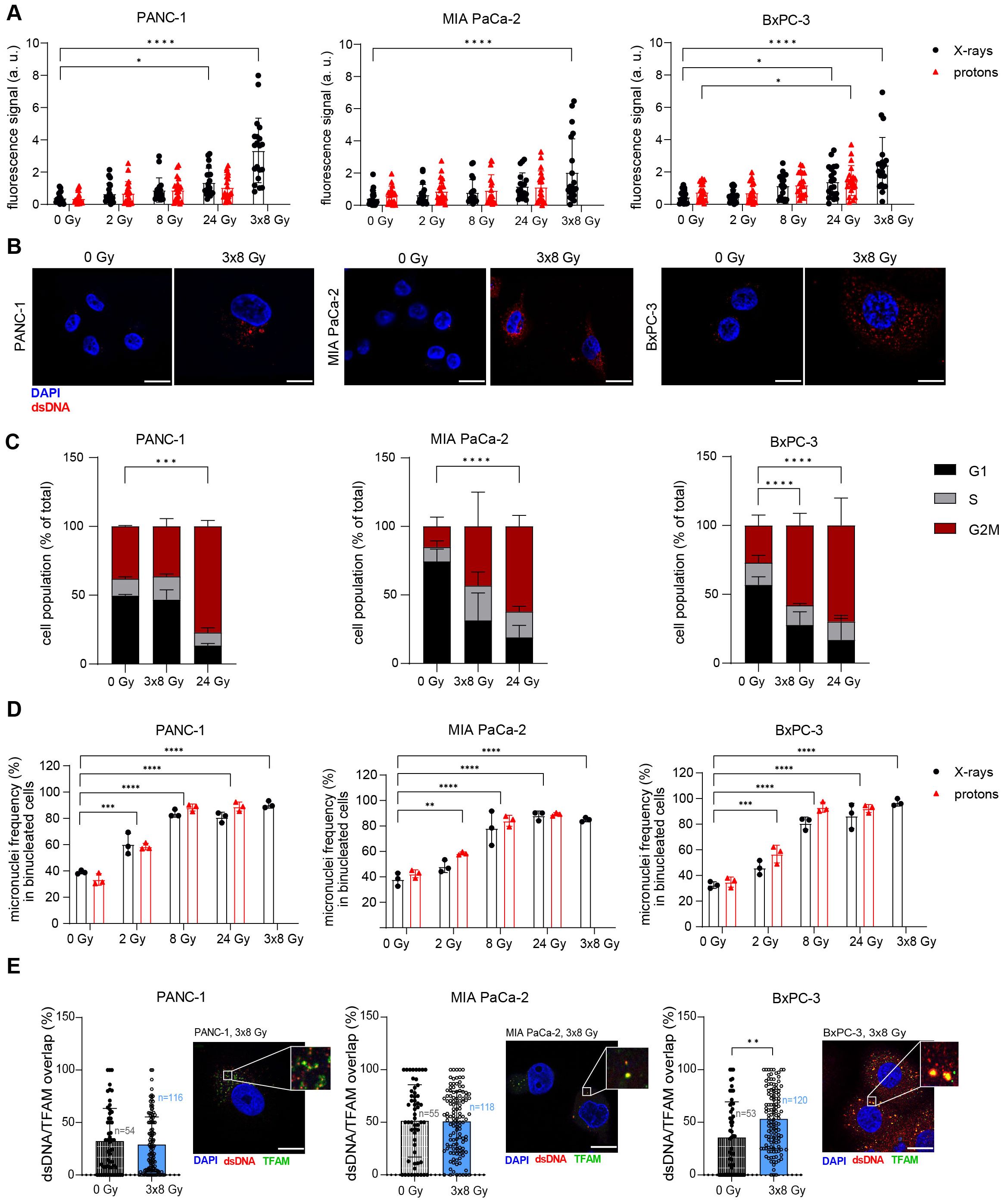
Figure 2. Radiation induces cytosolic dsDNA accumulation in human PDAC cell lines. (A, B) Cytosolic dsDNA content in PANC-1, MIA PaCa-2 and BxPC-3 cells 24 h post radiation with different doses of X-rays (black circles) or protons (red triangles). 5 cells were analysed per experiment, 4 independent experiments were performed. (A) Quantification and (B) representative images are shown. Scale bar = 20 µm. (C) Flow cytometry analysis of cell cycle distribution in untreated cells and 24 h after treatment with 3x8 Gy or 24 Gy X-rays (N = 4). (D) Percentage of micronuclei-positive PANC-1, MIA PaCa-2 and BxPC-3 cells exposed to different doses of X-rays. A cytokinesis block was induced with cytochalasin B 48 h after radiation for 24 h. Micronuclei-positive binucleated cells were scored. A minimum of 50 cells were analysed per experiment (N=3). (E) Colocalization analyses of dsDNA and TFAM in untreated cells and 24 h after treatment with 3x8 Gy X-rays using Airyscan high resolution imaging (N=3). Quantification and representative images are shown. Scale bar = 20 µm. Data points represent mean values ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001.
Radiation-induced immunostimulatory DNA may originate from micronuclei (34) or damaged mitochondria (35, 36). Micronuclei arise from mitotic defects in chromosome segregation, whereby lagging chromosomes are encapsulated by their own nuclear envelope. Cell cycle analyses revealed a G2/M arrest after 24 Gy or 3x8 Gy X-rays in all three PDAC cell lines (Figure 2C). The cytokinesis block assay (37) showed a pronounced increase in micronuclei in all three cell lines after 8 Gy, 24 Gy and 3x8 Gy, which was comparable for the same dose of photons and protons (Figure 2D). To evaluate the mitochondrial origin of cytosolic dsDNA, we performed a co-localization analysis with the mitochondrial transcription factor A (TFAM) and found radiation-induced increase in dsDNA/TFAM co-localization in BxPC-3 24 h after exposure to 3x8 Gy X-rays (Figure 2E). Subcellular fractionation followed by qPCR analysis of mitochondrial DNA (mtDNA) and genomic DNA (gDNA) in the cytosolic fraction showed an increase in gDNA for all cell lines and an increase in mtDNA for PANC-1 and MIA PaCa-2 on Day 3 after exposure to 3x8 Gy X-rays (Supplementary Figure 3). Overall, our results show that 3x8 Gy X-rays triggers cytosolic dsDNA accumulation, which can act as an immunostimulatory signal originating from micronuclei or mitochondria.
2.3 A hypofractionation regimen of 3x8 Gy X-rays induces the type I interferon and NF-κB response in PDAC cell lines
To probe whether radiation-induced dsDNA accumulation can activate the cGAS/STING signal transduction pathway and IFN-1 response, as previously shown in lung, colorectal and cervical cancer (19, 38, 39), we investigated the effects of different irradiation regimens on the signalling cascade 24 h after radiation exposure. Radiation induced cGAS dose-dependently in all three cell lines following X-rays or protons. No radiation-mediated changes were observed for the other proteins. The hypofractionation regimen of 3x8 Gy, but not the single doses, increased the expression of IFNB1 in PANC-1 and BxPC-3 (Figure 3). In MIA PaCa-2, IFNB1 could not be detected in any experimental condition. Upon release into extracellular space, IFNB1 binds to interferon-α/β receptor (IFNAR) on neighbouring cells, resulting in phosphorylation of STAT1. All three investigated cell lines express IFNAR (40). STAT1 phosphorylation was not observed 24 h post-irradiation in any of the three cell lines, despite significantly increased IFNB1 transcripts in PANC-1 and BxPC-3 (Figure 3). Interferon-gamma (IFNγ) was used as a positive control, demonstrating intact STAT1 phosphorylation capacity in all three cell lines.
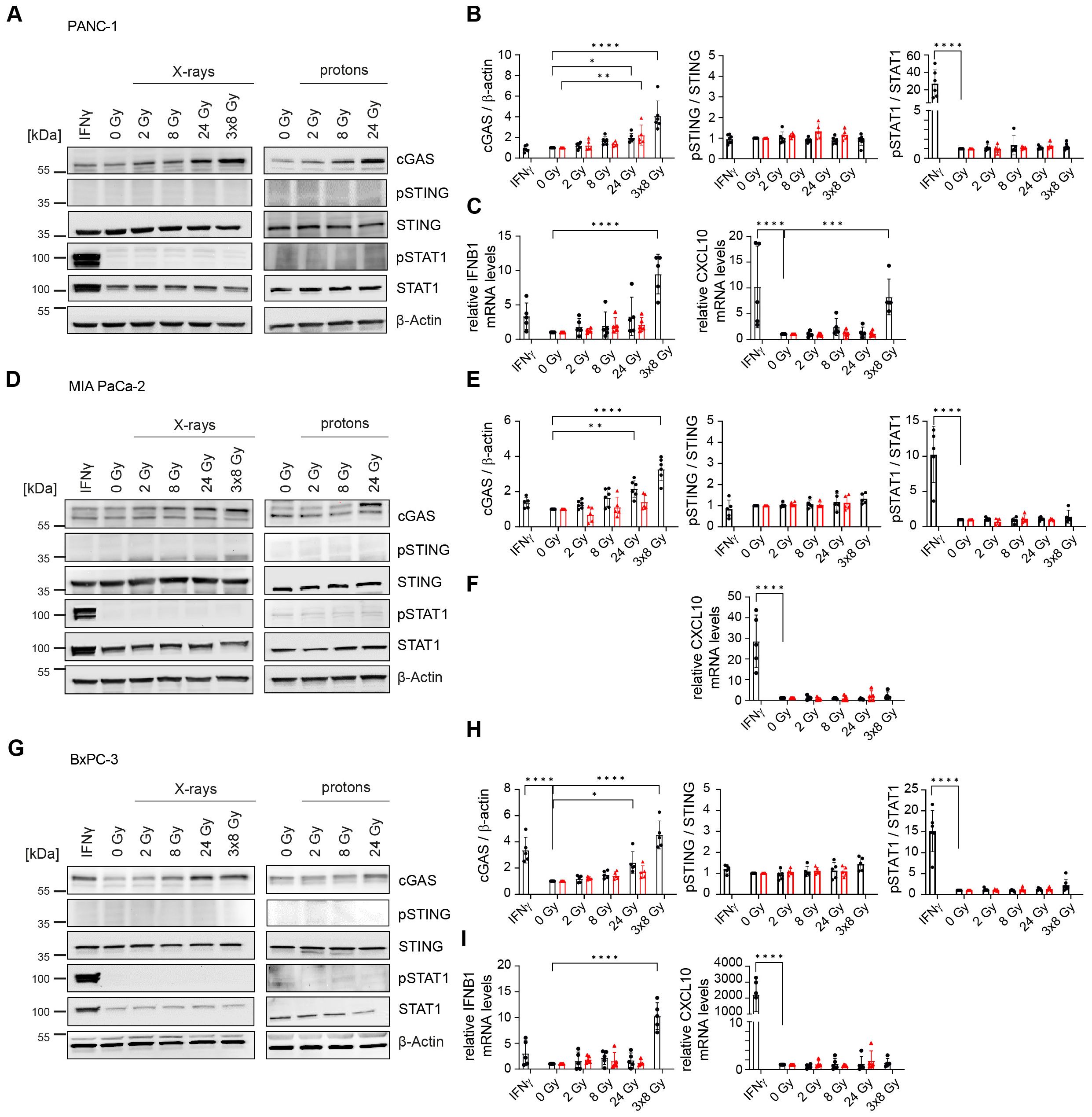
Figure 3. Hypofractionated irradiation (3x8 Gy) activates IFNB1 in human PDAC cells. Activation of the cGAS/STING pathway 24 h after X-ray (black circles) or proton (red triangles) irradiation was analysed by western blotting and RT-qPCR in (A-C) PANC-1, (D-F) MIA PaCa-2 and (G-I) BxPC-3. (A, D, G) Representative western blots and (B, E, H) quantification based on five replicates are shown. GAPDH was used as a loading control. (C, F, I) Relative expression of IFNB1 and the interferon-stimulated gene (ISG) CXCL10 compared to 0 Gy was determined by RT-qPCR (N=5). No qPCR Ct values could be determined for MIA PaCa-2 IFNB1. Data points represent mean values ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001.
