 Maciej Cedzyński
Maciej Cedzyński Anna S. Świerzko
Anna S. Świerzko- Laboratory of Immunobiology of Infections, Institute of Medical Biology, Polish Academy of Sciences, Łódź, Poland
The immune system starts to develop early in embryogenesis. However, at birth it is still immature and associated with high susceptibility to infection. Adaptation to extrauterine conditions requires a balance between colonization with normal flora and protection from pathogens. Infections, oxidative stress and invasive therapeutic procedures may lead to transient organ dysfunction or permanent damage and perhaps even death. Newborns are primarily protected by innate immune mechanisms. Collectins (mannose-binding lectin, collectin-10, collectin-11, collectin-12, surfactant protein A, surfactant protein D) and ficolins (ficolin-1, ficolin-2, ficolin-3) are oligomeric, collagen-related defence lectins, involved in innate immune response. In this review, we discuss the structure, specificity, genetics and role of collectins and ficolins in neonatal health and disease. Their clinical associations (protective or pathogenic influence) depend on a variety of variables, including genetic polymorphisms, gestational age, method of delivery, and maternal/environmental microflora.
1 Collectins and ficolins
The immune system starts to develop early in embryogenesis but at birth is still very immature, especially in prematurely born neonates. Therefore premature babies in particular are highly susceptible to infection. Adaptation to extrauterine conditions requires keeping a balance between colonization with commensal flora and protection from pathogens. Infections, oxidative stress and invasive procedures used for saving lives may lead to transient organ dysfunction, their permanent damage or even fatal outcome [reviewed in (1–4)]. In this review, the role of two distinct but structurally and functionally related molecular families, collectins and ficolins, is discussed in the context of neonatal health and disease.
Both collectins and ficolins are oligomers of basic subunits, consisting of three polypeptide chains. Each chain possesses four domains: a N-terminal cysteine-rich domain, followed by a collagen-like sequence, an α-helical neck region and a functional (pattern recognition) domain at the C-terminus. The functional domain differs between collectins and ficolins: it is a globular carbohydrate-recognition (lectin) domain (CRD) in collectins and a fibrinogen-like (FBG) domain in ficolins. The collagen-like region includes repeating Gly-X-Y triplets (where X and Y correspond to any amino acids). Both collectins and ficolins act as pattern-recognition molecules (PRM). They are able to interact with and participate in elimination of a variety of pathogens or altered autologous cells, by recognising pathogen- or danger-associated molecular patterns (PAMP, DAMP), through opsonisation or agglutination and (for all ficolins and some collectins) complement activation. In general, their activity requires the presence of calcium cations [reviewed in (5–7)]. The schematic overview of their activities, interactions with other immune factors and cross-talks is given in Figure 1.
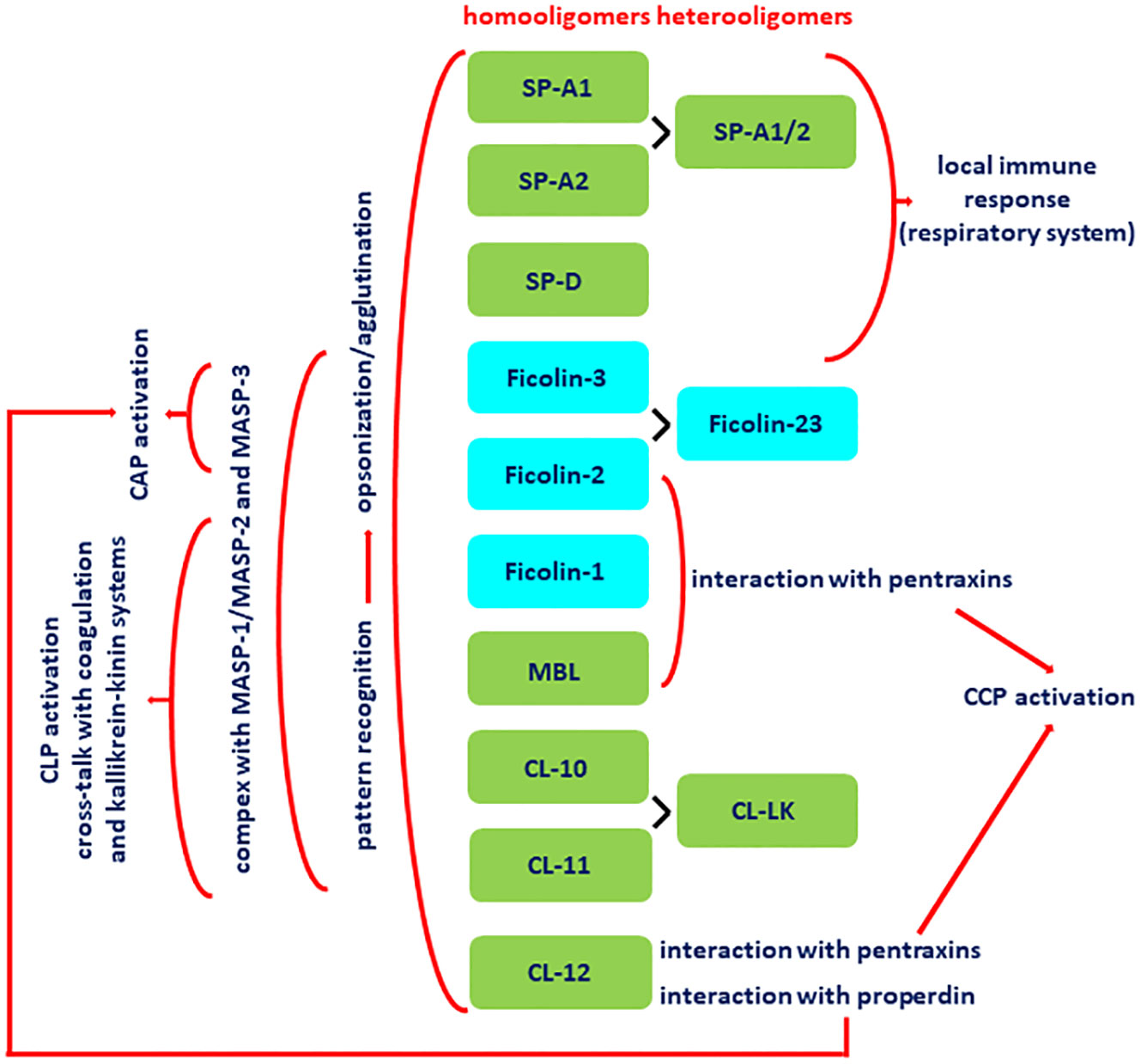
Figure 1 Basic activities, interactions and cross-talks of collectins (green boxes) and ficolins (blue boxes). All demonstrated collectins and ficolins are pattern-recognition molecules (PRM) able to opsonise and/or agglutinate pathogen cells. With an exception for SP-A and SP-D (pulmonary collectins) and CL-12, they form complexes with MASP-1, MASP-2 and MASP-3. Upon binding of PRM-MASP complex to the ligand, MASP-1 undergoes autoactivation and activates MASP-2, which substrates are C4 and C2 (that enables complement activation via the lectin pathway). Those enzymes contribute to cross-talk with coagulation system via cleavage of prothrombin (MASP-1 and -2) and factor XIII (MASP-1) as well as kallikrein-kinin system via cleavage of high molecular weight kininogen (HMWK) (MASP-1). MASP-3 substrate is pro-factor D (what enables contribution to complement activation via the alternative pathway). Collectin-12 due to interaction with pentraxins and properdin contributes to complement activation via classical and alternative pathways, respectively. MBL, ficolin-2 and ficolin-3 interacting with pentraxins are able to participate in complement activation via classical pathway. Pulmonary surfactant collectins and ficolin-3 are expressed within respiratory system and therefore participate in local immune response. CAP, complement alternative pathway; CCP, complement classical pathway; CLP, complement lectin pathway; CRP, C-reactive protein; SP-A1/2, heterocomplex of SP-A1 and SP-A2; CL-LK, heterocomplex of CL-10 and CL-11; ficolin-23, heterocomplex of ficolin-2 and ficolin-3; MASP, MBL-associated serine proteases.
1.1 Structure, specificity and genetics
1.1.1 Structure and specificity of collectins
The term “collectin” reflects the structure and function of proteins constituting a subfamily of C-type lectins: COLlagenLECTIN. Three human collectins, mannose-binding lectin (MBL) (or mannan-binding lectin), collectin-10 (CL-10) (or collectin-liver 1, CL-L1) and collectin-11 (CL-11) (or collectin-kidney 1, CL-K1), form complexes with enzymes called MASP (MBL-associated serine proteases) and share ability to initiate complement activation via the lectin pathway. Those collectins can also cross-talk with the alternative pathway of complement, the coagulation cascade and the kallikrein-kinin system (5–7) (Figure 1). Another member of this subfamily, collectin-12 (CL-12) (or collectin-placenta 1, CL-P1) is known to contribute to complement activation via the alternative (8, 9) and classical (10, 11) pathways, due to interaction with properdin and short (C-reactive protein, CRP; serum amyloid P, SAP) or long (pentraxin 3, PTX3) pentraxins, respectively. Furthermore, MBL interacts with SAP and PTX3, resulting in enhancement of opsonophagocytosis and/or amplification of complement activation via cross-talk between lectin and classical pathways (12). The two other collectins, surfactant protein A (SP-A) and surfactant protein D (SP-D) are unable to activate complement directly or indirectly (13) (Figure 1).
The single polypeptide (glycosylated and acetylated) chain of MBL (32 kDa) consists of 228 amino acid (AA) residues: 21 (including 3 Cys) in the cysteine-rich domain, 59 in the collagen-like region (including 19 Gly-X-Y triplet repeats, interrupted after 7th one by Gly-Gln), 30 in the neck domain and 118 in the CRD (Table 1; Figure 2A). The interruption gives rise to a kink that causes the “tulip bouquet” shape of the oligomer (up to hexamer) of subunits [reviewed in (6, 14, 15)]. The lectin domain binds to D-mannose (D-Man), and N-acetyl-D-mannosamine (D-ManNAc), N-acetyl-D-glucosamine (D-GlcNAc) and L-fucose (L-Fuc), leading to recognition of numerous pathogens via interaction with pathogen-associated molecular patterns (PAMP) decorating their cells (more details are given in Tables 2, 3). Furthermore, MBL contributes to the elimination of senescent fibroblasts, apoptotic, necrotic and certain cancerous cells (7, 16).
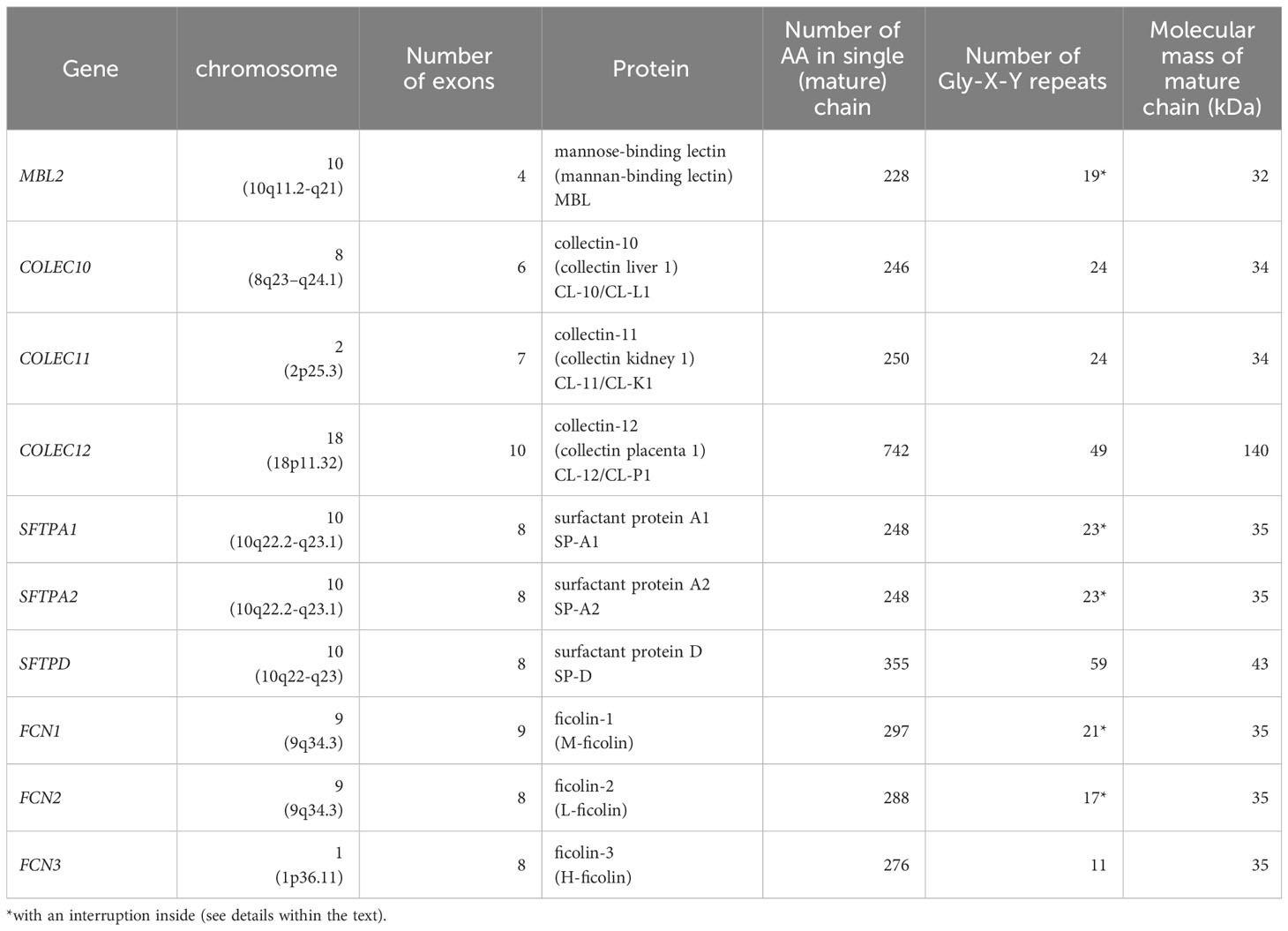
Table 1 Genes encoding for human collectins and ficolins and corresponding proteins.
