
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 January 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1258740
This article is part of the Research Topic Emerging Talents in Inflammation: 2022 View all 6 articles
 Rui Chen1,2,3†
Rui Chen1,2,3† Hui Zhang3,4†
Hui Zhang3,4† Linke Li1,2
Linke Li1,2 Jinsheng Li4
Jinsheng Li4 Jiang Xie5
Jiang Xie5 Jie Weng4Huan Tan1,3Yanjun Liu1,3*Tailin Guo1,3*Mengyuan Wang1,2,3*
Jie Weng4Huan Tan1,3Yanjun Liu1,3*Tailin Guo1,3*Mengyuan Wang1,2,3*Ubiquitin-specific proteases (USPs), as one of the deubiquitinating enzymes (DUBs) families, regulate the fate of proteins and signaling pathway transduction by removing ubiquitin chains from the target proteins. USPs are essential for the modulation of a variety of physiological processes, such as DNA repair, cell metabolism and differentiation, epigenetic modulations as well as protein stability. Recently, extensive research has demonstrated that USPs exert a significant impact on innate and adaptive immune reactions, metabolic syndromes, inflammatory disorders, and infection via post-translational modification processes. This review summarizes the important roles of the USPs in the onset and progression of inflammatory diseases, including periodontitis, pneumonia, atherosclerosis, inflammatory bowel disease, sepsis, hepatitis, diabetes, and obesity. Moreover, we highlight a comprehensive overview of the pathogenesis of USPs in these inflammatory diseases as well as post-translational modifications in the inflammatory responses and pave the way for future prospect of targeted therapies in these inflammatory diseases.
Deubiquitinating enzymes (DUBs), a large group of the ubiquitin system (1), have ubiquitin binding motifs (2), which ensure specific and rigorous regulation by assisting the recognition and recruitment of ubiquitinated proteins (3). Ubiquitin (Ub) attaches to proteins by cascades of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes and regulates protein interactions (4), while DUBs remove Ub from ubiquitinated proteins through splitting the peptide or isopeptide bonds between Ub and its substrates, thereby stabilizing the substrate proteins and reversing ubiquitination (5) (Figure 1). DUBs can avoid over-activation of the signaling pathway by modulating the signal transduction and have a significant impact on maintaining the balance of the ubiquitin system (6).
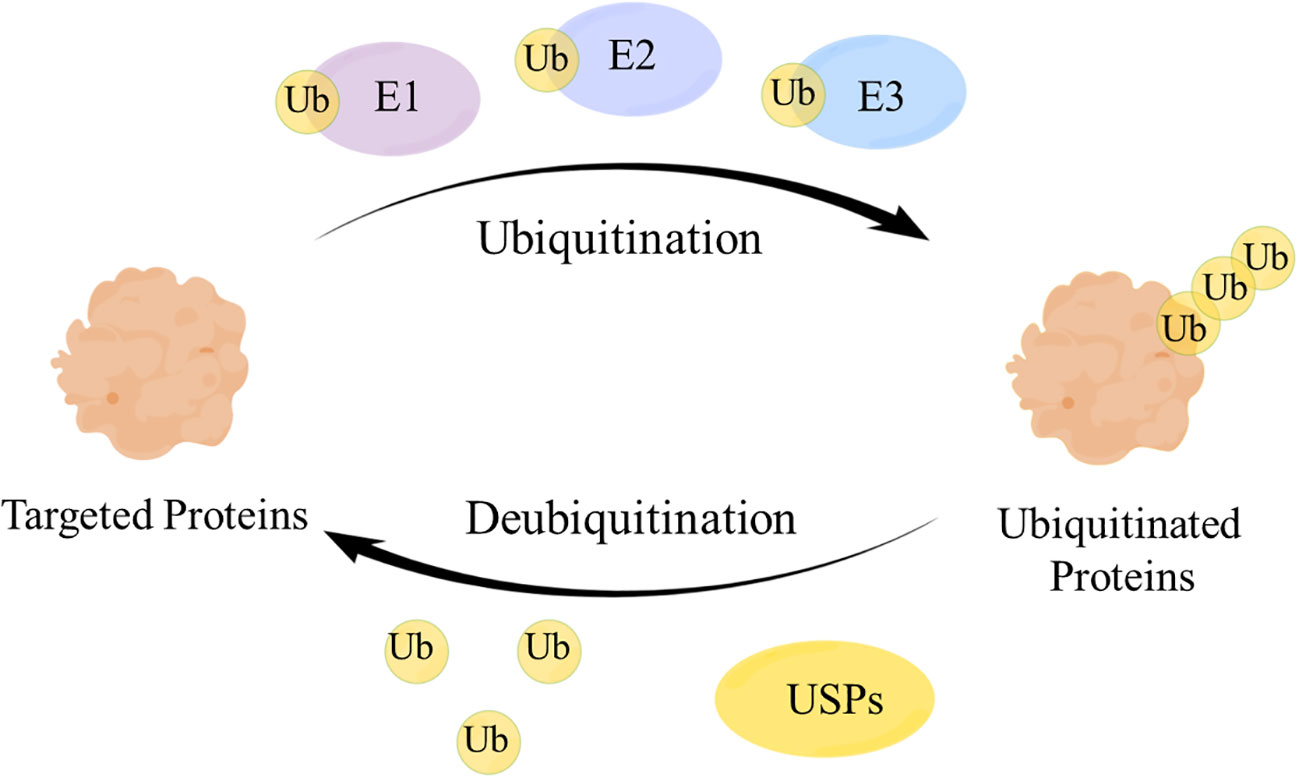
Figure 1 The mechanism of the ubiquitin-protease system. Ubiquitin (Ub) attaches to the targeted proteins by E1, E2, E3 enzymes to realize the ubiquitination of targeted proteins. Ubiquitin-specific proteases (USPs) remove Ub from the ubiquitinated proteins to regulate the fate of proteins and signaling pathway transduction.
The human genome contains nearly 100 DUBs. They are classified into seven families based on their sequence and structural similarity, including the Ubiquitin-specific proteases (USPs), Ovarian tumor proteases (OTUs), Josephins and JAB1/MPN/Mov34 metalloenzymes (JAMMs), Machado-Josephin disease proteins (MJDs), Ubiquitin carboxyl-terminal hydrolases (UCHs), motif interacting with ubiquitin-containing novel DUB family proteases (MINDYs) and zinc finger-containing ubiquitin peptidase 1 (ZUP1) (6, 7) (Figure 2). Among these families, USPs have the largest number of members and play crucial roles in regulating protein fate and signaling pathway transduction by removing Ub from targeted proteins (8). Furthermore, USPs are essential for the modulation of a variety of physiological processes, such as DNA repair, cell metabolism and differentiation, epigenetic modulations as well as protein stability (9, 10). Recent studies have highlighted the significant impact of USPs on innate and adaptive immune reactions, metabolic syndromes, inflammatory disorders, and infections through post-translational modification (11). Dysregulation of USPs has been observed in several inflammatory diseases, suggesting their potential involvement in the underlying mechanisms (12).
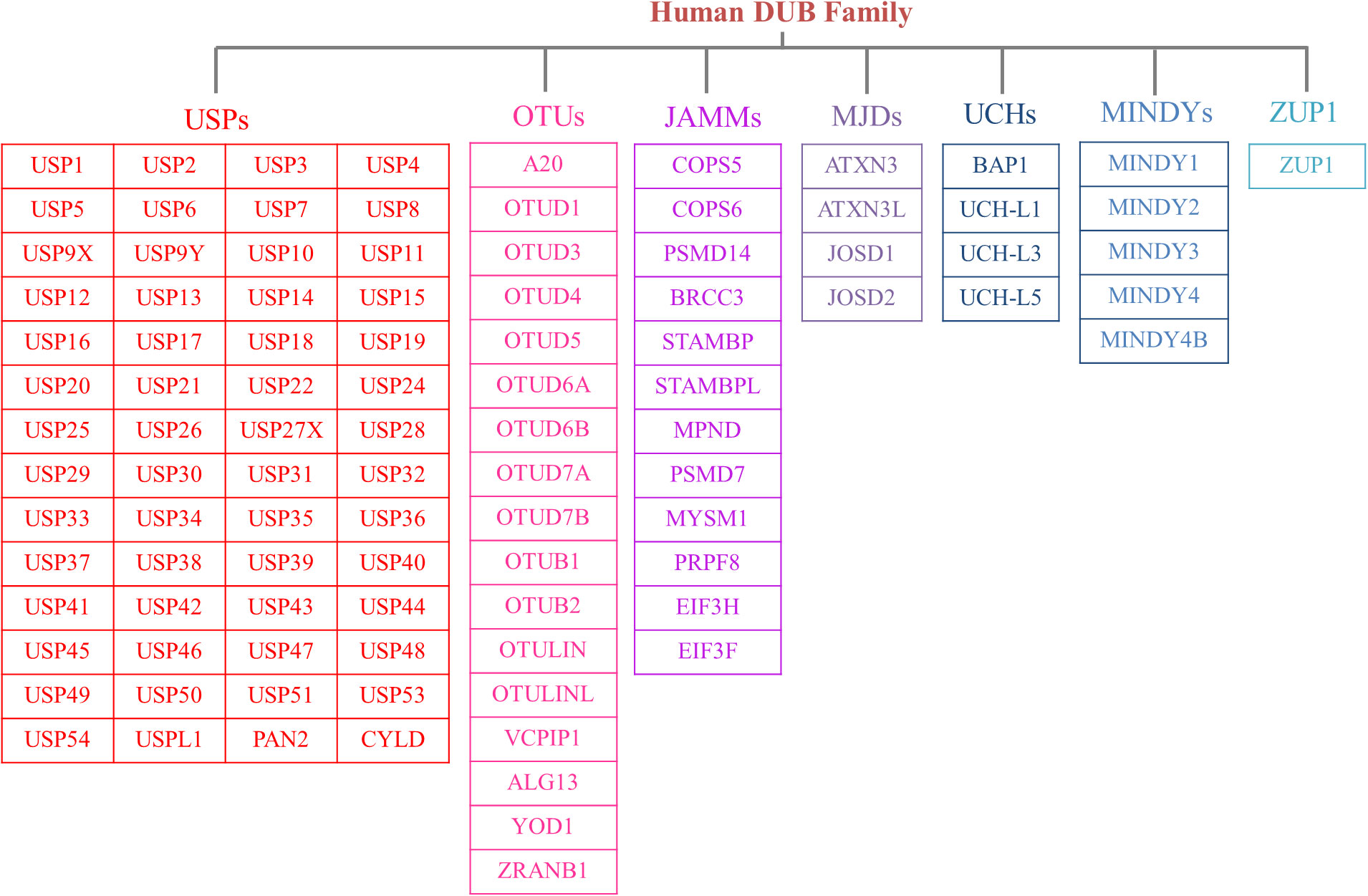
Figure 2 The members of DUB family. According to sequence and structural similarity, DUBs are classified into seven families, including the Ubiquitin-specific proteases (USPs), Ovarian tumor proteases (OTUs), Josephins and JAB1/MPN/Mov34 metalloenzymes (JAMMs), Machado-Josephin disease proteins (MJDs), Ubiquitin carboxyl-terminal hydrolases (UCHs), motif interacting with ubiquitin-containing novel DUB family proteases (MINDYs) and zinc finger-containing ubiquitin peptidase 1 (ZUP1). And the USP family is the largest group of the DUBs.
Inflammation, which is triggered by various molecules and signaling pathways, serves as a defense response of the immune system to pathogen infection and tissue damage (13). Inflammation is like a double-edged sword. Moderate inflammatory reaction can protect the human body, while excessive inflammatory response will be harmful (14). Numerous inflammatory diseases, such as periodontitis, pneumonia, inflammatory bowel disease, and hepatitis, pose significant challenges to maintaining tissue homeostasis and preventing damage (15). Traditional treatments for inflammation often involve extensive immune suppression or direct cell eradication, which can compromise the immune system and increase the risk of infections (13). The roles of USPs in inflammatory diseases have garnered increasing attention, with extensive research suggesting their involvement in the onset and progression of inflammatory responses (12). However, due to the diverse functions and importance of USPs, further exploration is required to fully understand their precise roles in inflammatory diseases (5). Therefore, gaining a comprehensive understanding of the contribution of USPs to inflammatory diseases may pave the way for the development of novel targeted therapies.
Recent reviews have notably underscored the correlation between USPs and metabolic disorders (16), as well as their involvement in bone-related inflammatory ailments such as osteoarthritis and rheumatoid arthritis (17). A multitude of studies have effectively elucidated the pivotal roles that USPs play in the landscape of inflammatory diseases. In this review, we choose the most prevailing and focal inflammatory diseases associated with USPs, including periodontitis, pneumonia, atherosclerosis, inflammatory bowel disease, sepsis, hepatitis, diabetes, and obesity (Figure 3). Moreover, we highlight a comprehensive overview of the pathogenesis of the USPs in these inflammatory diseases, emphasizing the involvement of post-translational modifications in inflammatory responses. Our review aims to pave the way for future prospect of targeted therapies in these inflammatory diseases.
