 Man Wang
Man Wang Fei Yu
Fei Yu Yuan Zhang
Yuan Zhang Peifeng Li
Peifeng Li- Institute for Translational Medicine, The Affiliated Hospital of Qingdao University, College of Medicine, Qingdao University, Qingdao, China
Programmed cell death (PCD) is an evolutionarily conserved mechanism of cell suicide that is controlled by various signaling pathways. PCD plays an important role in a multitude of biological processes, such as cell turnover, development, tissue homeostasis and immunity. Some forms of PCD, including apoptosis, autophagy-dependent cell death, pyroptosis, ferroptosis and necroptosis, contribute to carcinogenesis and cancer development, and thus have attracted increasing attention in the field of oncology. Recently, increasing research-based evidence has demonstrated that PCD acts as a critical modulator of tumor immunity. PCD can affect the function of innate and adaptive immune cells, which leads to distinct immunological consequences, such as the priming of tumor-specific T cells, immunosuppression and immune evasion. Targeting PCD alone or in combination with conventional immunotherapy may provide new options to enhance the clinical efficacy of anticancer therapeutics. In this review, we introduce the characteristics and mechanisms of ubiquitous PCD pathways (e.g., apoptosis, autophagy-dependent cell death, pyroptosis and ferroptosis) and explore the complex interaction between these cell death mechanisms and tumor immunity based on currently available evidence. We also discuss the therapeutic potential of PCD-based approaches by outlining clinical trials targeting PCD in cancer treatment. Elucidating the immune-related effects of PCD on cancer pathogenesis will likely contribute to an improved understanding of oncoimmunology and allow PCD to be exploited for cancer treatment.
1 Introduction
Programmed cell death (PCD) is a common process in living organisms that is critical for development, maintenance of cellular homeostasis, immunity and responses to stress (1). PCD is controlled by a wide range of evolutionarily conserved pathways and well-defined mechanisms of action (2). Based on distinct morphological, immunological and genetic features, PCD can be categorized into apoptosis, autophagy-dependent cell death, ferroptosis, necroptosis and pyroptosis (3–5). Conventional cancer treatments mainly rely on cell-suicide programs induced by genotoxic or nongenotoxic stress (6–8). Thus, PCD plays an important role in anticancer therapy-mediated tumor suppression. Moreover, accumulating evidence has demonstrated reciprocal communication between PCD and tumor immunity (9). Based on its ability to trigger an adaptive immune response, PCD can be further grouped into immunogenic and tolerogenic (nonimmunogenic) PCD (10). Immunogenic PCD (e.g., pyroptosis, necroptosis and ferroptosis) reprograms the tumor microenvironment (TME) through extravasation of cellular components, including proinflammatory cytokines and other damage-associated molecular patterns (DAMPs) (9). These signals can be recognized by pattern recognition receptors (PRRs) expressed on innate immune cells, which drives diverse downstream immune responses (11). However, some forms of PCD, such as apoptosis, do not cause the release of cellular contents, and instead culminate in phagocyte-mediated clearance of dead cells without triggering inflammation (12). Immunogenic PCD may finally induce a robust and persistent antitumor immune response, while tolerogenic PCD may facilitate immune tolerance and attenuate the efficacy of anticancer therapy (13). Autophagy is an important cellular mechanism that mediates unnecessary or dysfunctional cytoplasmic constituents to the lysosome for degradation and recycling (14). Autophagy is considered a fundamental prosurvival mechanism that enables cells to avoid excessive PCD. Autophagy can also act to induce cell death, namely, autophagy-dependent cell death, under some circumstances (15). Autophagy-dependent cell death is a type of regulated cell death that mechanistically relies on the molecular machinery of autophagy or its components (15). This form of PCD can be blocked by genetic suppression of at least two components of autophagy pathways (16). The crosstalk between cell death pathways and the TME shapes the complexity and heterogeneity of tumor immunity. Manipulation of PCD is emerging as an attractive approach to improve immunotherapy outcomes (17). Here, we review the molecular mechanisms of common forms of PCD, including apoptosis, autophagy-dependent cell death, pyroptosis and ferroptosis. Furthermore, we summarize the communication among these forms of PCD and the TME and discuss their potential clinical implications in cancer immunotherapy.
2 Cell death programs
Based on morphological criteria, cell death can be classified into three subtypes: apoptosis (type I cell death), autophagy-dependent cell death (type II) and necrosis (type III) (18). These cell death processes are executed through different and interlinked signaling cascades that are triggered by distinct stimuli. The term immunological cell death (ICD) was first proposed in 2005 (19). ICD, a form of PCD, is sufficient to activate adaptive immunity (20). Cells undergoing ICD release or expose DAMPs, which promote dendritic cell (DC)-mediated antigen presentation and culminates in the mobilization of cytotoxic T cell responses (21). Various cell death pathways including apoptosis, autophagy-dependent cell death, pyroptosis, ferroptosis and necroptosis have been classified as ICD (22). ICD can be induced by a plethora of therapeutic agents and modalities (23).