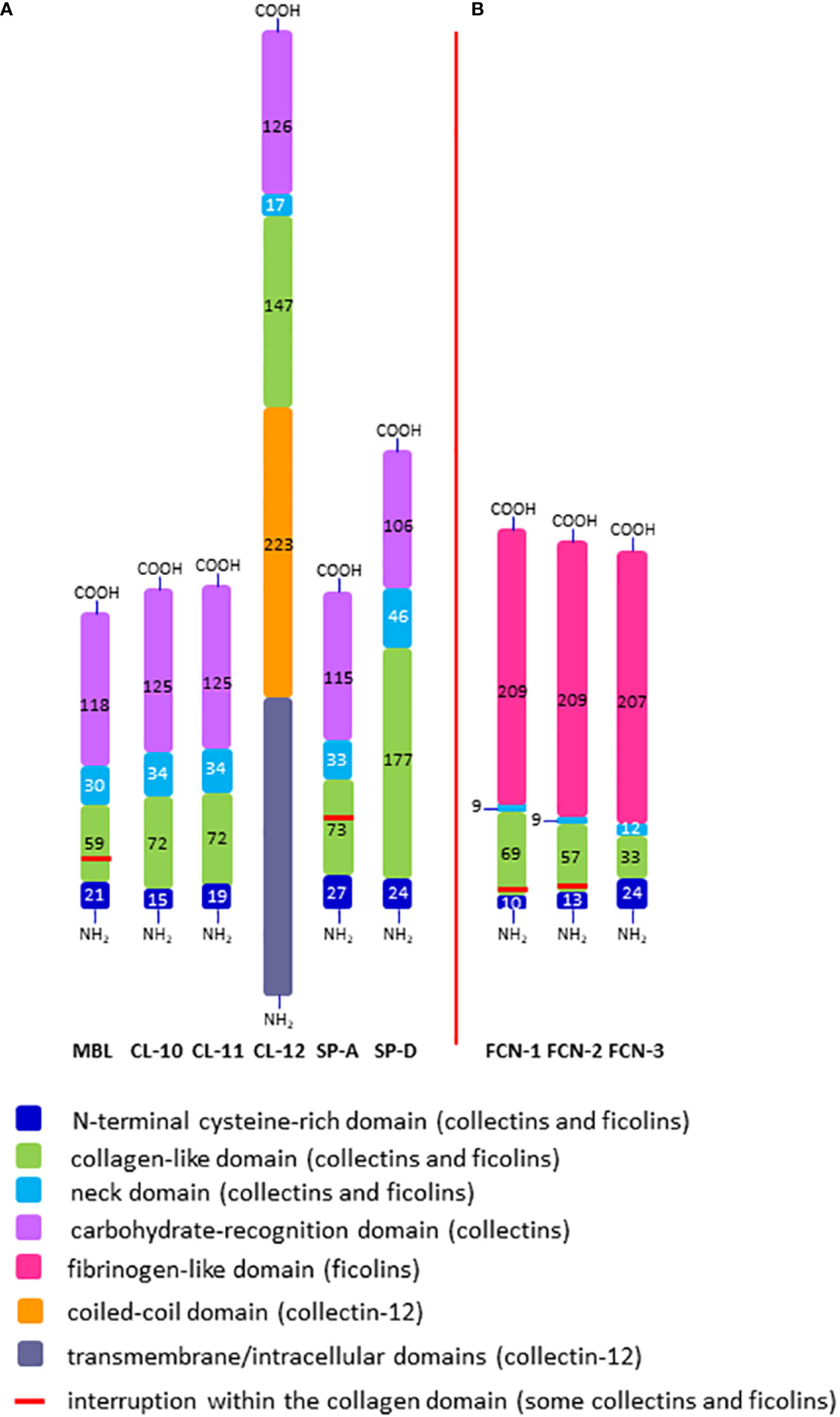
Figure 2 Domain organisation of polypeptide chains of collectins (A) and ficolins (B). The length of each schematic domain is proportional to the number of amino acid residues (no secondary, tertiary or quaternary structures like α-helices, globular heads, trimers, multimers, etc. are shown). For collectin-12, details in ectodomain (including lectin and collagen-like domains) are presented while transmembrane/intracellular domains are displayed as one distinct region. SP-A corresponds to SP-A1 and SP-A2; FCN-1, -2, -3 correspond to ficolin-1, ficolin-2 and ficolin-3, respectively; -NH2 and -COOH correspond to the N- and C- termini, respectively.

Table 2 Pathogens recognised by collectins and ficolins.
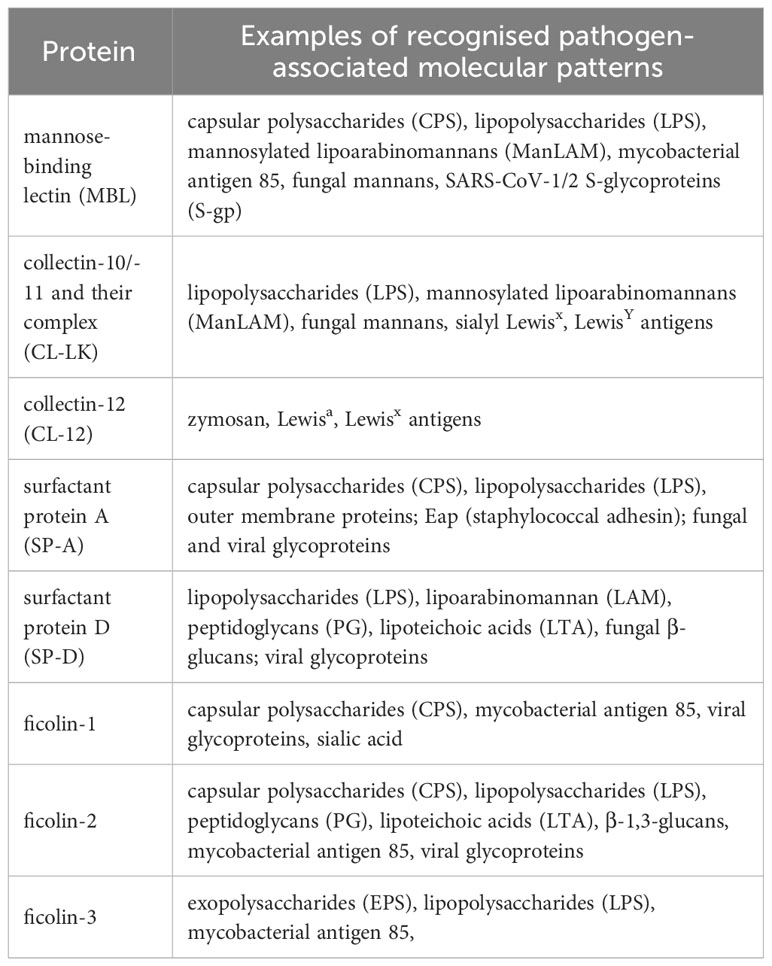
Table 3 Examples of major microbial ligands recognised by collectins and ficolins.
The chains of CL-10 and CL-11 (34 kDa) consist of 246 and 250 AA residues, respectively (Table 1; Figure 2A). Their N-terminal domains differ in length (15 and 19 AA, respectively) whilst the collagen-like, neck and carbohydrate recognition domains consist of an identical number of residues: 72 (24 Gly-X-Y repeats), 34, and 125, respectively, but with distinct sequences [reviewed in (17–19)]. The basic subunits may be homo- or heterotrimers (consisting of one CL-10 and two CL-11 polypeptides). In the latter case, the molecule is called CL-LK (20). Those trimers undergo further oligomerization (up to octadecamers) (17, 20). The ligand specificity of CL-LK is rather broad, and similar to some extent to that of MBL (21, 22). That probably reflects a synergistic effect between CRDs of CL-10 and CL-11. CL-10 recognises D-Man, L- and D-fucose, D-galactose (D-Gal), and D-GlcNAc (19, 23) while CL-11 recognises the D-Man-(α1,2)-D-Man disaccharide (24) as well as D-Man, L-Fuc and D-ManAc (19, 25). That enables CL-LK to bind to a variety of pathogens as well as apoptotic cells [reviewed in (19, 22)]. Furthermore, Kirketerp-Moller et al. (26) reported specific recruitment of CL-11 to ligands by C1q/TNF-related protein (CTRP) and subsequent complement activation. Details concerning those microbes and associated molecular patterns are listed in Tables 2, 3.
Collectin-12 was initially described as a scavenger transmembrane receptor, expressed in vascular endothelial cells and in placenta (27, 28). It was later found on other cells (including macrophages, astrocytes and microglia) as well as in a soluble form (8, 29). Its ectodomain includes carbohydrate recognition (126 AA), neck (17 AA) and collagen-like (147 AA) regions preceded by a long (223 AA) coiled-coil fragment (Table 1; Figure 2A). Due to that and additional transmembrane and intracellular domains, the MW of its single chain is higher than that of other members of the subfamily (140 kDa, 742 amino acid residues) Furthermore, it exists as lower oligomers (one to three subunits) (8, 26, 27). Collectin-12 recognises D-GalNAc, D-Gal, Lewisx (Lex), T-antigen and Tn-antigen. The Lex is the D-Gal-(β1,4)-L(Fuc-(α1,3)-D-GlcNAc trisaccharide motif, present in some self-cells but also in surface structures of such pathogens as Helicobacter pylori, Streptococcus sanguinis or Schistosoma mansoni eggs. It should however be stressed that CL-12 does not bind to its sialylated (commonly expressed) form (sLex). Tumour-associated T- and Tn-antigens contain both carbohydrate and amino acid residues: D-Gal(β1,3)-D-GalNAc-Ser/Thr and D-GalNAc-Ser/Thr, respectively (18, 30–34). For more details see Tables 2, 3.
Human surfactant protein A exists in two forms, SP-A1 and SP-A2, closely related but synthesized under the control of distinct genes (see below). The SP-A subunits may be homo- or heterotrimers. The chains of both forms are built up of 248 amino acid residues (35 kDa), with 27 in cysteine-rich, 73 in collagen-like, 33 in neck and 115 in carbohydrate recognition domains. The collagen-like region includes 23 Gly-X-Y repeats and, like MBL, an interrupting sequence (Pro-Cys-Pro-Pro, after the 13th triplet) (Table 1; Figure 2A), resulting in a “tulip bouquet” shape of the multimer (up to 6 subunits). SP-A has affinity for D-ManNAc, D-Man, L-Fuc, D-maltose (D-Mal), D-glucose (D-Glc) and D-GlcNAc (13, 35–40). It moreover recognises non-carbohydrate bacterial ligands, as extracellular adherence protein (Eap adhesin), a virulence factor of Staphylococcus aureus (41). Pathogens and corresponding molecular patterns recognised by SP-A are presented in Tables 2, 3.
The polypeptide chain of another surfactant protein, SP-D (355 amino acid residues, 43 kDa) is characterised by a long collagen-like region (59 Gly-X-Y repeats, no kink). The cysteine-rich region, coiled-coil segment and CRD are built-up from 24, 46 and 106 AA, respectively (Table 1; Figure 2A). Basic subunits oligomerise to form tetramers which may further multimerise (up to 8 tetramers/96 chains). The lectin domain of SP-D binds to D-Mal, D-Man, L-Fuc, D-GlcNAc, D-Glc and D-Gal. It furthermore is able to recognise inositol and a disaccharide, lactose (13, 37–40, 42, 43). Microbes and their basic structures that are ligands for this collectin are listed in Tables 2, 3.
1.1.2 Genes encoding collectins and their polymorphisms
Genes encoding MBL (MBL2), SP-A (SFTPA1, SFTPA2) and SP-D (SFTPD) are localised to chromosome 10 (10q21-24) within a cluster including also MBL- and SP-A-related pseudogenes. Genes encoding MBL and SP-A contain four exons while that for SP-D has eight (Table 1). All of them are highly polymorphic. The majority of disease-association studies of the MBL2 gene has been focused on 6 (or less) polymorphisms: [rs11003125 (-550 G>C, commonly termed H/L), rs76206 (-221 G>C known as Y/X)] (both localised to the promoter region), [rs7095891 (+4 C>T, P/Q] (at 5’untranslated region), rs5030737 (+223 C>T, R52C, commonly called A/D), rs1800450 (+230 G>A, G54D, A/B) and 1800451 (+239 G>A, G57E, A/C)], all from exon 1. Variant (minority) alleles D, B, and C are collectively named O [reviewed in (15)]. Due to strong linkage disequilibria, only seven haplotypes are considered common (HYPA, LYPA, LYQA, LXPA, LYPB, LYQC, HYPD), differently distributed among various ethnic groups. All those polymorphisms affect the concentration of MBL in blood: those in promoter via influencing the level of gene expression, those in exon 1 via influencing protein half-life (shortened for products related to O variants due to higher sensitivity to serum metalloproteases). Additionally, presence of D, B and/or C allele leads to lower MBL activity. The O/O and LXPA/O (or LXA/O, XA/O) genotypes are associated with MBL primary deficiency (considered the commonest human immunodeficiency) but in some individuals carrying other genotypes (including not only A/O but also A/A), this lectin may be undetectable or present in serum at a very low level. Generally, huge inter-genotype differences are not uncommon (15). Moreover, certain polymorphisms of the 5’UTR of exon 4 have been described which are believed to be clinically relevant, mainly in relation to cancer, therefore they will not be discussed here.
The genes encoding SP-A1 (SFTPA1) and SP-A2 (SFTPA2) are in linkage disequilibrium and are located in opposite transcriptional orientation. Combinations of their polymorphisms localised to the coding regions were used for establishing several frequent variants called 6A, 6A2, 6A3, 6A4, 6A5 (for SFTPA1) or 1A, 1A0, 1A1, 1A2, 1A3, 1A5 (for SFTPA2), affecting protein activity [reviewed in (40, 44, 45)]. Details are given in Table 4. Among 14 included polymorphisms, 8 are silent, associated with single nucleotide but not amino acid residue exchanges; 5 of them differentiate between SFTPA1 and SFTPA2 genes. Furthermore, both gene and AA sequences differ at 4 additional positions: codons 66 (rs1059049; ATG/Met for SP-A1 and ACA/Thr for SP-A2), 73 (rs1059052; GAT/Asp and AAT/Asn, respectively), 81 (rs1059053; ATC/Ile and GTC/Val, respectively), and 85 (rs1059053; TGT/Cys and CGT/Arg, respectively), all localised to the collagen-like domain. Interestingly, as SNP in codons 9 and 19 are related to the signal peptide-encoding sequence, there are several pairs of SFTPA1 or SFTPA2 variants giving identical mature proteins (6A and 6A3, 1A and 1A5, 1A0 and 1A2, 1A1 and 1A3) (40) (see Table 4). The most common variants are 6A2and 1A0. Additionally, some splicing (called A, B, B’, C, C’, D, D’) or sequence variations of both genes at their 5’ and 3’UTR, respectively, were reported to affect their expression (44, 45).