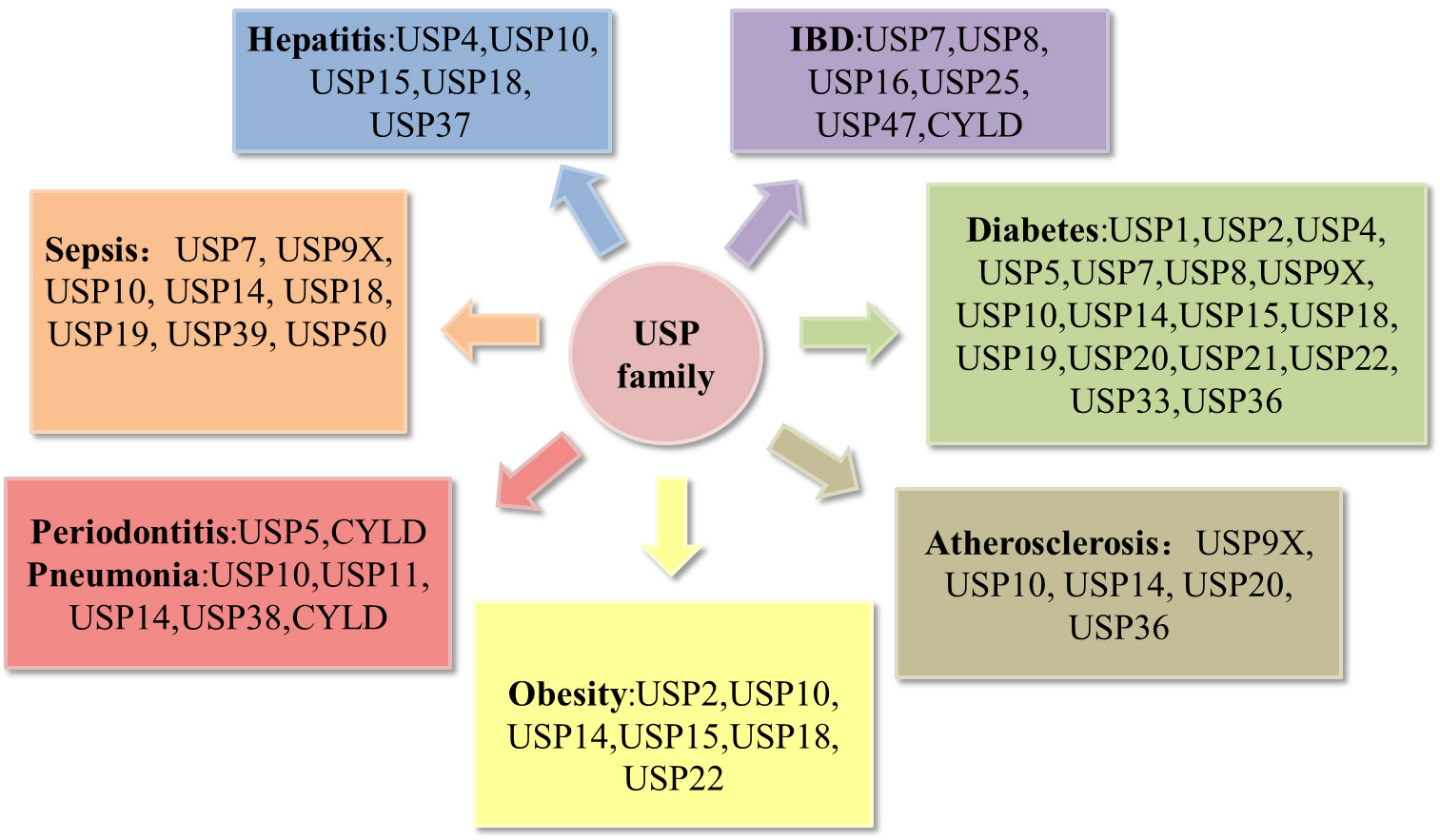
Figure 3 Deregulation of the USP family in inflammatory diseases. Pathological alterations of the USP family can affect the development of inflammatory diseases, including periodontitis, pneumonia, atherosclerosis, obesity, hepatitis, sepsis, inflammatory bowel disease (IBD), diabetes.
Epigenetic mechanisms exert control over gene expression at both the transcriptional and post-transcriptional levels, including DNA methylation, RNA methylation, RNA interference, and histone modifications (18–20). Ubiquitin-mediated degradation and the reverse process of deubiquitination regulate histones as part of the epigenetic machinery. Epigenetic mechanisms play an active role in reshaping the cellular transcriptome, thereby governing gene expression, cell differentiation, and development (20).
Innate and adaptive immune reactions are related to epigenetic modification (21). Remarkably, epigenetic regulation is a crucial player in the onset and development of inflammatory response (22). Numerous studies have highlighted the roles of USPs in various inflammatory diseases by modifying epigenetics. USPs play crucial roles in epigenetic mechanisms by regulating certain post-translationally modified epigenetic factors, such as NF-κB and p53, thereby influencing inflammation (23, 24). For example, USP9X can interact with Bcl10 of the Carma1-Bcl10-Malt1 (CBM) complex and remove the TCR-induced ubiquitin chain from Bcl10, thus promoting NF-κB activation induced by TCR signaling pathway, and then actively affecting the cytokine production, T cell proliferation and differentiation (24). Additionally, USPs can interfere with the stability of proteins involved in epigenetic mechanisms, such as histone deacetylases, regulating the termination of DNA repair and the reorganization of chromatin structure (25). USP11 can interfere with the chromatin remodeling NuRD complex, and coordinate with NuRD-associated histone deacetylation to regulate the DNA repair process and genomic stability (25). Overall, USPs play crucial roles in regulating the inflammatory response by removing Ub from targeted protein complexes and affecting the stability of epigenetic factors, thereby regulating inflammation-related gene expression and the transmission of epigenetic information.
Periodontitis, a chronic inflammatory disease, is characterized by the inflammation of the pocket wall, the resorption of alveolar bone, the formation of periodontal pocket, and the separation between gingiva and tooth, as well as the loss of tooth (26). Moreover, periodontitis is the primary cause of tooth loss in adults, and it leads to periodontal tissue damage and is associated with a variety of systemic complications, such as diabetes, obesity, cardiovascular disease, and so on (27–29). Numerous studies have stated periodontitis is mainly caused by the imbalance of the oral microbiota and host resistance (30), associated with environment factors, host factors and genetic factors (31). As one of the epigenetic modifications, USPs are key regulators of the inflammation responses in periodontitis (32).
Recent studies have shed light on the contrasting roles of USP5 and CYLD in periodontitis, offering potential avenues for therapeutic interventions (Table 1). It has been reported that USP5 is upregulated in the gingival crevicular fluid and gingival tissues of patients with periodontitis, showing a positive correlation with proinflammatory factors through the STAT3 signaling pathway, which exacerbates the inflammatory response in chronic periodontitis (33). Furthermore, CYLD has been reported to ameliorate alveolar bone loss in mouse models of periodontitis by regulating the number and activity of osteoclasts, as well as the genes related to osteogenesis and osteoclastogenesis in alveolar bones (34, 35). CYLD can also modulate the NF-κB signaling pathway, which plays a crucial role in the inflammatory response and osteoclast mediation in periodontitis (34).
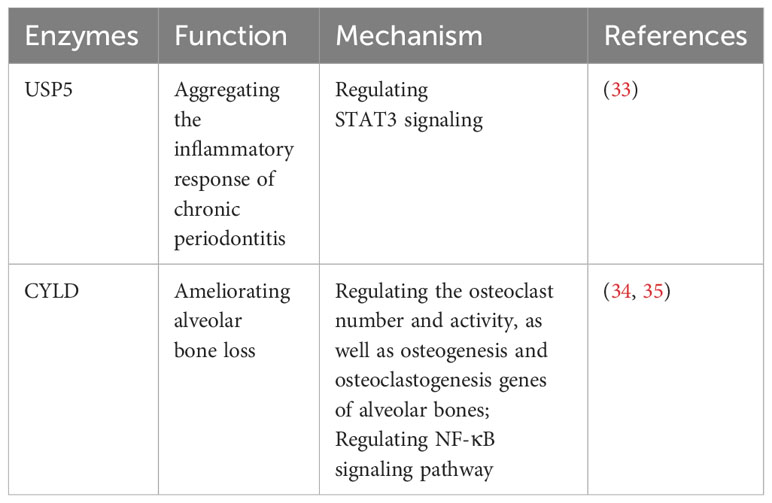
Table 1 USPs and periodontitis.
Pneumonia is a prevalent respiratory system inflammatory disease, which can be caused by a variety of pathogens. The clinical symptoms include cough, phlegm production, chest pain, fever, as well as severe complications such as acute lung injury (ALI) and pulmonary fibrosis (36, 37). However, there is still a lack of accurate treatment to cure pneumonia, necessitating the exploration of better therapeutic targets. Recently, it has been reported the key roles of USPs in pneumonia, which may provide novel insights that could have clinical implications for the management of the disease (Table 2).
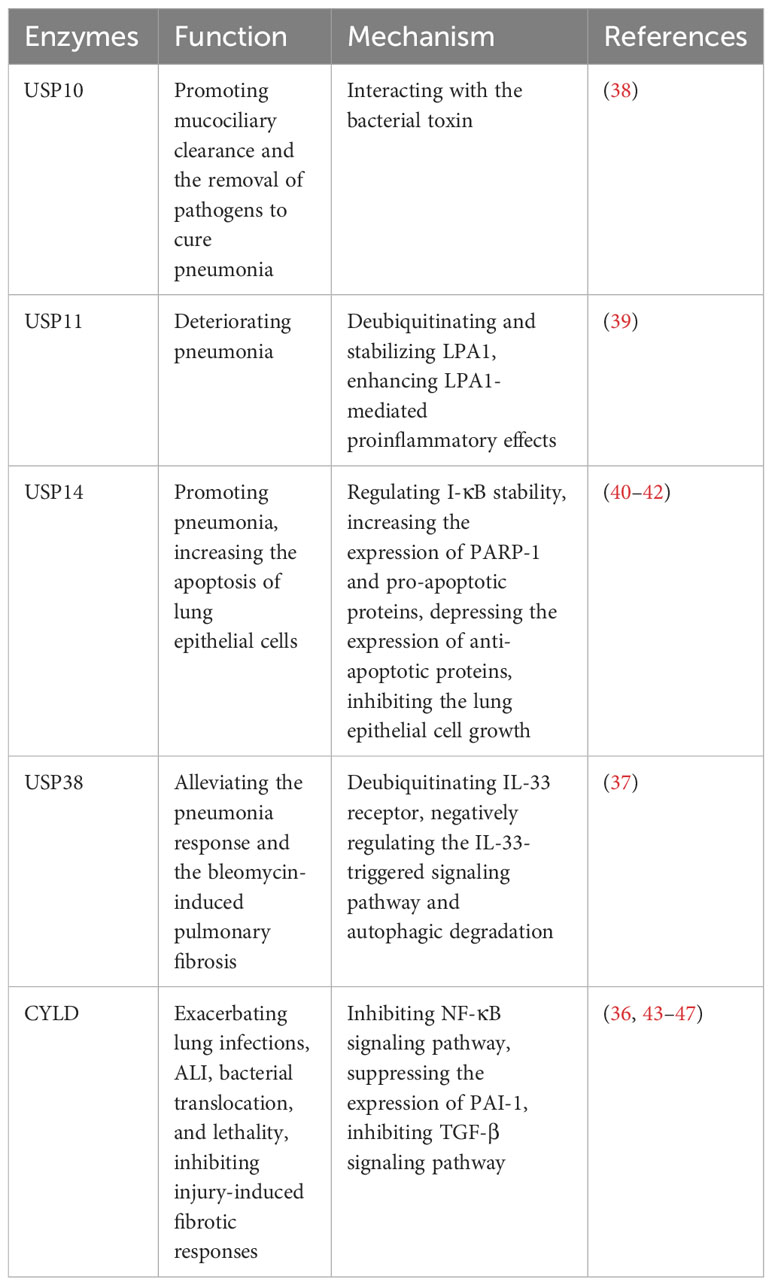
Table 2 USPs and pneumonia.
Several studies have reported that USP11 and USP14 contribute to the aggravation of inflammation in LPS-induced pneumonia (39–41). USP11 exacerbates pneumonia by deubiquitinating and stabilizing LPA1, a proinflammatory factor, enhancing LPA1-mediated proinflammatory effects (39). Knockdown or pharmaceutical inhibition of USP11 alleviates the lung damage induced by LPS in mice (39). Furthermore, USP14 plays a proinflammatory role in pneumonia (40, 41). The overexpression of USP14 in mouse models leads to the degradation of I-κB, increasing the release of factors such as TNF-α and IL-8 in lung epithelial cells (41). Besides, PARP-1 is an important binding enzyme of pneumonia signaling pathway (42). In human lung epithelial cells, it has been demonstrated that USP14 aggravates the inflammatory response by interacting with PARP-1 and increasing its expression (40). Furthermore, USP14 upregulates the expression of pro-apoptotic proteins while downregulating the anti-apoptotic protein (40). This leads to increased apoptosis of lung epithelial cells and inhibition of cell growth, which can contribute to severe complications in pneumonia (40).
In contrast, some USPs show the anti-inflammatory role in pneumonia. USP38 has been shown to alleviate the inflammatory response in lung and bleomycin-induced pulmonary fibrosis in mouse models (37). This effect is achieved through deubiquitinating the IL-33 receptor and negatively regulating the IL-33-triggered signaling pathway and autophagic degradation (37).
In the early stages of infection, Klebsiella can evade the host defense immunity system by causing a lack of inflammatory response (43). CYLD is known to suppress the NF-κB signaling pathway during the initial inflammatory response triggered by Escherichia coli, Klebsiella, and Streptococcus pneumoniae, which ultimately exacerbates subsequent lung infections (43–45). Notably, CYLD deficiency offers protection against Streptococcus pneumoniae pneumolysin (PLY)-induced ALI, bacterial translocation, and lethality (36). Furthermore, CYLD contributes to the worsening of ALI by suppressing the expression of plasminogen activator inhibitor 1 (PAI-1), a recognized biomarker of tissue injury that plays a crucial role in tissue repair (36, 46). However, it’s important to note that after infection with Streptococcus pneumoniae, CYLD serves as a critical negative regulator for injury-induced fibrotic response by inhibiting TGF-β signaling pathway (36, 47).
Pathogens can also impact host deubiquitinating enzymes to contribute to disease progression. For example, Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) is a secretory chloride channel, which plays an important role in mucociliary clearance by human airway epithelial cells and the innate immune response in the lung (38). Pseudomonas aeruginosa secretes the bacterial toxin Cif, which inhibits USP10, leading to decrease USP10-mediated deubiquitination of CFTR and increase CFTR degradation in lysosomes, causing the weaker mucociliary clearance and the harder removal of pathogens to cure pneumonia (38).
Furthermore, there are some drug discoveries about USP and pneumonia, such as QingFeiPaiDu decoction and wogonoside. Mechanically, it decreases the expression of USP14 to reduce the phosphorylation of LPS-stimulated transcription factor 2(ATF2), an important regulator of cytokines, to play an anti-inflammatory role (48).
Atherosclerosis, an inflammatory disease primarily situated within arterial walls, arises due to the aberrant accumulation of low-density lipids (LDL) and lipid proteins (49–51). This process involves the participation of diverse cell types, including smooth muscle cells (SMCs), macrophages, neutrophil granulocytes, endothelial cells (ECs), and other leukocytes (49–51). Existing research has demonstrated the pivotal roles of NLRP3 inflammasome, IL-1β, and TNF in atherosclerosis (49, 51). Consequently, there is an imperative to identify more precise upstream or downstream targets to facilitate a more accurate therapeutic approach for atherosclerosis (49, 51).
Recent investigations have significantly highlighted the substantial involvement of USPs in atherosclerosis progression. Studies indicate that USP9X, USP10, USP14, USP17, USP20, and USP36 play regulatory roles in the atherosclerotic process (Table 3). Macrophages intake an excess of lipids, leading to the proliferation of macrophages and secrete inflammatory factors, which causes the formation of foam cells, thus aggravating atherosclerosis (49, 52, 54). This process hinges on receptor activity, including CD36, SR-A, and SR-B1 (57).