Given that IFNB1 gene expression was induced, we hypothesized that IFN-1 signalling and STAT1 phosphorylation are delayed in irradiated PDAC cells. To test this, we performed a time course analysis of all doses of X-rays (Supplementary Figures 4, 5). Indeed, STAT1 phosphorylation, which is indicative of extracellular IFNB1 presence and thus active paracrine IFN-1 signalling, was observed from day 2 onwards after exposure to 3x8 Gy X-rays (Supplementary Figure 4). The increase in pSTAT1 was coupled with the induction of IFNB1 and the interferon-stimulated gene CXCL10 in PANC-1 and BxPC-3, whereas MIA PaCa-2 only showed CXCL10 induction (Supplementary Figure 4). Interestingly, a single dose of 8 Gy X-rays consistently showed the induction of IFNB1 and/or CXCL10 in all cell lines 4 days after irradiation (Supplementary Figure 5), suggesting that cell cycle progression is required for the activation of IFN-1 response (34, 41). Compared to 8 Gy single dose irradiation, the hypofractionation regimen of 3x8 Gy was more potent in inducing IFNB1 and/or CXCL10 expression (Figure 4).
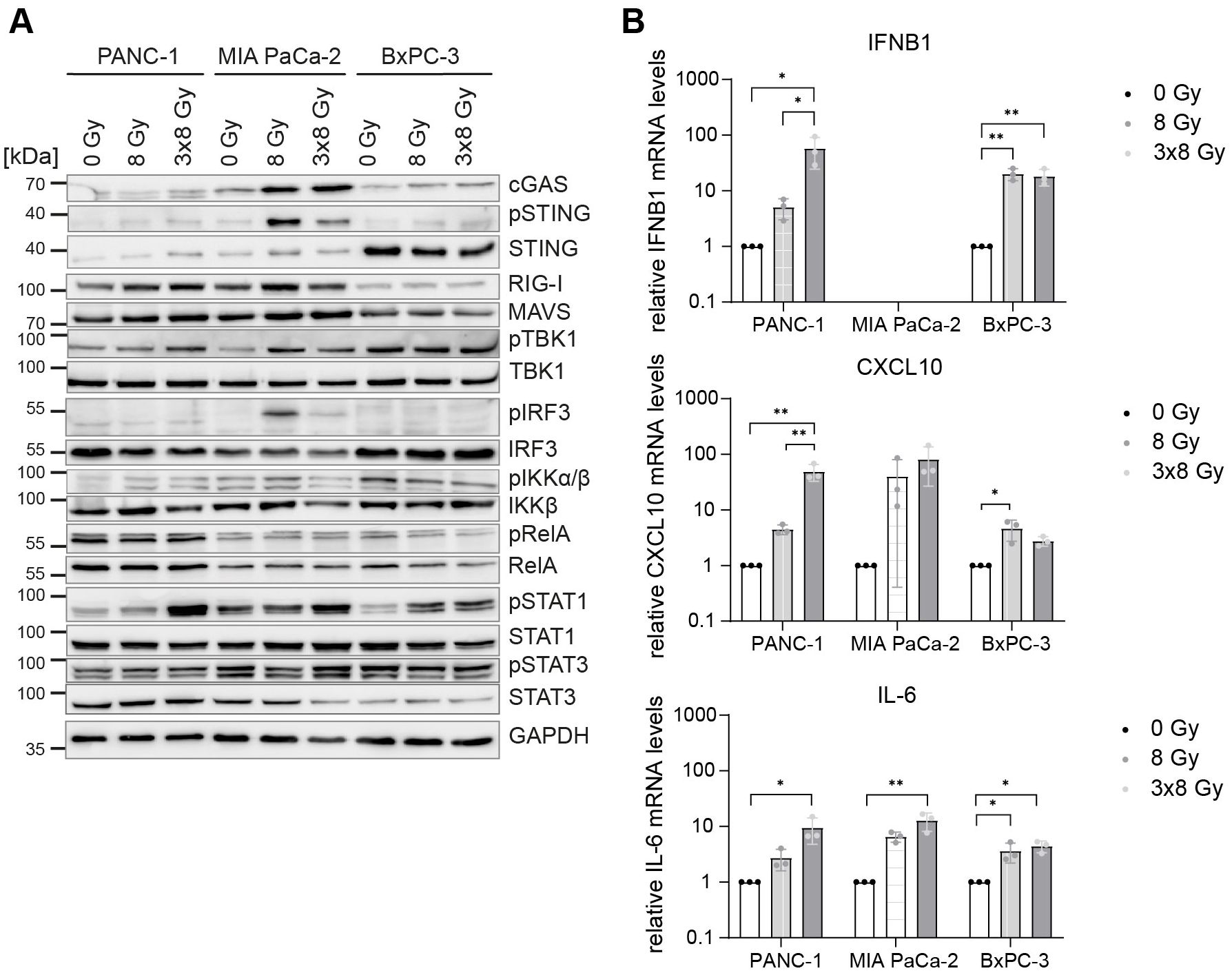
Figure 4. Comparative expression levels and activation of cGAS/STING, RIG-I/MAVS, NF-κB and STAT proteins in untreated and irradiated PDAC cells. PANC-1, MIA PaCa-2 and BxPC-3 cells were untreated or irradiated with 8 Gy or 3x8 Gy X-rays and harvested 72 h after radiation. (A) Representative western blots and (B) RT-qPCR analysis of IFNB1, CXCL10 and IL-6 compared to 0 Gy. Data points represent mean values of 5 independent experiments ± standard deviation. *≤ 0.05; **≤ 0.01.
Of the three PDAC cell lines, PANC-1 mounted the strongest IFN-1 response to 3x8 Gy X-rays as evidenced by the induction of pSTAT1, IFNB1 and CXCL10 (Figure 4). PANC-1 also showed the highest expression of RIG-I/MAVS and the canonical NF-κB subunit RelA (p65) (Figure 4A). Conversely, STING and IRF3 levels were the highest in BxPC-3 (Figure 4A). Despite a strong induction of pSTING and pIRF3 in MIA PaCa-2, this cell line failed to induce IFNB1 expression after radiation (Figure 4).
To examine radiation-induced changes in gene expression genome-wide, we performed RNA-seq in untreated cells and on day 1 and day 3 after 3x8 Gy (Figures 5A-F). We found upregulation of >700 genes (fold change >2; p-value <0.05) in all three PDAC cell lines on Day 3 after radiation exposure. Gene ontology (GO) analysis showed enrichment of genes implicated in IFN-1 signalling and canonical NF-κB signalling (Figure 5G). Genes involved in proliferation (MYC and E2F targets) and G2/M checkpoint were enriched among downregulated genes (Figure 5G). Among highly upregulated genes we identified many genes implicated in cytosolic nucleic acid sensing, non-canonical NF-κB complex (NFKB2/RELB), STAT1-STAT2-IRF9 (ISGF3 complex), ISGs and inflammatory cytokines (Figure 5H). Overall, our data show that radiation induces genome-wide IFN-1 and NF-κB response in human PDAC cells.
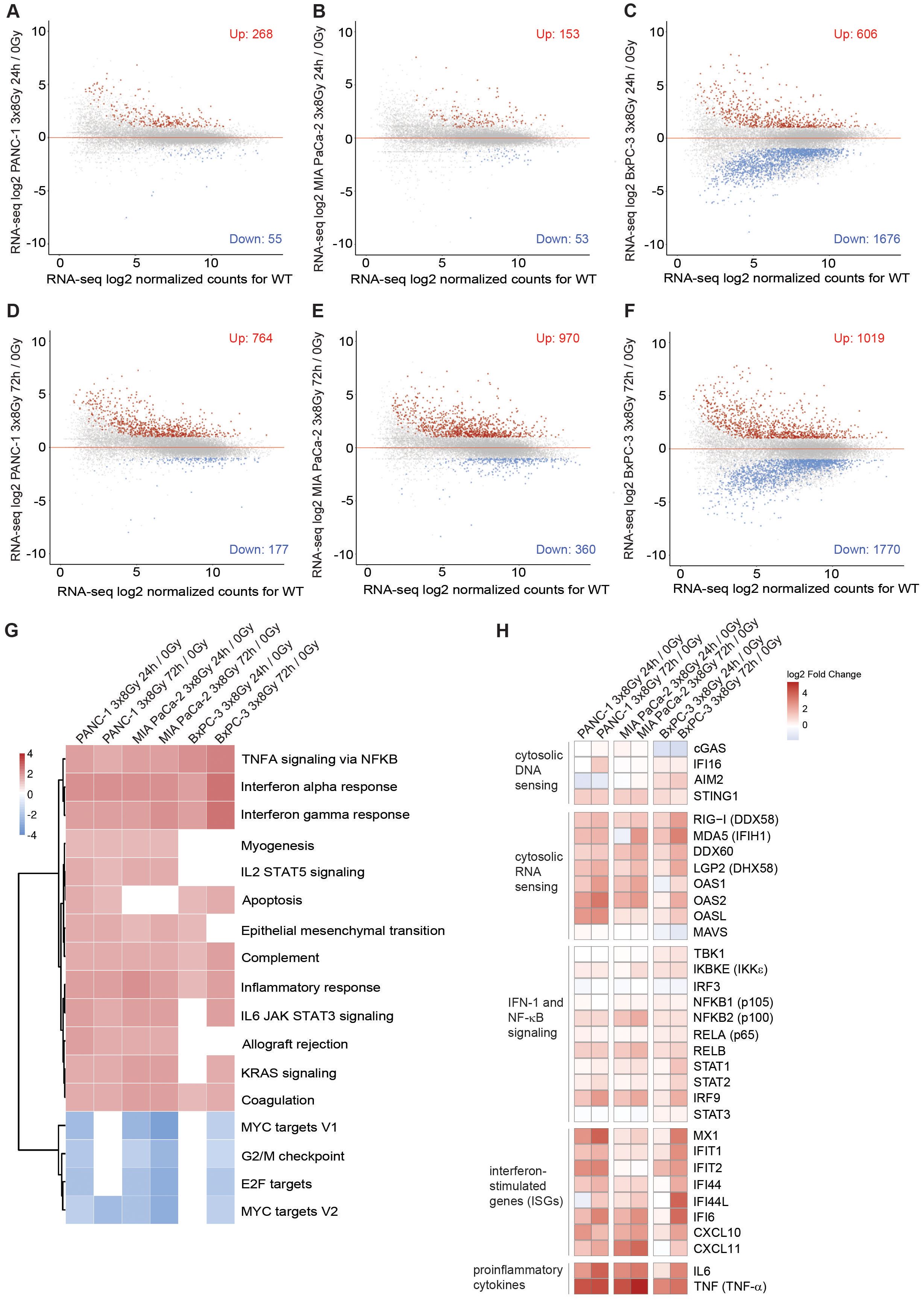
Figure 5. Type I interferon response and NF-κB signalling are globally upregulated in human PDAC cells after fractionated X-ray dose. (A-C) RNA-seq analysis in (A) PANC-1, (B) MIA PaCa-2 and (C) BxPC-3 cells 24 h after 3x8 Gy of X-rays compared to untreated. (D-F) RNA-seq analysis in (D) PANC-1, (E) MIA PaCa-2 and (F) BxPC-3 (A) PANC-1, (B) MIA PaCa-2 and (C) BxPC-3 cells 72 h after 3x8 Gy of X-rays compared to untreated. Genes with fold-change>2, p<0.05 are marked in red as upregulated or blue as downregulated (N=3). (G) GO analysis of upregulated genes shows enrichment of genes involved in type I interferon response and NF-κB signalling. Normalized enrichment scores (NESs) for Hallmark gene sets (MSigDB) between indicated irradiation treatments and controls in PANC-1, MIA PaCa-2 and BxPC-3 cells. Shown are pathways significant in at least 4 comparisons with significance level p < 0.05 adjusted for multiple testing. (H) Log2 fold changes in gene expression between indicated irradiation and control samples of chosen genes.