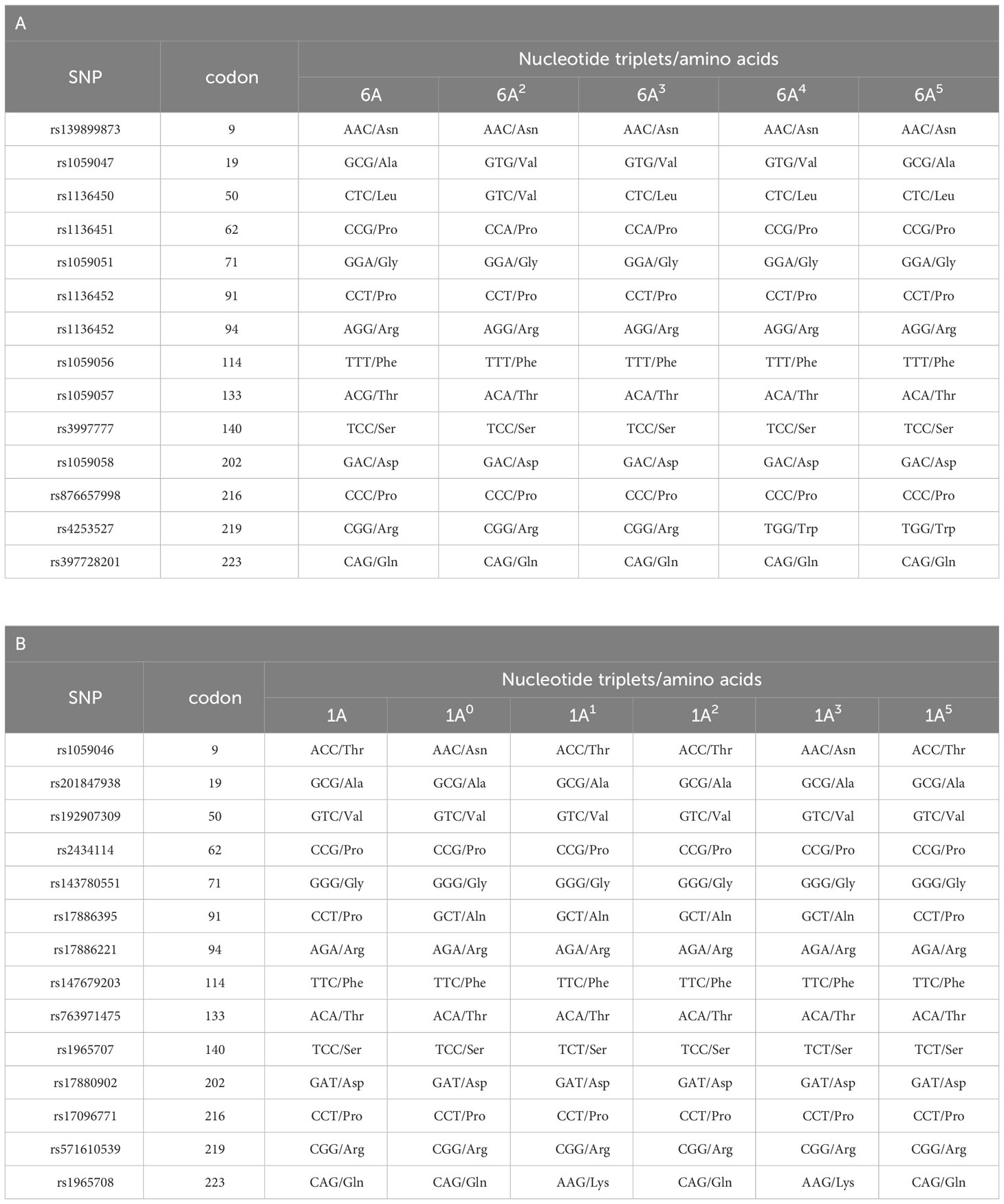
Table 4 Common polymorphisms localised to the coding region of the SFTPA1 (SP-A1) (A) and SFTPA2 (B) genes, and corresponding variants (related codons and amino acid residues are given).
The SFTPD (SP-D) gene is located in the same transcriptional orientation as SFTPA2. It contains 8 exons (7 coding) (Table 1). Its polymorphisms, located in 3’UTR, protein encoding region and 5’UTR are known to influence SP-D concentration/activity and/or to have a variety of disease associations (44, 46, 47). Some of those located within the protein encoding exons are silent [rs6413520 (+75 T>C, Ser25Ser), rs1051246 (+858 T>C, Ala286Ala)], others are related to amino acid substitutions [rs721917 (+32 T>C, Met11Thr), rs2243639 (+478 A>G, Thr160Ala), rs3088308 (+868 T>A, Ser270Thr)] (44).
The human COLEC10 gene (encoding for CL-10/CL-L1) is localised to chromosome 8 and contains 6 exons (Table 1). It is highly conserved (as is COLEC11, for CL-11/CL-K1) and the majority of its polymorphisms is located in non-coding regions (48). However, two COLEC10 polymorphisms are associated with amino acid exchange: rs150828850 (+23881 A>C, Glu78Asp, affecting structure of the collagen-like region) and rs149331285 (+36545 C>T, Arg125Trp, affecting structure of the neck region and CL-10 concentration in serum) (17, 18, 22, 48).
The COLEC11 gene (chromosome 2; 7 exons) (Table 1), in contrast to COLEC10, produces a variety of transcript isoforms. However, two only were confirmed to be translated and detected in circulation: a or f (full-length, 34 kDa chain) and d (lacking part of the collagen-like region encoded by exon 8; 31 kDa) (18, 20, 21). Like in the case of COLEC10, most of COLEC11 SNP is located within promoter or introns. One of them, rs3820897 (-9570 C>T, promoter region) was reported to influence serum level of CL-11. The non-synonymous polymorphism in exon 7, rs7567833 (+39618 C>G, His219Arg) is considered to affect ligand binding capacity but not concentration in blood (17, 22, 48).
The gene encoding collectin-12, COLEC12, is located on chromosome 18 and contains 10 exons (Table 1). Two major isoforms (I – full length and II – lacking CRD) and several others were found to be transcribed (17, 27, 33). Ohmori et al. (49) identified six polymorphisms, including three related to coding exons: one silent, in exon 5 (+800 C>T, Thr267Thr) and two non-synonymous, in exon 6 (+1563 C>T, Ser522Pro and +1815 A>G, Gly606Ser) and constructed 22 related haplotypes (49). However, the GeneCards database mentions several other SNP which influence amino acid sequence: +182 T>C (Val61Ala), +319 G>A (Glu 107Lys), +494 C>G (Thr165Ser), +707 G>A (Arg236Gln) (https://www.genecards.org/cgi-bin/carddisp.pl?gene=COLEC12).
1.1.3 Structure and specificity of ficolins
Unlike collectins, ficolins have a globular, fibrinogen-like domain (FBG) at their C-termini. Therefore, the name of this protein family comes from FIbrinogenCOllagenLectIN, although the FBG region is not a typical lectin domain. Three human ficolins have been described: ficolin-1 (or M-ficolin), ficolin-2 (or L-ficolin) and ficolin-3 (or H-ficolin), encoded by the FCN1, FCN2 and FCN3 genes, respectively. All of them, by forming complexes with MASP, are able to activate complement via the lectin pathway [reviewed in (50, 51)]. Importantly, ficolin-2 was demonstrated to interact with CRP and PTX3 leading to enhancement of recognition of some pathogens and boosting complement-mediated killing via lectin/classical pathways (52, 53). Similar cross-talks of ficolin-1 are associated with amplification of complement activation and a modulatory effect on secretion of inflammatory cytokines (54, 55) (Figure 1).
The mature polypeptide chains of ficolins consist of 297 (ficolin-1), 288 (ficolin-2) or 276 (ficolin-3) AA residues (molecular weight 34 - 35 kDa), including 21, 17 or 11 Gly-X-Y repeats in their collagen-like regions, respectively. After the second repeat in ficolin-1 and ficolin-2 there is a six amino acid interruption. The ficolin-1, ficolin-2 and ficolin-3 N-terminal regions encompass 10, 13 and 24 AA, neck domains of 9, 9 and 12 AA, respectively. The C-terminal FBG region has 209, 209 and 207 AA, respectively, in ficolins 1 to 3. (Table 1; Figure 2B). Single chains generally form homotrimeric basic subunits via disulphide bonds, due to the presence of cysteine residues in their N-terminal domains, which further oligomerize, usually into dodecamers or octadecamers (50, 56, 57). Moreover, it was evidenced that ficolin-2 and ficolin-3 may form heterocomplexes (called ficolin-23), supposed to include homotrimers of both or heterotrimers (58).
Ficolins generally exhibit affinity to acetylated sugars but also to some non-acetylated structures. Namely, ficolin-1 binds D-GlcNAc, N-acetyl-D-galactosamine (D-GalNAc), D-ManNAc and sialic acid; ficolin-2 binds D-GlcNAc, D-GalNAc, D-ManNAc, N-acetyl-neuraminic acid (NeuNAc), N-acetylated cysteine, acetylcholine and heparin; and ficolin-3 binds D-GlcNAc, D-GalNAc, D-Gal and D-Fuc [reviewed in (5, 50, 51)]. Unlike other proteins of this family, ficolin-2 has four distinct binding sites that give it a broad specificity (59–62), which is further extended by cross-talk with short and long pentraxins. Details concerning microbial ligands for human ficolins are presented in Tables 2, 3.
1.1.4 Genes encoding ficolins and their polymorphisms
The FCN1 (9 exons) and FCN2 (8 exons) genes are located on chromosome 9q34 (Table 1). Due to their localisation, exon organisation and high sequence homology (approx. 80% at the amino acid level) it is assumed that they originate from gene duplication (63, 64). The FCN3 gene, containing 8 exons, is located on chromosome 1p36.11) (Table 1). It was suggested to branch out by gene duplication in the early stage of evolution of Vertebrata. The amino acid sequence of its product, ficolin-3, shows approx. 48% homology with both other ficolins (63, 64).
All three genes are highly polymorphic and distribution of their variants differs in various populations/ethnic groups [discussed in details in (63, 64)]. Some of them were demonstrated to influence expression level or activity of ficolins, and to have clinical associations. Ammitzboll et al. (65) found minor alleles for three promoter [rs7857015 (-1524 T>C), rs10120023 (-542 G>A), rs10117466 (-144 C>A)] and one exon 1 [rs10858293 (+33 G>T, Gly11Gly) SNP of the FCN1 gene (all in strong linkage disequilibrium) to be associated with high concentrations of ficolin-1 in serum. Earlier, Munthe-Fog et al. (66) reported such a relationship for rs10120023 and rs10117466 (ficolin-1 levels were determined in plasma) and found moreover lower expression of FCN1 mRNA in granulocytes and monocytes in carriers of G and/or C alleles, respectively. Regarding non-synonymous polymorphisms, variant alleles at rs148649884 [+6658 G>A, Ala218Thr (exon 8)] and rs138055828 [+7959 A>G, Asn289Ser (exon 9)] were found associated with lower serum concentrations of ficolin-1 while those at rs147309328 [+4759 G>A, Arg124Gln (exon 6)] and rs56084543 [+4837 C>T, Thr150Met (exon 6)] had the opposite effect. Furthermore, the presence of threonine at position 218 and serine at 289 affected ligand binding capacity. Another SNP, localised to exon 9, rs150625869 (+7895 T>C, Ser268Pro) was deduced to cause total ficolin-1 deficiency in a homozygous state, however no such case has been identified to date (65).
Similarly, no primary (genetically-determined) instance of ficolin-2 deficiency has been found. Minor alleles corresponding to three promoter region SNP of the FCN2 gene were associated with low ficolin-2 concentration in blood: rs3124952 (-986 A>G), rs3811140 (-557 A>G) and rs7865453 (-64 A>C) whereas minor alleles at rs3124953 (-602 G>A) and rs17514136 (-4 A>G) were associated with higher levels (67–70). Moreover, variant alleles for rs17549193 (+6359 C>T, Thr236Met) and rs7851696 (+6424 G>T, Ala258Ser) (both located in exon 8) are related to higher and lower concentrations of the gene product, respectively, and also affect affinity to the ligand (D-GlcNAc) (67–70). Due to linkage disequilibria, their effect on ficolin-2 concentration is quite ambiguous and marked differences in the level of this protein in serum are observed among carriers of the same genotype, even between monozygotic twins (67, 68, 70). More recently, Świerzko et al. (71) found two FCN2 gene 3’UTR polymorphisms, rs4521835 (T>G) and rs73664188 (T>C), to influence ficolin-2 concentration significantly: the corresponding GC haplotype was associated with a low level of this protein in the cord serum (71).
Rare ficolin-3 deficiency is associated with a single nucleotide deletion in exon 5 (+1637delC; rs28357092), leading to a frame shift causing an early stop codon and lack of protein synthesis (63, 72). Described originally in heterozygotes, it was hypothesized to cause ficolin-3 deficiency in homozygotes (72). Indeed, Munthe-Fog et al. (73) identified the first case soon after. The patient had severe and recurrent infections (73). Later, several other cases were described, including newborns (discussed below). A dozen or so other FCN3 polymorphisms, within the promoter region, exons and introns have been described in various populations, mostly with low (<0.1) minor allele frequency (63, 64, 72). Interestingly, promoter SNPs appear to have no significant impact on the concentration of ficolin-3 in serum (72). The most common in European (Danish) population, rs41415450 (-325 G>A; MAF=0.11) was not observed in Africans (Ghana, Mozambique) or Japanese (63, 64).
2 Clinical associations of collectins in the neonate
2.1 Mannose-binding lectin
The most extensively investigated complement-activating collectin in the context of neonatal health and disease is MBL. Several, differently designed, studies have found significant correlations between cord blood MBL concentration and gestational age (74–79). Lau et al. (74) found that preterms having low MBL concentration in serum (defined as <400 ng/ml) were born at shorter gestational age than babies with higher levels of this collectin. They moreover observed generally lower MBL in preterm compared with term babies what was further confirmed by Hilgendorff et al. (76) and Sallenbach et al. (79). Van der Zwet et al. (78) reported higher frequency of low MBL-associated haplotypes in preterm compared with term neonates. However, Kilpatrick et al. (80) found no correlation with gestational age. Moreover, data from a large (n>1800) cohort of consecutively recruited newborns revealed neither significant correlation of MBL with GA nor a greater difference in median MBL concentration in cord sera between term and preterm newborns. No association of MBL2 genotypes or MBL concentrations in cord sera with birthweight was observed (81). These conflicting findings are difficult to reconcile. Complicating matters further, low MBL-dependent lectin pathway activity was found to be associated with prematurity (81).
Some contradictory data were also published in the context of associations of MBL2 genotypes in general and/or genetically-determined MBL deficiency with spontaneous preterm births. Bodamer et al. (82) reported the D allele (corresponding to codon 52 polymorphism, rs5030737) to be a risk factor and L/L homozygosity (related to promoter SNP at position -550, rs11003125) to have the opposite effect. Furthermore, O/O genotypes tended to be more common in preterm compared with term babies (82). In contrast, Swierzko et al. (81) found A/A MBL2 genotypes [where A corresponds to major alleles in codons 52 (A/D, rs5030737), 54 (A/B, rs1800450) and 57 (A/C, rs1800451)] to be significantly more frequent among preterms. That finding was later partially confirmed by Carvalho da Silva et al. (83) who genotyped neonates and their mothers. They found maternal/neonatal LYA haplotypes to be associated with preterm birth and the LYB haplotype to be protective. Chan et al. (84) related spontaneous preterm birth with depletion of Lactobacillus sp. from the vaginal microbiome leading to a maternal inflammatory response expressed through increased concentration of MBL, C3b, C5, IL-6, IL-8, IL-1β, IgM and IgG. Therefore, they suggested cross-talk between the innate and adaptive defence mechanisms, bridged by complement activation, to enhance the risk of preterm delivery. Earlier, Frakking et al. (77), Karjalainen et al. (85) and Grumach et al. (86) found no difference of frequency of MBL2 genotypes between preterm and term neonates. Moreover no impact of maternal genotype was reported by Karjalainen et al. (85).
One of the leading causes of prematurity is preterm premature rupture of membranes (pPROM). Modi et al. (87, 88), using whole genome sequencing, identified the rs74754826 mutation of the MBL2 gene [codon 210, + 628 G>T, Glu210Ter (introducing “stop” codon)] as a risk factor for pPROM in African Americans. This mutation introduces a stop codon, resulting in deletion of 38 terminal amino acid residues in the CRD.
Premature births are often associated with very low birthweight (VLBW). The earliest reports suggested MBL2 promoter region polymorphisms associated with low circulating MBL were linked to low body mass (89). However, Gibson et al. (90) observed a high prevalence of the LYPA haplotype (corresponding to relatively high MBL concentration in serum/activity) in small for gestational age, (SGA, birthweight <10 percentile) premature (born at GA <32 weeks) babies. In contrast, Cakmak et al. (91) reported the codon 54 variant (B) allele (commonly related to LYPB haplotype) and low MBL concentration to be associated with low birthweight in a corresponding gestational age group while Hartz et al. (92) reported no impact of MBL on incidence of VLBW.
