 Lihua Xu1
Lihua Xu1 Pengju Wang
Pengju Wang- 1Sino-British Research Centre for Molecular Oncology, National Centre for International Research in Cell and Gene Therapy, State Key Laboratory of Esophageal Cancer Prevention & Treatment, School of Basic Medical Sciences, Academy of Medical Sciences, Zhengzhou University, Zhengzhou, China
- 2Centre for Biomarkers & Biotherapeutics, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
Oncolytic virotherapy (OVT) is a promising form of cancer treatment that uses genetically engineered viruses to replicate within cancer cells and trigger anti-tumor immune response. In addition to killing cancer cells, oncolytic viruses can also remodel the tumor microenvironment and stimulate a long-term anti-tumor immune response. Despite achieving positive results in cellular and organismal studies, there are currently only a few approved oncolytic viruses for clinical use. Vaccinia virus (VACV) has emerged as a potential candidate due to its ability to infect a wide range of cancer cells. This review discusses the mechanisms, benefits, and clinical trials of oncolytic VACVs. The safety and efficacy of different viral backbones are explored, as well as the effects of oncolytic VACVs on the tumor microenvironment. The potential combination of oncolytic VACVs with immunotherapy or traditional therapies is also highlighted. The review concludes by addressing prospects and challenges in the field of oncolytic VACVs, with the aim of promoting further research and application in cancer therapy.
1 Introduction
Oncolytic virotherapy (OVT) is an emerging tumor treatment modality that is based on naturally or genetically engineered oncolytic viruses (OVs) to selectively lyse tumor cells while sparing healthy cells (1). The history of OVs include three stages: the discovery and application of wild-type viruses (before 1990), the research and development of genetically engineered viruses (1991–2000), and the insertion of therapeutic genes in viruses and synergistic therapies (21st century) (2). In 1991, Martuza et al. first reported a genetically engineered herpes simplex virus (HSV)-1 (dlsptk) for inhibiting glioma growth in nude mice, which accelerated the field of OVT (3). With deeper insight into the anti-tumor mechanisms, OVs have been deemed to not only lyse tumor cells selectively, but activate anti-tumor immunity and remodel the tumor microenvironment (TME), making them promising as oncologic therapeutic agents (4).
According to their genomic characteristics, OVs are divided into two categories, DNA viruses (e.g., adenovirus, vaccinia virus (VACV), herpesvirus) and RNA viruses [e.g., reovirus, measles virus, New Castle disease virus (NDV), vesicular stomatitis virus (VSV)], in which DNA oncolytic viruses are superior to other viruses due to their larger genome, genetic stability, and high replication ability (5). Larger genome allows for more flexibility for the expression of recombinant payloads. Most of the currently approved OVs that include Rigvir (enteric cytopathic human orphan virus approved in Latvia for melanoma in 2004), Oncorine (adenovirus approved in China for head and neck cancer in 2005), T-VEC (HSV approved in the United States for melanoma in 2015), and DELYTACT (HSV approved in Japan for glioblastoma in 2021) are DNA viruses. Besides the above approved OVs, many other candidate OVs show promising pre-clinical evidence of anti-tumor activity, and increasing clinical trials are currently underway to determine the efficacy of OVs in different cancer patients (6). However, the number of approved OVs and the applicable cancer types remain very rare in the clinic. Intratumor administration is one of the most important limiting factors. The presence of preexisting antiviral neutralizing antibodies or their development during viral therapy render repeat systemic treatments of OVs ineffective, limiting the application in some cancer types with metastases or unable to be administrated in situ. For instance, T-VEC is only approved in patient without visceral metastases. The development of novel and more potent oncolytic viruses is urgently needed.
As one kind of DNA virus, VACV is a large DNA prototypic poxvirus that replicates exclusively in the cytoplasm, and it is therefore fully nonintegrative. VACV has been used as a smallpox vaccine for many years with relatively low adverse reactions (7). Recent preclinical results and clinical data about different engineered oncolytic VACVs [e.g., Pexa-Vec (JX-594)] also show its potential for intravenous infusion and tumor therapy via highly and stably expressing many therapeutic genes (8). According to the phase 1b trial results of biweekly intravenous Pexa-Vec in colorectal cancer, no patients developed Pexa-Vec infusion-related reactions. The common adverse effects mainly include headache, nausea, anorexia, rash, and vomiting (9). Hence, VACVs are expected to be safe candidates for OVT.
This review outlines the characteristics, viral backbones and clinical trials of oncolytic VACVs. The anti-tumor mechanisms and effects on tumor microenvironment are also discussed. In addition, the potential combination treatments with immunotherapy or traditional therapies are highlighted. The prospects and challenges of oncolytic VACVs are also addressed to promote the further research and application in cancer treatment.
2 Characteristics and anti-tumor mechanisms of oncolytic VACVs
2.1 Characteristics of oncolytic VACVs
VACV is an enveloped double stranded DNA orthopoxvirus, in which the most widely used experimental strains include Lister, Western Reserve (WR), Wyeth strains, Copenhagen strains, Ankara (also known as MVA), and Tiantan stains etc (10). MVA and Tiantan stains are non-replicative strains of VACVs for cancer vaccine, while the other strains are replication competent viruses. During replication, VACVs produce four infectious forms which differ in their outer membranes: intracellular mature virion (IMV), the intracellular enveloped virion (IEV), the cell-associated enveloped virion (CEV) and the extracellular enveloped virion (EEV) (11). The IMV is the most abundant infectious form, responsible for spread between host, while the CEV and EEV are responsible for cell-to-cell transfer and long-range dissemination within the host organism, respectively (12, 13).
Compared with other virus vectors, VACVs have many advantages in OVT. 1) VACVs replicate exclusively in the cytoplasm, with no integration of viral DNA into the host genome, indicating its safety (14). 2) VACVs have a large genome (~190 kb), capable of inserting and stably expressing exogenous therapeutic genes of at least 25 kb in a single vector (15). 3) VACVs own natural tumor tropism, potential for systemic administration (16). 4) VACVs have a rapid and lytic replication cycle. The first viruses can be released from the cells within 8 h after infection, and the infected cells can be destroyed after 48-72 h of infection. 5) VACVs can replicate in hypoxic conditions (17). 6) Because of no limitation on receptors during entry, VACVs exhibit high infectivity not only in various host species but also in a large range of tissues, beneficial for preclinical researches (18, 19).
In addition, like other oncolytic viruses, oncolytic VACVs can exert anti-tumor effect via oncolysis and activation of anti-tumor immune responses. To date, several studies have shown that deletion of some endogenous genes enhances the oncolysis of oncolytic VACVs. The key VACV genes and corresponding oncolytic functions are listed in the Table 1. Deletion those genes can improve the antitumor efficacy through multiple ways such as increasing the tumor selectivity, safety and anti-tumor immune response.
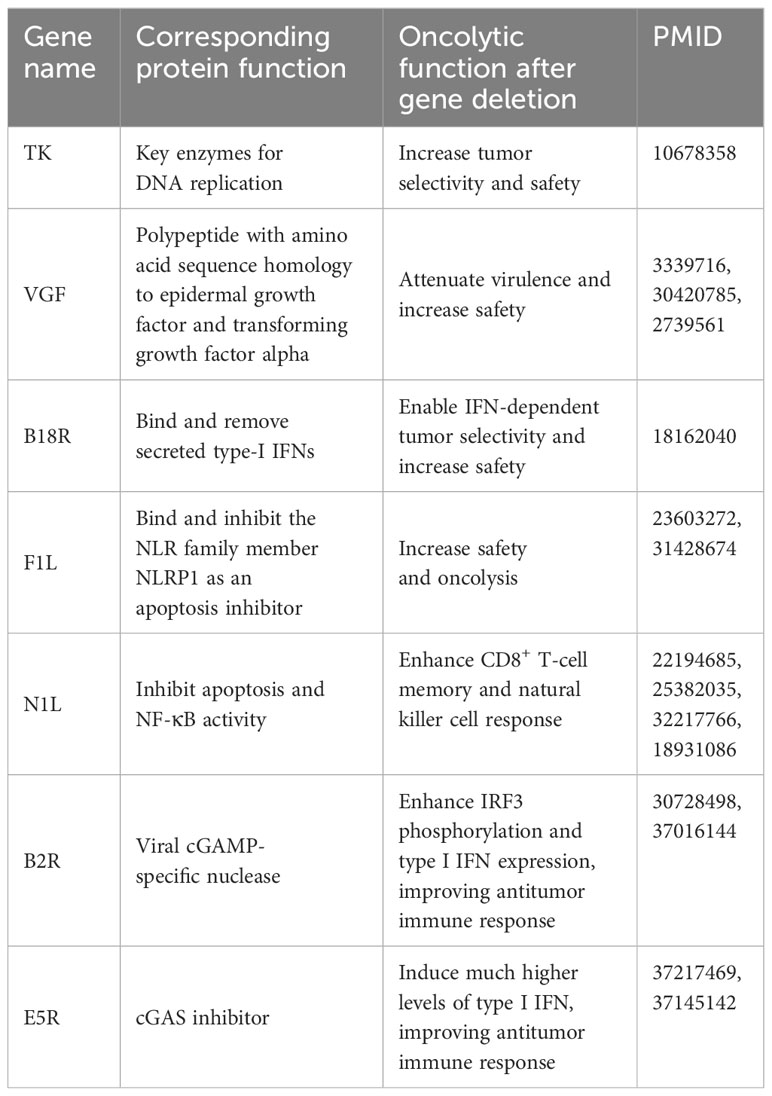
Table 1 Oncolytic functions of key VACV genes.
Based on the above advantages, oncolytic VACV is an alternative virus vector for OVT.
2.2 Anti-tumor mechanisms of oncolytic VACVs
Oncolytic VACVs primarily destroy tumor tissues via three mechanisms: direct oncolysis of tumor cells, disrupting tumor vasculature (tumor-associated endothelial cells lysis-mediated vascular collapse, neutrophils accumulation-mediated thrombosis) and activating anti-tumor immunity (Figure 1). In the following sections, we explore the detailed anti-tumor mechanisms of oncolytic VACVs.
