 Maria Kral
Maria Kral Emiel P.C. van der Vorst
Emiel P.C. van der Vorst Alexey Surnov
Alexey Surnov Christian Weber
Christian Weber Yvonne Döring
Yvonne Döring
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 December 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1326440
This article is part of the Research TopicAdaptive Immunity in AtherosclerosisView all 9 articles
Crosstalk between innate and adaptive immunity is pivotal for an efficient immune response and to maintain immune homeostasis under steady state conditions. As part of the innate immune system, type 2 innate lymphoid cells (ILC2s) have emerged as new important regulators of tissue homeostasis and repair by fine-tuning innate-adaptive immune cell crosstalk. ILC2s mediate either pro- or anti-inflammatory immune responses in a context dependent manner. Inflammation has proven to be a key driver of atherosclerosis, resembling the key underlying pathophysiology of cardiovascular disease (CVD). Notably, numerous studies point towards an atheroprotective role of ILC2s e.g., by mediating secretion of type-II cytokines (IL-5, IL-13, IL-9). Boosting these protective responses may be suitable for promising future therapy, although these protective cues are currently incompletely understood. Additionally, little is known about the mechanisms by which chemokine/chemokine receptor signaling shapes ILC2 functions in vascular inflammation and atherosclerosis. Hence, this review will focus on the latest findings regarding the protective and chemokine/chemokine receptor guided interplay between ILC2s and other immune cells like T and B cells, dendritic cells and macrophages in atherosclerosis. Further, we will elaborate on potential therapeutic implications which result or could be distilled from the dialogue of ILC2s with cells of the immune system in cardiovascular diseases.
The first line of defense in our immune system, known as innate immunity, promptly identifies invading pathogens through the immediate recognition of pathogen-associated molecular patterns (PAMPs) present on their surfaces (1) or damage-associated molecular patterns (DAMPs) released upon tissue injury or inflammation (2). Receptor activation upon PAMP or DAMP binding leads to the initiation of signaling cascades that result in the release of pro-inflammatory cytokines and chemokines produced by cells of the innate immune system. Effective communication between innate and adaptive immune cells is a fundamental requirement for a robust immune response. This communication is partially ensured by the innate immune system instructing the adaptive immune system through the presentation of pathogen-derived peptides to adaptive immune cells or via the signaling axis involving chemokines and their receptors. Chemokines serve as chemotactic cytokines and play a crucial role in recruiting leukocytes to sites of inflammation or injury (3).
Immune cell subsets are distributed among different sites in our body where they reside in specific tissue areas or circulate via the blood stream. Thus, cell-cell interactions across different locations add another layer of complexity to this cross-talk. Most of our knowledge about the human immune system is derived from studies from human peripheral blood. However, recent advances in computational modeling as well as better access to human tissue samples has enabled us to study immune reactions and functions of different immune cells across various locations in the body in addition to peripheral blood (4–9). Recent cell populations gaining attention are for example natural killer (NK) cells or innate lymphoid cells (ILCs), both of which mainly reside in tissues [e.g. in mucosal linings or adipose tissue (AT)] and adapt to changes in their environment. ILCs are an important source of cytokines for the initiation of immune reactions and have newly been described as an important bridge between the innate and adaptive immune system (10). ILCs have been discovered as the “innate immune cell pendant” to CD4+ T cells, since they develop from the common lymphoid progenitor and they express signature cytokines similar to CD4+ T cells. ILCs comprise of three subgroups: ILC1, ILC2, and ILC3 and their main common characteristics are the lack of antigen-specific receptors and lack of markers that are expressed on immune cells of the common hematopoietic cell lineages (11).
ILCs especially reside at mucosal barriers, where they are rapidly activated in response to invading pathogens. ILC2s secrete cytokines typical for T helper cell type 2 (Th2) cells (interleukin (IL)-5, IL-4, IL-9, and IL-13) in response to IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) and require expression of GATA binding protein 3 (GATA3). Importantly, ILC2s have been first discovered in AT and their critical role in regulating AT homeostasis by the release of type-2 cytokines has henceforth been intensively studied (12). In addition, ILC2s have been implicated in the development of several diseases, where they exert either pro- or anti-inflammatory properties in a tissue-dependent manner depending on the expression of specific markers. Moreover, it has been demonstrated by Huang et al. that tissue-resident natural ILC2s (nILC2) phenotypically differ from ILC2s that are activated upon infection, termed inflammatory ILC2s (iILC2S) (13). For example, they have been described to be involved in the pathophysiology of cardiovascular diseases (CVDs). CVDs consist of conditions affecting the heart or blood vessels including coronary heart disease and cerebrovascular disease. The dominant cause for this condition is atherosclerosis, the thickening of the arteries due to accumulation of lipids and immune cells resulting in plaque development. However, the exact mechanisms by which ILC2s mediate their pleitropic roles in different diseases including CVDs are not well understood.
Hence, this review will concentrate on elucidating the mechanisms through which ILC2s facilitate their multifaceted communication with various immune cells to orchestrate immune responses. We will describe their roles at different anatomical sites in the body in steady state and in diseases like asthma or skin fibrosis. Additionally, we will underscore their significant contributions to CVD and will shed light on the specific chemokine-chemokine receptor signaling pathways that play a pivotal role in mediating immune responses of ILC2s in CVD. As an outlook we will discuss how these cell-to-cell interactions during immune responses can serve as promising novel targets for therapeutic interventions in the management of chronic inflammatory disorders.
ILC2s express different surface markers depending on the tissue they inhabit which facilitates their tissue-specific identification. These cells are most prominently found in the skin, intestine, lung, and AT where they exert context specific functions in steady state (11, 14, 15).
Within visceral adipose tissue (VAT) for example, ILC2s primarily produce IL-5 and IL-13 in response to IL-33, leading to the recruitment of eosinophils (14, 15). These eosinophils, in conjunction with regulatory T cells (Tregs) and anti-inflammatory M2 macrophages, play crucial roles in reducing inflammation within the AT. The presence of IL-33 attracts these tissue-resident Tregs to sites of AT inflammation through ligation of its receptor interleukin 1 receptor-like 1 (IL1RL1), also known as ST2 (16). Therefore, ILC2s function as mediators for Treg recruitment via interaction with the co-stimulatory molecule OX40L expressed on ILC2s following IL-33 stimulation, a mechanism also observed in other tissues such as the lung and intestine (17). Conversely, it has been reported that during inflammatory conditions, such as obesity, the numbers of ILC2s decrease in both mouse and human adipose tissue (18, 19). This leads to a transition from anti-inflammatory M2 macrophages to pro-inflammatory M1 macrophages, marked by the release of pro-inflammatory cytokines. Consequently, studies utilizing mice deficient in ILC2s have demonstrated a significant reduction in eosinophils and M2 macrophages in VAT, ultimately leading to increased adiposity and insulin resistance in animal models (19).
Interestingly, ILC2s which are distributed along different tissues are also involved in mechanisms of tissue repair. In response to tissue damage, e.g. after cardiac stress, liver damage, or upon skin fibrosis, stromal cells release the alarmin IL-33 to recruit ILC2s via IL-33R/ST2 (20). This interaction triggers GATA3 phosphorylation, a transcription factor which is required for regulating type-2 cytokine genes (IL-4, IL-5, IL-13) (21). Moreover, the release of IL-13 by ILC2s recruits alternatively activated M2 macrophages, which have been shown to be important to initiate wound healing and tissue homeostasis (19, 22, 23).