There is more agreement, but by no means total consistency, in the extensive literature concerning MBL and neonatal sepsis. Frakking et al. (93) observed associations of low MBL and its genetically-determined deficiency not only with early onset sepsis, but also with culture-proven sepsis and pneumonia; in that study, both preterm and term newborns were recruited. Several other groups found lower serum MBL levels in cases of neonatal sepsis compared with controls as well (90, 94–99). Dzwonek et al. (100) based on data from preterms (GA 24-36 weeks) demonstrated that MBL2 A/O and O/O genotypes are risk factors for sepsis. Koroglu et al. (101) found codon 54 (A/B) and 57 (A/C) SNP to be associated with a higher risk of sepsis (especially early onset) but not culture-proven sepsis, while Ozdemir et al. (102) noted lower MBL levels related with sepsis in general and especially with culture-proven sepsis in neonates born at GA ≤34 weeks with fetal inflammatory response syndrome. Similarly, Wahab Mohamed and Saeed (103) published data from both term and preterm babies demonstrating significantly lower MBL levels in individuals with confirmed sepsis (and especially septic shock) than in the control (infection-free) group. Świerzko et al. (104) observed significantly lower median MBL concentrations in babies with diagnosed sepsis compared with the control group (no infection till hospital discharge). However, no significant differences were found between newborns who had a milder disease course (infection but not sepsis) and controls or between disease groups (104). Hartz et al. (105), investigating a large cohort of very low birthweight infants (VLBWI) found that total MBL deficiency (undetectable in serum) is related to sepsis in babies born at GA ≥32 ≤ 37 weeks only. Dogan et al. (106) suggested MBL concentrations <700 ng/ml enhanced the risk of late-onset sepsis in preterms. Both Dogan et al. (106) and Schlapbach et al. (107) found the strongest association of MBL deficiency to be with Gram-negative sepsis. Recently, Elhawary et al. (99) reported significant association of B allele with sepsis in preterms.
The association with deficiency of circulating MBL could be misleading, as the relationship is not as readily apparent at the DNA level. Auriti et al. (95) found no significant difference in the frequencies of MBL2 genotypes between infected and noninfected neonates, despite a marked difference in serum MBL concentrations. Their findings are supported by several other studies reporting no association between neonatal sepsis and either MBL2 genotype or the presence of the B allele (78, 97, 108–110). These findings are, of course, in contrast to some of the reports cited earlier, so collectively are hard to reconcile.
Meta-analyses of available data have concluded that the codon 54 SNP variant influences risk for development of neonatal sepsis in general (111) or culture-proven sepsis (112, 113). However MBL concentration was estimated to be of only moderate prognostic value (111).
The relationship of MBL to neonatal bronchopulmonary dysplasia (BPD) is also unclear. Hilgendorff et al. (109) reported that X and B MBL2 variants are risk factors for BPD. A harmful effect of B/B and protective influence of A/A genotype, respectively, were later postulated by Cakmak et al. (91). Furthermore, Capoulongo et al. (114) found the A/D genotype to be a BPD risk factor and the L allele to be beneficial. Later, Speletas et al. (115) reported a significant association of XA/O and O/O genotypes with transient tachypnea and respiratory distress syndrome (RDS) (their cohort comprised of both term and preterm neonates). On the other hand, Koroglu et al. (101) found no relationship between MBL2 genotype and BPD or RDS.
2.2 Collectin-10, collectin-11 and collectin-12
Both collectin-10 and collectin-11 are considered to be essential for fetal development: a variety of mutations of COLEC10, COLEC11 and MASP1/3 genes (the last one encodes MBL-associated serine proteases-1, -3 and non-enzymatic MAp-44, forming complexes with MBL, CL-10, CL-11 and ficolins) were reported in patients diagnosed with Malpuech, Michels, Mingarelli and Carnevale (3MC) syndrome, with craniofacial, renal or genital abnormalities, growth and intellectual disability [reviewed in (22)].
Although collectin-12 (or collectin placenta-1) was demonstrated to recognise certain pathogens, including Aspergillus fumigatus, Staphylococcus aureus and Escherichia coli (34, 116), to activate complement via both alternative and classical pathways (8–11) and its soluble form was detected in cord plasma (in contrast to plasma from adult donors) (8), there is no data published concerning its significance in neonatal infections. On the other hand, like CL-10 and CL-11, it is considered crucial for ontogenesis. McLennan et al. (117), based on computational analysis and data from an animal model (chick embryo), found CL-12 contributed to cranial neural crest cell trajectories and promoted collective cell migration. Recently, Ajami et al. (118) described siblings (3.5 year-old girl and 10 month-old boy) with derivative chromosome 9, resulting in partial 9p monosomy and 18p trisomy. Fifty-three and twenty-two (including COLEC12) affected genes were identified for the deletion and duplication, respectively. Both patients were born with dysmorphic features and congenital malformations. At the time of examination, besides craniofacial abnormalities, some intellectual/developmental disabilities were also found. Furthermore, a cryptorchidism and incorrect brain magnetic resonance image (demonstrating delayed myelination, enlarged sylvian cistern, cavum septum pellucidum, hypoplastic corpus callosum and trigonocephaly) was documented in the male sibling (118). Although COLEC12 was one of several translocated genes, the clinical manifestation reported may confirm its importance for embryo-/ontogenesis.
2.3 Surfactant protein A and surfactant protein D – collectins which do not initiate complement activation
The complement non-activating collectins, surfactant proteins A and D (SP-A, SP-D) are present in the female reproductive system and are considered to protect from intrauterine infections (which are risk factors for miscarriages, premature births and neonatal sepsis) [reviewed in (13, 119)]. During pregnancy, they are detectable in chorion, amnion, amniotic fluid and placenta (120–125). Snegovskikh et al. (126) postulated that decidual SP-A regulates expression of prostaglandin F2α (PGF2α) in the uterus and therefore contributes to the prevention of premature birth. They moreover excluded association of rs1136451 polymorphism of the SFTPA1 gene (codon 62, + 186 G>A, Pro62Pro) with prematurity (126). Lower SP-A (and concomitantly higher SP-D and MBL) expression was found in term placentas from spontaneous deliveries compared with caesarean section deliveries (127) which partially confirmed and extended the afore-mentioned data published by Snegovskikh et al. (126). Furthermore, Chaiworapongsa et al. (128) observed lower median SP-A in amniotic fluid samples from women at term in labour compared with those not in labour. Earlier, Pryhuber et al. (120) noticed an increase of amniotic fluid SP-A levels with duration of gestation, especially during third trimester. The SFTPA1 mRNA expression level in chorioamniotic membranes did not differ between similarly defined groups, but it was higher compared with membranes from preterm deliveries (123). Miyamura et al. (121) reported a marked increase of SP-A (and moderate SP-D) concentration in amniotic fluid from 32nd week of gestation towards term. Interestingly, data from a murine model suggested that SP-A produced by the fetal lung may act as a signal for initiation of delivery [reviewed in (129)]. In contrast, Karjalainen et al. (85) demonstrated that the Met allele (corresponding to SFTPD Met31Thr SNP) in babies (but not in their mothers) is associated with spontaneous preterm births and suggested it contributes to a parturition-inducing signal. They however found no similar relationship for SFTPA1, SFTPA2 or MBL2 polymorphisms.
SP-A may be detected in terminal bronchioles and alveolar spaces after the 35th week of gestation. Its amount in the alveoli was demonstrated to increase perinatally and to decrease after the 1st week of life (130). The SP-A concentration in umbilical cord serum did not differ depending on gestational age, birthweight, delivery mode or Apgar score (131).
The median concentration of SP-D in umbilical cord sera from term babies was reported to be higher than in preterms born at GA 28-32 weeks but not with those born at shorter GA. Levels of this lectin in tracheal aspirates positively correlated with gestational age and birthweight (76). Similarly, SP-D concentration in bronchoalveolar lavage fluid (BALF) from extremely preterm babies was found to rise with increasing GA (132). In contrast, Dahl et al. (133) found higher SP-D in cord blood from newborns with lower GA [further confirmed by Briana et al. (134)] and no significant correlation with BW. On the other hand, concentrations of this lectin in capillary blood sera (taken postnatally, at age 4-10 days) were decreased with increased body mass. Interestingly, SP-D levels in capillary serum were higher in babies born by caesarean section (CS) than in those born vaginally, whilst cord serum concentrations did not differ significantly. However, rupture of membranes for >1 h was associated with lower SP-D in cord blood within CS group. In neonates delivered vaginally, cord serum SP-D was higher when born within 1 h of labour. Furthermore, significant relationships with maternal age and smoking status (lower concentration in babies from the youngest women and from smokers) were noted (133). Later, analysing data from preterm newborns only, Dahl et al. (135) confirmed relationships of SP-D levels in cord serum with membrane rupture and observed significant association of capillary serum SP-D with that complication, mode of delivery and maternal smoking. Furthermore, concentration of this collectin in cord blood from babies diagnosed with intrauterine growth restriction (IUGR) was lower in comparison with those appropriate for GA (135). In contrast, Briana et al. (134) found an opposite relationship in term newborns. Another discrepancy to the above-mentioned report published by Dahl et al. (133) was higher levels of SP-D in newborns delivered vaginally compared with those born via CS. They furthermore observed an inverse correlation between cord serum SP-D and gestational age (134). Both Dahl et al. (133, 135) and Briana et al. (134) noted SP-D levels in sera taken postnatally were higher than those from cord blood. It has been postulated that concentration of this collectin in cord serum in IUGR patients may be a marker predicting lung function impairment in adulthood (136).
Preterm delivery is often a result of intrauterine infection, which may be manifested by chorioamnionitis. Han et al. (123) found SFTPA1-specific mRNA expression to be significantly higher in chorioamniotic membranes from preterm deliveries associated with chorioamnionitis compared with those not. Hilgendorff et al. (76) observed lower SP-D concentrations in sera from preterms born with symptoms of chorioamnionitis. Chaiworapongsa et al. (137) found amniotic fluid concentrations of both SP-A and SP-D to be unchanged. Interestingly, recent data published by De Luca et al. (132) demonstrated higher SP-D levels in BALF from extremely preterm babies affected by chorioamnionitis with fetal involvement compared with the corresponding reference group.
Although no difference in SP-D concentration in cord sera was noted between preterm babies diagnosed with congenital sepsis and without infection (76), the levels of this protein were higher in endotracheal aspirates from preterms aged 1-20 days in comparison with those from controls (aged 1-30 days) (138).
Surfactant deficiency, which is not uncommon among preterms, contributes to a variety of adverse effects, including high susceptibility to respiratory infections and neonatal respiratory distress syndrome (RDS) both of which may in turn lead to bronchopulmonary dysplasia (BPD) or chronic lung disease (CLD). However, those pathologies are not restricted to prematurity-associated impairment of synthesis of surfactant components. Hilgendorff et al. (109) observed elevated concentrations of SP-D in tracheal aspirates from neonates born between 28th and 32nd weeks of gestation but not from extremely preterm babies GA <28 weeks). Later, they found a protective role of minor allele homozygosity for rs1923537 polymorphism of the SFTPD gene (+11208 A>G, localised to 3’UTR) in newborns <32 weeks of GA. The variant allele in affected individuals was associated with lower prevalence for need for surfactant, oxygen supplementation and for diuretics (139). A relationship between the SNP and RDS in very preterm infants was further confirmed by Ryckman et al. (140). Several other SP-D variants, related to SNP localised to coding region or 5’UTR of the SFTPD gene have been considered as RDS risk/outcome modifying factors. Sorensen et al. (141), based on analysis of seven polymorphisms, proposed four of them to be associated with respiratory outcome in preterm babies: rs1923534 (5’UTR), rs721917 (+32 T>C, Met11Thr), rs2243639 (+478 G>A, Ala160Thr; referred also as rs17885900), and rs3088308 (+868 T>A, Ser270Thr). Carrying of the major alleles for rs1923534, rs721917 and rs3088308 was associated with higher risk of respiratory distress, need for oxygen supplementation and respiratory support. On the other hand, three of seventeen reconstructed haplotypes (with frequency >0.05), including minor alleles for rs1923534, rs721917 and rs3088308 were related to lower concentration of SP-D, lower risk of respiratory distress, need for oxygen supplementation and respiratory support (141). In contrast, Chang et al. (142) found no significance of rs721917 or rs2243639. That was later confirmed by Amatya et al. (143), however an intriguing intergenic SNP interactions model was established in their paper, based on investigation of 17 polymorphisms of the SFTPA1, SFTPA2, SFTPB, SFTPC and SFTPD genes. That led to the identification of a protective effect of rs17886395 (SFTPA2) and rs721917 (SFTPD) interaction, when both had a dominant effect. Analysis of three-SNP interactions revealed six with SFTPD rs721917 or rs2243639 dominant effect involvement. With one exception, combination of rs1965708 (SFTPA2, dominant) rs1136450 (SFTPA1, dominant) and rs2243639 (odds ratio of 1.62) they were considered protective (143). A similar approach, although restricted to genes encoding pulmonary collectins, was earlier proposed by Thomas et al. (144). They found 1 two-marker (A at SFTPD rs2243639 and 1A1 of SFTPA2), 2 three-marker [(T at rs721917, A at rs2243639 and 1A1) and (A at rs2243639, 1A2 of SFTPA2 and 6A4 of SFTPA1) and 2 four-marker [(T at rs721917, A at rs2243639, 1A1 and 6A3 of SFTPA1) and (T at rs721917, A at rs2243639, 1A2 and 6A4)] haplotypes (144). Later, an association of SFTPA2 1A1 and SFTPA1 6A4 haplotypes was confirmed by Jo et al. (145) and Tsitoura et al. (146), respectively. Moreover, Tsitoura et al. (146) considered SFTPA2 1A5 variant as RDS risk factor as well while Jo et al. (145) suggested SFTPA2 1A0 protective of preterm neonates. Some seemingly contradictory data were published with regard to role of SFTPA1 6A2 and 6A3 variants in RDS. Several reports demonstrated 6A2 haplotype/homozygosity alone or in cooperation with other SNP/haplotypes of surfactant proteins-encoding genes as a risk factor. Ramet et al. (147) found 6A2 haplotype and its combination with 1A0 more frequent among RDS neonatal (mostly but not only preterm) cases, compared with controls. The 6A3 haplotype, especially when accompanied by SFTPA2 1A1 or 1A2 were deemed protective (147). Marttila et al. (148), in a study recruiting premature singletons and twins/multiples, observed higher frequency of both 6A2 haplotype and 6A2/6A2 genotype among affected singletons when accompanied by Thr/Thr homozygosity for SFTPB Ile131Thr polymorphism (rs1130866). Surprisingly, the discussed SP-A1 gene variants were underrepresented among twins/multiples with RDS when associated with Ile/Thr or Thr/Thr SP-B genotypes. Furthermore, it was found that the role of 6A2/Thr combination in singletons depends on gestational age: it seemed to increase the RDS risk when shorter and to protect when close to term (148). In another report, concerning twins born at GA 32-36 weeks, they observed 6A2 variant (also homozygous) less frequently in affected compared with unaffected babies. Moreover, that haplotype was noted to be associated with a higher birthweight (149). Earlier, Haataja et al. (150) demonstrated 6A2 and 6A2/1A0 combination more frequently among SFTPB Thr allele carrying preterms born at GA <32 weeks, diagnosed with RDS compared with corresponding controls. An opposite effect was observed for 6A3 and 6A3/1A2 variants (150). Some reports underlined certain differences depending not only on gestational age but also on ethnicity. Although Kala et al. (151) observed a protective association of homozygosity for 6A3 independently of those factors, the 1A0 allele effect was noticeable in Caucasians born at GA >28 weeks (including term neonates) but not in Afro-Americans or babies born at shorter GA. Again, a synergistic effect with the SFTPB polymorphism was noted (151). Later, Floros et al. (152) extended those findings demonstrating 6A2 and 1A0 variants as RDS risk factors when accompanied by specific SFTPB variants in preterm Caucasians as well as 6A3 one to be protective in Afro-Americans born at GA 31-35 weeks, in association with another SP-B gene polymorphism (152). Another issue considered by Haataja et al. (153) and Floros et al. (154) was parent-offspring transmission of genotypes and its association with phenotype. Haataja et al. (153) investigated that problem recruiting mother-father-preterm baby Finnish trios (neonates affected with RDS born at GA <37 weeks and unaffected born at GA <32 weeks, termed hypernormal). The 6A2/1A0 haplotype was found excessively transmitted to babies with RDS. On the other hand, decreased transmission of the 6A2 variant to unaffected preterms was observed (153). In the second mentioned work (154), families of various ethnicity were recruited. They confirmed a higher frequency of 6A2/1A0 haplotype and found a lower frequency of 6A4/1A5. Again, the 6A3 haplotype seemed protective for Afro-American but not Caucasian preterms. This study confirmed also some differences in distribution of susceptibility alleles, depending on gestational age (154).