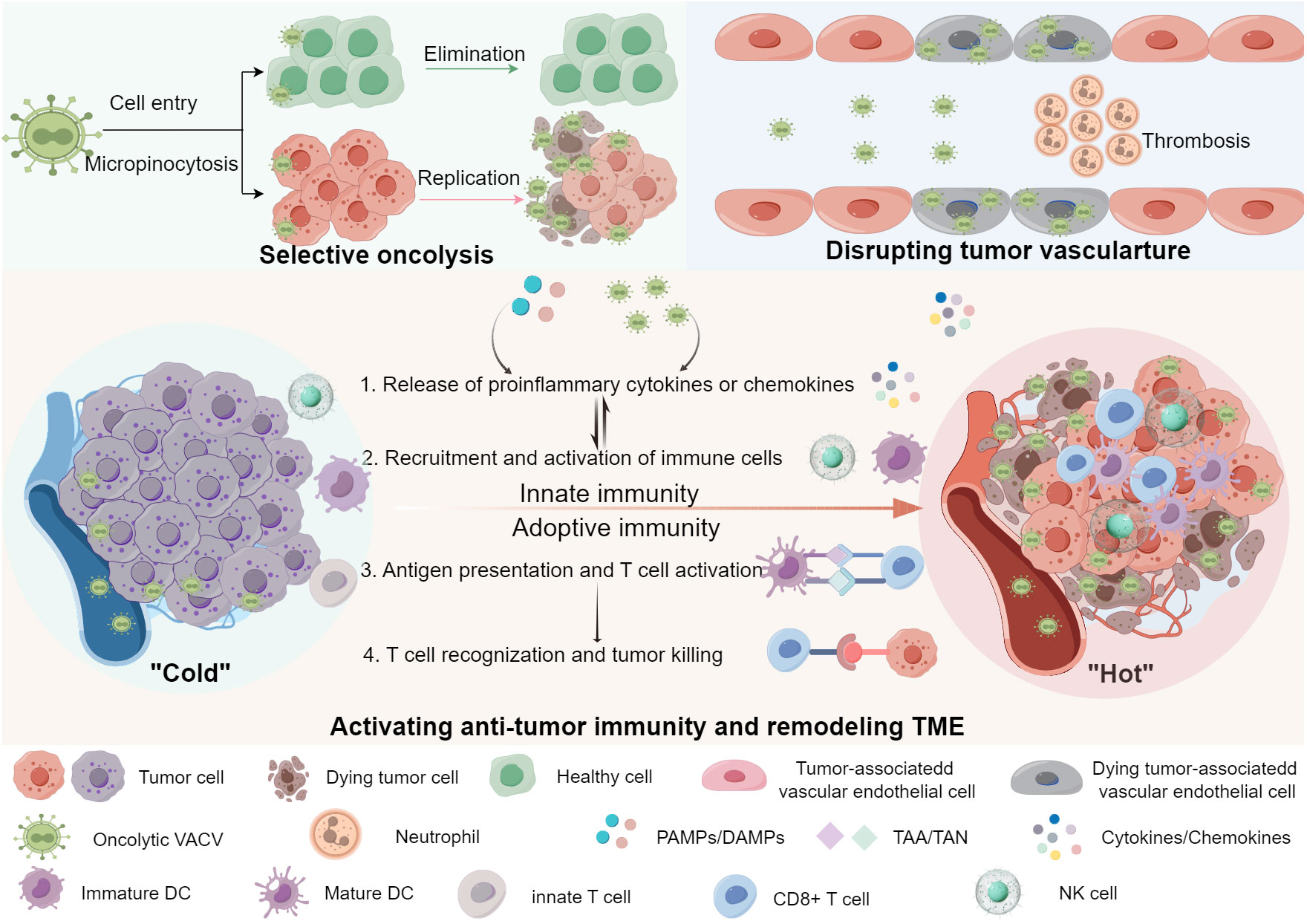
Figure 1 Anti-tumor mechanisms of oncolytic VACVs. Oncolytic VACVs can kill cancer cells via a variety of mechanisms. First, they directly infect, replicate and lyse tumor cells sparing normal cells. Released virions can infect neighbor tumor cells and so forth. Second, oncolytic VACVs can infect and lyse tumor associated vascular endothelial cells, meanwhile recruiting neutrophile cells and inducing thrombosis. Third, they can remodel the “cold” TME to “hot” by activating innate and adoptive anti-tumor immunity. The release of progeny viruses and PAMPs/DAMPs can promote the innate immune cells to produce proinflammatory cytokines and chemokines, which in turn lead to the recruitment and activation of more immune cells, thus innate immune responses are activated. With the activation of antigen presentation cells, DCs can present the released TAA/TAN to T-cells, enhancing the tumor recognition and killing ability of CD8+ T-cells, inducing a tumor-specific adoptive immune response. PAMPs, Pathogen-associated molecular patterns; DAMPs, Damage-associated molecular pattern; TAA, Tumor-associated antigens; TAN, Tumor-associated neoantigens; DC, Dendritic cell; NK cell, Natural killer cell. The figure is drawn by FigDraw.
2.2.1 Selective self-replication and oncolysis in tumor cells
Unlike some viruses such as adenoviruses, VACVs have inherent tumor tropism. As one kind of invasive viruses, oncolytic VACVs infect host tumor cells in several steps (20). Firstly, oncolytic VACVs use distinct forms of macropinocytosis for host-cell entry, independent on receptors. A study from Helenius’s group demonstrated that mature virions (MVs) from both the WR strain and the International Health Department-J (IHD-J) strain entered host cells by macropinocytosis due to virion-exposed phosphatidylserine. However, different macropinocytic mechanisms were possible in the same cell line through subtle differences in the activating ligand. The results showed that MVs from the WR strain entered HeLa cells by activating transient plasma membrane blebbing, while MVs from the IHD-J strain induced rapid formation (and lengthening) of filopodia (19). Next, DNA and proteins for oncolytic VACVs replication are synthesized in the cytoplasm instead of the cell nucleus. Last, progeny of VACVs released from lysed tumor cells spread to surrounding uninfected tumor cells, leading to amplification of their oncolytic activity and inducing tumor cell death. The selective oncolysis of oncolytic VACVs mainly lies on the differences between tumor cells and normal cells. On the one hand, various tumor suppressor genes (e.g., p53, RAS, and PTEN) and antiviral signals (such as type I interferon (IFN) pathway) are significantly downregulated, which make it easier for oncolytic VACVs to survive and replicate in the tumor cell (21). On the other hand, most of oncolytic VACVs are constructed by deletion of some viral genes [e.g., thymidine kinase (TK)] that overexpressed in tumor cells (22). In the healthy cell, oncolytic VACVs cannot replicate due to the deletion of some viral genes that are essential for VACV replication. Once exposure of oncolytic VACV genome in the cytoplasm, cyclic GMP-AMP synthase (cGAS)/stimulator of interferon gene (STING) pathway is activated followed by the production of antiviral cytokines such as type I IFN, further inhibiting oncolytic VACV replication. Subsequently, the genome and proteins of oncolytic VACVs are degraded by DNA and protein-degrading enzymes.
2.2.2 Disrupting tumor vasculature
In general, the peripheral region of a tumor contains enriched vasculature which is primarily composed of endothelial cells, and affects tumor growth and metastasis (23). Targeting the neovasculature is an alternative approach to eradication of tumor cells via starving and suffocating tumors. For the first time, Kirn and coworkers found that the oncolytic VACV (JX-594) could induce cytokines and chemokines-mediated neutrophils accumulation in blood vessels, leading to intravascular thrombosis (24). Soon afterwards, they revealed that JX-594, with deletion of TK genes, was able to specifically target and infect tumor-associated endothelial cells and efficiently replicate due to increased vascular endothelial growth factors (VEGF) signaling-mediated TK overexpression, contributing to vascular collapse (25). After intravenous injection of oncolytic VACVs, tumor perfusion in patient biopsy decreased from magnetic resonance imaging results, and no clinical signs of damage to normal vasculature were observed. Another research from Bell’s group revealed that VEGF/VEGFR2 signaling in remodeling vessels could also sensitize the tumor vasculature to oncolytic VACVs infection by PRD1-BF1/Blimp1-mediated antiviral immune suppression (26). However, Santry et al. considered that despite many potential benefits by oncolytic VACV-mediated tumor vascular collapse such as leukomonocyte recruitment, shutting off blood vessels may also limit oncolytic VACVs spread and obstruct the delivery of subsequent therapeutic agents, and the entry of immunological effector cells (27).
2.2.3 Activating anti-tumor immune response and remodeling TME
Early the anti-tumor mechanism of OVT was recognized as direct oncolysis of OVs. With the development of tumor immunotherapy, the impact of OVT on the immune response to tumors has become widely studied. It is widely acknowledged that oncolytic VACVs can activate innate and adaptive immune systems in tumors, and further remodel tumor immunosuppressive microenvironment.
Generally, after infection by oncolytic VACVs, pathogen-associated molecular patterns (PAMPs) and progeny viruses are released and sensed by pattern recognition receptors (PRRs) on innate immune cells. Subsequently, chemokines (e.g., CCL3, CCL5, CXCL8, CXCL9) and proinflammatory cytokines (e.g., type I IFNs, interleukin (IL)-12, GM-CSF, TNF-α) are released to recruit and activate more innate immune cells such as macrophages, neutrophils, dentritic cells (DCs) and T-cells in the TME to eliminate the infected tumor cells, which can further stimulate the production of proinflammatory cytokines and chemokines to amplify the initial innate response (28).
In addition, accumulating evidences suggest that oncolytic VACVs induce immunogenic cell death (ICD) in infected cancer cells with the release of damage-associated molecular pattern molecules (DAMPs), such as the “danger” signals high mobility group box 1 (HMGB1), the “find me” signal adenosine triphosphate (ATP) and the “eat me” signal calreticulin (CRT) (29). The DAMPs can activate the immature DC (iDC) into the mature DC (mDC). mDCs are the main antigen presentation cells that present the released tumor-associated antigens (TAAs), and tumor-associated neoantigens (TANs) after oncolysis to T-cells, resulting in activation, proliferation and differentiation of T-cells (30). Effector T-cells exert specific tumor killing, meanwhile memory T-cells are responsible for long-term and distant anti-tumor effect. Thus, adaptive immune responses to oncolytic VACV-infected tumor cells are activated.
It can be seen from the clinical data reported by Samson et al. that JX-594 infusion contributed to a significant increase of IFN and neutralizing antibodies in plasma for viral clearance. Meanwhile, many other proinflammatory and chemokines such as IL12, CXCL10, CXCL2 also increased, leading to the activation of natural killer (NK) cells, and the recruitment and activation of T-cells and DCs (31).
As stated above, oncolytic VACV acts as a bait in the tumor to increase the number of immune cells and proinflammatory cytokines and chemokines in the TME, resulting in the amplification of the anti-tumor immune response and remodeling immunologically “cold” tumors into “hot” tumors. However, immune cells in the TME may be detrimental to effectiveness of OVT by eliminating virally infected cells. Antiviral immunity is a double-edged sword that needs to be well-balanced in tumor therapy (32). It is reflected not only in the tumor tissues, but in the blood circulation when intravenously administrating reovirus or coxsackievirus according to the study from Berkeley et al. (33). They found that neutralizing antibodies had an actual beneficial effect on OVT by forming virus/neutralizing antibody complexes which can be internalized by monocytes and delivered to tumor sites in spite of viral clearance. This finding is very exciting and beneficial for the understanding and application of OVT in future. However, whether this phenomenon exist in the oncolytic VACVs treatment still needs to be explored.
3 Construction of oncolytic VACV backbones
Compared with non-replicative VACVs, replication competent VACVs retain their ability to lyse tumor cells and spread through tumor tissues while the lower safety and efficacy limited the application. With the ever-increasing knowledge in the fields of molecular virology and cancer cell biology, engineered oncolytic VACVs can be obtained through the DNA recombinant technology (e.g., CRISPR-Cas9 system) (34). The multiple safe and efficacious tumor-targeted oncolytic VACV backbones have been developed in recent years via genome editing. Based on the function of the deletion genes, they are mainly classified into three types: viral replication-related, viral dissemination-related and antiviral immune evasion-related. The functions of some VACV genes are listed in the Table 2.