These studies highlight that ILC2s differentially orchestrate immune responses in a tissue-specific manner under steady state and inflammatory conditions in order to maintain tissue homeostasis or to initiate tissue repair. However, depending on tissue type and disease context, ILC2s have been reported to exert dual roles of mediating either pro- or anti-inflammatory responses.
Initially, ILC2s have been described to exert type-2 immunity by secreting type-2 cytokines (IL-5, IL-13) upon helminth infection (24). A sustained type-2 immunity response, however, can also lead to tissue damage and chronic inflammation. For instance, in asthma and allergic lung inflammation, ILC2s are the main producers of type-2 immune cytokines, thereby driving these pathologies. Accordingly, it has been demonstrated that ILC2-mediated IL-13-IL-33 signaling induced airway hyper-reactivity (AHR) upon influenza infection in mice (25). Conversely, blocking the IL-33/IL-33R signaling pathway or the use of anti-CD90 monoclonal antibodies to block ILC2 expansion protected mice from the influenza-induced AHR, thereby proving an essential role of ILC2s in mediating airway inflammation (25). Importantly, Halim et al. could demonstrate that ILC2s are required to initiate a Th2 cell differentiation upon intranasal papain administration, thus building a bridge from innate to adaptive immunity in allergic lung inflammation (26).
Similarly, ILC2s have also been reported to mediate pro-inflammatory immune responses in inflamed liver tissue (27). In a murine model of Con A-induced immune-mediated hepatitis, evidence could be provided for a link between increased levels of IL-33 and enhanced numbers of ILC2s, which recruited eosinophils and the sustained release of type-2 cytokines, further promoting tissue damage, as seen in the AHR mouse model. Interestingly, the study found that a priori IL-33 treatment before Con A challenge led to an expansion of ST2+ Tregs and that this specific population regulated ILC2 activity and hence, led to a reduction of liver inflammation (27).
Conversely, in a model of arthritis, ILC2s are required for the resolution of inflammation. Mechanistically, it has been shown that IL-9 triggered the activation of ILC2s which in turn recruits Tregs via interaction of the co-stimulatory molecules Glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR; Tregs) - GITR ligand (GITRL; ILC2s) and Inducible T Cell Costimulator (ICOS; Tregs) - ICOS ligand (ICOSL; ILC2s) (28). Moreover, patients in remission phase of rheumatoid arthritis showed increased numbers of IL-9-expressing ILC2s in their joints and in the circulation (28).
The dual role of ILC2 subsets, i.e. promoting either pro- or anti-inflammatory responses depending on disease- and tissue-context based on the expression of specific markers as outlined earlier, is further emphasized by recent publications showing a regulatory function of ILC2s through the production of IL-10 upon exposure to the allergen papainin in the lung (29) and during intestinal inflammation (30). Treg-inducing factor, retinoic acid (RA), induced the IL-10-producing subset of ILC2s in vitro (31) and these regulatory ILC2s were able to mediate resolution of lung and intestinal inflammation in mouse models (29, 30). In humans, these cells were increased in human nasal tissue from healthy donors compared to patients suffering from chronic rhinosinusitis with nasal polyps (30). More recently, regulatory ILC2s have been associated with a better clinical response upon allergen-induced immunotherapy (32). In contrast to this, analysis of extracellular particles (EPs) derived from severe Coronavirus Disease of 2019 (COVID-19) patients revealed an activated phenotype of ILC2s, which led to a more aggressive disease score (33). Interestingly, EPs from severe COVID-19 patients also failed to dampen the cytokine production of ILC2s (33).
Taken together, these studies outline how environmental stimuli, the surrounding milieu, disease context and the release of specific cytokines shape ILC2 function in healthy and diseased states. Their pleiotropic function is further highlighted in low-grade inflammatory obesity (34). As outlined before, in obese patients or mice, numbers of ILC2s are reduced and thus, an inflammatory milieu prevails. However, the underlying mechanisms of an obesity-induced decrease in ILC2 numbers remain poorly understood. The pro-inflammatory environment in obesity is associated with an increased risk of developing cardiovascular disorders. Therefore, we will shed light on the consequence of ILC2 dysregulation in inflammatory-induced CVDs.
CVDs are the leading cause of death worldwide and they comprise of coronary heart diseases, peripheral artery disease, stroke, and cerebrovascular diseases, among others. Herein, atherosclerosis is the underlying pathological process which arises from the accumulation of lipids and immune cells in the intima of arteries, forming so called atherosclerotic plaques, leading to an increased thickness and stiffness of the vessels. Besides non-modifiable risk factors like increasing age, gender or genetic factors, several modifiable risk factors exist for atherosclerosis, like smoking, hypertension and hyperlipidemia. Therefore it is not surprising that obesity, characterized by excessive lipid accumulation and hyperlipidemia, is also a major risk factor for atherosclerotic cardiovascular diseases (ACVDs), although the underlying mechanisms explaining this association are still not completely understood.
Retention of lipids causes the modification of low-density-lipoprotein (LDL) into oxidized-LDL (oxLDL), which is targeted by the immune system as “non-self”. Moreover, obesity-induced chronic inflammation is caused by elevated serum levels of pro-inflammatory cytokines, which further attracts leukocytes to infiltrate the lesion (35).
The artery vessels are further composed of media and adventitia, which are imbedded into perivascular adipose tissue (PVAT). It has become clear, that the PVAT plays a major role in the pathogenesis of atherosclerosis. Under homeostatic conditions, PVAT regulates vascular tone and intravascular thermoregulation. However, chronic inflammation (as in atherosclerosis) leads to PVAT dysfunction (36, 37). Of interest, ILC2s have been shown to regulate PVAT homeostasis by secreting type-2 cytokines (IL-4, IL-5, IL-13) in order to maintain an anti-inflammatory milieu by induction of M2 macrophages and eosinophils (Figure 1).