2.4 Radiation-induced type I interferon response is STING- and MAVS-dependent
Radiation-induced IFN-1 response was previously shown to be MAVS-dependent in PANC-1 cells (21). To test the relative requirement of STING and MAVS pathways for the induction of IFNB1 after radiation, we generated CRISPR/Cas9 knock-outs (KOs) of STING and MAVS in PANC-1 and BxPC-3 cells and tested their effect on the induction of IFNB1 on day 3 after 8 Gy and 3x8 Gy X-ray radiation (Figures 6, 7). In all three cell lines we additionally compared the effect of the STING inhibitor (H-151) (42) and the inhibitor of NF-κB signalling (BI605906) (43), as this pathway was also induced after radiation based on RNA-seq analysis (Figure 5G). BI605906 inhibits IKKβ and thereby prevents activation of RelA (p65) (43). STING and MAVS KO abrogated pSTAT1, IFNB1 and CXCL10 induction in PANC-1 (Figure 6) and BxPC-3 (Figure 7) and IFNB1 protein levels in the supernatant as measured by ELISA (Figures 6C, 7C). Phosphorylation of TYK2, as a marker of IFNAR activation, was also abrogated in STING and MAVS KO cells (Figures 6A, 7A). STING KO showed a slightly stronger effect in BxPC-3, in line with a higher STING expression in this cell line, whereas MAVS KO showed a stronger effect in PANC-1 cells, which express higher RIG-I/MAVS levels (Figures 4, 6, 7). The importance of STING for radiation-induced IFN-1 response was corroborated by siRNA-mediated STING depletion in PANC-1 and BxPC-3 treated with 3x8 Gy X-rays (Supplementary Figure 6). STING depletion resulted in diminished phosphorylation of STAT1 (Supplementary Figures 6A, C) and diminished induction of IFNB1 and CXCL10 (Supplementary Figures 6B, D) compared to non-targeting siRNA controls. STING siRNA depletion reduced, but did not abrogate radiation-induced IFN-1 response most likely due to partial STING silencing. Similarly, STING inhibition reduced, but did not abrogate, IFNB1 and CXCL10 activation in PANC-1 and BxPC-3 (Figures 6, 7), and reduced CXCL10 activation in MIA PaCa-2 (Figure 8).
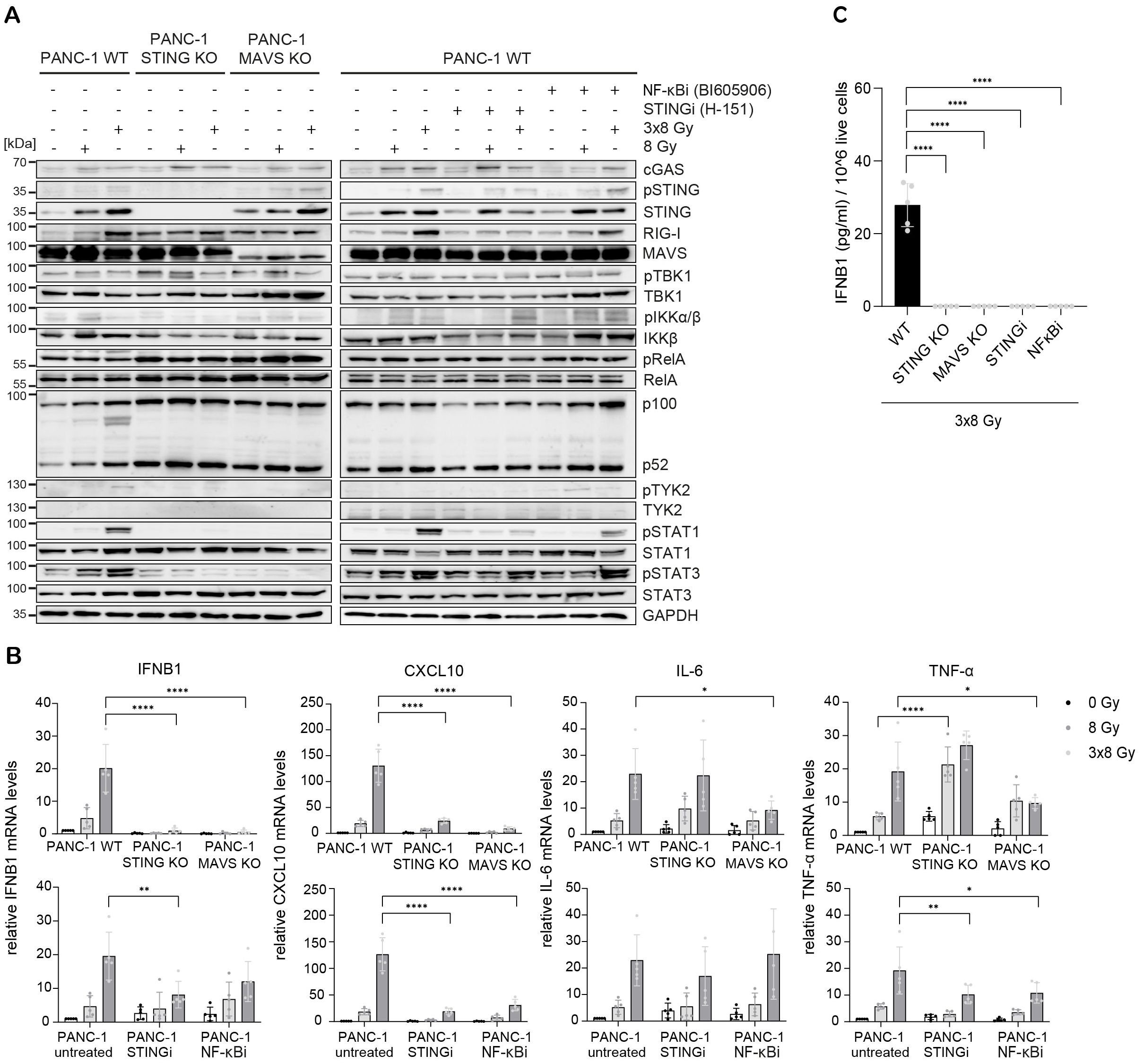
Figure 6. STING, MAVS and NF-κB drive radiation-induced immunogenic signalling in PANC-1. PANC-1 WT, STING KO and MAVS KO cells were untreated (0 Gy) or irradiated with 8 Gy or 3x8 Gy X-rays and harvested 72 h after radiation. WT cells were also treated with STINGi (10 µM H-151) or NF-κBi (10 µM BI-605906) without or with radiation. Cells were pretreated with inhibitors for 1 h before irradiation and kept until harvesting. (A) Representative western blot analysis of cGAS/STING, RIG-I/MAVS, IFN-1 and NF-κB signalling. (B) Relative mRNA levels of IFNB1, CXCL10, IL-6 and TNF-α compared to untreated WT cells. (C) IFNB1 was measured in the supernatant of PANC-1 cells by ELISA. Data points represent mean values of 5 independent experiments ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005.
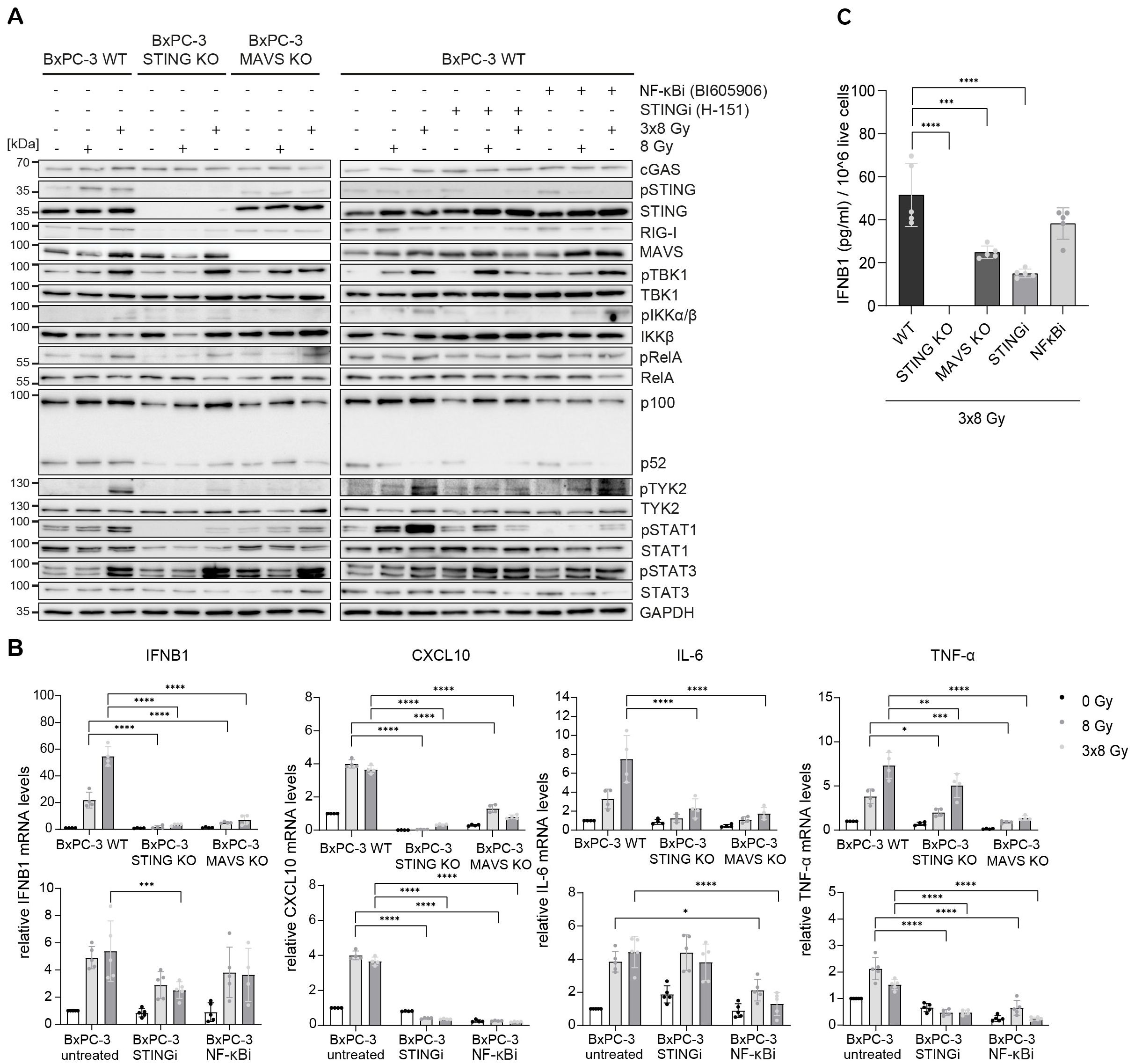
Figure 7. STING, MAVS and NF-κB drive radiation-induced immunogenic signalling in BxPC-3. BxPC-3 WT, STING KO and MAVS KO cells were untreated (0 Gy) or irradiated with 8 Gy or 3x8 Gy X-rays and harvested 72 h after radiation. WT cells were also treated with STINGi (10 µM H-151) or NF-κBi (10 µM BI-605906) without or with radiation. Cells were pretreated with inhibitors for 1 h before irradiation and kept until harvesting. (A) Representative western blot analysis of cGAS/STING, RIG-I/MAVS, IFN-1 and NF-κB signalling. (B) Relative mRNA levels of IFNB1, CXCL10, IL-6 and TNF-α compared to untreated WT cells. (C) IFNB1 was measured in the supernatant of BxPC-3 cells by ELISA. Data points represent mean values of 4-5 independent experiments ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001.