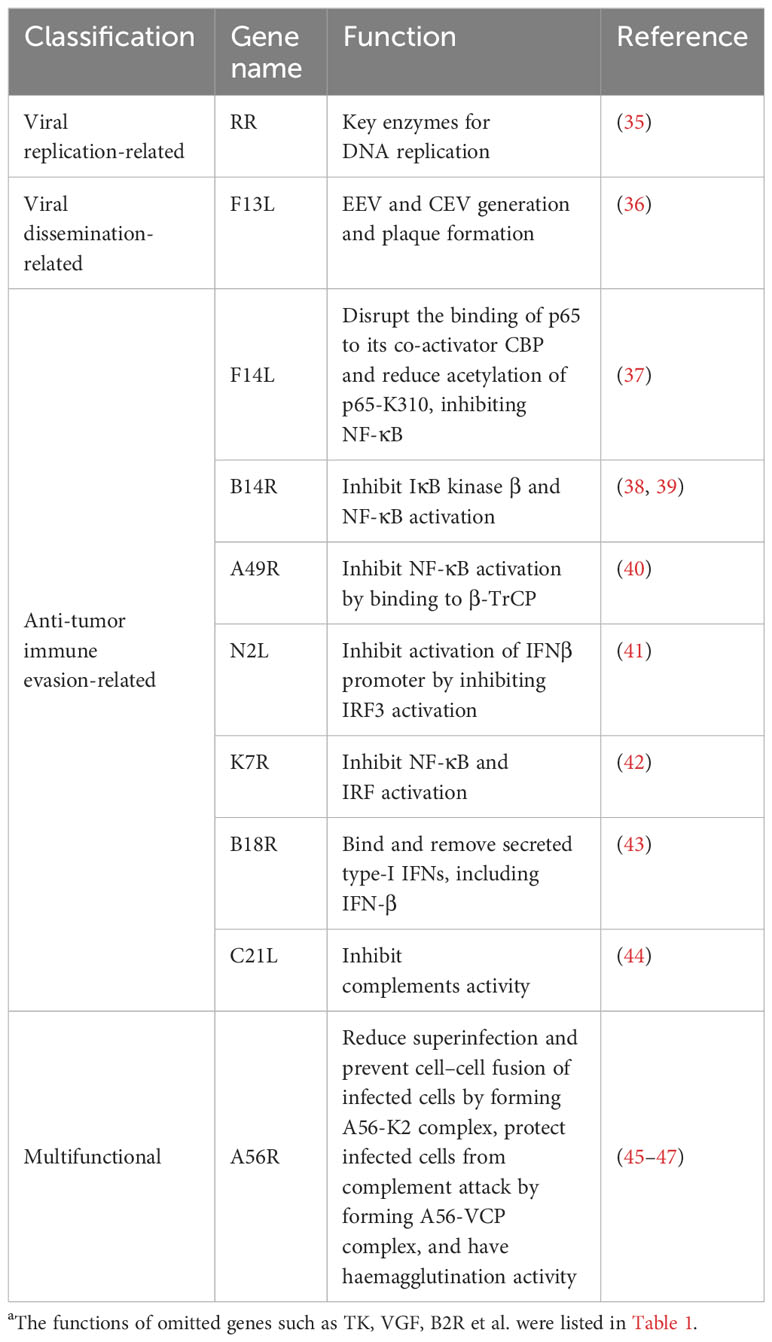
Table 2 Functions of some VACV genesa.
Targeting the overexpressed proteins [e.g., TK and ribonucleotide reductase (RR)] in tumor cells that are necessary for viral replication is the common method to increase the safety of oncolytic VACVs (22, 35). JX-594 is a typical TK-deleted oncolytic VACV with confirmed safety in phase I clinical trials (e.g., NCT02977156 and NCT01394939). Double deletion of TK and RR also showed enhanced safety and selective replication in the preclinical studies (48).
Deletion of viral virulence genes can further enhance the safety of oncolytic VACVs. VACV virulence is related to viral replication, dissemination, and antiviral immune evasion. Viral growth factors (VGF) is an EGF homologue that activates EGFR-Ras pathway to promote cell proliferation. Studies have shown that deletion of VGF contributes to low replication and virulence (49, 50). Double deletion of TK and VGF led to significantly attenuated virulence in resting cells in vitro and tumor-specific replication in vivo (51). As mentioned previously, EEV and CEV forms of VACVs are responsible for viral dissemination in vivo. Deletion of the vaccinia virus F13L gene results in a highly attenuated virus that is defective in EEV and CEV generation and plaque formation (36).
Antagonizing antiviral immunity is an important way for oncolytic VACVs to potentiate virulence. cGAS/STING pathway plays a central role in immune defense against tumors and viral infections, where TANK-binding kinase 1 (TBK1) recruitment to STING further activates both NF-κB and interferon regulatory factor 3 (IRF3) (52). cGAS is a DNA sensor for host defense against VACV infection. Vaccinia B2R gene was recently discovered to encode a cytosolic cGAMP nuclease that obstruct the cGAS signal. Vaccinia E5 is another virulence factor that inhibits cGAS by abolishing cGAMP production. Studies showed that deletion of B2R or E5R gene made the virulence of VACVs attenuated and anti-tumor immunity enhanced (53, 54).
In addition, inhibition of NF-κB signaling and/or IFR signaling cannot only reduce viral virulence, but enhance anti-tumor immunity. F14 is a selective NF-κB inhibitor in the nucleus by disrupting the binding of p65 to its co-activator CBP and reducing acetylation of p65-K310 and a VACV strain lacking F14 has reduced virulence in a mouse model (37). There are many other proteins that show antagonism of NF-κB activation with different action sites. For instance, B14 interacts directly with IκB kinase β to inhibit its activation, and A49 inhibits NF-κB activation by binding to β-TrCP, contributing to virus virulence (38–40). Additionally, N2 is a nuclear virulence factor that inhibits activation of IFNβ promoter by inhibiting IRF3 activation, deletion of which reduced virulence of VACVs (41). Interestingly, some proteins (e.g., N1 and K7) have dual functions of inhibiting NF-κB and IRF activation (42, 55). Studies proved that deletion of the N1L gene reduced virulence and inhibited VACV replication, meanwhile it improved the generation of immediate and long-term memory CD8+ T-cell responses and induced a stronger NK cell response to infection (56, 57). Moreover, deletion of B8R or B18R that directly binds to several species of IFN and neutralizes the antiviral activity also makes oncolytic VACV attenuated for mice (43, 58).
The activation of complement system is another key innate immune defense against viral infection through classical and alternative pathways. Isaacs and coworkers demonstrated that VACV complement-control protein (VCP) could prevent antibody-dependent complement-enhanced neutralization of infectivity and contribute to virulence, as VCP gene (C21L) knockout viruses were attenuated in an intradermal rabbit model (44).
Notably, one gene may be involved in complex biological processes. For example, the VACV A56 protein with haemagglutination activity is able to bind two other viral proteins, a serine protease inhibitor (K2) and VCP, and express them at the surface of the infected cell. The A56–K2 complex binds to the entry–fusion machinery of VACV; reducing superinfection and preventing cell–cell fusion of infected cells, while the A56–VCP complex protects infected cells from complement attack (45, 46). However, the deletion of A56R did not attenuate VACVs in some cases according to the summary from DeHaven and coworkers (47). Given that A56R gene is non-essential for viral replication, it still can be used as a region of VACV genome suitable for exogenous gene insertion. GL-ONC1 is a typical VACV that inserts β-glucuronidase into the A56R loci of VACV genome. The increasing understanding of the multiple functions of VACV genes is conducive to optimize the VACV backbones.
4 Oncolytic VACV-based immune-related combination therapies
OVT is an effective form of immunotherapy that has been used to treat cancer. As previously mentioned, OVs can selectively kill tumor cells, activate anti-tumor immunity and remodel the “cold” TME to “hot”. However, OV-based monotherapy has restricted ability to activate anti-tumor immunity, given the potential antiviral machinery induced by activation of the IFN signaling pathway and the highly variable heterogeneity of malignant cells. In this section, strategies augmenting anti-tumor immune responses via synergistic therapies are summarized (Figure 2).
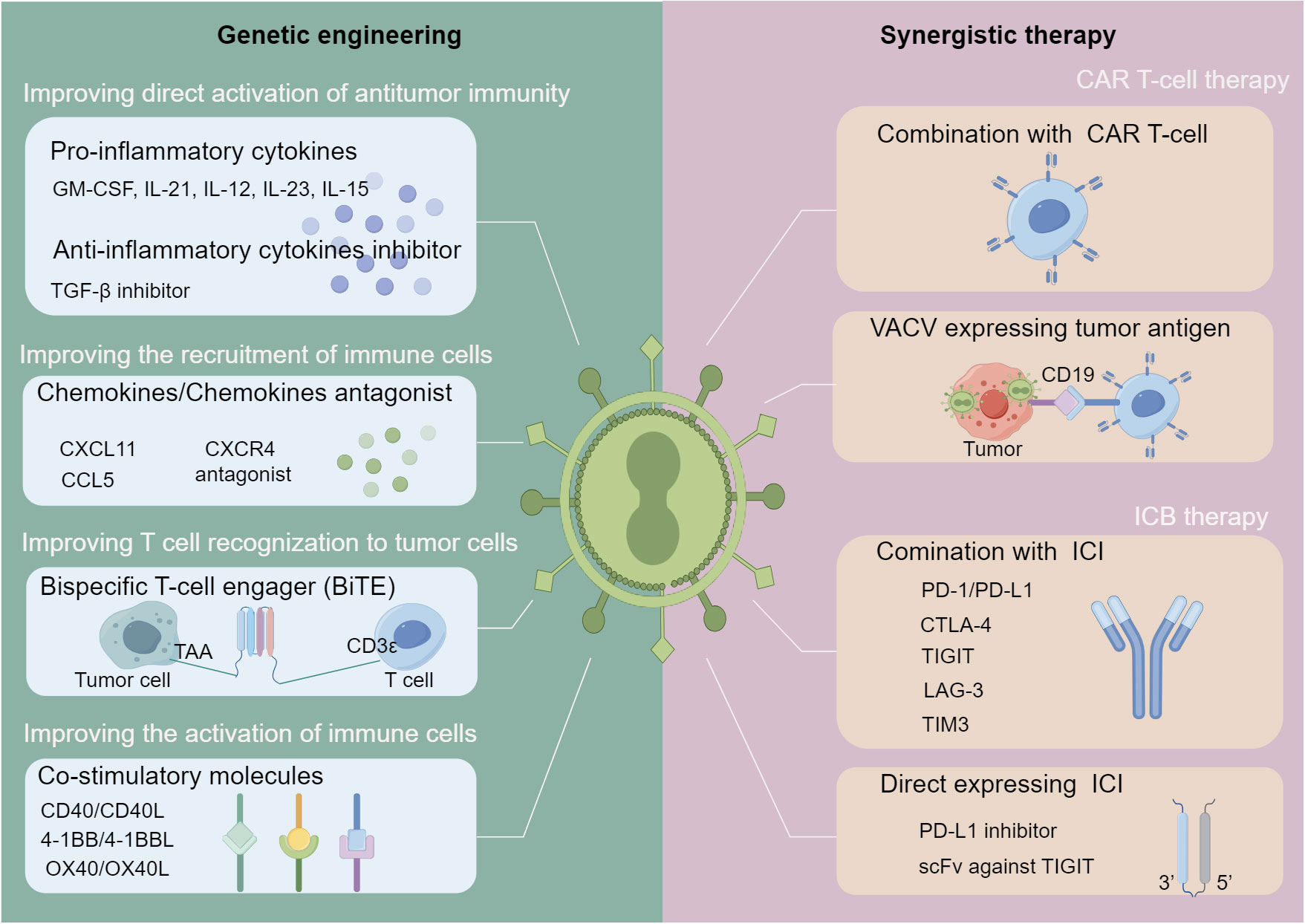
Figure 2 Strategies for oncolytic VACV-based immune-related combination therapies. Strategies are divided into two main categories: genetic engineering of oncolytic VACVs and combination with other immunotherapies. In terms of genome engineering (left), various immunostimulatory genes can be inserted into the genome of oncolytic VACVs, including those can activate the anti-tumor immunity directly, improve the T-cells recognition to cancer cells, recruit and activate immune cells. In terms of synergistic immunotherapies (right), oncolytic VACVs can be combined with CAR T-cell or ICI molecules directly or express CAR or ICI to enhance the synergistic therapies. oncolytic VACV, Oncolytic vaccinia virus; TAA, Tumor associated antigen; BiTE, Bispecific T-cell engager; CAR, Chimeric antigen receptor; ICI, Immune checkpoint inhibitor; scFv, Single chain variable fragment. The figure is drawn by FigDraw.
