 Heike R. Döppler
Heike R. Döppler Peter Storz
Peter Storz- Department of Cancer Biology, Mayo Clinic, Jacksonville, FL, United States
Pancreatic inflammation is a risk factor for the development of pancreatic cancer. Increased presence of inflammatory macrophages can be found in response to a KRAS mutation in acinar cells or in response to experimentally-induced pancreatitis. Inflammatory macrophages induce pancreatic acinar cells to undergo dedifferentiation to a duct-like progenitor stage, a process called acinar-to-ductal metaplasia (ADM). Occurrence of ADM lesions are believed to be the initiating event in tumorigenesis. Here we will discuss how macrophage-induced oxidative stress contributes to ADM and how ADM cells shape the fibrotic stroma needed for further progression.
Introduction
Chronic pancreatic inflammation (chronic pancreatitis) is a risk factor for pancreatic cancer (1, 2). Macrophages that rapidly increase in numbers after inflammation or in response to an oncogenic KRAS mutation may either originate from tissue resident populations (3, 4) or from external sources such as the peritoneum or blood monocytes (5). Presence of these inflammatory cells can initiate various processes that contribute to lesion formation and progression. For example, in response to macrophage infiltration pancreatic acinar cells can undergo acinar-to-ductal metaplasia (ADM), the dedifferentiation to a progenitor stage with duct-like features (6). ADM lesions are believed to be the initiating lesions for pancreatic intraepithelial neoplasia 1 (PanIN1) (7–10). PanIN1 are precancerous low-grade lesions that form in presence of a KRAS mutation, which occurs in 90-95% of all cases of pancreatic ductal adenocarcinoma (11). In absence of a KRAS mutation, ADM is a reversible process and may contribute to pancreas regeneration after the inflammation resolves (12–14). ADM is triggered by oxidative stress that is generated in acinar cells by macrophage-secreted factors (10, 15–17). ADM cells once formed then crosstalk with different macrophage populations to further drive generation of fibrotic stroma in the lesion microenvironment (18, 19). In the following we will highlight the role of ROS in driving ADM and in progression of ADM lesions with KRAS mutations to precancerous PanIN lesions, but also the contribution of this to the formation of the fibrotic stroma, thus setting the foundation for tumor development.
Mutant KRAS as an inducer of macrophage attraction
Pro-inflammatory macrophages are the major immune cell population driving the formation of ADM lesions (10, 16, 17), and their crucial role in this process was demonstrated in vivo (in mice) by genetic ablation and by chemical depletion (10, 20). Using the KC (p48Cre;LSL-KrasG12D) mouse model it was shown that during the development of pancreatic cancer these macrophages accumulate rapidly in ADM regions (21). In KC mice acinar cells with an oncogenic KRAS mutation can upregulate the expression of factors that function as chemoattractants for macrophages or monocytes (21–23). For example, KRAS induces expression of the cell surface glycoprotein Intercellular Adhesion Molecule-1 (ICAM-1, CD54) in acinar cells, and ICAM-1 in its soluble form (sICAM-1) acts as a chemoattractant for inflammatory macrophages (21) (Figure 1). Since this leads to focal inflammation, it seems plausible that the initiating macrophage population is recruited locally from a tissue resident population (4).