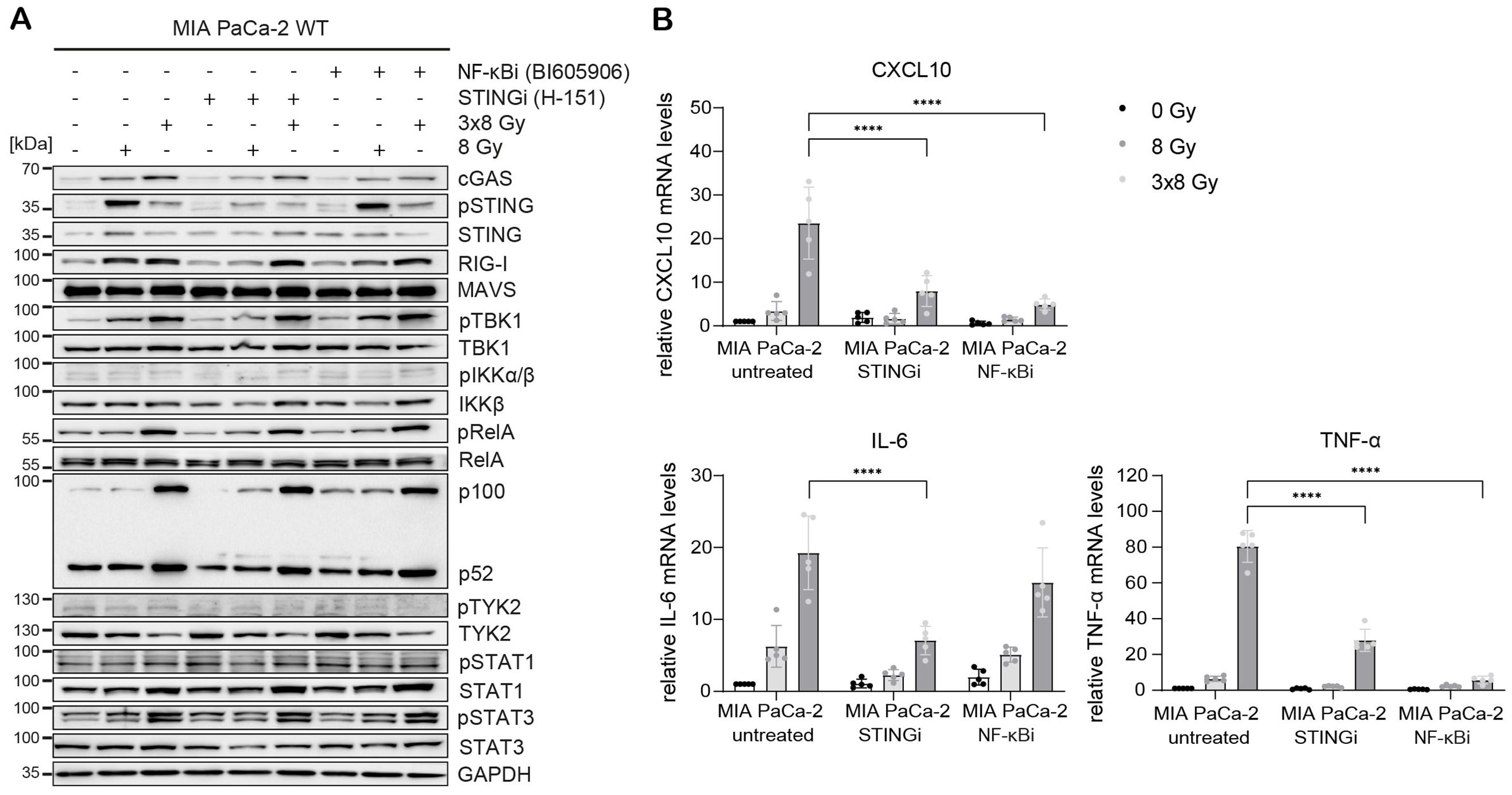
Figure 8. STING and NF-κB drive radiation-induced immunogenic signalling in MIA PaCa-2. MIA PaCa-2 cells were either untreated (0 Gy) or irradiated with 8 Gy or 3x8 Gy X-rays and harvested 72 h after radiation without or with STINGi (10 µM H-151) or NF-κBi (10 µM BI-605906). (A) Representative western blot analysis of cGAS/STING, RIG-I/MAVS, IFN-1 and NF-κB signalling. (B) Relative mRNA levels of CXCL10, IL-6 and TNF-α compared to untreated WT cells. Data points represent mean values of 5 independent experiments ± standard deviation. ****≤0.0001.
NF-κB signalling was assessed based on the expression of proinflammatory cytokines IL-6 and TNF-α. In PANC-1 cells, IL-6 expression was reduced only in MAVS KO, whereas TNF-α expression was reduced in MAVS KO and NF-κB inhibitor-treated cells (Figure 6), suggesting that in PANC-1 cells IL-6 expression is partly regulated through the MAVS pathway independent of NF-κB, whereas TNF-α expression is regulated by both MAVS-dependent and MAVS-independent NF-κB signalling. Interestingly, the expression of RelA (canonical NF-κB) and p100/p52 (non-canonical NF-κB) was increased in PANC-1 STING and MAVS KO cells, which may act as a compensatory mechanism to sustain the expression of proinflammatory cytokines independent of STING and MAVS.
In BxPC-3 cells, the expression of IL-6 and TNF-α was reduced, but not abrogated, in STING and MAVS KOs and in cells treated with the NF-κB inhibitor (Figure 7). This suggests that radiation-induced expression of IL-6 and TNF-α is regulated by both STING/MAVS-dependent and independent NF-κB signalling in BxPC-3 cells.
In MIA PaCa-2 cells, radiation-induced expression of CXCL10, IL-6 and TNF-α was reduced after STING or NF-κB inhibition (Figure 8), suggesting that the expression of these cytokines is regulated by STING-dependent and STING-independent NF-κB signalling. Interestingly, the non-canonical NF-κB pathway was highly upregulated after radiation irrespective of STING or canonical NF-κB (Figure 8A). This may explain the lack of IFNB1 expression in MIA PaCa-2, given that the non-canonical NF-κB pathway was shown to dampen IFNB1 expression by epigenetically regulating its promoter (44).
Taken together, our data show that radiation-induced IFNB1 expression is dependent on STING and MAVS pathways, whereas the expression of proinflammatory cytokines IL-6 and TNF-α is controlled by STING/MAVS-dependent and independent NF-κB signalling.
2.5 Radiation-mediated effects on T-cell modulating factors
Modulating the TME is expected to improve response to immunotherapy for pancreatic cancer. Hypofractionation with 3x8 Gy was found to elicit the IFN-1 response that aims at immune effector recruitment, including cytotoxic T-cells. Successful T-cell activation depends on various factors, for which radiation-mediated effects could either be beneficial or deleterious in the context of anti-tumour efficacy. Thus we examined proteins with known immune effector interactions such as Gal-1, MHC-I and PD-L1. Gal-1 was implicated in the development and maintenance of an immunosuppressive TME by inducing helper- and cytotoxic T-cell apoptosis and anergy, promoting tolerogenic dendritic cells and expanding regulatory T-cells (33). Radiation-induced upregulation of Gal-1 was reported in human glioma cells (45) but no data is available for PDAC. We detected increased Gal-1 secretion at 24 h and 72 h after 3x8 Gy, but not after single dose irradiation (Figure 9A).
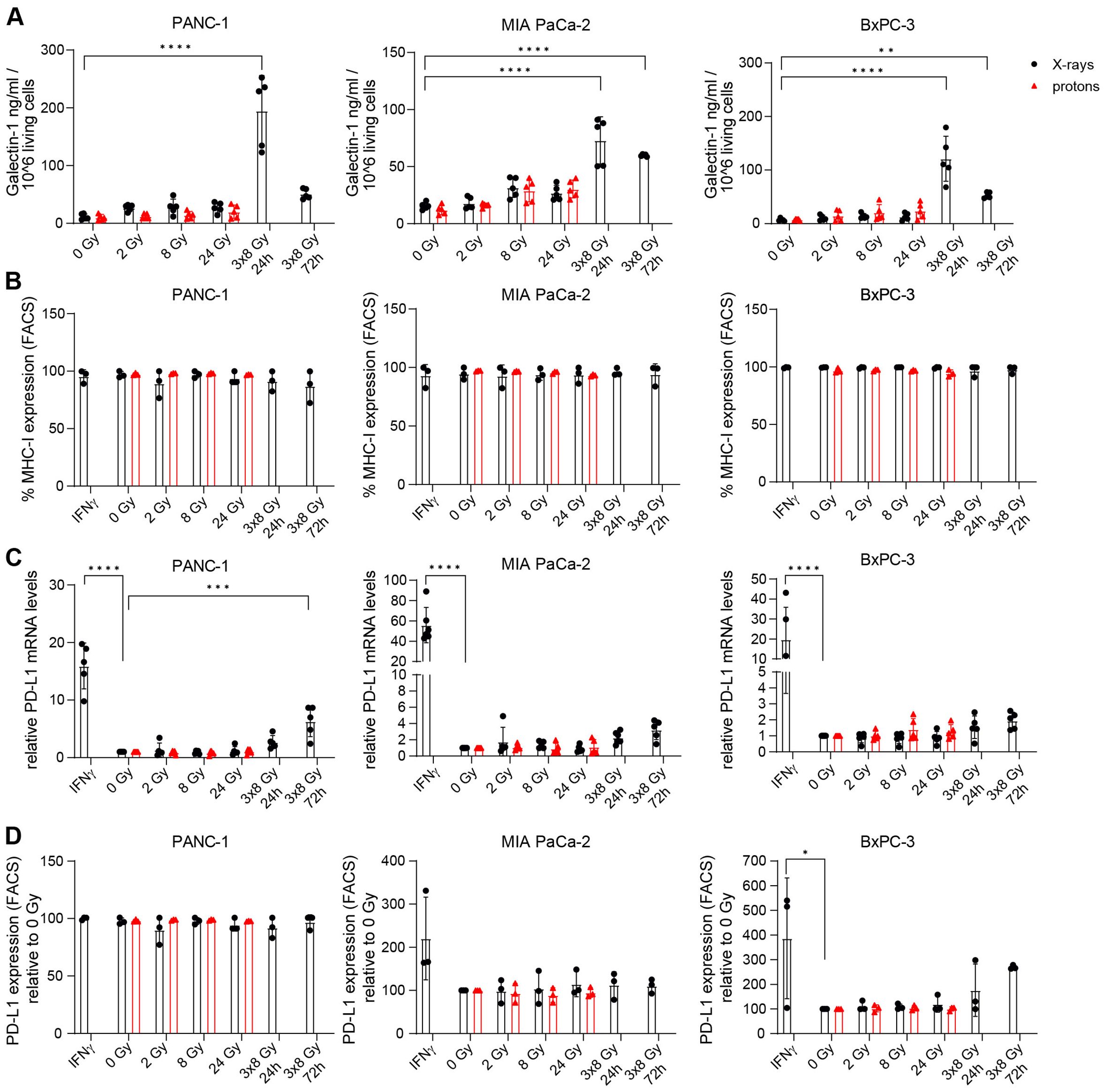
Figure 9. The effects of radiation on Galectin-1, MHC-I and PD-L1 in PDAC cells. (A) Galectin-1 was measured by ELISA in the supernatant of PDAC cells exposed to different doses of X-ray (black circles) or proton (red triangles) radiation 24 h post radiation and also 72 h after 3x8 Gy (N=5). (B) MHC-I expression was analysed by flow cytometry (N=3). (C,D) PD-L1 levels were measured by (C) RT-qPCR (N=5) and (D) FACS (N=3). Data points represent mean values of 3 to 5 independent experiments ± standard deviation. *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001.
