 Chloe S. Yang
Chloe S. Yang Craig M. Coopersmith
Craig M. Coopersmith John D. Lyons
John D. Lyons
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 11 January 2024
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1347401
This article is part of the Research Topic Adjuvant Immunotherapies and Regulation of Cell Death – A New Way Forward in Sepsis? View all 6 articles
Cell death proteins play a central role in host immune signaling during sepsis. These interconnected mechanisms trigger cell demise via apoptosis, necroptosis, and pyroptosis while also driving inflammatory signaling. Targeting cell death mediators with novel therapies may correct the dysregulated inflammation seen during sepsis and improve outcomes for septic patients.
Programmed cell death (PCD) networks are fundamental components of the host response to infection (1, 2). These front-line defense mechanisms both eliminate infected cells and trigger inflammation, and they are increasingly recognized as significant contributors to the dysregulated immune environment of sepsis (3–7). Importantly, these networks display significant overlap and interplay with one another, and attempts at manipulating PCD cascades must contend with diverse and complex signaling outcomes. This review will highlight core mediators of PCD pathways and examine current and potential future therapeutic strategies to target cell death proteins in sepsis.
The caspases are a family of highly evolutionarily conserved proteases that cleave peptide bonds within proteins by hydrolysis. Perhaps best known as mediators of apoptosis (Figure 1), caspases are now understood to also support inflammatory signaling, and distinct groups of pro-inflammatory and apoptotic caspase members have been well described (8, 9).
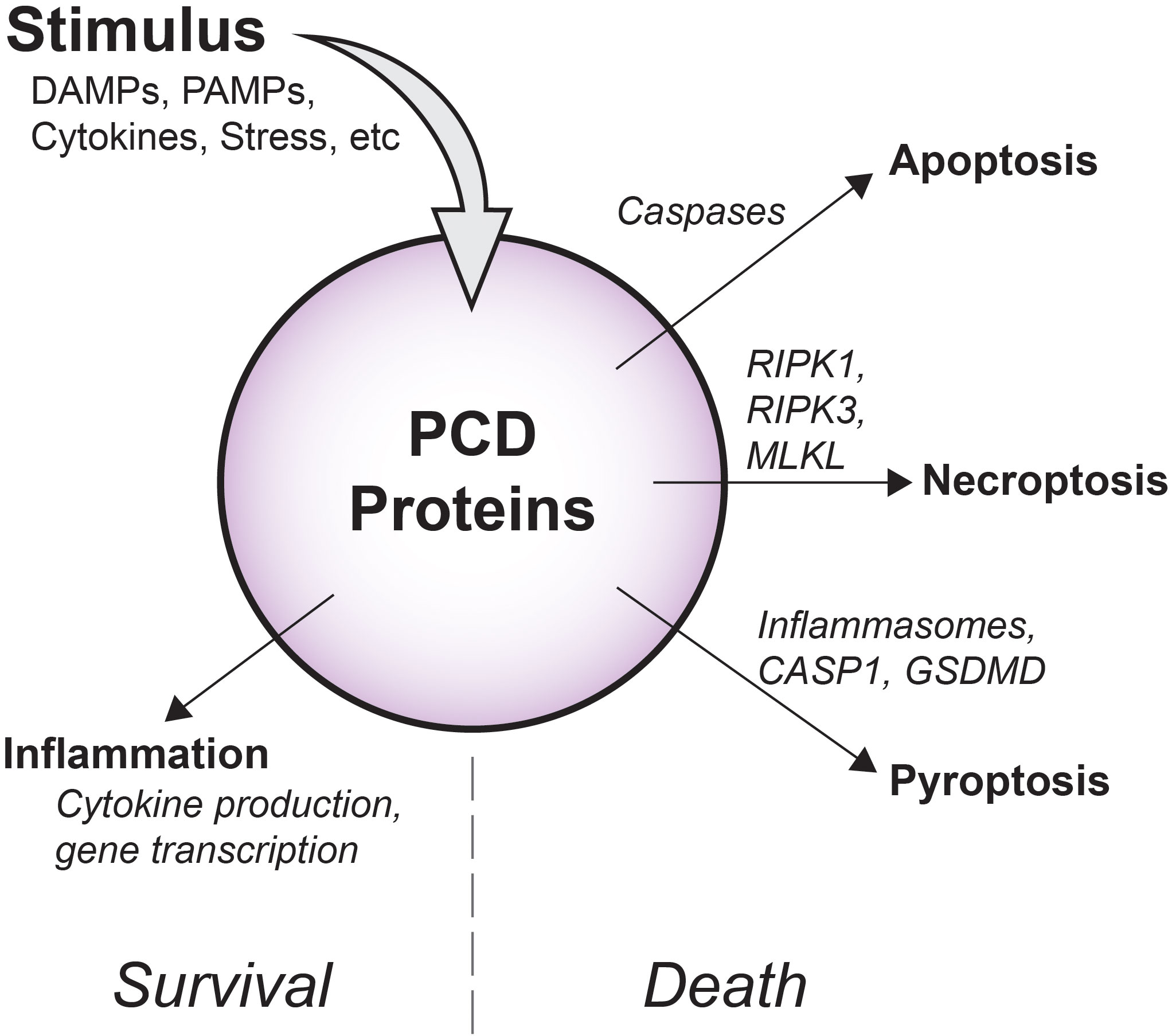
Figure 1 Overview of diverse PCD signaling outcomes. Depending on cellular conditions, flux through PCD signaling pathways has variable outcomes. Apoptotic caspases drive non-inflammatory cell death via apoptosis, while RIPK1, RIPK3, and MLKL support inflammatory membrane rupture via necroptosis. As part of inflammasomes, pro-inflammatory CASP1 promotes cleavage of GSDMD and cell death by pyroptosis. Importantly, in some contexts, these molecules may also induce inflammatory gene transcription and cytokine production that occurs without causing cell death.