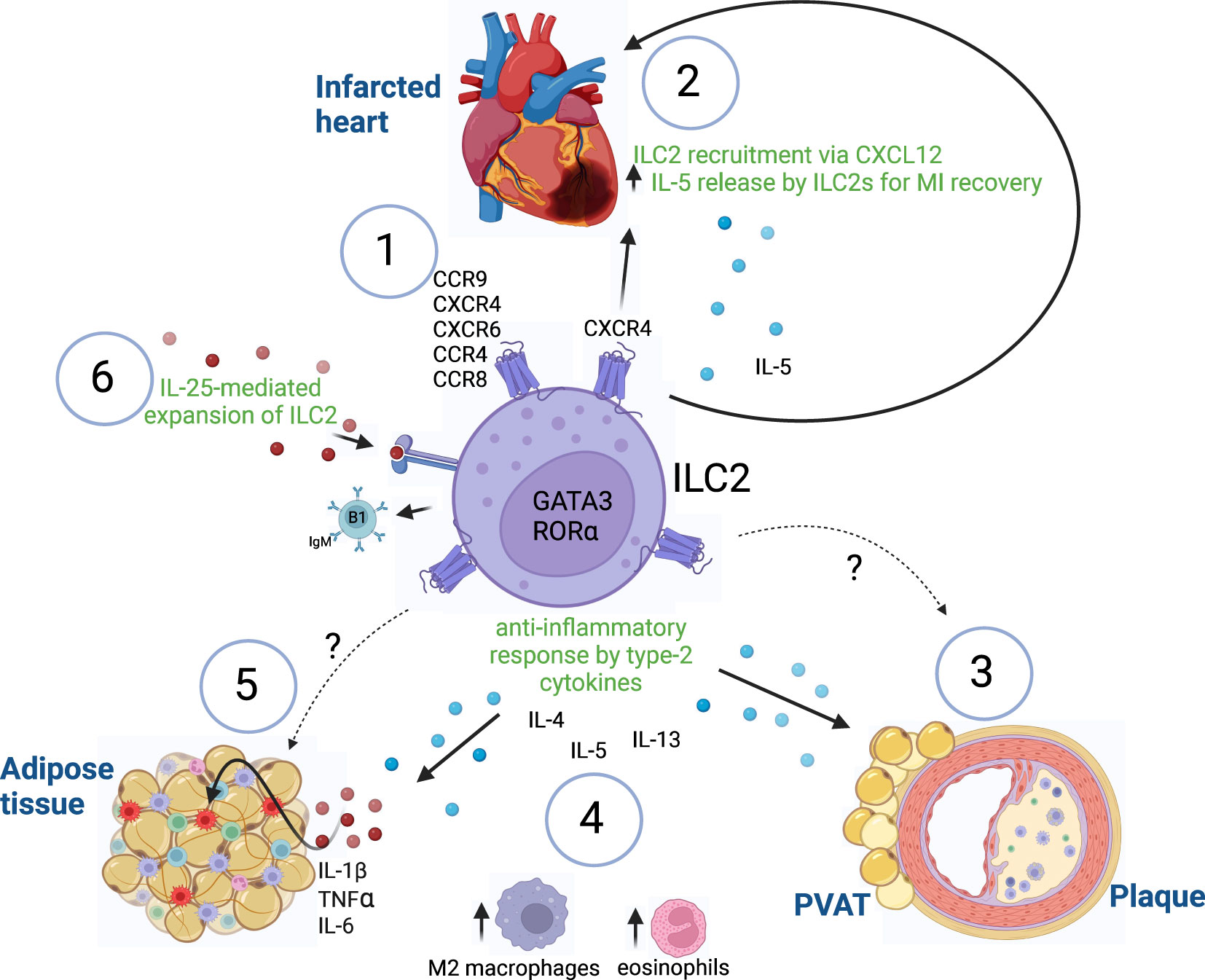
Figure 1 ILC2-mediated immune responses. (1) Expression of known chemokine receptors on ILC2s. (2) Recruitment of ILC2s after myocardial infarction via CXCR4/CXCL12 to initiate tissue repair by release of IL-5. (3) Atheroprotective functions of ILC2s in PVAT and atherosclerotic plaques are mediated by yet unidentified chemokine-chemokine receptor interactions. (4) ILC2s maintain an anti-inflammatory environment by the release of type-2 cytokines (IL-4, IL-5, IL-13), which leads to recruitment of alternatively activated M2 macrophages and eosinophils in adipose tissues. (5) Increase of pro-inflammatory cytokines (IL-1β, TNFα, IL-6) in adipose tissue inflammation leads to dysfunction of ILC2-mediated responses, which might be mediated by yet unknown chemokine-chemokine receptor signaling. (6) IL-25 treatment leads to expansion of ILC2 exerting atheroprotective functions by the production of B1-secreting IgM cells in atherosclerosis. Innate lymphoid cells type 2 (ILC2), GATA binding protein 3 (GATA3), RAR related orphan receptor A (RORα), myocardial infarction (MI), C-X-C chemokine receptor (CXCR), chemokine (C-X-C motif) ligand (CXCL), CC chemokine receptor (CCR), immunoglobulin M (IgM), Interleukin (IL), Tumor necrosis factor α (TNFα), perivascular adipose tissue (PVAT). Made with biorender.com. The "?" indicates that there is a yet unknown mechanism.
LDL receptor deficient (Ldlr−/−) mouse models are widely used as a model to study an altered cholesterol metabolism due to lack of endocytosis of circulating LDL caused by the LDL-R deletion. Consequently, circulating cholesterol levels are increased, which can be further enhanced when the mice are subjected to a high-fat diet (38). In this regard, first studies in 2015 using immunodeficient Ldlr−/−rag1−/− mouse models showed a protective role of ILC2s in atherosclerosis (39). Specifically, such mice treated with the IL-2/anti-IL-2 complex demonstrated expansion of CD25+ ILC2s and a reduction in very low-density-lipoprotein (VLDL) levels. Moreover, an increase in eosinophils in VAT and liver upon ILC2 expansion was observed (39). However, the use of an immunodeficient mouse model and the expansion of ILC2s by IL-2/anti-IL2 complexes rather created an artificial environment.
Therefore, to evaluate the role of naturally occurring ILC2s in atherosclerosis, another group made use of an apolipoprotein e-deficient (Apoe-/-) mouse model that is susceptible for atherosclerosis development (40). Intriguingly, ILC2s present in PVAT and lymph nodes phenotypically differed from those found in AT. Upon high-fat diet, ILC2s were not only reduced in numbers in PVAT but those remaining also altered their protective phenotype (40). PVAT-resident ILC2s resembled the inflammatory Killer cell Lectin-like receptor G1 (KLRG1)hi ST2- ILC2 (iILC2-like) phenotype and produced less IL-5 and IL-13 (41).
Collectively, these data suggest that ILC2s exerted their anti-inflammatory functions via the production of IL-5 and IL-13, which could be potentially a prerequisite for changes in macrophages and B1-dependent natural IgM production. Importantly, IgM antibodies produced by B1 cells exert atheroprotective properties by capturing lipids expressing oxidation-specific epitopes (OSEs) present on LDL, thereby hindering the uptake by macrophages (42–44).
A strong atheroprotective role of the ILC2-released cytokine IL-5 has been further demonstrated by showing an association of low levels of IL-5 with the presence of plaque development in the carotid bulb in humans (45). To confirm the previous finding in an experimental mouse model, the authors used Western diet-fed Apoe-/- mice lacking IL-5 and implanted a perivascular shear stress modifier on the right carotid artery to disrupt the pattern of hemodynamic flow. Interestingly, mice that lacked IL-5 displayed enlarged plaques at the location of the shear stress modifier implant, further supporting the importance of IL-5 production by local mediators in order to mediate atherprotective functions (45). The finding that IL-5 has strong atheroprotective functions and that it is mostly produced by ILC2s has been further supported by another study showing that ILC2 depletion in Ldlr−/− mice led to an acceleration in atherosclerosis progression. Conversely, only wild-type ILC2s could rescue this phenotype in these mice, whereas Il5−/− ILC2s did not (40).
Myocardial infarctions (MI) cause almost half of the cases of CVDs (46) and after acute MI, an increase of inflammation is induced in order to initiate cell repair. However, persistent inflammation can lead to adverse remodeling of the injured tissue. Several immune cell types migrate to the site of infarction. In this regard, one study found an increased accumulation of ILC2s at sites of MI, which released IL-5 and attracted eosinophils and dendritic cells (DCs) to induce tissue repair (47) (Figure 1). Mice with a diphtheria toxin-induced ILC2-specific deletion showed worsened cardiac dysfunction after MI induction, suggesting an important role of ILC2s in repairing tissue injury post-MI (47).