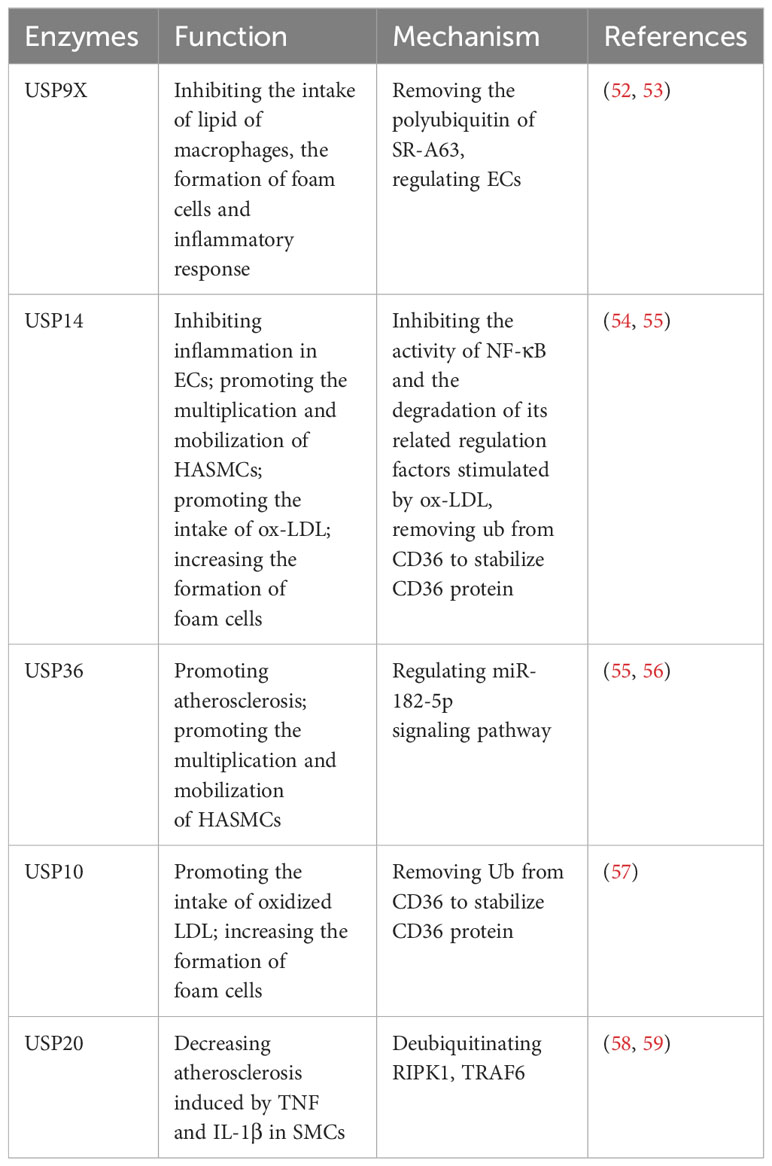
Table 3 USPs and atherosclerosis.
Notably, USP9X has been identified as a suppressor of macrophage lipid intake, exhibiting lower expression levels in atherosclerosis compared to normal cells. Mechanically, USP9X removes polyubiquitin chains from SR-A63, thus inhibiting lipid intake by macrophages, impeding foam cell formation, and reducing the ensuing inflammatory response (52). Furthermore, USP9X, alongside USP14 and USP36, contributes to the regulation of ECs (53, 54, 60). Respectively, in atherosclerosis patients, USP14 is observed to be downregulated in ECs, and it inhibits the inflammatory response in ECs (54). This downregulation hampers the activity of NF-κB and the degradation of its associated regulatory factors, which are stimulated by oxidized LDL (ox-LDL) (54). On the other hand, USP36 contributes to atherosclerosis progression through the exosomal microRNA-197-3p signaling pathway (60). Furthermore, both USP14 and USP36 play roles in the proliferation and mobilization of human aortic smooth muscle cells (HASMCs) (55, 56). Additionally, USP14 and USP10 can increase the formation of foam cells via removing Ub from CD36, thereby stabilizing the CD36 protein and promoting the uptake of ox-LDL (57, 61). To briefly sum up, USP14 has shown effects on ECs, SMCs and foam cells in the development of atherosclerosis (54, 55, 61). Moreover, USP20 alleviates inflammation in SMCs in TNF and IL-1β-induced atherosclerosis by deubiquitinating RIPK1, a vital factor of inflammation and cell death (58, 59).
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract, encompassing ulcerative colitis (UC) and Crohn’s disease (CD) (62). The etiology and mechanisms underlying IBD are complex, and extensive studies have implicated the involvement of USPs in the disease (Table 4).
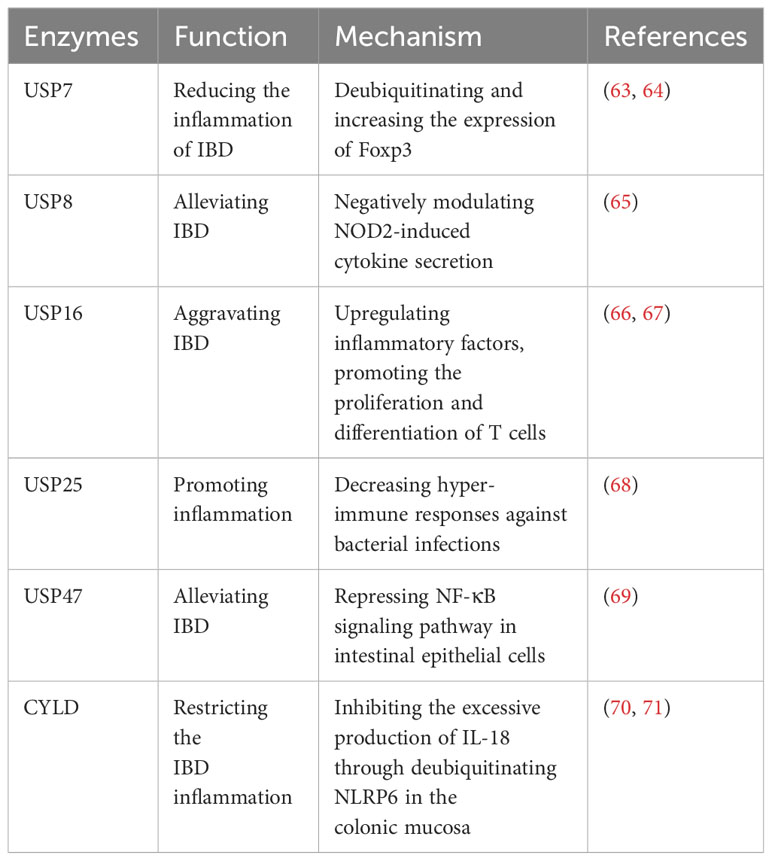
Table 4 USPs and inflammatory bowel disease.
Recent studies have highlighted the significant impact of USPs in IBD. USP7, USP47, USP8, and CYLD have been implicated in alleviating IBD. Foxp3 is an essential factor for the development of regulatory T cells (Tregs) (63). USP7 increases the quantity and function of Tregs to maintain self-tolerance and reduce the inflammation of IBD by directly deubiquitinating and enhancing the expression of Foxp3 (63). Cambogin, a potential drug, can enhance the effect of USP7 and holds promise as a future treatment for IBD based on this mechanism (64). Additionally, USP47 is observed to be downregulated in chronic inflammatory mucosal tissue of CD and UC in patients with IBD (69). Knocking down USP47 in mice makes it easier to induce IBD and results in a more severe inflammatory response and tissue damage (69). This occurs by suppressing the NF-κB signaling pathway in intestinal epithelial cells (69). NOD2 is reported to be a strong genetic factor associated with IBD, and USP8 can negatively regulate NOD2-induced IL-8 and IL-6 in bone marrow-derived macrophages to inhibit inflammation in IBD (65). CYLD, another USP, restricts IBD inflammation in the colonic mucosa by inhibiting excessive production of IL-18 through deubiquitinating NLRP6 (70). However, CYLD is significantly downregulated in IBD patients (71). This downregulation leads to severe infection by adherent-invasive Escherichia coli (AIEC), activation of NF-κB, and degradation of IκB-α, thus exacerbating IBD (71).
However, USP16 and USP25 has the contrary impact on IBD. USP16 exhibits increased expression in patients with IBD and contributes to disease inflammation (66, 67). It has demonstrated that USP16 promotes the proliferation and differentiation of T cells, thereby increasing CD4+ T cell infiltration and aggravating IBD (66). Furthermore, USP25 significantly regulates proinflammatory cytokines in the colon (68). Knockdown of USP25 leads to hyper-immune responses against bacterial infections, thereby restricting bacterial replication and inflammation (68).
The liver, being the body’s primary detoxifying organ, is susceptible to hepatitis caused by various factors. Hepatitis can be classified into different types, such as viral hepatitis, autoimmune hepatitis, nonalcoholic steatohepatitis (NASH), and others (72, 73). Viral hepatitis has the largest population of hepatitis in the world, including HAV, HBV, HCV, HDV and HEV (73). The development of hepatitis involves complex mechanisms, often characterized by liver inflammation, damage, scarring, and the potential progression to hepatocirrhosis, liver cancer, and even death (73). Recent research has shed light on the involvement of several USPs in the development and progression of hepatitis. These USPs regulate various cellular processes that are crucial to the pathogenesis of hepatitis, including inflammation, immune response, viral replication, and cell death (Table 5). By modulating these processes, USPs contribute to the intricate mechanisms underlying hepatitis.
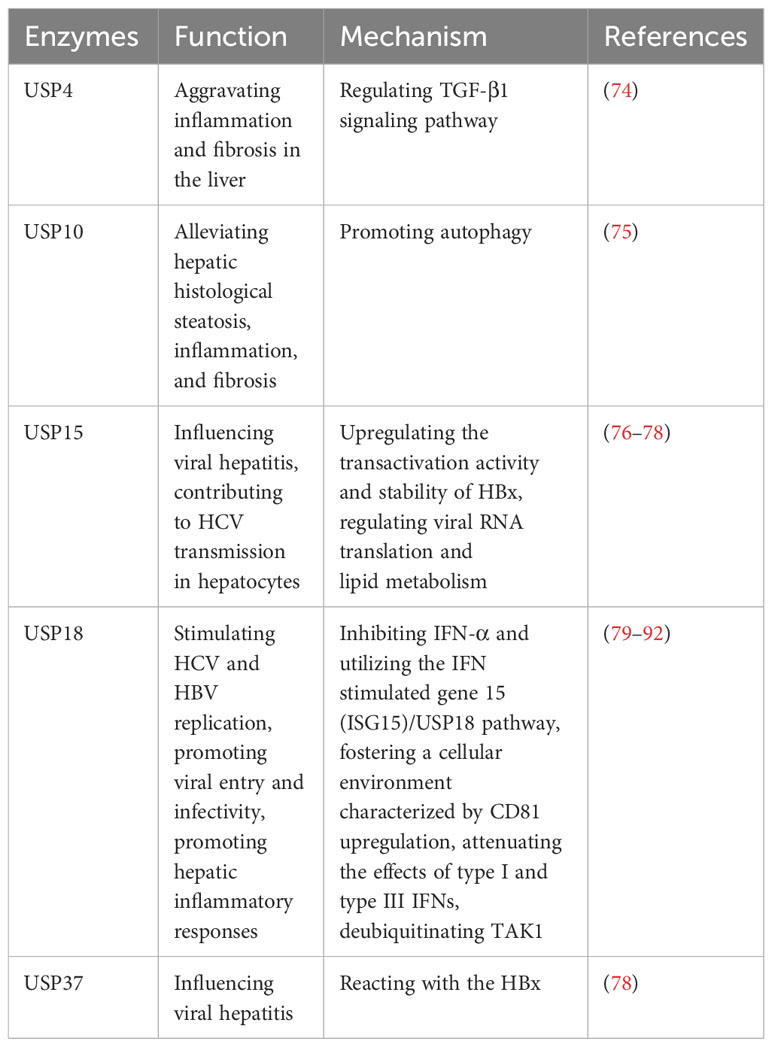
Table 5 USPs and hepatitis.
In viral hepatitis, various factors such as LPS, TNF-α, IL-17A, ISG15, Ach, and IFN-λ4 can increase the expression of USP18 in hepatocytes, leading to the inhibition of IFN signaling and attenuating the antiviral activity of IFN-α (79–83). The upregulation of USP18 in hepatocytes has been associated with poor outcomes in IFN-α therapy for chronic HBV and HCV patients (84–86). Conversely, silencing USP18 can enhance the effectiveness of IFN treatment by improving IFN-α2a signaling, inducing IFN-stimulated genes, and enhancing antiviral activity (84–86). Furthermore, USP18 promotes HCV and HBV replication by specifically inhibiting IFN-α and utilizing the IFN stimulated gene 15 (ISG15)/USP18 pathway, as well as fostering a cellular environment characterized by CD81 upregulation and promoting viral entry and infectivity (87, 88). Additionally, USP18 dampens the effects of type I and type III IFNs by exerting negative feedback control through its regulatory role (89). Moreover, USP18 also binds to and deubiquitinates TAK1, thereby promoting hepatic inflammatory responses (90–92).
In addition, USP15 and USP37 can react with the HBV X protein (HBx), which regulates viral replication in viral hepatitis (76–78). USP15 enhances the transactivation activity and stability of HBx, potentially influencing HCV transmission in hepatocytes by modulating viral RNA translation and lipid metabolism (76–78). Moreover, Qisheng Li et al. have employed functional genomics approaches and HCV model systems to demonstrate the involvement of USP11 in HCV-mediated translation, but the finer details of its regulatory mechanisms remain elusive (93).These findings highlight the involvement of specific USPs in viral hepatitis, particularly their impact on IFN signaling, viral replication, and hepatic inflammatory responses.
USP4 shows significant upregulation in autoimmune hepatitis (74). The intervention using Vialinin A and liver X receptor α (LXRα)-induced cannabinoid receptor 2 (CB2) has been found to reduce the level of USP4, leading to alleviate inflammation and fibrosis in the liver (74). Additionally, it has been reported that CB2 inhibited USP4, leading to the stabilization of TGF-β1R, and subsequently ameliorating hepatic autoimmune hepatitis (74). Furthermore, USP10 plays a role in attenuating hepatic steatosis in nonalcoholic steatohepatitis (NASH) through the promotion of autophagy, which alleviates hepatic steatosis, inflammation, and fibrosis (75).
Sepsis is characterized by the deadly dysfunction of the host’s immune system in response to infections (94). The serious systemic inflammation derives from localized infections or sterile inflammatory diseases (95). Current approaches mainly involve supportive measures such as antibiotics and oxygen therapy, lacking precise targeted therapies (95). Researchers have underscored the complexity of small molecule mechanisms, including molecules like TNF-α, which are being explored as potential targeted therapy (95). However, the more precise targeted molecules are necessary to improve sepsis treatment.
Recently, there have been some studies on the regulation of USPs in the development of sepsis (Table 6). In sepsis, the level of USP9X is upregulated in CD8+ T cells, and this upregulation contributes to the development of sepsis by causing dysfunction in CD8+ T cells (96). This dysfunction can be reversed by WP1130, an inhibitor of USP9X (96). In sepsis, the transcription factor SRY-box 9 (SOX9) is highly expressed in cardiac muscle cells, contributing to cardiac damage (97). USP7 exacerbates the adverse effects of sepsis on the heart by deubiquitinating SOX9 and leading to an upregulation of its expression (97). Moreover, USP10 has been identified as a protective factor for renal tubular epithelial cells against acute kidney injury in LPS-induced sepsis (98).