4.1 Oncolytic VACVs encoding immunostimulatory genes
With the ever-increasing knowledge of immune regulation mechanisms in tumor tissues, some immunostimulatory genes have been inserted into the genome of VACVs to amplify the anti-tumor immunity of oncolytic VACVs. Anti-tumor immunostimulatory factors include cytokines, chemokines, co-stimulatory factors and so on. Inflammatory cytokines are soluble proteins with the function of regulating innate and adoptive immunity, including pro-inflammatory and anti-inflammatory cytokines.
Arming pro-inflammatory cytokines is a usual way to enhance OVT by recruiting, activating immune cells and inhibiting immunosuppressive cells. GM-CSF is one of the most widely used cytokines that improve OVT. It recruits both DCs and NK cells to promote the maturation of DCs, which in turn, activates anti-tumor immunity (59). T-VEC, a GM-CSF armed HSV, has been approved in the United States for melanoma in 2015. JX-594, a genetically modified VACV that inserts the GM-CSF gene also has shown promising anti-tumor ability in clinical trials. IL-21 arming potentiated the anti-tumor activity of oncolytic VACVs by increasing effector CD8+ T-cell populations (60, 61). Some pro-inflammatory cytokines (e.g., IL-12, IL-23, IL-15) armed oncolytic VACVs showed stronger anti-tumor immune response by enhancing T-cell and NK cell activation and cytotoxicity in addition to increasing the production of IFN-γ (62–64). Other cytokines that have been used to engineer oncolytic viruses such as adenovirus and HSV may also enhance the anti-tumor effect of oncolytic VACVs through rational design (65). What is noteworthy is that assessment of actual exposure to potential payloads and management of safety issues that may arise from these payloads need to be considered when engineering oncolytic VACVs.
Owing to the important role of anti-inflammatory cytokines (e.g., TGF−β, IL-10) in host immune response, they are also designed to improve the therapeutic efficacy of OVT. Delgoffe and coworkers found that oncolytic VACV-delivered TGFβ inhibitor could overcome the immunosuppressive tumor microenvironment by blocking the immunosuppressive function of TGF−β, and increasing the sensitivity to INF-γ (66). The IL-10 is a recognized immunosuppressive cytokine that inhibits the production of pro-inflammatory cytokines such as IL-12 and INF-γ (67). Hickman and coworkers demonstrated that locally produced IL-10 after VACVs infection limited VACVs replication and dissemination (68). Another study from Wang and coworkers revealed IL-10 could also enhance the anti-tumor efficacy of oncolytic VACVs in pancreatic cancer by dampening antiviral immune response, and prolonging viral persistence in tumors (69).
Chemokines are secreted chemotactic cytokines that attract immune cells into tumor lesions and mediate anti-tumor immune effects. Bartlett’s group demonstrated that oncolytic VACVs encoding CCL5 or CXCL11 elicited potent anti-tumor immunity and enhanced therapeutic efficacy by attracting activated immune cells such as T (Th1) and NK cells (70, 71). Generally, chemokine receptors (e.g., CXCR4) help activate the immune system, while the regulation imbalance of signaling pathway promote the tumor growth and metastasis. CXCR4 antagonist armed oncolytic VACVs showed the ability of TME modulation such as inhibiting intratumoral accumulation of immunosuppressive myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (Tregs) (72).
In addition, oncolytic viruses can be modified to express co-stimulatory molecules of T-cell, thereby enhancing T-cell-mediated anti-tumor immunity. ONCOLYTIC VACVs expressing co-stimulatory molecules (e.g., CD40L, 4-1BBL, OX40L) boosted anti-tumor immune responses by activating antigen presentation cells and T lymphocytes, and reprogramming Treg (73–75). Bispecific T-cell engager (BiTE) further improved the anti-tumor activity of oncolytic VACVs by expressing tumor specific antigen in tumor cells and redirecting T-cells to the tumor (76).
Apart from the immune stimulators, some genes associated with other signal pathways are also found to activate anti-tumor immunity. For example, expression of DNA-dependent activator of IFN-regulatory factors (DAI, a cytosolic dsDNA sensor) by an oncolytic VACV boosted innate immune activation and enhanced anti-tumor immunity (77). Oncolytic VACVs expressing White-Spotted Charr Lectin not only induced type I IFN production to elicit anti-tumor activity, but also inhibited IFN-induced ISG production, helping oncolytic VACVs escape elimination (78).
Taken together, oncolytic VACVs encoding genes that induce anti-tumor immune responses have the potential to promote stronger anti-tumor effect.
4.2 Oncolytic VACVs and chimeric antigen receptor (CAR) T-cell therapy
CAR T-cell therapy is a kind of adoptive immunotherapy aimed at augmenting the anti-tumor immune responses of T-cell. In CAR T-cell therapy, one or more CARs are expressed for both antigen-binding and T-cell activating, bypassing the antigen processing and presentation (79). Currently, two products targeting CD19-CAR (Tisagenlecleucel and Axicabtagene Ciloleucel) have been approved to treat hematologic malignancies, and many other CARs are being developed for solid tumors (80). However, the application of CAR T-cells in solid tumors faces several challenges, such as the reduced persistence and expansion of CAR T-cells in immunosuppressive TME and the antigen loss/escape (81, 82).
It is now recognized that oncolytic VACVs can activate the anti-tumor immune response and remodel the “cold” TME to “hot”. The enhanced type I IFN signature and the released DAMPs and cytokines in the TME after oncolysis of oncolytic VACVs can further support the priming, proliferation, clonal expansion, effector function and/or memory formation of CD8+ T-cells, enabling the anti-tumor effect of CAR T-cells (83). For example, the combination of HER-2-CAR T-cell and oncolytic VACV with deletion of TK and VGF showed a significant enhancement of tumor killing in the murine breast cancer (D2F2) cell line (84).
To enhance the tumor targeting of CAR T-cells, Priceman’s group designed an oncolytic VACV (OV19t) encoding a non-signaling, truncated human CD19 (CD19t) protein in which highly expressed CD19t on the surface of tumor cells worked as a specific tumor antigen to enable the tumor targeting of CD19-CAR T-cells (85). The combination therapy of OV19t/oncolytic viruses encoding the murine CD19t (OVm19t) protein and CD19-CAR T-cells showed durable and effective anti-tumor effects in MDA-MB-468/U251T bearing human tumor xenograft models and a MC38 bearing immunocompetent murine syngeneic tumor models. Another study from Aalipour et al. also demonstrated that the combination therapy of CD19 encoding oncolytic VACVs with CD19-CAR T-cells significantly reduced tumor growth and improved median survival achieved the similar results in an immunocompetent model of B16 melanoma (86). In addition, oncolytic VACVs encoding CXCL11 can further enhance the anti-tumor effect of mesothelin-CAR T-cell therapy via CXCL11-mediated recruitment of T-cells (87).
Taken together, oncolytic VACVs can boost the CAR T-cell therapy by the expression of tumor antigen or immunostimulatory factors and activation of anti-tumor immune responses themselves.
4.3 Oncolytic VACVs and immune checkpoint blockade therapy(ICB)
ICB therapy is an emerging immunotherapy aiming at blocking immunosuppressive tumor signals and restoring anti-tumor immune responses by targeting checkpoint receptors or ligands such as PD-1/PD-L1, CTLA-4, TIGIT, LAG-3, TIM3, among which PD-1/PD-L1 is the most common research object. To date, several immune checkpoint inhibitors (ICIs) have been approved for tumor treatment, including PD-1 inhibitors (nivolumab, pembrolizumab, cemiplimab), PD-L1 inhibitors (avelumab, durvalumab, atezolizumab), and CTLA-4 inhibitors (ipilimumab). However, the efficiency of ICIs is restricted by the immunosuppressive TME and low expression of immune checkpoint molecules.
Oncolytic VACVs cannot only recruit immune cells into immunodeficient tumors and remodel the “cold” TME to “hot”, but increase the expression of ICIs such as PD-L1, CTLA-4 or TIGIT inhibitors, sensitizing tumors to ICB therapy. The combination of CXCL11 encoded oncolytic VACV and PD-L1 blockade synergistically enhanced the therapeutic efficacy by increasing T-cell infiltration into the tumor and upregulating the expression of PD-L1 (88). The manganese superoxide dismutase (MnSOD) or immunostimulatory cytokines (such as IL-21, IL-15) expressing oncolytic VACVs could also improve PD-1/PD-L1 inhibition outcome by remodeling the suppressive tumor microenvironment (89–91). In addition, oncolytic VACVs have been engineered to directly express ICIs. For instance, oncolytic VACVs encoding a single chain variable fragment against TIGIT induced effective anti-tumor immunity and achieved a profound remodeling of the inhibitory tumor microenvironment from a “cold” state to a “hot” state synergizing with PD-1 or LAG-3 blockade (92). Oncolytic VACVs co-expressing ICIs and immune stimulatory cytokines exhibited synergistic anti-tumor ability. Oncolytic VACVs expressing PD-L1 inhibitors and GM-CSF activated tumor neoantigen-specific T-cell responses by synergistic action of VACV replication, GM-CSF stimulation, and PD-L1 inhibition on tumor cells and immune cells (93). Oncolytic VACVs expressing CTLA-4 inhibitors and GM-CSF elicited robust systemic CD8+ T-cell-dependent anti-tumor immunity and long-lasting anti-tumor immunity by expanding peripheral effector CD8+ T-cells and reducing Tregs and exhausted CD8+ T-cells (94).
Despite potential synergistic effect of oncolytic VACVs and ICIs, it is worth noting that ICIs may stimulate OVs clearance by activating host immune response. Thus, the optimal administration timing should be considered. Nguyen and coworkers concluded that OV lead-in → anti-PD-1 or OV lead-in → concurrent therapy could be the treatment option for tumor regression and eradication according to PubMed literature search results (95).
