
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 January 2024
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1321228
This article is part of the Research TopicRegulatory T cells in Immune-mediated diseasesView all 14 articles
 Samikshya Santosh Nirmala1
Samikshya Santosh Nirmala1 Kayani Kayani2,3,4
Kayani Kayani2,3,4 Mateusz Gliwiński5
Mateusz Gliwiński5 Yueyuan Hu1
Yueyuan Hu1 Dorota Iwaszkiewicz-Grześ5Magdalena Piotrowska-Mieczkowska5
Dorota Iwaszkiewicz-Grześ5Magdalena Piotrowska-Mieczkowska5 Justyna Sakowska5
Justyna Sakowska5 Martyna Tomaszewicz5
Martyna Tomaszewicz5 José Manuel Marín Morales1
José Manuel Marín Morales1 Kavitha Lakshmi1
Kavitha Lakshmi1 Natalia Maria Marek-Trzonkowska6
Natalia Maria Marek-Trzonkowska6 Piotr Trzonkowski5
Piotr Trzonkowski5 Ye Htun Oo2,7,8,9
Ye Htun Oo2,7,8,9 Anke Fuchs1*
Anke Fuchs1*The initial idea of a distinct group of T-cells responsible for suppressing immune responses was first postulated half a century ago. However, it is only in the last three decades that we have identified what we now term regulatory T-cells (Tregs), and subsequently elucidated and crystallized our understanding of them. Human Tregs have emerged as essential to immune tolerance and the prevention of autoimmune diseases and are typically contemporaneously characterized by their CD3+CD4+CD25high CD127lowFOXP3+ phenotype. It is important to note that FOXP3+ Tregs exhibit substantial diversity in their origin, phenotypic characteristics, and function. Identifying reliable markers is crucial to the accurate identification, quantification, and assessment of Tregs in health and disease, as well as the enrichment and expansion of viable cells for adoptive cell therapy. In our comprehensive review, we address the contributions of various markers identified in the last two decades since the master transcriptional factor FOXP3 was identified in establishing and enriching purity, lineage stability, tissue homing and suppressive proficiency in CD4+ Tregs. Additionally, our review delves into recent breakthroughs in innovative Treg-based therapies, underscoring the significance of distinct markers in their therapeutic utilization. Understanding Treg subsets holds the key to effectively harnessing human Tregs for immunotherapeutic approaches.
FOXP3+ regulatory T-cells (Tregs) represent 4-7% of the CD4+ T-cell population and are essential for regulating peripheral tolerance and immune homeostasis. Early phase clinical trials have demonstrated proof-of-principle for the use of isolated and expanded polyclonal Tregs in the treatment of inflammatory disorders, transplant rejection and autoimmune diseases. Treg research traces its origins back more than 40 years. The first evidence for thymus-derived cells with suppressive function came from mouse thymectomy experiments conducted in the early 1970s (1). Mice thymectomised between day two and four of life developed severe autoimmunity, which did not occur if thymectomy was performed before 24 hours or after day five of life (2). This led to the hypothesis that autoreactive T-cells are exported from the thymus within the first few days of life, followed later by an anergising subset of T-cells. Importantly, autoimmunity could be reversed with thymic transplantation at day ten. In line with these findings, Gershon and Kondo showed thymus-derived lymphocytes were crucial for inducing tolerance (3), and tolerance could be adoptively transferred to naïve recipients (4). The presence of suppressor T-cells, however, was questioned when several other groups failed to verify the mechanisms of suppression postulated using the novel techniques, including monoclonal antibodies and Sanger sequencing. These negative findings together with a lack of specific markers led to a loss of interest in the suppressor T-cell theory in the mid-1980s.
In the 1990s, a significant breakthrough emerged with the identification of regulatory T-cells (Tregs), by Sakaguchi and colleagues following the work on tolerance induction by Hall (5–7). Sakaguchi et al. demonstrated that Tregs could be distinguished by the expression of the IL-2 receptor α-chain, CD25, which was exclusive to a minority of CD4+ cells, facilitating the isolation of relatively pure Tregs populations from mice. The Shevach group provided the first direct evidence of Tregs inhibiting CD4+ effector T-cell proliferation in culture (8). In 2001, several research groups independently succeeded in identifying Tregs in human peripheral blood utilizing CD25 (9–11). Given human conventional T-cells upregulate CD25 upon activation, it soon became evident that this early cell-surface marker had certain limitations, despite David Hafler’s group demonstrating CD25 expression directly correlates with suppressive capacity (12).
The discovery of Foxp3, a forkhead box transcription factor, as the master control gene in mouse Treg development and function, provided further progress in the field and crucial insights into Treg regulatory mechanisms (13–15). It also explained the fatal lymphoproliferative disease observed in scurfy mice, which carry a frameshift mutation resulting in scurfin lacking the forkhead domain, and consequently autoimmunity (16). Intranuclear FOXP3 expression was demonstrated in human Tregs in 2005 (17), and mutations in FOXP3 explained the immune dysregulation, polyendocrinopathy, enteropathy and X-linked clinical syndrome (IPEX) (18, 19). Due to its intranuclear localization necessitating cell fixation, this new marker could not be used for the isolation of live Tregs. The discovery that FOXP3 expression could be induced in conventional human T-cells upon activation further complicated the identification of pure human Tregs (20).
In 2006, the expression of the IL-7 receptor α-chain CD127 was found to inversely correlate with FOXP3 expression and suppressive function (21, 22). In combination with CD25, low CD127 expression levels facilitated the isolation of live human Tregs with high purity and FOXP3 expression in post-sort analyses. Shortly thereafter, a more specific method to determine Treg purity among FOXP3 expressing cells was identified. Methylation analysis of the FOXP3 conserved non-coding sequence 2 (CNS2) (also termed the Tregs-Specific Demethylated Region (TSDR)) is at least partially methylated in conventional T-cells, but is completely demethylated in bona fides FOXP3+ Tregs and remains the most specific method of identifying Tregs (23, 24).
Currently, CD3+CD4+CD25hiCD127lo remains the most widely used gating strategy for the isolation of viable Tregs when cell sorting, with intranuclear staining of FOXP3 used to confirm purity and function. Of note, however, FOXP3, whilst exclusive to Tregs in mice, does not provide unambiguous identification of human Tregs. Finding a specific marker for human Tregs remains an unmet need. Nevertheless, over the past two decades, a substantial number of novel Treg markers associated with their origin, maturity, stability, and function have been identified (Figure 1). This review gives a comprehensive analysis of human Treg markers, facilitating the precise identification of CD4+ Tregs and their subsets, enabling the characterization of Tregs in a variety of immunological disorders and physiological processes.
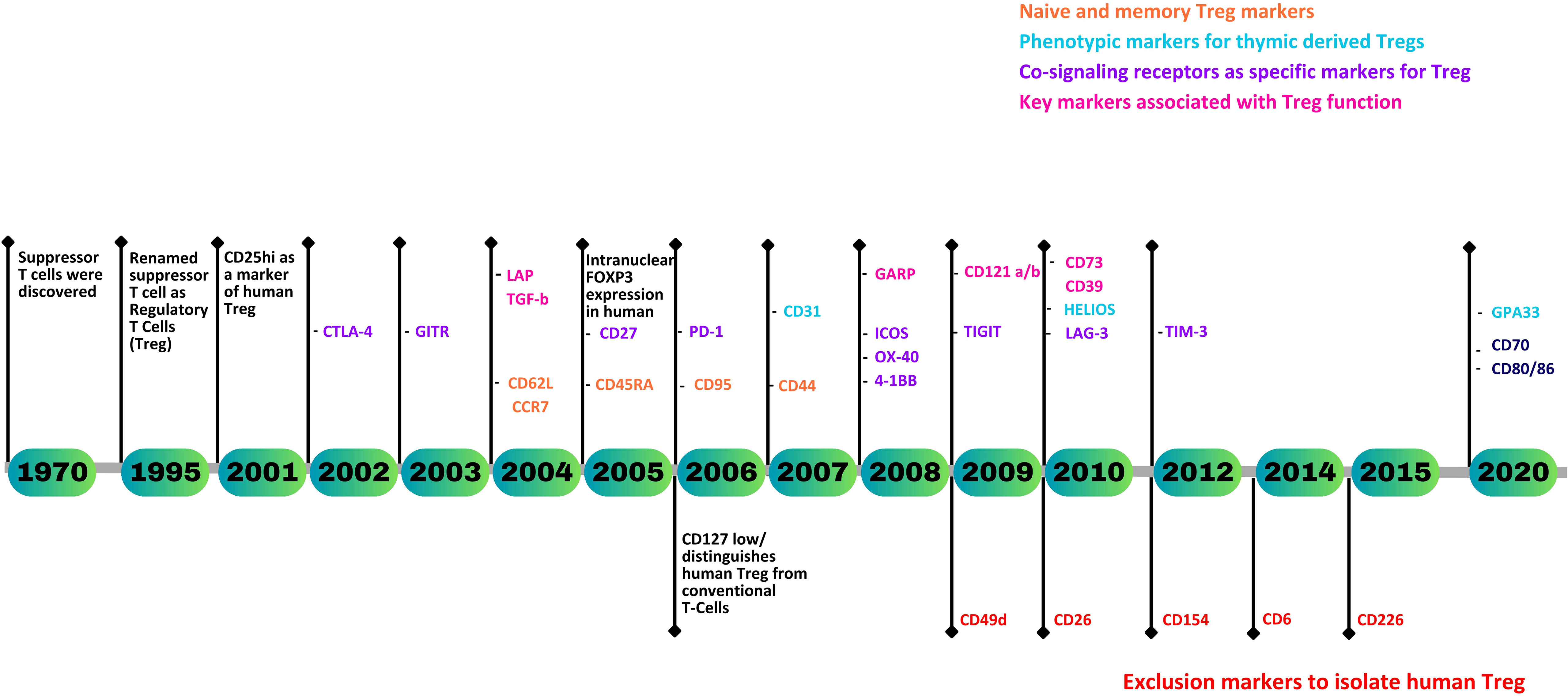
Figure 1 A timeline representing the discovery of human Treg markers.
Until 2005, Tregs in the peripheral blood were believed to predominantly exhibit a memory-like phenotype, characterized by the expression of CD45RO, until Valmori et al. described the existence of circulating Tregs expressing the naïve T-cell marker CD45RA (25). The prevalence of this subset inversely correlates with age, and concomitantly falls with the naïve CD4+ T-cell fraction, whilst the total Treg frequency remains constant throughout life. Naïve Tregs also have longer telomeres than their memory counterparts, similar to naïve conventional CD4+ T-cells. These naïve Tregs were postulated to be derived from CD4+CD25+ cells, selected in the thymus as precursors to antigen-experienced memory Treg subsets, awaiting TCR stimulation-mediated maturation, leading to an interest in umbilical cord blood Tregs.
A comparative study of CD45RA+ naïve Tregs from umbilical cord blood and adult blood found slightly lower levels of CD25 and FOXP3 expression in both when compared to memory Tregs, but similar in vitro suppressive capacities to memory Tregs (26). In 2006, Hofmann and Edinger showed the CD45RA+ naïve fraction of adult Tregs, which phenotypically resemble cord blood Tregs, gives rise to a more homogeneous Treg pool upon in vitro expansion as compared to bulk Tregs, thus proposing naïve Tregs as the optimal source of Tregs for adoptive cell therapies (27). They also demonstrated the superior stability of CD45RA+FOXP3+ cells. However, after three weeks of in vitro expansion with repetitive stimulation, CD45RA− Tregs preferentially down-regulated FOXP3 expression and were liable to produce pro-inflammatory cytokines (28). Miyara et al. confirmed the enhanced proliferative capacity of naïve Tregs and found that once activated, naïve Tregs upregulate FOXP3 and convert to an activated memory phenotype, all whilst exerting suppression during and after their proliferation and conversion (24).
Furthermore, both adult naïve and cord blood Tregs were shown to express significant levels of the secondary lymph node homing receptors CD62L (L-selectin) and CCR7 (25, 26). These markers, along with CD45RA/RO are commonly used to distinguish between naïve (CD45RA+/RO-, CD62Lhi, CCR7hi), effector memory (Tem) (CD45RA-/RO+, CD62Llo, CCR7lo), and central memory (Tcm) (CD45RA-/RO+, CD62Lhi, CCR7hi) conventional CD4+ T-cells. While studies have identified CCR7 as a differential marker defining Tem and Tcm subsets within the human CD4+CD25+ Treg population (29, 30), there is limited evidence for clear Tem and Tcm characteristics in Tregs based solely on these markers. Nevertheless, the expression of CD62L and CCR7 is crucial to the migration of Tregs to secondary lymphoid organs in murine models, proving essential to Treg-mediated suppression of autoimmune (30–33), allograft and other immune responses (34–38). Interestingly, even after undergoing extensive in vitro polyclonal expansion, human CD4+CD25high Tregs retain the expression of CD62L and CCR7 (39). It would be intriguing to monitor CD62L and CCR7 expression in Tregs used for human cell therapy trials, examining if, and how, these markers change with Treg migration to secondary lymphoid organs and inflamed tissues.
In contrast to conventional CD4+ T-cells, the majority of Tregs in adult peripheral blood exhibit elevated levels of CD45RO and CD95, making them susceptible to CD95L-mediated apoptosis (40). Interestingly, CD4+CD25+FOXP3+ T-cells present in cord blood are predominantly naïve, display low expression of CD95 like adult naïve Tregs, and remain resistant to FAS-mediated apoptosis. However, upon brief stimulation with anti-CD3/CD28 monoclonal antibodies, cord blood Tregs significantly upregulate CD95, sensitizing them to CD95L (40).
The expression of CD44, a cell surface molecule with important roles in activation, migration, and apoptosis, is linked to FOXP3 expression and Treg function in mice (41). Similar studies in human Tregs indicated that CD44 enhances the suppressive action of CD4+CD25+ Tregs (42). Further investigation in 2009 demonstrated that CD44 co-stimulation plays a crucial role in enhancing the persistence and function of FOXP3+ Tregs through the production of IL-2, IL-10, and TGF-β1 in both humans and mice (43).
It was formerly believed that natural Tregs are thymically derived CD4+ cells that constitutively display CD25. Work in mice demonstrating the capacity for naïve CD4+ T-cells to transform into FOXP3+ Tregs in the periphery led to a paradigm shift. Subsequent categorization of Treg subsets now includes thymically derived Tregs (tTregs) and peripherally induced Tregs (pTregs) (44). However, the presence of pTregs in humans remains a controversial topic. Multiple reports have indicated that pTregs are plastic and can produce inflammatory cytokines (45–47). Significant attention has been devoted to identifying phenotypic markers capable of distinguishing between pTregs and tTregs.
Helios (Ikzf2) is a member of the Ikaros transcription factor family and is restricted to T lymphocytes. Microarray studies characterizing unique gene signatures in FOXP3+ mouse Tregs provided the first indication of Helios as a Treg-specific gene (48, 49). In 2010, the Shevach group published work indicating Helios to be a potential marker specific to FOXP3+ tTregs in mice and humans (50). Approximately 70% of Tregs in human peripheral blood and mouse peripheral lymphoid tissues, as well as over 95% of Tregs in mouse thymi, were Helios positive. Additionally, Helios expression was not seen neither in murine nor human in vitro or in vivo induced Tregs. Interestingly, the majority of FOXP3+ Tregs producing IL2, IL17, and IFN-γ belong to the Helios negative subset. Additionally, more than 90% of Tregs in human cord blood and thymic samples express Helios (51). Crucially, the Helios-negative Treg subset, comprising less than 10% of the total, does not originate exclusively from the thymus. Studies have suggested, in the case of cord blood, Helios-negative Tregs are generated in the periphery during fetal gestation, while those found in the thymus are recirculated Tregs from the periphery. Subsequent studies have demonstrated that human FOXP3+Helios+ Tregs demonstrate less than 10% CpG methylation in the TSDR, while FOXP3+Helios− Tregs are more than 40% methylated (52, 53). Thus, the early consensus was Helios could serve as a marker for tTregs, with Tregs lacking Helios representing pTregs.
More recent studies have disputed this consensus (54, 55), demonstrating inducible Helios expression in pTregs (56), findig Helios expression with Treg activation and proliferation (57). The coexistence of both Helios positive and negative human FOXP3+ tTregs has also been demonstrated (58), with further studies supporting the notion that Helios expression cannot differentiate between tTregs or pTregs (59). Thus, the question of whether Helios is a reliable specific marker for tTregs remains open (60).
A 2007 study by Hass et al. on Treg dysfunction in multiple sclerosis demonstrated a significant correlation between Treg-mediated immunosuppression and the presence of recent thymic emigrants (RTE) Tregs (61). Naïve Tregs expressing CD31 (PECAM1) play a role in the functional characteristics of the entire Treg population. CD45RA+CD45RO−CD31+ naive Tregs have been observed to decline with age in healthy individuals, contrasting with their CD31− memory Tregs counterparts. Subsequent studies indicate that, unlike memory Tregs, a substantial proportion of CD45RA+ naive Tregs express CD31 (62, 63).
A novel marker, Neuropilin-1 (Nrp-1), was identified in 2012 by Bluestone and Lafaille. It was found to be expressed on tTregs, but not on in vivo or in vitro induced pTregs in mice (64, 65). While these murine studies are useful to our understanding of Treg development and putative tTreg biomarkers, the translation of these findings into the human context remains disputed (59, 66, 67).
Glycoprotein A33 (GPA33) is a cell surface antigen and a member of the immunoglobulin superfamily. While this marker is known to be expressed in approximately >95% of primary and metastatic colon cancers, its presence on normal tissues and cells was previously unreported (68). In a 2020 study, Opstelten et al. identified GPA33 on tTregs, reporting high levels of mRNA and surface protein expression in CD4+CD25+CD45RA+ naive Tregs (69). All CD4+CD25+CD127-GPA33+ Tregs were shown to express FOXP3 and Helios, whereas only 86% of CD4+CD25+CD127-CD45RA+ expressed FOXP3 and Helios, suggesting CD127-GPA33+ selects naïve Tregs with enhanced purity, lineage stability, and suppressive function. Subsequent single-cell transcriptomic studies have demonstrated that GPA33 is acquired by pre-Tregs in a late developmental stage, prior to the acquisition of CD45RA, validating GPA33 as a tTreg marker (70). Combined, this suggests GPA33+ is found on a highly pure and stable population of CD45RA+ Tregs originating from the thymus. It will be interesting to assess the applicability of this intriguing new marker for the isolation and expansion of functional Tregs, especially as the function of GPA33 is presently unclear.
Co-stimulatory and co-inhibitory receptors, collectively described as co-signaling receptors, are crucial to regulating the expansion and function of conventional CD4+ T-cells, modulating immune responses. Their discovery marked a turning point in our understanding of Treg function, eventually leading to various clinically employed therapeutic agents, called checkpoint inhibitors, which target Co-signaling receptors to restore desired immune responses (71, 72).
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), also known as CD152, is an immunoglobulin superfamily (Ig SF) member expressed by activated inhibitory conventional T-cells (73). CTLA-4 is often referred to as a “moving target” due to its rapid internalization and recycling to the plasma membrane of T-cells (74). Notably, while CTLA-4 is expressed in conventional T-cells upon activation, it is constitutively expressed in resting Tregs, representing one of the hallmarks of this cell subset (75, 76). Post TCR-mediated activation, human Tregs strongly upregulate CTLA-4.
In 2000, work by the Powrie and Sakaguchi groups provided compelling evidence of CTLA-4’s crucial role in contact-dependent suppressive function in mice (76, 77). Subsequent human Treg in vitro studies demonstrated conflicting results. Some supported the notion that blocking CTLA-4 using anti-CTLA-4 monoclonal antibodies (mAbs) or Fab fragments reversed the suppressive effect of CD4+CD25+ Tregs on Teff proliferation (78–80). Other studies came to the opposite conclusion (9, 10, 81, 82), highlighting the complex nature of CTLA-4’s role in Treg-mediated suppression. However, variations in assay conditions, including the type of antigen-presenting cells (APCs) used and the strength of the TCR stimulus may account for the differing results observed. CTLA-4 interacts with CD80/CD86 on APCs with higher affinity and avidity than its counterpart CD28, which competes to interact with CD80/86. As a target gene of FOXP3, CTLA-4 plays a pivotal role in regulating Treg homeostasis by acting as an intrinsic regulator of Treg proliferation. Deletion or blockade of CTLA-4 enhances Treg proliferation and affects the number and function of memory Tregs (83–86). Although the role of CTLA-4 in Treg biology has remained controversial for several years, its established use as a diagnostic marker and therapeutic target for various human pathologies confirms its critical role in Treg-mediated immunosuppression (72, 87–89).