Similarly, Yu et al. examined the role of ILC2s in restoring postischemic injury by using a mouse model of MI with a genetic deletion of ILC2s (48). In wildtype mice, the authors could find an increased number of ILC2s at the site of infarction which contributed to tissue repair as assessed by histology and echocardiography, while genetic deletion of ILC2s impeded this recovery. Importantly, low-dose IL-2 treatment in post-MI mice activated ILC2s and thus, improved the recovery rate of these mice. Of note, this therapeutic approach showed also promising results in a clinical trial of patients with acute coronary syndromes by increased production of IL-5 by ILC2s (48).
As outlined in the previous section, depending on the disease and tissue context, ILC2s express different markers. One study analyzed the role of IL-25-expanded ILC2s isolated from spleens of IL-25-treated Apoe-/- mice in an in vitro and in vivo model (49). High-fat diet-fed Apoe-/- mice were injected subcutaneously with 0.5 × 106 in vitro-expanded splenic ILC2s for a total of four transfers with two weeks breaks in between. With this experimental approach, the authors were able to show that the transfer of ILC2s led to a reduction of the lipid content in atherosclerotic lesions of Apoe-/- mice (Figure 1). Interestingly, an increased production of B1-dependent IgM antibodies was observed (49). These findings further highlight the importance of ILC2 interactions with other immune cells ranging from innate to adaptive immunity as well as humoral immunity in order to mediate atheroprotective responses in cardiovascular inflammation.
More recently, it has been proposed that atherosclerosis also has autoimmune-like features. In this regard, it was shown that Tregs and ILC2s directly interact with each other during cardiovascular inflammation (50). Specifically, the disruption of cell-to-cell contacts reduced the release of IL-13 by ILC2s in vitro and in vivo. The importance of IL-13 in protecting against cardiovascular inflammation has been highlighted in previous sections.
Intriguingly, ILC2 (18, 19) and Treg numbers [summarized in (51)] are reduced in atherosclerotic mouse models and humans with CVDs. Moreover, Tregs from Apoe-/- mice have been shown to have a reduced ability to suppress CD4+ T cell proliferation in vitro (52). Notably, under conditions of sustained inflammation such as in atherosclerosis, Tregs have been shown to acquire a pro-inflammatory phenotype (53). A similar ILC2 plasticity has also been observed in airway diseases (54–56). However, the exact mechanisms leading to reduced numbers of ILC2s in atherosclerosis remain unclear. Moreover, understanding mechanisms involved in the Treg-ILC2 are still in its infancy.
Most of the studies used the Rorafl/flIl7rCre/+mouse model for the generation of ILC2 deletion, however, other ILC subsets might be affected in this model as well (57). Therefore, new mouse lines that specifically target or delete ILC2s are currently being investigated in order to improve the specificity (58–60).
Overall, these studies could demonstrate that ILC2s play an important role in protecting against vascular inflammation. However, under conditions of sustained inflammation, ILC2s acquire an inflammatory phenotype in atherosclerosis. Therefore, a better understanding of underlying mechanisms that shape ILC2 responses under such conditions are needed.
Chemokines and chemokine receptors play an important role in recruiting immune cells to sites of inflammation and therefore significantly impact on disease progression. Importantly, while chemokine receptors can have multiple ligands and vice-versa chemokines can bind to more than one receptor, they only pair with the structurally identical cysteine residues (3). However, depending on the pairing, chemokines and their receptors can exert different effects. In the past years, it has become clear that chemokine-chemokine receptor signaling is not only essential for leukocyte recruitment in atherosclerosis, but that their signaling cascade also mediates other important biological functions. For instance, blockade of chemokine (C-C motif) ligand 19 (CCL19) and CCL21 binding to CC chemokine receptor 7 (CCR7) has been shown to preserve foam cell content (61). Moreover, depending on the cells they are expressed on, chemokine-chemokine receptor interactions contribute to their pleiotropic function of either mediating pro- or anti-inflammatory responses (62).
Some early studies in the 2000’s already describe how chemokine receptors are able to shape T cell subsets and their responses. During that time, the existence of ILCs had not been known yet. One could therefore speculate, that those studies describing “uncommitted, primed, precursor cells” (Thpp), misidentified first interactions of ILC2s as being Th2 precursors with chemokines (63, 64).
Currently, several chemokine receptors expressed on ILC2s have been identified: CCR9, C-X-C chemokine receptor 4 (CXCR4), CXCR6 (15, 65–67), CCR4 (68) and CCR8 (69) in mice, and the expression of some chemokine receptors (CCR2, CCR4, CCR10 CXCR4) on ILC2s have also been described in humans, although studies remain scarce (70, 71) (Figures 1, 2).
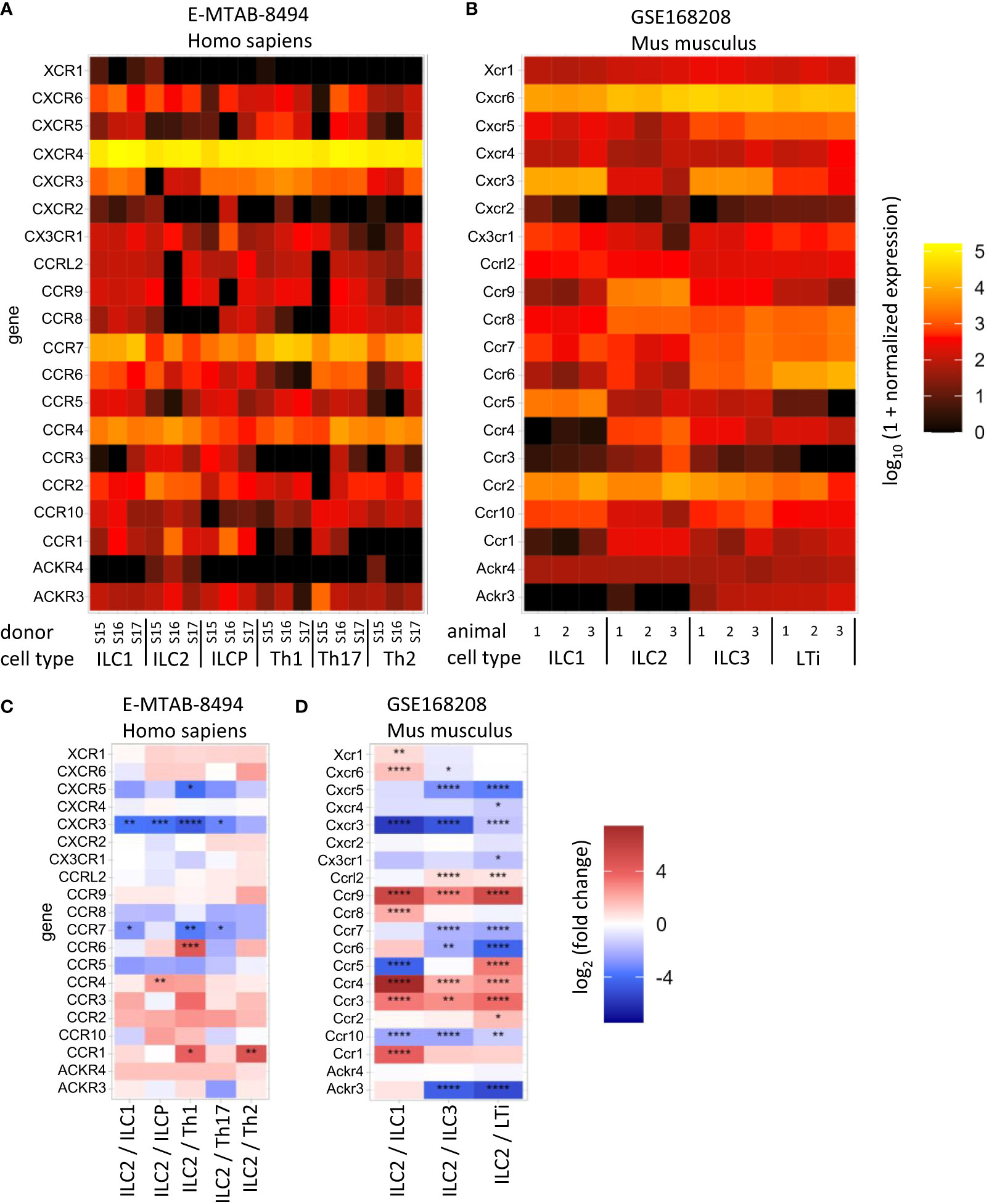
Figure 2 Heat maps show normalized mRNA copy number (A, B) or the expression difference in the indicated cell types (C, D) of the selected chemokine receptor-coding genes. The data from human (A, C) and mouse (B, D) cells are available at ArrayExpress E-MTAB-8494 (72) and GEO GSE168208 (73), respectively. Innate lymphoid cell (ILC), helper T cell (Th), Lymphoid tissue inducer (LTi), ILC precursor (ILCP). The analysis was performed as described in (73). P-value notation: * 0.01 ≤ p < 0.05, ** 0.001 ≤ p < 0.01, *** 0.0001 ≤ p < 0.001, ****p < 0.0001.