5 Other oncolytic VACV-based combination therapies
5.1 Oncolytic VACVs and radiotherapy (RT)
Radiotherapy (RT) is a type of conventional treatment for cancer by inducing DNA damage and apoptosis. With continuous exploration, combination of RT and oncolytic VACVs are recognized as a potent treatment modality for cancer. Focally irradiation in tumor tissues can facilitate the replication of systemically delivered oncolytic VACVs (96). In turn, VACVs encoding the sodium iodide symporter (NIS) can transfer radioactive iodine into virus-infected cancer cells, contributing to enhanced anti-tumor effects of RT in pancreatic cancer and breast cancer (97). Inspired by the theory of peptide-receptor radiotherapy (PRRT), and the ability of selected replication and exogenous gene expression of oncolytic VACVs, McCart and coworkers found that VACVs encoding somatostatin receptor (SSTR) led to virus-directed tumor-specific accumulation of radiopeptides, enabling the imaging and improved treatment of intraperitoneal CRC tumors using 177Lu-DOTATOC (98). In addition, studies have shown that radiation combined with oncolytic VACVs displayed considerably superior anti-tumor efficacy in several tumor models such as glioblastoma and pancreatic cancer (99, 100). For the synergistic mechanisms, a study from Chen et al. showed that RT combined with oncolytic VACVs could trigger tumor cell necroptosis and modify macrophages through the release of DAMPs, generating potent anti-tumor immunity and enhanced anti-tumor efficacy (101). Another study from Kyula et al. illustrated that synergistic cytotoxicity of radiation and oncolytic Lister strain VACV in V600D/E BRAF mutant melanoma depended on JNK and TNF-α signaling (102). Beside preclinical studies, phase II study about the combination of oncolytic VACV (GL-ONC1) with radiation and chemotherapy is warranted after phase I study in patients with locoregionally advanced head and neck carcinoma (103). All above studies proved the potential of the combination of RT and oncolytic VACVs.
5.2 Oncolytic VACVs and chemotherapy
Chemotherapy is the most widely used approach for tumor therapy due to the efficiency and broad spectrum. Chemotherapeutic drugs are cytotoxic agents that act primarily by inhibiting DNA replication or disrupting microtubule structures (104). Some of these cytotoxic agents have been extensively studied in combination with oncolytic VACVs and achieved many positive results. Yu and coworkers found that the anti-tumor efficacy of oncolytic VACV (GLV-1h68) was enhanced by cisplatin or gemcitabine with several potential mechanisms such as the changes in apoptosis, nucleotide pools and DNA repair pathways (105). As seen from the results of a phase II non-randomized clinical trial (NCT05281471) in patients with platinum-resistant or platinum-refractory ovarian cancer (PRROC), oncolytic VACV (Olvi-Vec) in combination with platinum-based chemotherapy demonstrated a favorable objective response rate (ORR) and progression-free survival (PFS) with a manageable safety profile in patients, and that patients are being recruited for phase III trial (NCT05281471) (106, 107). Cyclophosphamide (CPA) enhanced the replication of oncolytic VACVs by transiently suppressing the anti-viral immune response, while another study revealed that the enhanced anti-tumor efficacy of combining oncolytic VACV (GLV-1h68) with CPA was due to an effect on the vasculature rather than an immunosuppressive action of CPA (108, 109). In addition, Thorne’s group for the first time revealed that oncolytic VACVs could further sensitize tumor cells to chemotherapy such as anti-microtubule agent paclitaxel by inducing the release of several cytokines including type I IFN and HMGB1 (110). Some other chemotherapeutic drugs might boost OVT by synergistically activating ICD-mediated anti-tumor immunity such as doxorubicin (111).
Arming VACVs with a prodrug-activator gene is another approach to augment synergistic anti-tumor effects of OVT and chemotherapy with less systemic toxicity. Seubert and coworker demonstrated that GLV-1h68, an engineered oncolytic VACVs encoding β-galactosidase, exhibited enhanced oncolysis and tumor shrinkage with a β-galactosidase-activatable prodrug (112). GLV-1h94 encoding supercytosine deaminase (SCD) increased the cell specific-sensitivity of chemotherapeutic compound 5-fluorouracil (5-FU) by converting the prodrug 5-fluorocytosine (5-FC) to 5-FU only in the oncolytic VACV infected tumor cells (113). The addition of 5-FC made 85% of the cell lines highly sensitive to the combination treatment, none of which tested exhibited a “highly resistant” pattern. Nevertheless, the converted 5-FU reduced the replication of GLV-1h94 in tumor cells. The balance between cell line-specific susceptibility to GLV-1h94-induced oncolysis and 5-FU sensitivity should be taken into consideration.
5.3 Oncolytic VACVs and molecular targeted therapy
Different from chemotherapy, molecular targeted therapy, also known as “bio-missile”, is aimed at precisely killing tumor cells by targeted selection of blockers for certain key molecules that are overexpressed in tumor cells but not in normal cells. Given the lower toxicity, molecular targeted therapy has become a promising approach for tumor treatment.
Studies have shown that molecular targeted therapy can amplify the anti-tumor efficacy of oncolytic VACVs through several mechanisms such as evading antiviral immunity and increasing anti-tumor immunity. Inhibition of MEK-ERK pathway enhanced oncolytic VACVs accumulation in doxorubicin-resistant ovarian cancer by abrogating cytosolic DNA sensing and viral defense (114). Transient inhibition of PI3Kδ or COX-2 enabled repeated administration of oncolytic VACVs via inhibiting the monocyte uptake of VACVs or decreasing the production of neutralizing antibodies against oncolytic VACVs, with no effect on virus replication, respectively, enhancing the anti-tumor efficacy of IV-delivered oncolytic VACVs (115, 116). Histone deacetylase inhibitors (HDIs) and STAT3 inhibitors were found to enhance the spread and replication of oncolytic VACVs selectively and effectively in tumor cells by dampening innate antiviral immune response (117). What’s more, addition of a multitargeted receptor tyrosine kinase inhibitor sunitinib also amplified the anti-tumor effects of JX-594, by improving anti-tumor immune responses such as increasing CD8+ T-cell recruitment and decreasing Tregs and MDSCs (118). It is worth mentioning that the administration sequence should be considered because some drugs influence viral replication. For example, if given simultaneously in vitro, sorafenib could inhibit JX-594 replication (119). Hence, in the phase III trial of JX-594, sorafenib was administrated after JX-594 to avoid its antiviral effect. Oncolytic VACVs encoding GLAF-1 exhibited significantly enhanced therapeutic efficacy by directing against VEGF (120).
In a word, the combination of molecular targeted therapy and oncolytic VACVs will be a promising new anticancer therapy. More available molecules for tumor targeting and corresponding drugs need to be developed to enhance OVT.
6 Clinical trials with oncolytic VACVs
As the exciting anti-tumor research results increase, some potential engineered Oncolytic VACVs are warranted for clinical trials. The current clinical trials about replication-competent oncolytic VACVs are found from the website of https://clinicaltrials.gov, and listed in the Table 3. TK gene deletion is the most common way to increase the selectivity of oncolytic VACVs and different therapeutic genes are inserted for better anti-tumor effect. Pexa-Vec and GL-ONC1 are the only two oncolytic VACVs that are already in phase III clinical trials, indicating the safety and efficacy in some cancer patients. Unfortunately, the phase III trial of Pexa-Vec for hepatocellular carcinoma was declared a failure because the interim analysis of the study showed that it was unlikely to prolong the overall survival of patients with Pexa-Vec treatment. Perhaps other combination therapies such as ICB with OVT will exert promising anti-tumor effects.
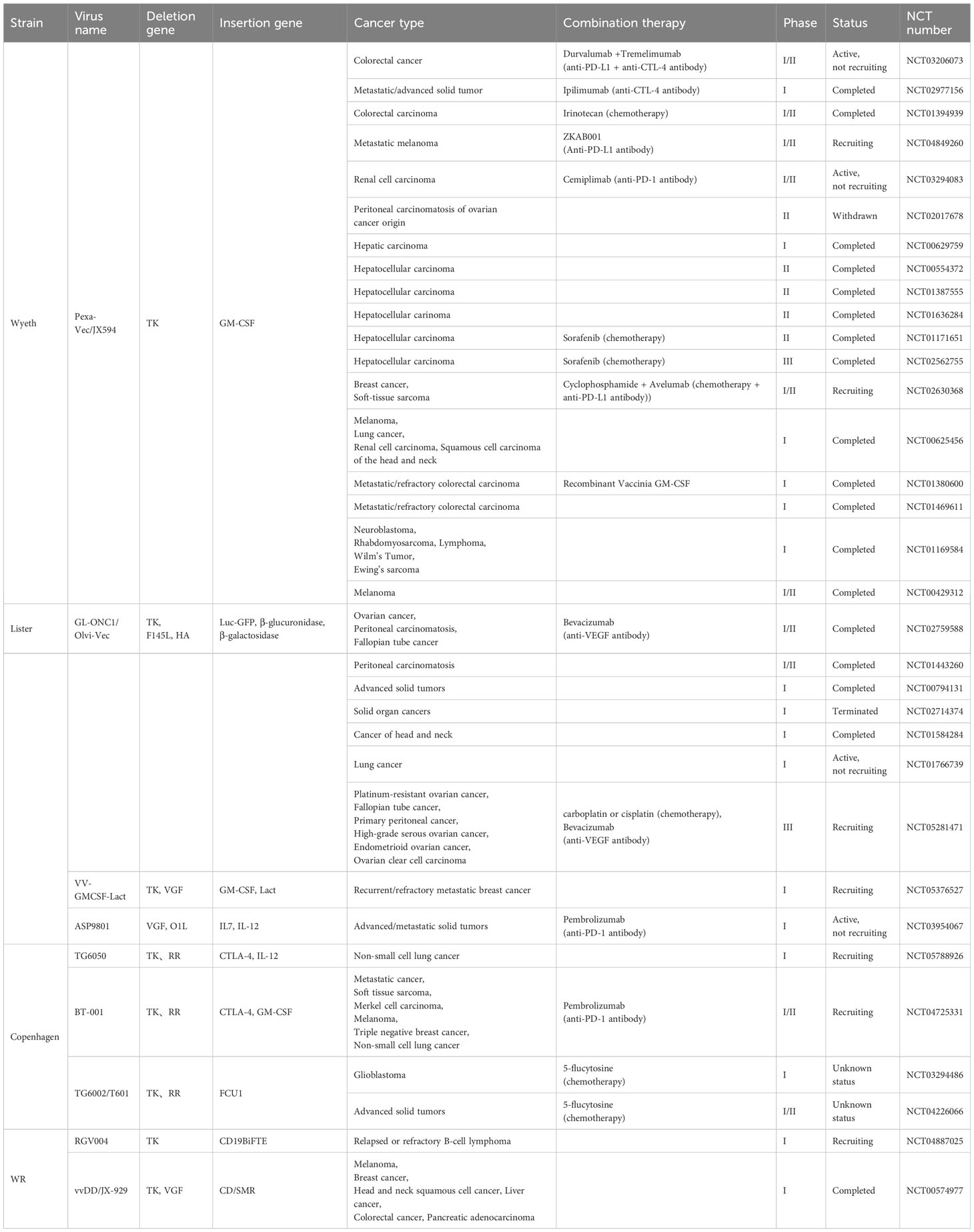
Table 3 Current clinical trials with replication-competent oncolytic VACVs.
7 Delivery route of oncolytic VACVs
Up to now, all the approved OVs are intratumorally injected, which renders them ineffective for cancers that are difficult for in situ administration or have been already metastatic. Developing intravenous OVs is essential to broaden the clinical applications of OVs. The data about 97 independent clinical trials reporting OV studies from 2000 to 2020 showed that the most common route was intratumoral delivery used in 48 of the clinical trials (49.5%) followed by intravenous delivery used in 34 of the clinical trials (35%) (121). In the clinical trials, intravenous oncolytic VACVs exhibit good safety and potential anti-tumor activity. Different from adenoviruses, oncolytic VACVs can be administrated once via intravenous injection because there are no preexisting neutralizing antibodies in most of human bodies. However, after oncolytic VACV treatment, neutralizing antibodies generation occurred, limiting the repeated systematic delivery.