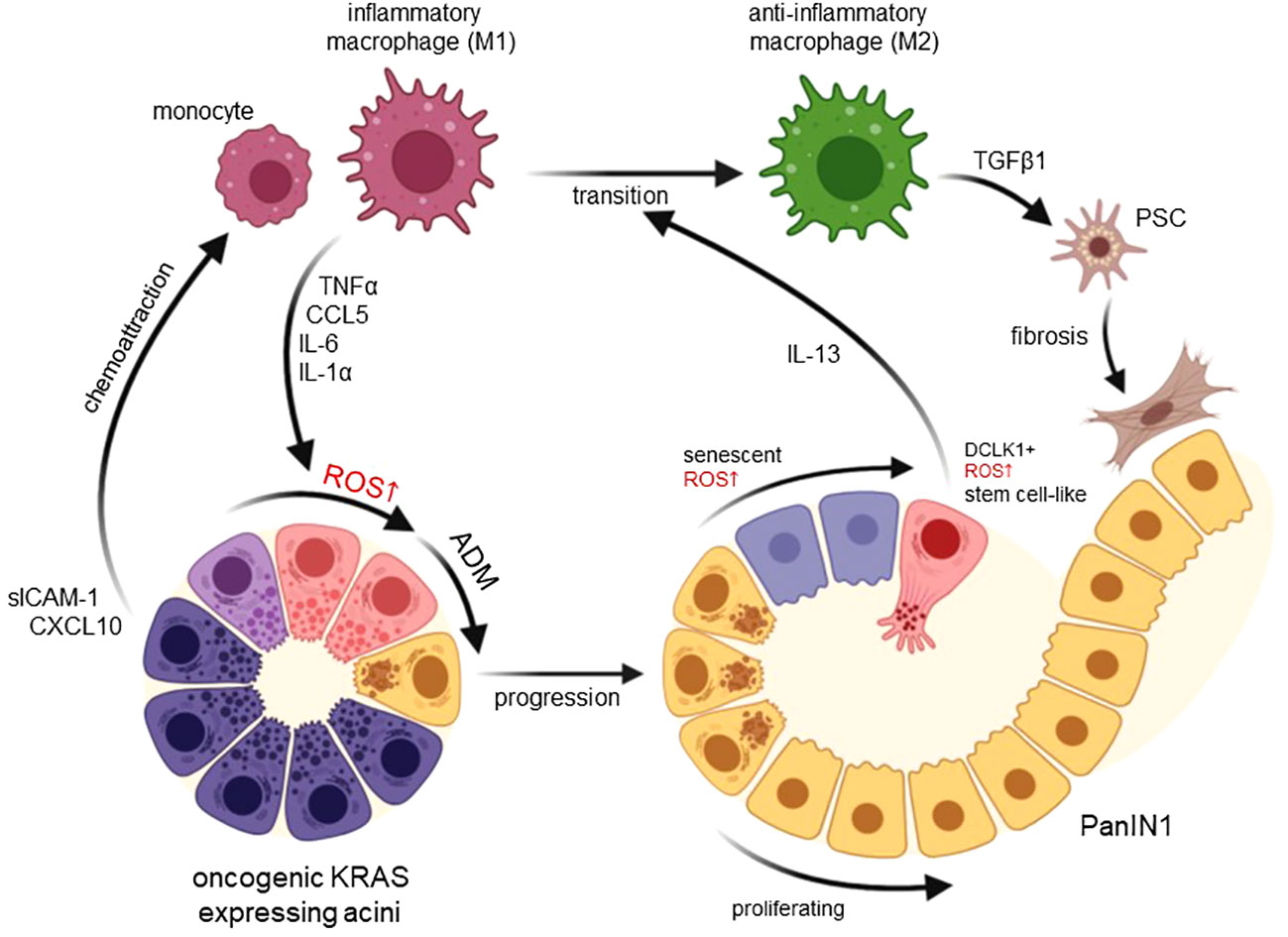
Figure 1 Functions of macrophage-induced ROS in developing pancreatic cancer. Expression of an oncogenic KRAS in pancreatic acinar cells upregulates chemoattractants for monocytes and inflammatory (M1) macrophages including CXCL10 and sICAM-1. M1-secreted factors such as TNFα, CCL5, IL-6 and IL-1α then increase ROS in acinar cells to drive their transdifferentiation to a duct-like phenotype. This process termed acinar-to-ductal metaplasia (ADM) in mice has been shown to be an inducing event for PDA formation when ADM lesions further progress to PanIN lesions. Increasing ROS in PanIN lesion cells can lead to senescence or increased occurrence of DCLK1+ cancer stem cells that produce IL-13. lL-13 mediates a phenotype switch from inflammatory to anti-inflammatory (M2) macrophages in the lesion microenvironment. M2 once abundant can activate pancreatic stellate cells (PSC) to increase fibrosis but also mediate proliferation of lesion cells. Created with BioRender.com.