In parallel to CTLA-4, programmed cell death 1 (PD-1) is another co-inhibitory molecule from the Ig SF found in Tregs. In 2003, it was found that blocking the interaction between PD-1 and its ligand, PD-L1, influences Treg suppressive function (82). A more comprehensive 2006 study demonstrated that freshly isolated murine and human Tregs retain PD-1 in the intracellular compartment (90). PD-1 appears to transmigrate to the cell surface upon TCR stimulation, indicating that purifying CD25+PD-1− T-cells may give a pure population of resting Tregs, prompting evaluation of PD-1 as a marker to distinguish Tregs from activated CD4+CD25+ T-cells. Francisco et al. showed that the expression of PD-1 minimally converts naive CD4+ T-cells into induced Tregs (iTregs) by promoting FOXP3 expression, enhancing iTreg suppressive activity (91). Our current understanding of PD-1/PD-L1 signaling in Tregs is complex and context-dependent, with effects including Treg expansion (92), survival and functionality (93) in addition to suppression of effector T-cells and autoreactive B-cells (94). These effects have implications for tumor immunity and autoimmunity, as reviewed by Cai et al. (95) and the clinical use of PD-1 inhibitors highlights the therapeutic value of targeting this receptor. Nevertheless, further research deciphering the intricate mechanisms of PD-1 signaling in Treg development and function is required.
Inducible T-cell costimulator (ICOS) is another CD28-related member of the immunoglobulin superfamily of molecules (IgSF) that stands apart from CTLA-4 and PD-1 by functioning as a costimulatory receptor. ICOS’s discovery on the surface of T-cells following TCR-mediated stimulation in 1999 represented a breakthrough in T-cell research (96). ICOS+ T-cells, although a minority among peripheral blood CD4+ T-cells, comprise approximately 20% of FOXP3+ Tregs (97). Ito et al. observed that both ICOS+ and ICOS– Tregs exist in cord blood and neonatal thymi (98) and that ICOS+ Tregs are able to produce IL-10 and suppress CD86 upregulation on DCs, while ICOS- Tregs produce TGFβ. Early investigations unveiled a link between elevated ICOS expression on CD4+ T-cells and IL-10 production in mice (99). This was later confirmed in humans (98) and demonstrated the central role ICOS has in the differentiation and function of FOXP3+ Tregs. Over the past decade, ICOS’s multifaceted roles, including involvement in the production, proliferation and survival of Tregs and enhancing Treg suppressive function have been unraveled (100–103).
CTLA4, PD1 and ICOS are now recognized targets in cancer immunotherapy, leading to an interest in novel Ig SF co-inhibitory receptors such as Lymphocyte activation gene-3 (Lag-3), T-cell (or transmembrane) immunoglobulin and mucin domain 3 (Tim-3), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), to address CTLA-4 and PD-1 checkpoint inhibitor non-responders.
Lag-3, also called CD223, belongs to the CD2/Signaling Lymphocytic Activation Molecule (SLAM) family of the Ig SF. It was initially discovered as a molecule upregulated on activated CD4+ and CD8+ T-cells (104) and has subsequently been reported to be highly expressed in both tTregs and pTregs upon activation (105). Lag-3 blockade on Tregs abolishes Treg suppressor function, while ectopic expression of Lag-3 in non-Treg CD4+ T-cells confers suppressive activity. Additionally, Lag-3 is necessary for Treg-mediated control of T-cell homeostasis (106). In subsequent years, Lag-3 has emerged as a useful Treg surface marker that can be cleaved into a soluble form (107, 108). Functional studies of Lag-3 in human Tregs report only small proportions of Tregs are LAG-3 positive, although this fraction increases in inflamed tissues such as in the lamina propria of ulcerative colitis (UC) patients, and various tumors (109–111). Thus, Lag-3 may serve as a diagnostic and treatment response biomarker in the future, however further research into the mechanism of action of Lag-3 in Tregs and its impact on T-cells is warranted.
TIGIT, a member of the poliovirus receptor (PVR)-like family of Ig SFs, is a co-inhibitory receptor expressed on many lymphocyte subsets. It is highly expressed on Tregs, where it promotes suppressive function. The first link between TIGIT and human Treg biology was reported in 2009 (112), however, its functional role in Tregs was not shown until 2014 (113). TIGIT is present on the surface of both human and murine Tregs, including tTregs and pTregs. TIGIT-positive Tregs exhibit increased expression of key Treg-associated genes, including FOXP3, CD25, and CTLA-4, and demonstrate enhanced demethylation in the TSDR, indicating lineage stability. Activation of TIGIT on Tregs triggers the production of suppressive molecules like IL-10 and Fgl2. Moreover, TIGIT expression defines a Treg subset that demonstrates selective suppression of Th1 and Th17 but not Th2 responses. Additional studies have reported TIGIT to identify highly suppressive Tregs, indicating the therapeutic potential of TIGIT Tregs in treating disease (114–116).
Tim-3 (HAVCR2) has attracted significant attention as a potential negative regulator of T-cell responses (117, 118). Tim-3 expression is limited to a small subset (2-5%) of Tregs in the periphery during steady-state conditions. However, during immune responses, Tim-3 is upregulated on Tregs, with elevated expression in Tregs infiltrating allografts, tissues, and tumors in mice (119). Subsequent studies have found that Tim-3 is predominantly expressed on tumor-infiltrating Tregs in both human and experimental tumor models (120–126), and Tim-3+ Tregs exhibit enhanced suppressive capacity in in vitro suppression assays compared to Tim-3- Tregs (122, 124). Moreover, Tim-3+ Tregs display increased expression of suppressive molecules (CTLA-4, Lag-3, and PD-1), and enhanced secretion of immunosuppressive cytokines (IL-10 and TGF-β). These, and more recent findings, strongly argue for Tim-3+ Tregs cells as promising therapeutic target in cancer immunotherapy (127). Furthermore, a substantial increase in Tim-3 levels are seen following the ex vivo expansion of clinical-grade human Tregs, increasing to 29% (128). Sorted Tim-3+ Treg populations show significant enhancements in IL-10 and Granzyme B production, indicating the suppressive capacity of this subset and its potential as a cell-based therapy to induce allograft tolerance.
The TNF receptor superfamily also includes several co-stimulatory molecules that have been identified on Tregs, including GITR, 4-1BB, OX40 and CD27.
Glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR), also known as TNFRSF18 and CD357, acts as a costimulatory molecule. While expressed at low levels on naïve T-cells, Tregs and activated T-cells show high surface expression of GITR (129, 130). Soon after the discovery of GITR on murine Tregs (131, 132), studies reported GITR to be co-expressed with CD25 and FOXP3 in human tTregs (133–135). GITR is now an established Treg marker with tTreg specificity, whose gene loci is demethylated in Tregs (136). Tregs upregulate GITR expression upon activation, with levels correlating with Treg immunosuppressive function, leading to the proposal of using GITR as a marker for selecting highly functional Tregs. Additionally, Tregs residence in the tumor microenvironment demonstrates GITR upregulation and GITR’s expression in Tregs has been extensively studied in autoimmune diseases, demonstrating its potential as an immunotherapy target having already shown promise in murine models (137, 138).
4-1BB (also known as CD137), was initially considered a proliferation and activation marker of CD8+ conventional T-cells, but was later found to be constitutively expressed in murine CD4+CD25+ Tregs in two separate DNA microarray studies (132, 139). 4-1BB expression on resting Tregs was subsequently identified, with upregulation demonstrated following 2 days of in vitro combined CD3 and IL-2 stimulation (140). Subsequently, in both in vitro and in vivo conditions, 4-1BB mediated enhanced Treg survival and proliferation was observed, without alteration to immunosuppressive function (140, 141). Additionally, animal studies have shown infusions of agonistic anti–4-1BB mAbs lead to increases in splenic Tregs, with suppression of chemically induced colitis (142). Furthermore, artificial APCs (aAPCs) expressing 4-1BBL have been shown to expand human umbilical cord blood Tregs and enhance their suppressive function (143). Most significantly, it has been found that gating on 4-1BB+CD40L- (CD137+CD154-) identifies both ex vivo, and in vitro activated Tregs, facilitating the isolation of epigenetically stable antigen-activated Tregs, enabling their rapid functional testing (144, 145).
Along with the discovery of 4-1BB on Tregs, the aforementioned DNA microarray studies also identified OX40 (CD134) to be highly expressed on Tregs, which had previously been recognized as a costimulatory molecule on activated CD4+ effector T-cells (132, 139). OX40 signaling is crucial to the development, homeostasis, and suppressive activity of mouse Tregs (146). Early studies of human cord blood Tregs demonstrated significantly higher Treg expansion using aAPCs modified to express OX40L compared to bead- or non-modified aAPCs (143). However, OX40 signaling has also shown the potential to oppose Treg-mediated suppression in antigen-engaged naive T-cells in in vivo mouse models (146). The contrasting functions of OX40 in Tregs, which has also been reported in a variety of human and mouse studies, make its exact role in Tregs unclear (147–151). While uncertainty continues regarding OX40 in Tregs, it remains recognized as a Treg activation marker and is believed to contribute to Treg proliferation.
CD27 is present on the majority of CD4+ T-cells, interacting with CD70 on APCs, and in vitro T-cell expansion can be enhanced using anti-CD27 antibodies (152). Initially discovered through DNA microarray studies (132), CD27 is now recognized for its crucial role in Treg function. Along with CD25, CD27 can identify highly suppressive FOXP3+ Tregs (153) and subsequent investigations have illustrated a novel use of CD27 to induce, expand, and select highly suppressive, Ag-specific human Treg subsets (154). Moreover, following the ex vivo expansion of human pTregs, CD27 can discriminate between regulatory and non-regulatory cells (155). With multiple studies highlighting CD27 as a reliable marker for the identification of highly functional Tregs (156, 157), CD27 demonstrates significant potential as a marker for the isolation of suppressive Tregs for clinical application.
Potential markers of Tregs exhaustion: Whilst the effects of chronic T-cell stimulation have been extensively studied, our understanding of Treg exhaustion is limited. In conventional T-cells, exhaustion from chronic antigen exposure is characterized by the expression of inhibitory receptors (PD-1, TIM-3, and LAG-3), reduced proliferation, cytokine production, and increased apoptosis (158–161). Studying Treg exhaustion in vitro is hampered by the contradictory roles of inhibitory receptors in Tregs, as discussed above. Tonic-signaling chimeric antigen receptors (TS-CARs) have facilitated the first comprehensive investigation of exhaustion in Tregs, revealing that Tregs rapidly acquire an exhaustion-like phenotype, with increased expression of inhibitory receptors and transcription factors including PD-1, TIM3, TOX and BLIMP1, akin to conventional T-cells (162). Epigenetic changes can also be observed with repetitive TCR-mediated stimulation, including those that may affect Treg functionality and lead to exhaustion, with TIM-3 and TIGIT implicated, among others (163). However, further clarification is needed on the implications of these recent studies on these markers for identifying exhausted Tregs.
One of the major obstacles hampering the clinical application of Tregs is the lack of suitable extracellular markers, complicating their identification and isolation. Due to a lack of clinical-grade CD4 negative isolation reagents, clinical grade GMP magnetic enrichment typically involves CD8 depletion, an optional CD19 depletion step, followed by CD25 enrichment. Where cell sorting is possible, Tregs are typically isolated as CD4+CD25+/hiCD127-/low. Over the past decade, numerous studies have attempted to identify additional negative selection markers to facilitate the efficient sorting of pure and highly functional Tregs, while eliminating contaminating effector T-cells. In this section, we explore cell surface markers that are specifically absent on Tregs and consider negative selection strategies for Treg isolation for both functional research and clinical application.
CD49d, the α chain of the integrin VLA-4 (α4β1), functions as a costimulatory and adhesion molecule for lymphocyte transmigration into inflamed tissues (164, 165). In 2009, Kleinewietfeld et al. demonstrated CD49d expression on more than 80% of human PBMCs, with expression predominantly on pro-inflammatory effector cells, including non-regulatory CD4+CD25low T-cells, cytokine-secreting CD4+ effector cells, and FOXP3 expressing Th1- and Th17-like cells. Significantly, approximately 70% of immunosuppressive FOXP3+ Tregs are CD49d-/low. Considering the differential distribution of CD49d, this marker has been proposed to effectively deplete cells that otherwise contaminate CD25+-based Treg preparations (166). This has led to novel CD49d- gating strategies, designed to obtain “untouched” FOXP3+ Tregs (i.e. cells that have not been targeted by an antibody during purification) in combination with CD127-. Tregs isolated in this manner are highly pure, can be expanded, and show effective in vitro and in vivo suppressive function (166). However, other data has shown CD49d-CD127- isolated Tregs after in vitro expansion are less suppressive, exhibit lower levels of FOXP3 and TSDR demethylation than CD4+CD25hiCD127- or CD4+CD25hiICOS+ isolated cells (167). Despite this conflicting evidence, untouched Treg isolation protocols have been scaled up for the generation of GMP cell therapy products from large-scale leukapheresis samples, with the final product demonstrating <10% contamination with CD4 effector T-cells and <2% of all other cell types (168). Singapore General Hospital is currently preparing to conduct T-cell therapy trials using untouched Tregs in graft-versus-host disease following stem cell transplantation.
Another putative negative selection marker for Tregs is CD26, a widely distributed, 110-kDa, membrane-bound glycoprotein with intrinsic dipeptidyl peptidase IV activity. The ectonucleotidases, CD39 and CD73, in combination with adenosine deaminase (ADA), which degrades adenosine into inosine, together regulate pericellular adenosine concentrations. In humans, ADA is associated with the extracellular domain of CD26, a complex not seen in mice (169). The lack of anti-ADA antibodies suitable for flow cytometry necessitates CD26 analysis as a surrogate for ADA expression in T-cells. Despite a large amount of data supporting the costimulatory role of CD26 in T-cells (170–172), its exact function in Tregs has not been confirmed. The absence of CD26-ADA expression in Tregs was first described in 2010 (173). More comprehensive work in 2012 reported the presence of CD26-ADA on FOXP3-expressing activated CD4+ Teff cells, but not suppressive Tregs (174). In combination with markers like CD25, FOXP3 and CD127, CD26 may facilitate quantitative evaluation of Tregs or Treg isolation from samples containing activated Teff cells. Other studies have also described the use of CD26 as a negative selection marker for Tregs (175–178). However, further investigation is required to determine whether the CD26-/low phenotype is a reliable method of isolating human Tregs and one that improves on current protocols.
CD6 and CD226 have not only facilitated the depletion of contaminants but also shed light on further subclassifications of tTregs. CD6 is a cell-surface glycoprotein predominantly expressed on T-cells (179, 180). It functions as a costimulatory molecule during TCR activation and plays a role in the responses of mature T-cells to both nominal antigens and autoantigens (181, 182). In 2014, Garcia Santana et al. identified CD4+CD25hiCD6lo/- Tregs in man as FOXP3+ natural Tregs (nTregs), exhibiting in vitro suppressive activity on CD8+ T-cell proliferation. CD6 in combination with CD127 was postulated to serve as a tool to identify and isolate nTregs (183). Although CD6 has been reported as a negative marker for human Tregs in different studies, further investigation is needed to validate the utility of CD6 as a Treg exclusion marker (175, 184).
Although CD226, also known as DNAM-1, plays a critical role in immunoregulation, little was known about its cellular distribution in human Tregs until 2015, when Fuhrman et al. demonstrated that isolating CD226- Tregs gave a highly pure and suppressive population. The CD226+ population is less pure and suppressive after expansion and demonstrated less demethylation in the TSDR locus, as well as significantly higher production of effector cytokines (185). Further work showed CD4+CD25+CD226- cells, after 14 days of expansion, are more suppressive, produce more TGF-β1 and fewer effector cytokines like IFN-γ, TNF, and IL-17A than CD4+CD25+CD127lo/- cells (186). Furthermore, CD4+ CD25+ CD226- Tregs may not only be long-lived but also potentially localize more readily to secondary lymphoid organs. These data argue for CD226 as an important negative phenotypic marker of Tregs and the excluding CD226-expressing cells during Tregs sorting yields a population with increased purity, lineage stability, and suppressive capabilities, which may benefit Treg adoptive cell therapy. However, this approach should be taken with caution, as CD226 is also highly expressed by IL-10-secreting Tr1-like T-cells (187).
In addition to the aforementioned, CD154, also known as CD40L, can also be used to negatively identify activated Tregs. This member of the TNF superfamily is a recognized activation marker of CD4+ Teff cells (188, 189). The absence of CD154 expression in the presence of 4-1BB (4-1BB+CD154-) has emerged as an ex vivo and in vitro Treg activation signature, allowing for the identification and isolation of epigenetically stable, antigen-activated Tregs for rapid functional assessment (144, 145).
Tregs exert their suppressive effects via a plethora of mechanisms, acting on various targets. These include modulating the cytokine microenvironment (85, 190, 191), metabolic disruption of target T-cells (8, 192), regulating the activation capacity of dendritic cells (193, 194), and direct cytolysis (195, 196). Building upon this, we next review previously discovered and newly identified markers of Tregs function.
Transforming growth factor-β (TGF-β), the most common isoform of which is TGF-β1, is a pleiotropic cytokine. First discovered in 1986, it was noted to induce anchorage-independent growth (197). TGF-β later became the primary focus of the immunosuppression and tolerance research field because of its potent inhibition of immune responses, impacting particularly upon T-cell proliferation and differentiation (198, 199). This was solidified when the connection between TGF-β and the suppressive function of murine Tregs was identified (77, 200), and the concept of TGF-β1 tethered to the cell surface of Tregs was introduced. Upon stimulation, CD4+CD25+ Tregs but not CD4+CD25- conventional T-cells express high and persistent levels of TGF-β1 on the cell surface. However, the role of TGF-β in the immunoregulatory function of Tregs has sparked controversy, as previous studies had shown that neutralizing TGF-β using anti-TGF-β antibodies failed to reverse the suppressive function of Tregs in vitro. This was addressed in 2004 when it was reported that Latency associated protein (LAP) binds latent TGF-β1 in Tregs (201). Thus, TGF-β1-mediated immunosuppression occurs in a two-step process: first, TGF-β1 dissociates from LAP, then free TGF-β1 can interact with its receptor. This multistep process explains why antibody mediated TGF-β inhibition requires the high antibody concentrations necessary for TGF-β1 quenching. Crucially, expression of LAP on activated human FOXP3+ Tregs has since been shown to identify highly pure and functional Tregs from ex vivo expansion cultures (202, 203). For reviews on TGF-β in Treg biology, and the associated debates, see reviews by Tran and Moreau et al. (204, 205).
The cell surface molecule Glycoprotein A repetitions predominant (GARP), also known as LRRC32, was first reported in human Tregs by Wang et al. and has since been identified to be involved in TGF-β1 mediated Treg immunosuppression. TCR-activated Tregs exhibit significant upregulation of GARP on their cell surface, which has been linked to enhanced Treg suppressive capacity (206). Several groups have independently reported that GARP on Tregs binds latent TGF-β1, promoting the release of the activated TGF-β, leading to TGF-β mediated immunosuppression (207–209). Studies of Tregs in human disease have confirmed that TGF-β, GARP and LAP are vital to Treg function in inflammatory diseases (210) and cancers (211), highlighting the potential value of these receptors as immunotherapy targets.
Tregs can also suppress effector T-cells through adenosine binding to A2A receptors (212, 213). ATP is cleaved in tandem by two Treg-associated ectonucleotidases, CD39 and CD73, leading to adenosine production. Of the two ectonucleotidases, CD39, which hydrolyses ATP and ADP into AMP, is the rate-limiting enzyme (212). CD73, an ecto-5′-nucleotidase, exists in both soluble and membrane-bound forms and catalyzes the dephosphorylation of AMP into adenosine (214, 215). Adenosine then binds the A2A receptor on T-cells, leading to cAMP-mediated suppression of TCR signaling via PKA.