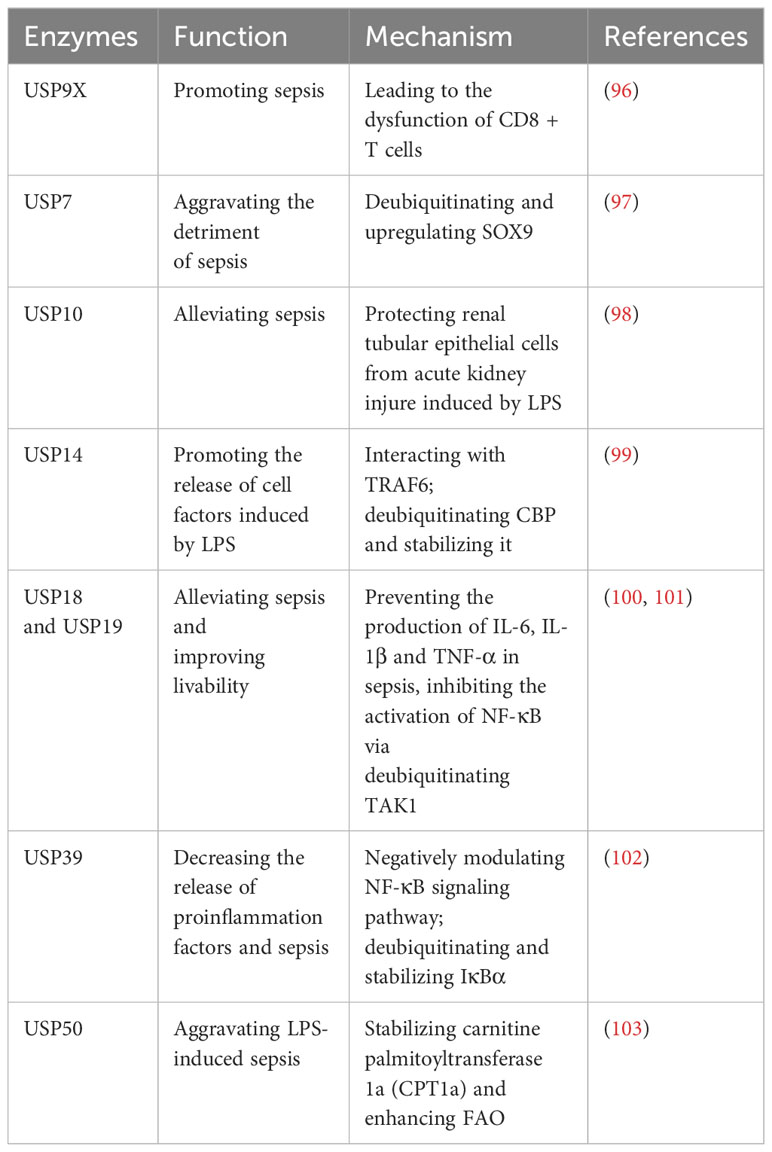
Table 6 USPs and sepsis.
CBP, responsible for the release of cellular factors during sepsis, undergoes degradation due to ubiquitination (99). USP14 facilitates the release of cell factors in response to LPS during sepsis by deubiquitinating CBP and thereby stabilizing it (99). Recently, it has been proposed that a novel USP14 inhibitor, Neochromine S5, binds to USP14, diminishing its deubiquitination activity and disrupting the interaction between USP14 and TRAF6, which effectively reduces proinflammatory factors, and downregulates NF-κB and STAT1 signaling pathways (104). As a result, Neochromine S5 shows higher efficacy and safety in alleviating sepsis (104).
Moreover, both USP18 and USP19 have shown potential in mitigating sepsis and enhancing survivability (100, 101). They can curb the production of IL-6, IL-1β, and TNF-α in sepsis by inhibiting the activation of NF-κB through deubiquitination of TAK1 (100, 101). Furthermore, USP39 is downregulated in LPS-induced sepsis and it can decrease the release of proinflammation factors and subsequently sepsis-related inflammation (102). Mechanically, USP39 is recognized as a negative modulator of NF-κB signaling pathway through deubiquitinating and stabilizing IκBα (102). In sepsis, energy consumption changes from C6H12O6 to fatty acid oxidation (FAO) (103). USP50 plays a role in aggravating sepsis by stabilizing carnitine palmitoyltransferase 1a (CPT1a) and enhancing FAO (103).
Diabetes is a chronic epidemic disease characterized by the presence of high levels of glucose in the blood, which can cause various complications, including diabetic retinopathy (DR), diabetic nephropathy (DN), diabetic neuropathic pain (DNP), diabetic foot (DF), diabetic cardiomyopathy (DCM), and so on (105–107). It is classified into type 1 diabetes (T1D) and type 2 diabetes (T2D). Recently, there have been numerous studies on USPs in diabetes, shedding light on the intricate roles of USPs in the disease. Further exploration of USPs in diabetes may provide insights into novel treatment strategies for the disease and its associated complications.
Type 1 diabetes (T1D) is an autoimmune disease characterized by the attack of T cells on pancreatic β cells, which are responsible for insulin production and the regulation of blood glucose levels (108). Several ubiquitin-specific proteases (USPs) have been implicated in the development of T1D (Table 7). DNA damage is highly expressed in diabetes and aggravates diabetes (109). Suppression of USP1 has been demonstrated to improve diabetes outcomes by the inhibition of DNA damage, preventing pancreatic β cell apoptosis, preserving insulin secretion, and enhancing β-cell maturation in human islets (109). In addition, the CLEC16a-NRDP1-USP8 complex mediates ubiquitin-dependent signaling that promotes mitophagy and maintains mitochondrial quality in β cells (110). This process contributes to the preservation of precise β-cell function and helps regulate blood glucose levels (110). McL-1 is one of the anti-apoptotic Bcl-2 protein family, which is reduced in islets in T1D patients, and USP9X modulates McL-1 protein turnover mediated by cytokines to prevent islet cell death in β cells (111). Moreover, the dysregulation of secretagogue-dependent USP9X deubiquitinase activity can lead to decreased insulin utilization (112).
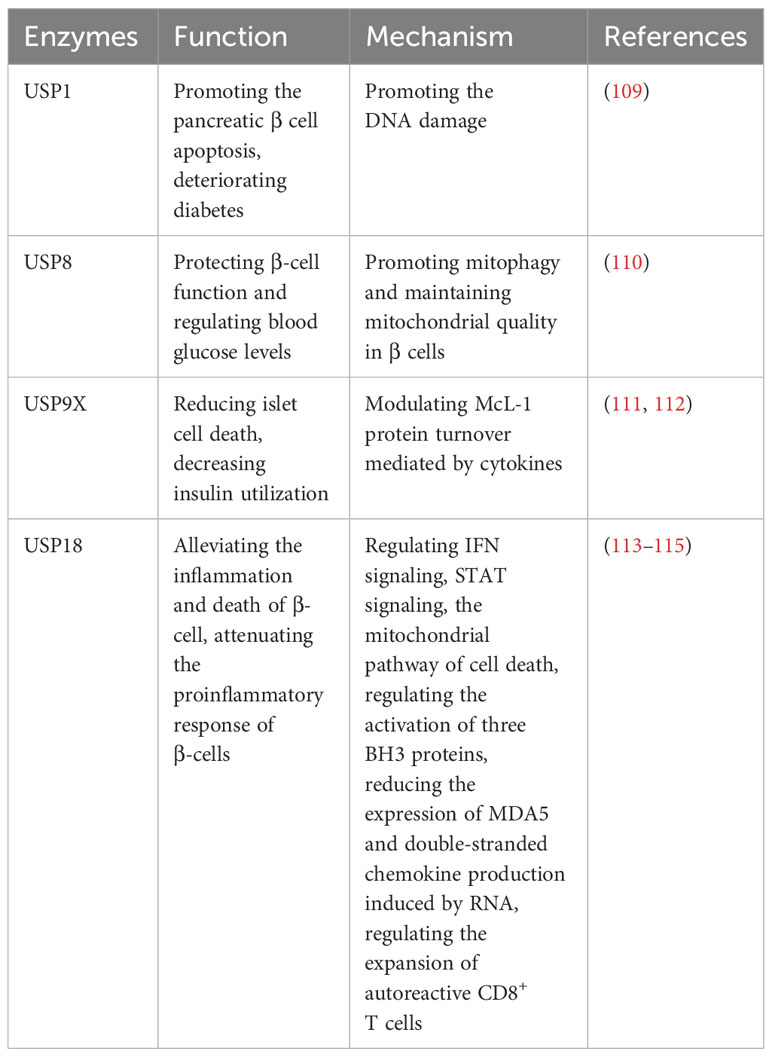
Table 7 USPs and type 1 diabetes.
USP18-driven dendritic cells play a significant role in breaking immune tolerance in autoimmune diabetes (113). The genetic deletion of USP18 can mitigate the expansion of autoreactive CD8+ T cells and provide protection against autoimmune diabetes (113). In pancreatic β cells, USP18 acts as a crucial modulator of the IFN signaling pathway and three BH3 proteins, which exerts a significant impact on the inflammation and death of β cells (114). Suppression of USP18 promotes inflammation by STAT signaling and aggravates β-cell apoptosis induced by IFN through the cell death in mitochondria (114). Moreover, USP18 reduces the expression of MDA5, the T1D candidate gene, leading to the downregulation of double-stranded chemokine production induced by RNA and attenuate the proinflammatory response of β cells (115).
Type 2 diabetes (T2D) is characterized by chronic inflammation, immune factor activation, and impaired insulin secretion and sensitivity, leading to elevated blood glucose levels (116). Numerous studies have identified several USPs that play roles in T2D (Table 8). A study indicates that USP2A and USP2 can alter insulin sensitivity, and USP2A blocks obesity-induced insulin resistance through adipocyte-dependent mechanisms (117). Additionally, USP4 decreases the ubiquitination and degradation of insulin receptors, consequently leading to reduced insulin resistance (118). Gastrodin, a compound derived from Gastrodia elata, has been reported to enhance the expression of USP4 and may represent a novel targeted treatment for T2D (118).
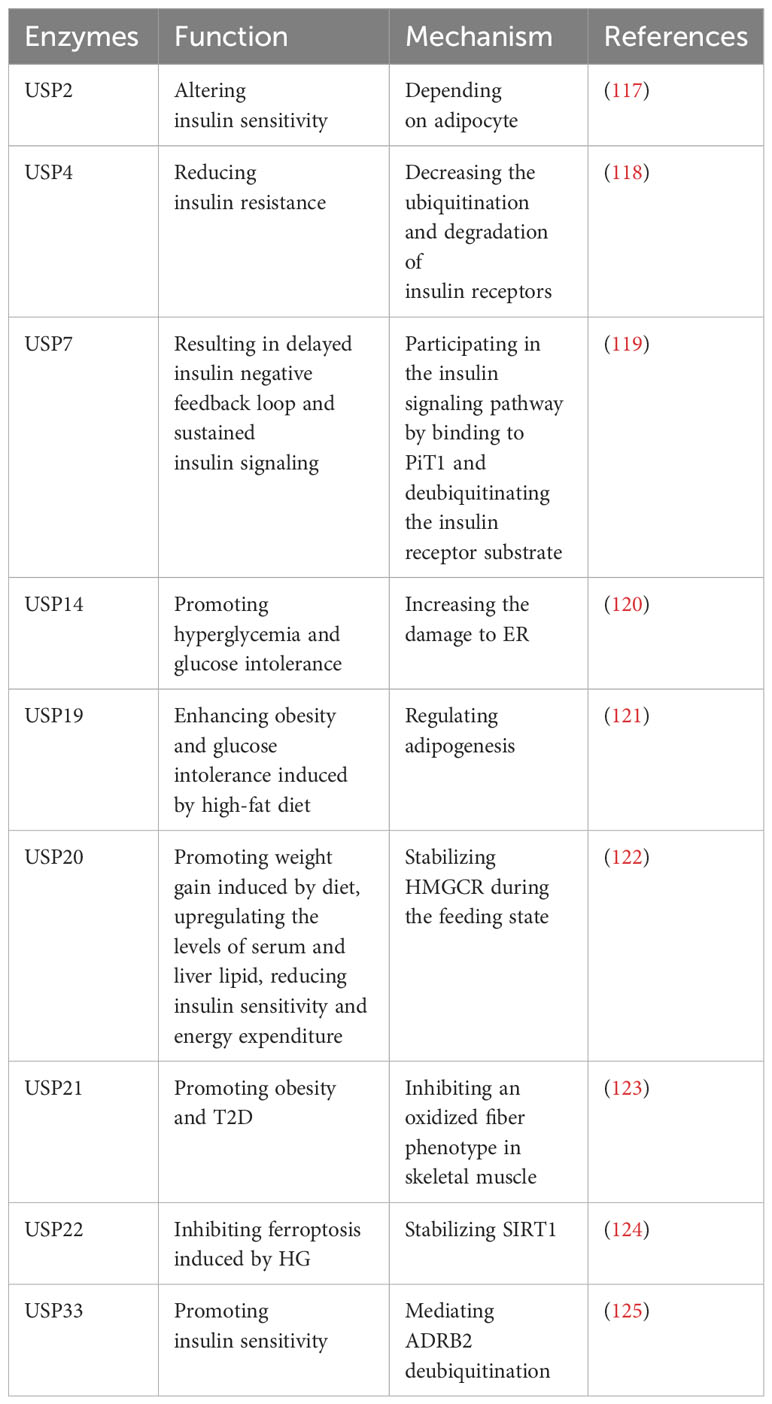
Table 8 USPs and type 2 diabetes.
However, certain USPs can aggravate T2D. It has been reported that USP7 can delay the insulin negative feedback loop, leading to sustained insulin signaling by binding to PiT1 and deubiquitinating the insulin receptor substrate (119). In addition, endoplasmic reticulum (ER) stress plays a significant role in T2D, and sustained ER stress upregulates USP14 (120). The knockdown of USP14 reduces the damage to ER associated with glucose metabolism and improves hyperglycemia and glucose intolerance in obese mice (120). Moreover, inhibition of USP19 has shown promise in regulating adipogenesis and improving glucose intolerance and obesity induced by a high-fat diet (121). Subsequently, the upregulation of postprandial glucose and insulin levels promotes the phosphorylation of USP20 (122). Suppression of USP20 significantly reduces weight gain, lipid levels in serum and liver, and improves insulin sensitivity and energy expenditure (122). This is achieved by stabilizing HMG-CoA reductase (HMGCR), which serves as the rate-limiting enzyme in the cholesterol biosynthesis pathway (122). In addition, ablation of USP21 in skeletal muscle enhances energy expenditure by stimulating an oxidative fiber phenotype that inhibits obesity and T2D (123). In T2DM mice models, ferroptosis is observed in pancreatic β cells (124). USP22 stabilizes Sirt1 to inhibit ferroptosis induced by HG (124). Furthermore, USP33 participates in the deubiquitination of β2-adrenergic receptor (ADRB2), stimulating insulin sensitivity in skeletal muscle (125).