Cancer immune recognition is contingent on tumour peptide antigen presentation via MHC-I. Limited MHC-I expression in PDAC patients contributes to immunosuppressive TME by hindering tumour antigen presentation (46). Radiation-mediated stimulation of MHC-I expression was observed in murine pancreatic cancer models (47). However, MHC-I expression was not affected by radiation in our study (Figure 9B).
PD-L1 is an immune checkpoint protein that supresses antigen-independent T-cell activation and is often upregulated in cancers to evade immune surveillance. PD-L1 is commonly found in PDAC patients but PD-L1 blockade monotherapy failed in clinical studies due to the lack of intratumoral T-cells and the presence of myeloid suppressor cell populations. Radiation-mediated upregulation of PD-L1 cell surface expression was described in murine PDAC cell lines (8, 48). For human cell lines, no data is available and thus we assessed the effect of radiation on PD-L1 expression. PD-L1 expression was induced after IFNγ treatment in all three cell lines and at 72 h after 3x8 Gy in PANC-1 (Figure 9C). IFNγ-mediated PD-L1 stimulation was confirmed on the protein level in BxPC-3 (Figure 9D). Radiation with either X-rays or protons did not affect PD-L1 protein levels in any of the three investigated PDAC cell lines (Figure 9D).
3 Discussion
The majority of pancreatic cancer patients did not benefit from recent advances in cancer immunotherapy due to immunosuppressive TME (4). Relapse affects up to 80% of pancreatic cancer patients within a few months after therapy (49). Remodelling the immunosuppressive TME by enhancing T-cell priming and preventing T-cell exhaustion may be the only road to success for this early metastasizing malignancy that has proven highly refractory to any treatment. Radiotherapy can shift immunologically unresponsive tumours into an immunologically more favourable phenotype by inducing an anti-tumour innate immune response through IFN-1 signalling (19, 38, 39). By converting tumours into an in situ vaccine, radiotherapy could activate an organism-wide tumour control mechanism and establish anti-tumour immune memory to prevent relapse. The likelihood of achieving immune-mediated disease control was shown to depend on optimal radiation dose/fractionation regimen (26) and may vary between malignancy types.
In this study, we assessed a variety of immunogenic radiation response parameters in three human PDAC cell lines. We used photon and proton single doses and a clinically relevant photon hypofractionation scheme to decipher whether (i) PDAC cells undergo immunogenic alterations in response to radiation, (ii) a specific radiation dose or fractionation regimen is more immunostimulatory, (iii) IFN-1 response is temporally modulated, and (iv) protons yield differential responses compared to photons at equal physical doses. We investigated the anti-tumour immunity conferred by the STING/MAVS/IFN-1 axis in a multifaceted approach and provide evidence for radiation-induced immunogenic cell death and immunogenicity in PDAC cells exposed to a hypofractionation radiation regimen of 3x8 Gy. We found that protons are slightly more effective compared to photons with RBE10 values around 1.1 for all three investigated cell lines, which supports the continued use of a uniform RBE assuming 10% increased effectiveness in the clinical setting.
We furthermore show that hypofractionated radiation treatment elicits accumulation of cytosolic dsDNA, phosphorylation of STING and STAT1, and induction of IFNB1 and ISG expression in a time-dependent manner. In line with previous reports (34, 41), radiation-induced IFN-1 response is delayed and most likely dependent on mitotic progression and accumulation of DNA damage. Mitochondrial DNA was reported as a significant contributing factor in mouse mammary carcinoma cells and may also play a role in the context of PDAC, based on the observed co-localization of cytosolic DNA with the mitochondrial transcription factor TFAM and assessment of cytosolic mitochondrial DNA via qPCR. Although micronuclei accumulate after irradiation, recent studies show that micronuclei do not activate cGAS/STING (50, 51).
The three investigated PDAC cell lines responded similarly to radiation with regard to survival and immunogenic cell death marker secretion but diverged in their ability to activate the IFN-1 axis (Figure 10). Despite a strong induction of pSTING and pIRF3, IFNB1 was not detectable in MIA PaCa-2, neither by RT-qPCR nor by ELISA. This may be due to a strong induction of non-canonical NF-κB signalling in irradiated MIA PaCa-2 based on RNA-seq analysis (NFKB2 in Figure 5H) and western blot analysis (p100/p52 in Figure 8A). Non-canonical NF-κB signalling was previously shown to negatively regulate IFNB1 expression (44) and may therefore account for the lack of IFNB1 induction in MIA PaCa-2.
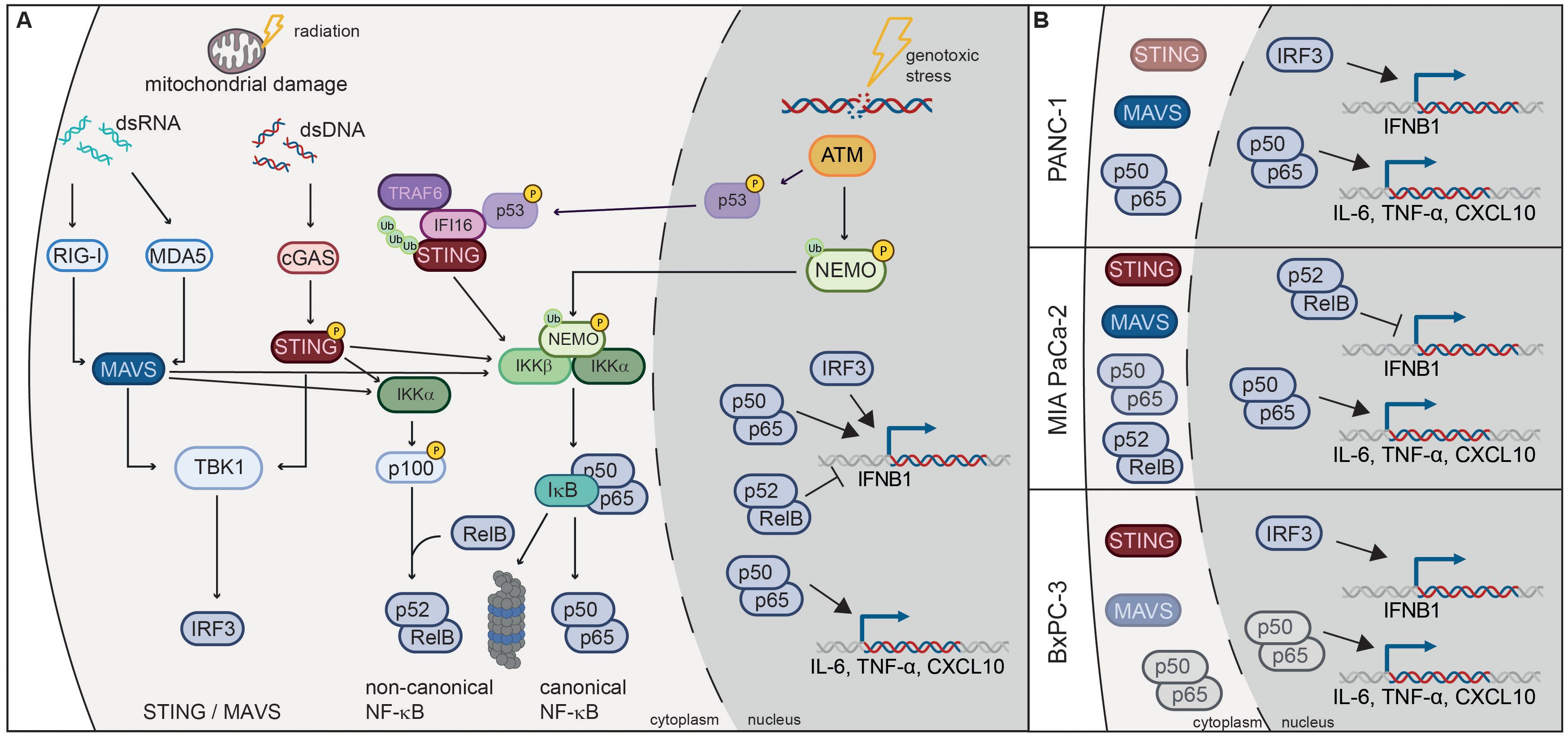
Figure 10. Radiation-induced immunogenic signalling in PDAC cell lines. (A) Damaged mitochondria release dsDNA and dsRNA, which activate cGAS/STING and RIG-I/MAVS signalling pathways respectively. Activation of these pathways results in the translocation of transcription factors IRF3 and NF-κB into the nucleus, where they induce the expression of IFNB1 and proinflammatory cytokines such as CXCL10, IL-6 and TNF-α. Activation of the non-canonical NF-κB pathway dampens the induction of IFNB1. Canonical NF-κB signalling can also be induced by the ATM kinase, which is activated by double-strand DNA breaks (DSBs) induced by genotoxic stress. ATM phosphorylates NEMO and promotes its monoubiquitination, which is required for translocation into the cytoplasm where it activates the IKK complex. ATM also induces K63-linked ubiquitination of STING through the p53-IFI16-TRAF6 complex, which activates canonical NF-κB. (B) STING, MAVS and NF-κB are responsible for radiation-induced expression of IFNB1 and proinflammatory cytokines in PDAC cell lines. MAVS is the dominant pathway in PANC-1, STING in BxPC-3, while non-canonical NF-κB is induced in MIA PaCa-2 and dampens IFNB1 expression. NF-κB signalling is more pronounced in PANC-1 and MIA PaCa-2, which harbour oncogenic KRAS mutations G12D and G12C respectively.
Of the three PDAC cell lines, PANC-1 showed the highest expression of RIG-I/MAVS and the NF-κB subunit RelA (p65), and mounted the strongest IFN-1 response to 3x8 Gy X-rays as evidenced by the induction of pSTAT1, IFNB1, CXCL10 and other ISGs (Figures 4, 5H). Conversely, STING and IRF3 levels were the highest in BxPC-3 (Figure 4A). Accordingly, MAVS KO and STING KO showed somewhat stronger reduction in IFNB1 and CXCL10 expression in PANC-1 and BxPC-3 cells respectively (Figures 6, 7), suggesting that RIG-I/MAVS is the dominant IFN-1 pathway in PANC-1, while cGAS/STING is more relevant for BxPC-3 cells (Figure 10B). Nevertheless, the expression of IFNB1 and CXCL10 was strongly reduced in both STING and MAVS KO PANC-1 and BxPC-3 cells, suggesting that cytosolic nucleic acid sensing through both STING and MAVS pathways is essential for the IFN-1 response. Our results also suggest that STING and MAVS act co-dependently as knockout of either of the two genes abrogates IFNB1 expression. Indeed, STING and MAVS were previously shown to interact and form a complex (52, 53). However, our results are at variance with published data showing no effect of STING depletion on pSTAT1 or IFNB1 expression in PANC-1 (21, 54).
The high basal activity of NF-κB p65/p50 in PANC-1 cells (Figure 4) may be due to the KRAS G12D oncogenic driver mutation, which was shown to induce the expression of IL-1 and thereby activate NF-κB signalling (55). BxPC-3 cells do not have a KRAS mutation, while MIA PaCa-2 harbour the oncogenic G12C mutation. In MIA PaCa-2 phosphorylation of RelA (p65) was induced after 3x8 Gy, but not in BxPC-3 (Figures 7, 8), reinforcing the link between oncogenic KRAS mutations and NF-κB signalling. Radiation-induced expression of IL-6 and TNF-α as NF-κB target cytokines was partly dependent on STING and MAVS in all three PDAC cell lines (Figure 10B), which is concordant with previous studies showing that STING and MAVS can activate NF-κB signalling (56–59). Apart from cytosolic nucleic acid sensing pathways, NF-κB can be activated after genotoxic stress by ATM signalling of double-strand DNA breaks (DSBs), resulting in NEMO phosphorylation in the nucleus, followed by its monoubiquitination and export into the cytoplasm where it activates the IKK complex (60). Alternatively, ATM and PARP1 were shown to induce the formation of the p53-IFI16-TRAF6 complex, which ubiquitinates STING and promotes STING-dependent NF-κB activation (61) (Figure 10A).