Inflammatory caspases (also called “Group I caspases”) cleave inactive mediators into their functional forms (9). Caspase-1 (CASP1) was first identified as the enzyme responsible for the conversion of pro-interleukin-1β (IL-1β) to IL-1β, and it plays a similar role in the maturation of IL-18. Group I also includes CASP4 and CASP5. These proteases do not participate in IL-1β production, but rather, in concert with CASP1, hydrolyze and activate gasdermin-D (GSDMD), a pore-forming molecule that perforates cell membranes to release cytokines and damage associated molecular patterns (DAMPs) in the lytic cell death mode of pyroptosis (Figure 1). Importantly, CASP4/5 (and the murine equivalent, CASP11), also bind directly to cytosolic lipopolysaccharide (LPS) which causes auto-activation, demonstrating that inflammatory caspases also function as direct pathogen sensors (10).
Apoptotic caspases include both initiator and effector varieties (“Group II” and “Group III” caspases, respectively) (9). When activated, these enzymes cleave hundreds of target sequences within cell proteins, facilitating homeostatic tissue turnover and non-inflammatory cell death by apoptosis (9). Unlike pyroptosis, apoptosis does not disrupt cell membrane integrity, and cell components are broken down and packaged into apoptotic bodies for orderly clearance by phagocytes. A plethora of stimuli promote apoptotic caspase signaling, including mitochondrial stress, DNA damage, and infection (11).
Sepsis induces broad and diverse caspase activity, and the resulting impact on septic immune dysfunction is complex and context-dependent. Initial descriptions of apoptotic caspases in sepsis noted pronounced death of immune cells and gut epithelial cells (12), and later studies revealed that transgenic animals with inhibited apoptotic machinery consistently display improved sepsis survival (13–15). Inflammatory caspases support robust cytokine production early in sepsis and may trigger pyroptosis in infected patients, though whether these signaling outcomes are beneficial or detrimental to the host cannot be uniformly stated (16–19). Moreover, it must be emphasized that substantial crosstalk occurs between caspase subfamilies and other cell death machinery, and any therapeutic approach based on caspase modulation must consider complicated signaling dynamics (8). For instance, caspase-8 actively protects cells against stimulation of receptor interacting protein kinase 3 (RIPK3) which would cause cell death by necroptosis, and correspondingly, inhibition of RIPK3 function may predispose cells to death by caspase-mediated apoptosis (Figure 2) (20–22). Such trap door mechanisms abound in PCD signaling and must be carefully anticipated to avoid unintended cellular toxicity.
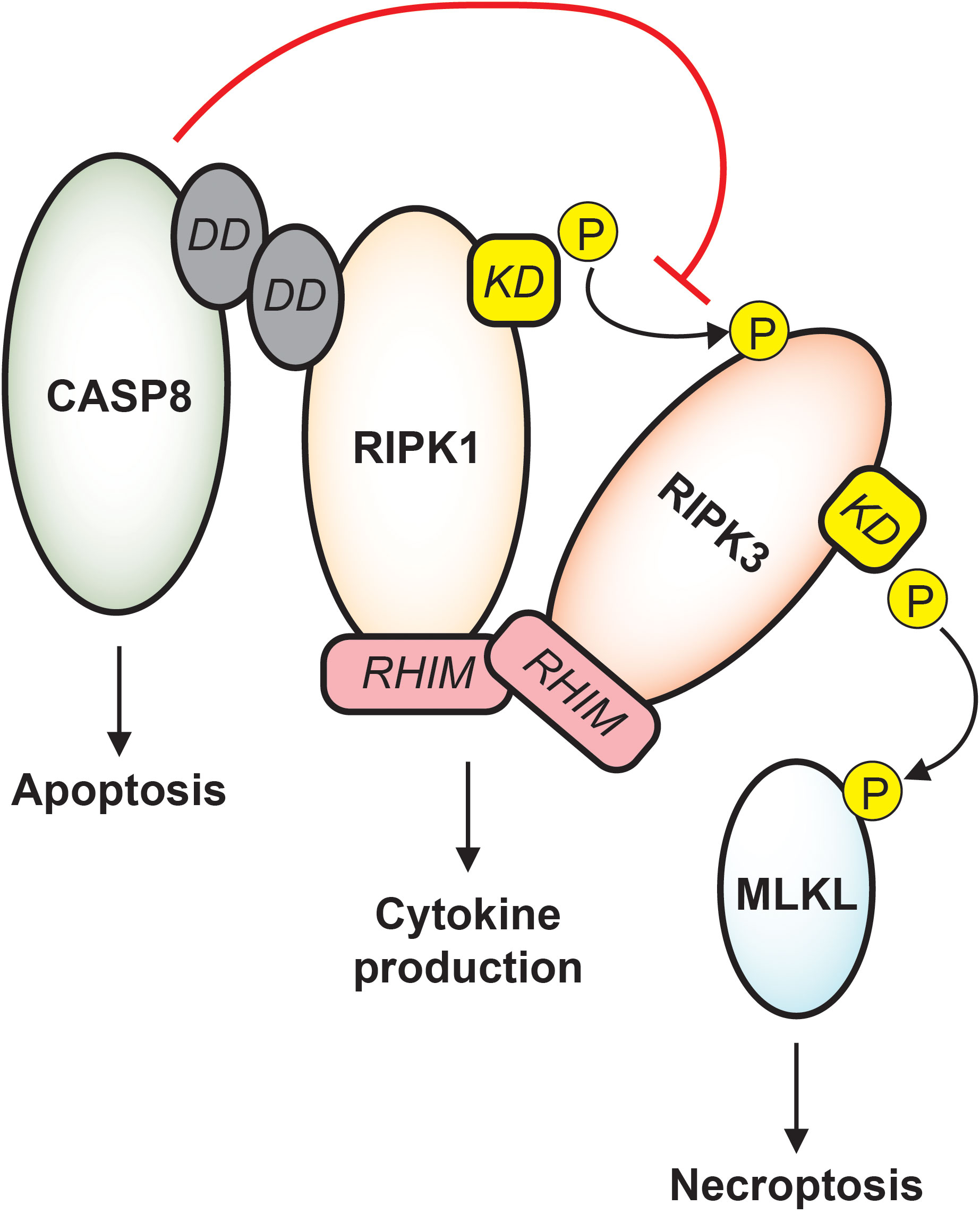
Figure 2 Apoptotic and necroptotic crosstalk. CASP8 interacts with RIPK1 via death domain (DD) binding. Downstream of TNF receptor signaling, CASP8 drives apoptosis while inhibiting necroptosis. RIPK1 and RIPK3 binding is stabilized by RHIM-RHIM interactions, and under conditions of CASP8 inhibition, the RIPK1 kinase domain (KD) phosphorylates RIPK3 which phosphorylates MLKL, causing cell death by necroptosis. RIPK1/RIPK3 RHIM interactions also support cytokine production that is independent of both apoptosis and necroptosis, and destabilization of RIPK1 or RIPK3 may trigger caspase-mediated apoptosis.