Diabetic retinopathy (DR) is a common microvascular complication of diabetes, and endothelial barrier integrity is important for vascular steady (126). Several pharmacological inhibitors of USP1 have been identified as potential treatments for DR, such as Primaquine Diphosphate, as they can alleviate β-cell death or inhibit vascular endothelial growth factor (VEGF)-induced leakage, thus improving DR outcomes (109, 127, 128). In DR patients, USP14 can modulate inflammatory response through the TGF-β1 signal transduction, IκBα and NF-κB signaling pathway s in HG-treated Müller cells, as well as reactive oxygen species (ROS) (126).This modulation plays a role in mediating the progression of DR (126) (Table 9).
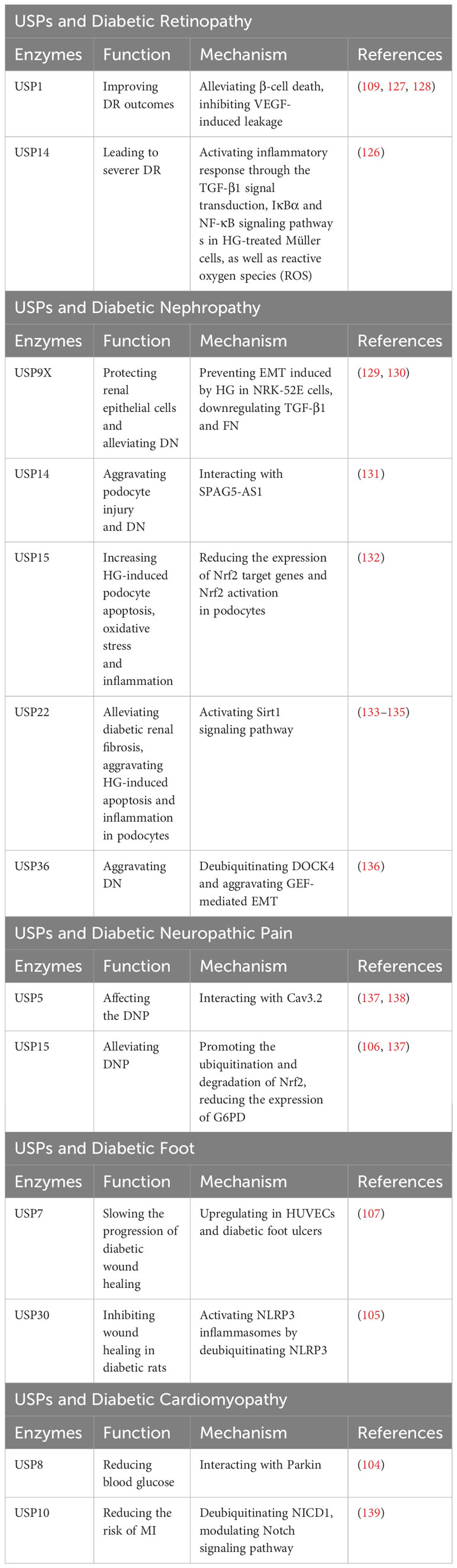
Table 9 USPs and diabetic retinopathy, diabetic nephropathy, diabetic neuropathic pain, diabetic foot, diabetic cardiomyopathy.
Epithelial-mesenchymal transition (EMT) has a remarkable influence on diabetic nephropathy (DN) (129). In the kidney tissues of db/db mice and HG-induced NRK-52E cells, the expression of USP9X protein was dramatically decreased (129). USP9X has been identified as a key regulator in preventing HG-induced EMT and protecting renal epithelial cells in DN, while knockdown of USP9X aggravates the EMT process in HG-induced NRK-52E cell (129). Moreover, USP9X alleviates the development of diabetic renal fibrosis by downregulating TGF-β1 and fibronectin (FN), two markers of fibrosis in glomerular mesangial cells (GMCs) (130).
In contrast, it has been indicated that some USPs may exacerbate DN. The development of podocytes serves as a clinical marker for DN (131). Dysfunctional autophagy plays a significant role in podocyte injury (131). In human podocytes, USP14 can negatively regulate autophagy and aggravate podocyte injury by deubiquitinating and stabilizing SPAG5, which inhibits autophagy (131). Furthermore, USP15 is increased in podocytes upon HG stimulation, and the inhibition of USP15 reduces HG-induced podocyte apoptosis, oxidative stress and inflammation by enhancing the expression of Nrf2 target genes and Nrf2 activation (132). HeQing Huang et al. have reported that USP22 alleviates diabetic renal fibrosis, while Qin Huang et al. have indicated that USP22 aggravates HG-induced apoptosis and inflammation in podocytes (133, 134). Therefore, USP22 may affect the progression of DN from different aspects with different environments, more researches are needed to reveal the roles of USP22 in the progression of DN. Teneligliptin, a drug for T2D treatment, can stabilize USP22 to delay the progression of DN through activating Sirt1 signaling pathway (135). Besides, the expression of USP36 is increased in diabetic renal tubular epithelial cells, which has a great relationship with upregulated EMT (136). USP36 contributes to DN progression by directly deubiquitinating cytokinin 4 (DOCK4) and aggravating guanine nucleotide exchange factor (GEF)-mediated EMT (136) (Table 9).
Cav3.2 calcium channels are proteins involved in nociceptive transmission, which are increased in response to nerve damage and peripheral neuroinflammation (137). Disrupting the interaction between Cav3.2 and USP5 using the TAT-cUBP1-USP5 peptide has been shown to reduce the levels of the Cav3.2 calcium channel in vitro, which attenuates thermal hyperalgesia in diabetic neuropathy animals (137, 138). Furthermore, USP15 has been found to enhance the current of Cav3.2 in afferent neurons by mediating the deubiquitination of the channel (137). Additionally, USP15 promotes the degradation of Nrf2 and upregulates the expression of glucose-6-phosphate dehydrogenase (G6PD) (137). In contrast, Mir-497 has been shown to downregulate the expression of USP15, thereby alleviating diabetic neuropathic pain (106) (Table 9).
The expression of USP7 has been reported to be upregulated in human umbilical vein endothelial cells and in diabetic foot ulcers (107). Small molecule inhibitors of USP7, such as Quiazolin-4-one scaffold and HBX 41108, have demonstrated the ability to accelerate wound healing (107). Furthermore, USP30 has been identified as a regulator of NLRP3 inflammasomes by deubiquitinating NLRP3 and activating its function (105). In diabetic rats, Mf-094, an inhibitor of USP30, can reduce NLRP3 expression and its downstream target caspase-1 p20, therefore alleviating the inflammation and promoting wound healing (105) (Table 9).
In diabetic conditions, excessive production of reactive oxygen species (ROS) can cause mitochondrial damage, leading to myocardial damage (104). Parkin facilitates clearance of damaged mitochondria (104). Studies have shown that in diabetic heart tissue, USP8 promotes mitophagy by deubiquitinating Parkin and facilitating the recruitment of Parkin to mitochondria, thereby protecting from myocardial injury (104). Patients with T2D are at increased risk for myocardial infarction (MI) (139). USP10, a deubiquitinating enzyme, modulates the Notch signaling pathway by targeting NICD1, the receptor of Notch1 (139). This pathway plays a vital role in the regulation of myocardial fibrosis (139). Follistatin-like protein 1 (FSTL1) has been found to protect cardiac fibroblasts from injury induced by diabetes mellitus-associated myocardial infarction (DM-MI). FSTL1 exerts its protective effects by downregulating fibrosis markers and upregulating USP10/Notch1 signaling, thereby inhibiting myocardial fibrosis and apoptosis (139) (Table 9).
Obesity, a significant health issue of the 21st century, is associated with the activation of the innate immune system, expansion of adipose tissue, and various metabolic disturbances (140). It gives rise to numerous long-term complications, including diabetes, cardiovascular disease, and non-alcoholic fatty liver disease (NAFLD) (141). There are several causes of obesity, such as food intake, adipogenesis and the activation of inflammation (142). In recent years, there has been a growing interest in the roles of USPs in research related to obesity (Table 10). Several USPs, including USP2, USP10, USP14, USP15, USP18, and USP22, have been associated with obesity and related metabolic disorders.
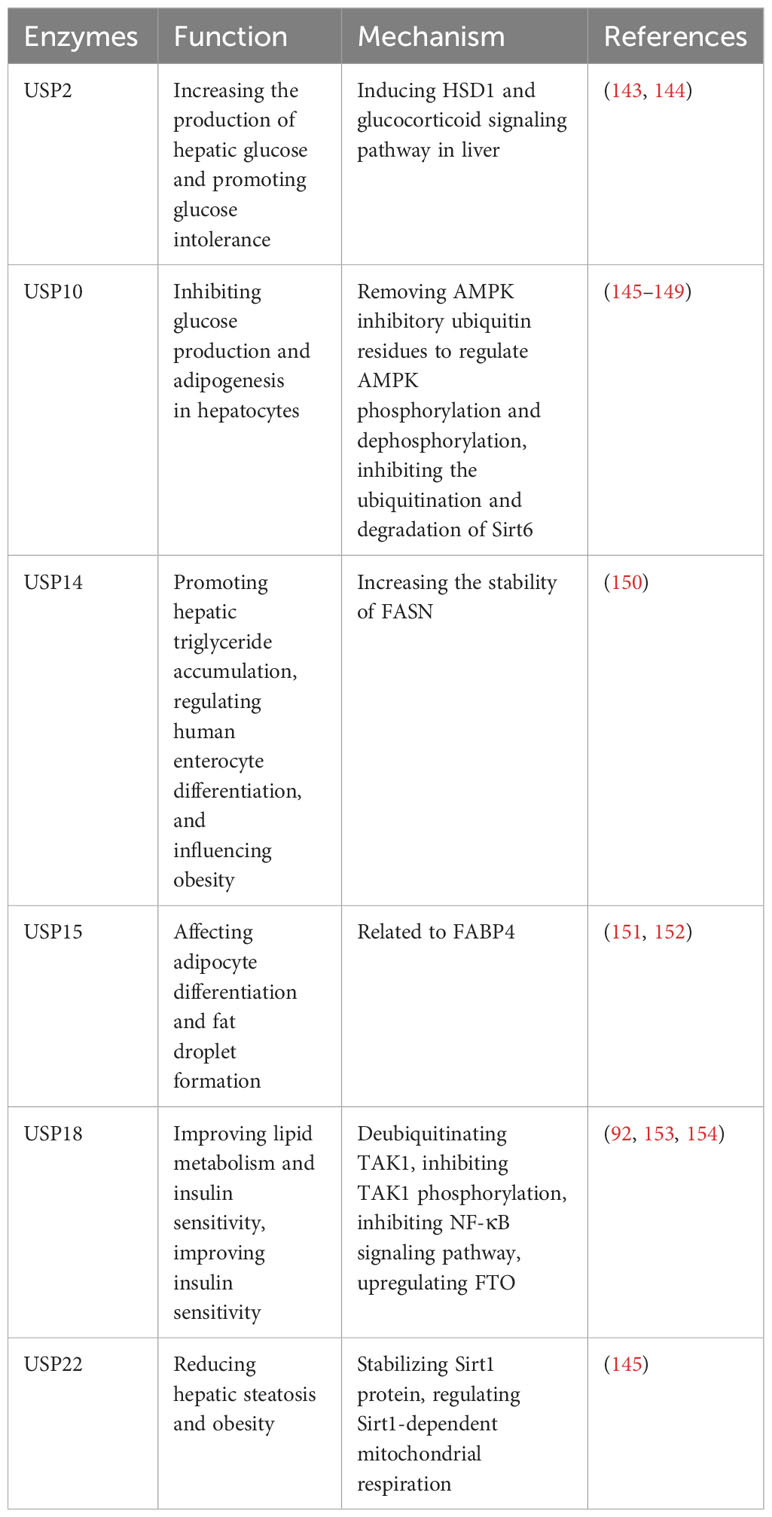
Table 10 USPs and obesity.
In diet-induced obese mice, it is reported that USP2 can increase the production of hepatic glucose and promote glucose intolerance (143). USP2 is regulated by peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and influences hepatic glucose metabolism by inducing 11β-hydroxysteroid dehydrogenase 1 (HSD1) and glucocorticoid signaling pathways (143). Furthermore, 3,3’-diindolylmethane, derived from cruciferous vegetables, inhibits adipogenesis in preadipocytes by targeting USP2 activity, thereby inhibiting high-fat diet-induced obesity (144).
NAFLD is caused by mitochondrial dysfunction with persistent imbalance between energy intake and expenditure, and USP10 is a negative regulator of NAFLD (145). However, the protein level of USP10 in liver is decreased in obese and NAFLD patients (146). USP10 inhibits glucose production and adipogenesis in hepatocytes, and its activity modulated by long noncoding RNA myocardial infarct-related transcript 2 (Mirt2) (147). Besides, USP10 improves metabolic dysfunction associated with obesity by regulating liver steatosis, inflammation, and insulin resistance (146, 148, 149). Mechanically, USP10 regulates AMPK phosphorylation and dephosphorylation by removing inhibitory ubiquitin residues (146, 148, 149). Additionally, it inhibits the ubiquitination and degradation of Sirt6 (146, 148, 149).
USP14 can promote hepatic triglyceride accumulation by increasing the stability of fatty acid synthase (FASN), which is the crucial enzyme for hepatic adipogenesis (150). Besides, human intestinal cell differentiation is altered during bariatric surgery, and USP14 can regulate human enterocyte differentiation and influence obesity (155).
USP15 plays a significant role in adipocyte differentiation and the formation of fat droplets. It has been reported that USP15 is closely and functionally related to fatty acid binding protein 4 (FABP4), which is abundant in mature adipocytes (151, 152).
USP18 is downregulated in obese mice, which can improve lipid metabolism and insulin sensitivity (92). In principle, TAK1-dependent signaling is vital for the development of hepatic insulin resistance (153). USP18 can improve insulin sensitivity by deubiquitinating TAK1 and inhibiting TAK1 phosphorylation to downregulate NF-κB signaling pathway and upregulate fat mass and obesity-related protein (FTO) (153, 154).
Furthermore, USP22 reduces hepatic steatosis and obesity by stabilizing Sirt1 protein and regulating Sirt1-dependent mitochondrial respiration (145).
In recent years, extensive research has shed light on the mechanisms of ubiquitin-specific proteases (USPs) in inflammatory diseases. This review summarizes the important roles of the USPs in the onset and progression of inflammatory diseases, including periodontitis, pneumonia, atherosclerosis, inflammatory bowel disease, sepsis, hepatitis, diabetes, and obesity (40, 75). However, the functions of the USP family have not been fully elucidated (15).
While some USPs have similar functions in inflammation, their underlying mechanisms can differ. The inflammatory response is complex, and the interactions between USPs and inflammatory diseases vary depending on their substrates, tissue types, and environmental conditions (156). USPs participate in various signaling pathways due to the multitude of their substrates (157). Understanding how USPs select their substrates and express themselves specifically in targeted tissues may enable the development of accurate therapies for specific inflammatory diseases. Thus, exploring the role of USPs in inflammatory diseases represents a vast domain for further investigation (158).