Regarding RDS-associated SNP of genes for SP-A, as well as intra- or intergenic SNP combinations previously discussed, some single polymorphism relationships were described. Amatya et al. (143) demonstrated C allele at rs17886395 (SFTPA2; +271 G>C, Ala91Pro) to be protective from RDS. Earlier, Abuelhamed et al. (155) found A allele and A/A homozygosity for another SFTPA2 polymorphism (+751 G>A) to be RDS risk factors. In the cited above study, Chang et al. (142) reported A/A genotype/A allele and C/C genotype/C allele related to rs1136451 (+186 A>G, Pro62Pro) and rs4253527 (+655 C>T, Arg219Trp) SFTPA2 SNP, respectively, to be associated with neonatal RDS. Both mentioned variants corresponded to lower SP-A2 levels as well. Furthermore, analysis of haplotypes encompassing rs1136451, rs4253527 (SFTPA2), -18 A>C (5’UTR), rs1130866 (+1580 C>T, Thr131Ile) (SFTPB, encoding a hydrophobic lung surfactant component, SP-B), rs721917 and rs17885900 (SFTPD) led to the conclusion that ACACCA and ACACCG patterns may increase RDS risk in preterms while ACACTG seems to be protective (142). That finding again demonstrates the significance of inter- and intergenic associations.
Eguchi et al. (156) and Stevens et al. (157) observed lower SP-A levels in tracheal aspirates from babies with RDS in comparison with infants ventilated for other reasons. Furthermore, the latter investigation revealed no change in SP-A concentration within four days in non-survivors and a significant increase in survivors. Recently, Ghitoi et al. (158) found a significant correlation between expression of SP-A protein in the lung sections from fatal preterm RDS cases and bronchopneumonia. The association of lung SP-A expression and death from neonatal respiratory distress syndrome was however noticed over two decades ago by DeMello et al. (159, 160). Furthermore, low concentration of this lectin in the amniotic fluid was considered as predictor of fetal lung immaturity, and therefore RDS (although with low specificity) (120). Higher specificity was reached when SP-A levels were determined in cord sera from preterms (161, 162).
Another, already mentioned, severe clinical condition is bronchopulmonary dysplasia (BPD), affecting mainly preterm babies with extremely/very low birthweight, requiring mechanical ventilation and oxygen supplementation. It is often one of the consequences of RDS and invasive procedures used for treatment. Pavlovic et al. (163) investigated a variety of polymorphisms of surfactant protein genes in preterm infants suffering from BPD (majority of them were diagnosed with RDS as well). They suggested G- (SFTPD rs721917) -1A2 (SFTPA2) and C- (SFTPD rs17885900) -1A2 (SFTPA2) to be protective. Later, Vinod et al. (164) found no greater association of SP-D concentrations in serum (taken at 3rd and 7th days of life) with BPD at 36th week of postmenstrual age in neonates born at GA ≤32 weeks. In a similar but not identical GA group (<32 weeks), Weber et al. (165) identified SFTPA1 6A6 haplotype as a BPD risk factor. Recently, De Luca et al. (132) observed lower concentrations of SP-D in BALF samples from extremely preterm newborns who developed that disease compared with those who did not or died with no diagnosis.
Neonatal RDS may also contribute to the development of chronic lung disease (CLD). Kotecha et al. (166) reported low activity (associated with presence of low oligomeric molecules) of SP-D in bronchoalveolar lavage fluid samples from preterm babies diagnosed with CLD. They furthermore observed lower SP-D expression at 1st day of life in individuals who developed disease compared with those who did not (166).
Pulmonary morbidity may result from congenital diaphragmatic hernia (CDH) itself and related therapeutic procedures including mechanical ventilation. Marks et al. (167) noticed a marked increase of SP-D concentrations within the 1st month in sera from infants who had more severe CHD. SP-D levels increased also in response to termination of extracorporeal membrane oxygenation (ECMO) as well as to diaphragm surgical repair. Those changes were suspected to reflect the lung injury (167).
Surfactant protein A is detectable in the blood-brain barrier and cerebrospinal fluid (CSF). Its elevated concentration, in comparison with reference group, was found in CSF from premature babies suffering from intraventricular haemorrhage. That was concluded to be associated with posthaemorrhagic CSF flow impairment (168). Recently, based on murine model, immunomodulatory role of SP-A in local and systemic neonatal neuroinflammation (intraventricular haemorrhage, hypoxic-ischemic encephalopathy, systemic sepsis) was demonstrated (169).
3 Clinical associations of ficolins in the neonate
3.1 Ficolin-1
Data concerning ficolin-1 in neonatal health and disease are still rather scarce. Its concentration in serum was demonstrated to increase with gestational age (79). Low levels of that protein were found associated with a requirement for mechanical ventilation and increased mortality in preterms with necrotising enterocolitis (170). Schlapbach et al. (171) reported also an association of high ficolin-1 with early-onset (but not late-onset) sepsis and its correlation with the absolute phagocyte count and immature/total neutrophil ratio. Later, Świerzko et al. (104) found higher median ficolin-1 concentrations in sera from babies suffering from perinatal infections who developed sepsis in comparison with healthy controls. A similar association was found with less severe infections, but no significant difference was noted between disease groups. Furthermore, heterozygosity for the rare FCN1 gene mutation, rs148649884 (+6658 G>A, Ala218Thr) was reported in one case of neonatal systemic inflammatory response syndrome (SIRS) (104).
3.2 Ficolin-2
Ficolin-2 has been the most extensively investigated member of that family in relation to neonates. Several reports describe strong associations of its low concentrations in serum with prematurity and/or low birthweight (also independently of gestational age) (70, 79, 81, 172). However, Briana et al. (173) found no significant relationship between ficolin-2 levels in cord blood and intrauterine growth restriction in term newborns. Recently, Szala-Poździej et al. (174) reported associations of SNPs localised to the FCN2 gene promoter with birthweight and/or gestational age in Polish preterm babies. Major alleles for rs3124952 (-986 A>G), rs7865453 (-64 A>C) and A/A homozygosity for both mentioned SNP were found less frequently among newborns with very low birthweight (VLBW, ≤1500 g) compared with the corresponding reference group. A similar relationship was observed for minor allele homozygosity (G/G) for rs17514136 (-4 A>G) polymorphism. That genotype seemed to protect from birth at GA <33 weeks as well. Moreover, reconstructed haplo- and diplotypes, including data concerning rs3124953 (-602 G>A) SNP were analysed in the context of GA and BW. The AGA (-986/-602/-64/-4) haplotype and AGAG/AGAG homozygosity were protective for VLBW. The GGCA haplotype had the opposite effect, independently of GA. Finally, the AGAG/GGAA diplotype was associated with both shorter (<33 weeks) GA and body mass at birth <1500 g (174). Earlier, Kilpatrick et al. (70) found a lower frequency of the A/A-A/A-A/A-A/A-A/A-C/C-G/G genotype [related to following FCN2 SNP: rs3124952 (-986 A>G), rs3124953 (-602 G>A), rs3811140 (-557 A>G), rs7865453 (-64 A>C), rs17514136 (-4 A>G), rs17549193 (+6359 C>T, Thr236Met) and rs7851696 (+6424 G>T, Ala258Ser)] in preterm, compared with term neonates. Those findings seem to confirm a significant relationship between low ficolin-2 and prematurity as well as low BW. Furthermore, two FCN2 SNP localised to 3’UTR: rs73664188 (T>C) and rs11103564 (T>C) were found to influence the risk of VLBW: the C allele of the first was found to increase the chance of BW <1500 g, while that of the second was protective in preterm newborns (71).
Low ficolin-2 concentration in cord serum was also found associated with a higher risk of perinatal infections, including sepsis (70, 81, 104). In those studies, data from consecutively taken samples (both term and preterm babies) were analysed. Low (<1 µg/ml) ficolin-2 levels were found significantly more often in disease groups (babies with perinatal infections who developed sepsis or with milder disease course) compared with infection-free controls (104). However, in another investigation, Schlapbach et al. (107) observed no significant difference between infants suffering from culture-proven sepsis and matched controls. Furthermore, no impact of the FCN2 promoter (-64 A>C and -4 A>G) or exon 8 (+6359 C>T and +6424 G>T] SNPs on risk of perinatal sepsis (104) or the promoter (-986/-602/-64/-4) SNP on early-onset infection in preterms (174) was found. On the other hand, Świerzko et al. (71) demonstrated diplotypes GTTTGT/GGTCGT and GTTTGT/GGTCGA [corresponding to FCN2 3’UTR polymorphisms: rs7851696 (G>T), rs4521835 (T>G), rs73664188 (T>C), rs11103564 (T>C), rs11103565 (G>A) and rs6537959 (T>A)] to be risk factors for early onset infections (EOI) in preterm babies. The protective influence of ficolin-2 on certain perinatal infections was strongly supported from evidence by Fuijeda et al. (175). They demonstrated its contribution to opsonophagocytosis and killing of group B streptococci (GBS, a common agent of neonatal infections, including pneumonia, meningitis and sepsis) of III, V and VIII serotypes (175).
Recently, Gajek et al. (176), investigating the previously-described cohort of preterm babies (71, 174), observed significantly lower concentrations of ficolin-2 in cord serum samples (independently of FCN2 promoter/3’UTR SNP) from individuals who developed RDS. Ficolin-2 concentrations were lower in babies who required intubation in the delivery room. Moreover, ficolin-2 levels correlated positively with Apgar score and inversely with duration of respiratory support and length of NICU stay. When GA did not exceed 32 weeks, ficolin-2 concentration partially distinguished those with and without RDS and differentiated between mild and moderate/severe disease (176).
3.3 Ficolin-3
Like other ficolins, ficolin-3 concentration increases with gestational age (77, 177), and correlates significantly with both GA and BW as well as with ficolin-2 (177). Low levels are associated with low birthweight independently of GA (177), but there was no relationship with IUGR in term babies (173). Schlapbach et al. (178) suggested that inherited deficiency of ficolin-3 may be responsible for necrotising enterocolitis as well as repeated staphylococcal skin infections (178). Michalski et al. (179) described a patient with severe Streptococcus agalactiae infection, but that individual had multiple immune defects so the contribution of ficolin-3 was uncertain. Additionally, for both those patients, no more severe infections were noted within 4-year and 8-year follow-ups, respectively (178, 179). Low ficolin-3 was found associated with neonatal sepsis by Świerzko et al. (104), and specifically Gram-positive sepsis by Schlapbach et al. (107). Although the median value determined for newborns with proven infections who had not developed sepsis did not differ from that of controls (104, 177), the frequency of low (<7.9 µg/ml) concentrations was significantly higher (104).
4 Concluding remarks
Collectins and ficolins create an universal group of pattern-recognising receptors having collectively broad specificity towards non-self or abnormal-self glycoconjugates (but not limited to saccharide structures). Their affinity to the target is enhanced by multimerization, enabling more efficient opsonisation or agglutination of pathogens. The majority is expressed mainly in hepatocytes and secreted into blood, but some (synthesized also within respiratory system) play an important role in local immunity (SP-A, SP-D, ficolin-3). CL-12 and ficolin-1 are produced by other cells and may be found as soluble or cell-associated forms. With the exception of SP-A and SP-D, they are capable of initiating complement activation, mostly via the lectin pathway, although CL-12 is known to initiate alternative and classical pathways. In this paper, we described the genetics, structure, and specificity of these collagen-related lectins, and reviewed their complex and multiple clinical associations in neonates. Their protective role or contribution to the pathogenesis, depend on a variety of variables, including genetic polymorphisms, gestational age, way of delivery, maternal/environmental (i.e. hospital/home) microflora, and so on.
Author contributions
MC: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. AS: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We are very grateful to Dr. David C. Kilpatrick for critical reading of the manuscript and helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sharma AA, Jen R, Butler A, Lavoie PM. The developing human preterm neonatal immune system: a case for more research in this area. Clin Immunol (2012) 145:61–8. doi: 10.1016/j.clim.2012.08.006
2. Stasiak-Barmuta A, Stankiewicz W, Dąbrowski MI, Jurkiewicz B, Dąbrowski MP. Distinctions of the neonatal immune system. Centr Eur J Immunol (2010) 35:252–8.
3. Gonzalez CA, Gonzalez S. Fetal and neonatal allo-immune response. Transfus Apher Sci (2020) 59:102945. doi: 10.1016/j.transci.2020.102945
4. Cedzynski M, Swierzko ASt, Kilpatrick DC. Factors of the lectin pathway of complement activation and their clinical associations in neonates. J BioMed Biotechnol (2012) 2012:363246. doi: 10.1155/2012/363246
5. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement – MBL and beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
6. Casals C, Garcia-Fojeda B, Minutti CM. Soluble defense collagens: sweeping up immune threats. Mol Immunol (2019) 112:291–304. doi: 10.1016/j.molimm.2019.06.007
7. Świerzko AS, Cedzyński M. The influence of the lectin pathway of complement activation on infections of the respiratory system. Front Immunol (2020) 11:585243. doi: 10.3389/fimmu.2020.585243
8. Ma YJ, Hein E, Munthe-Fog L, Skjoedt M-O, Bayarri-Olmos R, Romani L, et al. Soluble collecctin-12 (CL-12) is a pattern recognition molecule initiating complement via the alternative pathway. J Immunol (2015) 195:3365–73. doi: 10.4049/jimmunol.1500493
9. Zhang J, Song L, Pedersen DV, Li A, Lambris JD, Andersen GM, et al. Soluble collectin-12 mediates C3-independent docking of properdin that activates the alternative pathway of complement. eLife (2020) 9:e60908. doi: 10.7554/eLife.60908
10. Roy N, Ohtani K, Matsuda Y, Mori K, Hwang I, Suzuki Y, et al. Collectin CL-P1 utilizes C-reactive protein for complement activation. Biochim Biophys Acta (2016) 1860:1118–28. doi: 10.1016/j.bbagen.2016.02.012
11. Roy N, Ohtani K, Hidaka Y, Amano Y, Matsuda Y, Mori K, et al. Three pentraxins C-reactive protein, serum amyloid P component and pentraxin 3 mediate complement activation using collectin CL-P1. Biochim Biophys Acta (2017) 1861:1–14. doi: 10.1016/j.bbagen.2016.11.023
12. Ma YJ, Doni A, Skjoedt MO, Honore C, Arendrup M, Mantovani A, et al. Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J Biol Chem (2011) 286:3405–17. doi: 10.1074/jbc.M110.190637
13. Vieira F, Kung JW, Bhatti F. Structure, genetics and function of the pulmonary associated surfactant proteins A and D: The extra-pulmonary role of these C type lectins. Ann Anat. (2017) 211:184–201. doi: 10.1016/j.aanat.2017.03.002
14. Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol Immunol (2007) 44:3875–88. doi: 10.1016/j.molimm.2007.06.005
15. Cedzyński M, Kilpatrick DC, Świerzko AS. Mannose-binding lectin. In: Barnum S, Chein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 33–43.