However, the rapid increase in population density suggests that additional macrophages may be recruited from external sources (5). These could include macrophages from the peritoneum or circulating blood monocytes. Factors to attract both have been demonstrated to be produced in the ADM lesion microenvironment. For example, C-X-C motif chemokine ligand 10 (CXCL10) is produced by ADM lesions in mice and mediates the chemoattraction of inflammatory macrophages to the pancreas, but also enhances their proliferation and maintains their inflammatory identity (22). Other chemoattractants for macrophage are macrophage inflammatory protein-1 (MIP-1) and macrophage inflammatory protein-2 (MIP-2), produced by isolated PanIN lesion cells (22). Further, activated pancreatic stellate cells produce CXCL12 (24), which can act as a chemoattractant for a variety of immune cells, including macrophages. Moreover, the presence of C-C motif chemokine ligand 2 (CCL2), produced by a multitude of cells in the precancerous environment, attracts bone marrow-derived monocytes (23). Once attracted to the ADM/PanIN lesion regions, several cells in their microenvironment produce macrophage proliferation factors. These include M-CSF, produced by activated pancreatic stellate cells (24) and by lesion cells (22).
Macrophage secreted factors and oxidative stress as drivers of ADM
Pancreatic macrophages can induce acinar-to-ductal metaplasia (10, 17), and ADM lesions that originate from wildtype acinar cells are believed to revert to acini to regenerate the pancreas after the inflammatory stimulus resolves (12, 13, 20). However, in presence of an oncogenic KRAS mutation, ADM lesions progress to low-grade lesions (pancreatic intraepithelial neoplasia, PanIN), which are precursor lesions for PDA (6, 7, 25). The two extreme polarization phenotypes that have been described in vitro are pro-inflammatory M1-polarized, classically-activated macrophages (M1, IM) and anti-inflammatory M2-polarized, alternatively-activated macrophages (M2, AAM). In vivo, in the inflamed pancreas there is more of a continuous spectrum, however, the majority of macrophages show markers of these two groups, and for simplification we will adhere to these terms (M1 and M2).
Several factors secreted by M1 macrophages have been shown to induce ADM in explant culture. These include TNFα, CCL5 (RANTES), IL-6, and IL-1α (10, 15, 17) (Figure 1). Moreover, M2 macrophages can drive ADM through CCL2 (17). All these factors have in common that they induce hydrogen peroxide in acinar cells, which was shown to be the major driver of ADM (16, 17). Hydrogen peroxide most likely originates at the mitochondria, since mitochondrially-targeted catalase or the antioxidant MitoQ can block the initiation of ADM (16).
While several signaling pathways were implicated in driving ADM (reviewed in (6)), a critical event seems to be the upregulation of expression of epidermal growth factor receptor (EGFR) and its ligands TGFα and EGF via ROS-initiated NF-κB signaling in acinar cells (26). Such auto- or paracrine EGFR signaling then is the key event driving both the ADM process and ADM lesion growth (27, 28). Here it also should be noted that macrophages produce other ligands for EGFR including heparin-binding epidermal growth factor-like growth factor (HB-EGF) and amphiregulin (AR), which both may potentiate these effects (29–32).
Although it is still unclear how above macrophage-secreted factors induce ROS at the mitochondria, studies with mitochondrially-targeted antioxidants link ROS responsive signaling cascades and transcriptional activation to ADM. One of the main inducers of ADM downstream of mitochondrial ROS is the PKD1/NF-κB signaling cascade (16, 26, 33). Other transcription factors that have been demonstrated to be predominant drivers of ADM are Notch, Signal Transducer and Activator of Transcription 3 (STAT3), Kruppel-like factor 4 (KLF4) and Nuclear Factor of Activated T Cells 1/4 (NFAT1/4) (34–38). Some of these factors can be activated by macrophage-secreted inducers of oxidative stress. For example, STAT3 activity is upregulated by IL-6 (39) and regulates ADM (35). Others, such as Notch are activated downstream of oncogenic KRAS and the ROS-responsive kinase PKD1 (26, 34) and cooperates with NF-κB to induce ADM (40). However, it should be noted that for most of them a role of direct activation by ROS to drive the ADM processes was formally demonstrated.