Inflammatory diseases exhibit varying expressions of USPs in different cells and tissues. Most studies focus on immune cells or corresponding organ tissues, with only a few examining the expression of USPs in peripheral blood cells (159, 160). Therefore, further research is needed to investigate the role of USPs in other cells involved in inflammatory diseases, which may provide insights into disease phenotypes based on USP expression patterns.
USPs influence protein stability and can be regulated by small molecule inhibitors. Efforts have been made to develop drugs that target USPs for clinical applications (161, 162). For example, QingFeiPaiDu decoction and wogonoside have been reported to reduce USP14 levels, thereby alleviating pneumonia (48). These compounds are shown to reduce the phosphorylation of ATF2 and the NF-κB signaling pathway stimulated by LPS (48). However, due to the complex composition of these compounds, investigating the mechanisms presents a significant challenge (48). Furthermore, Gastrodin has been shown to upregulate the expression of USP4 and enhance the interaction between USP4 and insulin receptors to treat diabetes (118). Gastrodin has also been reported to increase the activity of pancreatic β-cells and stimulate insulin secretion, ultimately improving diabetes (118).
Some USP inhibitors have shown better pharmacological efficacy than previously known drugs and may overcome drug resistance through combination therapy. For example, it has been reported that administering USP7 alone or in combination with synergistic pathways significantly enhances DNA damage effects and overcomes treatment resistance (163).
Nonetheless, due to the high homology among members of the USP family, achieving specificity with USP inhibitors poses a significant challenge (164). This results in the existing inhibitors lacking specificity and often failing to exclusively target a single member of USPs, thereby somewhat limiting their utility. For instance, drugs like PR619 target a range of enzymes including USP2, USP4, USP5, USP7, USP8, USP15, USP20, USP28, USP47, UCHL1, UCHL3, UCHL5, highlighting the need to craft more selective agents (164). A prime example is the subsequent USP1 inhibitor ML323, exhibiting higher specificity compared to previous compounds like Pimozide and GW7647, thereby widening its scope of application (164). To achieve enhanced specificity in inhibitors, careful consideration of the chemical structure of USPs is needed (164). Nowadays, it is crucial to search for and apply suitable biological testing methods to screen and identify small molecule inhibitors of USPs. Currently, the existing methods include high-throughput screening (HTS), bioinformatics approaches, virtual screening and so on (165).
Furthermore, the diversity of pathways and physiological activities regulated by USPs has led to potential toxicity concerns (164). Deubiquitination virtually controls numerous aspects of human cellular biology and physiology, and any defects in these processes may lead to diseases (164). For instance, VLX1570 is an inhibitor of USP14. The clinical trial of VLX1570 in combination with dexamethasone for multiple myeloma patients was halted due to pulmonary toxicity (164). Besides, USP30 controls the import of mitochondrial proteins, suggesting potential toxic effects of the inhibitor of USP30 (166). Although numerous reagents have displayed promise in vitro and animal experiments, clinical trials validating their specific effects and side effects are scarce. It is vital to bridge the gap between experimental findings and clinical relevance through further investigations, including clinical trials, to establish the significance of USPs in diverse inflammatory conditions. Additionally, the solubility and stability of inhibitors in aqueous solutions significantly impact their pharmaceutical potential in clinical applications (164). These underscore the need for extensive exploration.
In summary, as we discover more phenotypes and mechanisms, USPs are increasingly recognized as essential modulators of inflammatory diseases. Further research will identify novel directions and effective approaches to improve therapeutic treatments. It is important to continue investigating the role of USPs in inflammatory diseases to enhance our understanding and develop targeted therapies for improved patient outcomes.
RC: Writing – original draft. HZ: Writing – review & editing. LL: Writing – review & editing. JL: Writing – review & editing. JX: Writing – review & editing. JW: Writing – review & editing. HT: Writing – review & editing. YL: Writing – review & editing. TG: Funding acquisition, Writing – review & editing. MW: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from National Natural Science Foundation of China (82101012, 82104069, 22172120); Sichuan Science and Technology Program (2021JDRC0158, 2022089); Southwest Jiaotong University Fundamental Research Cultivation Support Program for Medicine-Engineering Integration (2682023ZTPY046); Medical Youth Innovation Project of Sichuan Province (Q20019).
The figure technology was supported by Figdraw (www.figdraw.com) (ID: SWARU4654f).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J (1997) 11(14):1245–56. doi: 10.1096/fasebj.11.14.9409543
2. Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol (2009) 10(8):550–63. doi: 10.1038/nrm2731
3. Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol (2012) 9(2):123–30. doi: 10.1038/cmi.2011.59
4. Henneberg LT, Schulman BA. Decoding the messaging of the ubiquitin system using chemical and protein probes. Cell Chem Biol (2021) 28(7):889–902. doi: 10.1016/j.chembiol.2021.03.009
5. Cai J, Culley MK, Zhao Y, Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell (2018) 9(9):754–69. doi: 10.1007/s13238-017-0486-3
6. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol (2019) 20(6):338–52. doi: 10.1038/s41580-019-0099-1
7. Magin RS, Liu X, Felix A, Bratt AS, Chan WC, Buhrlage SJ. Small molecules as tools for functional assessment of deubiquitinating enzyme function. Cell Chem Biol (2021) 28(7):1090–100. doi: 10.1016/j.chembiol.2021.04.021
8. Woo B, Baek KH. Regulatory interplay between deubiquitinating enzymes and cytokines. Cytokine Growth Factor Rev (2019) 48:40–51. doi: 10.1016/j.cytogfr.2019.06.001
9. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer (2018) 18(2):69–88. doi: 10.1038/nrc.2017.105
10. Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer (2006) 6(10):776–88. doi: 10.1038/nrc1994
11. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
12. Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol (2011) 18(5):520–8. doi: 10.1038/nsmb.2066
13. Wright KT, Giardina C, Vella AT. Therapeutic targeting of the inflammome. Biochem Pharmacol (2014) 92(2):184–91. doi: 10.1016/j.bcp.2014.08.027
14. Herold K, Mrowka R. Inflammation-Dysregulated inflammatory response and strategies for treatment. Acta Physiol (Oxf) (2019) 226(3):e13284. doi: 10.1111/apha.13284
15. Liu J, Cheng Y, Zheng M, Yuan B, Wang Z, Li X, et al. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduct Target Ther (2021) 6(1):28. doi: 10.1038/s41392-020-00418-x
16. Kitamura H. Ubiquitin-specific proteases (USPs) and metabolic disorders. Int J Mol Sci (2023) 24(4):3219. doi: 10.3390/ijms24043219
17. Luo W, Zhang G, Wang Z, Wu Y, Xiong Y. Ubiquitin-specific proteases: Vital regulatory molecules in bone and bone-related diseases. Int Immunopharmacol (2023) 118:110075. doi: 10.1016/j.intimp.2023.110075
18. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab (2019) 29(5):1028–44. doi: 10.1016/j.cmet.2019.03.009
19. Li X, Liu Z, Xia C, Yan K, Fang Z, Fan Y. SETD8 stabilized by USP17 epigenetically activates SREBP1 pathway to drive lipogenesis and oncogenesis of ccRCC. Cancer Lett (2022) 527:150–63. doi: 10.1016/j.canlet.2021.12.018
20. Wu YL, Lin ZJ, Li CC, Lin X, Shan SK, Guo B, et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduction Targeted Ther (2023) 8(1):98. doi: 10.1038/s41392-023-01333-7
21. Lau CM, Adams NM, Geary CD, Weizman OE, Rapp M, Pritykin Y, et al. Epigenetic control of innate and adaptive immune memory. Nat Immunol (2018) 19(9):963–72. doi: 10.1038/s41590-018-0176-1
22. Naik S, Fuchs E. Inflammatory memory and tissue adaptation in sickness and in health. Nature (2022) 607(7918):249–55. doi: 10.1038/s41586-022-04919-3
23. Zhang X, Berger FG, Yang J, Lu X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J (2011) 30(11):2177–89. doi: 10.1038/emboj.2011.125
24. Park Y, Jin HS, Liu YC. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc Natl Acad Sci U.S.A. (2013) 110(23):9433–8. doi: 10.1073/pnas.1221925110
25. Ting X, Xia L, Yang J, He L, Si W, Shang Y, et al. USP11 acts as a histone deubiquitinase functioning in chromatin reorganization during DNA repair. Nucleic Acids Res (2019) 47(18):9721–40. doi: 10.1093/nar/gkz726
26. Wang M, Xie J, Wang C, Zhong D, Xie L, Fang H. Immunomodulatory properties of stem cells in periodontitis: current status and future prospective. Stem Cells Int (2020) 2020:9836518. doi: 10.1155/2020/9836518
27. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol (2015) 15(1):30–44. doi: 10.1038/nri3785
28. Genco RJ, Van Dyke TE. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol (2010) 7(9):479–80. doi: 10.1038/nrcardio.2010.120
29. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol (2011) 7(12):738–48. doi: 10.1038/nrendo.2011.106
30. Bhuyan R, Bhuyan SK, Mohanty JN, Das S, Juliana N, Juliana IF. Periodontitis and its inflammatory changes linked to various systemic diseases: A review of its underlying mechanisms. Biomedicines (2022) 10(10):2659. doi: 10.3390/biomedicines10102659
31. Brodzikowska A, Górski B. Polymorphisms in genes involved in inflammation and periodontitis: A narrative review. Biomolecules (2022) 12(4):552. doi: 10.3390/biom12040552
32. Jurdziński KT, Potempa J, Grabiec AM. Epigenetic regulation of inflammation in periodontitis: cellular mechanisms and therapeutic potential. Clin Epigenet (2020) 12(1):186. doi: 10.1186/s13148-020-00982-7
33. Chen Q, Su J, Chen X. Role of ubiquitin-specific protease 5 in the inflammatory response of chronic periodontitis. Oral Dis (2023) 29(3):1234–41. doi: 10.1111/odi.14114
34. Xu J, Wu HF, Ang ES, Yip K, Woloszyn M, Zheng MH, et al. NF-kappaB modulators in osteolytic bone diseases. Cytokine Growth Factor Rev (2009) 20(1):7–17. doi: 10.1016/j.cytogfr.2008.11.007
35. Cao Y, Zhang X, Hu M, Yang S, Li X, Han R, et al. CYLD inhibits osteoclastogenesis to ameliorate alveolar bone loss in mice with periodontitis. J Cell Physiol (2023) 238(5):1036–45. doi: 10.1002/jcp.30990
36. Lim JH, Stirling B, Derry J, Koga T, Jono H, Woo CH, et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity (2007) 27(2):349–60. doi: 10.1016/j.immuni.2007.07.011
37. Yi XM, Li M, Chen YD, Shu HB, Li S. Reciprocal regulation of IL-33 receptor-mediated inflammatory response and pulmonary fibrosis by TRAF6 and USP38. Proc Natl Acad Sci U.S.A. (2022) 119(10):e2116279119. doi: 10.1073/pnas.2116279119
38. Bomberger JM, Ye S, Maceachran DP, Koeppen K, Barnaby RL, O'Toole GA, et al. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PloS Pathog (2011) 7(3):e1001325. doi: 10.1371/journal.ppat.1001325
39. Zhao J, Wei J, Dong S, Bowser RK, Zhang L, Jacko AM, et al. Destabilization of lysophosphatidic acid receptor 1 reduces cytokine release and protects against lung injury. EBioMedicine (2016) 10:195–203. doi: 10.1016/j.ebiom.2016.07.020
40. Huang C, Cao H, Qin J, Xu L, Hu F, Gu Y, et al. Ubiquitin-specific protease 14 (USP14) aggravates inflammatory response and apoptosis of lung epithelial cells in pneumonia by modulating poly (ADP-ribose) polymerase-1 (PARP-1). Inflammation (2021) 44(5):2054–64. doi: 10.1007/s10753-021-01482-3
41. Mialki RK, Zhao J, Wei J, Mallampalli DF, Zhao Y. Overexpression of USP14 protease reduces I-κB protein levels and increases cytokine release in lung epithelial cells. J Biol Chem (2013) 288(22):15437–41. doi: 10.1074/jbc.C112.446682
42. Nie Y, Nirujogi TS, Ranjan R, Reader BF, Chung S, Ballinger MN, et al. PolyADP-ribosylation of NFATc3 and NF-κB transcription factors modulate macrophage inflammatory gene expression in LPS-induced acute lung injury. J Innate Immun (2021) 13(2):83–93. doi: 10.1159/000510269
43. Regueiro V, Moranta D, Frank CG, Larrarte E, Margareto J, March C, et al. Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner. Cell Microbiol (2011) 13(1):135–53. doi: 10.1111/j.1462-5822.2010.01526.x
44. Lim JH, Ha UH, Woo CH, Xu H, Li JD. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol (2008) 10(11):2247–56. doi: 10.1111/j.1462-5822.2008.01204.x
45. Frank CG, Reguerio V, Rother M, Moranta D, Maeurer AP, Garmendia J, et al. Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation. Cell Microbiol (2013) 15(7):1212–33. doi: 10.1111/cmi.12110
46. Wu J, Zhao J, Yu J, Zhang W, Huang Y. Cylindromatosis (CYLD) inhibits Streptococcus pneumonia-induced plasminogen activator inhibitor-1 expression via interacting with TRAF-6. Biochem Biophys Res Commun (2015) 463(4):942–7. doi: 10.1016/j.bbrc.2015.06.039
47. Lim JH, Jono H, Komatsu K, Woo CH, Lee J, Miyata M, et al. CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat Commun (2012) 3:771. doi: 10.1038/ncomms1776
48. Xu X, Xia J, Zhao S, Wang Q, Ge G, Xu F, et al. Qing-Fei-Pai-Du decoction and wogonoside exert anti-inflammatory action through down-regulating USP14 to promote the degradation of activating transcription factor 2. FASEB J (2021) 35(9):e21870.52–57. doi: 10.1096/fj.202100370RR
49. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis from experimental insights to the clinic. Nat Rev Drug Discovery (2021) 20(8):589–610. doi: 10.1038/s41573-021-00198-1
50. Gisterå A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol (2017) 13(6):368–80. doi: 10.1038/nrneph.2017.51
51. Weber C, Habenicht AJR, von Hundelshausen P. Novel mechanisms and therapeutic targets in atherosclerosis: inflammation and beyond. Eur Heart J (2023) 44(29):2672–81. doi: 10.1093/eurheartj/ehad304
52. Wang B, Tang X, Yao L, Wang Y, Chen Z, Li M, et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J Clin Invest (2022) 132(10):e154217. doi: 10.1172/JCI154217
53. Xu S, Ge Y, Wang X, Yin W, Zhu X, Wang J, et al. Circ-USP9X interacts with EIF4A3 to promote endothelial cell pyroptosis by regulating GSDMD stability in atherosclerosis. Clin Exp Hypertens (2023) 45(1):2186319. doi: 10.1080/10641963.2023.2186319
54. Fu Y, Qiu J, Wu J, Zhang L, Wei F, Lu L, et al. USP14-mediated NLRC5 upregulation inhibits endothelial cell activation and inflammation in atherosclerosis. Biochim Biophys Acta Mol Cell Biol Lipids (2023) 1868(5):159258. doi: 10.1016/j.bbalip.2022.159258
55. Liu H, Li X, Yan G, Lun R. Knockdown of USP14 inhibits PDGF-BB-induced vascular smooth muscle cell dedifferentiation via inhibiting mTOR/P70S6K signaling pathway. RSC Adv (2019) 9(63):36649–57. doi: 10.1039/C9RA04726C
56. Zhao Q, Lu YH, Wang X, Zhang XJ. Circ_USP36/miR-182-5p/KLF5 axis regulates the ox-LDL-induced injury in human umbilical vein smooth muscle cells. Am J Transl Res (2020) 12(12):7855–69.