The role of adenosine in the function of human Tregs was discovered in 2010, introducing CD39 as a novel phenotypic and functional marker specific to human Tregs (173). In contrast to mice, which express CD39 constitutively on virtually all CD4+CD25+ T-cells, expression on human T-cells is restricted to a subset of FOXP3+ effector/memory-like Tregs (216, 217). Nearly all (>90%) CD4+CD25hiFOXP3+ adult human Tregs are CD39 positive, distinguishing human Tregs from other T-cell subsets. Due to its expression on the cell surface, CD39 has facilitated the successful and reliable isolation of functionally active human natural Tregs from peripheral blood (218). However, this approach should be taken with caution, as CD39 is not exclusive to Tregs and the CD4+CD39+ fraction of PBMCs also includes CD25-FOXP3- T-cells (219, 220). Nevertheless, several studies have revealed that CD39+ Tregs demonstrate stronger stability and function under inflammatory conditions and superior suppressive capacity in vitro and in vivo (221–224), as well as an ability to suppress pathogenic Th17 responses (225).
CD73 was initially described as a characteristic surface marker of murine Tregs, however, it was only observed in a small proportion of human Tregs. Over 70% of CD4+CD25hi Tregs express CD73 intracellularly, while the expression is limited to 20% of CD4+CD25- T-cells (173). These data indicate that CD73 is readily internalized from the surface of human lymphocytes (226, 227) and is predominantly cytosolic in human Tregs. However, expanded human CD4+CD25hiCD127lo cells show a higher surface expression of CD73 (~35%) as compared to unstimulated memory Tregs (CD4+CD25hiCD127loCD45RA-) (~5%) (228). These expanded Tregs are highly suppressive, attributable to the surface expression of CD73 along with CD39. Additionally, CD73 on murine Tregs has been shown to suppress proliferation and cytokine secretion by T helper 1 (Th1) and Th2 cells (229). Taken together, these results strongly indicate the physiological importance of CD39 and CD73 expression in Tregs (228, 230).
As previously discussed, a multitude of Treg markers are associated with their origin, maturation, stability, and functional characteristics, as concisely delineated in Figure 2. In recent years, several emerging markers have garnered attention for their potential contributions to Treg-mediated immunosuppression. Although the precise roles and mechanisms of action remain a subject of ongoing investigation, preliminary research suggests their potential significance in modulating immune responses.
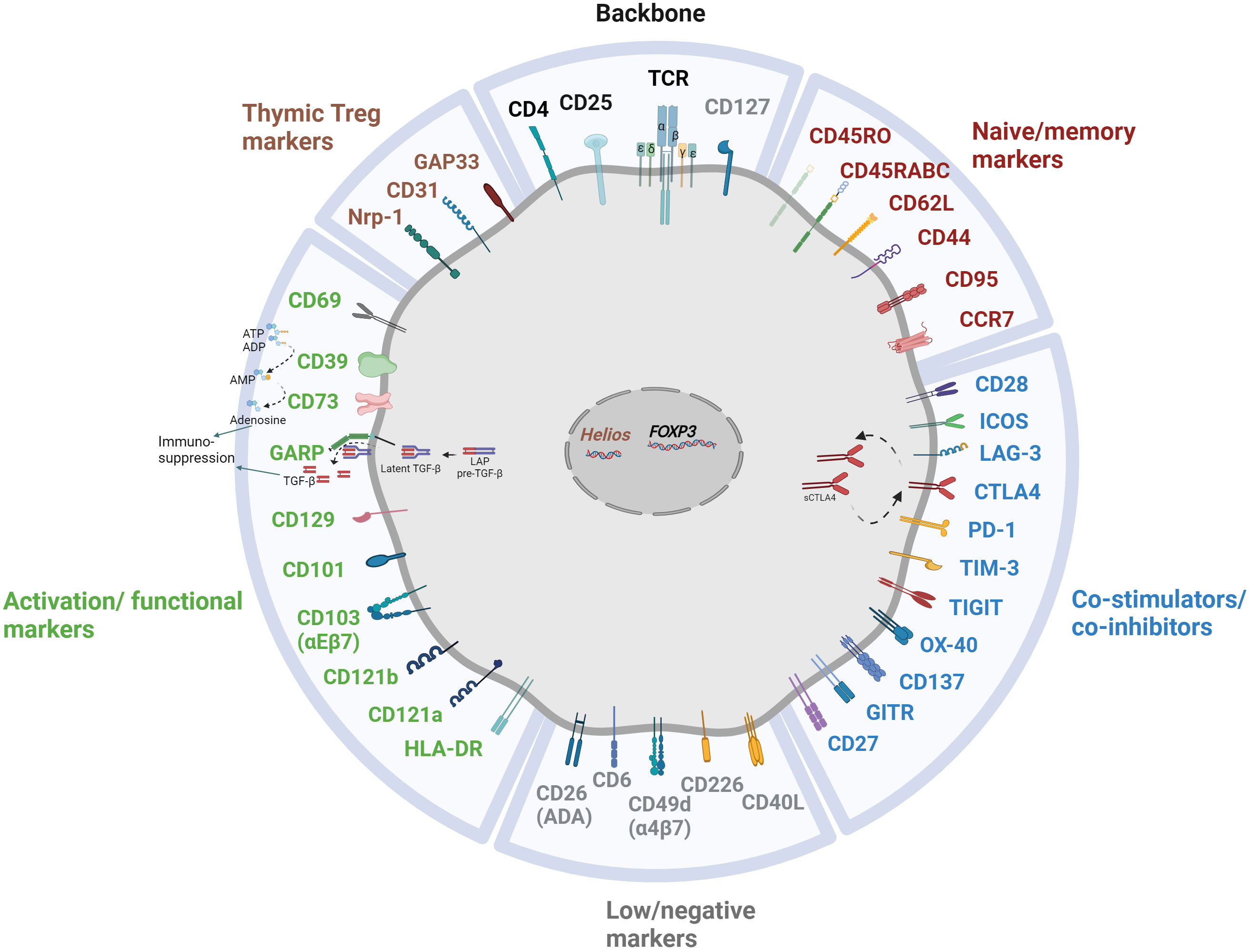
Figure 2 Multitude of markers illuminating the heterogeneity of Treg subpopulation.
CD121a and CD121b are two unique cell-surface antigens on human Tregs that are transiently expressed upon TCR-mediated Treg activation, distinguishing functional Tregs from activated FOXP3- and FOXP3+ non-Tregs, alongside LAP (202). These two highly specific surface markers were recommended for high yield, high purity Treg isolation, promising a rapid advancement in the therapeutic application and functional analysis of Tregs in human disease. However, the popularity of these two markers has waned due to a lack of subsequent studies that could verify these properties.
CD69 and HLA-DR are widely recognized as activation-induced cell surface markers in both conventional T-cells and Tregs, as well as markers of Treg differentiation and immunosuppression. CD69, upon binding with its ligand the S100A8/S100A9 complex, regulates Treg differentiation (231). Further reports indicate CD69 expression may be essential for tTreg development (232). Moreover, FOXP3+CD69+ Tregs express higher surface levels of suppression-associated markers and display enhanced suppressive activity as compared to FOXP3+CD69- Tregs (233, 234). CD69 also enhances the immunosuppressive function of Tregs by prompting IL-10 production (235). However, due to the lack of knowledge regarding the functional roles of CD69 in human Tregs, no definitive conclusions can be drawn at this time.
Approximately one-third of adult human peripheral blood CD4+ effector Tregs express HLA-DR, identifying a distinct and highly functional subset of terminally differentiated Tregs (236). HLA-DR+ Tregs exhibit higher levels of FOXP3 and employ contact-dependent immunosuppression, indicating superior suppressive capabilities (237, 238). However, despite HLA-DR+ Tregs being regarded as superior in functionality (239–241), there is insufficient data demonstrating HLA-DR as a marker of Treg function.
Two other cell surface receptors, CD101 and CD129, have been linked with Treg suppressive activity (242). Earlier mouse studies revealed that CD101 surface expression is strongly correlated with suppressive activity in CD4+CD25+ Tregs, both in vitro and in vivo (243). However, studies in human Tregs have not replicated this observation (244). Similarly, IL-9 generated by activated T-cells promotes the proliferation of Th clones and enhances the suppressive function of CD129 (the IL-9 receptor) expressing Tregs (245, 246). However, these two markers have lost popularity given the lack of evidence for them in man.
CD103, also known as integrin αEβ7 (ITGAE), is expressed on approximately 10-30% of mouse pTregs. Whilst not indicative of activation status, its levels can be upregulated in inflamed tissues (247–249). CD103+ Tregs have been found to exhibit slightly higher suppressive capacity and express higher levels of IL-10, contributing to their anti-inflammatory activity (132). Other findings in mice include the ability of CD103+ Tregs to restrain CD8+ T-cell activation (250). Human CD25+FOXP3+ Tregs, however, show limited expression of CD103 in various tissues, including blood (<5%) (251–254). Nevertheless, human CD4 T-cells can be induced to express CD103 through various stimuli in vitro (255, 256) and the percentage of CD103+ cells among CD4+ CD25+ T-cells is significantly higher than CD4+CD25− T-cells in patients with multiple sclerosis (251). Further investigation is needed to determine whether CD103 is upregulated on human Tregs in other inflammatory conditions.
Recent studies have identified the potential expression of CD70 (157), CD80, and CD86 (257) in Tregs, opening new possibilities for identifying novel Tregs subsets and markers of their potential therapeutic efficacy.
The summary of all the markers discussed above with their alternative designations, and their patterns of expression in human Tregs is presented in Table 1.
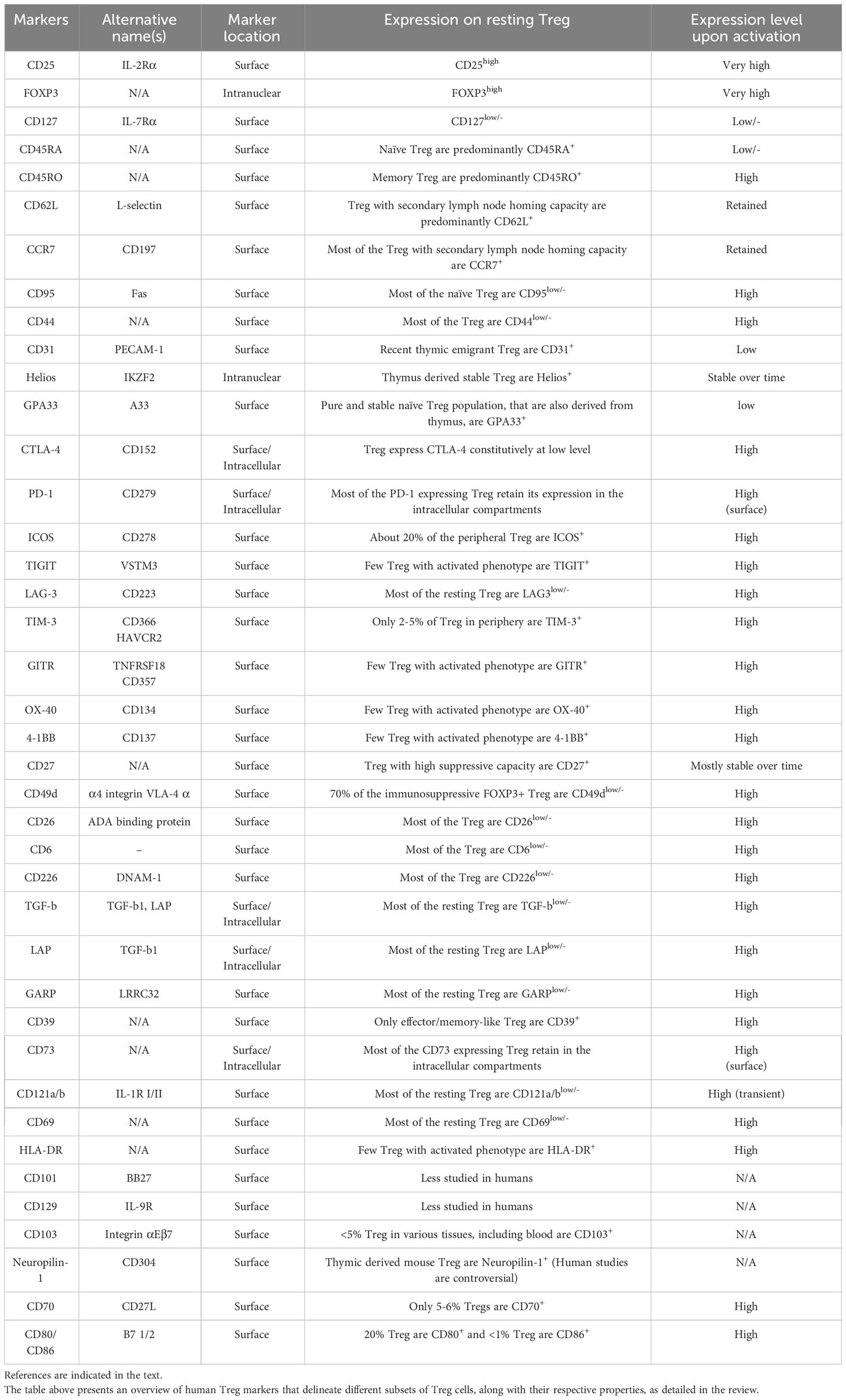
Table 1 Markers of human functional FOXP3+ Tregs.
Chemokines are a group of small heparin-binding proteins that direct the movement of circulating immune cells, influencing their migration within inflamed tissues (258, 259). When initially identified, chemokines were described to be associated with inflammatory diseases (260–262). Later, their involvement in the migration of immune cells was identified (263), followed by production by immune cells themselves (264, 265). A plethora of studies have shown how perturbations in the distribution of Tregs can lead to organ-specific inflammatory diseases (258, 266–275). Chemokines govern the trafficking and homing of Tregs, and understanding how Tregs reach their site of action sets the ground for targeting the pathogenic Treg distribution in the context of cancer and may facilitate tissue-specific targeting in the context of Treg therapy. Variations in chemokine expressions across different human organs is summarized in Figure 3.
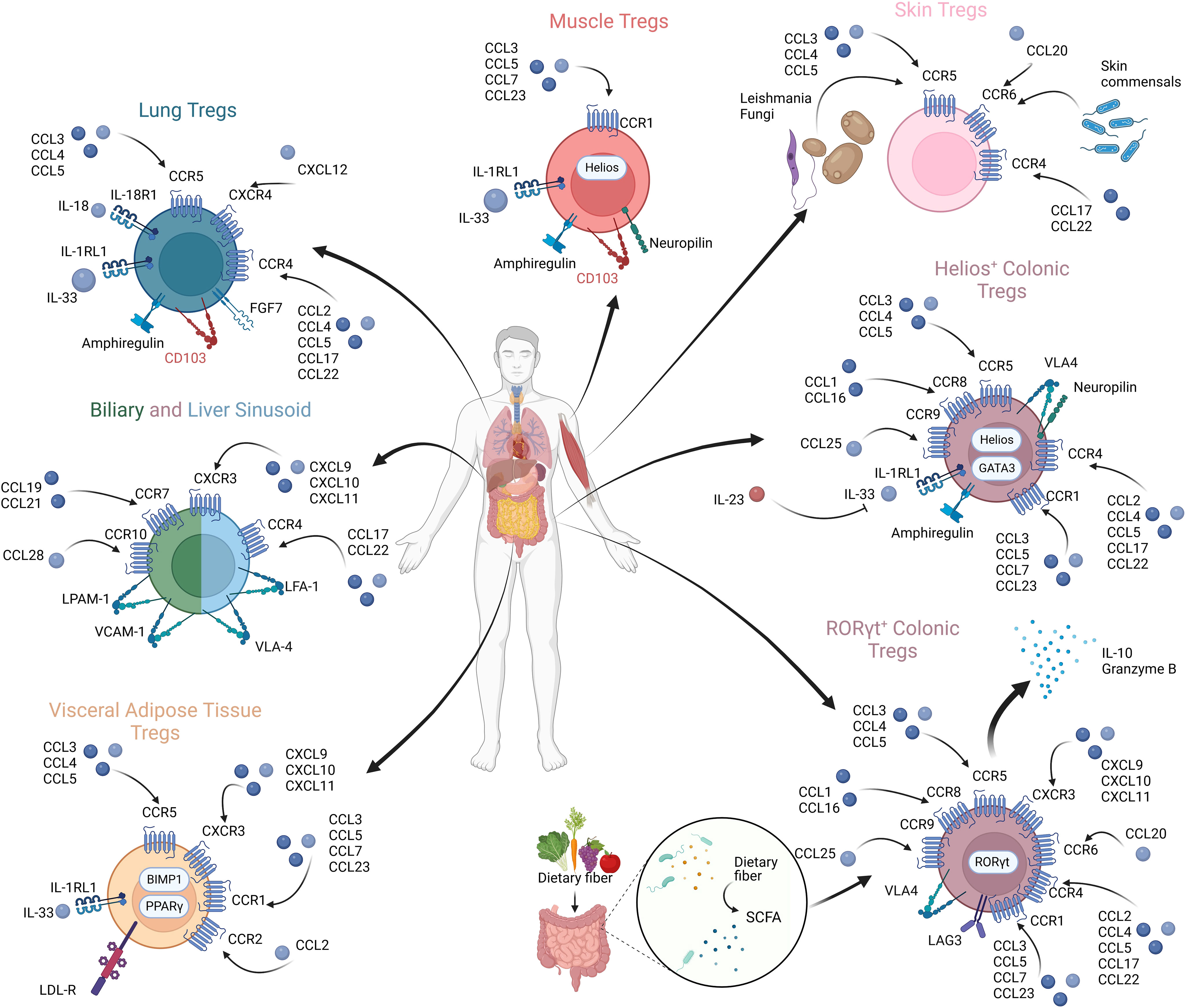
Figure 3 Tregs in diverse anatomical compartments exhibiting differential chemokine expressions and chemokine receptor profiles.
Several integrins have been identified to be crucial to Treg function and localization. The αvβ8 integrin is key to activating latent TGF-β and Tregs lacking αvβ8 are unable to suppress T-cell-mediated inflammation in vivo (276, 277). The α4β7 integrin (also known as LPAM-1 and whose classical ligand is MAD-CAM1) is recognized to play a crucial role in the migration of Tregs to the intestine and associated lymphoid tissues. Loss of the β7 has been shown to lead to disrupted migration, with subsequent colitis observed (266, 278, 279). The expression of the α4β7 integrin and CCR9 enables Tregs to home to the gut’s lamina propria, allowing for tolerance to food antigens, crucial to maintaining gut health (266, 267).
Tregs play a major role in constraining immune response in secondary lymphoid organs, with CCR7 being one of the first chemokine receptors identified on Tregs (30, 280). CD62LhiCCR7+ naïve Tregs favor migration into secondary lymphoid organs where professional antigen presentation allows for antigen-dependent stimulation, modifying the pattern of receptor expression to enable tissue homing (30, 30, 33, 281, 282). Lymph nodes draining these organs act with local dendritic cells to induce Treg activation and action, while CCR7 is downregulated, promoting accumulation in inflamed tissues (38, 283–285). Notably, the loss of CCR7 results in impaired in vivo Treg suppressive activity, while their in vitro activity remains intact. This highlights the significance of CCR7 to Treg function through facilitating localization (33, 38, 286, 287).
Native Th1 responses are dependent on CXCR3-mediated signals. Tregs inhibit stable contacts between CD4+ T-cells and dendritic cells and can influence the expression of CXCR3 ligands in draining lymph nodes. Early data demonstrated that Treg localization in the lymph node, when suppressing CD4+ T-cell responses, was via localization with dendritic cells, rather than the CD4+ T-cells, suggesting they act by inhibiting the dendritic cell-CD4+ T-cell interaction (193, 194). Further data has suggested, Tregs can also prevent the expression of CXCR3 on effector T-cells, inhibiting their migration and trafficking (288, 289). Tregs can also upregulate T-bet and express CXCR3 in response to INF-γ. These CXCR3 expressing Tregs not only resemble Th1 subsets of effector T-cells, expressing T-bet in addition to FOXP3 but also demonstrate an ability to produce INF-γ and IL-10. Crucially, they are essential to the suppression of Th1-mediated inflammation, and imaging data suggests their function is mediated by colocalization with effector Th1 CD4+ and CD8+ T-cells in secondary lymphoid organs (36, 290–292).