With respect to ILC2s, for instance, in response to allergens, IL-13 release by ILC2s leads to the production of DC-derived CCL17, thereby stimulating Th2 cell responses (26). Conversely, binding of CCL1 to CCR8 expressed on ILC2s protected mice from intestinal damage in a model of experimental colitis (74). Moreover, it has been shown that CCL1-CCR8 signaling on ILC2s triggered their proliferation and increased their ability to respond to helminth infections (68). In humans, CCR10+ ILC2s have been shown to expand upon severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pneumonia to trigger recovery and tissue repair and their increased frequency was negatively correlated with pro-inflammatory markers (75).
With respect to vascular inflammation, CXCR4 expressed on vascular cells as well as on B cells exerts protective functions in atherosclerosis (44, 76). In this regard, it has been shown that ILC2s are recruited via CXCR4-CXCL12 interaction and aid in the recovery from experimental MI in mice by increasing the levels of IL-5 (48) (Figure 1).
Further, CCR9-CCL25 signaling has been shown to be pro-atherogenic, since inhibition of CCR9 led to a delay in the development of atherosclerosis in Apoe-/- mice (77). However, the exact functions of CCR9 expressed on ILC2s in the context of ACVDs remain elusive. So far, it has only been reported that CCR9 might serve as trafficking receptor for ILC2s to several organs (65). Similarly, CXCR6 is an important T-cell homing receptor to the aortic wall where it accelerates atherosclerosis (78) but its role on ILC2s in ACVDs still needs to be explored. So far, it has only been shown that CXCR6 is crucial for ILC progenitors to egress from the bone marrow at steady state conditions (79).
Similarly, disrupted CCL1-CCR8 signaling in Tregs has been shown to exacerbate atherosclerosis in mice (80). Specifically, a reduction of the anti-inflammatory cytokine IL-10, and reduced Tregs could be observed in aorta and spleen of fat-fed Apoe-/- mice deficient in CCL1. Importantly, this effect could also be mimicked by blocking CCR8 in high-fat-fed ldlr−/− mice, as evidenced by decreased recruitment of Tregs to the aorta (80).
Importantly, CCR8 is also highly expressed on ILC2s (69). However, studies investigating the role of CCR8 on ILC2s in atherosclerosis are still lacking.
A list of known shared chemokine receptors between ILC2s and other immune cell subsets is shown in Table 1.
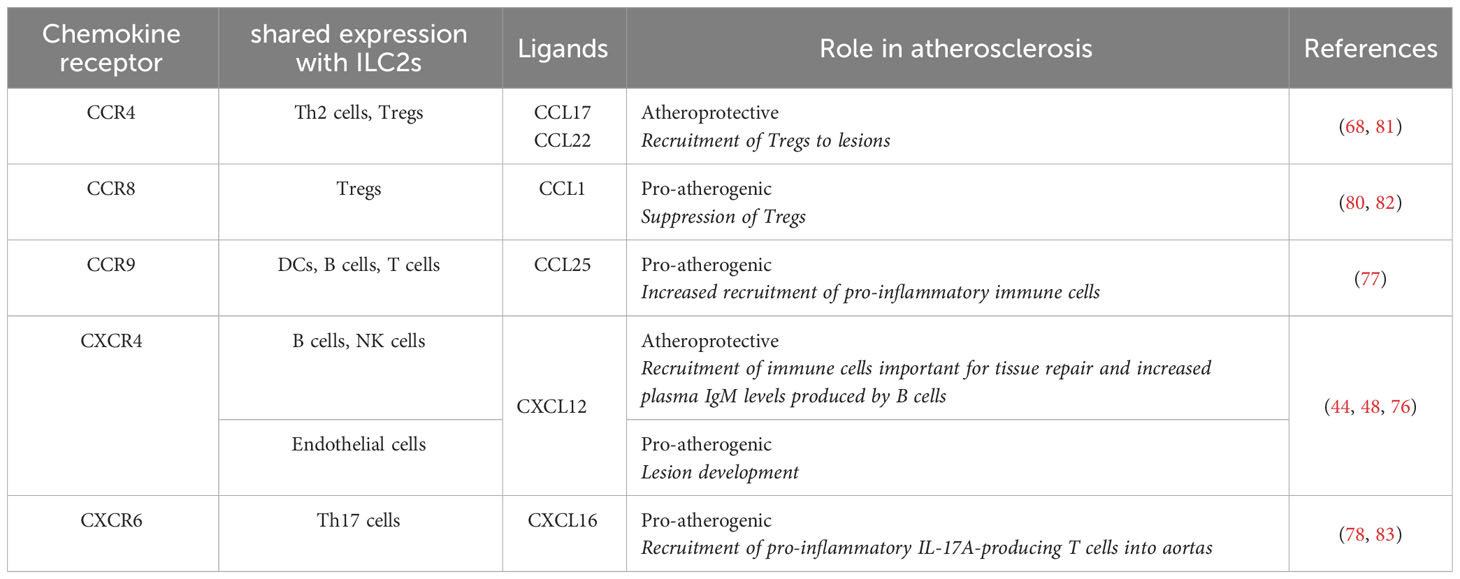
Table 1 ILC2-immune cell crosstalk and their function in atherosclerosis.
Due to their local availability, ILC2s are an attractive target for the development of novel therapies. Accordingly, some strategies to therapeutically manipulate ILC2s in disease have already been developed or are currently being investigated in clinical trials [see (84, 85)]. For instance, for the treatment of asthma there are several FDA-approved drugs like mepolizumab, benralizumab, and reslizumab that target the cytokine IL-5, which is expressed by ILC2s, among others (86). Current therapies using monoclonal antibodies targeting IL-5 are in clinical use as add-on therapy for the treatment of uncontrolled asthma (87). In this setting, IL-5 blockade is required to dampen hyperactivity in asthma by reducing recruitment of eosinophils and therefore, it is not specific to a given cell type but it is applied systemically. For the treatment of specific diseases where the role of ILC2s is implicated (e.g. CVDs), future studies are required that aim at specifically targeting ILC2-mediated IL-5 secretion.