16. Cedzyński M, Świerzko AS. Components of the lectin pathway of complement in haematologic malignancies. Cancers (Basel). (2020) 12:1792. doi: 10.3390/cancers12071792
17. Selman L, Hansen S. Structure and function of collectin liver 1 (CL-L1) and collectin 11 (CL-11, CL-K1). Immunobiology (2012) 217:851–63. doi: 10.1016/j.imbio.2011.12.008
18. Hansen SW, Ohtani K, Roy N, Wakamiya N. The collectins CL-L1, CL-K1 and CL-P1, and their roles in complement and innate immunity. Immunobiology (2016) 228:1058–67. doi: 10.1016/j.imbio.2016.05.012
19. Ohtani K, Wakamiya N. The collectins. In: Barnum S, Chein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 57–65.
20. Henriksen ML, Brandt J, Andrieu J-P, Nielsen C, Jensen PH, Holmskov U, et al. Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol (2013) 191:6117–27. doi: 10.4049/jimmunol.1302121
21. Hansen SWK, Aagaard JB, Bjerrum KB, Hejbol EK, Nielsen O, Schroder HD, et al. CL-L1 and CL-K1 exhibit widespread tissue distribution with high and co-localized expression in secretory epithelia and mucosa. Front Immunol (2018) 9:1757. doi: 10.3389/fimmu.2018.01757
22. Gajek G, Świerzko AS, Cedzyński M. Association of polymorphisms of MASP1, COLEC10 and COLEC11 genes with 3MC syndrome. Int J Mol Sci (2020) 21:5483. doi: 10.3390/ijms21155483
23. Axelgaard E, Jensen L, Dyrlund TF, Nielsen HJ, Enghild JJ, Thiel S, et al. Investigations on collectin liver 1. J Biol Chem (2013) 288:23407–20. doi: 10.1074/jbc.M113.492603
24. Venkatraman Grija U, Furze CM, Gingras AR, Yoshizaki T, Ohtani K, Marshall JE, et al. Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol (2015) 13:1–14. doi: 10.1186/s12915-015-0136-2
25. Hansen S, Selman L, Palaniyar N, Ziegler K, Brandt J, Kliem A, et al. Collectin 11 (CL-11, CL-K1) is a MASP1/3-associated plasma collectin with microbial binding activity. J Immunol (2010) 185:6096–104. doi: 10.4049/jimmunol.1002185
26. Kirketerp-Moller N, Bayarri-Olmos R, Krogfelt KA, Garred P. C1q/TNF-related protein 6 is a pattern recognition molecule that recruits collectin-11 from the complement system to ligands. J Immunol (2020) 204:1598–606. doi: 10.4049/jimmunol.1901316
27. Nakamura K, Funakoshi H, Miyamoto K, Tokunaga F, Nakamura T. Molecular cloning and functional characterization of a human scavenger receptor with C-type lectin (SRCL), a novel member of a scavenger receptor family. Biochem Biophys Res Commun (2001) 280:1028–35. doi: 10.1006/bbrc.2000.4210
28. Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Keshi H, et al. The membrane-type collectin CL-P1 is a scavenger receptor on vascular endothelial cells. J Biol Chem (2001) 276:44222–8. doi: 10.1074/jbc.M103942200
29. Selman L, Skjodt K, Nielsen O, Floridon C, Holmskov U, Hansen S. Expression and tissue localization of collectin placenta 1 (CL-P1, SRCL) in human tissues. Mol Immunol (2008) 45:3278–88. doi: 10.1016/j.molimm.2008.02.018
30. Yoshida T, Tsuruta Y, Iwasaki M, Yamane S, Ochi T, Suzuki R. SRCL/CL-P1 recognizes GalNAc and a carcinoma-associated antigen, Tn antigen. J Biol Chem (2003) 133:271–7. doi: 10.1093/jb/mvg037
31. Coombs PJ, Graham SA, Drickamer K, Taylor ME. Selective binding of the scavenger receptor C-type lectin to Lewisx trisaccharide and related glycan ligands. J Biol Chem (2005) 280:22993–9. doi: 10.1074/jbc.M504197200
32. Elola MT, Capurro MI, Barrio MM, Coombs PJ, Taylor ME, Drickamer K, et al. Lewis x antigen mediates adhesion of human breast carcinoma cells to activated endothelium. Possible involvement of the endothelial scavenger receptor C-type lectin. Breast Cancer Res Treat (2007) 101:167–71. doi: 10.1007/s10549-006-9286-9
33. Ohtani K, Suzuki Y, Wakamiya N. Biological functions of novel collectins CL-L1, CL-K1, and CL-P1. J BioMed Biotechnol (2012) 2012:493945. doi: 10.1155/2012/493945
34. Mori K, Ohtani K, Jang S, Kim Y, Hwang I, Roy N, et al. Scavenger receptor CL-P1 mainly utilizes a collagen-like domain to uptake microbes and modified LDL. Biochim Biophys Acta (2014) 1840:3345–56. doi: 10.1016/j.bbagen.2014.08.015
35. McCormack FX. Structure, processing and properties of surfactant protein A. Biochim Biophys Acta (1998) 1408:109–31. doi: 10.1016/s0925-4439(98)00062-3
36. Childs RA, Wright JR, Ross GF, Yuen CT, Lawson AM, Chai W, et al. Specificity of lung surfactant protein SP-A for both the carbohydrate and the lipid moieties of certain neutral glycolipids. J Biol Chem (1992) 267:9992–9. doi: 10.1016/S0021-9258(19)50187-9
37. Haagsman HP, Diemel RV. Surfactant-associated proteins: functions and structural variations. Comp Biochem Physiol A Mol Integr Physiol (2001) 129:91–108. doi: 10.1016/s1095-6433(01)00308-7
38. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol (2006) 43:1293–315. doi: 10.1016/j.molimm.2005.08.004
39. Jakel A, Qaseem AS, Kishore U, Sim RB. Ligands and receptors of lung surfactant proteins SP-A and SP-D. Front Biosci (2013) 18:1129–40. doi: 10.2741/4168
40. Floros J, Thorenoor N, Tsotakos N, Phelps DS. Human surfactant protein SP-A1 and SP-A2 variants differentially affect the alveolar microenvironment, surfactant structure, regulation and function of the alveolar macrophage, and animal and human survival under various conditions. Front Immunol (2021) 12:681639. doi: 10.3389/fimmu.2021.681639
41. Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang CH, Szeliga J, et al. (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem (2011) 286:4854–70. doi: 10.1074/jbc.M110.125567
42. Persson A, Chang D, Crouch E. Surfactant protein D is divalent cation-dependent carbohydrate-binding protein. J Biol Chem (1990) 265:5755–60. doi: 10.1016/S0021-9258(19)39427-X
43. Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res (2000) 1:93–108. doi: 10.1186/rr19
44. Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci (2012) 17:407–29. doi: 10.2741/3935
45. Silveyra P, Floros J. Genetic complexity of the human surfactant-associated proteins SP-A1 and SP-A2. Genes (2013) 531:126–32. doi: 10.1016/j.gene.2012.09.111
46. Heidinger K, Konig IR, Bohnert A, Kleinsteiber A, Hilgendorff A, Gortner L, et al. Polymorphisms in the human surfactant protein-D (SFTPD) gene: strong evidence that serum levels of surfactant protein-D (SP-D) are genetically influenced. Immunogenetics (2005) 57:1–7. doi: 10.1007/s00251-005-0775-5
47. Sorensen GL, Hjelmborg J, Kyvik KO, Fenger M, Hoj A, Bendixen C, et al. Genetic and environmental influences of surfactant protein D serum levels. Am J Physiol Lung Cell Mol Physiol (2006) 290:L1010–7. doi: 10.1152/ajplung.00487.2005
48. Bayarri-Olmos R, Hansen S, Henriksen ML, Storm L, Thiel S, Garred P, et al. Genetic variation of COLEC10 and COLEC11 and association with serum levels of collectin liver 1 (CL-L1) and collectin kidney 1 (CL-K1). PloS One (2015) 10(2):e0114883. doi: 10.1371/journal.pone.0114883
49. Ohmori H, Makita Y, Funamizo M, Chiba S, Ohtani K, Suzuki Y, et al. Haplotype analysis of the human collectin placenta 1 (hCL-P1) gene. J Hum Genet (2003) 48:82–5. doi: 10.1007/s100380300011
50. Endo Y, Matsushita M, Fujita T. New insights into the role of ficolins in the lectin pathway of innate immunity. Int Rev Cell Mol Biol (2015) 316:49–110. doi: 10.1016/bs.ircmb.2015.01.003
51. Matsushita M. Ficolins. In: Barnum S, Chein T, editors. The Complement Factsbook, 2nd edition. London, UK: Academic Press, Elsevier (2018). p. 45–56.
52. Ma YJ, Doni A, Hummelshoj T, Honore C, Bastone A, Mantovani A, et al. Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J Biol Chem (2009) 284:28263–75. doi: 10.1074/jbc.M109.009225
53. Ma YJ, Lee BL, Garred P. An overview of the synergy and crosstalk between pentraxins and collecttins/ficolins: their functional relevance in complement activation. Exp Mol Med (2017) 49:e320. doi: 10.1038/emm.2017.51
54. Gout E, Moriscot C, Doni A, Dumestre-Pérard C, Lacroix M, Perard J, et al. M-ficolin interacts with the long pentraxin PTX3: a novel case of cross-talk between soluble pattern-recognition molecules. J Immunol (2011) 186:5815–22. doi: 10.4049/jimmunol.1100180
55. Ma YJ, Doni A, Romani L, Jürgensen HJ, Behrendt N, Mantovani A, et al. Ficolin-1-PTX3 complex formation promotes clearance of altered self-cells and modulates IL-8 production. J Immunol (2013) 191:1324–33. doi: 10.4049/jimmunol.1300382
56. Fujita T, Matsushita M, Endo Y. The lectin-complement pathway – its role in innate immunity and evolution. Immunol Rev (2004) 198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x
57. Degn SE, Thiel S. Humoral pattern recognition and the complement system. Scand J Immunol (2013) 78:181–93. doi: 10.1111/sji.12070
58. Jarlhelt I, Pilely K, Clausen JB, Skjoedt MO, Bayarri-Olmos R, Garred P. Circulating ficolin-2 and ficolin-3 form heterocomplexes. J Immunol (2020) 204:1919–28. doi: 10.4049/jimmunol.1900694
59. Garlatti V, Belloy N, Martin L, Lacroix M, Matsushita M, Endo Y, et al. Structural insights into the innate immune recognition specificities of L- and H-ficolins. EMBO J (2007) 26:623–33. doi: 10.1038/sj.emboj.7601500
60. Garlatti V, Martin L, Lacroix M, Gout E, Arlaud GJ, Thielens NM, et al. Structural insights into the recognition properties of human ficolins. J Innate Immun (2010) 2:17–23. doi: 10.1159/000233475
61. Laffly E, Lacroix M, Martin L, Vassal-Stermann E, Thielens NM, Gaboriaud C. Human ficolin-2 recognition versatility extended: an update of the binding of ficolin-2 to sulphated/phosphated carbohydrates. FEBS Lett (2014) 588:4694–700. doi: 10.1016/j.febslet.2014.10.042
62. Thielens NM, Gout E, Lacroix M, Reiser JB, Gaboriaud C. Analysis of the ligand recognition specificities of human ficolins using surface plasmon resonance. Methods Mol Biol (2021) 2227:205–26. doi: 10.1007/978-1-0716-1016-9_19
63. Hummelshoj T, Munthe-Fog L, Madsen HO, Garred P. Functional SNPs in the human ficolin (FCN) genes reveal distinct geographical patterns. Mol Immunol (2008) 45:2508–20. doi: 10.1016/j.molimm.2008.01.003
64. Garred P, Honore C, Ma YJ, Rorvig S, Cowland J, Borregaard N, et al. The genetics of ficolins. J Innate Immun (2010) 2:3–16. doi: 10.1159/000242419
65. Ammitzboll CG, Kjaer TR, Steffensen R, Stengaard-Pedersen K, Nielsen HJ, Thiel S, et al. Non-synonymous polymorphisms in the FCN1 gene determine ligand-binding ability and serum levels of M-ficolin. PloS One (2012) 7:e50585. doi: 10.1371/journal.pone.0050585
66. Munthe-Fog L, Hummelshoj T, Honore C, Moller ME, Skjoedt MO, Palsgaard I, et al. Variation in FCN1 affects biosynthesis of ficolin-1 and is associated with outcome of systemic inflammation. Genes Immun (2012) 13:515–22. doi: 10.1038/gene.2012.27
67. Hummelshoj T, Munthe-Fog L, Madsen HO, Fujita T, Matsushita M, Garred P. Polymorphisms in the FCN2 gene determine serum variation and function of ficolin-2. Hum Mol Genet (2005) 14:1651–8. doi: 10.1093/hmg/ddi173
68. Munthe-Fog L, Hummelshoj T, Hansen BE, Koch C, Madsen HO, Skjodt K, et al. The impact of FCN2 polymorphisms and haplotypes on the ficolin-2 serum levels. Scand J Immunol (2007) 65:383–92. doi: 10.1111/j.1365-3083.2007.01915.x
69. Cedzynski M, Nuytinck L, Atkinson AP, Swierzko ASt, Zeman K, Szemraj J, et al. Extremes of L-ficolin concentration in children with recurrent infections are associated with single nucleotide polymorphisms in the FCN2 gene. Clin Exp Immunol (2007) 150:99–104. doi: 10.1111/j.1365-2249.2007.03471.x
70. Kilpatrick DC, Swierzko A, Matsushita M, Domzalska-Popadiuk I, Borkowska-Klos M, Szczapa J, et al. The relationship between FCN2 genotypes and serum ficolin-2 (L-ficolin) concentrations from a large cohort of Polish neonates. Hum Immunol (2013) 74:867–71. doi: 10.1016/j.humimm.2013.04.011
71. Świerzko AS, Jarych D, Gajek G, Chojnacka K, Kobiela P, Kufelnicka-Babout M, et al. Polymorphisms of the FCN2 gene 3’UTR and their clinical associations in preterm newborns. Front Immunol (2021) 12:741140. doi: 10.3389/fimmu.2021.741140
72. Munthe-Fog L, Hummelshoj T, Ma YJ, Hansen BE, Koch C, Madsen HO, et al. Characterization of the polymorphism in the coding sequence of FCN3 resulting in a Ficolin-3 (Hakata antigen) deficiency state. Mol Immunol (2008) 45:2660–6. doi: 10.1016/j.molimm.2007.12.012
73. Munthe-Fog L, Hummelshoj T, Honore C, Madsen HO, Permin H, Garred P. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med (2009) 360:2637–44. doi: 10.1056/NEJMoa0900381
74. Lau YL, Chan SY, Turner MW, Fong J, Karlberg J. Mannose-binding protein in preterm infants: developmental profile and clinical significance. Clin Exp Immunol (1995) 102:649–54. doi: 10.1111/j.1365-2249.1995.tb03866.x
75. Kielgast S, Thiel S, Henriksen TB, Bjerke T, Olsen J, Jensenius JC. Umbilical cord mannan-binding lectin and infections in early childhood. Scand J Immunol (2003) 57:167–72. doi: 10.1046/j.1365-3083.2003.01202.x
76. Hilgendorff A, Schmidt R, Bohnert A, Merz C, Gortner J. Host defence lectins in preterm neonates. Acta Pediatr (2005) 94:794–9. doi: 10.1111/j.1651-2227.2005.tb01987.x
77. Frakking FNJ, Brouwer N, Zweers D, Merkus MP, Kuijpers TW, Offringa M, et al. High prevalence of mannose-binding lectin deficiency in premature neonates. Clin Exp Immunol (2006) 145:5–12. doi: 10.1111/j.1365-2249.2006.03093.x
78. van der Zwet WC, Catsburg A, van Elburg RM, Savelkoul PH, Vandenbroucke-Grauls CMJE. Mannose-binding lectin (MBL) genotype in relation to risk of nosocomial infection in pre-term neonates in the neonatal intensive care unit. Clin Microbiol Infect (2008) 14:130–5. doi: 10.1111/j.1469-0691.2007.01886.x
79. Sallenbach S, Thiel S, Aebi C, Otth M, Bigler S, Jensenius JC, et al. Serum concentrations of lectin-pathway components in healthy neonates, children and adults: mannan-binding lectin (MBL), M-, L-, and H-ficolin, and MBL-associated serine protease-2 (MASP-2). Pediatr Allergy Immunol (2011) 22:424–30. doi: 10.1111/j.1399-3038.2010.01104.x
80. Kilpatrick DC, Liston WA, Midgley PC. Mannan-binding protein in human umbilical cord blood. Nat Immun (1996) 15:234–40.