57. Xia X, Hu T, He J, Xu Q, Yu C, Liu X, et al. USP10 deletion inhibits macrophage-derived foam cell formation and cellular-oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging (Albany NY) (2020) 12(22):22892–905. doi: 10.18632/aging.104003
58. Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discovery (2020) 19(8):553–71. doi: 10.1038/s41573-020-0071-y
59. Jean-Charles PY, Wu JH, Zhang L, Kaur S, Nepliouev I, Stiber JA, et al. USP20 (Ubiquitin-specific protease 20) inhibits TNF (Tumor necrosis factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler Thromb Vasc Biol (2018) 38(10):2295–305. doi: 10.1161/ATVBAHA.118.311071
60. Zhang Y, Li W, Li H, Zhou M, Zhang J, Fu Y, et al. Circ_USP36 silencing attenuates oxidized low-density lipoprotein-induced dysfunction in endothelial cells in atherosclerosis through mediating miR-197-3p/ROBO1 axis. J Cardiovasc Pharmacol (2021) 78(5):e761–72. doi: 10.1097/FJC.0000000000001124
61. Zhang F, Xia X, Chai R, Xu R, Xu Q, Liu M, et al. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J Cell Mol Med (2020) 24(6):3292–302. doi: 10.1111/jcmm.15002
62. Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun Rev (2017) 16(4):416–26. doi: 10.1016/j.autrev.2017.02.013
63. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity (2013) 39(2):259–71. doi: 10.1016/j.immuni.2013.05.018
64. Lu Y, Kim NM, Jiang YW, Zhang H, Zheng D, Zhu FX, et al. Cambogin suppresses dextran sulphate sodium-induced colitis by enhancing Treg cell stability and function. Br J Pharmacol (2018) 175(7):1085–99. doi: 10.1111/bph.14150
65. Warner N, Burberry A, Pliakas M, McDonald C, Núñez G. A genome-wide small interfering RNA (siRNA) screen reveals nuclear factor-κB (NF-κB)-independent regulators of NOD2-induced interleukin-8 (IL-8) secretion. J Biol Chem (2014) 289(41):28213–24. doi: 10.1074/jbc.M114.574756
66. Zhang Y, Liu RB, Cao Q, Fan KQ, Huang LJ, Yu JS, et al. USP16-mediated deubiquitination of calcineurin A controls peripheral T cell maintenance. J Clin Invest (2019) 129(7):2856–71. doi: 10.1172/JCI123801
67. Yu JS, Huang T, Zhang Y, Mao XT, Huang LJ, Li YN, et al. Substrate-specific recognition of IKKs mediated by USP16 facilitates autoimmune inflammation. Sci Adv (2021) 7(3):eabc4009. doi: 10.1126/sciadv.abc4009
68. Wang XM, Yang C, Zhao Y, Xu ZG, Yang W, Wang P, et al. The deubiquitinase USP25 supports colonic inflammation and bacterial infection and promotes colorectal cancer. Nat Cancer (2020) 1(8):811–25. doi: 10.1038/s43018-020-0089-4
69. Lei H, Yang L, Xu H, Wang Z, Li X, Liu M, et al. Ubiquitin-specific protease 47 regulates intestinal inflammation through deubiquitination of TRAF6 in epithelial cells. Sci China Life Sci (2022) 65(8):1624–35. doi: 10.1007/s11427-021-2040-8
70. Mukherjee S, Kumar R, Tsakem Lenou E, Basrur V, Kontoyiannis DL, Ioakeimidis F, et al. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat Immunol (2020) 21(6):626–35. doi: 10.1038/s41590-020-0681-x
71. Cleynen I, Vazeille E, Artieda M, Verspaget HW, Szczypiorska M, Bringer MA, et al. Genetic and microbial factors modulating the ubiquitin proteasome system in inflammatory bowel disease. Gut (2014) 63(8):1265–74. doi: 10.1136/gutjnl-2012-303205
72. Brown GT, Kleiner DE. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism (2016) 65(8):1080–6. doi: 10.1016/j.metabol.2015.11.008
73. Poortahmasebi V, Baghi HB. Living in the shadows of hepatitis. Lancet Infect Dis (2019) 19(11):1171–2. doi: 10.1016/S1473-3099(19)30534-1
74. Xu J, Chen D, Jin L, Chen Z, Tu Y, Huang X, et al. Ubiquitously specific protease 4 inhibitor-Vialinin A attenuates inflammation and fibrosis in S100-induced hepatitis mice through Rheb/mTOR signalling. J Cell Mol Med (2021) 25(2):1140–50. doi: 10.1111/jcmm.16180
75. Xin SL, Yu YY. Ubiquitin-specific peptidase 10 ameliorates hepatic steatosis in nonalcoholic steatohepatitis model by restoring autophagic activity. Dig Liver Dis (2022) 54(8):1021–9. doi: 10.1016/j.dld.2022.02.009
76. Kusakabe S, Suzuki T, Sugiyama Y, Haga S, Horike K, Tokunaga M, et al. USP15 participates in hepatitis C virus propagation through regulation of viral RNA translation and lipid droplet formation. J Virol (2019) 93(6):e01708–18. doi: 10.1128/JVI.01708-18
77. Su ZJ, Cao JS, Wu YF, Chen WN, Lin X, Wu YL, et al. Deubiquitylation of hepatitis B virus X protein (HBx) by ubiquitin-specific peptidase 15 (USP15) increases HBx stability and its transactivation activity. Sci Rep (2017) 7:40246. doi: 10.1038/srep40246
78. Saxena N, Kumar V. The HBx oncoprotein of hepatitis B virus deregulates the cell cycle by promoting the intracellular accumulation and re-compartmentalization of the cellular deubiquitinase USP37. PloS One (2014) 9(10):e111256. doi: 10.1371/journal.pone.0111256
79. Ganesan M, Poluektova LY, Tuma DJ, Kharbanda KK, Osna NA. Acetaldehyde disrupts interferon alpha signaling in hepatitis C virus-infected liver cells by up-regulating USP18. Alcohol Clin Exp Res (2016) 40(11):2329–38. doi: 10.1111/acer.13226
80. Sung PS, Hong SH, Chung JH, Kim S, Park SH, Kim HM, et al. IFN-λ4 potently blocks IFN-α signalling by ISG15 and USP18 in hepatitis C virus infection. Sci Rep (2017) 7(1):3821. doi: 10.1038/s41598-017-04186-7
81. Zhang J, Liu K, Zhang G, Ling N, Chen M. Interleukin-17A pretreatment attenuates the anti-hepatitis B virus efficacy of interferon-alpha by reducing activation of the interferon-stimulated gene factor 3 transcriptional complex in hepatitis B virus-expressing HepG2 cells. Virol J (2022) 19(1):28. doi: 10.1186/s12985-022-01753-x
82. Sung PS, Cheon H, Cho CH, Hong SH, Park DY, Seo HI, et al. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc Natl Acad Sci U.S.A. (2015) 112(33):10443–8. doi: 10.1073/pnas.1513341112
83. MacParland SA, Ma XZ, Chen L, Khattar R, Cherepanov V, Selzner M, et al. Lipopolysaccharide and tumor necrosis factor alpha inhibit interferon signaling in hepatocytes by increasing ubiquitin-like protease 18 (USP18) expression. J Virol (2016) 90(12):5549–60. doi: 10.1128/JVI.02557-15
84. Murray EJ, Burden F, Horscroft N, Smith-Burchnell C, Westby M. Knockdown of USP18 increases α 2a interferon signaling and induction of interferon-stimulating genes but does not increase antiviral activity in Huh7 cells. Antimicrob Agents Chemother (2011) 55(9):4311–9. doi: 10.1128/AAC.00644-11
85. Asahina Y, Izumi N, Hirayama I, Tanaka T, Sato M, Yasui Y, et al. Potential relevance of cytoplasmic viral sensors and related regulators involving innate immunity in antiviral response. Gastroenterology (2008) 134(5):1396–405. doi: 10.1053/j.gastro.2008.02.019
86. Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology (2005) 128(5):1437–44. doi: 10.1053/j.gastro.2005.01.059
87. Li Y, Ma MX, Qin B, Lin LT, Richardson CD, Feld J, et al. The ubiquitin-specific protease 18 promotes hepatitis C virus production by increasing viral infectivity. Mediators Inflamm 2019. (2019), 3124745. doi: 10.1155/2019/3124745
88. Chen L, Li S, McGilvray I. The ISG15/USP18 ubiquitin-like pathway (ISGylation system) in hepatitis C virus infection and resistance to interferon therapy. Int J Biochem Cell Biol (2011) 43(10):1427–31. doi: 10.1016/j.biocel.2011.06.006
89. François-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PloS One (2011) 6(7):e22200. doi: 10.1371/journal.pone.0022200
90. Li L, Lei QS, Kong LN, Zhang SJ, Qin B. Gene expression profile after knockdown of USP18 in Hepg2.2.15 cells. J Med Virol (2017) 89(11):1920–30. doi: 10.1002/jmv.24819
91. Li Y, Yao M, Duan X, Ye H, Li S, Chen L, et al. The USP18 cysteine protease promotes HBV production independent of its protease activity. Virol J (2020) 17(1):47. doi: 10.1186/s12985-020-01304-2
92. An S, Zhao LP, Shen LJ, Wang S, Zhang K, Qi Y, et al. USP18 protects against hepatic steatosis and insulin resistance through its deubiquitinating activity. Hepatology (2017) 66(6):1866–84. doi: 10.1002/hep.29375
93. Li Q, Zhang YY, Chiu S, Hu Z, Lan KH, Cha H, et al. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PloS Pathog (2014) 10(5):e1004163. doi: 10.1371/journal.ppat.1004163
94. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. Bmj (2016) 353:i1585. doi: 10.1136/bmj.i1585
95. Vincent JL, Mira JP, Antonelli M. Sepsis: older and newer concepts. Lancet Respir Med (2016) 4(3):237–40. doi: 10.1016/S2213-2600(15)00522-6
96. Sheng L, Chen J, Tong Y, Zhang Y, Feng Q, Tang Z. USP9x promotes CD8 (+) T-cell dysfunction in association with autophagy inhibition in septic liver injury. Acta Biochim Biophys Sin (Shanghai) (2022) 54(12):1–10. doi: 10.3724/abbs.2022174
97. Gong X, Li Y, He Y, Zhou F. USP7-SOX9-miR-96-5p-NLRP3 network regulates myocardial injury and cardiomyocyte pyroptosis in sepsis. Hum Gene Ther (2022) 33(19-20):1073–90. doi: 10.1089/hum.2022.078
98. Gao F, Qian M, Liu G, Ao W, Dai D, Yin C. USP10 alleviates sepsis-induced acute kidney injury by regulating Sirt6-mediated Nrf2/ARE signaling pathway. J Inflammation (Lond) (2021) 18(1):25. doi: 10.1186/s12950-021-00291-7
99. Wei J, Dong S, Bowser RK, Khoo A, Zhang L, Jacko AM, et al. Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci Signal (2017) 10(483):eaak9660. doi: 10.1126/scisignal.aak9660
100. Hu B, Ge C, Zhu C. Ubiquitin-specific peptidase 18 negatively regulates and inhibits lipopolysaccharide-induced sepsis by targeting transforming growth factor-β-activated kinase 1 activity. Int Immunol (2021) 33(9):461–8. doi: 10.1093/intimm/dxab029
101. Lei CQ, Wu X, Zhong X, Jiang L, Zhong B, Shu HB. USP19 inhibits TNF-α- and IL-1β-triggered NF-κB activation by deubiquitinating TAK1. J Immunol (2019) 203(1):259–68. doi: 10.4049/jimmunol.1900083
102. Quan J, Zhao X, Xiao Y, Wu H, Di Q, Wu Z, et al. USP39 regulates NF-κB-mediated inflammatory responses through deubiquitinating K48-linked IκBα. J Immunol (2023) 210(5):640–52. doi: 10.4049/jimmunol.2200603
103. Li R, Li X, Zhao J, Meng F, Yao C, Bao E, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics (2022) 12(2):976–98. doi: 10.7150/thno.63751
104. Sun Y, Lu F, Yu X, Wang B, Chen J, Lu F, et al. Exogenous H(2)S promoted USP8 sulfhydration to regulate mitophagy in the hearts of db/db mice. Aging Dis (2020) 11(2):269–85. doi: 10.14336/AD.2019.0524
105. Li X, Wang T, Tao Y, Wang X, Li L, Liu J. MF-094, a potent and selective USP30 inhibitor, accelerates diabetic wound healing by inhibiting the NLRP3 inflammasome. Exp Cell Res (2022) 410(2):112967. doi: 10.1016/j.yexcr.2021.112967
106. Zhang T, Wang L, Chen L. Alleviative effect of microRNA-497 on diabetic neuropathic pain in rats in relation to decreased USP15. Cell Biol Toxicol (2023) 39(5):1–16. doi: 10.1007/s10565-022-09702-8
107. Li X, Wang T, Tao Y, Wang X, Li L, Liu J. Inhibition of USP7 suppresses advanced glycation end-induced cell cycle arrest and senescence of human umbilical vein endothelial cells through ubiquitination of p53. Acta Biochim Biophys Sin (Shanghai) (2022) 54(3):311–20. doi: 10.3724/abbs.2022003
108. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science (2021) 373(6554):510–6. doi: 10.1126/science.abh1654
109. Gorrepati KDD, Lupse B, Annamalai K, Yuan T, Maedler K, Ardestani A. Loss of deubiquitinase USP1 blocks pancreatic β-cell apoptosis by inhibiting DNA damage response. iScience (2018) 1:72–86. doi: 10.1016/j.isci.2018.02.003
110. Pearson G, Chai B, Vozheiko T, Liu X, Kandarpa M, Piper RC, et al. Clec16a, nrdp1, and USP8 form a ubiquitin-dependent tripartite complex that regulates β-cell mitophagy. Diabetes (2018) 67(2):265–77. doi: 10.2337/db17-0321
111. Meyerovich K, Violato NM, Fukaya M, Dirix V, Pachera N, Marselli L, et al. MCL-1 is a key antiapoptotic protein in human and rodent pancreatic β-cells. Diabetes (2017) 66(9):2446–58. doi: 10.2337/db16-1252
112. Malenczyk K, Girach F, Szodorai E, Storm P, Segerstolpe Å, Tortoriello G, et al. A TRPV1-to-secretagogin regulatory axis controls pancreatic β-cell survival by modulating protein turnover. EMBO J (2017) 36(14):2107–25. doi: 10.15252/embj.201695347