The receptor interacting protein (RIP) kinases are a group of ubiquitously expressed proteins that regulate diverse cell functions. Each member of the RIPK family contains a homologous kinase domain and variable structural domains that dictate involvement with a range of cell signaling networks. RIPK1, for example, was first identified by virtue of death domain interactions with the cell surface receptor CD95 and was therefore referred to as “receptor interacting protein”. RIPK1 and RIPK3 are directly relevant to septic immune dysfunction, as each play a central role in mediating cell inflammation as well as death by necroptosis (23).
Necroptosis is a lytic and highly inflammatory mode of cell death and is similar to pyroptosis in its explosive release of intracellular contents (24). However, necroptotic membrane pores are formed by the pseudokinase mixed lineage kinase domain like (MLKL), a protein with general kinase structure that lacks ability to perform phosphorylation (25). Importantly, the two most well characterized upstream activators of MLKL are RIPK1 and RIPK3. In the context of TNF signaling, RIPK1 phosphorylates RIPK3, and RIPK3 in turn phosphorylates MLKL which then oligomerizes into channels that perforate the cell (Figure 2) (24). This RIPK1-RIPK3-MLKL sequence is but one example of many paths that lead to necroptosis, some of which proceed directly through RIPK3 and do not require RIPK1 (26). Additionally, activation of upstream regulators does not guarantee RIPK3-mediated cell death. Functional caspase-8 restricts necroptosis, highlighting the interconnectedness of these pathways and emphasizing that ultimate signaling outcomes are highly dependent on cellular conditions (22, 24).
Crucially, RIPK1 and RIPK3 also each facilitate signaling responses that are independent of cell death. The somewhat confusingly titled “death domain” on RIPK1 allows participation in protein complexes that govern signaling outcomes independent of cell death, such as pro-survival cytokine production (23). Furthermore, RIPK1 and RIPK3 each contain a RIP homotypic interaction motif (RHIM), a domain that forms β-amyloid-like binding with other RHIM-expressing proteins. This motif expands the function of these proteins as scaffolds in other death-independent signaling cascades including cytokine production and cell cycle progression (Figure 2) (23, 27).
Given the complex array of potential signaling outcomes downstream of RIPK1 and RIPK3, the understanding of their role in sepsis remains incomplete and is actively evolving. Though necroptosis was initially posited as a driver of multiple inflammatory pathologies, including sepsis, evidence of widespread necroptosis in septic humans is lacking, and the hope of limiting septic inflammation by narrowly targeting the RIPK1 or RIPK3 kinase domain is likely misplaced (27, 28). However, cohorts of septic patients do display increased expression of RIPK1, RIPK3 and MLKL (29–31), implying these mediators may be involved in sepsis progression. Additionally, animal data suggests the RIPK3 RHIM is a potent driver of septic inflammation independent of its cell death function (3), and loss of RIPK3, but not MLKL, improves control of viral infection (32). Thus, receptor interacting proteins are likely to contribute to host inflammatory signaling during sepsis, though perhaps via mechanisms that extend beyond necroptosis.