81. Swierzko A, Atkinson APM, Cedzynski M, MacDonald SL, Szala A, Domzalska-Popadiuk I, et al. Two factors of the lectin pathway of complement, L-ficolin and mannan-binding lectin, and their associations with prematurity, low birthweight and infections in a large cohort of Polish neonates. Mol Immunol (2009) 46:551–8. doi: 10.1016/j.molimm.2008.07.025
82. Bodamer OA, Mitterer G, Maurer W, Pollak A, Mueller MW, Schmidt WM. Evidence for an association between mannose-binding lectin 2 (MBL2) gene polymorphisms and pre-term birth. Genet Med (2006) 8:518–24. doi: 10.1097/01.gim.0000232478.43335.19
83. Carvahlo da Silva LV, Javorski N, Cavalcanti Brandao LA, de Carvalho Lima M, Crovella S, Eickmann SH. Influence of MBL2 and NOS3 polymorphisms on spontaneous preterm birth in North East Brazil: genetics and preterm birth. J Matern Fetal Neonatal Med (2020) 33:127–35. doi: 10.1080/14767058.2018.1487938
84. Chan D, Bennett PR, Lee YS, Kundu S, Teoh TG, Adan M, et al. Microbial-driven preterm labour involves crosstalk between the innate and adaptive immune response. Nat Commun (2022) 13:975. doi: 10.1038/s41467-022-286-20-1
85. Karjalainen MK, Huusko JM, Tuohimaa A, Luukkonen A, Haataja R, Hallman M. A study of collectin genes in spontaneous preterm birth reveals an association with a common surfactant protein D polymorphism. Pediatr Res (2012) 71:93–9. doi: 10.1038/pr.2011.2
86. Grumach AS, Ceccon ME, Rutz R, Fertig A, Kirschfink M. Complement profile in neonates of different gestational ages. Scand J Immunol (2014) 79:276–81. doi: 10.1111/sji.12154
87. Modi BP, Teves ME, Pearson LN, Parikh HI, Haymond-Thornburg H, Tucker JL, et al. Mutations in fetal genes involved in innate immunity and host defense against microbes increase risk of preterm premature rupture of membranes (PPROM). Mol Genet Genom Med (2017) 5:720–9. doi: 10.1002/mgg3.330
88. Modi BP, Parikh HI, Teves ME, Kulkarni R, Liyu J, Romero R, et al. Discovery of rare ancestry-specific variants in the fetal genome that confer risk of preterm premature rupture of membranes (PPROM) and preterm birth. BMC Med Genet (2018) 19:181. doi: 10.1186/s12881-018-0696-4
89. Rossner P Jr, Tabashidze N, Dostal M, Novakova Z, Chvatalova I, Spatova M, et al. Genetic, biochemical and environmental factors associated with pregnancy outcomes in newborns from the Czech Republic. Environ Health Perspect (2011) 119:265–71. doi: 10.1289/ehp.1002470
90. Gibson CS, Maclennan AH, Haan EH, Priest K, Dekker GA. Fetal MBL2 haplotypes combined with viral exposure are associated with adverse pregnancy outcomes. J Matern Fetal Neonatal Med (2011) 24:847–54. doi: 10.3109/14767058.2010.531324
91. Cakmak BC, Calkavur S, Ozkinay F, Koroglu OA, Onay H, Itirli G, et al. Association between bronchopulmonary dysplasia and MBL2 and IL1-RN polymorphisms. Pediatr Int (2012) 54:863–8. doi: 10.1111/j.1442-200X.2012.03714.x
92. Hartz A, Schreiter L, Pagel J, Moser K, Wieg C, Grotheer A, et al. Mannose-binding lectin and mannose-binding protein-associated sserine protease 2 levels and infection in very-low-birth-weight infants. Pediatr Res (2018) 84:134–8. doi: 10.1038/s41390-018-0017-9
93. Frakking FN, Brouwer N, van Eijkelenburg NK, Merkus MP, Kuijpers TW, Offringa M, et al. Low mannose-binding lectin (MBL) levels in neonates with pneumonia and sepsis. Clin Exp Immunol (2007) 150:255–62. doi: 10.1111/j.1365-2249.2007.03479.x
94. de Benedetti F, Auriti C, D’Urbano LE, Ronchetti MP, Ravà L, Tozzi A, et al. low serum levels of mannose-binding lectin are a risk factor for neonatal sepsis. Pediatr Res (2007) 61:325–8. doi: 10.1203/pdr.0b013e318030d12f
95. Auriti C, Prencipe G, Inglese R, Azzari C, Ronchetti MP, Tozzi A, et al. Role of mannose-binding lectin in nosocomial sepsis in critically ill neonates. Hum Immunol (2010) 71(11):1084–8. doi: 10.1016/j.humimm.2010.08.012
96. Özkan H, Köksal N, Çetinkaya M, Kiliç Ş, Çelebi S, Oral B, et al. Serum mannose-binding lectin (MBL) gene polymorphism and low MBL levels are associated with neonatal sepsis and pneumonia. J Perinatol. (2012) 32:210–7. doi: 10.1038/jp.2011.79
97. Badawy M, Mosallam DS, Saber D, Madani H. Use of mannose-binding lectin gene polymorphisms and the serum MBL level for the early detection of neonatal sepsis. J Pediatr Genet (2018) 7:150–7. doi: 10.1055/s-0038-1675801
98. El-Behedy EM, Akeel N, El-Maghraby HM, Shawky A. Serum level and genetic polymorphism of mannose-binding lectin in infants with neonatal sepsis at Zagazig University hospitals. Egypt J Immunol (2019) 26:91–9.
99. Elhawary IMB, Hussain NF, Ali AA, Samy RM, Abdeltawab MA. The role of mannose binding lectin (MBL2) gene polymorphism in incidence of neonatal sepsis. Eur Chem Bull (2023) 12:4605–13.
100. Dzwonek AB, Neth OW, Thiebaut R, Gulczynska E, Chilton M, Hellwig T, et al. The role of mannose-binding lectin in susceptibility to infection in preterm neonates. Pediatr Res (2008) 63:680–5. doi: 10.1203/PDR.0b013e31816fdbff
101. Koroglu OA, Onay H, Erdemir G, Yalaz M, Cakmak B, Akisu M, et al. Mannose-binding lectin polymorphism and early neonatal outcome in preterm infants. Neonatology (2010) 98:305–12. doi: 10.1159/000291487
102. Ozdemir O, Dinleyici EC, Tekin N, Colak O, Aksit MA. Low-mannose-binding lectin levels in susceptibility to neonatal sepsis in preterm neonates with fetal inflammatory response syndrome. J Matern Fetal Neonatal Med (2010) 23:1009–13. doi: 10.3109/14767050903551418
103. Wahab Mohamed WA, Saeed MA. Mannose-binding lectin serum levels in neonatal sepsis and septic shock. J Matern Fetal Neonatal Med (2012) 25:411–4. doi: 10.3109/14767058.2011.582903
104. Świerzko AS, Szala-Poździej A, Kilpatrick DC, Sobociński M, Chojnacka K, Sokołowska A, et al. Components of the lectin pathway of complement activation in paediatric patients of intensive care units. Immunobiology (2016) 221:657–69. doi: 10.1016/j.imbio.2016.01.003
105. Hartz A, Pagel J, Humberg A, Preuss M, Schreiter L, Rupp J, et al. The association of mannose-binding lectin 2 polymorphisms with outcome in very low birth weight infants. PloS One (2017) 12:e0178032. doi: 10.1371/journal.pone.0178032
106. Dogan P, Ozkan H, Koksal N, Oral. HB, Celebi S, Bagci O, Guney Varal I. Mannose-binding lectin levels in late-onset sepsis in preterm infants. Results from a prospective study in a tertiary care center. Fetal Pediatr Pathol (2020) 39:363–72. doi: 10.1080/15513815.2019.1652374
107. Schlapbach LJ, Mattmann M, Thiel S, Boillat C, Otth M, Nelle M, et al. Differential role of the lectin pathway of complement activation in susceptibility to neonatal sepsis. Clin Infect Dis (2010) 51:153–62. doi: 10.1086/653531
108. Aydemir C, Onay H, Oguz SS, Ozdemir TR, Erdeve O, Ozkinay F, et al. Mannose-binding lectin codon 54 gene polymorphism in relation to risk of nosocomial invasive fungal infections in preterm neonates in the neonatal invasive care unit. J Matern Fetal Neonatal Med (2011) 24:1124–7. doi: 10.3109/14767058.2010.536865
109. Hilgendorff A, Heidinger K, Pfeiffer A, Bohnert A, König IR, Ziegler A, et al. Association of polymorphisms in the mannose-binding lectin gene and pulmonary morbidity in preterm infants. Genes Immun (2007) 8:671–7. doi: 10.1038/sj.gene.6364432
110. Mashaly GE-S, El-Sabbagh AM, El-Kazzaz SS, Nour I. MBL2 gene polymorphism and the association with neonatal sepsis in Egyptian neonates, a case control study. Open J Immunol (2016) 6:111–9. doi: 10.4236/oji.2016.63012
111. Luo J, Xu F, Lu GJ, Lin HC, Feng ZC. Low mannose-binding lectin (MBL) levels and MBL genetic polymorphisms associated with the risk of neonatal sepsis: an updated meta-analysis. Early Hum Dev (2014) 90:557–64. doi: 10.1016/j.earlhumdev.2014.07.007
112. Sljivancanin Jakovljevic T, Martic J, Jacimovic J, Nikolic N, Milasin J, Mitrović TL. Association between innate immunity gene polymorphisms and neonatal sepsis development: a systematic review and meta-analysis. World J Pediatr (2022) 18:654–70. doi: 10.1007/s12519-022-00569-7
113. Ma J, Xu R, Xie Y, Liang J, Han W, Chen X, et al. The association between mannose binding lectin gene polymorphisms and the risk of neonatal sepsis: an updated meta-analysis. Heliyon (2023) 9:e14905. doi: 10.1016/j.heliyon.2023.e14905
114. Capoulongo E, Vento G, Rocchetti S, Giardina E, Concolino P, Sinibaldi C, et al. Mannose-binding lectin polymorphisms and pulmonary outcome in premature neonates: a pilot study. Intensive Care Med (2007) 33:1787–94. doi: 10.1007/s00134-007-0793-x
115. Speletas M, Gounaris A, Sevdali E, Kompoti M, Konstantinidi K, Sokou R, et al. MBL2 genotypes and their associations with MBL levels and NICU morbidity in a cohort of Greek neonates. J Immunol Res (2015) 2015:478412. doi: 10.1155/2015/478412
116. Zhang J, Li A, Yang C, Garred P, Ma YJ. Rapid and efficient purification of functional collecttin-12 and its opsonic activity against fungal pathogens. J Immunol Res (2019) 2019:9164202. doi: 10.1155/2019/9164202
117. McLennan R, Giniunaite R, Hildebrand K, Teddy JM, Kasemeier-Kulesa JC, Bolanos L, et al. Colec12 and Trail signaling confine cranial neural crest cell trajectories and promote collective cell migration. Dev Dyn. (2023) 252:629–46. doi: 10.1002/dvdy.569
118. Ajami N, Kerachian MA, Toosi MB, Ashrafzadeh F, Hosseini S, Robinson PN, et al. Inherited deletion of 9p22.3-p24.3 and duplication of 18p11.31-p11.32 associated with neurodevelopmental delay: phenotypic matching of involved genes. J Cell Mol Med (2023) 27:496–505. doi: 10.1111/jcmm.17662
119. Ujma S, Horsnell WGC, Katz AA, Clark HW, Schafer G. Non-pulmonary immune functions of surfactant proteins A and D. J Innate Immun (2017) 9:3–11. doi: 10.1159/000451026
120. Pryhuber GS, Hull WM, Fink I, McMahan M, Whitsett JA. Ontogeny of surfactant proteins A and B in human amniotic fluid as indices of fetal lung maturity. Pediatr Res (1991) 30:597–605. doi: 10.1203/00006450-199112000-00023
121. Miyamura K, Malhotra R, Hoppe HJ, Reid KB, Phizackerley PJ, Macpherson P, et al. Surfactant proteins A (SP-A) and D (SP-D): levels in human amniotic fluid and localization in the fetal membranes. Biochim Biophys Acta (1994) 1210:303–7. doi: 10.1016/0005-2760(94)90233-x
122. Leth-Larsen R, Floridon C, Nielsen O, Holmskov U. Surfactant protein D in the female genital tract. Mol Hum Reprod (2004) 10:149–54. doi: 10.1093/molehr/gah022
123. Han YM, Romero R, Kim YM, Kim J-S, Richani K, Friel LA, et al. Surfactant protein-A mRNA expression by human fetal membranes is increased in histological chorioamnionitis but not in spontaneous labour at term. J Pathol (2007) 211:489–96. doi: 10.1002/path.2131
124. Yadav AK, Madan T, Bernal AL. Surfactant proteins A and D in pregnancy and parturition. Front Biosci (Elite Ed). (2011) 3:291–300. doi: 10.2741/e244
125. Madhukaran SP, Kishore U, Jamil K, Choolani M, Lu J. Decidual expression and localization of human surfactant protein SP-A and SP-D, and complement protein C1q. Mol Immunol (2015) 66:197–207. doi: 10.1016/j.molimm.2015.03.001
126. Snegovskikh VV, Bhandari V, Wright JR, Tadesse S, Morgan T, Macneill C, et al. Surfactant protein-A (SP-A) selectively inhibits prostaglandin F2alpha (PGF2alpha) production in term decidua: implications for the onset of labor. J Clin Endocrinol Metab (2011) 96:E624–32. doi: 10.1210/jc.2010-1496
127. Yadav AK, Chaudhari H, Warke H, Shah PK, Dodagatta-Marri E, Kishore U, et al. Differential expression of collectins in human placenta and role in inflammation during spontaneous labor. PloS One (2014) 9:e108815. doi: 10.1371/journal.pone.0108815
128. Chaiworapongsa T, Hong J-S, Hull WM, Kim CJ, Gomez R, Mazor M, et al. The concentration of surfactant protein-A in amniotic fluid decreases in spontaneous human parturition at term. J Matern Fetal Neonatal Med (2008) 21:652–9. doi: 10.1080/14767050802215193
129. Mendelson CR, Montalbano AP, Gao L. Fetal-to maternal signaling in the timing of birth. J Steroid Biochem Mol Biol (2017) 170:19–27. doi: 10.1016/j.jsbmb.2016.09.006
130. Stray-Pedersen A, Vege A, Stray-Pedersen A, Holmskov U, Rognum TO. Post-neonatal drop in alveolar SP-A expression: Biological significance for increased vulnerability to SIDS? Pediatr Pulmonol (2008) 43:160–8. doi: 10.1002/ppul.20750