In addition to immunostimulatory effects, radiation-induced IFN-1 response may also upregulate T-cell inhibitory ligands such as PD-L1 and Gal-1. Gal-1 secretion was significantly upregulated following 3x8 Gy at 24 h post irradiation in all three cell lines and at 72 h post irradiation in MIA PaCa-2 and BxPC-3. However, we did not observe radiation-induced PD-L1 expression changes, neither in the two cell lines with no baseline PD-L1 expression (PANC-1 and MIA PaCa-2), nor in the cell line with baseline PD-L1 expression (BxPC-3). Similarly, MHC-I expression was not affected in any of the three investigated cell lines (Figure 7B), thus allowing unhindered antigen presentation to T-cells. Therefore, our results suggest that radiation as a means of stimulating anti-tumour immune activation may be considered for PDAC, and Gal-1 specific inhibitors may be of synergistic therapeutic benefit.
The activation of cGAS/STING signalling via STING agonists such as ADU-V16, ADU-S100 and DMXAA was shown to boost immunotherapy approaches in murine models of PDAC by stimulating the production of IFN-regulated chemokines, by promoting infiltration of cytotoxic T-cells and repolarization of M2 immunosuppressive into M1 pro-inflammatory macrophages, and by decreasing the levels of regulatory T-cells (Tregs) (62–64). STING agonists were also shown to enhance the efficacy of radiotherapy by enhancing anti-tumour immunity in PDAC. However, STING agonists act systemically and may induce adverse effects by promoting apoptosis of T-cells (65). Tumour cell-specific activation of cGAS/STING through accumulation of cytosolic dsDNA due to radiation-induced genomic instability of cancer cells may therefore prove advantageous compared to STING agonists. We furthermore demonstrate that protons have similar immunomodulatory effects compared to photons for single doses, supporting the use of particle therapy for PDAC treatment, based on the inverted depth-dose profile that would allow precise, tumour-conformal damage induction and STING axis activation with less risk for healthy surrounding organs.
Moreover, combinatorial treatments with DNA damage response (DDR) inhibitors may sensitize cancer cells to radiation treatment by activating the cGAS/STING and RIG-I/MAVS pathways. Indeed, a previous study showed that ATM inhibition can induce IFN signalling in PDAC cells (PANC-1 and Capan-1) in cGAS/STING-independent but TBK1-dependent manner, which is potentiated by radiation and sensitizes PDAC cells to ICB treatment (66). Further studies will reveal whether other DDR inhibitors can synergize with radiation to induce anti-tumour immunity in PDAC.
4 Methods and materials
4.1 Cell culture
The human pancreatic cancer cell lines PANC-1, MIA PaCa-2 and BxPc-3 were purchased from ATCC. PANC-1 and MIA PaCa-2 have KRAS mutations (G12D in PANC-1 and G12C in MIA PaCa-2) and are classified as mesenchymal, whereas BxPC-3 is classified as classical. PANC-1 and MIA PaCa-2 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) medium (Gibco, 41965-039), supplemented with 10 % fetal bovine serum (FBS) (Sigma, F7524-500 ml) and 1% penicillin/streptomycin (P/S) (Gibco, 15140-122). BxPC-3 were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, 21875-034), supplemented with 10 % FBS, 1% P/S and 1 % L-Glutamine (Gibco, 25030-024). All cell lines were maintained at 37°C in a humidified atmosphere with 95 % air and 5 % CO2. Cells were grown to 70-80 % of confluency at time of irradiation. Prior to irradiation, cell culture flasks were filled air-bubble free with unsupplemented medium to accommodate the vertical flask positioning in both irradiation workflows, which was replaced immediately after irradiation with fresh, supplemented medium.
4.2 Cell line generation
To generate CRISPR/Cas9 STING and MAVS KOs, gRNAs targeting exon 3 and 5 respectively were cloned between BbsI sites under the U6 promoter in plasmids encoding Cas9-EGFP or Cas9-Cherry (pX458). gRNA sequences were 5′-CACTGCAGAGATCTCAGCTG-3′ for STING and 5′-GTAGATACAACTGACCCTGT-3′ for MAVS. After 72 h of transfection with polyethylenimine (PEI; Polysciences), GFP- and mCherry-positive BxPc-3 or PANC-1 cells were FACS-sorted and allowed to recover in culture for several days. Cells were subsequently FACS-sorted and GFP- and mCherry-negative cells were seeded 1 cell/well in 96-wells plates. After 14 to 20 days, surviving clones were expanded in culture, genomic DNA was isolated and PCR-amplified Cas9 target region was sequenced. Positive clones were validated by western blotting.
4.3 RNA interference
Small interfering RNA (siRNA) oligonucleotides targeting STING as well as control siRNA were purchased from Dharmacon (ON-TARGETplus SMARTpool). 8 x 105 cells (all three cell lines) were seeded in T25 cell culture flasks one day before transfection. Cells were transfected with 150 pmol STING siRNA using Lipofectamine RNAiMax (Invitrogen, 13778-150) and Opti-Mem (Gibco, 31985-062) media. 24 h after transfection cells were irradiated with 8 Gy on three consecutive days (3x8 Gy) and harvested either 24 or 72 h after the last irradiation. Non-irradiated samples were used as a control (0 Gy). Samples were then used for western blotting and RT-qPCR.
4.4 Interferon gamma and inhibitor treatments
Recombinant human IFNγ protein (R&D Systems, 285-IF-100) was added at a concentration of 50 ng/µl to the cell cultures 24 h before cell harvest. The STING inhibitor (InvivoGen, H-151) and the NF-κB inhibitor (Cayman Chemical, BI-605906) were both used at a 10 µM concentration continuously 1 h before irradiation until 72 h after the last irradiation.
4.5 Irradiation procedures
X-ray irradiations were administered using a horizontal irradiation cabinet (YXLON, TU32-D03, 20 mA, 5.5FOC, filtration: 3 mm Be + 3 mm Al + 0.5 mm Cu), proton irradiations were all performed using the particle synchrotron at MedAustron. For both X-ray and proton irradiation, dedicated irradiation set-ups for all flask types used (Nunc™ Lab-Tek™ ChamberFlasks (170920), T25 (169900) and T75 (156800), all Thermo Fisher) were developed to accommodate for the horizontal beam geometry and ensure standardized positioning. Comprehensive dosimetric verification of all set-ups was performed prior to experiments (Supplementary Material and Methods, Supplementary Figure 7), according to recent recommendations for particle beam therapy (67, 68). Cells were irradiated with 0 Gy, 2 Gy, 8 Gy and 24 Gy of physical dose 200 kV X-rays or protons. Additionally, a fractionation regimen of 3x8 Gy was implemented for the X-ray experiments but not for protons due to limited beam time availability.
4.6 Clonogenic survival assay
Clonogenic survival assays were performed after 200 kV X-rays or proton irradiations. 2.5 x 105 cells were seeded in Nunc™ Lab-Tek™ ChamberFlasks (170920) one day prior to irradiation. Cells were harvested immediately after irradiation, diluted with supplemented medium appropriate for the cell line and seeded on 6-well plates in previously established, cell line specific concentrations. Following an incubation period of 12 days, cells were fixed with 96% methanol and stained with 0.5% crystal violet solution. Colonies of more than 50 cells were regarded as surviving clones. The linear-quadratic (LQ) formalism was used for survival curve fitting. Surviving fractions in relation to the plating efficiency of non-irradiated control samples were calculated for each value of the delivered physical dose in Gy. The mean and standard deviation result from a minimum of 4 independent experiments. Standard errors result from error propagation. A 1/σ-weighted minimum chi-square estimation was applied to the linear-quadratic model for survival curve fitting. Proton-relative biological effectiveness was calculated for the physical doses that reduced cell survival to 10% (RBE10).
4.7 Cytokinesis-block micronucleus assay
7 x 105 cells were seeded in Nunc™ Lab-Tek™ ChamberFlasks (170920) one day prior to irradiation. 3 µg/ml cytochalasin B (ThermoFisher, 228090010) was added 48 h after irradiation and after 24 h cells were fixed in 4% paraformaldehyde (Carl Roth), mounted with DAPI-containing mounting medium (VectaShield, H-2000) and evaluated using fluorescence microscopy. Binucleated cells were used for micronuclei quantification.
4.8 Western blotting
8 x 105 (PANC-1 and BxPC-3) and 1.2 x 106 cells (BxPC-3) were seeded in T25 flasks one day prior to irradiation. Cells were harvested and lysed with a lysis buffer containing NP40 buffer (Invitrogen, J60766-AP), 1% of protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78440), 1 mM PMSF (Thermo Fisher Scientific, 78431) in DMSO (Thermo Fisher Scientific, D12345) and 1X PhosStop (Merck, 4906837001). The protein concentration was determined with a protein assay kit (Pierce, 10678484). 30 µg of total protein for each sample were separated using SDS-PAGE gels. Proteins were transferred overnight onto PVDF membranes. The membranes were blocked in 5% non-fat milk (Roth, T145.1) for 1 h at room temperature and subsequently incubated with the primary antibody diluted 1:1000 in 5% non-fat milk over night at 4 °C. Washing steps were performed with TBST and membranes were probed with the horseradish peroxidase-conjugated secondary anti-rabbit antibody (7074S, Cell Signaling) for 2 h at room temperature. Membranes were incubated with ECL detection reagents and bands were visualized with ChemiDoc gel imager (Bio-Rad) and quantified using ImageLab software (Bio-Rad). All antibodies are listed in Supplementary Table 1.
4.9 Subcellular fractionation and whole-cell DNA extraction for mtDNA quantification
Non-irradiated (0 Gy) and 3x8 Gy irradiated cells harvested at 24 h and 72 h after the last irradiation were used for subcellular fractionation followed by isolation of the cytosolic fraction according to Sato et al, 2024 (50). 1 million cells were lysed in digitonin buffer (25 µg/ml digitonin, 150 mM NaCl, 50 mM Hepes, pH 7.4) for 10 min on ice. Lysed samples were centrifuged at 2000 g for 10 min (4 °C). The supernatants were transferred into new tubes and centrifuged at 2000 g for 20 min (4 °C). The two centrifugation steps were repeated three more times. The supernatant after the final centrifugation was used for qPCR-mediated detection of mitochondrial genes. This fractionation procedure does not allow separation of mitochondria from the rest of the cytoplasm. Whole-cell DNA was extracted from 1 million cells using the QIAmp DNA Mini kit (QIAGEN), following the manufacturer´s instructions. An intergenic region on chromosome 5 was analysed in addition to mitochondrial genes and is referred to as genomic DNA (gDNA). Primer sequences are listed in Supplementary Table 1.
4.10 Real-time quantitative PCR
RNA was extracted using RNeasy Plus extraction kit (Qiagen, 74136) according to the manufacturer’s instructions. Residual gDNA removal and subsequent cDNA synthesis was performed using iScript™ gDNA Clear cDNA Synthesis Kit (Bio-Rad, 1725034) according to the manufacturer’s instructions. IFNB1, CXCL10, IL-6 and TNF-α levels relative to TBP were quantified by multiplexing RT-PCR based on commercial primer sets (human IFNB1 UniqueAssayID: qHsaCEP0054112, human CXCL10 UniqueAssayID: qHsaCEP0053880, human IL-6 UniqueAssayID: qHsaCEP0051939, human TNF UniqueAssayID: qHsaCEP0040184, human TBP UniqueAssayID: qHsaCIP0036255). PCRs were run on a CFX Opus machine (Bio-Rad) operated by the embedded CFX Maestro software (Bio-Rad) using iQ™ Multiplex Powermix (Bio-Rad). RT-PCR data were analzyed using the ΔΔCt method with TBP as a housekeeping gene.