In CVD, IL-2 treatment has led to promising results in mice by the expansion of ILC2s and hence, promoting their atheroprotective function (39) and recovery rate in post-MI mice (48). Since data from murine studies point towards an atheroprotective role of ILC2s and Tregs, using low-dose IL-2 treatment with the goal to expand both immune cell subsets could be a promising approach to dampen inflammatory responses in early atherosclerosis. The current literature focused on the effects of IL-2/anti-IL-2 treatment in expanding either Tregs (88, 89) or ILC2s (40, 90) in a specific disease setting. These studies did not investigate the expansion of both cell types or others that might be expanded upon this treatment strategy. It would be therefore crucial to understand which cell types become activated upon that treatment and how IL-2-mediated expansion affects their phenotypes.
In this regard, the phase 2 clinical trial called IVORY has been started to investigate the beneficial effect of low-dose IL-2 therapy in patients with acute coronary syndromes (ACS), which will be completed in 2024 (91).
Interestingly, whereas treatment of high fat-fed Apoe-/- mice with IL-33 reduced atherosclerosis development (92), a more recent study showed that ILC2s can also mediate the IL-33-induced eosinophilic pericarditis (93). Here, anti-IL-5 treatment led to a reduction of infiltrating eosinophils into the heart during pericarditis (93).
These studies suggest that therapeutical targeting of the IL-33/ST2/ILC2s signaling axis could be used as potential treatment for several CVDs and highlight the importance to investigate the function of ILC2-mediated signaling pathways in a disease-dependent context.
Notably, systemically administered drugs have general safety concerns due to off-target effects. Thus, targeting tissue-resident ILC2s and/or their signaling cascade via chemokine-chemokine receptor interactions enables a more specific and efficient approach for therapeutic manipulation. Hence, a deep phenotypic characterization of ILC2 is required for a better understanding of their mode of action in a disease context such as ACVDs.
Along these lines new technologies such as single cell RNA (scRNA) or nuclei RNA (snRNA) sequencing could improve our understanding of the state of ILC2 in a disease- and tissue-specific manner. ScRNA sequencing has identified ILC2 cells among human lymphocytes (94), and characterized them based on their microenvironmental localization in steady state and disease in several human tissues (95, 96). Importantly, Jiang et al. identified new ILC2 subclusters in myocardial ischemia of mice (97) and another study using scRNA sequencing revealed heterogeneity in the immune phenotypes of ILC2s from lesional atopic dermatitis skin in humans (98). These studies highlight the potential of scRNA sequencing technologies to advance our understanding in the heterogeneity of ILC2 subset phenotypes in healthy and diseased conditions. Moreover, the identification of such subsets will critically contribute to the development of therapeutic applications.
MK: Writing – original draft. EV: Writing – review & editing. AS: Writing – review & editing. CW: Writing – review & editing. YD: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by funding to YD and CW through the Deutsche Forschungsgemeinschaft (SFB1123-A1) and was supported by a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University to EV.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Medzhitov R, Janeway CA Jr. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol (1997) 9(1):4–9. doi: 10.1016/S0952-7915(97)80152-5
2. Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol (2015) 6:422. doi: 10.3389/fimmu.2015.00422
3. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J (2018) 285(16):2944–71. doi: 10.1111/febs.14466
4. Boor PPC, Bosma BM, Tran KTC, van der Laan LJW, Hagenaars H, Ijzermans JNM, et al. Characterization of antigen-presenting cell subsets in human liver-draining lymph nodes. Front Immunol (2019) 10:441. doi: 10.3389/fimmu.2019.00441
5. James KR, Gomes T, Elmentaite R, Kumar N, Gulliver EL, King HW, et al. Distinct microbial and immune niches of the human colon. Nat Immunol (2020) 21(3):343–53. doi: 10.1038/s41590-020-0602-z
6. Schoettler N, Hrusch CL, Blaine KM, Sperling AI, Ober C. Transcriptional programming and T cell receptor repertoires distinguish human lung and lymph node memory T cells. Commun Biol (2019) 2:411. doi: 10.1038/s42003-019-0657-2
7. Senda T, Dogra P, Granot T, Furuhashi K, Snyder ME, Carpenter DJ, et al. Microanatomical dissection of human intestinal T-cell immunity reveals site-specific changes in gut-associated lymphoid tissues over life. Mucosal Immunol (2019) 12(2):378–89. doi: 10.1038/s41385-018-0110-8
8. Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E, et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol (2019) 4(33). doi: 10.1126/sciimmunol.aav5581
9. Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, et al. Spatiotemporal immune zonation of the human kidney. Science (2019) 365(6460):1461–6. doi: 10.1126/science.aat5031
10. Symowski C, Voehringer D. Interactions between innate lymphoid cells and cells of the innate and adaptive immune system. Front Immunol (2017) 8:1422. doi: 10.3389/fimmu.2017.01422
11. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells — a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
12. Odegaard JI, Chawla A. Type 2 responses at the interface between immunity and fat metabolism. Curr Opin Immunol (2015) 36:67–72. doi: 10.1016/j.coi.2015.07.003
13. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16(2):161–9. doi: 10.1038/ni.3078
14. Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity (2012) 37(4):601–10. doi: 10.1016/j.immuni.2012.10.003
15. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi: 10.1038/nri3349
16. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol (2015) 16(3):276–85. doi: 10.1038/ni.3085
17. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity (2018) 48(6):1195–1207.e6. doi: 10.1016/j.immuni.2018.05.003
18. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature (2015) 519(7542):242–6. doi: 10.1038/nature14115
19. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med (2013) 210(3):535–49. doi: 10.1084/jem.20121964
20. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity (2015) 42(6):1005–19. doi: 10.1016/j.immuni.2015.06.006
21. Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev (2018) 281(1):154–68. doi: 10.1111/imr.12619
22. Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med (2012) 18(2):260–6. doi: 10.1038/nm.2628
23. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
24. Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol (2010) 31(11):407–13. doi: 10.1016/j.it.2010.09.001
25. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12(7):631–8. doi: 10.1038/ni.2045
26. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40(3):425–35. doi: 10.1016/j.immuni.2014.01.011
27. Neumann K, Karimi K, Meiners J, Voetlause R, Steinmann S, Dammermann W, et al. A proinflammatory role of type 2 innate lymphoid cells in murine immune-mediated hepatitis. J Immunol (2017) 198(1):128–37. doi: 10.4049/jimmunol.1600418
28. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med (2017) 23(8):938–44. doi: 10.1038/nm.4373
29. Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun (2017) 8(1):1900. doi: 10.1038/s41467-017-02023-z
30. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell (2017) 171(1):201–216.e18. doi: 10.1016/j.cell.2017.07.027
31. Morita H, Kubo T, Ruckert B, Ravindran A, Soyka MB, Rinaldi AO, et al. Induction of human regulatory innate lymphoid cells from group 2 innate lymphoid cells by retinoic acid. J Allergy Clin Immunol (2019) 143(6):2190–2201.e9. doi: 10.1016/j.jaci.2018.12.1018
32. Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Lenormand MM, Li RCY, et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity (2021) 54(2):291–307.e7. doi: 10.1016/j.immuni.2020.12.013
33. Forte D, Pellegrino RM, Trabanelli S, Tonetti T, Ricci F, Cenerenti M, et al. Circulating extracellular particles from severe COVID-19 patients show altered profiling and innate lymphoid cell-modulating ability. Front Immunol (2023) 14:1085610. doi: 10.3389/fimmu.2023.1085610
34. Michailidou Z, Gomez-Salazar M, Alexaki VI. Innate immune cells in the adipose tissue in health and metabolic disease. J Innate Immun (2022) 14(1):4–30. doi: 10.1159/000515117
35. Bulló M, García-Lorda P, Megias I, Salas-Salvadó J. Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res (2003) 11(4):525–31. doi: 10.1038/oby.2003.74
36. Guzik TJ, Marvar PJ, Czesnikiewicz-Guzik M, Korbut R. Perivascular adipose tissue as a messenger of the brain-vessel axis: role in vascular inflammation and dysfunction. J Physiol Pharmacol (2007) 58(4):591–610.