Inflammasomes are cytosolic multi-protein complexes that facilitate inflammatory caspase activity, thus stimulating IL-1β production and pyroptosis (33). Inflammasome formation is based upon recognition of distinct danger signals through various pattern recognition receptors (PRRs). These PRRs bind to distinct pathogen-associated molecular patterns (PAMPs) or DAMPs, thus triggering inflammasome protein oligomerization and the recruitment of an adapter termed “apoptosis-associated speck-like protein containing CARD” (ASC). “CARD” refers to “caspase recruitment domain”, and it is this region of ASC that ultimately allows recruitment and activation of CASP1 (33). Inflammasomes can therefore be viewed as having several variable building block components: families of receptor proteins to detect danger signals, adapter proteins to form signaling complexes, and caspase-1 to cleave downstream targets (34). As mentioned previously, CASP4/5 may function as a direct sensor of LPS and is therefore termed a “non-canonical” inflammasome (35). As with other PCD mediators, the outcome of inflammasome signaling is dependent on cell conditions, and inflammasome activity cannot be assumed to result exclusively in IL-1β production or GSDMD cleavage and subsequent pyroprotis (36).
When activated during infection, inflammasomes represent a key component of the host immune response and are often protective, as loss of inflammasome function worsens bacterial and fungal infections in mice (37, 38). Likewise, in some human sepsis patients, sustained inflammasome activation is associated with improved survival (39). However, this finding is not consistent across sepsis models or patient cohorts, as enhanced inflammasome signaling in some instances correlates with increased mortality (40, 41), and deletion of inflammasome components may actually improve septic animal survival (42). The variability in these data may well reflect the heterogeneity within septic cohorts themselves. Broad analyses of inflammatory activity within septic patient populations have identified both hyper- and hypo-inflammatory endotypes, and these findings support the intuitive hypothesis that it is possible to have both too much and too little inflammation when battling infection (43–45). Thus, while increased inflammasome activity may be detrimental to a hyper-inflammatory endotype, it may prove beneficial in patients with more hypo-inflammatory or immunosuppressed phenotypes. The impact of inflammasome signaling on septic outcomes, as with other PCD mediators, is likely context dependent.
Detection of non-self nucleic acid sequences is a highly conserved form of innate immunity, and PRRs that bind DNA and RNA are intimately involved with host PCD signaling mechanisms (46, 47). Recognition of immunostimulatory nucleic acids by these receptors triggers both cytokine production and host cell death, depending on cell conditions. Circulating levels of DNA and RNA – released either from pathogens or from damaged host nuclei and mitochondria – increase during sepsis, and the impact of this antigen load on the overall immune response during sepsis is increasingly appreciated (48–50).
Nucleic acid sensors with particular relevance to bacterial sepsis include the cGAS-STING pathway, Toll-like receptor (TLR) 9, and z-DNA binding protein 1 (ZBP1). Cyclic GMP-AMP synthase (cGAS) is a sensor found in both the cytoplasm and sub-cellular compartments that binds to DNA and activates an endoplasmic reticulum protein termed “stimulator of interferon genes” (STING) (51). STING activation causes production of Type I interferons and inflammatory mediators and may trigger an array of cell death outcomes including apoptosis, pyroptosis, necroptosis, and autophagy, the breakdown of cellular components mediated by lysosomes (47, 51, 52). TLR9 recognizes unmethylated CpG DNA common in bacterial pathogens to stimulate inflammatory responses (53, 54), while RHIM-containing ZBP1 binds z-form nucleic acid structures and mediates diverse signaling outcomes including cell death (52). Each of these sensors and pathways are now understood to support inflammatory responses during bacterial infections, though how they might contribute to immune dysregulation in overt sepsis remains an open question. One intriguing possibility is that these mechanisms perpetuate a feed-forward cycle of host damage and continuous immune stimulation. In this scenario, PCD signaling causes cell death or damage and release of DNA and RNA molecules, and these are then sensed by PRRs, creating even more inflammation perpetuating host damage. Interestingly, knockout of TLR9 or treatment with circulating DNA scavengers reduces inflammation and improves septic animal survival, suggesting these pathways may indeed contribute to dysregulated immune signaling in sepsis and merit ongoing investigation (48, 55).
Several synthetic caspase inhibitors have been developed with the hope of treating diverse disease processes. Both specific and non-specific inhibitors have been evaluated in clinical trials and have previously been reviewed in detail (56). The pan-caspase inhibitor z-VAD-FMK effectively suppresses broad caspase activity and has been instrumental in detailing necroptotic signaling that arises when CASP8 function is compromised (56, 57). Treatment of septic mice with z-VAD-FMK limits sepsis-induced lymphocyte apoptosis and improves survival (5), as does local thymus injection with another broad caspase inhibitor z-LEHD-FMK (58). More specific CASP1 inhibitors have also shown promise in inflammatory disease models (56). Despite these findings, limited animal data and toxicities in human trials have withheld caspase inhibitors from widespread clinical adoption (56).
Given their emerging role in host inflammatory responses, RIPK1 and RIPK3 have also been targeted with the development of several small molecule kinase inhibitors. Necrostatin-1 (Nec-1) is an allosteric RIPK1 kinase inhibitor that prevents RIPK1-RIPK3 complex formation and inhibits kinase-mediated necroptotic signaling through RIPK1 (59). Though Nec-1 blocks phosphorylation of RIPK3 by RIPK1, it does not block RIPK3 autophosphorylation or necroptotic signaling that proceeds directly through RIPK3-MLKL (60–62). Nec-1 limits inflammation and cell death following LPS challenge (63), but treatment of polymicrobial sepsis in mice with Nec-1 surprisingly resulted in worsened mortality and increased inflammation (64). Such seemingly paradoxical findings have also been documented with use RIPK3 kinase inhibitors. Though pharmaceutical blockade of the RIPK3 kinase domain efficiently restricts necroptosis, it also unleashes caspase-mediated apoptosis, counteracting protective effects of necroptosis inhibition (21, 62). Genetic mutation of the RIPK3 kinase domain may also trigger unintended apoptosis via RHIM signaling, indicating manipulation of these mediators must be approached with care (21). Additionally, it should be emphasized that RIP kinase inhibition would not necessarily limit RHIM-dependent signaling outcomes and as evidenced may actually enhance these signal flux through kinase-independent pathways (3, 4). Ultimately, our limited understanding of the discrete mechanistic contributions of the RIP molecules to septic inflammation limits the therapeutic potential of isolated kinase domain inhibition in sepsis.