Effects of ROS in low grade lesion cells and role in further lesion progression
Established pancreatic low grade lesion cells (PanIN1) show relatively high levels of oxidative stress as indirectly measured by an increase in 4-hydroxynonenal (4-HNE), a α,β-unsaturated hydroxyalkenal that is produced by lipid peroxidation (41). This increase in oxidative stress correlates well with markers for cellular senescence (42). ROS as a driver of oncogene-induced senescence is established and was implicated in pancreatic lesion cells (42, 43). However, it was also shown that progressing lesion cells increasingly express nuclear factor erythroid 2-related factor 2 (NRF2) (26, 44, 45). NRF2 is a stress-responsive transcription factor that regulates a multitude of genes mediating the antioxidant response and metabolic changes (44, 46, 47). Upregulation of this factor is an important mechanism to overcome ROS-induced damage and senescence in lesion cells and to mediate further progression (Figure 1).
A small percentage of lesion cells, however, show exuberant high levels of ROS (48). These cells are positive for DCLK1 and show defective EGFR signaling due to ROS-mediated blockage of vesicle transport (48). DCLK1+ cells express stemness markers such as CD133 and OCT4 (48). Therefore, they have been discussed as stem cells for pancreatic cancer (49) or progenitor cells that promote tumorigenesis (50). Indeed, DCLK1+ cells when isolated and reintroduced in mice, form pancreatic tumors at a faster rate than other lesion cells (49). This may be supported by their secretion of factors that alter macrophage polarity and contribute to generation of the fibrotic microenvironment.
Alternatively activated macrophages and roles in lesion progression
In presence of an oncogenic KRAS mutation, cells that underwent ADM further progress to PanIN lesion. ADM and PanIN lesion cells can produce IL-13, which induces a polarization switch in inflammatory M1 macrophages to an anti-inflammatory M2 phenotype (19). Major producers of IL-13 are DCLK1+ lesion cells (19). Resulting M2 macrophages, best characterized in mice by expressing arginase (Arg1), YM1 (Chil3), Fizz1, CD206 and Trem2 as markers (18, 24), have multiple functions in the progression of lesions. They secrete factors such as TGFβ1 to activate pancreatic stellate cells (PSC) and establish the lesion microenvironment (24), and their ablation either chemically or via neutralization of IL-13 in KC mice leads to a drastic decrease in fibrosis (18, 19). In addition, they secret factors such as TIMP1, IL-4, IL1-ra and CCL2 that act on lesion cells to stimulate their growth via activation of ERK1/2 signaling (17–19, 24). By secretion of CCL2, which generates ROS in acinar cells (17), M2 macrophages also induce ADM in neighboring acinar cells and thus increase abnormal areas in the pancreata of mice. During further progression to PDA, M2 macrophages regulate additional hallmarks of immune escape such as the exclusion of cytotoxic T lymphocytes and fibrosis (51). They also crosstalk with dendritic cells and myeloid derived suppressor cells (MDSC), which inhibit T cell proliferation and induce of T cell death, to further enhance the immune suppressive environment (52, 53). Therefore, targeting this immunosuppressive macrophage population, or initiating their repolarization to an inflammatory phenotype, both are valid strategies to explore for prevention of PDA or to sensitize pancreatic tumors to T cell immunotherapy (19, 51, 54).
