 Yael Gernez1*
Yael Gernez1* Mansi Narula2
Mansi Narula2 Alma-Martina Cepika2
Alma-Martina Cepika2 Juanita Valdes Camacho3
Juanita Valdes Camacho3 Elisabeth G. Hoyte1
Elisabeth G. Hoyte1 Kirsten Mouradian2Bertil Glader2
Kirsten Mouradian2Bertil Glader2 Deepika Singh4
Deepika Singh4 Bindu Sathi5Latha Rao5
Bindu Sathi5Latha Rao5 Ana L. Tolin6
Ana L. Tolin6 Kenneth I. Weinberg2David B. Lewis1
Kenneth I. Weinberg2David B. Lewis1 Rosa Bacchetta2
Rosa Bacchetta2 Katja G. Weinacht2*
Katja G. Weinacht2*- 1Division of Allergy, Immunology and Rheumatology, Department of Pediatrics, Stanford School of Medicine, Stanford, CA, United States
- 2Division of Hematology, Oncology, Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA, United States
- 3Division of Allergy and Immunology, Department of Pediatrics, Louisiana State University (LSU) Health, Shreveport, LA, United States
- 4Division of Rheumatology, Department of Pediatrics, Valley Children Hospital, Madera, CA, United States
- 5Division of Hematology, Department of Pediatrics, Valley Children Hospital, Madera, CA, United States
- 6Division of Immunology, Department of Pediatrics, Hospital Pediatrico Dr. Humberto Notti, Mendoza, Argentina
Biallelic mutations in the ACP5 gene cause spondyloenchondrodysplasia with immune dysregulation (SPENCDI). SPENCDI is characterized by the phenotypic triad of skeletal dysplasia, innate and adaptive immune dysfunction, and variable neurologic findings ranging from asymptomatic brain calcifications to severe developmental delay with spasticity. Immune dysregulation in SPENCDI is often refractory to standard immunosuppressive treatments. Here, we present the cases of two patients with SPENCDI and recalcitrant autoimmune cytopenias who demonstrated a favorable clinical response to targeted JAK inhibition over a period of more than 3 years. One of the patients exhibited steadily rising IgG levels and a bone marrow biopsy revealed smoldering multiple myeloma. A review of the literature uncovered that approximately half of the SPENCDI patients reported to date exhibited increased IgG levels. Screening for multiple myeloma in SPENCDI patients with rising IgG levels should therefore be considered.
Introduction
SPENCDI (OMIM 607944) is a rare immuno-osseous dysplasia, with an increasing number of affected individuals reported in the literature (1–9). Clinically, the syndrome exhibits a broad phenotypic variability, but most patients present with skeletal and immune manifestations. Adaptative immune dysregulation and autoinflammation comprise the prevailing immune phenotype (1–4, 6–8) although immunodeficiency has also been reported (5, 9). In addition, SPENCDI can manifest with neurologic symptoms (1, 2, 4, 6, 8). Immune dyscrasias, most commonly manifesting as cytopenias and SLE-like symptoms (10–12), are often recalcitrant and refractory to conventional immunomodulatory therapies (1, 2, 6, 10, 13). SPENCDI is an autosomal recessive disease secondary to biallelic pathogenic variants in the ACP5 gene. ACP5 encodes for tartrate-resistant acid phosphatase (TRAP). In patients with SPENCDI, ACP5 mutations lead to abolished TRAP activity in the serum and increased phosphorylation of osteopontin (OPN). OPN has emerged as a possible unifying mediator, as it is expressed in osteoclasts and in cells of the immune system (Figure 1). Increased OPN phosphorylation has been linked to increased osteoclast activity, which is believed to contribute to the observed bone and cartilage defects in this disease (6). In antigen presenting cells of the immune system, increased OPN phosphorylation has been associated with increased interferon (IFN)-α release (6). Increased levels of IFN-α may contribute to both, cell autonomous largely autoinflammatory manifestations propagated by innate immune cells as well as non-cell-autonomous systemic autoimmunity in which deregulatory inflammatory signals adversely affect adaptive immune responses.