RNA interference (RNAi) is a conserved phenomenon across multicellular organisms whereby specific non-coding nucleic acid molecules inhibit translation of messenger RNA (mRNA) into functional protein, thus silencing gene expression (65). This highly-specific process serves not only as a homeostatic mechanism, but also as an arm of host defense, preventing translation of exogenous genetic material from invading microbes (66). Since its discovery, the concept of RNAi has been harnessed as a therapeutic tool to allow silencing of unwanted gene expression in disease states. This approach often utilizes small interfering RNA (siRNA), which are short, double-stranded RNA sequences that engage native RNAi machinery and suppress expression of the gene of interest (67).
Several siRNA-based therapies have been developed, and at least 10 are either approved or in late-stage clinical trials, treating conditions ranging from amyloidosis to acute kidney injury (68). These agents are not intended to treat sepsis, though preclinical models suggest targeting host PCD machinery may be beneficial, as si-RNA inhibition of BIM, a pro-apoptotic protein, limits lymphocyte apoptosis and improves overall survival in septic mice, and similar results are achieved by repressing CASP8 (69–71). Inhibition of RIPK3 with siRNA limits gut epithelial necroptosis and protects against colitis, an approach that could theoretically limit the gut barrier damage of sepsis and prevent ongoing immune stimulation (72, 73). Additionally, siRNA knockdown of high mobility group box protein 1 (HMGB1), a key DAMP released during necroptosis and pyroptosis (57, 74), rescues mice from septic mortality (75). These data indicate that targeting signal transduction distal to actual cell death events may be a useful strategy to combat PCD-driven inflammation. Despite these promising findings, significant hurdles remain in developing viable siRNA treatments for human septic patients, and no siRNA approaches are being studied in active sepsis clinical trials. Importantly, siRNA therapy would not eliminate existing proteins or prevent them from relaying signals, but rather would only stop new gene expression sometime after administration. Thus, therapies intended to limit early PCD events in sepsis like lymphocyte apoptosis would only be effective if given prior to the septic event (71, 75), and siRNA-mediated inhibition of the initial inflammatory surge of sepsis may not be feasible. Rather, siRNA-based therapies may be better directed at immunosuppressive proteins acting later in the course of sepsis, stopping their production before it occurs. Knowledge regarding the role of PCD proteins in late-phase sepsis is limited, and it remains to be seen if previous success of septic siRNA therapies can be translated to clinical applications.
In contrast to therapies using RNAi, which prevents expression of a given protein, therapies based on messenger RNA (mRNA) delivery induce protein expression by directing mRNA sequences into cells for translation by host ribosomes (76). This approach permits expression of essentially any chosen protein, creating substantial opportunity to modulate cell signaling for therapeutic benefit. Numerous clinical trials evaluating mRNA-based vaccines against viruses and malignancies are actively underway, and mRNA strategies have already proven effective and well tolerated in the widespread use of vaccines against SARS-CoV-2 (77). Considering this success, future mRNA-based sepsis therapies could conceivably be directed at host PCD machinery.
Given their central role in host defense, PCD proteins have been evolutionary targets of invading pathogens seeking to aid infection (78–80). As a result, several microbe-derived molecules that directly inhibit PCD signaling have been identified. E. coli expresses a protease that specifically cleaves RHIM domains, limiting RIPK1 and RIPK3-mediated necroptosis, and cytomegalovirus (CMV) infection produces proteins that restrict both RHIM interactions and caspase-8 function (79, 81–83). In theory, these PCD inhibitors could be coded into mRNA and delivered as a therapy. mRNA for the CMV protein M45, a RHIM inhibitor, does reduce cytokine production from infected macrophages in vitro, though it is unknown how such an approach would impact the widespread inflammation associated with sepsis (3). mRNA technology is rapidly evolving, with novel refinements being made to improve the efficacy and specificity of delivery, and preclinical animal models demonstrate the feasibility of combating infection with mRNA-based tools (84). Considering exogenous mRNA can be translated into protein within hours of delivery, it seems feasible that deleterious effects of host PCD signaling could be interrupted by timely administration of future mRNA therapies. However, mRNA therapies for sepsis have yet to be evaluated in trials, and ongoing studies will need to more clearly define how possible immunostimulatory effects of mRNA delivery will impact the dysregulated immune state of sepsis.