Conclusions
The crosstalk of pancreatic acinar cells with cells of the innate immune system are initiating events in the development of pancreatic cancer (summarized in Figure 1). Specifically, the presence of inflammatory macrophages at acinar cells is tightly linked to intracellular ROS generation and is prerequisite to ADM. Dependent of their resistance to ROS, ADM cells can further progress to PanIN lesions, in which they may proliferate or show a senescent phenotype, or to DCLK1+ cells that can withstand high levels of intracellular ROS. At PanIN lesions and their surrounding microenvironment macrophages are mainly anti-inflammatory and drive lesion growth and fibrosis (10, 18, 24). But they also produce factors that induce ROS in neighboring acinar cells. Therefore, it is fair to say that macrophage-caused oxidative stress is a key driver of events that occur during initiation of pancreatic cancer. However, while a role for ROS in initiation of lesion formation is well established, the roles of ROS in further lesion progression to a more aggressive phenotype is less defined, and it is not fully clear if, and how they contribute to further development of PDA.
Author contributions
PS: Visualization, Writing – review & editing. HD: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the NIH grants CA229560 and CA244167 to PS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. All authors have no conflict of interest. We also thank the Champions For Hope (Funk-Zitiello Foundation) and the Gary Chartrand Foundation for supporting our studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bansal P, Sonnenberg A. Pancreatitis is a risk factor for pancreatic cancer. Gastroenterology. (1995) 109:247–51. doi: 10.1016/0016-5085(95)90291-0
2. Gandhi S, de la Fuente J, Murad MH, Majumder S. Chronic pancreatitis is a risk factor for pancreatic cancer, and incidence increases with duration of disease: A systematic review and meta-analysis. Clin Transl Gastroenterol. (2022) 13:e00463. doi: 10.14309/ctg.0000000000000463
3. Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med. (2015) 212:1497–512. doi: 10.1084/jem.20150496
4. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38 e6. doi: 10.1016/j.immuni.2017.07.014
5. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
6. Storz P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol. (2017) 14:296–304. doi: 10.1038/nrgastro.2017.12
7. Storz P, Crawford HC. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology. (2020) 158:2072–81. doi: 10.1053/j.gastro.2020.02.059
8. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris J, et al. Identification of sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 22:737–50. doi: 10.1016/j.ccr.2012.10.025
9. Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. (2011) 19:728–39. doi: 10.1016/j.ccr.2011.05.011
10. Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through nf-kappab and mmps. J Cell Biol. (2013) 202:563–77. doi: 10.1083/jcb.201301001
11. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
12. Criscimanna A, Coudriet GM, Gittes GK, Piganelli JD, Esni F. Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and beta-cell regeneration in mice. Gastroenterology. (2014) 147:1106–18 e11. doi: 10.1053/j.gastro.2014.08.008
13. Stanger BZ, Hebrok M. Control of cell identity in pancreas development and regeneration. Gastroenterology. (2013) 144:1170–9. doi: 10.1053/j.gastro.2013.01.074
14. Wu J, Zhang L, Shi J, He R, Yang W, Habtezion A, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine. (2020) 58:102920. doi: 10.1016/j.ebiom.2020.102920
15. Deschenes-Simard X, Mizukami Y, Bardeesy N. Macrophages in pancreatic cancer: starting things off on the wrong track. J Cell Biol. (2013) 202:403–5. doi: 10.1083/jcb.201307066
16. Doppler HR, Liou GY, Storz P. Generation of hydrogen peroxide and downstream protein kinase D1 signaling is a common feature of inducers of pancreatic acinar-to-ductal metaplasia. Antioxid (Basel). (2022) 137:1–15. doi: 10.3390/antiox11010137