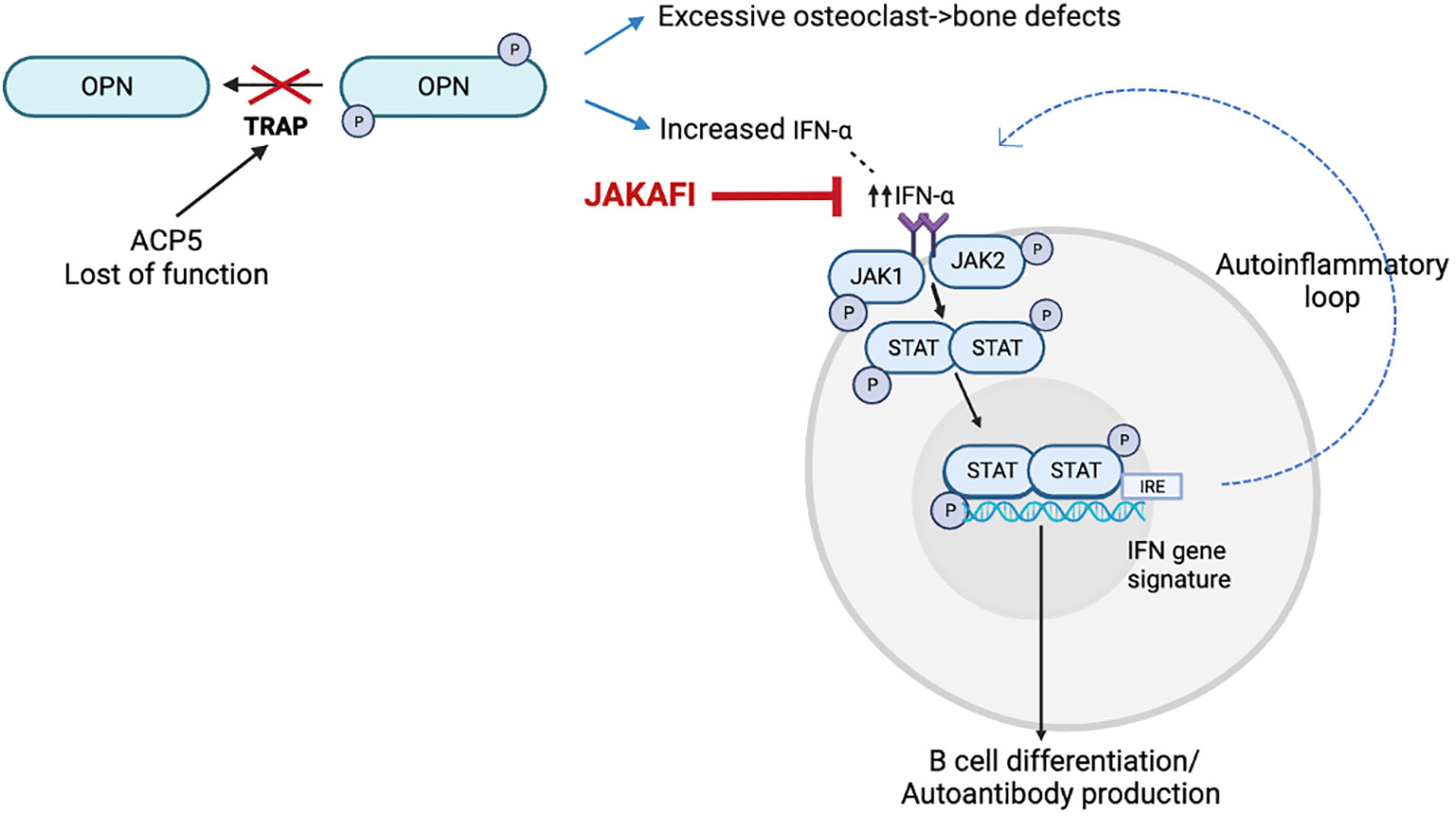
Figure 1 Model for the disease mechanism in SPENCDI, adapted from (6). ACP5 encodes for tartrate-resistant acid phosphatase (TRAP). Lack of TRAP phosphatase activity results in hyperphosphorylation of OPN. Increased OPN phosphorylation has been linked to increased osteoclast activity, which is believed to contribute to the observed bone and cartilage defects in this disease (6). In antigen presenting cells of the immune system, increased OPN phosphorylation has been associated with increased interferon (IFN)-α release (6). This figure was created using BioRender.com.
The constellation of clinical symptoms and biomarkers, i.e. increased type I interferon (IFN) levels and upregulated expression of interferon stimulated genes (ISGs) suggest that SPENCDI is a type I interferonopathy (1, 2, 6, 14, 15). Janus kinase (JAK) inhibitors, also known as jakinibs, are immunomodulators that inhibit the activity of one or more of the JAK enzyme family. The JAK inhibitors ruxolitinib and baricitinib inhibit JAK1 and JAK2 activity downstream of the type I interferon receptor. JAK inhibitors have shown therapeutic benefits in patients with type I interferonopathy, such as SAVI (stimulator of IFN genes–associated vasculopathy with onset in infancy), CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature), and Aicardi Goutières syndrome (AGS), as well as patients with undefined interferonopathies (16–18). Two of the patients with higher initial IFN signatures and undefined interferonopathies improved significantly, suggesting that genotype was not necessary to tailor treatment decisions (6). We therefore reasoned that our two patients with SPENCDI may also benefit from targeted JAK inhibition. Here, we report the favorable clinical and biological response of two patients with SPENCDI and recalcitrant cytopenias to ruxolinitb therapy.
Results
Patient 1
is a 19-year-old male who initially presented at 3-years of age with short stature, hypothyroidism (Hashimoto’s disease), and Evans syndrome, manifesting with autoimmune hemolytic anemia (AIHA), thrombocytopenia and neutropenia (Table 1). He had been glucocorticoid-dependent since diagnosis. Although his AIHA had initially been responsive to prednisone, the subsequent discovery of multiple vertebral compression fractures (Figure 2A) led to discontinuation of glucocorticoids due to concerns for bone metabolism side effects. These therapeutic considerations preceded the diagnosis of SPENCDI with osseous dysplasia as a possible alternative explanation for the vertebral abnormalities. Various other combinations of immunosuppressive therapies including immunoglobulins (IVIG), rituximab, sirolimus, bortezomib and mycophenolate mofetil (MMF) failed to improve his cytopenias or caused severe side effects, such as transaminitis (sirolimus) and anaphylaxis (bortezomib), ultimately necessitating their discontinuation. Further details on presentation and treatment are listed in the Supplementary Materials. After a brief period of clinical stability on MMF, the patient’s AIHA eventually recurred, requiring prolonged courses of oral steroids, and prompting referral to immunology for evaluation of an underlying immune dysregulation syndrome.