CCL3 and CCL4 interactions with CCR5 are recognized to be important to CD4+ and CD8+ T-cell interactions in the lymph node for generating memory CD8+ T-cells (293). The production of CCL3 and CCL4 by Tregs has been observed, attracting CD4+ and CD8+ T-cells via CCR5 expressed on these cells; bringing them into proximity and allowing Tregs to exert their suppressive function. Tregs deficient in CCL3 and CCL4 fail to prevent the progression of experimental autoimmune encephalomyelitis or islet allograft rejection in murine models and Tregs from individuals with type 1 diabetes demonstrate an impaired ability to produce CCL3 and CCL4 (268). Additionally, Tregs can interfere with the priming of CD8+ T-cells by modulating the expression of the CCR5 ligands CCL3, CCL4 and CCL5. The deletion of Tregs leads to the stabilization of interactions between dendritic cells and low-avidity T-cells, compromising the responses of high-avidity memory T-cells and memory responses (289, 294).
CXCR5 is key to the function of Tregs in modulating B cell and humoral responses. Chung et al. were able to show Tregs expressing CXCR5 and Bcl-6 localize to germinal centers, with CXCR5 expression being Bcl-6 dependent (295). These follicular Tregs, are absent in the thymus. Their depletion leads to exaggerated germinal center reactions, including germinal center B cells, affinity maturation of antibodies and the differentiation of plasma cells (289, 295). These results were confirmed by work by Linterman et al., which showed follicular Tregs regulate B cell responses, controlling germinal center responses (289, 296–299). Follicular Tregs are hypothesized to exert their effect via various mechanisms, including the production of IL-10, and TGF-β (300–302). Intriguingly, CXCR5 is not necessary for the localization of follicular Tregs, but the transcription factor Bcl-6 is necessary for function and localization (36, 297, 298, 303, 304).
Finally, a CCR6+ Treg subset has recently been demonstrated to be significant to thymic Treg development and function. These IL-1R2-positive Tregs express CCR6, but not CCR7, which suggests they recirculate from the periphery with an activated phenotype. These Tregs can quench IL-1, indicating they can maintain tTreg development even in inflammatory conditions (305).
Tregs in various tissues demonstrate specificity to site and function. Here we describe selected examples of organ-specific Tregs and the chemokine receptor profiles that facilitate and define their tissue tropism, allowing immunosuppression within their unique environments (285, 306).
Visceral Adipose Tissue: Tregs in visceral adipose tissue (VAT) show enrichment in CCR1, CCR2, CCR5 and CXCR3, as well as the IL-33 receptor, ST2 (also known as IL-1RL1) (307–309). Tregs in visceral adipose tissue migrate under the influence of the IL-33/ST2 axis, with the majority of IL-33 arising from mesenchymal stromal cells (310, 311). Hydroxyprostaglandin dehydrogenase expressed by VAT Tregs exerts an inhibitory effect on conventional T-cells and inflammation (312). Most interestingly, however, is the limited range and repertoire in Treg TCRs present on VAT Tregs, suggesting they respond to specific antigens found in the tissue of residence (275, 309, 313). This may relate to their function in regulating inflammation within VAT, and thus having a restricted repertoire of antigens which they may need to respond to, as it appears VAT Tregs regulate the inflammatory state and promote insulin sensitivity (314). Knockout of this subset of Tregs leads to impaired function in the insulin receptor and chronic inflammation (314).
Skin Tregs: A significant subset of peripheral blood Tregs appear to express CCR4, but Tregs in certain tissues seem to upregulate the expression of this receptor, with its presence noted in skin, liver, lung and intestinal Tregs (315–317). The loss of CCR4 on murine Tregs impairs Treg accumulation in skin and lung tissues, with complete loss of CCR4 in the Treg cell compartment leading to severe dermatitis, pneumonitis and lymphadenopathy (316).
CCR5 has been shown to be key to the function of many tissue resident Tregs, as well as potentially having a role in persistent infections. In a Leishmania major model, CCR5 was essential to the homing of Tregs to Leishmania infected dermal sites, promoting infection and parasite survival (318). Similar results have been observed in chronic fungal granulomata, with chronic inflammation mediated by CCR5 expressing Tregs, as pathogens generate CCR5 ligands, leading to Treg recruitment and dampened immune responses (270).
In neonates, CCR6 is crucial to Treg tropism to the skin. Interactions in early life between commensals and CCR6 expressing Tregs induce Treg accumulation at this site, facilitating tolerance to normal skin commensals, mediated by CCL20 and CCR6 interactions (319, 320).
Skeletal muscle and regulatory T-cells: Tregs in muscle are characterized in part by the presence of CCR1 on the surface, as well as ST2 and the key growth factor Amphiregulin (271). These Tregs seem to demonstrate a key role in promoting muscle repair and regeneration (271, 272, 321–323). Tregs in muscle are predominantly recruited from the circulation via the IL-33/ST2 axis, as is seen in VAT Tregs (321). Once in the muscle, CD103 plays a key role in adherence and retention of these Tregs (324). Interestingly, muscle Tregs seem to show a high level of Helios and Neuropilin, suggesting a thymic origin, however, data clearly shows their recruitment from the circulation. Similar to VAT Tregs, muscle Tregs demonstrate a limited repertoire of TCRs, suggesting specific and limited targets (271). They are recognized to be involved in the repair and regeneration of muscle tissue, having been identified in the inflammatory infiltrate of injured muscle, both in acute and chronic muscle injury, expressing IL-10 where observed (271, 272). Indeed, muscle Tregs were shown to decline with age which likely contributes to defects in muscle regeneration seen in aging (325). Amphiregulin seems to play a crucial role in the regeneration of muscle cells, acting directly on satellite cells, promoting myocyte differentiation in vitro and enhancing muscle repair in vivo (271, 272, 275, 308, 326).
Lung Tregs: Tregs found in the lung, as discussed above, predominantly demonstrate upregulation in CCR4 (327). Similar to muscle Tregs, Tregs in the lung also appear to have a high expression of amphiregulin. Distinct to these Tregs however is the expression of IL18R1, ST2, CCR5, CXCR4, and KGF with downregulation of CXCR5 upon activation (307, 328). Tregs are recruited to the lung via TGFβ expression by a resident IFNAR1hi TNFR2+ conventional dendritic cell 2 (iR2D2) population (329). Tregs play a crucial role in the resolution of lung injury, modulating immune responses and enhancing alveolar epithelial proliferation and tissue repair through the expression of amphiregulin and keratinocyte growth factor (KFG) (330). Inflammatory responses are also dampened by the upregulation of Mmp12, inhibiting neutrophil recruitment. Sik1, meanwhile, has been shown to be downregulated on activation of lung Tregs, leading to an increase in the expression of CD103 (αEβ7) in response, facilitating Treg adhesion and tissue retention (328). Furthermore, Tregs have been highlighted to play a role in asthma. The Notch4 pathway has been found to be upregulated in Tregs in asthmatics. Defective Hippo signaling gives rise to FOXP3 CN2 methylation in Tregs, impairing Treg stability and thus their ability to regulate inflammation in this environment. Thus, the rescue of Tregs in this environment and restabilizing them presents a future therapeutic direction in asthma (273).
Tregs in the gut: The gut provides a dynamic environment, containing a complex interplay of food antigens and resident microbial antigens. As the principal digestive and absorptive site in mammals, an immense range and variety of peptides are present. There is a complex interplay in the gut of various resident and migrating immune cells, from both innate and adaptive systems. Regulating these interactions is essential, and Tregs play a crucial role in orchestrating responses (331). The ablation of colonic Tregs has been demonstrated to lead to aberrant Th1 and Th17 mucosal responses, which are rescued with adoptive transfer (274, 275, 332). Chemokine receptors in gut resident Tregs include CCR1, CCR4, CCR5, CCR8, CCR9 and CXCR3, in addition to the integrin α4β7 (discussed above) (306, 307, 333). This subset of Tregs play a key role in the regulation of complex immune interactions and have been found to perhaps represent two distinct populations, one derived from the thymus and another from peripheral Tregs.
Single-cell transcriptomic data has since shown the colon has populations of tTregs, reflecting those seen in non-lymphoid tissues (expressing GATA-3, neuropillin-1, amphiregulin, ST2 and Helios), and suppressive pTregs (expressing IL-10, Granzyme B, LAG3 and CXCR3) defined by the expression of the transcriptional factor RORγt (306). Interestingly, the microbiome seems to play a crucial role in the recruitment of the latter, being totally absent in germ free mice, and bile acid metabolites impact their function and proliferation (334–337). The two first reports of RORγt Tregs were published together in 2015 (335, 336). Crucially, the data on the function of these cells was divergent, with one group finding that they inhibit Th1/17 responses, while the other demonstrated they respond to Th2 cells. Others have shown that RORγt expression in Tregs may represent a step in the process towards a Th17 Treg phenotype, expressing CCR6 and sharing phenotypic features usually observed in Th17 T-cells (97, 338).
IL-33 has been noted to be present in high quantities in human inflammatory bowel disease, as well as colonic inflammatory lesions. This binds ST2 on Tregs, stimulating responses by enhancing TGF- β1-mediated differentiation and providing signals for the accumulation and maintenance of Tregs in inflamed tissues. Conversely, IL-23, inhibits IL-33 responses. These data suggest that ST2-mediated responses and the balance between IL-33 and IL-23 may contribute to the control of intestinal immune responses (339). Additionally, Blimp-1 has been shown to control the differentiation and function of Tregs, promoting IL-10 generation and ICOS expression in Tregs and suppressing colitis via an IRF4-dependent pathway (340, 341).
Treg/Th17 imbalances are well established to be at play in the pathogenesis of ulcerative colitis. CCR6 Tregs have been shown to be enriched in ulcerative colitis patients and correlated with disease activity. CCR6+ Tregs also demonstrated a higher propensity to secrete IL-17A, suggesting that they may play a part in the ongoing colitis (269). RORγt expressing Tregs have been noted to express CCR6 and this may be a marker of Th17 biased Tregs (338).
In addition to the above, a novel chemokine receptor, GPR15, an orphan GPCR which binds to its own ligand (GPR15L) has been shown to play a role in the trafficking of Tregs to barrier tissues, including skin and colon, where it is involved in the suppression of colitis and graft rejection. GPR15 has been shown to have its expression modulated by both gut microbiota and TGF-β, with knockout of this receptor giving rise to colitis, rescued by transfer of Tregs with intact GPR15, suggesting GPR15 has a crucial role to play in mucosal health (36, 342, 343).
Liver Tregs: The liver, is a key organ to gut health and homeostasis. With a dual blood supply, and 75% of this from the portal system, it attracts a high burden of various food and microbial antigens and consequently, a tolerogenic environment has to be maintained. However, appropriate responses must be mounted to remove pathological antigens and particulates, preventing them from entering the systemic circulation (344).
CXCR3 is critical to Treg migration into the liver and liver sinusoids. This chemokine receptor allows for the colocalization of Tregs within the liver, with CCR4 cognate ligands (CCL-17 and -22) allowing for colocalization with infiltrating dendritic cells within inflammatory infiltrates in liver diseases (344, 345). Diseased livers demonstrate elevated expression of CXCR3 on Tregs (258). This adhesion is mediated by CXCL-9, -10 and -11 expression. In vitro models have demonstrated the upregulation of this chemokine under stress (346). Contacts made by lymphocytes and sinusoidal epithelial cells activates integrins, including LFA-1 and VLA-4 (also known as α4β1) on lymphocyte. These integrins engage adhesion molecules, including ICAM, VCAM and VAP-1 on epithelial cells, facilitating Treg transmigration into the hepatic parenchyma (347). This process enables Tregs to access the hepatic sinusoidal environment and may be utilized for clinical applications, as described below (258).
Additionally, CCL28 production in the biliary epithelium recruits CCR10 expressing Tregs. CCL28 is secreted by primary human cholangiocytes in response to LPS, IL-1 and bile acids. The CCL28 - CCR10+ interaction induces adhesion molecule expression, including VCAM-1, and α4β7. Furthermore, CCR10+ Tregs exhibit low expression of CCR7 (indicating a memory phenotype) and high levels of CXCR3 (348).
In inflamed bile ducts, CCL20 is secreted by biliary epithelial cells. In a Th17 inflammatory environment, replicated in vitro by a cocktail of IL1β, TNFα, IFNγ and IL-17, CCL20 is secreted by human biliary epithelium and leads to recruitment of CCR6 chemokine receptor expressing effector T-cells to sites of biliary inflammation (97, 349, 350). Additionally, CCR6 is a common Treg marker, associated with Th17 skewing. However, in the liver, CCR6 Tregs are liable to phenotypic instability and conversion to a Th17 inflammatory phenotype. This instability challenges their viablility as a candidate for clinical application or cell therapeutic (258). Additionally, it has been noted that CCR6 expressing Tregs in the colon can mount Th17 responses, regulated by IL-10 production in CCR6 expressing Tregs (97, 351, 352).
By demonstrating tissue selectivity and specificity through chemokine receptors, Treg cell products can be designed for organ-specific cell therapy. Liver diseases such as primary sclerosing cholangitis, primary biliary cirrhosis, autoimmune hepatitis and liver transplant rejection all demonstrate a significant contribution from T-cell immune responses to autoantigens expressed in either the biliary epithelium or hepatocytes (353–365). The proof-of-concept AUTUMN study (Autologous regUlatory T-cells infUsion and tracking in autoiMmuNe hepatitis) employed GMP-manufactured Tregs and found infused Tregs favoured migration to the liver (366).
Tregs in the Bone Marrow: Tregs expressing CXCR4 have been shown to collect in the bone marrow, with loss of CXCR4 impairing accumulation in the bone marrow. The depletion of CXCR4 Tregs has also been shown to lead to an increase in B1 mature B cells, exclusively within the bone marrow, with no observed changes in plasma cells or hematopoietic stem cells and no signs of immune activation elsewhere. However, the loss of these CXCR4 Tregs does lead to an increase in total serum IgM levels and a highly specific increase in IgM autoantibodies (367).
Tregs are enriched in the tumour microenvironment, and their enrichment has been correlated with worse outcomes (368). Tregs in the tumour microenvironment supress host immune responses, subverting immune destruction and thus supporting tumor growth (369). Consequently, Tregs have become a focus of cancer immunotherapy (370). However, specifically targeting cancer facilitating Tregs and Tregs in the tumour microenvironment remains a barrier to effective cancer immunotherapy with limited offsite side effects (371, 372).
CCR4 is predominantly expressed in tumor resident Tregs, enabling tumor immune evasion, and is thus regarded as a potential candidate target for cancer immunotherapy (71, 373, 374). CCR8’s more recent identification as a chemokine receptor specifically upregulated in cancer Tregs and it’s correlation with poor outcomes in patients has garnered significant interest (36, 375, 376). This cytokine receptor has been shown to be upregulated in colorectal, non-small cell lung, breast, and other cancers. Additionally, some data has shown CCR8 to be upregulated upon Treg activation, with autocrine production of CCR8’s ligand CCL1 (377). Data on targeting CCR8 in cancer thus far, however, has been conflicting, but some preclinical models have shown promise (378–381).
Nevertheless, both CCR4 and CCR8 appear to be expressed under the control of GATA3 and IRF4, both of which are canonical Th2 transcription factors (36). The expression of these transcription factors on activated Tregs hints at a Th2 skewed Treg population and suggests that CCR4 and CCR8 may be chemokines specific to Th2 Treg function (382–384). GATA-3, meanwhile, has been shown to be essential to Treg function, binding to the CNS2 in Foxp3 promoting Foxp3; with loss of GATA-3 leading to a 50% reduction in FOXP3 mRNA transcripts compared to baseline, with the same seen in other Tregs signature genes (CD25, CTLA-4, GITR). Furthermore, GATA-3 deficient Foxp3 Tregs acquire Th17 cytokine expression profiles (383, 385).
CCR6 has been shown to be a target of certain cancer-derived factors, promoting Treg migration. Eomesodermin has been shown to be generated by oesophageal adenocarcinoma, and this has been shown to drive CCL20 secretion, which binds to CCR6 on Tregs driving Treg chemotaxis and residency intratumorally, promoting cancer growth (386).
GVHD is a complication affecting every second stem cell transplanted patient. Both acute and chronic GvHD are characterised by a systemic inflammation of recipient tissue caused by donor cells in the graft. Severe treatment refractory GvHD can often be lethal. Polyclonal Tregs have successfully been used in named patient programs and clinical trials by us (Trzonkowski; Fuchs(Theil)) and others, both to prevent and to treat GVHD (387–393).
In a murine model of GVHD, the knock-out of CCR5 impaired the migration of donor Tregs to GVHD target organs, leading to lethal outcomes (394). Additionally, CCR8 in GVHD has been shown to promote Treg maintenance, by allowing tolerogenic interactions with donor CD11c+ antigen presenters. In a murine model of islet transplantation, CCR5 along with CCR2, CCR4 and P- and E-selectins were essential to the movement of Tregs from blood into pancreatic islet allografts. Subsequent migration to lymph nodes was found to be CCR2, CCR4 and CCR7 dependent, from which they could inhibit effector T-cell responses in both the allograft and lymph node (395).
Deep understanding of Treg biology, mechanisms of immunosuppression, as well as identification of chemokine receptor patterns responsible for tissue tropism are cornerstones for the generation of genetically modified cells with improved function and enhanced infiltration of the target tissue. Polyclonal autologous Tregs have demonstrated efficacy in several trials, but accumulating evidence from preclinical models demonstrate increased potency of antigen-targeted approaches (1) (388, 390, 396–399). Recent success and FDA/EMA approval of chimeric antigen receptor (CAR)-T-cells in hematological cancers has caused a paradigm shift (400), with increased interest in CAR-Tregs. For example the technology enables generation of Tregs that will precisely target HLA mismatches in transplantation or antigens within inflamed tissues, facilitating direct local suppressive effects. CAR constructs in general are built from: 1) an extracellular ligand binding domain which consists of the antigen-specific variable fragment of heavy and light antibody chains (single-chain variable fragment; scFv), 2) hinge region (providing scFv flexibility), 3) transmemberane domain (that anchors the receptor in plasmalemma) and finally 3) an intracellular costimulatory domain derived from T-cells (401, 402). Until now five generations of CARs have been developed and tested with the main difference in the intracellular domain. The first-generation CARs comprised CD3ζ, a part of the T-cell receptor-CD3 complex, while second-generation CARs combine CD3ζ with costimulatory molecules such as CD28, CD137, CD27 or CD134 delivering a second signal and were the most extensively studied approach. The third-generation CARs comprise of two costimulatory molecules (403–405), fourth generation CARs co-express cytokine or chemokine genes, While the fifth generation receptors contain intracellular domain of a cytokine receptor (e.g., IL2RB chain) that interacts with STAT3 (406–409). In case of genetic Treg modifications, mostly 2nd generation CARs were tested. The Levings´group conducted a comprehensive study exploring the Treg immunosuppressive capacity and stability after incorporation of various costimulatory domains into the CAR (including CD28, ICOS, CTLA4, PD1, GITR, OX40, CD137, and TNFR2) (410). Interestingy, in contrast to anti-tumor conventional CAR-T cells, Tregs with CD28-encoding CAR exhibited superior in vitro and in vivo performance in terms of proliferation, suppression, and delaying GvHD symptoms. In addition, both CD137- and TNFR2-CARs were found to negatively affect Treg function and stability, leading to FOXP3 locus methylation, decreased Helios expression, and reduced suppressive function in vitro and in vivo. Interesting the group published very recently, that Tregs expressing CARs encoding CD28, ICOS, PD1, and GITR, but not 4-1BB or OX40, all extended skin allograft survival in an immunocompetent transplant model (411). With other studies (412, 413) consistently reporting adverse consequences of using CD137 in Tregs, one may conclude that this co-stimulatory domain is suboptimal for CAR-Tregs. However, in the case of flexible modular chimeric antigen receptor technology called universal CAR (UniCAR-Tregs), both CD137 and CD28 costimulation induced robust suppressive capabilities. Nevertheless, due to higher background activation of Treg in case of CD28, UniCARs featuring a CD137-CD3ζ signaling domain were discussed as the preferred constructs for the clinical application of redirected Tregs (414). An intriguing avenue for exploration involves combining CD28 and CD137 domains to potentially optimize CAR-Treg suppression, as reviewed by Zhang, Qunfang et al. (415).