37. Guzik TJ, Mangalat D, Korbut R. Adipocytokines - novel link between inflammation and vascular function? J Physiol Pharmacol (2006) 57(4):505–28.
38. Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest (1994) 93(5):1885–93. doi: 10.1172/JCI117179
39. Engelbertsen D, Foks AC, Alberts-Grill N, Kuperwaser F, Chen T, Lederer JA, et al. Expansion of CD25+ Innate lymphoid cells reduces atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35(12):2526–35. doi: 10.1161/ATVBAHA.115.306048
40. Newland SA, Mohanta S, Clement M, Taleb S, Walker JA, Nus M, et al. Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat Commun (2017) 8:15781. doi: 10.1038/ncomms15781
41. Huang Y, Paul WE. Inflammatory group 2 innate lymphoid cells. Int Immunol (2016) 28(1):23–8. doi: 10.1093/intimm/dxv044
42. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res (2014) 114(11):1743–56. doi: 10.1161/CIRCRESAHA.113.301145
43. Tsiantoulas D, Gruber S, Binder C. B-1 cell immunoglobulin directed against oxidation-specific epitopes. Front Immunol (2013) 3. doi: 10.3389/fimmu.2012.00415
44. Doring Y, Jansen Y, Cimen I, Aslani M, Gencer S, Peters LJF, et al. B-cell-specific CXCR4 protects against atherosclerosis development and increases plasma IgM levels. Circ Res (2020) 126(6):787–8. doi: 10.1161/CIRCRESAHA.119.316142
45. Knutsson A, Bjorkbacka H, Duner P, Engstrom G, Binder CJ, Nilsson AH, et al. Associations of interleukin-5 with plaque development and cardiovascular events. JACC Basic Transl Sci (2019) 4(8):891–902. doi: 10.1016/j.jacbts.2019.07.002
46. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: A report from the American heart association. Circulation (2018) 137(12):e67–e492. doi: 10.1161/CIR.0000000000000558
47. Liu T, Meng Z, Liu J, Li J, Zhang Y, Deng Z, et al. Group 2 innate lymphoid cells protect mouse heart from myocardial infarction injury via interleukin 5, eosinophils, and dendritic cells. Cardiovasc Res (2023) 119(4):1046–61. doi: 10.1093/cvr/cvac144
48. Yu X, Newland SA, Zhao TX, Lu Y, Sage AS, Sun Y, et al. Innate lymphoid cells promote recovery of ventricular function after myocardial infarction. J Am Coll Cardiol (2021) 78(11):1127–42. doi: 10.1016/j.jacc.2021.07.018
49. Mantani PT, Duner P, Ljungcrantz I, Nilsson J, Bjorkbacka H, Fredrikson GN. ILC2 transfers to apolipoprotein E deficient mice reduce the lipid content of atherosclerotic lesions. BMC Immunol (2019) 20(1):47. doi: 10.1186/s12865-019-0330-z
50. Gao X, Lin J, Zheng Y, Liu S, Liu C, Liu T, et al. Type 2 innate lymphoid cells regulation by regulatory T cells attenuates atherosclerosis. J Mol Cell Cardiol (2020) 145:99–111. doi: 10.1016/j.yjmcc.2020.05.017
51. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol (2015) 35(2):280–7. doi: 10.1161/ATVBAHA.114.303568
52. Mor A, Planer D, Luboshits G, Afek A, Metzger S, Chajek-Shaul T, et al. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arterioscler Thromb Vasc Biol (2007) 27(4):893–900. doi: 10.1161/01.ATV.0000259365.31469.89
53. Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, et al. Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNgamma+ Th1/Tregs. Circ Res (2016) 119(11):1190–203. doi: 10.1161/CIRCRESAHA.116.309764
54. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17(6):636–45. doi: 10.1038/ni.3444
55. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol (2016) 17(6):646–55. doi: 10.1038/ni.3447
56. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol (2016) 17(6):626–35. doi: 10.1038/ni.3443
57. Oliphant CJ, Hwang You Y, Walker Jennifer JA, Salimi M, Wong See H, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi: 10.1016/j.immuni.2014.06.016
58. Gurram RK, Wei D, Yu Q, Butcher MJ, Chen X, Cui K, et al. Crosstalk between ILC2s and Th2 cells varies among mouse models. Cell Rep (2023) 42(2):112073. doi: 10.1016/j.celrep.2023.112073
59. Liang H-E, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol (2012) 13(1):58–66. doi: 10.1038/ni.2182
60. Pai SY, Truitt ML, Ting CN, Leiden JM, Glimcher LH, Ho IC. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity (2003) 19(6):863–75. doi: 10.1016/S1074-7613(03)00328-5
61. Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U.S.A. (2006) 103(10):3781–6. doi: 10.1073/pnas.0511043103
62. Wallrapp A, Riesenfeld SJ, Burkett PR, Kuchroo VK. Type 2 innate lymphoid cells in the induction and resolution of tissue inflammation. Immunol Rev (2018) 286(1):53–73. doi: 10.1111/imr.12702
63. Ward SG, Westwick J. Chemokines: understanding their role in T-lymphocyte biology. Biochem J (1998) 333(Pt 3):457–70. doi: 10.1042/bj3330457
64. Yang L, Mosmann T. Synthesis of several chemokines but few cytokines by primed uncommitted precursor CD4 T cells suggests that these cells recruit other immune cells without exerting direct effector functions. Eur J Immunol (2004) 34(6):1617–26. doi: 10.1002/eji.200424939
65. Kim MH, Taparowsky EJ, Kim CH. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity (2015) 43(1):107–19. doi: 10.1016/j.immuni.2015.06.009
66. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol (2013) 14(6):564–73. doi: 10.1038/ni.2584
67. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med (2018) 215(1):263–81. doi: 10.1084/jem.20170449
68. Knipfer L, Schulz-Kuhnt A, Kindermann M, Greif V, Symowski C, Voehringer D, et al. A CCL1/CCR8-dependent feed-forward mechanism drives ILC2 functions in type 2-mediated inflammation. J Exp Med (2019) 216(12):2763–77. doi: 10.1084/jem.20182111
69. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16(3):306–17. doi: 10.1038/ni.3094
70. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med (2013) 210(13):2939–50. doi: 10.1084/jem.20130351
71. Weston CA, Rana BMJ, Cousins DJ. Differential expression of functional chemokine receptors on human blood and lung group 2 innate lymphoid cells. J Allergy Clin Immunol (2019) 143(1):410–413.e9. doi: 10.1016/j.jaci.2018.08.030
72. Ercolano G, Wyss T, Salome B, Romero P, Trabanelli S, Jandus C. Distinct and shared gene expression for human innate versus adaptive helper lymphoid cells. J Leukoc Biol (2020) 108(2):723–37. doi: 10.1002/JLB.5MA0120-209R