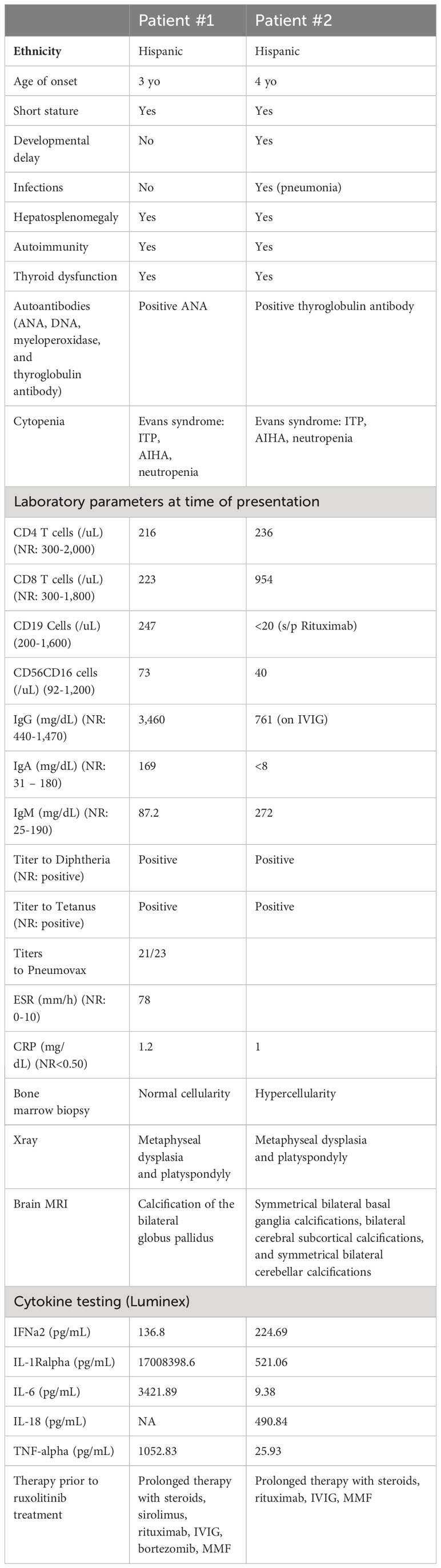
Table 1 Clinical features, laboratory parameters and treatments rendered prior to ruxolitinib therapy for patients #1 and #2.
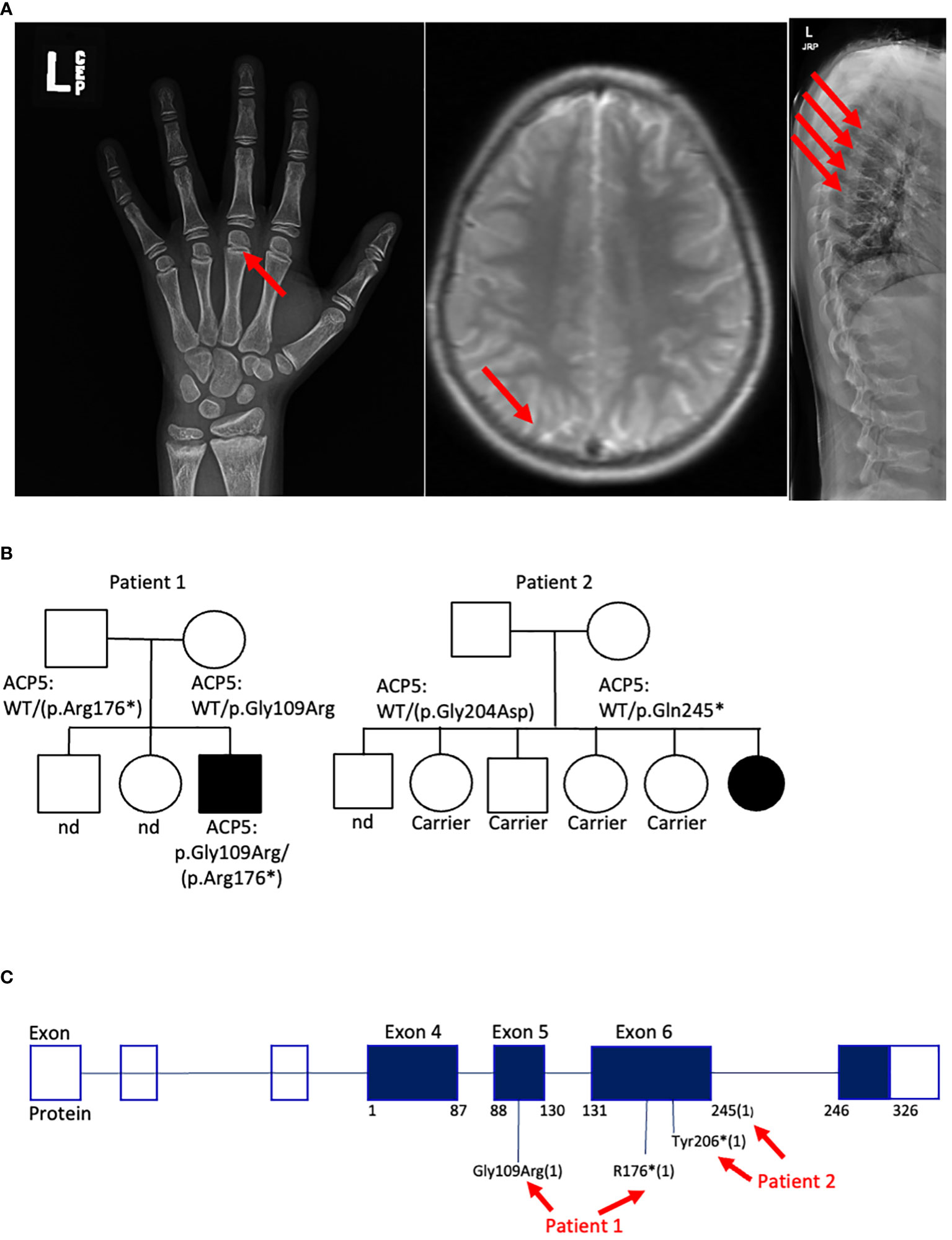
Figure 2 Genetic and clinical characteristics of SPENCDI patients. (A) Xray of the left hand (patient #1). Well-defined metaphyseal irregularity (indicated by red arrow) and sclerosis of the distal radius and ulna consistent with bone growth abnormality. Brain MRI (patient #1): nonspecific calcifications of the globus pallidus bilaterally without acute intracranial abnormality (indicated by red arrow). Xray of spine (patient #1): Multilevel compression deformities of the thoraco-lumbar spine (indicated by red arrows). Nd: not determined. (B) Family pedigree for patient #1 and patient #2. (C) Diagram illustrates the distribution of the reported ACP5 variants for patient #1 and patient #2. * : stop codon.