113. Honke N, Shaabani N, Zhang DE, Iliakis G, Xu HC, Häussinger D, et al. Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PloS Pathog (2013) 9(10):e1003650. doi: 10.1371/journal.ppat.1003650
114. Santin I, Moore F, Grieco FA, Marchetti P, Brancolini C, Eizirik DL. USP18 is a key regulator of the interferon-driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis (2012) 3(11):e419. doi: 10.1038/cddis.2012.158
115. Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and β-cell apoptosis. Diabetes Obes Metab (2013) 15 Suppl 3:71–81. doi: 10.1111/dom.12162
116. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol (2011) 11(2):98–107. doi: 10.1038/nri2925
117. Saito N, Kimura S, Miyamoto T, Fukushima S, Amagasa M, Shimamoto Y, et al. Macrophage ubiquitin-specific protease 2 modifies insulin sensitivity in obese mice. Biochem Biophys Rep (2017) 9:322–9. doi: 10.1016/j.bbrep.2017.01.009
118. Bai Y, Mo K, Wang G, Chen W, Zhang W, Guo Y, et al. Intervention of gastrodin in type 2 diabetes mellitus and its mechanism. Front Pharmacol (2021) 12:710722. doi: 10.3389/fphar.2021.710722
119. Forand A, Koumakis E, Rousseau A, Sassier Y, Journe C, Merlin JF, et al. Disruption of the phosphate transporter pit1 in hepatocytes improves glucose metabolism and insulin signaling by modulating the USP7/IRS1 interaction. Cell Rep (2016) 16(10):2736–48. doi: 10.1016/j.celrep.2016.08.012
120. Liu B, Zhang Z, Hu Y, Lu Y, Li D, Liu J, et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc Natl Acad Sci U.S.A. (2019) 116(43):21732–8. doi: 10.1073/pnas.1907288116
121. Coyne ES, Bédard N, Gong YJ, Faraj M, Tchernof A, Wing SS. The deubiquitinating enzyme USP19 modulates adipogenesis and potentiates high-fat-diet-induced obesity and glucose intolerance in mice. Diabetologia (2019) 62(1):136–46. doi: 10.1007/s00125-018-4754-4
122. Lu XY, Shi XJ, Hu A, Wang JQ, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature (2020) 588(7838):479–84. doi: 10.1038/s41586-020-2928-y
123. Kim A, Koo JH, Jin X, Kim W, Park SY, Park S, et al. Ablation of USP21 in skeletal muscle promotes oxidative fibre phenotype, inhibiting obesity and type 2 diabetes. J Cachexia Sarcopenia Muscle (2021) 12(6):1669–89. doi: 10.1002/jcsm.12777
124. Zhang S, Liu X, Wang J, Yuan F, Liu Y. Targeting ferroptosis with miR-144-3p to attenuate pancreatic β cells dysfunction via regulating USP22/SIRT1 in type 2 diabetes. Diabetol Metab Syndr (2022) 14(1):89. doi: 10.1186/s13098-022-00852-7
125. Niu Y, Jiang H, Yin H, Wang F, Hu R, Hu X, et al. Hepatokine ERAP1 disturbs skeletal muscle insulin sensitivity via inhibiting USP33-mediated ADRB2 deubiquitination. Diabetes (2022) 71(5):921–33. doi: 10.2337/db21-0857
126. Fu S, Zheng Y, Sun Y, Lai M, Qiu J, Gui F, et al. Suppressing long noncoding RNA OGRU ameliorates diabetic retinopathy by inhibition of oxidative stress and inflammation via miR-320/USP14 axis. Free Radic Biol Med (2021) 169:361–81. doi: 10.1016/j.freeradbiomed.2021.03.016
127. Noh M, Zhang H, Kim H, Park S, Kim YM, Kwon YG. Primaquine diphosphate, a known antimalarial drug, blocks vascular leakage acting through junction stabilization. Front Pharmacol (2021) 12:695009. doi: 10.3389/fphar.2021.695009
128. Kato M, Natarajan R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat Rev Nephrol (2014) 10(9):517–30. doi: 10.1038/nrneph.2014.116
129. Sun XH, Xiao HM, Zhang M, Lin ZY, Yang Y, Chen R, et al. USP9X deubiquitinates connexin43 to prevent high glucose-induced epithelial-to-mesenchymal transition in NRK-52E cells. Biochem Pharmacol (2021) 188:114562. doi: 10.1016/j.bcp.2021.114562
130. Huang K, Zhao X. USP9X prevents AGEs-induced upregulation of FN and TGF-β1 through activating Nrf2-ARE pathway in rat glomerular mesangial cells. Exp Cell Res (2020) 393(2):112100. doi: 10.1016/j.yexcr.2020.112100
131. Xu J, Deng Y, Wang Y, Sun X, Chen S, Fu G. SPAG5-AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/AKT/mTOR pathway. Cell Prolif (2020) 53(2):e12738. doi: 10.1111/cpr.12738
132. Xu E, Yin C, Yi X, Liu Y. Inhibition of USP15 ameliorates high-glucose-induced oxidative stress and inflammatory injury in podocytes through regulation of the Keap1/Nrf2 signaling. Environ Toxicol (2022) 37(4):765–75. doi: 10.1002/tox.23441
133. Huang KP, Chen C, Hao J, Huang JY, Liu PQ, Huang HQ. AGEs-RAGE system down-regulates Sirt1 through the ubiquitin-proteasome pathway to promote FN and TGF-β1 expression in male rat glomerular mesangial cells. Endocrinology (2015) 156(1):268–79. doi: 10.1210/en.2014-1381
134. Shi JX, Wang QJ, Li H, Huang Q. Silencing of USP22 suppresses high glucose-induced apoptosis, ROS production and inflammation in podocytes. Mol Biosyst (2016) 12(5):1445–56. doi: 10.1039/C5MB00722D
135. Elumalai S, Karunakaran U, Moon JS, Won KC. High glucose-induced PRDX3 acetylation contributes to glucotoxicity in pancreatic β-cells: Prevention by Teneligliptin. Free Radic Biol Med (2020) 160:618–29. doi: 10.1016/j.freeradbiomed.2020.07.030
136. Zhu S, Hou S, Lu Y, Sheng W, Cui Z, Dong T, et al. USP36-mediated deubiquitination of DOCK4 contributes to the diabetic renal tubular epithelial cell injury via wnt/β-catenin signaling pathway. Front Cell Dev Biol (2021) 9:638477. doi: 10.3389/fcell.2021.638477
137. Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA, et al. Small organic molecule disruptors of Cav3.2 - USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain (2015) 11:12. doi: 10.1186/s12990-015-0011-8
138. Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain (2016) 12:1744806916642444. doi: 10.1177/1744806916642444
139. Lu L, Ma J, Liu Y, Shao Y, Xiong X, Duan W, et al. FSTL1-USP10-notch1 signaling axis protects against cardiac dysfunction through inhibition of myocardial fibrosis in diabetic mice. Front Cell Dev Biol (2021) 9:757068. doi: 10.3389/fcell.2021.757068
140. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127(1):1–4. doi: 10.1172/JCI92035
141. Li DT, Habtemichael EN, Julca O, Sales CI, Westergaard XO, DeVries SG, et al. GLUT4 storage vesicles: specialized organelles for regulated trafficking. Yale J Biol Med (2019) 92(3):453–70.
142. Wen X, Zhang B, Wu B, Xiao H, Li Z, Li R, et al. Signaling pathways in obesity: mechanisms and therapeutic interventions. Signal Transduct Target Ther (2022) 7(1):298. doi: 10.1038/s41392-022-01149-x
143. Molusky MM, Li S, Ma D, Yu L, Lin JD. Ubiquitin-specific protease 2 regulates hepatic gluconeogenesis and diurnal glucose metabolism through 11β-hydroxysteroid dehydrogenase 1. Diabetes (2012) 61(5):1025–35. doi: 10.2337/db11-0970
144. Yang H, Seo SG, Shin SH, Min S, Kang MJ, Yoo R, et al. 3,3’-Diindolylmethane suppresses high-fat diet-induced obesity through inhibiting adipogenesis of pre-adipocytes by targeting USP2 activity. Mol Nutr Food Res (2017) 61(10). doi: 10.1002/mnfr.201700119
145. Kim TH, Yang YM, Han CY, Koo JH, Oh H, Kim SS, et al. Gα12 ablation exacerbates liver steatosis and obesity by suppressing USP22/SIRT1-regulated mitochondrial respiration. J Clin Invest (2018) 128(12):5587–602. doi: 10.1172/JCI97831
146. Luo P, Qin C, Zhu L, Fang C, Zhang Y, Zhang H, et al. Ubiquitin-specific peptidase 10 (USP10) inhibits hepatic steatosis, insulin resistance, and inflammation through sirt6. Hepatology (2018) 68(5):1786–803. doi: 10.1002/hep.30062
147. Zhang B, Li H, Li D, Sun H, Li M, Hu H. Long noncoding RNA Mirt2 upregulates USP10 expression to suppress hepatic steatosis by sponging miR-34a-5p. Gene (2019) 700:139–48. doi: 10.1016/j.gene.2019.02.096
148. Zhang Y, Yang Z, Xu Z, Wan J, Hua T, Sun Q. Exercise ameliorates insulin resistance and improves SIRT6-mediated insulin signaling transduction in liver of obese rats. Can J Physiol Pharmacol (2021) 99(5):506–11. doi: 10.1139/cjpp-2020-0083
149. Liu H, Ding J, Köhnlein K, Urban N, Ori A, Villavicencio-Lorini P, et al. The GID ubiquitin ligase complex is a regulator of AMPK activity and organismal lifespan. Autophagy (2020) 16(9):1618–34. doi: 10.1080/15548627.2019.1695399
150. Liu B, Jiang S, Li M, Xiong X, Zhu M, Li D, et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat Commun (2018) 9(1):4770. doi: 10.1038/s41467-018-07185-y
151. Bag S, Ramaiah S, Anbarasu A. fabp4 is central to eight obesity associated genes: a functional gene network-based polymorphic study. J Theor Biol (2015) 364:344–54. doi: 10.1016/j.jtbi.2014.09.034
152. Huang X, Ordemann J, Müller JM, Dubiel W. The COP9 signalosome, cullin 3 and Keap1 supercomplex regulates CHOP stability and adipogenesis. Biol Open (2012) 1(8):705–10. doi: 10.1242/bio.20121875
153. Zhang Y, Wan J, Xu Z, Hua T, Sun Q. Exercise ameliorates insulin resistance via regulating TGFβ-activated kinase 1 (TAK1)-mediated insulin signaling in liver of high-fat diet-induced obese rats. J Cell Physiol (2019) 234(5):7467–74. doi: 10.1002/jcp.27508
154. Song W, Yang K, Luo J, Gao Z, Gao Y. Dysregulation of USP18/FTO/PYCR1 signaling network promotes bladder cancer development and progression. Aging (Albany NY) (2021) 13(3):3909–25. doi: 10.18632/aging.202359
155. Basson MD, Wang Q, Chaturvedi LS, More S, Vomhof-DeKrey EE, Al-Marsoummi S, et al. Schlafen 12 interaction with serpinB12 and deubiquitylases drives human enterocyte differentiation. Cell Physiol Biochem (2018) 48(3):1274–90. doi: 10.1159/000492019
156. Sun T, Liu Z, Yang Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol Cancer (2020) 19(1):146. doi: 10.1186/s12943-020-01262-x
157. Bonacci T, Emanuele MJ. Dissenting degradation: Deubiquitinases in cell cycle and cancer. Semin Cancer Biol (2020) 67(Pt 2):145–58. doi: 10.1016/j.semcancer.2020.03.008
158. Zhao Z, Su Z, Liang P, Liu D, Yang S, Wu Y, et al. USP38 couples histone ubiquitination and methylation via KDM5B to resolve inflammation. Adv Sci (Weinh) (2020) 7(22):2002680. doi: 10.1002/advs.202002680
159. Hou X, Zhu F, Ni Y, Chen T, Du J, Liu X, et al. USP4 is pathogenic in allergic airway inflammation by inhibiting regulatory T cell response. Life Sci (2021) 281:119720. doi: 10.1016/j.lfs.2021.119720
160. Yang J, Wei P, Barbi J, Huang Q, Yang E, Bai Y, et al. The deubiquitinase USP44 promotes Treg function during inflammation by preventing FOXP3 degradation. EMBO Rep (2020) 21(9):e50308. doi: 10.15252/embr.202050308
161. Cockram PE, Kist M, Prakash S, Chen SH, Wertz IE, Vucic D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ (2021) 28(2):591–605. doi: 10.1038/s41418-020-00708-5
162. Chen S, Liu Y, Zhou H. Advances in the development ubiquitin-specific peptidase (USP) inhibitors. Int J Mol Sci (2021) 22(9):4546. doi: 10.3390/ijms22094546
163. Oliveira RI, Guedes RA, Salvador JAR. Highlights in USP7 inhibitors for cancer treatment. Front Chem (2022) 10:1005727. doi: 10.3389/fchem.2022.1005727
164. Zou M, Zeng QS, Nie J, Yang JH, Luo ZY, Gan HT. The role of E3 ubiquitin ligases and deubiquitinases in inflammatory bowel disease: friend or foe? Front Immunol (2021) 12:769167. doi: 10.3389/fimmu.2021.769167
165. Gao H, Yin J, Ji C, Yu X, Xue J, Guan X, et al. Targeting ubiquitin specific proteases (USPs) in cancer immunotherapy: from basic research to preclinical application. J Exp Clin Cancer Res (2023) 42(1):225. doi: 10.1186/s13046-023-02805-y
Keywords: deubiquitination, ubiquitin-specific proteases, inflammatory diseases, protein stability, targeted therapies
Citation: Chen R, Zhang H, Li L, Li J, Xie J, Weng J, Tan H, Liu Y, Guo T and Wang M (2024) Roles of ubiquitin-specific proteases in inflammatory diseases. Front. Immunol. 15:1258740. doi: 10.3389/fimmu.2024.1258740
Received: 14 July 2023; Accepted: 02 January 2024;
Published: 23 January 2024.
Edited by:
Eva Turley, Lawson Health Research Institute, CanadaReviewed by:
Min Fan, East Tennessee State University, United StatesCopyright © 2024 Chen, Zhang, Li, Li, Xie, Weng, Tan, Liu, Guo and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanjun Liu, bGl1eWFuanVuXzAwMUAxNjMuY29t; Tailin Guo, dGxndW9AaG9tZS5zd2p0dS5lZHUuY24=; Mengyuan Wang, d2FuZ21lbmd5dWFuQHN3anR1LmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.