4.11 RNA isolation and RNA-seq library preparation
Cells were harvested and counted, 8 million PDAC cells were mixed with 2 million Drosophila S2 cells as a spike-in control. Cell pellets were resuspended in 1 ml TRI reagent (Sigma). 200 µL chloroform (Applichem) was added, samples were mixed and centrifuged at 4 °C maximum speed for 15 min. The upper phase was transferred to a new tube and subjected to isopropanol precipitation. 20 µg of RNA were treated with 40 U DNase I (Roche, 50-100-3290) at 37 °C for 30 min and purified by phenol-chloroform extraction and ethanol precipitation. rRNA was depleted and RNA-seq libraries were prepared with NEBNext rRNA depletion kit v2 (Human/Mouse/Rat) and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760S) according to the manufacturer’s instructions using 600 ng total RNA input. Sequencing was performed on an Illumina NovaSeq 6000 instrument in readmode SR100 by the Next Generation Sequencing facility at Vienna BioCenter Core Facilities (VBCF).
4.12 RNA-seq data analysis
RNA-seq data from HEK293 cells from different genetic background was processed using PiGx-RNA-seqc pipeline (69). In short, the data was quantified using the GRCh38/hg38, and the dm6 versions of the human and Drosophila spike-in transcriptome (downloaded from the ENSEMBL database (70) using SALMON) (71) with default parameters. For visualization purposes, the data was mapped to the GRCh38/hg38, and dm6 versions of the human, and Drosophila genomes using STAR, with the following parameters: –limitOutSJcollapsed 20000000 –limitIObufferSize=1500000000 –outFilterMultimapNmax 10 –seedPerWindowNmax 5. The quantified data was processed using tximport (72) and the differential expression analysis was done using DESeq2 (73). Genes with less than 5 reads in all biological replicates of one condition were filtered out before the differential analysis. The data was normalized by taking the ratio of reads mapping to the human and the Drosophila transcriptome. Genes were defined as differentially expressed if they had a minimum absolute log2 fold change of 1, and a BH adjusted p value less than 0.05. Genes from Hallmark gene sets from Molecular Signatures Database (MSigDB) (74) were tested for enrichment among differentially expressed genes using Gene set enrichment analysis (GSEA) algorithm (75) implemented in clusterProfiler R package (76). Gene sets with adjusted p-value < 0.05 were defined as enriched. RNA-seq data have been deposited in ArrayExpress under accession code: E-MTAB-13096.
4.13 ELISA
2.5 x 105 cells (all cell lines) were seeded in T25 flasks one day prior to irradiation. Galectin-1, HMGB1 and IFNB1 levels in cell culture supernatants were quantified with ELISA kits (Galectin-1: DGAL10, R&D Systems; HMGB1: ST51011, IBL International; IFNB1: 41415-1, PBL Assay Science) as per the manufacturer´s recommendations. Photometric assessment was performed on a Varioskan Flash Microplate Reader (Thermo Scientific) using SkanIt Software 2.4.5 (Thermo Scientific).
4.14 Immunofluorescence microscopy
For immunofluorescence analyses, 1 x 105 cells (all cell lines) were seeded in Nunc™ Lab-Tek™ ChamberFlasks one day prior to irradiation treatment. Immunofluorescence experiments were performed 24 h post irradiation. Cells were fixed in 4% paraformaldehyde (Carl Roth), permeabilized with 0.1 % Tween20 (Sigma Aldrich) and 0.01 % Triton X-100 (Sigma Aldrich) in PBS and blocked with 1% BSA (Sigma Aldrich). Primary antibodies targeting dsDNA (Abcam, ab27156, 1:1000 dilution), dsRNA (Cell Signaling, 28764, 1:1000 dilution) or TFAM (Genetex, GTX103231, 1:500 dilution) were applied over night at 4 °C, followed by Rhodamine (TRITC)-conjugated AffiniPure goat anti-mouse IgG (Jackson ImmunoResearch, 115-025-072, 1:400 dilution) or Alexa Fluor 488-conjugated goat anti-rabbit IgG (Abcam, ab150077, 1:400 dilution) for 1 h at room temperature. Cells were mounted with DAPI-containing mounting medium (VectaShield, H-2000) and evaluated using fluorescence microscopy. Images were acquired with an Axio Z2 fluorescence microscope (Zeiss) or the LSM980 inverse confocal microscope (Zeiss) equipped with a Plan-Apochromat 63x/1.4 Oil DIC (WD 0.13 mm), a 405 nm laser diode (30mW), a 488 nm laser diode (30mW) and a 561 nm DPSS laser (25mW). Appropriate laser power, excitation wavelength range and pinhole size were chosen for each channel in alignment with negative controls. Images were acquired with Zen software (version 3.3, Zeiss) and processed with Fiji (ImageJ). The threshold was set to 15000 (dsDNA) and 10000 (TFAM). Fluorescence and co-localization signals were analysed with the “Analyze Particles” option in Fiji. For co-localization the number of overlapping objects with specifications of 0.1-infinity/0.1-1.0 (TFAM) and 0.05-1/0.2-1.0 (dsDNA) size/circularity were counted. Micronuclei were assessed using DAPI nuclear counterstain in the same samples used for dsDNA evaluation.
4.15 Flow cytometry
1.5 x 106 cells (PANC-1 and BxPC-3) or 2 x 106 (BxPC-3) were seeded in T75 flasks one day prior to irradiation. Cells were harvested with 0.5 mM EDTA and then stained for surface markers using fluorochrome-conjugated antibodies against HLA-A,B,C-APC-Cy7 (Biolegend, 311426) or PD-L1-APC (Biolegend, 329708). As positive control antibody, CD59-FITC (BD Pharmingen, 555763) was used. 5 x 105 cells in 50 µl were incubated with optimal amounts of diluted antibodies (20 µl) on ice for 30 minutes. Subsequentially, cells were washed with PBS+0.5 % BSA+0.05 % NaN3 and 10 µl of 1:4000 diluted DAPI was added. Samples were acquired on Cytoflex S flow cytometer (Beckmann Coulter, Brea, CA) and results were analyzed using the FlowJo™ software (BD Biosciences). Live cells were distinguished according to their forward- and side scatter characteristics and as being DAPI negative. Positivity was confirmed by isotype control antibodies and fluorescence minus one controls. Treatment-induced autofluorescence was accounted for by correcting the mean fluorescence intensity (MFI) of antibody-stained cells for the mean fluorescence of unstained cells for each individual sample.
4.16 Statistical analysis
All experiments were performed in 3-5 independent biological replicates and statistical significance was determined using two-way analysis of variance (ANOVA) with multiple comparisons correction: *≤ 0.05; **≤ 0.01; ***≤ 0.005, ****≤0.0001.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ArrayExpress under accession code: E-MTAB-13096.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
SK-G: Conceptualization, Writing – original draft, Writing – review & editing, Investigation, Methodology. AKl: Writing – review & editing, Data Curation, Methodology, Investigation, Visualization. KP: Writing – review & editing, Data Curation, Investigation. AKn: Writing – review & editing, Data Curation, Investigation. KS: Investigation, Methodology, Writing – original draft. AR: Investigation, Methodology, Writing – original draft. JB: Investigation, Methodology, Writing – original draft. AD: Writing – review & editing, Data Curation, Investigation. L-MA: Investigation, Methodology, Writing – original draft. LW: Investigation, Methodology, Writing – original draft. AP: Investigation, Methodology, Writing – original draft. SB: Investigation, Methodology, Writing – original draft. BK: Investigation, Methodology, Writing – original draft. WP: Supervision, Writing – review & editing. AA: Supervision, Writing – review & editing. FH: Writing – review & editing, Methodology, Investigation. VF: Data curation, Investigation, Methodology, Writing – original draft. JW: Supervision, Writing – review & editing. DG: Supervision, Writing – review & editing. DS: Supervision, Writing – review & editing, Conceptualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was funded by FWF doc.funds.connect PAIR ‘Pre-clinical ion beam therapy’ (DFH 13) and supported by the Danube Allergy Research Cluster (DANUBE-ARC) grant (project no. 10) of the country of Lower Austria and the Medical University of Vienna (WP and BK).