CRISPR/Cas technology has revolutionized gene editing and has been extensively reviewed elsewhere (85–87).While the CRISPR-associated (Cas) protein Cas9 cleaves DNA at specific sites to allow permanent gene editing (which would not be a viable approach for human sepsis patients), mutated “dead” Cas9 proteins (dCas9) have been developed that lack enzymatic activity and cannot cut DNA but still bind to target sequences (86). Coupled with promotor or suppressor proteins, dCas9 fusion proteins can function as specific and temporary silencers or inducers of gene expression (86, 88). Cas9 proteins can be delivered as intact, functional proteins, but they can also be delivered in the form of mRNA precursors that are then translated into active dCas9 molecules (89, 90). Though this novel technology is still in its infancy, Cas9 systems have been utilized to suppress inflammasome function in skin disease (90), and it is conceivable that similar approaches could someday be incorporated into clinical trials aiming to fine-tune pathologic PCD signaling in septic patients.
Programmed cell death mediators and their associated networks are central components of the host immune response during sepsis, dictating cell fate and directing inflammatory cascades. Several PCD mechanisms show promise as therapeutic targets in sepsis, though the substantial interconnectedness within PCD signaling arms requires ongoing analysis and a more complete description before preclinical findings can be translated to clinical use. As our understanding of these pathways increases, novel biotechnologies will offer unprecedented an ability to manipulate elements of PCD machinery and guide inflammatory signaling toward improved outcomes for sepsis patients.
CY: Investigation, Writing – original draft. CC: Conceptualization, Supervision, Writing – review & editing. JL: Conceptualization, Investigation, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. CC is funded by NIH R35GM148217. JDL is funded by NIH R35GM151220 and ARPA-H CUREIT: Curing the Uncurable via RNA-Encoded Immunogene Tuning.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol (2017) 17(5):333–40. doi: 10.1038/nri.2016.153
2. Place DE, Lee S, Kanneganti TD. PANoptosis in microbial infection. Curr Opin Microbiol (2021) 59:42–9. doi: 10.1016/j.mib.2020.07.012
3. Lyons JD, Mandal P, Otani S, Chihade DB, Easley KF, Swift DA, et al. The RIPK3 scaffold regulates lung inflammation during pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol (2023) 68(2):150–60. doi: 10.1165/rcmb.2021-0474OC
4. Orozco S, Oberst A. RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol Rev (2017) 277(1):102–12. doi: 10.1111/imr.12536
5. Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol (2000) 1(6):496–501. doi: 10.1038/82741
6. Mallarpu CS, Ponnana M, Prasad S, Singarapu M, Kim J, Haririparsa N, et al. Distinct cell death markers identified in critical care patient survivors diagnosed with sepsis. Immunol Lett (2021) 231:1–10. doi: 10.1016/j.imlet.2020.12.009
7. Zheng X, Chen W, Gong F, Chen Y, Chen E. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: A review. Front Immunol (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
8. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity (2019) 50(6):1352–64. doi: 10.1016/j.immuni.2019.05.020
9. Julien O, Wells JA. Caspases and their substrates. Cell Death Differ (2017) 24(8):1380–9. doi: 10.1038/cdd.2017.44
10. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/s41580-018-0089-8
11. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol (2019) 20(3):175–93. doi: 10.1038/s41580-018-0089-8
12. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med (1999) 27(7):1230–51. doi: 10.1097/00003246-199907000-00002
13. Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci United States Am (1999) 96(25):14541–6. doi: 10.1073/pnas.96.25.14541
14. Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM 2nd, Buchman TG, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. Jama (2002) 287(13):1716–21. doi: 10.1001/jama.287.13.1716
15. Bommhardt U, Chang KC, Swanson PE, Wagner TH, Tinsley KW, Karl IE, et al. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol (2004) 172(12):7583–91. doi: 10.4049/jimmunol.172.12.7583
16. Zhu Q, Zheng M, Balakrishnan A, Karki R, Kanneganti TD. Gasdermin D Promotes AIM2 Inflammasome Activation and Is Required for Host Protection against Francisella novicida. J Immunol (2018) 201(12):3662–8. doi: 10.4049/jimmunol.1800788
17. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep (2018) 22(11):2924–36. doi: 10.1016/j.celrep.2018.02.067
18. Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PloS Pathogens (2019) 15(4):e1007709. doi: 10.1371/journal.ppat.1007709
19. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res (2022) 1(8):732–47. doi: 10.1038/s44161-022-00108-7
20. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature (2011) 471(7338):368–72. doi: 10.1038/nature09857
21. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell (2014) 56(4):481–95. doi: 10.1016/j.molcel.2014.10.021
22. Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev (2017) 277(1):76–89. doi: 10.1111/imr.12541
23. Clucas J, Meier P. Roles of RIPK1 as a stress sentinel coordinating cell survival and immunogenic cell death. Nat Rev Mol Cell Biol (2023) 24(11):835–52. doi: 10.1038/s41580-023-00623-w
24. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
25. Zhan C, Huang M, Yang X, Hou J. MLKL: Functions beyond serving as the Executioner of Necroptosis. Theranostics (2021) 11(10):4759–69. doi: 10.7150/thno.54072
26. Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci United States Am (2017) 114(43):11506–11. doi: 10.1073/pnas.1700999114
27. Wegner KW, Saleh D, Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci (2017) 38(3):202–25. doi: 10.1016/j.tips.2016.12.005
28. Linkermann A, Green DR. Necroptosis. New Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
29. Yu L, Yang K, He X, Li M, Gao L, Zha Y. Repositioning linifanib as a potent anti-necroptosis agent for sepsis. Cell Death Discovery (2023) 9(1):57. doi: 10.1038/s41420-023-01351-y
30. Sureshbabu A, Patino E, Ma KC, Laursen K, Finkelsztein EJ, Akchurin O, et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight (2018) 3(11). doi: 10.1172/jci.insight.98411
31. Shashaty MGS, Reilly JP, Faust HE, Forker CM, Ittner CAG, Zhang PX, et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: a cohort study. Crit Care (2019) 23(1):235. doi: 10.1186/s13054-019-2482-x
32. Preston SP, Allison CC, Schaefer J, Clow W, Bader SM, Collard S, et al. A necroptosis-independent function of RIPK3 promotes immune dysfunction and prevents control of chronic LCMV infection. Cell Death Dis (2023) 14(2):123. doi: 10.1038/s41419-023-05635-0
33. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
34. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
35. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
36. Gaidt MM, Hornung V. Alternative inflammasome activation enables IL-1beta release from living cells. Curr Opin Immunol (2017) 44:7–13. doi: 10.1016/j.coi.2016.10.007
37. Goncalves AC, Ferreira LS, Manente FA, de Faria C, Polesi MC, de Andrade CR, et al. The NLRP3 inflammasome contributes to host protection during Sporothrix schenckii infection. Immunology (2017) 151(2):154–66. doi: 10.1111/imm.12719
38. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol (2018) 3(26). doi: 10.1126/sciimmunol.aar6676
39. Garnacho-Montero J, Palacios-Garcia I, Diaz-Martin A, Gutierrez-Pizarraya A, Lopez-Sanchez JM, Gomez EA, et al. Sequential changes of NLRP3 inflammasome activation in sepsis and its relationship with death. Shock (2020) 54(3):294–300. doi: 10.1097/SHK.0000000000001521
40. Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy (2016) 12(8):1272–91. doi: 10.1080/15548627.2016.1183081
41. Esquerdo KF, Sharma NK, Brunialti MKC, Baggio-Zappia GL, Assuncao M, Azevedo LCP, et al. Inflammasome gene profile is modulated in septic patients, with a greater magnitude in non-survivors. Clin Exp Immunol (2017) 189(2):232–40. doi: 10.1111/cei.12971
42. Jin L, Batra S, Jeyaseelan S. Deletion of nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol (2017) 198(3):1253–62. doi: 10.4049/jimmunol.1601745
43. Leligdowicz A, Matthay MA. Heterogeneity in sepsis: new biological evidence with clinical applications. Crit Care (2019) 23(1):80. doi: 10.1007/978-3-030-06067-1_40
44. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med (2016) 4(4):259–71. doi: 10.1016/S2213-2600(16)00046-1
45. Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, Henao R, et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit Care Med (2018) 46(6):915–25. doi: 10.1097/CCM.0000000000003084
46. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
47. Paludan SR, Reinert LS, Hornung V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat Rev Immunol (2019) 19(3):141–53. doi: 10.1038/s41577-018-0117-0
48. Dawulieti J, Sun M, Zhao Y, Shao D, Yan H, Lao YH, et al. Treatment of severe sepsis with nanoparticulate cell-free DNA scavengers. Sci Adv (2020) 6(22):eaay7148. doi: 10.1126/sciadv.aay7148
49. Jing Q, Leung CHC, Wu AR. Cell-free DNA as biomarker for sepsis by integration of microbial and host information. Clin Chem (2022) 68(9):1184–95. doi: 10.1093/clinchem/hvac097
50. Harrington JS, Choi AMK, Nakahira K. Mitochondrial DNA in sepsis. Curr Opin Crit Care (2017) 23(4):284–90. doi: 10.1097/MCC.0000000000000427
51. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21(9):548–69. doi: 10.1038/s41577-021-00524-z
52. Kuriakose T, Kanneganti TD. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol (2018) 39(2):123–34. doi: 10.1016/j.it.2017.11.002
53. Kim TH, Kim D, Gautam A, Lee H, Kwak MH, Park MC, et al. CpG-DNA exerts antibacterial effects by protecting immune cells and producing bacteria-reactive antibodies. Sci Rep (2018) 8(1):16236. doi: 10.1038/s41598-018-34722-y
54. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
55. Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med (2008) 205(6):1277–83. doi: 10.1084/jem.20080162
56. Dhani S, Zhao Y, Zhivotovsky B. A long way to go: caspase inhibitors in clinical use. Cell Death Dis (2021) 12(10):949. doi: 10.1038/s41419-021-04240-3
57. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
58. Oberholzer C, Tschoeke SK, Moldawer LL, Oberholzer A. Local thymic caspase-9 inhibition improves survival during polymicrobial sepsis in mice. J Mol Med (Berl) (2006) 84(5):389–95. doi: 10.1007/s00109-005-0017-1
59. Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol Res (2021) 163:105297. doi: 10.1016/j.phrs.2020.105297
60. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis (2012) 3(11):e437. doi: 10.1038/cddis.2012.176