Trio whole-exome-sequencing (WES) revealed biallelic compound heterozygous pathogenic variants in the ACP5 gene (c.325G>A, p.Gly109Arg/c.526C>T, p.Arg176*) establishing the diagnosis of spondyloenchondrodysplasia with immune dysregulation (SPENCDI). Both parents were identified as heterozygous asymptomatic carriers of one of the variants (Figures 2B, C; Supplementary Materials; Supplementary Table 1). Based on the favorable therapeutic response of other patients with type I interferonopathies to JAK inhibitors (3, 4, 12, 19–21), and the shared pathophysiologic hallmarks of SPENCDI with other type I interferonopathies (14, 22), a therapeutic trial with the JAK1/JAK2 inhibitor ruxolitinib was initiated (0.4 mg/kg/day). Within a week from initiation of therapy, the patient reported significantly improved energy levels. Within three months, the patient’s anemia and thrombocytopenia were brought into remission and he has remained clinically stable with red blood cell indices in the normal range and platelet counts of approximately 100,000 per microliter of blood without any other additional therapies for the following three-and-a-half years (Figure 3A; Supplementary Table 2).
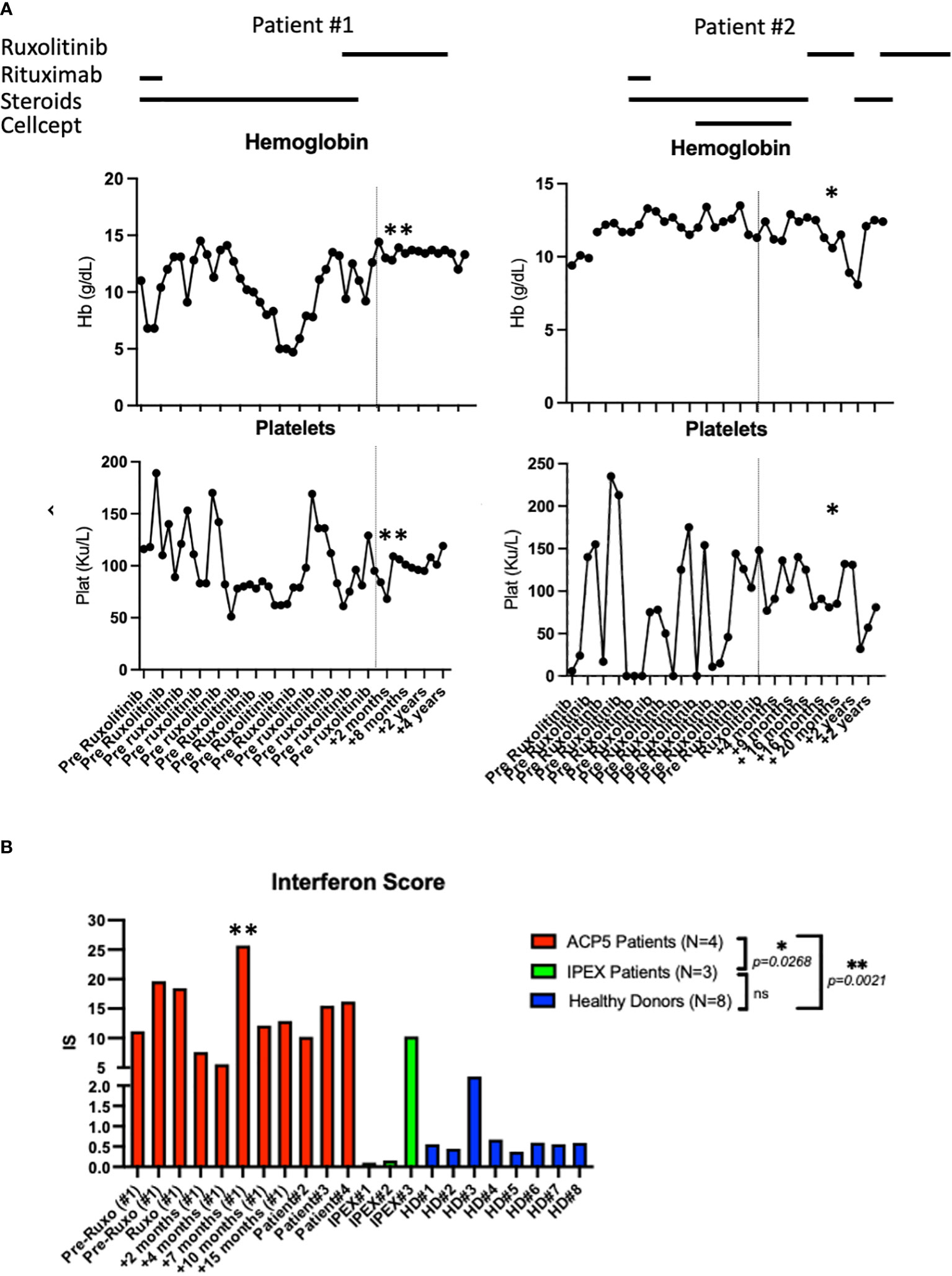
Figure 3 Laboratory parameters and interferon scores pre- and post-ruxolitinib therapy. (A) Hemoglobin and platelet count pre and post therapy with ruxolitinib in patient #1 and patient #2. Asterix (*) indicates time of hospital admission for acute viral and bacterial pneumonia complicated by acute SPENCDI flare. Asterix (**) indicates ISG expression in patient #1 transiently spiked during an infectious trigger while he remained clinically symptom free (B) Interferon Score of a panel of interferon stimulated genes in 4 SPENCDI patients at various time points throughout treatment, three subjects with IPEX and eight healthy donors. Dashed lines indicate the start of ruxolitinib. HD: healthy donors, Pat: patient, pre: pre ruxolitinib, post: post ruxolitinib, ruxo: ruxolitinib, IS: Interferon Score. Grouped statistical analysis performed using One-Way ANOVA (Kruskal-Wallis test) with Dunn’s multiple comparison test. p-value between groups indicated on the graph. Overall p-value: <0.0001. Level of significance (p)=0.05. Additional information on ACP patient #3 and #4 is detailed in Supplementary Table 3.