17. Liou GY, Fleming Martinez AK, Doppler HR, Bastea LI, Storz P. Inflammatory and alternatively activated macrophages independently induce metaplasia but cooperatively drive pancreatic precancerous lesion growth. iScience. (2023) 26:106820. doi: 10.1016/j.isci.2023.106820
18. Bastea LI, Liou GY, Pandey V, Fleming AK, von Roemeling CA, Doeppler H, et al. Pomalidomide alters pancreatic macrophage populations to generate an immune-responsive environment at precancerous and cancerous lesions. Cancer Res. (2019) 79:1535–48. doi: 10.1158/0008-5472.CAN-18-1153
19. Liou GY, Bastea L, Fleming A, Doppler H, Edenfield BH, Dawson DW, et al. The presence of interleukin-13 at pancreatic adm/panin lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep. (2017) 19:1322–33. doi: 10.1016/j.celrep.2017.04.052
20. Folias AE, Penaranda C, Su AL, Bluestone JA, Hebrok M. Aberrant innate immune activation following tissue injury impairs pancreatic regeneration. PloS One. (2014) 9:e102125. doi: 10.1371/journal.pone.0102125
21. Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, et al. Mutant kras-induced expression of icam-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. (2015) 5:52–63. doi: 10.1158/2159-8290.CD-14-0474
22. Pandey V, Fleming-Martinez A, Bastea L, Doeppler HR, Eisenhauer J, Le T, et al. Cxcl10/cxcr3 signaling contributes to an inflammatory microenvironment and its blockade enhances progression of murine pancreatic precancerous lesions. Elife. (2021) 10:1–24. doi: 10.7554/eLife.60646
23. Teper Y, Eibl G. Pancreatic macrophages: critical players in obesity-promoted pancreatic cancer. Cancers (Basel). (2020) 1946:1–16. doi: 10.3390/cancers12071946
24. Fleming Martinez AK, Doppler HR, Bastea LI, Edenfield BH, Liou GY, Storz P. Ym1(+) macrophages orchestrate fibrosis, lesion growth, and progression during development of murine pancreatic cancer. iScience. (2022) 25:104327. doi: 10.1016/j.isci.2022.104327
25. Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. (2006) 66:95–106. doi: 10.1158/0008-5472.CAN-05-2168
26. Liou GY, Doppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, et al. Mutant kras-induced mitochondrial oxidative stress in acinar cells upregulates egfr signaling to drive formation of pancreatic precancerous lesions. Cell Rep. (2016) 14:2325–36. doi: 10.1016/j.celrep.2016.02.029
27. Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. Egf receptor is required for kras-induced pancreatic tumorigenesis. Cancer Cell. (2012) 22:304–17. doi: 10.1016/j.ccr.2012.07.024
28. Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. Egf receptor signaling is essential for K-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 22:318–30. doi: 10.1016/j.ccr.2012.08.001
29. Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ Jr., et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. (2005) 132:3767–76. doi: 10.1242/dev.01925
30. Means AL, Ray KC, Singh AB, Washington MK, Whitehead RH, Harris RC Jr., et al. Overexpression of heparin-binding egf-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology. (2003) 124:1020–36. doi: 10.1053/gast.2003.50150
31. Song SY, Gannon M, Washington MK, Scoggins CR, Meszoely IM, Goldenring JR, et al. Expansion of pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology. (1999) 117:1416–26. doi: 10.1016/s0016-5085(99)70292-1
32. Wagner M, Weber CK, Bressau F, Greten FR, Stagge V, Ebert M, et al. Transgenic overexpression of amphiregulin induces a mitogenic response selectively in pancreatic duct cells. Gastroenterology. (2002) 122:1898–912. doi: 10.1053/gast.2002.33594
33. Storz P, Toker A. Protein kinase D mediates a stress-induced nf-kappab activation and survival pathway. EMBO J. (2003) 22:109–20. doi: 10.1093/emboj/cdg009
34. Liou GY, Doppler H, Braun UB, Panayiotou R, Scotti Buzhardt M, Radisky DC, et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat Commun. (2015) 6:6200. doi: 10.1038/ncomms7200
35. Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka TA, Yamamoto K, Kato K, et al. Persistent expression of pdx-1 in the pancreas causes acinar-to-ductal metaplasia through stat3 activation. Genes Dev. (2006) 20:1435–40. doi: 10.1101/gad.1412806
36. Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, et al. Nfatc1 links egfr signaling to induction of sox9 transcription and acinar-ductal transdifferentiation in the pancreas. Gastroenterology. (2015) 148:1024–34 e9. doi: 10.1053/j.gastro.2015.01.033