Recently, researchers are dedicated to developing diverse approaches to realize the efficient tumor treatment by repeated administration of intravenous oncolytic VACVs. Ferguson et al. found that transient inhibition of PI3Kδ by the PI3Kδ-selective inhibitor IC87114 or the clinically approved idelalisib (CAL-101) prior to intravenous delivery of a tumor-tropic VACV could inhibit viral attachment to, but not internalization by, systemic macrophages through perturbation of signaling pathways involving RhoA/ROCK, AKT, and Rac, thus potentiating intravenous delivery of oncolytic VACVs to tumors (115). In addition, COX-2 inhibitor treatment can enhance the long-term protective anti-tumor effects generated by oncolytic VACVs via inhibiting the generation of neutralizing antibodies against oncolytic VACVs infection, enabling the repeated administration of oncolytic VACVs (116). Another study from McCart’s group demonstrated that CP40 (a complement inhibitor) pretreatment elicited an average 10-fold increase in infectious titer in the blood early after the JX-594 infusion by preventing oncolytic VACVs neutralization, beneficial for repeated intravenous administration of oncolytic VACVs (122). The expression of human CD55 protein by oncolytic VACVs could also prolonged viral survival by protecting against complement-mediated lysis and evading neutralization by VACV-specific antibodies, improving intravenous efficacy (123). All above approaches are theoretically and clinically feasible via antiviral inhibitors treatment prior to intravenous injection of oncolytic VACVs.
Besides inhibition of antiviral immunity, direct oncolytic VACVs shielding is another alternative to enhance the anti-tumor efficacy of intravenous oncolytic VACVs. As a non-enveloped virus, adenovirus has been delivered by many kinds of cell carriers (e.g., mesenchymal stem cells, erythrocytes), nanomaterials (e.g., liposomes, exosomes) or polymers (e.g., poly(ethylene glycol) (PEG), polyamidoamine) to improves the pharmacokinetics (124, 125). Hill et al. found that polymer (Chol-PEG(10K)-NHS) coating reduced the binding of neutralizing anti-VACV antibodies and oncolytic VACVs and increased the circulation time in vivo, although VACV is an enveloped virus (126). These results showed that VACV coating may be an effective way to improve the systematic delivery of oncolytic VACVs. Many other kinds of methods for VACV coating need to be developed.
8 Conclusions and prospects
With the continuous improvement of gene function and anti-tumor mechanism of oncolytic VACVs, a variety of genetically functional VACVs have been constructed and shown safety and efficacy in preclinical and clinical studies. In addition, VACV-based OVT has been acknowledged as a potential adjuvant immunotherapy when combining with traditional anti-tumor therapies. However, challenges remain in the clinical application of engineered oncolytic VACVs. Firstly, failure of the phase III trial about JX-594 reminds us the importance of selecting proper combination therapy, and ensuring the administration sequence and timing. In addition, the two sides of oncolytic VACV-mediated antiviral immunity should be overall considered during the construction of VACV backbones and gene engineering of oncolytic VACVs. Another challenge is repeated intravenous injection of oncolytic VACVs with lower dosages. It is acknowledged that natural tumor tropism and none of initial neutralizing antibodies make VACVs potential for a single intravenous injection, while repeated administration can activate strong antiviral immunity to eliminate VACVs, indicating that higher dosage is needed to achieve anti-tumor effect. With the discovery of diverse signal pathways that are related to antiviral immunity, some drugs show the positive effect on the VACVs circulation in vivo, facilitating the progress of VACVs systematic administration. While much work remains, lots of studies have shown the enormous potential of VACV-based OVT. We believe it will not be long before oncolytic VACVs are approved for clinical anti-tumor therapy.
Author contributions
LX: Conceptualization, Writing – original draft. HS: Writing – review & editing. NL: Writing – review & editing. YX: Writing – review & editing, Supervision. PW: Supervision, Writing – review & editing, Conceptualization, Funding acquisition, Project administration.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Natural Science Foundation of Henan Province for Excellent Young Scholars (202300410360, PW), the National Natural Science Foundation of China (81872486, PW), and the National Key Research and Development Program of China (2019YFC1316101, PW).
Acknowledgments
The authors would like to thank Home for Researcher for preparation of Figures 1, 2 (www.home-for-researchers.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer (2023) 9(2):122–39. doi: 10.1016/j.trecan.2022.10.003
2. Tian Y, Xie D, Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduction Targeted Ther (2022) 7(1):117. doi: 10.1038/s41392-022-00951-x
3. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (1991) 252(5007):854–6. doi: 10.1126/science.1851332
4. Wang L, Chard Dunmall LS, Cheng Z, Wang Y. Remodeling the tumor microenvironment by oncolytic viruses: beyond oncolysis of tumor cells for cancer treatment. J Immunother Cancer (2022) 10(5):e004167. doi: 10.1136/jitc-2021-004167
5. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug Discovery (2015) 14(9):642–62. doi: 10.1038/nrd4663
6. Duan S, Wang S, Qiao L, Yu X, Wang N, Chen L, et al. Oncolytic virus-driven biotherapies from bench to bedside. Small (2023) 19(23):e2206948. doi: 10.1002/smll.202206948
7. Thèves C, Crubézy E, Biagini P. History of smallpox and its spread in human populations. Microbiol Spectr (2016) 4(4). doi: 10.1128/microbiolspec.PoH-0004-2014
8. Haddad D. Genetically engineered vaccinia viruses as agents for cancer treatment, imaging, and transgene delivery. Front Oncol (2017) 7:96. doi: 10.3389/fonc.2017.00096
9. Park SH, Breitbach CJ, Lee J, Park JO, Lim HY, Kang WK, et al. Phase 1b trial of biweekly intravenous pexa-vec (Jx-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther (2015) 23(9):1532–40. doi: 10.1038/mt.2015.109
10. Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer (2009) 9(1):64–71. doi: 10.1038/nrc2545
11. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer (2019) 7(1):6. doi: 10.1186/s40425-018-0495-7
12. Blasco R, Moss B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol (1992) 66(7):4170–9. doi: 10.1128/jvi.66.7.4170-4179.1992
13. Payne LG. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J Gen Virol (1980) 50(1):89–100. doi: 10.1099/0022-1317-50-1-89
14. Greseth MD, Czarnecki MW, Bluma MS, Traktman P, Sandri-Goldin RM. Isolation and characterization of Vδi3 confirm that vaccinia virus ssb plays an essential role in viral replication. J Virol (2018) 92(2):e01719–17. doi: 10.1128/jvi.01719-17
15. Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene (1983) 25(1):21–8. doi: 10.1016/0378-1119(83)90163-4
16. Yu YA, Shabahang S, Timiryasova TM, Zhang Q, Beltz R, Gentschev I, et al. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol (2004) 22(3):313–20. doi: 10.1038/nbt937
17. Hiley CT, Yuan M, Lemoine NR, Wang Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther (2009) 17(2):281–7. doi: 10.1038/gt.2009.132
18. Moss B. Poxvirus cell entry: how many proteins does it take? Viruses (2012) 4(5):688–707. doi: 10.3390/v4050688
19. Mercer J, Knébel S, Schmidt FI, Crouse J, Burkard C, Helenius A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc Natl Acad Sci (2010) 107(20):9346–51. doi: 10.1073/pnas.1004618107
20. Greseth MD, Traktman P. The life cycle of the vaccinia virus genome. Annu Rev Virol (2022) 9(1):239–59. doi: 10.1146/annurev-virology-091919-104752
21. de Queiroz NMGP, Xia T, Konno H, Barber GN. Ovarian cancer cells commonly exhibit defective sting signaling which affects sensitivity to viral oncolysis. Mol Cancer Res (2019) 17(4):974–86. doi: 10.1158/1541-7786.Mcr-18-0504
22. Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther (2000) 7(1):66–73. doi: 10.1038/sj.cgt.7700075
23. Nasri D, Manwar R, Kaushik A, Er EE, Avanaki K. Photoacoustic imaging for investigating tumor hypoxia: A strategic assessment. Theranostics (2023) 13(10):3346–67. doi: 10.7150/thno.84253
24. Liu T-C, Hwang T, Park B-H, Bell J, Kirn DH, Hu Y-C. The targeted oncolytic poxvirus jx-594 demonstrates antitumoral, antivascular, and anti-hbv activities in patients with hepatocellular carcinoma. Mol Ther (2008) 16(9):1637–42. doi: 10.1038/sj.mt.6300236
25. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res (2013) 73(4):1265–75. doi: 10.1158/0008-5472.Can-12-2687
26. Arulanandam R, Batenchuk C, Angarita Fernando A, Ottolino-Perry K, Cousineau S, Mottashed A, et al. Vegf-mediated induction of prd1-bf1/blimp1 expression sensitizes tumor vasculature to oncolytic virus infection. Cancer Cell (2015) 28(2):210–24. doi: 10.1016/j.ccell.2015.06.009
27. Santry LA, van Vloten JP, Knapp JP, Matuszewska K, McAusland TM, Minott JA, et al. Tumour vasculature: friend or foe of oncolytic viruses? Cytokine Growth Factor Rev (2020) 56:69–82. doi: 10.1016/j.cytogfr.2020.07.007
28. Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol (2018) 39(3):209–21. doi: 10.1016/j.it.2017.11.006
29. Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis (2020) 11(1):48. doi: 10.1038/s41419-020-2236-3
30. Yaghchi CA, Zhang Z, Alusi G, Lemoine NR, Wang Y. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy (2015) 7(12):1249–58. doi: 10.2217/imt.15.90
31. Samson A, West EJ, Carmichael J, Scott KJ, Turnbull S, Kuszlewicz B, et al. Neoadjuvant intravenous oncolytic vaccinia virus therapy promotes anticancer immunity in patients. Cancer Immunol Res (2022) 10(6):745–56. doi: 10.1158/2326-6066.Cir-21-0171
32. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front Immunol (2018) 9:866. doi: 10.3389/fimmu.2018.00866
33. Berkeley RA, Steele LP, Mulder AA, van den Wollenberg DJM, Kottke TJ, Thompson J, et al. Antibody-neutralized reovirus is effective in oncolytic virotherapy. Cancer Immunol Res (2018) 6(10):1161–73. doi: 10.1158/2326-6066.Cir-18-0309
34. Yuan M, Zhang W, Wang J, Al Yaghchi C, Ahmed J, Chard L, et al. Efficiently editing the vaccinia virus genome by using the crispr-cas9 system. J Virol (2015) 89(9):5176–9. doi: 10.1128/jvi.00339-15
35. Aye Y, Li M, Long MJC, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene (2015) 34(16):2011–21. doi: 10.1038/onc.2014.155
36. Vliegen I, Yang G, Hruby D, Jordan R, Neyts J. Deletion of the vaccinia virus F13l gene results in a highly attenuated virus that mounts a protective immune response against subsequent vaccinia virus challenge. Antiviral Res (2012) 93(1):160–6. doi: 10.1016/j.antiviral.2011.11.010
37. Albarnaz JD, Ren H, Torres AA, Shmeleva EV, Melo CA, Bannister AJ, et al. Molecular mimicry of nf-κb by vaccinia virus protein enables selective inhibition of antiviral responses. Nat Microbiol (2022) 7(1):154–68. doi: 10.1038/s41564-021-01004-9
38. Chen RAJ, Jacobs N, Smith GL. Vaccinia virus strain western reserve protein B14 is an intracellular virulence factor. J Gen Virol (2006) 87(6):1451–8. doi: 10.1099/vir.0.81736-0
39. Buller ML, Chen RAJ, Ryzhakov G, Cooray S, Randow F, Smith GL. Inhibition of Iκb kinase by vaccinia virus virulence factor B14. PloS Pathog (2008) 4(2):e22. doi: 10.1371/journal.ppat.0040022
40. Neidel S, Ren H, Torres AA, Smith GL. Nf-κb activation is a turn on for vaccinia virus phosphoprotein A49 to turn off nf-κb activation. Proc Natl Acad Sci (2019) 116(12):5699–704. doi: 10.1073/pnas.1813504116
41. Ferguson BJ, Benfield CTO, Ren H, Lee VH, Frazer GL, Strnadova P, et al. Vaccinia virus protein N2 is a nuclear irf3 inhibitor that promotes virulence. J Gen Virol (2013) 94(9):2070–81. doi: 10.1099/vir.0.054114-0
42. Benfield CTO, Ren H, Lucas SJ, Bahsoun B, Smith GL. Vaccinia virus protein K7 is a virulence factor that alters the acute immune response to infection. J Gen Virol (2013) 94(7):1647–57. doi: 10.1099/vir.0.052670-0
43. Kirn DH, Wang Y, Boeuf FL, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PloS Med (2007) 4(12):e353. doi: 10.1371/journal
44. Isaacs SN, Kotwal GJ, Moss B. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Proc Nati Acad Sci (1992) 89:628–32. doi: 10.1073/pnas.89.2.628
45. Wagenaar TR, Moss B. Expression of the A56 and K2 proteins is sufficient to inhibit vaccinia virus entry and cell fusion. J Virol (2009) 83(4):1546–54. doi: 10.1128/jvi.01684-08
46. DeHaven BC, Girgis NM, Xiao Y, Hudson PN, Olson VA, Damon IK, et al. Poxvirus complement control proteins are expressed on the cell surface through an intermolecular disulfide bridge with the viral A56 protein. J Virol (2010) 84(21):11245–54. doi: 10.1128/jvi.00372-10
47. DeHaven BC, Gupta K, Isaacs SN. The vaccinia virus A56 protein: A multifunctional transmembrane glycoprotein that anchors two secreted viral proteins. J Gen Virol (2011) 92:1971–80. doi: 10.1099/vir.0.030460-0
48. Delaunay T, Nader J, Grard M, Farine I, Hedwig V, Foloppe J, et al. High oncolytic activity of a double-deleted vaccinia virus copenhagen strain against Malignant pleural mesothelioma. Mol Ther–Oncolytics (2020) 18:573–8. doi: 10.1016/j.omto.2020.08.011
49. Beerli C, Yakimovich A, Kilcher S, Reynoso GV, Fläschner G, Müller DJ, et al. Vaccinia virus hijacks egfr signalling to enhance virus spread through rapid and directed infected cell motility. Nat Microbiol (2019) 4(2):216–25. doi: 10.1038/s41564-018-0288-2
50. Lai ACK, Pogo BGT. Attenuated deletion mutants of vaccinia virus lacking the vaccinia growth factor are defective in replication in vivo. Microb Pathog (1989) 6(3):219–26. doi: 10.1016/0882-4010(89)90071-5
51. McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res (2001) 61(24):8751–7.
52. Yum S, Li M, Fang Y, Chen ZJ. Tbk1 recruitment to sting activates both irf3 and nf-κb that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci (2021) 118(14):e2100225118. doi: 10.1073/pnas.2100225118
53. Riederer S, del Canizo A, Navas J, Peter MG, Link EK, Sutter G, et al. Improving poxvirus-mediated antitumor immune responses by deleting viral cgamp-specific nuclease. Cancer Gene Ther (2023) 30(7):1029–39. doi: 10.1038/s41417-023-00610-5
54. Yang N, Wang Y, Dai P, Li T, Zierhut C, Tan A, et al. Vaccinia E5 is a major inhibitor of the DNA sensor cgas. Nat Commun (2023) 14(1):2898. doi: 10.1038/s41467-023-38514-5
55. Barry M, Maluquer de Motes C, Cooray S, Ren H, Almeida GMF, McGourty K, et al. Inhibition of apoptosis and nf-κb activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PloS Pathog (2011) 7(12):e1002430. doi: 10.1371/journal.ppat.1002430
56. Jacobs N, Bartlett NW, Clark RH, Smith GL. Vaccinia virus lacking the bcl-2-like protein N1 induces a stronger natural killer cell response to infection. J Gen Virol (2008) 89(11):2877–81. doi: 10.1099/vir.0.2008/004119-0
57. Ahmed J, Chard LS, Yuan M, Wang J, Howells A, Li Y, et al. A new oncolytic vaccinia virus augments antitumor immune responses to prevent tumor recurrence and metastasis after surgery. J Immunother Cancer (2020) 8(1):e000415. doi: 10.1136/jitc-2019-000415
58. Verardi PH, Jones LA, Aziz FH, Ahmad S, Yilma TD. Vaccinia virus vectors with an inactivated gamma interferon receptor homolog gene (B8r) are attenuated in vivo without a concomitant reduction in immunogenicity. J Virol (2001) 75(1):11–8. doi: 10.1128/jvi.75.1.11-18.2001
60. Wang N, Wang J, Zhang Z, Cao H, Yan W, Chu Y, et al. A novel vaccinia virus enhances anti-tumor efficacy and promotes a long-term anti-tumor response in a murine model of colorectal cancer. Mol Ther–Oncolytics (2021) 20:71–81. doi: 10.1016/j.omto.2020.11.002
61. Chen T, Ding X, Liao Q, Gao N, Chen Y, Zhao C, et al. Il-21 arming potentiates the anti-tumor activity of an oncolytic vaccinia virus in monotherapy and combination therapy. J Immunother Cancer (2021) 9(1):e001647. doi: 10.1136/jitc-2020-001647
62. Chen L, Chen H, Ye J, Ge Y, Wang H, Dai E, et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics (2021) 11(14):6668–81. doi: 10.7150/thno.56494
63. Shakiba Y, Vorobyev PO, Yusubalieva GM, Kochetkov DV, Zajtseva KV, Valikhov MP, et al. Oncolytic therapy with recombinant vaccinia viruses targeting the interleukin-15 pathway elicits a synergistic response. Mol Ther–Oncolytics (2023) 29:158–68. doi: 10.1016/j.omto.2023.05.002
64. Ge Y, Wang H, Ren J, Liu W, Chen L, Chen H, et al. Oncolytic vaccinia virus delivering tethered il-12 enhances antitumor effects with improved safety. J Immunother Cancer (2020) 8(1):e000710. doi: 10.1136/jitc-2020-000710
65. Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther–Oncolytics (2019) 13:14–21. doi: 10.1016/j.omto.2019.03.001
66. DePeaux K, Rivadeneira DB, Lontos K, Dean VG, Gunn WG, Watson MJ, et al. An oncolytic virus–delivered tgfβ Inhibitor overcomes the immunosuppressive tumor microenvironment. J Exp Med (2023) 220(10):e20230053. doi: 10.1084/jem.20230053
67. Ouyang W, O’Garra A. Il-10 family cytokines il-10 and il-22: from basic science to clinical translation. Immunity (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
68. Sigal LJ, Cush SS, Reynoso GV, Kamenyeva O, Bennink JR, Yewdell JW, et al. Locally produced il-10 limits cutaneous vaccinia virus spread. PloS Pathog (2016) 12(3):e1005493. doi: 10.1371/journal.ppat.1005493
69. Chard LS, Maniati E, Wang P, Zhang Z, Gao D, Wang J, et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res (2015) 21(2):405–16. doi: 10.1158/1078-0432.Ccr-14-0464
70. Li J, O'Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther (2011) 19(4):650–7. doi: 10.1038/mt.2010.312
71. Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. Cxcl11-armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. OncoImmunology (2015) 5(3):e1091554. doi: 10.1080/2162402x.2015.1091554
72. Komorowski M, Tisonczyk J, Kolakowska A, Drozdz R, Kozbor D. Modulation of the tumor microenvironment by cxcr4 antagonist-armed viral oncotherapy enhances the antitumor efficacy of dendritic cell vaccines against neuroblastoma in syngeneic mice. Viruses (2018) 10(9):455. doi: 10.3390/v10090455
73. Parviainen S, Ahonen M, Diaconu I, Hirvinen M, Karttunen Å, Vähä-Koskela M, et al. Cd40 ligand and tdtomato-armed vaccinia virus for induction of antitumor immune response and tumor imaging. Gene Ther (2013) 21(2):195–204. doi: 10.1038/gt.2013.73
74. Hinterberger M, Giessel R, Fiore G, Graebnitz F, Bathke B, Wennier S, et al. Intratumoral virotherapy with 4-1bbl armed modified vaccinia ankara eradicates solid tumors and promotes protective immune memory. J Immunother Cancer (2021) 9(2):e001586. doi: 10.1136/jitc-2020-001586
75. Yang N, Wang Y, Liu S, Tariq SB, Luna JM, Mazo G, et al. Ox40l-expressing recombinant modified vaccinia virus ankara induces potent antitumor immunity via reprogramming tregs. J Exp Med (2023) 220(8):e20221166. doi: 10.1084/jem.20221166
76. Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song X-T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther (2014) 22(1):102–11. doi: 10.1038/mt.2013.240
77. Hirvinen M, Capasso C, Guse K, Garofalo M, Vitale A, Ahonen M, et al. Expression of dai by an oncolytic vaccinia virus boosts the immunogenicity of the virus and enhances antitumor immunity. Mol Ther–Oncolytics (2016) 3:16002. doi: 10.1038/mto.2016.2
78. Wang X, Zhou N, Liu T, Jia X, Ye T, Chen K, et al. Oncolytic vaccinia virus expressing white-spotted charr lectin regulates antiviral response in tumor cells and inhibits tumor growth in vitro and in vivo. Mar Drugs (2021) 19(6):292. doi: 10.3390/md19060292
79. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.Cd-12-0548
80. Choi T, Kang Y. Chimeric antigen receptor (Car) T-cell therapy for multiple myeloma. Pharmacol Ther (2022) 232:108007. doi: 10.1016/j.pharmthera.2021.108007
81. Brown CE, Mackall CL. Car T cell therapy: inroads to response and resistance. Nat Rev Immunol (2019) 19(2):73–4. doi: 10.1038/s41577-018-0119-y
82. Liu L, Qu Y, Cheng L, Yoon CW, He P, Monther A, et al. Engineering chimeric antigen receptor T cells for solid tumour therapy. Clin Transl Med (2022) 12(12):e1141. doi: 10.1002/ctm2.1141
83. Miyakoda M, Honma K, Kimura D, Akbari M, Kimura K, Matsuyama T, et al. Differential requirements for irf4 in the clonal expansion and homeostatic proliferation of naive and memory murine cd8+ T cells. Eur J Immunol (2018) 48(8):1319–28. doi: 10.1002/eji.201747120
84. VanSeggelen H, Tantalo DGM, Afsahi A, Hammill JA, Bramson JL. Chimeric antigen receptor–engineered T cells as oncolytic virus carriers. Mol Ther–Oncolytics (2015) 2:15014. doi: 10.1038/mto.2015.14
85. Park AK, Fong Y, Kim S-I, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver car targets to solid tumors. Sci Transl Med (2020) 12(559):eaaz1863. doi: 10.1126/scitranslmed.aaz1863
86. Aalipour A, Le Boeuf F, Tang M, Murty S, Simonetta F, Lozano AX, et al. Viral delivery of car targets to solid tumors enables effective cell therapy. Mol Ther–Oncolytics (2020) 17:232–40. doi: 10.1016/j.omto.2020.03.018
87. Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of cxcl11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology (2018) 7(3):e1395997. doi: 10.1080/2162402x.2017.1395997
88. Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and pd-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun (2017) 8(1):14754. doi: 10.1038/ncomms14754
89. Lou J, Dong J, Xu R, Zeng H, Fang L, Wu Y, et al. Remodeling of the tumor microenvironment using an engineered oncolytic vaccinia virus improves pd-L1 inhibition outcomes. Biosci Rep (2021) 41(6):BSR20204186. doi: 10.1042/bsr20204186
90. Sun Y, Zhang Z, Zhang C, Zhang N, Wang P, Chu Y, et al. An effective therapeutic regime for treatment of glioma using oncolytic vaccinia virus expressing il-21 in combination with immune checkpoint inhibition. Mol Ther–Oncolytics (2022) 26:105–19. doi: 10.1016/j.omto.2022.05.008
91. Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist il-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with pd-1 blockade. Mol Ther (2018) 26(10):2476–86. doi: 10.1016/j.ymthe.2018.07.013
92. Zuo S, Wei M, Xu T, Kong L, He B, Wang S, et al. An engineered oncolytic vaccinia virus encoding a single-chain variable fragment against tigit induces effective antitumor immunity and synergizes with pd-1 or lag-3 blockade. J Immunother Cancer (2021) 9(12):e002843. doi: 10.1136/jitc-2021-002843
93. Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, et al. An engineered oncolytic virus expressing pd-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun (2020) 11(1):1395. doi: 10.1038/s41467-020-15229-5
94. Semmrich M, Marchand J-B, Fend L, Rehn M, Remy C, Holmkvist P, et al. Vectorized treg-depleting αctla-4 elicits antigen cross-presentation and cd8+ T cell immunity to reject ‘Cold’ Tumors. J Immunother Cancer (2022) 10(1):e003488. doi: 10.1136/jitc-2021-003488
95. Nguyen H-M, Bommareddy PK, Silk AW, Saha D. Optimal timing of pd-1 blockade in combination with oncolytic virus therapy. Semin Cancer Biol (2022) 86:971–80. doi: 10.1016/j.semcancer.2021.05.019
96. Advani SJ, Buckel L, Chen NG, Scanderbeg DJ, Geissinger U, Zhang Q, et al. Preferential replication of systemically delivered oncolytic vaccinia virus in focally irradiated glioma xenografts. Clin Cancer Res (2012) 18(9):2579–90. doi: 10.1158/1078-0432.Ccr-11-2394
97. Mansfield DC, Kyula JN, Rosenfelder N, Chao-Chu J, Kramer-Marek G, Khan AA, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther (2016) 23(4):357–68. doi: 10.1038/gt.2016.5
98. Ottolino-Perry K, Mealiea D, Sellers C, Acuna SA, Angarita FA, Okamoto L, et al. Vaccinia virus and peptide-receptor radiotherapy synergize to improve treatment of peritoneal carcinomatosis. Mol Ther–Oncolytics (2023) 29:44–58. doi: 10.1016/j.omto.2023.04.001
99. Storozynsky QT, Agopsowicz KC, Noyce RS, Bukhari AB, Han X, Snyder N, et al. Radiation combined with oncolytic vaccinia virus provides pronounced antitumor efficacy and induces immune protection in an aggressive glioblastoma model. Cancer Lett (2023) 562:216169. doi: 10.1016/j.canlet.2023.216169
100. Dai MH, Liu SL, Chen NG, Zhang TP, You L, Q. Zhang F, et al. Oncolytic vaccinia virus in combination with radiation shows synergistic antitumor efficacy in pancreatic cancer. Cancer Lett (2014) 344(2):282–90. doi: 10.1016/j.canlet.2013.11.007
101. Chen W-Y, Chen Y-L, Lin H-W, Chang C-F, Huang B-S, Sun W-Z, et al. Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and damps. Cancer Lett (2021) 523:149–61. doi: 10.1016/j.canlet.2021.09.040
102. Kyula JN, Khan AA, Mansfield D, Karapanagiotou EM, McLaughlin M, Roulstone V, et al. Synergistic cytotoxicity of radiation and oncolytic lister strain vaccinia in V600d/ebraf mutant melanoma depends on jnk and tnf-α Signaling. Oncogene (2014) 33(13):1700–12. doi: 10.1038/onc.2013.112
103. Mell LK, Brumund KT, Daniels GA, Advani SJ, Zakeri K, Wright ME, et al. Phase I trial of intravenous oncolytic vaccinia virus (Gl-onc1) with cisplatin and radiotherapy in patients with locoregionally advanced head and neck carcinoma. Clin Cancer Res (2017) 23(19):5696–702. doi: 10.1158/1078-0432.Ccr-16-3232
104. Wennier ST, Liu J, McFadden G. Bugs and drugs: oncolytic virotherapy in combination with chemotherapy. Curr Pharm Biotechnol (2012) 13(9):1817–33. doi: 10.2174/138920112800958850
105. Yu YA, Galanis C, Woo Y, Chen N, Zhang Q, Fong Y, et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus glv-1h68. Mol Cancer Ther (2009) 8(1):141–51. doi: 10.1158/1535-7163.Mct-08-0533
106. Holloway RW, Thaker P, Mendivil AA, Ahmad S, Al-Niaimi AN, Barter J, et al. A phase iii, multicenter, randomized study of olvimulogene nanivacirepvec followed by platinum-doublet chemotherapy and bevacizumab compared with platinum-doublet chemotherapy and bevacizumab in women with platinum-resistant/refractory ovarian cancer. Intl J Gynecol Cancer (2023) 33(9):1458–63. doi: 10.1136/ijgc-2023-004812
107. Holloway RW, Mendivil AA, Kendrick JE, Abaid LN, Brown JV, LeBlanc J, et al. Clinical activity of olvimulogene nanivacirepvec–primed immunochemotherapy in heavily pretreated patients with platinum-resistant or platinum-refractory ovarian cancer: the nonrandomized phase 2 viro-15 clinical trial. JAMA Oncol (2023) 9(7):903–8. doi: 10.1001/jamaoncol.2023.1007
108. Lun XQ, Jang J-H, Tang N, Deng H, Head R, Bell JC, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for Malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res (2009) 15(8):2777–88. doi: 10.1158/1078-0432.Ccr-08-2342
109. Hofmann E, Weibel S, Szalay AA. Combination treatment with oncolytic vaccinia virus and cyclophosphamide results in synergistic antitumor effects in human lung adenocarcinoma bearing mice. J Transl Med (2014) 12:197. doi: 10.1186/1479-5876-12-197
110. Huang B, Sikorski R, Kirn DH, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the ifn response and hmgb1. Gene Ther (2010) 18(2):164–72. doi: 10.1038/gt.2010.121
111. Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X, et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials (2016) 102:187–97. doi: 10.1016/j.biomaterials.2016.06.032
112. Seubert CM, Stritzker J, Hess M, Donat U, Sturm JB, Chen N, et al. Enhanced tumor therapy using vaccinia virus strain glv-1h68 in combination with a β-galactosidase-activatable prodrug seco-analog of duocarmycin sa. Cancer Gene Ther (2010) 18(1):42–52. doi: 10.1038/cgt.2010.49
113. Berchtold S, Beil J, Raff C, Smirnow I, Schell M, D’Alvise J, et al. Assessing and overcoming resistance phenomena against a genetically modified vaccinia virus in selected cancer cell lines. Int J Mol Sci (2020) 21(20):7618. doi: 10.3390/ijms21207618
114. Lee S, Yang W, Kim DK, Kim H, Shin M, Choi KU, et al. Inhibition of mek-erk pathway enhances oncolytic vaccinia virus replication in doxorubicin-resistant ovarian cancer. Mol Ther–Oncolytics (2022) 25:211–24. doi: 10.1016/j.omto.2022.04.006
115. Ferguson MS, Chard Dunmall LS, Gangeswaran R, Marelli G, Tysome JR, Burns E, et al. Transient inhibition of pi3kδ Enhances the therapeutic effect of intravenous delivery of oncolytic vaccinia virus. Mol Ther (2020) 28(5):1263–75. doi: 10.1016/j.ymthe.2020.02.017
116. Chang C-L, Ma B, Pang X, Wu TC, Hung C-F. Treatment with cyclooxygenase-2 inhibitors enables repeated administration of vaccinia virus for control of ovarian cancer. Mol Ther (2009) 17(8):1365–72. doi: 10.1038/mt.2009.118
117. Jin D-Y, MacTavish H, Diallo J-S, Huang B, Stanford M, Le Boeuf F, et al. Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PloS One (2010) 5(12):e14462. doi: 10.1371/journal.pone.0014462
118. Kim M, Nitschké M, Sennino B, Murer P, Schriver BJ, Bell A, et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by sunitinib through multiple mechanisms. Cancer Res (2018) 78(4):922–37. doi: 10.1158/0008-5472.Can-15-3308
119. Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, et al. Sequential therapy with jx-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther (2011) 19(6):1170–9. doi: 10.1038/mt.2011.39
120. Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, et al. Anti-vegf single-chain antibody glaf-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci (2009) 106(31):12915–20. doi: 10.1073/pnas.0900660106
121. Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer (2020) 8:e001486. doi: 10.1136/jitc-2020-001486
122. Evgin L, Acuna SA, de Souza CT, Marguerie M, Lemay CG, Ilkow CS, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther (2015) 23(6):1066–76. doi: 10.1038/mt.2015.49
123. Lee N, Jeon Y-H, Yoo J, Shin S-k, Lee S, Park M-J, et al. Generation of Novel Oncolytic Vaccinia Virus with Improved Intravenous Efficacy through Protection against Complement-Mediated Lysis and Evasion of Neutralization by Vaccinia Virus-Specific Antibodies. J ImmunoTherapy Cancer (2023) 11(1):e006024. doi: 10.1136/jitc-2022-006024
124. Ban W, Guan J, Huang H, He Z, Sun M, Liu F, et al. Emerging systemic delivery strategies of oncolytic viruses: A key step toward cancer immunotherapy. Nano Res (2022) 15(5):4137–53. doi: 10.1007/s12274-021-4031-6
125. Yokoda R, Nagalo BM, Vernon B, Oklu R, Albadawi H, DeLeon TT, et al. Oncolytic virus delivery: from nanopharmacodynamics to enhanced oncolytic effect. Oncolytic Virother (2017) 6:39–49. doi: 10.2147/OV.S145262
Keywords: oncolytic virotherapy, vaccinia virus, cancer immunotherapy, combination therapy, tumor microenvironment
Citation: Xu L, Sun H, Lemoine NR, Xuan Y and Wang P (2024) Oncolytic vaccinia virus and cancer immunotherapy. Front. Immunol. 14:1324744. doi: 10.3389/fimmu.2023.1324744
Received: 20 October 2023; Accepted: 29 December 2023;
Published: 12 January 2024.
Edited by:
Björn L. Frendéus, BioInvent, SwedenReviewed by:
Kaïdre Bendjama, Transgene, FranceLance Hellman, Nevada State College, United States
Arash Arashkia, Pasteur Institute of Iran (PII), Iran
Copyright © 2024 Xu, Sun, Lemoine, Xuan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengju Wang, d2FuZ3BlbmdqdUB6enUuZWR1LmNu; Yujing Xuan, eXVqaW5nX3h1YW5AMTI2LmNvbQ==