While on treatment with ruxolitinib, the patient has not experienced any significant infections. His shorter stature compared to his siblings is likely a sequela of his underlying disease, possibly confounded by early and prolonged glucocorticoid use. He remains neurologically asymptomatic and is performing well academically. Upon transfer to our care, an initial screening MRI brain was obtained which showed nonspecific findings, including calcifications of the globus pallidus bilaterally, but no acute intracranial abnormalities. A subsequent brain MRI three years later showed stable calcifications but no additional abnormal findings. He was immunized with the mRNA COVID-19 vaccine and did not experience any side effects. Despite being fully vaccinated and boosted against COVID-19, he contracted the infection but remained asymptomatic without flaring of his autoimmunity.
In the context of a slowly but steadily rising serum IgG level over 36 months, a bone marrow biopsy screening was obtained which revealed a monotypic population of kappa light-chain restricted plasma cells without an overall increase in blasts. This monotypic population, which lacked cytogenetic abnormalities, encompassed 20% of all bone marrow cells. The patient was subsequently referred to an oncologist and diagnosed with smoldering multiple myeloma. These findings prompted a review of the literature that revealed that approximately half of the patients with SPENCDI also report hypergammaglobulinemia (2, 4, 12, 13, 20, 22).
Patient 2
is an 8-year-old female who was diagnosed with systemic lupus erythematosus (SLE) at the age of 5 years old. The patient presented with short stature and developmental delay. At diagnosis, the patient suffered from mucocutaneous disease (petechia, purpuric rash), anemia, thrombocytopenia, and splenomegaly. The patient also had, elevated inflammatory markers [ESR: 78 mm/h (normal range (NR):0-15mm/h); CRP:1.2 mg/dL(NR:<0.3 mg/dL)], severely decreased complement levels [CH50: 0 (NR: 42-95 U/mL); C3: 41 mg/dL (NR: 89-173 mg/dL); C4: <2.9mg/dL (NR: 17.0-42.0 mg/dL)], hypergammaglobulinemia, positive ANA titer (>1:1280), elevated dsDNA antibodies (>300 IU/mL), and elevated myeloid peroxidase (MPO) antibodies (Table 1). There was no evidence of renal involvement. Cytopenias were moderately responsive to glucocorticoid therapy, but refractory to rituximab and IVIG. At the time of initial assessment in immunology clinic, the patient was treated with glucocorticoids and MMF. Further details on presentation and treatment are listed in the Supplementary Materials.
Genetic analysis revealed that the ACP5 gene had a pathogenic variant with a premature stop codon (c.733C>T, p.Gln245*) and a variant of unknown significance (VUS) resulting in an amino acid substitution (c.611G>A, p.Gly204Asp). Both parents were identified as heterozygous asymptomatic carriers of one of these variants establishing that the patient was compound heterozygote for these two variants (Figures 2B, C; Supplementary Materials; Supplementary Table 1). Given these findings, the patient was assessed for the presence of metaphyseal dysplasia. X-rays of hands and spine showed metaphyseal dysplasia and platyspondyly. Additionally, characteristic radiolucent metaphyseal and vertebral lesions were identified. Apart from an episode of pneumonia at age 4 years, there was no significant history of infections. Due to unclear developmental delay, the patient underwent a screening MRI of the brain which was normal. Considering the positive outcome observed in patient #1, we initiated treatment with ruxolitinib at a dosage of 0.4 mg/kg/day and this patient also showed significantly improved energy levels within one week and improved platelet counts within less than one month from starting treatment. She remained clinically stable and was steroid-free for the next 2-and-a-half years until she developed acute respiratory failure due to parainfluenza and rhinoviral pneumonia with bacterial superinfection and sepsis. Ruxolitinib was discontinued and the patient experienced, after a few days, an acute flare of ITP treated with glucocorticoids. After resolution of sepsis and following the reinitiation of ruxolitinib therapy, the patient’s platelet count improved within 4 weeks (Figure 3A).
There is no specific biomarker for tartrate-resistant acid phosphatase activity, however, increased expression in interferon stimulated genes (ISG) has consistently been found in SPENCDI patients. The interferon score of both patients with SPENCDI in our study (patient 1 and 2) was significantly elevated at baseline compared to healthy controls and patients with IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, where autoimmunity is caused by a regulatory T cell defect (Figure 3B; Supplementary Figure 1). As previously reported by Fremond et al., we only observed a minimal decrease in interferon score in patient #1 following the initiation of ruxolitinib therapy (Figure 3B) (23). However, the resolution of cytopenias, combined with the increase in energy, academic performance and general wellbeing associated with ruxolitinib treatment in both patients suggests that interferon scores fail to adequately capture the clinical response. Notably, ISG expression in patient #1 transiently spiked during an infectious trigger while he remained clinically symptom free (Figures 3A, B; Supplementary Figure 1).
Discussion
There is growing evidence that SPENCDI is a type I interferonopathy that shares clinical features with other interferonopathies, including autoimmune and autoinflammatory manifestations (1–9). Specifically, a clinical presentation consistent with SLE, in patient #2, should prompt consideration for further genetic testing as monogenic forms of SLE have been described in association with variants in C1Q, C1R, C1S, CA, DNASE1, TREX1 and ACP5 (11, 12, 24). Genetic testing was pursued for this patient because of the severe and refractory nature of Evans’s syndrome (25–28).