Acknowledgments
We thank the VBCF Next Generation Sequencing facility; the Max Perutz Labs Biooptics and FACS facilities; MedAustron operations; Prof. Thomas Decker for helpful advice; Prof. Birgit Strobl for sharing antibodies; Prof. Sandra Demaria for comments on the manuscript. The ministry of Education, Science and Research is gratefully acknowledged for the financial support providing beam time at MedAustron.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1286942/full#supplementary-material
References
1. National Cancer Institue. Cancer Stat Facts: Pancreatic Cancer . Available online at: https://seer.cancer.gov/statfacts/html/pancreas.html. (Accessed April 04, 2022)
2. Kolbeinsson HM, Chandana S, Wright GP, Chung M. Pancreatic cancer: A review of current treatment and novel therapies. J Invest Surg. (2023) 36:2129884. doi: 10.1080/08941939.2022.2129884
3. Mukherji R, Debnath D, Hartley ML, Noel MS. The role of immunotherapy in pancreatic cancer. Curr Oncol. (2022) 29:6864–92. doi: 10.3390/curroncol29100541
4. Henriksen A, Dyhl-Polk A, Chen I, Nielsen D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat Rev. (2019) 78:17–30. doi: 10.1016/j.ctrv.2019.06.005
5. Karamitopoulou E. Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br J Cancer. (2019) 121:5–14. doi: 10.1038/s41416-019-0479-5
6. Bockorny B, Grossman JE, Hidalgo M. Facts and hopes in immunotherapy of pancreatic cancer. Clin Cancer Res. (2022) 28:4606–17. doi: 10.1158/1078-0432.CCR-21-3452
7. Galluzzi L, Aryankalayil MJ, Coleman CN, Formenti SC. Emerging evidence for adapting radiotherapy to immunotherapy. Nat Rev Clin Oncol. (2023) 20:543–57. doi: 10.1038/s41571-023-00782-x
8. Azad A, Yin Lim S, D’Costa Z, Jones K, Diana A, Sansom OJ, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. (2017) 9:167–80. doi: 10.15252/emmm.201606674
9. Yasmin-Karim S, Bruck PT, Moreau M, Kunjachan S, Chen GZ, Kumar R, et al. Radiation and local anti-CD40 generate an effective in situ vaccine in preclinical models of pancreatic cancer. Front Immunol. (2018) 9:2030. doi: 10.3389/fimmu.2018.02030
10. Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, et al. Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res. (2018) 78:4282–91. doi: 10.1158/0008-5472.CAN-17-3821
11. Stump CT, Roehle K, Manjarrez Orduno N, Dougan SK. Radiation combines with immune checkpoint blockade to enhance T cell priming in a murine model of poorly immunogenic pancreatic cancer. Open Biol. (2021) 11:210245. doi: 10.1098/rsob.210245
12. Fujiwara K, Saung MT, Jing H, Herbst B, Zarecki M, Muth S, et al. Interrogating the immune-modulating roles of radiation therapy for a rational combination with immune-checkpoint inhibitors in treating pancreatic cancer. J Immunother Cancer. (2020) 8. doi: 10.1136/jitc-2019-000351
13. Xie C, Duffy AG, Brar G, Fioravanti S, Mabry-Hrones D, Walker M, et al. Immune checkpoint blockade in combination with stereotactic body radiotherapy in patients with metastatic pancreatic ductal adenocarcinoma. Clin Cancer Res. (2020) 26:2318–26. doi: 10.1158/1078-0432.CCR-19-3624
14. Parikh AR, Szabolcs A, Allen JN, Clark JW, Wo JY, Raabe M, et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat Cancer. (2021) 2:1124–35. doi: 10.1038/s43018-021-00269-7
15. Chen IM, Donia M, Chamberlain CA, Jensen AWP, Draghi A, Theile S, et al. Phase 2 study of ipilimumab, nivolumab, and tocilizumab combined with stereotactic body radiotherapy in patients with refractory pancreatic cancer (TRIPLE-R). Eur J Cancer. (2023) 180:125–33. doi: 10.1016/j.ejca.2022.11.035
16. Wang J, Gai J, Zhang T, Niu N, Qi H, Thomas DL 2nd, et al. Neoadjuvant radioimmunotherapy in pancreatic cancer enhances effector T cell infiltration and shortens their distances to tumor cells. Sci Adv. (2024) 10:eadk1827. doi: 10.1126/sciadv.adk1827
17. Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. (2022) 23:487–500. doi: 10.1038/s41590-022-01132-2
18. Hopfner K-P, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
19. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
20. Du SS, Chen GW, Yang P, Chen YX, Hu Y, Zhao QQ, et al. Radiation Therapy Promotes Hepatocellular Carcinoma Immune Cloaking via PD-L1 Upregulation Induced by cGAS-STING Activation. Int J Radiat Oncol. Biol. Phys. (2022) 112:1243–55. doi: 10.1016/j.ijrobp.2021.12.162
21. Feng X, Tubbs A, Zhang C, Tang M, Sridharan S, Wang C, et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. (2020) 39:e104036. doi: 10.15252/embj.2019104036
22. Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget. (2016) 7:26496–515. doi: 10.18632/oncotarget.v7i18
23. Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M Jr., et al. Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection. J Virol. (2014) 88:1582–90. doi: 10.1128/JVI.02007-13
24. Zhang T, Ma C, Zhang Z, Zhang H, Hu H. NF-κB signaling in inflammation and cancer. MedComm. (2020). (2021) 2:618–53. doi: 10.1002/mco2.104
25. Brandmaier A, Formenti SC. The impact of radiation therapy on innate and adaptive tumor immunity. Semin Radiat Oncol. (2020) 30:139–44. doi: 10.1016/j.semradonc.2019.12.005
26. Mohseni G, Li J, Ariston Gabriel AN, Du L, Wang YS, Wang C. The function of cGAS-STING pathway in treatment of pancreatic cancer. Front Immunol. (2021) 12:781032. doi: 10.3389/fimmu.2021.781032
27. Kabashima A, Matsuo Y, Ito S, Akiyama Y, Ishii T, Shimada S, et al. cGAS-STING signaling encourages immune cell overcoming of fibroblast barricades in pancreatic cancer. Sci Rep. (2022) 12:10466. doi: 10.1038/s41598-022-14297-5
28. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
29. Sia J, Szmyd R, Hau E, Gee HE. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front Cell Dev Biol. (2020) 8:41. doi: 10.3389/fcell.2020.00041
30. Kim TH, Lee WJ, Woo SM, Oh ES, Youn SH, Jang HY, et al. Efficacy and feasibility of proton beam radiotherapy using the simultaneous integrated boost technique for locally advanced pancreatic cancer. Sci Rep. (2020) 10:21712. doi: 10.1038/s41598-020-78875-1
31. Helm A, Totis C, Durante M, Fournier C. Are charged particles a good match for combination with immunotherapy? Current knowledge and perspectives. Int Rev Cell Mol Biol. (2023) 376:1–36. doi: 10.1016/bs.ircmb.2023.01.001
32. Orozco CA, Martinez-Bosch N, Guerrero PE, Vinaixa J, Dalotto-Moreno T, Iglesias M, et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc Natl Acad Sci United. States America. (2018) 115:E3769–e78. doi: 10.1073/pnas.1722434115
33. Tang D, Gao J, Wang S, Yuan Z, Ye N, Chong Y, et al. Apoptosis and anergy of T cell induced by pancreatic stellate cells-derived galectin-1 in pancreatic cancer. Tumour. Biol. (2015) 36:5617–26. doi: 10.1007/s13277-015-3233-5
34. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. (2017) 548:466–70. doi: 10.1038/nature23470
35. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
36. Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. (2020) 21:1160–71. doi: 10.1038/s41590-020-0751-0
37. Fenech M, Morley AA. Measurement of micronuclei in lymphocytes. Mutat Res. (1985) 147:29–36. doi: 10.1016/0165-1161(85)90015-9
38. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. (2018) 24:1845–51. doi: 10.1038/s41591-018-0232-2
39. Chen J, Chen C, Zhan Y, Zhou L, Chen J, Cai Q, et al. Heterogeneity of IFN-mediated responses and tumor immunogenicity in patients with cervical cancer receiving concurrent chemoradiotherapy. Clin Cancer Res. (2021) 27:3990–4002. doi: 10.1158/1078-0432.CCR-20-4521
40. Vitale G, van Eijck CH, van Koetsveld Ing PM, Erdmann JI, Speel EJ, van der Wansem Ing K, et al. Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann Surg. (2007) 246:259–68. doi: 10.1097/01.sla.0000261460.07110.f2
41. Chen J, Harding SM, Natesan R, Tian L, Benci JL, Li W, et al. Cell cycle checkpoints cooperate to suppress DNA- and RNA-associated molecular pattern recognition and anti-tumor immune responses. Cell Rep. (2020) 32:108080. doi: 10.1016/j.celrep.2020.108080
42. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature. (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
43. Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, et al. Novel cross-talk within the IKK family controls innate immunity. Biochem J. (2011) 434:93–104. doi: 10.1042/BJ20101701
44. Jin J, Hu H, Li HS, Yu J, Xiao Y, Brittain GC, et al. Noncanonical NF-κB pathway controls the production of type I interferons in antiviral innate immunity. Immunity. (2014) 40:342–54. doi: 10.1016/j.immuni.2014.02.006
45. Strik HM, Schmidt K, Lingor P, Tönges L, Kugler W, Nitsche M, et al. Galectin-1 expression in human glioma cells: modulation by ionizing radiation and effects on tumor cell proliferation and migration. Oncol Rep. (2007) 18:483–8. doi: 10.3892/or
46. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
47. Zebertavage LK, Alice A, Crittenden MR, Gough MJ. Transcriptional upregulation of NLRC5 by radiation drives STING- and interferon-independent MHC-I expression on cancer cells and T cell cytotoxicity. Sci Rep. (2020) 10:7376. doi: 10.1038/s41598-020-64408-3
48. Hartmann L, Schröter P, Osen W, Baumann D, Offringa R, Moustafa M, et al. Photon versus carbon ion irradiation: immunomodulatory effects exerted on murine tumor cell lines. Sci Rep. (2020) 10:21517. doi: 10.1038/s41598-020-78577-8
49. Gbolahan OB, Tong Y, Sehdev A, O’Neil B, Shahda S. Overall survival of patients with recurrent pancreatic cancer treated with systemic therapy: a retrospective study. BMC Cancer. (2019) 19:468. doi: 10.1186/s12885-019-5630-4
50. Sato Y, Hayashi MT. Micronucleus is not a potent inducer of the cGAS/STING pathway. Life Sci Alliance. (2024) 7. doi: 10.26508/lsa.202302424
51. Takaki T, Millar R, Hiley CT, Boulton SJ. Micronuclei induced by radiation, replication stress, or chromosome segregation errors do not activate cGAS-STING. Mol Cell. (2024) 84(11):2203-13.e5. doi: 10.1016/j.molcel.2024.04.017
52. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
53. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
54. Zhang Q, Jiang L, Wang W, Huber AK, Valvo VM, Jungles KM, et al. Potentiating the radiation-induced type I interferon antitumoral immune response by ATM inhibition in pancreatic cancer. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.168824
55. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:105–20. doi: 10.1016/j.ccr.2011.12.006
56. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
57. Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKϵ Act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. (2020) 31:107492. doi: 10.1016/j.celrep.2020.03.056
58. Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci United. States America. (2021) 118. doi: 10.1073/pnas.2100225118
59. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
60. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Sci (New. York. NY). (2006) 311:1141–6. doi: 10.1126/science.1121513
61. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. (2018) 71:745–60.e5. doi: 10.1016/j.molcel.2018.07.034
62. Vonderhaar EP, Barnekow NS, McAllister D, McOlash L, Eid MA, Riese MJ, et al. STING activated tumor-intrinsic type I interferon signaling promotes CXCR3 dependent antitumor immunity in pancreatic cancer. Cell Mol Gastroenterol Hepatol. (2021) 12:41–58. doi: 10.1016/j.jcmgh.2021.01.018
63. Jing W, McAllister D, Vonderhaar EP, Palen K, Riese MJ, Gershan J, et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer. (2019) 7:115. doi: 10.1186/s40425-019-0573-5
64. Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.122857
65. Baird JR, Friedman D, Cottam B, Dubensky TW Jr., Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. (2016) 76:50–61. doi: 10.1158/0008-5472.CAN-14-3619
66. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. (2019) 79:3940–51. doi: 10.1158/0008-5472.CAN-19-0761
67. Durante M, Paganetti H, Pompos A, Kry SF, Wu X, Grosshans DR. Report of a National Cancer Institute special panel: Characterization of the physical parameters of particle beams for biological research. Med Phys. (2019) 46:e37–52. doi: 10.1002/mp.13324
68. Magrin G, Palmans H, Stock M, Georg D. State-of-the-art and potential of experimental microdosimetry in ion-beam therapy. Radiother. Oncol. (2023) 182:109586. doi: 10.1016/j.radonc.2023.109586
69. Wurmus R, Uyar B, Osberg B, Franke V, Gosdschan A, Wreczycka K, et al. PiGx: reproducible genomics analysis pipelines with GNU Guix. GigaScience. (2018) 7:1-14. doi: 10.1093/gigascience/giy123
70. Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. (2018) 46:D754–d61. doi: 10.1093/nar/gkx1098
71. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. (2017) 14:417–9. doi: 10.1038/nmeth.4197
72. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. (2015) 4:1521. doi: 10.12688/f1000research
73. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
74. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. (2015) 1:417–25. doi: 10.1016/j.cels.2015.12.004
75. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci United States America. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
Keywords: interferon, radiation, protons, pancreatic cancer, STING, MAVS, NF-κB
Citation: Kerschbaum-Gruber S, Kleinwächter A, Popova K, Kneringer A, Appel L-M, Stasny K, Röhrer A, Dias AB, Benedum J, Walch L, Postl A, Barna S, Kratzer B, Pickl WF, Akalin A, Horvat F, Franke V, Widder J, Georg D and Slade D (2024) Cytosolic nucleic acid sensors and interferon beta-1 activation drive radiation-induced anti-tumour immune effects in human pancreatic cancer cells. Front. Immunol. 15:1286942. doi: 10.3389/fimmu.2024.1286942
Received: 31 August 2023; Accepted: 05 August 2024;
Published: 20 September 2024.
Edited by:
Wei Wang, Jiangsu Institute of Parasitic Diseases (JIPD), ChinaReviewed by:
Claudia Fournier, Helmholtz Association of German Research Centres (HZ), GermanyChristian Zierhut, Institute of Cancer Research (ICR), United Kingdom
Copyright © 2024 Kerschbaum-Gruber, Kleinwächter, Popova, Kneringer, Appel, Stasny, Röhrer, Dias, Benedum, Walch, Postl, Barna, Kratzer, Pickl, Akalin, Horvat, Franke, Widder, Georg and Slade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia Kerschbaum-Gruber, c3lsdmlhLmtlcnNjaGJhdW0tZ3J1YmVyQG1lZHVuaXdpZW4uYWMuYXQ=; Dea Slade, ZGVhLnNsYWRlQG1lZHVuaXdpZW4uYWMuYXQ=