37. Hessmann E, Zhang JS, Chen NM, Hasselluhn M, Liou GY, Storz P, et al. Nfatc4 regulates sox9 gene expression in acinar cell plasticity and pancreatic cancer initiation. Stem Cells Int. (2016) 2016:5272498. doi: 10.1155/2016/5272498
38. Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z, et al. Klf4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell. (2016) 29:324–38. doi: 10.1016/j.ccell.2016.02.005
39. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. Stat3 plays a critical role in kras-induced pancreatic tumorigenesis. Cancer Res. (2011) 71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908
40. Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, et al. Crosstalk between the canonical nf-kappab and notch signaling pathways inhibits ppargamma expression and promotes pancreatic cancer progression in mice. J Clin Invest. (2011) 121:4685–99. doi: 10.1172/JCI45797
41. Liou GY, Storz P. Detecting reactive oxygen species by immunohistochemistry. Methods Mol Biol. (2015) 1292:97–104. doi: 10.1007/978-1-4939-2522-3_7
42. Wang P, Han L, Shen H, Wang P, Lv C, Zhao G, et al. Protein kinase D1 is essential for ras-induced senescence and tumor suppression by regulating senescence-associated inflammation. Proc Natl Acad Sci USA. (2014) 111:7683–8. doi: 10.1073/pnas.1310972111
43. Su Y, Wang P, Shen H, Sun Z, Xu C, Li G, et al. The protein kinase D1-mediated classical protein secretory pathway regulates the ras oncogene-induced senescence response. J Cell Sci. (2018) 131:1–14. doi: 10.1242/jcs.207217
44. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced nrf2 transcription promotes ros detoxification and tumorigenesis. Nature. (2011) 475:106–9. doi: 10.1038/nature10189
45. DeNicola GM, Tuveson DA. Ras in cellular transformation and senescence. Eur J Cancer. (2009) 45 Suppl 1:211–6. doi: 10.1016/S0959-8049(09)70036-X
46. Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. Nrf2 promotes tumor maintenance by modulating mrna translation in pancreatic cancer. Cell. (2016) 166:963–76. doi: 10.1016/j.cell.2016.06.056
47. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. Nrf2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. (2015) 47:1475–81. doi: 10.1038/ng.3421
48. Fleming Martinez AK, Doppler HR, Bastea LI, Edenfield B, Patel T, Leitges M, et al. Dysfunctional egfr and oxidative stress-induced pkd1 signaling drive formation of dclk1+ Pancreatic stem cells. iScience. (2021) 24:102019. doi: 10.1016/j.isci.2020.102019
49. Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, et al. Dclk1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. (2014) 146:245–56. doi: 10.1053/j.gastro.2013.09.050
50. Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, et al. Dclk1 defines quiescent pancreatic progenitors that promote injury-induced regeneration and tumorigenesis. Cell Stem Cell. (2016) 18:441–55. doi: 10.1016/j.stem.2016.03.016
51. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by ly6c(Low) F4/80(+) extratumoral macrophages. Gastroenterology. (2015) 149:201–10. doi: 10.1053/j.gastro.2015.04.010
52. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for pd-1/pd-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. (2017) 66:124–36. doi: 10.1136/gutjnl-2016-312078
53. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (Mdsc), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
Keywords: reactive oxygen species, pancreatic cancer, macrophage, initiation, inflammatory
Citation: Döppler HR and Storz P (2024) Macrophage-induced reactive oxygen species in the initiation of pancreatic cancer: a mini-review. Front. Immunol. 15:1278807. doi: 10.3389/fimmu.2024.1278807
Received: 16 August 2023; Accepted: 12 March 2024;
Published: 21 March 2024.
Edited by:
Yaqing Zhang, University of Michigan, United StatesReviewed by:
Mehrdad Zarei, Case Western Reserve University, United StatesKaren Liby, Indiana University Bloomington, United States
Copyright © 2024 Döppler and Storz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Storz, c3RvcnoucGV0ZXJAbWF5by5lZHU=