The long-term prognosis and quality of life in patients with SPENCDI is influenced by multiple factors. The severity of the immune dysregulation and the side effects of conventional immunosuppressive therapies play an important role. The use of JAK inhibition in interferonopathies (14) aims at achieving immunomodulation, i.e., a normalization of the augmented immune response by downregulating the signal that is downstream of the type I interferon receptor; which is expected to ameliorate both, autoinflammatory and adaptive autoimmune manifestations alike. Patient #1 has experienced multiple vertebral compression fractures, likely associated with his primary disease but possibly confounded by the long-term glucocorticoid use. The two patients reported in our study were refractory to conventional immunosuppressive therapies but responded to ruxolitinib within only a few weeks from starting treatment, while their interferon score remained elevated. One plausible explanation for this discrepancy could be the consequence of JAK inhibitors on the B cell differentiation by blocking the effects of pro-inflammatory cytokines. JAK inhibitors are known to inhibit the differentiation of human B cells into plasmablasts in response to type I interferon stimuli, thereby reducing the levels of autoantibodies (29, 30). While JAK inhibitor treatment did yield significant improvements in autoimmune cytopenias, it is possible that genes associated with type I interferonopathy remain activated in immune cells with cell-intrinsic defects. Importantly, both patients are tolerating ruxolitinib well, with no significant adverse effects, except for mild neutropenia in patient #1 which constitutes a manageable dose limiting toxicity. Ruxolitinib has been associated with various side-effects including myelosuppression, increased risk of viral infections, transaminitis and non-melanoma skin malignancy. Patients on prolonged ruxolitinib treatment can also develop treatment resistance (31). Both patients were regularly monitored for Epstein-Barr virus, cytomegalovirus, JC, and BK virus reactivation by PCR. which remained negative throughout treatment with ruxolitinib. Ruxolitinib discontinuation syndrome has been documented among myelofibrosis patients, manifesting after the cessation of ruxolitinib treatment. The reported rebound effects vary significantly in severity, ranging from mild to potentially life-threatening symptoms. Abrupt discontinuation of ruxolitinib in myelofibrosis patients may lead to conditions such as worsening of cytopenia, splenomegaly, spleen rupture or acute respiratory syndrome, affecting approximately 15% of individuals (32). The patients and their families have been educated about the risk of rebound if ruxolitinib is abruptly discontinued.
An increase in IgG levels in patients with SPENCDI has been observed by us and others (2) and ultimately lead to the diagnosis of smoldering multiple myeloma in patient #1. We therefore recommend SPENCDI patients should be monitored prospectively for the development of monoclonal gammopathy and regular assessment of immunoglobulin levels, free kappa/lambda light chain ratio and 24 hours urine analysis for Bence-Jones proteinuria should be considered. Any abnormal findings may warrant a referral to the hematologist to assess the need for a bone marrow biopsy.
To the best of our knowledge, there have been no reports of multiple myeloma in patients with SPENCDI. It’s important to highlight that MM and MGUS have also been observed in individuals with VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) syndrome, a condition propelled by somatic myeloid mutations resulting in autoinflammatory responses (33, 34). In a comprehensive study involving 116 French patients diagnosed with VEXAS syndrome, MDS (myelodysplastic syndrome) was detected in 58 out of 116 cases (50%). Out of these 58 MDS cases, 12 exhibited MGUS (17%) (35). Nevertheless, there remains uncertainty regarding whether somatic UBA1 variants directly instigate MM’s development or whether these variants contribute to conditions like MGUS and plasma cell disorders.
We carefully assessed the potential involvement of the JAK inhibitor therapy in the pathogenesis of multiple myeloma in patient #1. Notably, the patient’s hyperglobulinemia predated the initiation of ruxolitinib treatment. Furthermore, multiple myeloma is not a malignancy associated with either primary T-cell immunodeficiency or prolonged immunosuppressive therapy. There is no documented association between STAT-1 loss-of-function and multiple myeloma (36). In contrast, recent clinical approaches have demonstrated some success in treating refractory multiple myeloma using a combination of ruxolitinib, corticosteroids, and lenalidomide (37). Given the scarcity of literature specifically addressing baricitinib in the context of multiple myeloma, we have elected to continue treatment with ruxolitinib in patient #1.
Another important consideration for the long-term prognosis in patients with SPENDCI is the development of neurologic symptoms in the context of nonspecific brain calcifications. Despite exhibiting bilateral calcification of the globulus pallidus on the brain MRI, patient #1 did not display any neurological symptoms. By contrast, patient #2 exhibited developmental delay but no calcifications noted in the brain MRI. Notably, the neurological status of both patients has remained stable on ruxolitinib treatment for more than 2 years. One might speculate that mitigating systemic inflammation with ruxolitinib could also have beneficial effects on the neurologic manifestations in SPENCDI as has been shown for patients with type I interferonopathy due to Aicardi-Goutières syndrome (38).
The presented cases by us and others (3, 4, 12, 19–21) support the use of JAK-inhibitors as an effective targeted therapy for patients with SPENCDI and severe cytopenias. Lack of effective biomarkers to capture the clinical response to JAK-inhibitors (e.g. IFN scores) highlight the need for further studies to delineate the exact underlying disease mechanism in SPENCDI. SPENCDI patients should be monitored prospectively for the development of monoclonal gammopathy. Further randomized controlled trials will be needed to confirm these findings in larger patient cohorts and evaluate possible neuroprotective benefits of targeted JAK-STAT inhibition in patients with SPENCDI and other interferonopathies.
Methods
Laboratory testing
T, B and NK subsets as well as immunoglobulin levels (IgA, IgM and IgG levels) were measured by flow cytometry in the CLIA-certified clinical core laboratory at Stanford (Stanford, CA, USA).
Cytokines and chemokine testing
IL-18 serum level and CXCL9 were measured by ELISA in a CLIA-certified clinical core laboratory (Cincinnati Children’s hospital, Cincinnati, OH, USA). In Table 1, IFNa2, IL-1Ralpha, IL-6, IL-18 and TNF-alpha were measured via Luminex (Human Immune Monitoring Center Stanford, CA, USA). In Supplementary Table 2: IL-1β, IL-2 receptor, IL-6, IL-10, IL-18, IFN-gamma, IFN-α were measured by ELISA method (ARUP, Salt Lake City, UT, USA).