Noteworthy, IL-10 secretion was successfully induced into 4th generation CAR-Tregs, enhancing suppressive function (416). However, no reports on fifth- generation CAR-Tregs have been available yet and their specific effects on CAR-Tregs needs further investigation. The state of knowledge on CAR-Treg generation is summarized in a review by M. Levings, 2020 (417). Further innovative adaptations of CAR designs have emerged with the potential to enhance the durability, stability, proliferative capacity, and function of CAR-Tregs. Additionally, novel CAR-Tregs specific for E.coli-derived flagellin have been designed and introduced into humanized murine models. These FliC-CAR-Tregs were directed to the colon (which confirmed improved homing abilities) and were characterized by higher expression of PD-1, highlighting higher immunosuppressive capacity and potential for application in inflammatory bowel diseases (418). Adoptive Treg therapy is limited by the plasticity of Tregs, potentially transforming into conventional T-cells. To overcome this challenge, additional genetic engineering has been suggested to enhance Treg stability and robustness. Examples include engineered epigenetic and post-transcriptional changes in FOXP3 and metabolic stabilization by enhancing CD39 and CD73 expression. Elevating IL-10, TGFβ, IL-34, IL-35, and FGL-2 can boost Treg function, while suicide genes help manage Treg adverse effects. This idea of the next generation of Super-Tregs with increased function, stability, redirected specificity and survival is summarized by Amini et al. (419).
Engineered antigen-specificity has demonstrated improved responses in mouse models, emphasizing utility (420, 421). In mouse models, researchers demonstrated that lentiviral transduction did not alter the Treg phenotype and HLA-A2 CAR-Tregs were effective in suppression. CAR-Tregs produced small amounts of IFN-ɣ, but more importantly, secreted the highest amounts of IL-10. Finally, transduced cells migrated more rapidly through HLA-A2+ HUVECs and contributed to the survival of human skin grafts (416).
Currently, CAR-Tregs remain under investigation (422), and two clinical trials focused on transplantation are registered on clinicaltrials.gov. Sangamo Therapeutics in 2021 initiated the first trial using CAR-Tregs (NCT04817774), infusing TX200-TR101 into HLA-A02-negative patients awaiting kidney transplants from HLA-A02-positive donors. Encouraging preclinical studies demonstrated specific activation and potent allogeneic Tconv suppression by TX200-TR101, both in vitro and vivo. No side effects have been reported underscoring its potential as a safe and effective therapeutic alternative (423–425). Quell Therapeutics’s QEL-001, similar to TX200-TR101, utilizes an HLA-A2-specific CAR to abrogate immunosuppression (426, 427)Tregs, and the LIBERATE clinical trial (NCT05234190) is currently enrolling patients who have received HLA A2-mismatched liver transplants 12 months to 5 years prior the time of enrolment.
Sonoma Biotechnologies’s pipeline includes SBT-77-7101, a CAR-Treg product specific to citrullinated vimentin (CV), a known antigen present in the synovial fluid (SF) of Rheumatoid arthritis (RA) patients (428). Preclinical studies (429–432) have shown promise in using Treg therapy for Systemic lupus erythematosus (SLE), demonstrating the as-yet unrealized potential benefit of these novel technologies.
Additionally, an inventive way of using Tregs in autoimmune disease treatment is planning to be introduced into clinical trials on Multiple sclerosis (MS) and type 1 diabetes. The company “Abata Therapeutics” started to engineer Tregs expressing TCRs recognizing tissue-restricted antigens. Similar to the CAR concept, their TCR-based targeted methods reduce systemic suppressive response limiting it to the specific tissue to which autoimmune response is directed. The company PolTREG develops TCR-engineered Tregs in type 1 diabetes. Interestingly, this company works in neuroinflammatory conditions, such as MS and amyotrophic lateral sclerosis, using a CAR-based concept in which CAR-Tregs are designed to strengthen the blood-brain barrier. As the engineered Tregs landscape expands, continued investigation will unveil the most effective strategies to unlock the full potential of engineered Tregs.
Tregs constitute a minor fraction of the broader population of CD4+ T-cells. The lack of an exclusive Treg marker together with phenotypical similarities to activated CD4+ effector cells present challenges in the context of cell therapy. However, a variety of markers, often applied in combinatorial approaches identifies functional suppressive Tregs and distinct subsets, and contributed to an enhanced understanding of biological processes. As our knowledge improves and we enter the era of engineered Tregs, significant strides addressing pathological immune-mediated processes can be anticipated. Learnings from ongoing clinical trials, advancement in methods to determine Treg function, together with means to identify patients who are most likely to benefit from Treg therapy, will determine the fate of Treg cell therapy.
SS: Conceptualization, Writing – original draft, Writing – review & editing, Validation. KK: Writing – original draft, Writing – review & editing, Validation. MG: Writing – original draft, Validation. YH: Writing – review & editing, Validation. DI-G: Writing – original draft, Validation. MP-M: Writing – original draft, Validation. JS: Writing – original draft, Validation. MT: Writing – original draft, Validation. JM: Writing – review & editing, Validation. KL: Writing – review & editing, Validation. NM-T: Supervision, Writing – review & editing, Validation. PT: Supervision, Writing – review & editing, Validation. YO: Supervision, Writing – review & editing. AF: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding for publication was received from ‘‘The Saxon State and University Library (SLUB), Dresden’’. SS and YH are funded by the German Federal Ministry of Education and Research under the funding code 03ZU1111AA as part of the Cluster4Future, SaxoCell. JM, KL, and AF are funded by Mildred Scheel Early Career Center (MSNZ), German Cancer Aid (Deutsche Krebshilfe, DKH). KK and YO are funded by Sir Jules Thorn Biomedical Research Charity, London. KK is funded by the National Institute for Health and Care Research (NIHR). NM-T is supported by the project “International Centre for Cancer Vaccine Science” that is carried out within the International Research Agendas Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund. NM-T is supported by the project “International Centre for Cancer Vaccine Science” that is carried out within the International Research Agendas Programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (MAB/2017/3).
We would like to express our sincere appreciation to all the contributors and reviewers for their valuable insights and contributions to this review.
The authors MG, Dl, MP-M, JS, MT and PT are employed by Poltreg S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) PT and NM-T declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nishizuka Y, Tanaka Y, Sakakura T, Kojima A. Murine thyroiditis induced by neonatal thymectomy. Experientia (1973) 29(11):1396–8. doi: 10.1007/BF01922839
2. Kojima A, Tanaka-Kojima Y, Sakakura T, Nishizuka Y. Spontaneous development of autoimmune thyroiditis in neonatally thymectomized mice. Lab Invest (1976) 34(6):550–7.
3. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology (1970) 18(5):723–37.
5. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64. doi: 10.4049/jimmunol.155.3.1151
6. Hall BM, Pearce NW, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. III. Further characterization of the CD4+ suppressor cell and its mechanisms of action. J Exp Med (1990) 171(1):141–57. doi: 10.1084/jem.171.1.141
7. Pearce NW, Spinelli A, Gurley KE, Hall BM. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. V. Dependence of CD4+ suppressor cells on the presence of alloantigen and cytokines, including interleukin 2. Transplantation (1993) 55(2):374–80. doi: 10.1097/00007890-199302000-00027
8. Thornton AM, Shevach EM. CD4+CD25+ Immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188(2):287–96. doi: 10.1084/jem.188.2.287
9. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human cd4+Cd25+ T cells with regulatory properties isolated from peripheral blood. J Exp Med (2001) 193(11):1285–94. doi: 10.1084/jem.193.11.1285
10. Levings MK, Sangregorio R, Roncarolo MG. Human cd25+Cd4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med (2001) 193(11):1295–302. doi: 10.1084/jem.193.11.1295
11. Ng WF, Duggan PJ, Ponchel F, Matarese G, Lombardi G, Edwards AD, et al. Human CD4(+)CD25(+) cells: a naturally occurring population of regulatory T cells. Blood (2001) 98(9):2736–44. doi: 10.1182/blood.V98.9.2736
12. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol (2001) 167(3):1245–53. doi: 10.4049/jimmunol.167.3.1245
13. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
14. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4(4):337–42. doi: 10.1038/ni909
15. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
16. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
17. Roncador G, Brown PJ, Maestre L, Hue S, Martínez-Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol (2005) 35(6):1681–91. doi: 10.1002/eji.200526189
18. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
19. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
20. Wang J, Ioan-Facsinay A, van der Voort EIH, Huizinga TWJ, Toes REM. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol (2007) 37(1):129–38. doi: 10.1002/eji.200636435
21. Liu W, Putnam AL, Xu-yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
22. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203(7):1693–700. doi: 10.1084/jem.20060468
23. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol (2007) 37(9):2378–89. doi: 10.1002/eji.200737594
24. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
25. Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4+ Tregs. J Clin Invest (2005) 115(7):1953–62. doi: 10.1172/JCI23963
26. Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, et al. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood (2006) 107(7):2830–8. doi: 10.1182/blood-2005-06-2403
27. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108(13):4260–7. doi: 10.1182/blood-2006-06-027409
28. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol (2009) 39(4):1088–97. doi: 10.1002/eji.200838904
29. Tosello V, Odunsi K, Souleimanian NE, Lele S, Shrikant P, Old LJ, et al. Differential expression of CCR7 defines two distinct subsets of human memory CD4+CD25+ Tregs. Clin Immunol (2008) 126(3):291–302. doi: 10.1016/j.clim.2007.11.008
30. Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses L-selectin and high levels of CCR7. J Immunol (2002) 169(5):2461–5. doi: 10.4049/jimmunol.169.5.2461
31. Samy ET, Parker LA, Sharp CP, Tung KSK. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J Exp Med (2005) 202(6):771–81. doi: 10.1084/jem.20041033
32. Fu S, Yopp AC, Mao X, Chen D, Zhang N, Chen D, et al. CD4+ CD25+ CD62+ T-regulatory cell subset has optimal suppressive and proliferative potential. Am J Transplant (2004) 4(1):65–78. doi: 10.1046/j.1600-6143.2003.00293.x
33. Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med (2007) 204(4):735–45. doi: 10.1084/jem.20061405
34. Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, Levine BL, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood (2004) 104(12):3804–12. doi: 10.1182/blood-2004-05-1850
35. Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, et al. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood (2005) 105(5):2220–6. doi: 10.1182/blood-2004-05-2044
36. Dikiy S, Rudensky AY. Principles of regulatory T cell function. Immunity (2023) 56(2):240–55. doi: 10.1016/j.immuni.2023.01.004
37. Huehn J, Siegmund K, Lehmann JCU, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ Regulatory T cells. J Exp Med (2004) 199(3):303–13. doi: 10.1084/jem.20031562
38. Menning A, Höpken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol (2007) 37(6):1575–83. doi: 10.1002/eji.200737201
39. Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood (2004) 104(3):895–903. doi: 10.1182/blood-2004-01-0086
40. Fritzsching B, Oberle N, Pauly E, Geffers R, Buer J, Poschl J, et al. Naive regulatory T cells: a novel subpopulation defined by resistance toward CD95L-mediated cell death. Blood (2006) 108(10):3371–8. doi: 10.1182/blood-2006-02-005660
41. Firan M, Dhillon S, Estess P, Siegelman MH. Suppressor activity and potency among regulatory T cells is discriminated by functionally active CD44. Blood (2006) 107(2):619–27. doi: 10.1182/blood-2005-06-2277
42. Bollyky PL, Lord JD, Masewicz SA, Evanko SP, Buckner JH, Wight TN, et al. Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J Immunol (2007) 179(2):744–7. doi: 10.4049/jimmunol.179.2.744
43. Bollyky PL, Falk BA, Long A, Preisinger A, Braun KR, Wu RP, et al. CD44 co-stimulation promotes FoxP3+ regulatory T-cell persistence and function via production of IL-2, IL-10 and TGF-beta. J Immunol (2009) 183(4):2232–41. doi: 10.4049/jimmunol.0900191
44. Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol (2004) 173(12):7259–68. doi: 10.4049/jimmunol.173.12.7259
45. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4+ FoxP3+ T cells. Curr Opin Immunol (2009) 21(3):281–5. doi: 10.1016/j.coi.2009.05.007
46. Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30(5):646–55. doi: 10.1016/j.immuni.2009.05.001
47. da Silva Martins M, Piccirillo CA. Functional stability of Foxp3+ regulatory T cells. Trends Mol Med (2012) 18(8):454–62. doi: 10.1016/j.molmed.2012.06.001
48. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol (2006) 18(8):1197–209. doi: 10.1093/intimm/dxl060
49. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity (2007) 27(5):786–800. doi: 10.1016/j.immuni.2007.09.010
50. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of helios, an ikaros transcription factor family member, differentiates thymic-derived from peripherally induced foxp3+ T regulatory cells. J Immunol (2010) 184(7):3433–41. doi: 10.4049/jimmunol.0904028
51. MacDonald KG, Han JM, Himmel ME, Huang Q, Kan B, Campbell AIM, et al. Response to comment on ‘helios+ and helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans’. J Immunol (2013) 190(9):4440–1. doi: 10.4049/jimmunol.1390019
52. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186(7):3918–26. doi: 10.4049/jimmunol.1003099
53. Kim YC, Bhairavabhotla R, Yoon J, Golding A, Thornton AM, Tran DQ, et al. Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood (2012) 119(12):2810–8. doi: 10.1182/blood-2011-09-377895
54. Thornton AM, Shevach EM. Helios: still behind the clouds. Immunology (2019) 158(3):161–70. doi: 10.1111/imm.13115
55. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259(1):88–102. doi: 10.1111/imr.12160
56. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol (2012) 188(3):976–80. doi: 10.4049/jimmunol.1102964
57. Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PloS One (2011) 6(8):e24226. doi: 10.1371/journal.pone.0024226
58. Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol (2013) 190(5):2001–8. doi: 10.4049/jimmunol.1201379
59. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, Kisielow P, et al. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ Regulatory T cells. PloS One (2015) 10(10):e0141161. doi: 10.1371/journal.pone.0141161
60. Elkord E. Helios should not be cited as a marker of human thymus-derived tregs. Commentary: helios(+) and helios(-) cells coexist within the natural FOXP3(+) T regulatory cell subset in humans. Front Immunol (2016) 7:276. doi: 10.3389/fimmu.2016.00276
61. Haas J, Fritzsching B, Trübswetter P, Korporal M, Milkova L, Fritz B, et al. Prevalence of newly generated naive regulatory T cells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol (2007) 179(2):1322–30. doi: 10.4049/jimmunol.179.2.1322
62. Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol (2010) 184(8):4317–26. doi: 10.4049/jimmunol.0903781
63. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
64. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209(10):1713–22. doi: 10.1084/jem.20120822
65. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209(10):1723–42. doi: 10.1084/jem.20120914
66. Battaglia A, Buzzonetti A, Monego G, Peri L, Ferrandina G, Fanfani F, et al. Neuropilin-1 expression identifies a subset of regulatory T cells in human lymph nodes that is modulated by preoperative chemoradiation therapy in cervical cancer. Immunology (2008) 123(1):129–38. doi: 10.1111/j.1365-2567.2007.02737.x
67. Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, et al. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol (2009) 39(6):1466–71. doi: 10.1002/eji.200839040
68. Frey D, Coelho V, Petrausch U, Schaefer M, Keilholz U, Thiel E, et al. Surface expression of gpA33 is dependent on culture density and cell-cycle phase and is regulated by intracellular traffic rather than gene transcription. Cancer Biother Radiopharm (2008) 23(1):65–73. doi: 10.1089/cbr.2007.0407
69. Opstelten R, de Kivit S, Slot MC, van den Biggelaar M, Iwaszkiewicz-Grześ D, Gliwiński M, et al. GPA33: A marker to identify stable human regulatory T cells. J Immunol (2020) 204(12):3139–48. doi: 10.4049/jimmunol.1901250
70. Morgana F, Opstelten R, Slot MC, Scott AM, van Lier RAW, Blom B, et al. Single-cell transcriptomics reveals discrete steps in regulatory T cell development in the human thymus. J Immunol (2022) 208(2):384–95. doi: 10.4049/jimmunol.2100506
71. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
72. Marin-Acevedo JA, Kimbrough EO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol (2021) 14(1):45. doi: 10.1186/s13045-021-01056-8
73. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-X
74. Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol (1997) 159(1):144–51. doi: 10.4049/jimmunol.159.1.144
75. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12(4):431–40. doi: 10.1016/S1074-7613(00)80195-8
76. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by cd25+Cd4+Regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J Exp Med (2000) 192(2):303–10. doi: 10.1084/jem.192.2.303
77. Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med (2000) 192(2):295–302. doi: 10.1084/jem.192.2.295
78. Birebent B, Lorho R, Lechartier H, de Guibert S, Alizadeh M, Vu N, et al. Suppressive properties of human CD4+CD25+ regulatory T cells are dependent on CTLA-4 expression. Eur J Immunol (2004) 34(12):3485–96. doi: 10.1002/eji.200324632
79. Manzotti CN, Tipping H, Perry LCA, Mead KI, Blair PJ, Zheng Y, et al. Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol (2002) 32(10):2888–96. doi: 10.1002/1521-4141(2002010)32:10<2888::AID-IMMU2888>3.0.CO;2-F
80. Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34(11):2996–3005. doi: 10.1002/eji.200425143
81. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10–producing, nonproliferating cd4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med (2000) 192(9):1213–22. doi: 10.1084/jem.192.9.1213
82. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis Found Symp (2003) 252:67–88; discussion 88-91, 106–14. doi: 10.1002/0470871628.ch6
83. Schmidt EM, Wang CJ, Ryan GA, Clough LE, Qureshi OS, Goodall M, et al. Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J Immunol (2009) 182(1):274–82. doi: 10.4049/jimmunol.182.1.274
84. Tang AL, Teijaro JR, Njau MN, Chandran SS, Azimzadeh A, Nadler SG, et al. CTLA4 expression is an indicator and regulator of steady-state CD4+ FoxP3+ T cell homeostasis. J Immunol (2008) 181(3):1806–13. doi: 10.4049/jimmunol.181.3.1806
85. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
86. Holt MP, Punkosdy GA, Glass DD, Shevach EM, Signaling TCR. and CD28/CTLA-4 signaling cooperatively modulate T regulatory cell homeostasis. J Immunol (2017) 198(4):1503–11. doi: 10.4049/jimmunol.1601670
87. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
88. Kim GR, Kim WJ, Lim S, Lee HG, Koo JH, Nam KH, et al. In vivo induction of regulatory T cells via CTLA-4 signaling peptide to control autoimmune encephalomyelitis and prevent disease relapse. Adv Sci (Weinh). (2021) 8(14):2004973. doi: 10.1002/advs.202004973
89. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A. (2019) 116(2):609–18. doi: 10.1073/pnas.1812186116
90. Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J Immunol (2006) 176(5):2808–16. doi: 10.4049/jimmunol.176.5.2808
91. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
92. Lin C, Huang H, Hsieh C, Fan C, Lee Y. Jagged1-expressing adenovirus-infected dendritic cells induce expansion of Foxp3+ regulatory T cells and alleviate T helper type 2-mediated allergic asthma in mice. Immunology (2019) 156(2):199–212. doi: 10.1111/imm.13021
93. Woods DM, Ramakrishnan R, Sodré AL, Berglund A, Weber J. PD-1 blockade induces phosphorylated STAT3 and results in an increase of Tregs with reduced suppressive function. J Immunol (2017) 198(1_Supplement):56.7–7. doi: 10.4049/jimmunol.198.Supp.56.7
94. Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, et al. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proc Natl Acad Sci U.S.A. (2012) 109(26):10468–73. doi: 10.1073/pnas.1201131109
95. Cai J, Wang D, Zhang G, Guo X. The role of PD-1/PD-L1 axis in treg development and function: implications for cancer immunotherapy. Onco Targets Ther (2019) 12:8437–45. doi: 10.2147/OTT.S221340
96. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
97. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi: 10.1182/blood-2011-11-392324
98. Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two functional subsets of Foxp3+ regulatory T cells in human thymus and periphery. Immunity (2008) 28(6):870–80. doi: 10.1016/j.immuni.2008.03.018
99. Löhning M, Hutloff A, Kallinich T, Mages HW, Bonhagen K, Radbruch A, et al. Expression of ICOS in vivo defines CD4+ Effector T cells with high inflammatory potential and a strong bias for secretion of interleukin 10. J Exp Med (2003) 197(2):181–93. doi: 10.1084/jem.20020632
100. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, et al. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol (2008) 180(2):774–82. doi: 10.4049/jimmunol.180.2.774
101. Busse M, Krech M, Meyer-Bahlburg A, Hennig C, Hansen G. ICOS mediates the generation and function of CD4+CD25+Foxp3+ regulatory T cells conveying respiratory tolerance. J Immunol (2012) 189(4):1975–82. doi: 10.4049/jimmunol.1103581
102. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci (2018) 15(7):666–73. doi: 10.7150/ijms.23940
103. Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL. ICOS-deficient regulatory T cells display normal induction of Il10 but readily downregulate expression of Foxp3. J Immunol (2019) 202(4):1039–44. doi: 10.4049/jimmunol.1801266
104. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
105. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
106. Workman CJ, Vignali DAA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol (2005) 174(2):688–95. doi: 10.4049/jimmunol.174.2.688
107. Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J (2007) 26(2):494–504. doi: 10.1038/sj.emboj.7601520
108. Woo SR, Li N, Bruno TC, Forbes K, Brown S, Workman C, et al. Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. Eur J Immunol (2010) 40(6):1768–77. doi: 10.1002/eji.200939874
109. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
110. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol (2011) 186(9):5173–83. doi: 10.4049/jimmunol.1002050
111. Slevin SM, Garner LC, Lahiff C, Tan M, Wang LM, Ferry H, et al. Lymphocyte activation gene (LAG)-3 is associated with mucosal inflammation and disease activity in ulcerative colitis. J Crohns Colitis (2020) 14(10):1446–61. doi: 10.1093/ecco-jcc/jjaa054
112. Yu X, Harden K C, Gonzalez L, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
113. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the co-inhibitory molecule TIGIT selectively inhibit pro-inflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
114. Li S, Zhang P, Li A, Bao J, Pan Z, Jie Y. TIGIT-fc prolongs corneal allograft survival in mice by upregulating TIGIT/CD226 expression and the proportion of helios + Foxp3 + Treg cells. Transplantation (2023) 107(2):372–81. doi: 10.1097/TP.0000000000004257
115. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MWL, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest (2015) 125(11):4053–62. doi: 10.1172/JCI81187
116. Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.699895
117. Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res (2014) 2(5):393–8. doi: 10.1158/2326-6066.CIR-14-0039
118. Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, et al. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci (2017) 18(3):645. doi: 10.3390/ijms18030645
119. Gupta S, Thornley TB, Gao W, Larocca R, Turka LA, Kuchroo VK, et al. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J Clin Invest (2012) 122(7):2395–404. doi: 10.1172/JCI45138
120. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One (2012) 7(2):e30676. doi: 10.1371/journal.pone.0030676
121. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PloS One (2013) 8(3):e58006. doi: 10.1371/journal.pone.0058006
122. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ Regulatory T cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
123. Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer (2013) 109(10):2629–35. doi: 10.1038/bjc.2013.645
124. Liu Z, McMichael EL, Shayan G, Li J, Chen K, Srivastava R, et al. Novel effector phenotype of Tim-3+ regulatory T cells leads to enhanced suppressive function in head and neck cancer patients. Clin Cancer Res (2018) 24(18):4529–38. doi: 10.1158/1078-0432.CCR-17-1350
125. Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology (2013) 2(4):e23849. doi: 10.4161/onci.23849
126. Shen P, Yue R, Tang J, Si H, Shen L, Guo C, et al. Preferential Tim-3 expression on Treg and CD8+ T cells, supported by tumor-associated macrophages, is associated with worse prognosis in gastric cancer. Am J Transl Res (2016) 8(8):3419–28.
127. Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep (2021) 36(11):109699. doi: 10.1016/j.celrep.2021.109699
128. Ulbar F, Villanova I, Giancola R, Baldoni S, Guardalupi F, Fabi B, et al. Clinical-grade expanded regulatory T cells are enriched with highly suppressive cells producing IL-10, granzyme B, and IL-35. Biol Blood Marrow Transplant (2020) 26(12):2204–10. doi: 10.1016/j.bbmt.2020.08.034
129. Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, Sakaguchi S, et al. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol (2004) 172(12):7306–14. doi: 10.4049/jimmunol.172.12.7306
130. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity (2016) 44(5):1005–19. doi: 10.1016/j.immuni.2016.04.019
131. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
132. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16(2):311–23. doi: 10.1016/S1074-7613(02)00280-7
133. Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, et al. Functional assay for human CD4+CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neurosci Res (2003) 74(2):296–308. doi: 10.1002/jnr.10766
134. de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol (2004) 172(10):6435–43. doi: 10.4049/jimmunol.172.10.6435
135. Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, et al. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol (2004) 173(5):3119–30. doi: 10.4049/jimmunol.173.5.3119
136. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
137. Vence LM, Bucktrout SL, Fernandez I, Blando JM, Smith BM, Mahne AE, et al. Characterization and comparison of GITR expression in solid tumors. Clin Cancer Res (2019) 25(21):6501–10. doi: 10.1158/1078-0432.CCR-19-0289
138. Pedroza-Gonzalez A, Zhou G, Singh SP, Boor PP, Pan Q, Grunhagen D, et al. GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology (2015) 4(12):e1051297. doi: 10.1080/2162402X.2015.1051297
139. Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol (2002) 3(1):33–41. doi: 10.1038/ni743
140. Zheng G, Wang B, Chen A. The 4-1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J Immunol (2004) 173(4):2428–34. doi: 10.4049/jimmunol.173.4.2428
141. Zhang P, Gao F, Wang Q, Wang X, Zhu F, Ma C, et al. Agonistic anti-4-1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol (2007) 66(4):435–40. doi: 10.1111/j.1365-3083.2007.01994.x
142. Lee J, Lee EN, Kim EY, Park HJ, Chang CY, Jung DY, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol Lett (2005) 101(2):210–6. doi: 10.1016/j.imlet.2005.06.001
143. Hippen KL, Harker-Murray P, Porter SB, Merkel SC, Londer A, Taylor DK, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood (2008) 112(7):2847–57. doi: 10.1182/blood-2008-01-132951
144. Schoenbrunn A, Frentsch M, Kohler S, Keye J, Dooms H, Moewes B, et al. A converse 4-1BB and CD40 ligand expression pattern delineates activated regulatory T cells (Treg) and conventional T cells enabling direct isolation of alloantigen-reactive natural Foxp3+ Treg. J Immunol (2012) 189(12):5985–94. doi: 10.4049/jimmunol.1201090
145. Nowak A, Lock D, Bacher P, Hohnstein T, Vogt K, Gottfreund J, et al. CD137+CD154– expression as a regulatory T cell (Treg)-specific activation signature for identification and sorting of stable human tregs from in vitro expansion cultures. Front Immunol (2018) 9:199. doi: 10.3389/fimmu.2018.00199
146. Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol (2004) 172(6):3580–9. doi: 10.4049/jimmunol.172.6.3580
147. Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood (2007) 110(7):2501–10. doi: 10.1182/blood-2007-01-070748
148. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood (2005) 105(7):2845–51. doi: 10.1182/blood-2004-07-2959
149. Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 costimulation inhibits foxp3 expression and treg induction via BATF3-dependent and independent mechanisms. Cell Rep (2018) 24(3):607–18. doi: 10.1016/j.celrep.2018.06.052
150. Deng J, Zhao S, Zhang X, Jia K, Wang H, Zhou C, et al. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Onco Targets Ther (2019) 12:7347–53. doi: 10.2147/OTT.S214211
151. Polesso F, Sarker M, Weinberg AD, Murray SE, Moran AE. OX40 agonist tumor immunotherapy does not impact regulatory T cell suppressive function. J Immunol (2019) 203(7):2011–9. doi: 10.4049/jimmunol.1900696
152. Gravestein LA, Nieland JD, Kruisbeek AM, Borst J. Novel mAbs reveal potent co-stimulatory activity of murine CD27. Int Immunol (1995) 7(4):551–7. doi: 10.1093/intimm/7.4.551
153. Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, et al. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med (2005) 201(11):1793–803. doi: 10.1084/jem.20050085
154. Koenen HJPM, Fasse E, Joosten I. CD27/CFSE-based ex vivo selection of highly suppressive alloantigen-specific human regulatory T cells. J Immunol (2005) 174(12):7573–83. doi: 10.4049/jimmunol.174.12.7573
155. Duggleby RC, Shaw TNF, Jarvis LB, Kaur G, Hill Gaston JS. CD27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood CD4+ CD25+ cells. Immunology (2007) 121(1):129–39. doi: 10.1111/j.1365-2567.2006.02550.x
156. Muth S, Klaric A, Radsak M, Schild H, Probst HC. CD27 expression on Treg cells limits immune responses against tumors. J Mol Med (Berl). (2022) 100(3):439–49. doi: 10.1007/s00109-021-02116-9
157. Arroyo Hornero R, Georgiadis C, Hua P, Trzupek D, He LZ, Qasim W, et al. CD70 expression determines the therapeutic efficacy of expanded human regulatory T cells. Commun Biol (2020) 3(1):1–17. doi: 10.1038/s42003-020-1097-8
158. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
159. Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, et al. T-cell exhaustion in chronic infections: reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol (2018) 9:2569. doi: 10.3389/fimmu.2018.02569
160. Fribourg M, Anderson L, Fischman C, Cantarelli C, Perin L, La Manna G, et al. T-cell exhaustion correlates with improved outcomes in kidney transplant recipients. Kidney Int (2019) 96(2):436–49. doi: 10.1016/j.kint.2019.01.040
161. Thorp EB, Stehlik C, Ansari MJ. T-cell exhaustion in allograft rejection and tolerance. Curr Opin Organ Transplant (2015) 20(1):37–42. doi: 10.1097/MOT.0000000000000153
162. Lamarche C, Ward-Hartstonge K, Mi T, Lin DTS, Huang Q, Brown A, et al. Tonic-signaling chimeric antigen receptors drive human regulatory T cell exhaustion. Proc Natl Acad Sci (2023) 120(14):e2219086120. doi: 10.1073/pnas.2219086120
163. Ou K, Hamo D, Schulze A, Roemhild A, Kaiser D, Gasparoni G, et al. Strong expansion of human regulatory T cells for adoptive cell therapy results in epigenetic changes which may impact their survival and function. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.751590
164. Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature (1992) 356(6364):63–6. doi: 10.1038/356063a0
165. Waldrop SL, Davis KA, Maino VC, Picker LJ. Normal human CD4+ memory T cells display broad heterogeneity in their activation threshold for cytokine synthesis. J Immunol (1998) 161(10):5284–95. doi: 10.4049/jimmunol.161.10.5284
166. Kleinewietfeld M, Starke M, Di Mitri D, Borsellino G, Battistini L, Rötzschke O, et al. CD49d provides access to ‘untouched’ human Foxp3+ Treg free of contaminating effector cells. Blood (2009) 113(4):827–36. doi: 10.1182/blood-2008-04-150524
167. Ukena SN, Höpting M, Velaga S, Ivanyi P, Grosse J, Baron U, et al. Isolation strategies of regulatory T cells for clinical trials: phenotype, function, stability, and expansion capacity. Exp Hematol (2011) 39(12):1152–60. doi: 10.1016/j.exphem.2011.08.010
168. Haase D, Puan KJ, Starke M, Lai TS, Soh MYL, Karunanithi I, et al. Large-scale isolation of highly pure ‘Untouched’ Regulatory T cells in a GMP environment for adoptive cell therapy. J Immunother (2015) 38(6):250–8. doi: 10.1097/CJI.0000000000000083
169. Schrader WP, West CA, Miczek AD, Norton EK. Characterization of the adenosine deaminase-adenosine deaminase complexing protein binding reaction. J Biol Chem (1990) 265(31):19312–8. doi: 10.1016/S0021-9258(17)30659-2
170. Richard E, Arredondo-Vega FX, Santisteban I, Kelly SJ, Patel DD, Hershfield MS. The binding site of human adenosine deaminase for CD26/Dipeptidyl peptidase IV: the Arg142Gln mutation impairs binding to cd26 but does not cause immune deficiency. J Exp Med (2000) 192(9):1223–36. doi: 10.1084/jem.192.9.1223
171. Ohnuma K, Dang NH, Morimoto C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol (2008) 29(6):295–301. doi: 10.1016/j.it.2008.02.010
172. Cordero OJ, Salgado FJ, Viñuela JE, Nogueira M. Interleukin-12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology (1997) 197(5):522–33. doi: 10.1016/S0171-2985(97)80084-8
173. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem (2010) 285(10):7176–86. doi: 10.1074/jbc.M109.047423
174. Salgado FJ, Pérez-Díaz A, Villanueva NM, Lamas O, Arias P, Nogueira M. CD26: a negative selection marker for human Treg cells. Cytometry A. (2012) 81(10):843–55. doi: 10.1002/cyto.a.22117
175. Piekarska A, Pérès M, Toton M, Kulczycka M, Lewandowski K, Vergez F. Identification of circulating regulatory T lymphocytes with membrane markers - a new multiparameter flow cytometry protocol. Folia Histochem Cytobiol (2021) 59(2):75–85. doi: 10.5603/FHC.a2021.0014
176. Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26high T cells elicit tumor immunity against multiple Malignancies via enhanced migration and persistence. Nat Commun (2017) 8(1):1961. doi: 10.1038/s41467-017-01867-9
177. Mandapathil M, Szczepanski M, Harasymczuk M, Ren J, Cheng D, Jackson EK, et al. CD26 expression and adenosine deaminase activity in regulatory T cells (Treg) and CD4(+) T effector cells in patients with head and neck squamous cell carcinoma. Oncoimmunology (2012) 1(5):659–69. doi: 10.4161/onci.20387
178. Zhao X, Wang W, Zhang K, Yang J, Fuchs H, Fan H. Involvement of CD26 in differentiation and functions of th1 and th17 subpopulations of T lymphocytes. J Immunol Res (2021) 2021:6671410. doi: 10.1155/2021/6671410
179. Gangemi RM, Swack JA, Gaviria DM, Romain PL. Anti-T12, an anti-CD6 monoclonal antibody, can activate human T lymphocytes. J Immunol (1989) 143(8):2439–47. doi: 10.4049/jimmunol.143.8.2439
180. Dockrell HM. Leucocyte typing IV. White cell differentiation antigens. Immunology (1991) 72(1):159–60.
181. Osorio LM, Garcia CA, Jondal M, Chow SC. The anti-CD6 mAb, IOR-T1, defined a new epitope on the human CD6 molecule that induces greater responsiveness in T cell receptor/CD3-mediated T cell proliferation. Cell Immunol (1994) 154(1):123–33. doi: 10.1006/cimm.1994.1062
182. Singer NG, Richardson BC, Powers D, Hooper F, Lialios F, Endres J, et al. Role of the CD6 glycoprotein in antigen-specific and autoreactive responses of cloned human T lymphocytes. Immunology (1996) 88(4):537–43.
183. Garcia Santana CA, Tung JW, Gulnik S. Human treg cells are characterized by low/negative CD6 expression. Cytometry A. (2014) 85(10):901–8. doi: 10.1002/cyto.a.22513
184. Consuegra-Fernández M, Martínez-Florensa M, Aranda F, de Salort J, Armiger-Borràs N, Lozano T, et al. Relevance of CD6-mediated interactions in the regulation of peripheral T-cell responses and tolerance. Front Immunol (2017) 8:594. doi: 10.3389/fimmu.2017.00594
185. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi: 10.4049/jimmunol.1402381
186. Brown ME, Peters LD, Hanbali SR, Arnoletti JM, Sachs LK, Nguyen KQ, et al. Human CD4+CD25+CD226- tregs demonstrate increased purity, lineage stability, and suppressive capacity versus CD4+CD25+CD127lo/- tregs for adoptive cell therapy. Front Immunol (2022) 13:873560. doi: 10.3389/fimmu.2022.873560
187. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol (2011) 41(6):1652–62. doi: 10.1002/eji.201041120
188. Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, et al. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med (2005) 11(10):1118–24. doi: 10.1038/nm1292
189. Chattopadhyay PK, Yu J, Roederer M. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat Med (2005) 11(10):1113–7. doi: 10.1038/nm1293
190. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13(15 Pt 1):4345–54. doi: 10.1158/1078-0432.CCR-07-0472
191. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi: 10.1038/nature06306
192. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
193. Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med (2006) 203(3):505–11. doi: 10.1084/jem.20050783
194. Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol (2006) 7(1):83–92. doi: 10.1038/ni1289
195. Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood (2006) 107(10):3925–32. doi: 10.1182/blood-2005-11-4502
196. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
197. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U.S.A. (1986) 83(12):4167–71. doi: 10.1073/pnas.83.12.4167
198. Wahl SM, Hunt DA, Wong HL, Dougherty S, McCartney-Francis N, Wahl LM, et al. Transforming growth factor-beta is a potent immunosuppressive agent that inhibits IL-1-dependent lymphocyte proliferation. J Immunol (1988) 140(9):3026–32. doi: 10.4049/jimmunol.140.9.3026
199. Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med (1986) 163(5):1037–50. doi: 10.1084/jem.163.5.1037
200. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med (2001) 194(5):629–44. doi: 10.1084/jem.194.5.629
201. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol (2004) 172(2):834–42. doi: 10.4049/jimmunol.172.2.834
202. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood (2009) 113(21):5125–33. doi: 10.1182/blood-2009-01-199950
203. Sun J, Tang DN, Fu T, Sharma P. Identification of human regulatory T cells in the setting of T-cell activation and anti-CTLA-4 immunotherapy on the basis of expression of latency-associated peptide. Cancer Discovery (2012) 2(2):122–30. doi: 10.1158/2159-8290.CD-11-0236
204. Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4(1):29–37. doi: 10.1093/jmcb/mjr033
205. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol (2022) 7(69):eabi4613. doi: 10.1126/sciimmunol.abi4613
206. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces foxp3 expression. PloS One (2008) 3(7):e2705. doi: 10.1371/journal.pone.0002705
207. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol (2009) 39(12):3315–22. doi: 10.1002/eji.200939684
208. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. (2009) 106(32):13445–50. doi: 10.1073/pnas.0901944106
209. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. (2009) 106(32):13439–44. doi: 10.1073/pnas.0901965106
210. Cai H, Liu Y, Dong X, Jiang F, Li H, Ouyang S, et al. Analysis of LAP+ and GARP+ Treg subsets in peripheral blood of patients with neuromyelitis optica spectrum disorders. Neurol Sci (2023) 44(5):1739–47. doi: 10.1007/s10072-023-06629-8
211. Jin H, Sun L, Tang L, Yu W, Li H. Expression of GARP is increased in tumor-infiltrating regulatory T cells and is correlated to clinicopathology of lung cancer patients. Front Immunol (2017) 8:138. doi: 10.3389/fimmu.2017.00138
212. Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112(2):358–404. doi: 10.1016/j.pharmthera.2005.04.013
213. Thiel M, Caldwell CC, Sitkovsky MV. The critical role of adenosine A2A receptors in downregulation of inflammation and immunity in the pathogenesis of infectious diseases. Microbes Infect (2003) 5(6):515–26. doi: 10.1016/S1286-4579(03)00068-6
214. Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, et al. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Exp Med (1995) 182(5):1603–8. doi: 10.1084/jem.182.5.1603
215. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev (1998) 161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x
216. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110(4):1225–32. doi: 10.1182/blood-2006-12-064527
217. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
218. Mandapathil M, Lang S, Gorelik E, Whiteside TL. Isolation of functional human regulatory T cells (Treg) from the peripheral blood based on the CD39 expression. J Immunol Methods (2009) 346(1–2):55–63. doi: 10.1016/j.jim.2009.05.004
219. Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant (2010) 10(11):2410–20. doi: 10.1111/j.1600-6143.2010.03291.x
220. Schuler PJ, Harasymczuk M, Schilling B, Lang S, Whiteside TL. Separation of human CD4+CD39+ T cells by magnetic beads reveals two phenotypically and functionally different subsets. J Immunol Methods (2011) 369(1–2):59–68. doi: 10.1016/j.jim.2011.04.004
221. Huang Hh, Wang S, Wang Hf, Fu Jl, Han P, Wang Fs. [Phenotypical and functional characteristic of FoxP3(+);CD39(+); regulatory T cells in humans]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2010) 26(6):536–8.