73. Beckstette M, Lu CW, Herppich S, Diem EC, Ntalli A, Ochel A, et al. Profiling of epigenetic marker regions in murine ILCs under homeostatic and inflammatory conditions. J Exp Med (2022) 219(10). doi: 10.1084/jem.20210663
74. Kang L, Schmalzl A, Leupold T, Gonzalez-Acera M, Atreya R, Neurath MF, et al. CCR8 signaling via CCL1 regulates responses of intestinal IFN-gamma producing innate lymphoid celIs and protects from experimental colitis. Front Immunol (2020) 11:609400. doi: 10.3389/fimmu.2020.609400
75. Gomes AMC, Farias GB, Dias-Silva M, Laia J, Trombetta AC, Godinho-Santos A, et al. SARS-CoV2 pneumonia recovery is linked to expansion of innate lymphoid cells type 2 expressing CCR10. Eur J Immunol (2021) 51(12):3194–201. doi: 10.1002/eji.202149311
76. Doring Y, Noels H, van der Vorst EPC, Neideck C, Egea V, Drechsler M, et al. Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity: evidence from mouse and human studies. Circulation (2017) 136(4):388–403. doi: 10.1161/CIRCULATIONAHA.117.027646
77. Abd Alla J, Langer A, Elzahwy SS, Arman-Kalcek G, Streichert T, Quitterer U. Angiotensin-converting enzyme inhibition down-regulates the pro-atherogenic chemokine receptor 9 (CCR9)-chemokine ligand 25 (CCL25) axis. J Biol Chem (2010) 285(30):23496–505. doi: 10.1074/jbc.M110.117481
78. Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, Bruce A, et al. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation (2007) 116(16):1801–11. doi: 10.1161/CIRCULATIONAHA.106.678474
79. Chea S, Possot C, Perchet T, Petit M, Cumano A, Golub R. CXCR6 expression is important for retention and circulation of ILC precursors. Mediators Inflammation (2015) 2015:368427. doi: 10.1155/2015/368427
80. Vila-Caballer M, Gonzalez-Granado JM, Zorita V, Abu Nabah YN, Silvestre-Roig C, Del Monte-Monge A, et al. Disruption of the CCL1-CCR8 axis inhibits vascular Treg recruitment and function and promotes atherosclerosis in mice. J Mol Cell Cardiol (2019) 132:154–63. doi: 10.1016/j.yjmcc.2019.05.009
81. Amin H, Sasaki N, Horibe S, Kawauchi S, Hirata K, Rikitake Y. Pharmacological inhibition of C-C chemokine receptor 4 aggravates atherosclerosis through prevention of regulatory T cell recruitment to the lesions. Eur Heart J (2020) 41(Supplement_2). doi: 10.1093/ehjci/ehaa946.3727
82. Weber C, Meiler S, Doring Y, Koch M, Drechsler M, Megens RT, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest (2011) 121(7):2898–910. doi: 10.1172/JCI44925
83. Butcher MJ, Wu CI, Waseem T, Galkina EV. CXCR6 regulates the recruitment of pro-inflammatory IL-17A-producing T cells into atherosclerotic aortas. Int Immunol (2016) 28(5):255–61. doi: 10.1093/intimm/dxv068
84. Cobb LM, Verneris MR. Therapeutic manipulation of innate lymphoid cells. JCI Insight (2021) 6(6). doi: 10.1172/jci.insight.146006
85. Huang YA, Strohm A, Doherty T. Modulating ILC2 function for treatment of type 2 airway diseases. Curr Trends Immunol (2022) 23:85–90.
86. Ridolo E, Pucciarini F, Nizi MC, Makri E, Kihlgren P, Panella L, et al. Mabs for treating asthma: omalizumab, mepolizumab, reslizumab, benralizumab, dupilumab. Hum Vaccin Immunother (2020) 16(10):2349–56. doi: 10.1080/21645515.2020.1753440
87. FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2016) 388(10056):2128–41. doi: 10.1016/S0140-6736(16)31322-8
88. Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi FD. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur J Immunol (2010) 40(6):1577–89. doi: 10.1002/eji.200939792
89. Zhang H, Xia Y, Ye Q, Yu F, Zhu W, Li P, et al. In vivo expansion of regulatory T cells with IL-2/IL-2 antibody complex protects against transient ischemic stroke. J Neurosci (2018) 38(47):10168–79. doi: 10.1523/JNEUROSCI.3411-17.2018
90. Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol (2015) 136(6):1653–1663.e7. doi: 10.1016/j.jaci.2015.03.043
91. Zhao TX, Newland SA, Mallat Z. 2019 ATVB plenary lecture: interleukin-2 therapy in cardiovascular disease: the potential to regulate innate and adaptive immunity. Arterioscler Thromb Vasc Biol (2020) 40(4):853–64. doi: 10.1161/ATVBAHA.119.312287
92. Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med (2008) 205(2):339–46. doi: 10.1084/jem.20071868
93. Choi HS, Won T, Hou X, Chen G, Bracamonte-Baran W, Talor MV, et al. Innate lymphoid cells play a pathogenic role in pericarditis. Cell Rep (2020) 30(9):2989–3003.e6. doi: 10.1016/j.celrep.2020.02.040
94. Suffiotti M, Carmona SJ, Jandus C, Gfeller D. Identification of innate lymphoid cells in single-cell RNA-Seq data. Immunogenetics (2017) 69(7):439–50. doi: 10.1007/s00251-017-1002-x
95. Mazzurana L, Czarnewski P, Jonsson V, Wigge L, Ringner M, Williams TC, et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res (2021) 31(5):554–68. doi: 10.1038/s41422-020-00445-x
96. Song P, Cao K, Mao Y, Ai S, Sun F, Hu Q, et al. Tissue specific imprinting on innate lymphoid cells during homeostasis and disease process revealed by integrative inference of single-cell transcriptomics. Front Immunol (2023) 14:1127413. doi: 10.3389/fimmu.2023.1127413
97. Jiang S, Zheng Y, Lv B, He S, Yang W, Wang B, et al. Single-cell landscape dissecting the transcription and heterogeneity of innate lymphoid cells in ischemic heart. Front Immunol (2023) 14:1129007. doi: 10.3389/fimmu.2023.1129007
Keywords: ILC2, atherosclerosis, chemokine receptors, inflammation, cardiovascular disease, tissue repair
Citation: Kral M, van der Vorst EPC, Surnov A, Weber C and Döring Y (2023) ILC2-mediated immune crosstalk in chronic (vascular) inflammation. Front. Immunol. 14:1326440. doi: 10.3389/fimmu.2023.1326440
Received: 23 October 2023; Accepted: 05 December 2023;
Published: 20 December 2023.
Edited by:
Bram Slütter, Leiden University, NetherlandsReviewed by:
Takeshi Kawabe, Tohoku University, JapanCopyright © 2023 Kral, van der Vorst, Surnov, Weber and Döring. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yvonne Döring, eXZvbm5lLmRvZXJpbmdAdW5pYmUuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.