Interferon score
Six out of 15 ISGs were selected based on their expression level in a previous study (15) and measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Supplementary Figure 1. In brief, 2.5 mL of blood was collected into PAXgene tubes (PreAnalytix), and total RNA was extracted from whole blood using a PAXgene RNA isolation kit (MagMAX). qRT-PCR analysis was performed using the TaqMan Universal PCR Master Mix (Applied Biosystems) and cDNA derived from 40 ng of total RNA and TaqMan probes for the ISGs IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1), and SIGLEC1 (Hs00988063_m1). Taqman probes for HPRT1 (Hs03929096_g1) and 18S (Hs999999001_s1) ribosomal RNA was used for internal normalization of the PCR assay (Applied Biosystems qpcr analysis module). Delta delta Ct method was used for relative quantification (RQ) of gene expression for each of the six probes, against a single reference control sample. Subsequently, Interferon Score (IS) was calculated by the fold change in the median of the six probes per sample compared to the median of all the healthy controls (n=8).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Stanford, Research of compliance, CA, USA. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) (patient 1) and minor(s)’ legal guardian (patient 2) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YG: Data curation, Supervision, Writing – original draft, Writing – review & editing. MN: Writing – review & editing, Data curation. JV: Writing – review & editing. A-MC: Writing – review & editing. EH: Writing – review & editing. KM: Writing – review & editing. BG: Writing – review & editing. DS: Writing – review & editing. BS: Writing – review & editing. LR: Writing – review & editing. AT: Writing – review & editing. KIW: Writing – original draft. DL: Writing – review & editing. RB: Writing – review & editing. KGW: Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the donors of the Center for Genetic Immune Diseases for their generous philanthropic support. We thank Alexander Goldhammer for his technical support. We thank Ellen Tsudama, N.P, Tandy Aye, M.D., and Michaela Liedtke for direct support of patient care and the patients and their families for participating in our research study.
Conflict of interest
KGW is a scientific advisory board member for Garuda Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1328005/full#supplementary-material
Supplementary Figure 1 | Interferon signature per gene for 8 healthy controls, 3 IPEX patients, and 4 SPENCDI patients.
Supplementary Table 1 | ACP5 variants for patients #1 and #2.
Supplementary Table 2 | Clinical phenotype and biological parameters of patient #1 pre and post ruxolitinib therapy.
Supplementary Table 3 | Clinical phenotype and biological parameters for ACP5 patient #3 and #4.
References
1. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet (2011) 43(2):127–31. doi: 10.1038/ng.748
2. Briggs TA, Rice GI, Adib N, Ades L, Barete S, Baskar K, et al. Spondyloenchondrodysplasia due to mutations in ACP5: A comprehensive survey. J Clin Immunol (2016) 36(3):220–34. doi: 10.1007/s10875-016-0252-y
3. Dri J, Dos Santos E, Pereyra M, Guillamondegui MJ, Ballester C, Tolin A, et al. Spondyloenchondrodysplasia with immune dysregulation related to ACP5. A report of 4 cases. Arch Argent Pediatr (2024) 122(1):e202303031. doi: 10.5546/aap.2023-03031.eng
4. Elhossini RM, Elbendary HM, Rafat K, Ghorab RM, Abdel-Hamid MS. Spondyloenchondrodysplasia in five new patients: identification of three novel ACP5 variants with variable neurological presentations. Mol Genet Genomics (2023) 298(3):709–20. doi: 10.1007/s00438-023-02009-1
5. Kulkarni ML, Baskar K, Kulkarni PM. A syndrome of immunodeficiency, autoimmunity, and spondylometaphyseal dysplasia. Am J Med Genet A (2007) 143A(1):69–75. doi: 10.1002/ajmg.a.31526
6. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet (2011) 43(2):132–7. doi: 10.1038/ng.749
7. Navarro V, Scott C, Briggs TA, Barete S, Frances C, Lebon P, et al. Two further cases of spondyloenchondrodysplasia (SPENCD) with immune dysregulation. Am J Med Genet A (2008) 146A(21):2810–5. doi: 10.1002/ajmg.a.32518
8. Renella R, Schaefer E, LeMerrer M, Alanay Y, Kandemir N, Eich G, et al. Spondyloenchondrodysplasia with spasticity, cerebral calcifications, and immune dysregulation: clinical and radiographic delineation of a pleiotropic disorder. Am J Med Genet A (2006) 140(6):541–50. doi: 10.1002/ajmg.a.31081
9. Roifman CM, Melamed I. A novel syndrome of combined immunodeficiency, autoimmunity and spondylometaphyseal dysplasia. Clin Genet (2003) 63(6):522–9. doi: 10.1034/j.1399-0004.2003.00033.x
10. An J, Briggs TA, Dumax-Vorzet A, Alarcon-Riquelme ME, Belot A, Beresford M, et al. Tartrate-resistant acid phosphatase deficiency in the predisposition to systemic lupus erythematosus. Arthritis Rheumatol (2017) 69(1):131–42. doi: 10.1002/art.39810
11. Baglan E, Ozdel S, Gungor T, Karakus R, Bagriacik EU, Yucel AA, et al. Spondylenchondrodysplasia mimicking a systemic lupus erythematosus: A diagnostic challenge in a pediatric patient. Eur J Med Genet (2021) 64(9):104286. doi: 10.1016/j.ejmg.2021.104286
12. Sait H, Gangadharan H, Gupta A, Aggarwal A, Jain M, Phadke SR. Monogenic lupus with igA nephropathy caused by spondyloenchondrodysplasia with immune dysregulation. Indian J Pediatr (2021) 88(8):819–23. doi: 10.1007/s12098-020-03636-x
13. Sacri AS, Bruwier A, Baujat G, Breton S, Blanche S, Briggs TA, et al. Childhood-onset autoimmune cytopenia as the presenting feature of biallelic ACP5 mutations. Pediatr Blood Cancer (2017) 64(2):306–10. doi: 10.1002/pbc.26195