131. Nam M, Hur M, Kim H, Lee G-H, Park M, Kwon H-S, et al. Distribution of presepsin, Krebs von den Lungen 6, and surfactant protein A in umbilical cord blood. Diagnostics (2022) 12:2213. doi: 10.3390/diagnostics12092213
132. De Luca D, Arroyo R, Foligno S, Autilio C, Touqui L, Kingma PS. Early life surfactant protein-D levels in bronchoalveolar lavage fluids of extremely preterm neonates. Am J Physiol Lung Cell Mol Physiol (2023) 325:L411–8. doi: 10.1152/ajplung.00079.2023
133. Dahl M, Juvonen PO, Holmskov U, Husby S. Surfactant protein D in newborn infants: factors influencing surfactant protein D levels in umbilical cord blood and capillary blood. Pediatr Res (2005) 58:908–12. doi: 10.1203/01.PDR.0000181379.72900.EC
134. Briana DD, Gourgiotis D, Baka S, Boutsikou M, Vraila V-M, Boutsikou T, et al. The effect of intrauterine growth restriction on circulating surfactant protein D concentrations in the perinatal period. Reprod Sci (2010) 17:653–8. doi: 10.1177/1933719110366165
135. Dahl M, Holmskov U, Husby S, Juvonen PO. Surfactant protein D levels in umbilical cord blood and capillary blood of premature infants. The influence of perinatal factors. Pediatr Res (2006) 59:806–10. doi: 10.1203/01.pdr.0000219122.81734.03
136. Briana DD, Malamitsi-Puchner A. Perinatal biomarkers implying ‘Developmental Origins of Health and Disease’ consequences in intrauterine growth restriction. Acta Paediatr (2020) 109:1317–22. doi: 10.1111/apa.15022
137. Chaiworapongsa T, Hong J-S, Hull WM, Romero R, Whitsett JA. Amniotic fluid concentration of surfactant proteins in amniotic in intra-amniotic infection. J Matern Fetal Neonatal Med (2008) 21:663–70. doi: 10.1080/14767050802215664
138. Mackay R-MA, Townsend JP, Calvert J, Anthony M, Wilkinson AR, Postle AD, et al. Increased surfactant protein-D levels in the airways of preterm neonates with sepsis indicated responses to infectious challenges. Acta Paediatr (2019) 108:870–6. doi: 10.1111/apa.14630
139. Hilgendorff A, Heidinger K, Bohnert A, Kleinsteiber A, König IR, Ziegler A, et al. Association of polymorphisms in the human surfactant protein-D (SFTPD) gene and postnatal pulmonary adaptation in the preterm infant. Acta Paediatr (2009) 98:112–7. doi: 10.1111/j.1651-2227.2008.01014.x
140. Ryckman KK, Dagle JM, Kelsey K, Momany AM, Murray JC. Genetic associations of surfactant protein D and angiotensin-converting enzyme with lung disease in preterm neonates. J Perinatol. (2012) 32:349–55. doi: 10.1038/jp.2011.104
141. Sorensen GL, Dahl M, Tan Q, Bendixen C, Holmskov U, Husby S. Surfactant protein-D-encoding gene variant polymorphisms are linked to respiratory outcome in premature infants. J Pediatr (2014) 165:683–9. doi: 10.1016/j.jpeds.2014.05.042
142. Chang HY, Li F, Li FS, Zheng CZ, Lei YZ, Wang J. Genetic polymorphism of SP-A, SP-B, and SP-D and risk of respiratory distress syndrome in preterm neonates. Med Sci Monit (2016) 22:5091–100. doi: 10.12659/msm.898553
143. Amatya S, Ye M, Yang L, Gandhi CK, Wu R, Nagourney B, et al. Single nucleotide polymorphisms interactions of the surfactant protein genes associated with respiratory distress syndrome susceptibility in preterm infants. Front Pediatr (2021) 9:682160. doi: 10.3389/fped.2021.682160
144. Thomas NJ, Fan R, DiAngelo S, Hess JC, Floros J. Haplotypes of the surfactant protein genes A and D as susceptibility factors of respiratory distress syndrome. Acta Paediatr (2007) 96:985–9. doi: 10.1111/j.1651-2227.2007.00319.x
145. Jo HS, Cho S-I, Chang YH, Kim BI, Choi J-H. Surfactant protein A associated with respiratory distress syndrome in Korean preterm infants: evidence of ethnic difference. Neonatology (2013) 103:44–7. doi: 10.1159/000342498
146. Tsitoura M-E, Stavrou EF, Maraziotis JA, Sarafidis K, Athanassiadou A, Dimitriou D. Surfactant protein A and B gene polymorphisms and risk of respiratory distress syndrome in late-preterm neonates. PloS One (2016) 11:e0166516. doi: 10.1371/journal.pone.0166516
147. Ramet M, Haataja M, Marttila R, Floros J, Hallman M. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am J Hum Genet (2000) 66:1569–79. doi: 10.1086/302906
148. Marttila R, Haataja M, Guttentag S, Hallman M. Surfactant protein A and B genetic variants in respiratory distress syndrome in singletons and twins. Am J Respir Crit Care Med (2003) 168:1216–22. doi: 10.1164/rccm.200304-524OC
149. Marttila R, Haataja M, Ramet M, Pokela M-L, Tammela O, Hallman M. Surfactant protein A gene locus and respiratory distress syndrome in Finnish premature twin pairs. Ann Med (2003) 35:344–52. doi: 10.1080/07853890310006389
150. Haataja M, Ramet M, Marttila R, Hallman M. Surfactant proteins A and B as interactive genetic determinants of neonatal respiratory distress syndrome. Hum Mol Genet (2000) 9(18):2751–60. doi: 10.1093/hmg/9.18.2751
151. Kala P, Ten Have T, Nielsen H, Dunn M, Floros J. Association of pulmonary surfactant protein A (SP-A) gene and respiratory distress syndrome: interaction with SP-B. Pediatr Res (1998) 43:169–77. doi: 10.1203/00006450-199802000-00003
152. Floros J, Fan R, DiAngelo S, Guo X, Wert J, Luo J. Surfactant protein (SP) B associations and interactions with SP-A in white and black subjects with respiratory distress syndrome. Pediatr Int (2001) 43:567–76. doi: 10.1046/j.1442-200x.2001.01474.x
153. Haataja M, Ramet M, Marttila R, Hallman M. Respiratory distress syndrome: evaluation of genetic susceptibility and protection by transmission disequilibrium test. Hum Genet (2001) 109:351–5. doi: 10.1007/s004390100574
154. Floros J, Fan R, Matthews A, DiAngelo S, Luo J, Nielsen H, et al. Family-based transmission disequilibrium test (TDT) and case-control association studies reveal surfactant protein A (SP-A) susceptibility alleles for respiratory distress syndrome (RDS) and possible race differences. Clin Genet (2001) 60:178–87. doi: 10.1034/j.1399-0004.2001.600303.x
155. Abuelhamed WA, Zeidan N, Shahin WA, Rizk HI, Rabie WA. Human surfactant proteins A2 (SP-A2) and B (SP-B) genes as determinants of respiratory distress syndrome. Indian Pedr. (2015) 52:391–4. doi: 10.1007/s13312-015-0643-9
156. Eguchi K, Koyama N, Tanaka T, Kamiya K, Ogawa Y. Surfactant apoprotein A (SP-A) in tracheal aspirates of newborn infants with RDS. Acta Paediatr Jpn (1991) 33:649–54. doi: 10.1111/j.1442-200x.1991.tb01880.x
157. Stevens PA, Shadow B, Bartholain S, Segerer H, Obladen M. Surfactant protein A in the course of respiratory distress syndrome. Eur J Pediatr (1992) 151:596–600. doi: 10.1007/BF01957730
158. Ghitoi S-A, Ashie M, Cozaru GC, Enciu M, Matei E, Nicolau A-A, et al. Surfactant proteins analysis in perinatal deceased preterm twins among the Romanian population. Med (Baltimore). (2022) 101:e29701. doi: 10.1097/MD.0000000000029701
159. DeMello DE, Phelps DS, Patel G, Floros J, Lagunoff D. Expression of the 35kDA and low molecular weight surfactant-associated proteins in the lungs of infants dying with respiratory distress syndrome. Am J Pathol (1989) 134:1285–93.
160. DeMello DE, Heyman S, Phelps DS, Floros J. Immunogold localization of SP-A in lungs of infants dying from respiratory distress syndrome. Am J Pathol (1993) 142:1631–40. doi: 10.1159/000014198
161. Cho K, Matsuda T, Okajima S, Matsumoto Y, Sagawa T, Fujimoto S, et al. Prediction of respiratory distress syndrome by the level of pulmonary surfactant protein A in cord blood sera. Biol Neonate. (2000) 77:83–7. doi: 10.1159/000014198
162. Kaneko K, Shimizu H, Arakawa H, Ogawa Y. Pulmonary surfactant protein A in sera for assessing neonatal lung maturation. Early Hum Dev (2001) 62:11–21. doi: 10.1016/s0378-3782(00)00133-x
163. Pavlovic J, Papagaroufalis C, Xanthou M, Liu W, Fan R, Thomas NJ, et al. Genetic variants of surfactant proteins A, B, C, and D in bronchopulmonary dysplasia. Dis Markers. (2006) 22:277–91. doi: 10.1155/2006/817805
164. Vinod S, Gow A, Weinberger B, Potak D, Hiatt M, Chandra S, et al. Serum surfactant protein D as a marker for bronchopulmonary dysplasia. J Matern Fetal Neonatal Med (2019) 32:815–19. doi: 10.1080/14767058.2017.1392506
165. Weber B, Borkhardt A, Stoll-Becker S, Reiss I, Gortner L. Polymorphisms of surfactant protein A genes and the risk of bronchopulmonary dysplasia in premature infants. Turk J Pediatr (2000) 42:181–5. doi: 10.1136/thoraxjnl-2012-202729
166. Kotecha S, Davies PL, Clark HW, McGreal EP. Increased prevalence of low oligomeric state surfactant protein D with restricted lectin activity in bronchoalveolar lavage fluid from preterm infants. Thorax (2013) 68:460–7. doi: 10.1136/thoraxjnl-2012-202729
167. Marks KT, Landis MW, Lim FY, Haberman B, Kingma PS. Evaluation of lung injury in infants with congenital diaphragmatic hernia. J Pediatr Surg (2019) 54:2443–7. doi: 10.1016/j.jpedsurg.2019.06.016
168. Krause M, Hartig W, Mahr CV, Richter C, Schob J, Puchta J, et al. CSF surfactant protein changes in preterm infants after intraventricular hemorrhage. Front Pediatr (2020) 8:572851. doi: 10.3389/fped.2020.572851
169. Crocker CE, Sharmeen R, Tran TT, Khan AM, Li W, Alcorn JL. Surfactant protein a attenuates generalized and localized neuroinflammation in neonatal mice. Brain Res (2023) 1807:148308. doi: 10.1016/j.brainres.2023.148308
170. Schlapbach LJ, Kessler U, Thiel S, Hansen AG, Nelle M, Ammann RA, et al. M-ficolin in the neonatal period: Associations with need for mechanical ventilation and mortality in premature infants with necrotising enterocolitis. Mol Immunol (2009) 46:2597–603. doi: 10.1016/j.molimm.2009.05.003
171. Schlapbach LJ, Kjaer TR, Thiel S, Mattmann M, Nelle M, Wagner BP, et al. M-ficolin concentrations in cord blood are related to circulating phagocytes and to early-onset sepsis. Pediatr Res (2012) 71:368–74. doi: 10.1038/pr.2011.71
172. Kilpatrick DC, Fujita T, Matsushita M. P35, an opsonic lectin of the ficolin family, in human blood from neonates, normal adults, and recurrent miscarriage patients. Immunol Lett (1999) 67:109–12. doi: 10.1016/s0165-2478(98)00147-3
173. Briana DD, Liosi S, Gourgiotis D, Boutsikou M, Baka S, Marmarinos A, et al. The potential role of the lectin pathway of complement in the host defense of full-term intrauterine growth restricted neonates at birth. J Matern Fetal Neonatal Med (2012) 25:531–4. doi: 10.3109/14767058.2011.636108
174. Szala-Poździej A, Świerzko AS, Gajek G, Kufelnicka-Babout M, Chojnacka K, Kobiela P, et al. Association of the FCN2 gene promoter polymorphisms with very low birthweight in preterm neonates. Int J Mol Sci (2022) 23:15336. doi: 10.3390/ijms232315336
175. Fujieda M, Aoyagi Y, Matsubara K, Takeuchi Y, Fujimaki W, Matsushita M, et al. L-ficolin and capsular polysaccharide-specific IgG in cord serum act synergistically to opsonophagocytic killing of serotype III and V group B streptococci. Infect Immun (2012) 80:2053–60. doi: 10.1128/IAI.06232-11
176. Gajek G, Świerzko AS, Jarych D, Mikulski D, Kobiela P, Chojnacka K, et al. Association of low ficolin-2 concentration in cord serum with respiratory distress syndrome in preterm newborns. Front Immunol (2023) 14:1107063. doi: 10.3389/fimmu.2023.1107063
177. Michalski M, Szala A, Swierzko A, Lukasiewicz J, Maciejewska A, Kilpatrick DC, et al. H-ficolin (ficolin-3) concentrations and FCN3 gene polymorphism in neonates. Immunobiology (2012) 217:730–7. doi: 10.1016/j.imbio.2011.12.004
178. Schlapbach LJ, Thiel S, Kessler U, Ammann RA, Aebi C, Jensenius JC. Congenital H-ficolin deficiency in premature infants with severe necrotising enterocolitis. Gut (2011) 60:1438–9. doi: 10.1136/gut.2010.226027
Keywords: collectin, ficolin, mannose-binding lectin (MBL), neonate, prematurity, perinatal infection, pulmonary surfactant, respiratory distress syndrome (RDS)
Citation: Cedzyński M and Świerzko AS (2023) Collectins and ficolins in neonatal health and disease. Front. Immunol. 14:1328658. doi: 10.3389/fimmu.2023.1328658
Received: 27 October 2023; Accepted: 04 December 2023;
Published: 19 December 2023.
Edited by:
Guirong Wang, Upstate Medical University, United StatesReviewed by:
Zissis C. Chroneos, The Pennsylvania State University, United StatesJulie Gunnells Ledford, University of Arizona, United States
Copyright © 2023 Cedzyński and Świerzko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maciej Cedzyński, bWNlZHp5bnNraUBjYm0ucGFuLnBs