61. Deng XX, Li SS, Sun FY. Necrostatin-1 Prevents Necroptosis in Brains after Ischemic Stroke via Inhibition of RIPK1-Mediated RIPK3/MLKL Signaling. Aging Dis (2019) 10(4):807–17. doi: 10.14336/AD.2018.0728
62. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
63. Xu Q, Guo J, Li X, Wang Y, Wang D, Xiao K, et al. Necroptosis underlies hepatic damage in a piglet model of lipopolysaccharide-induced sepsis. Front Immunol (2021) 12:633830. doi: 10.3389/fimmu.2021.633830
64. Zhang Q, Wei S, Lu J, Fu W, Chen H, Huang Q, et al. Necrostatin-1 accelerates time to death in a rat model of cecal ligation and puncture and massively increases hepatocyte caspase-3 cleavage. Am J Physiol Gastrointest Liver Physiol (2019) 316(4):G551–G61. doi: 10.1152/ajpgi.00175.2018
65. Dana H, Chalbatani GM, Mahmoodzadeh H, Karimloo R, Rezaiean O, Moradzadeh A, et al. Molecular mechanisms and biological functions of siRNA. Int J BioMed Sci (2017) 13(2):48–57. doi: 10.59566/IJBS.2017.13048
66. Zhou R, Rana TM. RNA-based mechanisms regulating host-virus interactions. Immunol Rev (2013) 253(1):97–111. doi: 10.1111/imr.12053
67. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discovery (2019) 18(6):421–46. doi: 10.1038/s41573-019-0017-4
68. Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem Pharmacol (2021) 189:114432. doi: 10.1016/j.bcp.2021.114432
69. Brahmamdam P, Watanabe E, Unsinger J, Chang KC, Schierding W, Hoekzema AS, et al. Targeted delivery of siRNA to cell death proteins in sepsis. Shock (2009) 32(2):131–9. doi: 10.1097/SHK.0b013e318194bcee
70. Wesche-Soldato DE, Chung CS, Lomas-Neira J, Doughty LA, Gregory SH, Ayala A. In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood (2005) 106(7):2295–301. doi: 10.1182/blood-2004-10-4086
71. Schwulst SJ, Muenzer JT, Peck-Palmer OM, Chang KC, Davis CG, McDonough JS, et al. Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock (2008) 30(2):127–34. doi: 10.1097/SHK.0b013e318162cf17
72. Duan C, Xu X, Lu X, Wang L, Lu Z. RIP3 knockdown inhibits necroptosis of human intestinal epithelial cells via TLR4/MyD88/NF-kappaB signaling and ameliorates murine colitis. BMC Gastroenterol (2022) 22(1):137. doi: 10.1186/s12876-022-02208-x
73. Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med (2013). doi: 10.1016/j.molmed.2013.08.004
74. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun (2020) 11(1):4561. doi: 10.1038/s41467-020-18443-3
75. Ye C, Choi JG, Abraham S, Wu H, Diaz D, Terreros D, et al. Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci United States Am (2012) 109(51):21052–7. doi: 10.1073/pnas.1216195109
76. Rotolo L, Vanover D, Bruno NC, Peck HE, Zurla C, Murray J, et al. Species-agnostic polymeric formulations for inhalable messenger RNA delivery to the lung. Nat Mater (2022). doi: 10.1038/s41563-022-01404-0
77. Wang YS, Kumari M, Chen GH, Hong MH, Yuan JP, Tsai JL, et al. mRNA-based vaccines and therapeutics: an in-depth survey of current and upcoming clinical applications. J BioMed Sci (2023) 30(1):84. doi: 10.1186/s12929-023-00977-5
78. Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Delivery Rev (2014) 66:110–6. doi: 10.1016/j.addr.2013.12.008
79. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discovery (2014) 13(10):759–80. doi: 10.1038/nrd4278
80. Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol (2014) 192(5):2019–26. doi: 10.4049/jimmunol.1302426
81. Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol (2017) 2:16258. doi: 10.1038/nmicrobiol.2016.258
82. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11(3):290–7. doi: 10.1016/j.chom.2012.01.016
83. Daley-Bauer LP, Roback L, Crosby LN, McCormick AL, Feng Y, Kaiser WJ, et al. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci United States Am (2017) 114(13):E2786–E95. doi: 10.1073/pnas.1616829114
84. Blanchard EL, Vanover D, Bawage SS, Tiwari PM, Rotolo L, Beyersdorf J, et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat Biotechnol (2021) 39(6):717–26. doi: 10.1038/s41587-021-00822-w
85. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell (2014) 157(6):1262–78. doi: 10.1016/j.cell.2014.05.010
86. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun (2018) 9(1):1911. doi: 10.1038/s41467-018-04252-2
87. Song M. The CRISPR/Cas9 system: Their delivery, in vivo and ex vivo applications and clinical development by startups. Biotechnol Prog (2017) 33(4):1035–45. doi: 10.1002/btpr.2484
88. Li T, Yang Y, Qi H, Cui W, Zhang L, Fu X, et al. CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduct Target Ther (2023) 8(1):36. doi: 10.1038/s41392-023-01309-7
89. Beyersdorf JP, Bawage S, Iglesias N, Peck HE, Hobbs RA, Wroe JA, et al. Robust, Durable Gene Activation In Vivo via mRNA-Encoded Activators. ACS Nano (2022) 16(4):5660–71. doi: 10.1021/acsnano.1c10631
Keywords: sepsis, cell death, apoptosis, necroptosis, pyroptosis, inflammation
Citation: Yang CS, Coopersmith CM and Lyons JD (2024) Cell death proteins in sepsis: key players and modern therapeutic approaches. Front. Immunol. 14:1347401. doi: 10.3389/fimmu.2023.1347401
Received: 30 November 2023; Accepted: 21 December 2023;
Published: 11 January 2024.
Edited by:
Satoru Ito, Aichi Medical University, JapanReviewed by:
Xin Mu, Tianjin University, ChinaCopyright © 2024 Yang, Coopersmith and Lyons. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John D. Lyons, amRseW9uc0BlbW9yeS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.