222. Ye L, Goodall JC, Zhang L, Putintseva EV, Lam B, Jiang L, et al. TCR usage, gene expression and function of two distinct FOXP3(+)Treg subsets within CD4(+)CD25(hi) T cells identified by expression of CD39 and CD45RO. Immunol Cell Biol (2016) 94(3):293–305. doi: 10.1038/icb.2015.90
223. Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39hi regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol (2017) 14(6):521–8. doi: 10.1038/cmi.2016.30
224. Lu Y, Wang X, Gu J, Lu H, Zhang F, Li X, et al. iTreg induced from CD39(+) naive T cells demonstrate enhanced proliferate and suppressive ability. Int Immunopharmacol (2015) 28(2):925–30. doi: 10.1016/j.intimp.2015.03.039
225. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183(11):7602–10. doi: 10.4049/jimmunol.0901881
226. Thomson LF, Ruedi JM, Glass A, Moldenhauer G, Moller P, Low MG, et al. Production and characterization of monoclonal antibodies to the glycosyl phosphatidylinositol-anchored lymphocyte differentiation antigen ecto-5’-nucleotidase (CD73). Tissue Antigens (1990) 35(1):9–19. doi: 10.1111/j.1399-0039.1990.tb01750.x
227. Airas L, Niemelä J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol (1997) 136(2):421–31. doi: 10.1083/jcb.136.2.421
228. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically expanded human regulatory T-cells are super-suppressive due to HIF1A induced expression of CD73. Commun Biol (2021) 4(1):1186. doi: 10.1038/s42003-021-02721-x
229. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177(10):6780–6. doi: 10.4049/jimmunol.177.10.6780
230. Ye H, Zhao J, Xu X, Zhang D, Shen H, Wang S. Role of adenosine A2a receptor in cancers and autoimmune diseases. Immun Inflammation Dis (2023) 11(4):e826. doi: 10.1002/iid3.826
231. Lin CR, Wei TYW, Tsai HY, Wu YT, Wu PY, Chen ST. Glycosylation-dependent interaction between CD69 and S100A8/S100A9 complex is required for regulatory T-cell differentiation. FASEB J (2015) 29(12):5006–17. doi: 10.1096/fj.15-273987
232. Sánchez-Díaz R, Blanco-Dominguez R, Lasarte S, Tsilingiri K, Martín-Gayo E, Linillos-Pradillo B, et al. Thymus-derived regulatory T cell development is regulated by C-type lectin-mediated BIC/microRNA 155 expression. Mol Cell Biol (2017) 37(9):e00341–16. doi: 10.1128/MCB.00341-16
233. Cortés JR, Sánchez-Díaz R, Bovolenta ER, Barreiro O, Lasarte S, Matesanz-Marín A, et al. Maintenance of immune tolerance by Foxp3+ regulatory T cells requires CD69 expression. J Autoimmun (2014) 55:51–62. doi: 10.1016/j.jaut.2014.05.007
234. Piekarska K, Urban-Wójciuk Z, Kurkowiak M, Pelikant-Małecka I, Schumacher A, Sakowska J, et al. Mesenchymal stem cells transfer mitochondria to allogeneic Tregs in an HLA-dependent manner improving their immunosuppressive activity. Nat Commun (2022) 13(1):856. doi: 10.1038/s41467-022-28338-0
235. Yu L, Yang F, Zhang F, Guo D, Li L, Wang X, et al. CD69 enhances immunosuppressive function of regulatory T-cells and attenuates colitis by prompting IL-10 production. Cell Death Dis (2018) 9(9):1–14. doi: 10.1038/s41419-018-0927-9
236. Chang CH, Hong SC, Hughes CC, Janeway CA, Flavell RA. CIITA activates the expression of MHC class II genes in mouse T cells. Int Immunol (1995) 7(9):1515–8. doi: 10.1093/intimm/7.9.1515
237. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58(3):652–62. doi: 10.2337/db08-1168
238. Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol (2006) 176(8):4622–31. doi: 10.4049/jimmunol.176.8.4622
239. Cao L, Huang D, Burns H, Hu M, Hawthorne W, Yi S, et al. Human HLA-DR+CD27+ Memory-type regulatory T cells show potent xenoantigen-specific suppression in vitro. Transplantation (2018) 102:S392. doi: 10.1097/01.tp.0000543159.58254.47
240. Schaier M, Seissler N, Becker LE, Schaefer SM, Schmitt E, Meuer S, et al. The extent of HLA-DR expression on HLA-DR(+) Tregs allows the identification of patients with clinically relevant borderline rejection. Transpl Int (2013) 26(3):290–9. doi: 10.1111/tri.12032
241. Yang H, Ye S, Goswami S, Li T, Wu J, Cao C, et al. Highly immunosuppressive HLADRhi regulatory T cells are associated with unfavorable outcomes in cervical squamous cell carcinoma. Int J Cancer (2020) 146(7):1993–2006. doi: 10.1002/ijc.32782
242. Åkesson K, Tompa A, Rydén A, Faresjö M. Low expression of CD39+/CD45RA+ on regulatory T cells (Treg) cells in type 1 diabetic children in contrast to high expression of CD101+/CD129+ on Treg cells in children with coeliac disease. Clin Exp Immunol (2015) 180(1):70–82. doi: 10.1111/cei.12559
243. Fernandez I, Zeiser R, Karsunky H, Kambham N, Beilhack A, Soderstrom K, et al. CD101 surface expression discriminates potency among murine FoxP3+ regulatory T cells. J Immunol (2007) 179(5):2808–14. doi: 10.4049/jimmunol.179.5.2808
244. Jovanovic DV, Boumsell L, Bensussan A, Chevalier X, Mancini A, Battista J a. D. CD101 expression and function in normal and rheumatoid arthritis-affected human T cells and monocytes/macrophages. J Rheumatol (2011) 38(3):419–28. doi: 10.3899/jrheum.100676
245. De Smedt M, Verhasselt B, Kerre T, Vanhecke D, Naessens E, Leclercq G, et al. Signals from the IL-9 receptor are critical for the early stages of human intrathymic T cell development. J Immunol (2000) 164(4):1761–7. doi: 10.4049/jimmunol.164.4.1761
246. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci (2009) 106(31):12885–90. doi: 10.1073/pnas.0812530106
247. Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, et al. Expression of the integrin αEβ7 identifies unique subsets of CD25+ as well as CD25– regulatory T cells. Proc Natl Acad Sci U S A. (2002) 99(20):13031–6. doi: 10.1073/pnas.192162899
248. Banz A, Peixoto A, Pontoux C, Cordier C, Rocha B, Papiernik M. A unique subpopulation of CD4+ regulatory T cells controls wasting disease, IL-10 secretion and T cell homeostasis. Eur J Immunol (2003) 33(9):2419–28. doi: 10.1002/eji.200324205
249. Zhao D, Zhang C, Yi T, Lin CL, Todorov I, Kandeel F, et al. In vivo–activated CD103+CD4+ regulatory T cells ameliorate ongoing chronic graft-versus-host disease. Blood (2008) 112(5):2129–38. doi: 10.1182/blood-2008-02-140277
250. van Hooren L, Handgraaf SM, Kloosterman DJ, Karimi E, van Mil LWHG, Gassama AA, et al. CD103+ regulatory T cells underlie resistance to radio-immunotherapy and impair CD8+ T cell activation in glioblastoma. Nat Cancer (2023) 4(5):665–81. doi: 10.1038/s43018-023-00547-6
251. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens JL, et al. Compromised CD4+ CD25high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology (2008) 123(1):79–89. doi: 10.1111/j.1365-2567.2007.02690.x
252. Rötzschke O, Borsellino G, Battistini L, Falk K, Kleinewietfeld M. In vivo-activated CD103+ Foxp3+ Tregs: of men and mice. Blood (2009) 113(9):2119–20. doi: 10.1182/blood-2008-11-188847
253. Lim HW, Broxmeyer HE, Kim CH. Regulation of trafficking receptor expression in human forkhead box P3+ regulatory T cells. J Immunol (2006) 177(2):840–51. doi: 10.4049/jimmunol.177.2.840
254. Iellem A, Colantonio L, D’Ambrosio D. Skin-versus gut-skewed homing receptor expression and intrinsic CCR4 expression on human peripheral blood CD4+CD25+ suppressor T cells. Eur J Immunol (2003) 33(6):1488–96. doi: 10.1002/eji.200323658
255. Allakhverdi Z, Fitzpatrick D, Boisvert A, Baba N, Bouguermouh S, Sarfati M, et al. Expression of CD103 identifies human regulatory T-cell subsets. J Allergy Clin Immunol (2006) 118(6):1342–9. doi: 10.1016/j.jaci.2006.07.034
256. Rao PE, Petrone AL, Ponath PD. Differentiation and expansion of T cells with regulatory function from human peripheral lymphocytes by stimulation in the presence of TGF-{beta}. J Immunol (2005) 174(3):1446–55. doi: 10.4049/jimmunol.174.3.1446
257. Trzupek D, Dunstan M, Cutler AJ, Lee M, Godfrey L, Jarvis L, et al. Discovery of CD80 and CD86 as recent activation markers on regulatory T cells by protein-RNA single-cell analysis. Genome Med (2020) 12(1):55. doi: 10.1186/s13073-020-00756-z
258. Richardson N, Wootton GE, Bozward AG, Oo YH. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin Immunopathol (2022) 44(4):461–74. doi: 10.1007/s00281-022-00940-w
259. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. New Engl J Med (2006) 354(6):610–21. doi: 10.1056/NEJMra052723
260. Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med (1998) 338(7):436–45. doi: 10.1056/NEJM199802123380706
261. Deuel TF, Keim PS, Farmer M, Heinrikson RL. Amino acid sequence of human platelet factor 4. Proc Natl Acad Sci U S A. (1977) 74(6):2256–8. doi: 10.1073/pnas.74.6.2256
262. Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature (1985) 315(6021):672–6. doi: 10.1038/315672a0
263. Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, et al. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci U S A. (1987) 84(24):9233–7. doi: 10.1073/pnas.84.24.9233
264. Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med (1988) 167(2):570–81. doi: 10.1084/jem.167.2.570
265. Luster AD, Leder P. IP-10, a -C-X-C- chemokine, elicits a potent thymus-dependent antitumor response in vivo. J Exp Med (1993) 178(3):1057–65. doi: 10.1084/jem.178.3.1057
266. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity (2011) 34(2):237–46. doi: 10.1016/j.immuni.2011.01.016
267. Cassani B, Villablanca EJ, Quintana FJ, Love PE, Lacy-Hulbert A, Blaner WS, et al. Gut-tropic T cells that express integrin α4β7 and CCR9 are required for induction of oral immune tolerance in mice. Gastroenterology (2011) 141(6):2109–18. doi: 10.1053/j.gastro.2011.09.015
268. Patterson SJ, Pesenacker AM, Wang AY, Gillies J, Mojibian M, Morishita K, et al. T regulatory cell chemokine production mediates pathogenic T cell attraction and suppression. J Clin Invest (2016) 126(3):1039–51. doi: 10.1172/JCI83987
269. Long Y, Zhao X, Xia C, Li X, Fan C, Liu C, et al. Upregulated IL-17A secretion and CCR6 co-expression in Treg subsets are related to the imbalance of Treg/Th17 cells in active UC patients. Scandinavian J Immunol (2020) 91(6):e12842. doi: 10.1111/sji.12842
270. Moreira AP, Cavassani KA, Tristão FSM, Campanelli AP, Martinez R, Rossi MA, et al. CCR5-dependent regulatory T cell migration mediates fungal survival and severe immunosuppression1. J Immunol (2008) 180(5):3049–56. doi: 10.4049/jimmunol.180.5.3049
271. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155(6):1282–95. doi: 10.1016/j.cell.2013.10.054
272. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Trans Med (2014) 6(258):258ra142–258ra142. doi: 10.1126/scitranslmed.3009925
273. Harb H, Stephen-Victor E, Crestani E, Benamar M, Massoud A, Cui Y, et al. A regulatory T cell Notch4-GDF15 axis licenses tissue inflammation in asthma. Nat Immunol (2020) 21(11):1359–70. doi: 10.1038/s41590-020-0777-3
274. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331(6015):337–41. doi: 10.1126/science.1198469
275. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol (2016) 34(1):609–33. doi: 10.1146/annurev-immunol-032712-095948
276. Worthington JJ, Kelly A, Smedley C, Bauché D, Campbell S, Marie JC, et al. Integrin αvβ8-mediated TGF-β activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity (2015) 42(5):903–15. doi: 10.1016/j.immuni.2015.04.012
277. Edwards JP, Thornton AM, Shevach EM. Release of active TGF-β1 from the latent TGF-β1/GARP complex on T regulatory cells is mediated by integrin β8. J Immunol (2014) 193(6):2843–9. doi: 10.4049/jimmunol.1401102
278. Zhang HL, Zheng YJ, Pan YD, Xie C, Sun H, Zhang YH, et al. Regulatory T-cell depletion in the gut caused by integrin β7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal Immunol (2016) 9(2):391–400. doi: 10.1038/mi.2015.68
279. Wagner N, Löhler J, Kunkel EJ, Ley K, Leung E, Krissansen G, et al. Critical role for β7 integrins in formation of the gut-associated lymphoid tissue. Nature (1996) 382(6589):366–70. doi: 10.1038/382366a0
280. Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J Immunol (2000) 164(1):240–7. doi: 10.4049/jimmunol.164.1.240
281. Chow Z, Banerjee A, Hickey MJ. Controlling the fire—tissue-specific mechanisms of effector regulatory T-cell homing. Immunol Cell Biol (2015) 93(4):355–63. doi: 10.1038/icb.2014.117
282. Hirahara K, Liu L, Clark RA, Yamanaka Ki, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+ CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol (2006) 177(7):4488–94. doi: 10.4049/jimmunol.177.7.4488
283. Lee JH, Kang SG, Kim CH. FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues1. J Immunol (2007) 178(1):301–11. doi: 10.4049/jimmunol.178.1.301
284. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2020) 10. doi: 10.3389/fimmu.2019.03100
285. Mempel TR, Marangoni F. Guidance factors orchestrating regulatory T cell positioning in tissues during development, homeostasis, and response. Immunol Rev (2019) 289(1):129–41. doi: 10.1111/imr.12761
286. Kocks JR, Davalos-Misslitz AC, Hintzen G, Ohl L, Förster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J Exp Med (2007) 204(4):723–34. doi: 10.1084/jem.20061424
287. Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol (2008) 8(5):362–71. doi: 10.1038/nri2297
288. Sarween N, Chodos A, Raykundalia C, Khan M, Abbas AK, Walker LSK. CD4+CD25+ Cells controlling a pathogenic CD4 response inhibit cytokine differentiation, CXCR-3 expression, and tissue invasion1. J Immunol (2004) 173(5):2942–51. doi: 10.4049/jimmunol.173.5.2942
289. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32(1):659–702. doi: 10.1146/annurev-immunol-032713-120145
290. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10(6):595–602. doi: 10.1038/ni.1731
291. Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol (2015) 16(2):197–206. doi: 10.1038/ni.3053
292. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature (2017) 546(7658):421–5. doi: 10.1038/nature22360
293. Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature (2006) 440(7086):890–5. doi: 10.1038/nature04651
294. Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, et al. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science (2012) 338(6106):532–6. doi: 10.1126/science.1227049
295. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
296. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
297. Sage PT, Sharpe AH. The multifaceted functions of follicular regulatory T cells. Curr Opin Immunol (2020) 67:68–74. doi: 10.1016/j.coi.2020.10.009
298. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
299. Lian J, Luster AD. Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr Opin Cell Biol (2015) 36:1–6. doi: 10.1016/j.ceb.2015.05.003
300. Sungnak W, Wagner A, Kowalczyk MS, Bod L, Kye YC, Sage PT, et al. T follicular regulatory cell–derived fibrinogen-like protein 2 regulates production of autoantibodies and induction of systemic autoimmunity. J Immunol (2020) 205(12):3247–62. doi: 10.4049/jimmunol.2000748
301. Lu Y, Jiang R, Freyn AW, Wang J, Strohmeier S, Lederer K, et al. CD4+ follicular regulatory T cells optimize the influenza virus–specific B cell response. J Exp Med (2021) 218(3):e20200547. doi: 10.1084/jem.20200547
302. Gonzalez-Figueroa P, Roco JA, Papa I, Núñez Villacís L, Stanley M, Linterman MA, et al. Follicular regulatory T cells produce neuritin to regulate B cells. Cell (2021) 184(7):1775–1789.e19. doi: 10.1016/j.cell.2021.02.027
303. Vanderleyden I, Fra-Bido SC, Innocentin S, Stebegg M, Okkenhaug H, Evans-Bailey N, et al. Follicular regulatory T cells can access the germinal center independently of CXCR5. Cell Rep (2020) 30(3):611–619.e4. doi: 10.1016/j.celrep.2019.12.076
304. Clement RL, Daccache J, Mohammed MT, Diallo A, Blazar BR, Kuchroo VK, et al. Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat Immunol (2019) 20(10):1360–71. doi: 10.1038/s41590-019-0472-4
305. Nikolouli E, Elfaki Y, Herppich S, Schelmbauer C, Delacher M, Falk C, et al. Recirculating IL-1R2+ Tregs fine-tune intrathymic Treg development under inflammatory conditions. Cell Mol Immunol (2021) 18(1):182–93. doi: 10.1038/s41423-019-0352-8
306. Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity (2019) 50(2):493–504.e7. doi: 10.1016/j.immuni.2019.01.001
307. Ni H, Chen Y. Differentiation, regulation and function of regulatory T cells in non-lymphoid tissues and tumors. Int Immunopharmacol (2023) 121:110429. doi: 10.1016/j.intimp.2023.110429
308. Rocha VZ, Folco EJ, Ozdemir C, Sheikine Y, Christen T, Sukhova GK, et al. CXCR3 controls T-cell accumulation in fat inflammation. Arterioscler Thromb Vasc Biol (2014) 34(7):1374–81. doi: 10.1161/ATVBAHA.113.303133
309. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi: 10.1038/nm.2002
310. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue–resident regulatory T cells. Nat Immunol (2015) 16(3):276–85. doi: 10.1038/ni.3085
311. Spallanzani RG, Zemmour D, Xiao T, Jayewickreme T, Li C, Bryce PJ, et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol (2019) 4(35):eaaw3658. doi: 10.1126/sciimmunol.aaw3658
312. Schmidleithner L, Thabet Y, Schönfeld E, Köhne M, Sommer D, Abdullah Z, et al. Enzymatic activity of HPGD in treg cells suppresses tconv cells to maintain adipose tissue homeostasis and prevent metabolic dysfunction. Immunity (2019) 50(5):1232–1248.e14. doi: 10.1016/j.immuni.2019.03.014
313. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab (2015) 21(4):543–57. doi: 10.1016/j.cmet.2015.03.005
314. Fernandes RA, Li C, Wang G, Yang X, Savvides CS, Glassman CR, et al. Discovery of surrogate agonists for visceral fat Treg cells that modulate metabolic indices in vivo. eLife (2020) 9:e58463. doi: 10.7554/eLife.58463.sa2
315. Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med (2001) 194(6):847–53. doi: 10.1084/jem.194.6.847
316. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J Exp Med (2007) 204(6):1335–47. doi: 10.1084/jem.20070081
317. Baatar D, Olkhanud P, Sumitomo K, Taub D, Gress R, Biragyn A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4- Tregs, regulate effector T cells using FasL. J Immunol (2007) 178(8):4891–900. doi: 10.4049/jimmunol.178.8.4891
318. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med (2006) 203(11):2451–60. doi: 10.1084/jem.20060956
319. Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong HA, et al. Commensal microbes and hair follicle morphogenesis coordinately drive treg migration into neonatal skin. Cell Host Microbe (2017) 21(4):467–477.e5. doi: 10.1016/j.chom.2017.03.001
320. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity (2015) 43(5):1011–21. doi: 10.1016/j.immuni.2015.10.016
321. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity (2016) 44(2):355–67. doi: 10.1016/j.immuni.2016.01.009
322. Gazzerro E, Baldassari S, Assereto S, Fruscione F, Pistorio A, Panicucci C, et al. Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X axis. Am J Pathol (2015) 185(12):3349–60. doi: 10.1016/j.ajpath.2015.08.010
323. Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, et al. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signaling (2011) 4(162):ra12–2. doi: 10.1126/scisignal.2001270