14. Crow YJ. Type I interferonopathies: mendelian type I interferon up-regulation. Curr Opin Immunol (2015) 32:7–12. doi: 10.1016/j.coi.2014.10.005
15. Rice GI, Melki I, Fremond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol (2017) 37(2):123–32. doi: 10.1007/s10875-016-0359-1
16. Fremond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol (2016) 138(6):1752–5. doi: 10.1016/j.jaci.2016.07.015
17. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest (2018) 128(7):3041–52. doi: 10.1172/JCI98814
18. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol (2017) 139(5):1629–40 e2. doi: 10.1016/j.jaci.2016.11.022
19. Chougule A, Taur P, Gowri V, Consortium COE, Desai MM. SPENCD presenting with evans phenotype and clinical response to JAK1/2 inhibitors-a report of 2 cases. J Clin Immunol (2023) 43(2):331–4. doi: 10.1007/s10875-022-01400-8
20. Li W, Wang W, Wang W, Zhong L, Gou L, Wang C, et al. Janus kinase inhibitors in the treatment of type I interferonopathies: A case series from a single center in China. Front Immunol (2022) 13:825367. doi: 10.3389/fimmu.2022.825367
21. Melaccio A, Reale A, Saltarella I, Desantis V, Lamanuzzi A, Cicco S, et al. Pathways of angiogenic and inflammatory cytokines in multiple myeloma: role in plasma cell clonal expansion and drug resistance. J Clin Med (2022) 11(21):6491. doi: 10.3390/jcm11216491
22. Briggs TA, Rice GI, Adib N, Ades L, Barete S, Baskar K, et al. Erratum to: spondyloenchondrodysplasia due to mutations in ACP5: A comprehensive survey. J Clin Immunol (2016) 36(5):529–30. doi: 10.1007/s10875-016-0287-0
23. Fremond ML, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients. J Allergy Clin Immunol Pract (2021) 9(2):803–18 e11. doi: 10.1016/j.jaip.2020.11.007
24. Kara B, Ekinci Z, Sahin S, Gungor M, Gunes AS, Ozturk K, et al. Monogenic lupus due to spondyloenchondrodysplasia with spastic paraparesis and intracranial calcification: case-based review. Rheumatol Int (2020) 40(11):1903–10. doi: 10.1007/s00296-020-04653-x
25. Besnard C, Levy E, Aladjidi N, Stolzenberg MC, Magerus-Chatinet A, Alibeu O, et al. Pediatric-onset Evans syndrome: Heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol (2018) 188:52–7. doi: 10.1016/j.clim.2017.12.009
26. Grimes AB, Kim TO, Kirk SE, Flanagan J, Lambert MP, Grace RF, et al. Refractory autoimmune cytopenias in pediatric Evans syndrome with underlying systemic immune dysregulation. Eur J Haematol (2021) 106(6):783–7. doi: 10.1111/ejh.13600
27. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magerus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood (2019) 134(1):9–21. doi: 10.1182/blood-2018-11-887141
28. Miano M, Guardo D, Grossi A, Palmisani E, Fioredda F, Terranova P, et al. Underlying inborn errors of immunity in patients with evans syndrome and multilineage cytopenias: A single-centre analysis. Front Immunol (2022) 13:869033. doi: 10.3389/fimmu.2022.869033
29. Kubo S, Nakayamada S, Sakata K, Kitanaga Y, Ma X, Lee S, et al. Janus kinase inhibitor baricitinib modulates human innate and adaptive immune system. Front Immunol (2018) 9:1510. doi: 10.3389/fimmu.2018.01510
30. Rizzi M, Lorenzetti R, Fischer K, Staniek J, Janowska I, Troilo A, et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J Autoimmun (2017) 77:55–66. doi: 10.1016/j.jaut.2016.10.005
31. Gupta V, Cerquozzi S, Foltz L, Hillis C, Devlin R, Elsawy M, et al. Patterns of ruxolitinib therapy failure and its management in myelofibrosis: perspectives of the canadian myeloproliferative neoplasm group. JCO Oncol Pract (2020) 16(7):351–9. doi: 10.1200/JOP.19.00506
32. Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc (2011) 86(12):1188–91. doi: 10.4065/mcp.2011.0518
33. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med (2020) 383(27):2628–38. doi: 10.1056/NEJMoa2026834
34. Poulter JA, Collins JC, Cargo C, De Tute RM, Evans P, Ospina Cardona D, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood (2021) 137(26):3676–81. doi: 10.1182/blood.2020010286
35. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol (2022) 186(3):564–74. doi: 10.1111/bjd.20805
36. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical relevance of gain- and loss-of-function germline mutations in STAT1: A systematic review. Front Immunol (2021) 12:654406. doi: 10.3389/fimmu.2021.654406
37. Berenson JR, Martinez D, Safaie T, Boccia R, Yang H, Moezi M, et al. Ruxolitinib and methylprednisolone for treatment of patients with relapsed/refractory multiple myeloma. Br J Haematol (2023) 200(6):722–30. doi: 10.1111/bjh.18593
Keywords: spondyloenchondrodysplasia, ACP5, tartrate-resistant acid phosphatase, autoimmunity, interferonopathy, JAK inhibitor, myeloma
Citation: Gernez Y, Narula M, Cepika A-M, Valdes Camacho J, Hoyte EG, Mouradian K, Glader B, Singh D, Sathi B, Rao L, Tolin AL, Weinberg KI, Lewis DB, Bacchetta R and Weinacht KG (2024) Case report: Refractory Evans syndrome in two patients with spondyloenchondrodysplasia with immune dysregulation treated successfully with JAK1/JAK2 inhibition. Front. Immunol. 14:1328005. doi: 10.3389/fimmu.2023.1328005
Received: 25 October 2023; Accepted: 28 December 2023;
Published: 29 January 2024.
Edited by:
Jemima Albayda, Johns Hopkins University, United StatesReviewed by:
Xiao Peng, Johns Hopkins University, United StatesSnehal Shabrish, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), India
Copyright © 2024 Gernez, Narula, Cepika, Valdes Camacho, Hoyte, Mouradian, Glader, Singh, Sathi, Rao, Tolin, Weinberg, Lewis, Bacchetta and Weinacht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yael Gernez, eWFlbGdAc3RhbmZvcmQuZWR1; Katja G. Weinacht, a2d3MUBzdGFuZm9yZC5lZHU=