324. Cho J, Kuswanto W, Benoist C, Mathis D. T cell receptor specificity drives accumulation of a reparative population of regulatory T cells within acutely injured skeletal muscle. Proc Natl Acad Sci (2019) 116(52):26727–33. doi: 10.1073/pnas.1914848116
325. Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med (2015) 21(8):854–62. doi: 10.1038/nm.3918
326. Shao Q, Gu J, Zhou J, Wang Q, Li X, Deng Z, et al. Tissue tregs and maintenance of tissue homeostasis. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.717903
327. Grindebacke H, Stenstad H, Quiding-Järbrink M, Waldenström J, Adlerberth I, Wold AE, et al. Dynamic development of homing receptor expression and memory cell differentiation of infant CD4+CD25high regulatory T cells1. J Immunol (2009) 183(7):4360–70. doi: 10.4049/jimmunol.0901091
328. Mock JR, Dial CF, Tune MK, Norton DL, Martin JR, Gomez JC, et al. Transcriptional analysis of Foxp3+ Tregs and functions of two identified molecules during resolution of ALI. JCI Insight (2019) 4(6):e124958. doi: 10.1172/jci.insight.124958
329. Mansouri S, Katikaneni DS, Gogoi H, Pipkin M, Machuca TN, Emtiazjoo AM, et al. Lung IFNAR1hi TNFR2+ cDC2 promotes lung regulatory T cells induction and maintains lung mucosal tolerance at steady state. Mucosal Immunol (2020) 13(4):595–608. doi: 10.1038/s41385-020-0254-1
330. Dial CF, Tune MK, Doerschuk CM, Mock JR. Foxp31 regulatory t cell expression of keratinocyte growth factor enhances lung epithelial proliferation. Am J Respir Cell Mol Biol (2017) 57(2):162–73. doi: 10.1165/rcmb.2017-0019OC
331. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157(1):121–41. doi: 10.1016/j.cell.2014.03.011
332. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity (2011) 34(5):794–806. doi: 10.1016/j.immuni.2011.03.021
333. Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, et al. Human G protein–coupled receptor gpr-9-6/cc chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine–mediated chemotaxis. J Exp Med (1999) 190(9):1241–56. doi: 10.1084/jem.190.9.1241
334. Hegazy AN, Powrie F. Microbiota RORgulates intestinal suppressor T cells. Science (2015) 349(6251):929–30. doi: 10.1126/science.aad0865
335. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORγt+ T cells. Science (2015) 349(6251):989–93. doi: 10.1126/science.aac4263
336. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science (2015) 349(6251):993–7. doi: 10.1126/science.aaa9420
337. Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature (2020) 577(7790):410–5. doi: 10.1038/s41586-019-1865-0
338. Kim BS, Lu H, Ichiyama K, Chen X, Zhang YB, Mistry NA, et al. Generation of RORγt+ Antigen-specific T regulatory 17 cells from foxp3+ Precursors in autoimmunity. Cell Rep (2017) 21(1):195–207. doi: 10.1016/j.celrep.2017.09.021
339. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513(7519):564–8. doi: 10.1038/nature13577
340. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol (2011) 12(4):304–11. doi: 10.1038/ni.2006
341. Martins GA, Cimmino L, Shapiro-Shelef M, Szabolcs M, Herron A, Magnusdottir E, et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol (2006) 7(5):457–65. doi: 10.1038/ni1320
342. Suply T, Hannedouche S, Carte N, Li J, Grosshans B, Schaefer M, et al. A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci Signaling (2017) 10(496):eaal0180. doi: 10.1126/scisignal.aal0180
343. Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science (2013) 340(6139):1456–9. doi: 10.1126/science.1237013
344. Oo YH, Shetty S, Adams DH. The role of chemokines in the recruitment of lymphocytes to the liver. Digest Diseases (2010) 28(1):31–44. doi: 10.1159/000282062
345. Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol (2010) 184(6):2886–98. doi: 10.4049/jimmunol.0901216
346. Curbishley SM, Eksteen B, Gladue RP, Lalor P, Adams DH. CXCR 3 activation promotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am J Pathol (2005) 167(3):887–99. doi: 10.1016/S0002-9440(10)62060-3
347. Lalor PF, Sun PJ, Weston CJ, Martin-Santos A, Wakelam MJ, Adams DH. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-kappaB-dependent increase in lymphocyte adhesion. Hepatology (2007) 45(2):465–74. doi: 10.1002/hep.21497
348. Eksteen B, Miles A, Curbishley SM, Tselepis C, Grant AJ, Walker LS, et al. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J Immunol (2006) 177(1):593–603. doi: 10.4049/jimmunol.177.1.593
349. Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, et al. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J Hepatol (2012) 57(5):1044–51. doi: 10.1016/j.jhep.2012.07.008
350. Jeffery HC, Hunter S, Humphreys EH, Bhogal R, Wawman RE, Birtwistle J, et al. Bidirectional cross-talk between biliary epithelium and th17 cells promotes local th17 expansion and bile duct proliferation in biliary liver diseases. J Immunol (2019) 203(5):1151–9. doi: 10.4049/jimmunol.1800455
351. Kitamura K, Farber JM, Kelsall BL. CCR6 marks regulatory T cells as a colon-tropic, IL-10–producing phenotype. J Immunol (2010) 185(6):3295–304. doi: 10.4049/jimmunol.1001156
352. Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORγt+ T cells. J Exp Med (2008) 205(6):1381–93. doi: 10.1084/jem.20080034
353. Oo YH, Hubscher SG, Adams DH. Autoimmune hepatitis: new paradigms in the pathogenesis, diagnosis, and management. Hepatol Int (2010) 4(2):475–93. doi: 10.1007/s12072-010-9183-5
354. Ronca V, Mancuso C, Milani C, Carbone M, Oo YH, Invernizzi P. Immune system and cholangiocytes: A puzzling affair in primary biliary cholangitis. J Leukoc Biol (2020) 108(2):659–71. doi: 10.1002/JLB.5MR0320-200R
355. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet (2013) 382(9904):1587–99. doi: 10.1016/S0140-6736(13)60096-3
356. Si L, Whiteside TL, SChade RR, Starzl TE, Van Thiel DH. T-lymphocyte subsets in liver tissues of patients with primary biliary cirrhosis (PBC), patients with primary sclerosing cholangitis (PSC), and normal controls. J Clin Immunol (1984) 4(4):262–72. doi: 10.1007/BF00915293
357. Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol (2006) 6(3):244–51. doi: 10.1038/nri1784
358. Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheumatol (2008) 58(8):2498–510. doi: 10.1002/art.23674
359. Ichiki Y, Aoki CA, Bowlus CL, Shimoda S, Ishibashi H, Gershwin ME. T cell immunity in autoimmune hepatitis. Autoimmun Rev (2005) 4(5):315–21. doi: 10.1016/j.autrev.2005.01.005
360. Li Y, Li B, You Z, Zhang J, Wei Y, Li Y, et al. Cytotoxic KLRG1 expressing lymphocytes invade portal tracts in primary biliary cholangitis. J Autoimmun (2019) 103:102293. doi: 10.1016/j.jaut.2019.06.004
361. Poch T, Krause J, Casar C, Liwinski T, Glau L, Kaufmann M, et al. Single-cell atlas of hepatic T cells reveals expansion of liver-resident naive-like CD4(+) T cells in primary sclerosing cholangitis. J Hepatol (2021) 75(2):414–23. doi: 10.1016/j.jhep.2021.03.016
362. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575(7783):512–8. doi: 10.1038/s41586-019-1631-3
363. Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol (2001) 166(2):973–81. doi: 10.4049/jimmunol.166.2.973
364. Issa F, Schiopu A, Wood KJ. Role of T cells in graft rejection and transplantation tolerance. Expert Rev Clin Immunol (2010) 6(1):155–69. doi: 10.1586/eci.09.64
365. Herrera OB, Golshayan D, Tibbott R, Salcido Ochoa F, James MJ, Marelli-Berg FM, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol (2004) 173(8):4828–37. doi: 10.4049/jimmunol.173.8.4828
366. Oo YH, Ackrill S, Cole R, Jenkins L, Anderson P, Jeffery HC, et al. Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Rep (2019) 1(4):286–96. doi: 10.1016/j.jhepr.2019.08.001
367. Elias S, Sharma R, Schizas M, Valdez I, Rampersaud S, Park SM, et al. CXCR4+ Treg cells control serum IgM levels and natural IgM autoantibody production by B-1 cells in the bone marrow. J Exp Med (2022) 219(7):e20220047. doi: 10.1084/jem.20220047
368. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1158/1557-3125.HIPPO19-B11
369. Cinier J, Hubert M, Besson L, Di Roio A, Rodriguez C, Lombardi V, et al. Recruitment and expansion of tregs cells in the tumor environment—How to target them? Cancers (2021) 13(8):1850. doi: 10.3390/cancers13081850
370. Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res (2010) 107:57–117. doi: 10.1016/S0065-230X(10)07003-X
371. Li Q, Lu J, Li J, Zhang B, Wu Y, Ying T. Antibody-based cancer immunotherapy by targeting regulatory T cells. Front Oncol (2023) 13:1157345. doi: 10.3389/fonc.2023.1157345
372. Koyama S, Nishikawa H. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer (2021) 9(7):e002591. doi: 10.1136/jitc-2021-002591
373. Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer science (2006) 97(11):1139–46. doi: 10.1111/j.1349-7006.2006.00307.x
374. Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res (2009) 69(14):5996–6004. doi: 10.1158/0008-5472.CAN-08-4619
375. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity (2016) 45(5):1135–47. doi: 10.1016/j.immuni.2016.10.021
376. Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity (2016) 45(5):1122–34. doi: 10.1016/j.immuni.2016.10.032
377. Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, et al. CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc Natl Acad Sci (2017) 114(23):6086–91. doi: 10.1073/pnas.1621280114
378. Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, et al. CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology (2021) 163(4):512–20. doi: 10.1111/imm.13337
379. Villarreal DO, L’Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, et al. Targeting CCR8 induces protective antitumor immunity and enhances vaccine-induced responses in colon cancer. Cancer Res (2018) 78(18):5340–8. doi: 10.1158/0008-5472.CAN-18-1119
380. Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, et al. Fc-optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res (2021) 81(11):2983–94. doi: 10.1158/0008-5472.CAN-20-3585
381. Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci (2022) 119(7):e2114282119. doi: 10.1073/pnas.2114282119
382. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature (2009) 458(7236):351–6. doi: 10.1038/nature07674
383. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest (2011) 121(11):4503–15. doi: 10.1172/JCI57456
384. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13(10):1010–9. doi: 10.1038/ni.2402
385. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity (2011) 35(3):337–48. doi: 10.1016/j.immuni.2011.08.012
386. Lian J, Liu S, Yue Y, Yang Q, Zhang Z, Yang S, et al. Eomes promotes esophageal carcinoma progression by recruiting Treg cells through the CCL20-CCR6 pathway. Cancer science (2021) 112(1):144–54. doi: 10.1111/cas.14712
387. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117(14):3921–8. doi: 10.1182/blood-2010-10-311894
388. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127– T regulatory cells. Clin Immunol (2009) 133(1):22–6. doi: 10.1016/j.clim.2009.06.001
389. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
390. Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy (2015) 17(4):473–86. doi: 10.1016/j.jcyt.2014.11.005
391. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
392. Theil A, Wilhelm C, Kuhn M, Petzold A, Tuve S, Oelschlägel U, et al. T cell receptor repertoires after adoptive transfer of expanded allogeneic regulatory T cells. Clin Exp Immunol (2017) 187(2):316–24. doi: 10.1111/cei.12887
393. Landwehr-Kenzel S, Müller-Jensen L, Kuehl JS, Abou-El-Enein M, Hoffmann H, Muench S, et al. Adoptive transfer of ex vivo expanded regulatory T cells improves immune cell engraftment and therapy-refractory chronic GvHD. Mol Ther (2022) 30(6):2298–314. doi: 10.1016/j.ymthe.2022.02.025
394. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood (2005) 106(9):3300–7. doi: 10.1182/blood-2005-04-1632
395. Zhang N, Schröppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity (2009) 30(3):458–69. doi: 10.1016/j.immuni.2008.12.022
396. Zieliński M, Żalińska M, Iwaszkiewicz-Grześ D, Gliwiński M, Hennig M, Jaźwińska-Curyłło A, et al. Combined therapy with CD4+CD25highCD127– T regulatory cells and anti-CD20 antibody in recent-onset type 1 diabetes is superior to monotherapy: Randomized phase I/ II trial. Diabetes Obes Metab (2022) 24(8):1534–43. doi: 10.1111/dom.14723
397. Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, Ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med (2015) 7(304):304ps18. doi: 10.1126/scitranslmed.aaa7721
398. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — Results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
399. Selck C, Dominguez-Villar M. Antigen-specific regulatory T cell therapy in autoimmune diseases and transplantation. Front Immunol (2021) 12:661875. doi: 10.3389/fimmu.2021.661875
400. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
401. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257(1):107–26. doi: 10.1111/imr.12131
402. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev (2019) 290(1):127–47. doi: 10.1111/imr.12772
403. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
404. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18(4):676–84. doi: 10.1038/sj.leu.2403302
405. Schubert ML, Schmitt A, Neuber B, Hückelhoven-Krauss A, Kunz A, Wang L, et al. Third-generation CAR T cells targeting CD19 are associated with an excellent safety profile and might improve persistence of CAR T cells in treated patients. Blood (2019) 134:51. doi: 10.1182/blood-2019-125423
406. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat Med (2018) 24(3):352–9. doi: 10.1038/nm.4478
407. Kim D, Cho JY. Recent advances in allogeneic CAR-T cells. Biomolecules (2020) 10(2):263. doi: 10.3390/biom10020263
408. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer (2019) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
409. Bai Y, Kan S, Zhou S, Wang Y, Xu J, Cooke JP, et al. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discovery (2015) 1(1):1–15. doi: 10.1038/celldisc.2015.40
410. Dawson NAJ, Rosado-Sánchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med (2020) 12(557):eaaz3866. doi: 10.1126/scitranslmed.aaz3866
411. Rosado-Sánchez I, Haque M, Salim K, Speck M, Fung VC, Boardman DA, et al. Tregs integrate native and CAR-mediated costimulatory signals for control of allograft rejection. JCI Insight (2023) 8(19):e167215. doi: 10.1172/jci.insight.167215
412. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight (2019) 5(8):e126194. doi: 10.1172/jci.insight.126194
413. Imura Y, Ando M, Kondo T, Ito M, Yoshimura A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight (2020) 5(14):e136185. doi: 10.1172/jci.insight.136185
414. Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun (2018) 90:116–31. doi: 10.1016/j.jaut.2018.02.006
415. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) treg: A promising approach to inducing immunological tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
416. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim M a. A, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant (2017) 17(4):931–43. doi: 10.1111/ajt.14185
417. Rosado-Sánchez I, Levings MK. Building a CAR-Treg: Going from the basic to the luxury model. Cell Immunol (2020) 358:104220. doi: 10.1016/j.cellimm.2020.104220
418. Boardman DA, Wong MQ, Rees WD, Wu D, Himmel ME, Orban PC, et al. Flagellin-specific human CAR Tregs for immune regulation in IBD. J Autoimmun (2023) 134:102961. doi: 10.1016/j.jaut.2022.102961
419. Amini L, Greig J, Schmueck-Henneresse M, Volk HD, Bézie S, Reinke P, et al. Super-treg: toward a new era of adoptive treg therapy enabled by genetic modifications. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.611638
420. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Trans Med (2011) 3(83):83ra42–2. doi: 10.1126/scitranslmed.3002076
421. Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, Lombardi G. The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Front Immunol (2020) 11:1608. doi: 10.3389/fimmu.2020.01608
422. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-treg) cells: engineering and applications. Biomedicines (2022) 10(2):287. doi: 10.3390/biomedicines10020287
423. Katabathina V, Menias CO, Pickhardt P, Lubner M, Prasad SR. Complications of immunosuppressive therapy in solid organ transplantation. Radiol Clin North Am (2016) 54(2):303–19. doi: 10.1016/j.rcl.2015.09.009
424. Schreeb K, Culme-Seymour E, Ridha E, Dumont C, Atkinson G, Hsu B, et al. Study design: human leukocyte antigen class I molecule A∗02-chimeric antigen receptor regulatory T cells in renal transplantation. Kidney Int Rep (2022) 7(6):1258–67. doi: 10.1016/j.ekir.2022.03.030
425. Proics E, David M, Mojibian M, Speck M, Lounnas-Mourey N, Govehovitch A, et al. Preclinical assessment of antigen-specific chimeric antigen receptor regulatory T cells for use in solid organ transplantation. Gene Ther (2023) 30(3–4):309–22. doi: 10.1038/s41434-022-00358-x
426. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7(7):7563–77. doi: 10.18632/oncotarget.6927
427. Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant (2020) 20(4):1125–36. doi: 10.1111/ajt.15700
428. Van Steendam K, Tilleman K, De Ceuleneer M, De Keyser F, Elewaut D, Deforce D. Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res Ther (2010) 12(4):R132. doi: 10.1186/ar3070
429. Yaniv G, Twig G, Shor DBA, Furer A, Sherer Y, Mozes O, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev (2015) 14(1):75–9. doi: 10.1016/j.autrev.2014.10.003
430. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis (2020) 79(1):141–9. doi: 10.1136/annrheumdis-2019-215396
431. Peng L, Wu C, Hong R, Sun Y, Qian J, Zhao J, et al. Clinical efficacy and safety of sirolimus in systemic lupus erythematosus: a real-world study and meta-analysis. Ther Adv Musculoskelet Dis (2020) 12:1759720X20953336. doi: 10.1177/1759720X20953336
Keywords: regulatory T cells, FOXP3, Treg markers, Treg heterogeneity, Treg function, Treg therapy, Treg chemokine receptors
Citation: Santosh Nirmala S, Kayani K, Gliwiński M, Hu Y, Iwaszkiewicz-Grześ D, Piotrowska-Mieczkowska M, Sakowska J, Tomaszewicz M, Marín Morales JM, Lakshmi K, Marek-Trzonkowska NM, Trzonkowski P, Oo YH and Fuchs A (2024) Beyond FOXP3: a 20-year journey unravelling human regulatory T-cell heterogeneity. Front. Immunol. 14:1321228. doi: 10.3389/fimmu.2023.1321228
Received: 13 October 2023; Accepted: 19 December 2023;
Published: 12 January 2024.
Edited by:
Giang Tran, University of New South Wales, AustraliaReviewed by:
Anette S. B. Wolff, University of Bergen, NorwayCopyright © 2024 Santosh Nirmala, Kayani, Gliwiński, Hu, Iwaszkiewicz-Grześ, Piotrowska-Mieczkowska, Sakowska, Tomaszewicz, Marín Morales, Lakshmi, Marek-Trzonkowska, Trzonkowski, Oo and Fuchs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anke Fuchs, YW5rZS5mdWNoczFAdHUtZHJlc2Rlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.