2.1 Overview of apoptosis
Apoptosis is the most extensively studied form of PCD and was first described by Kerr et al. in 1972 (24). It is morphologically characterized by cell shrinkage, membrane blebbing, destruction of cell organelles, nuclear and cytoplasmic condensation, DNA fragmentation and rapid phagocytosis of the cell and cell components by neighboring cells (25). Apoptosis relies on a cascade of caspase proteases and is triggered through either extrinsic or intrinsic pathways (26) (Figure 1).
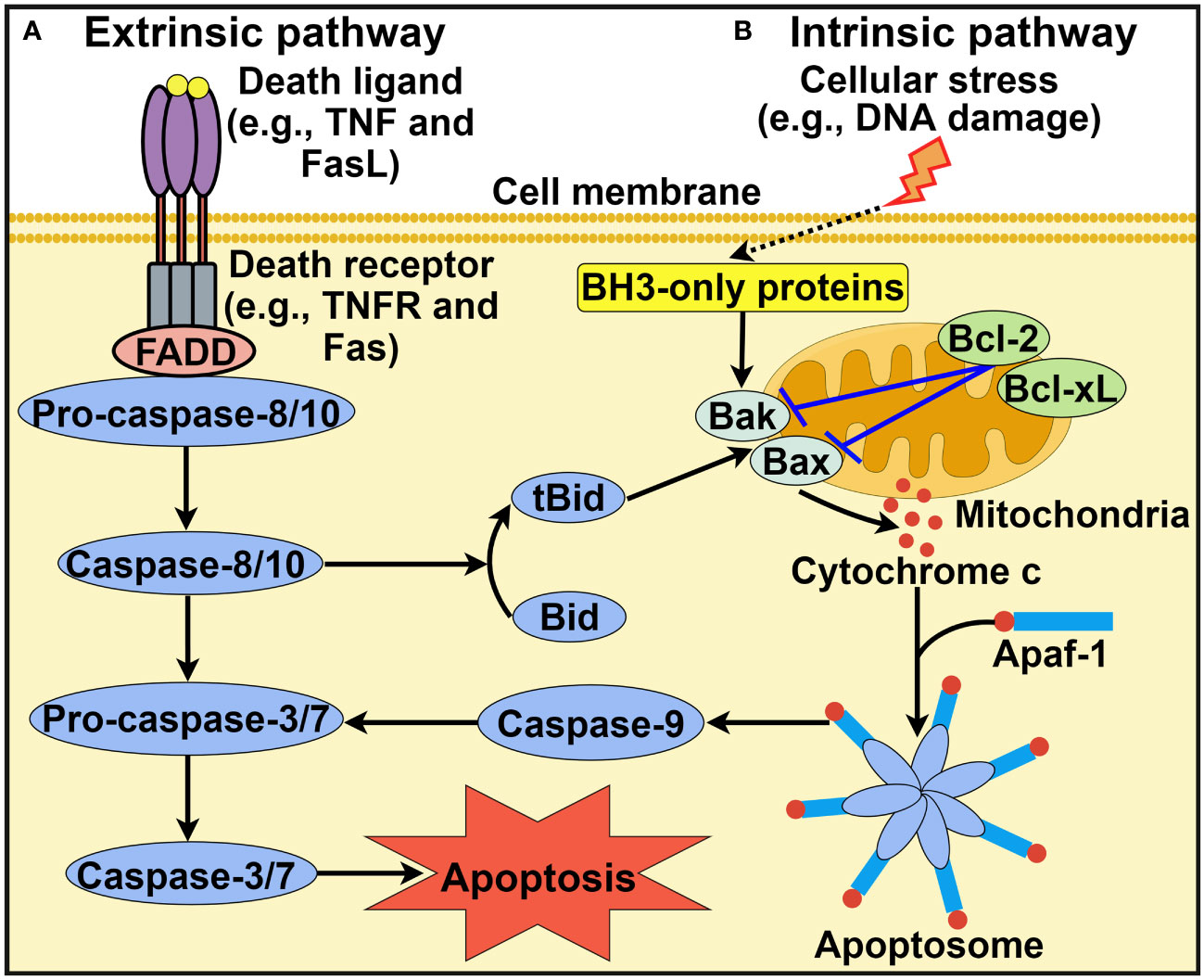
Figure 1 Schematic representation of intrinsic and extrinsic apoptotic pathways. (A) Extrinsic apoptotic pathway. Death ligands (e.g., TNF and FasL) bind to their corresponding receptors. This leads to the recruitment of the adaptor protein FADD and pro-caspase-8 or pro-caspase-10. Active caspase-8 or caspase-10 then activates caspase-3 and caspase-7 to driven cell apoptosis. (B) Intrinsic apoptotic pathway. This pathway is induced by diverse stress stimuli (e.g., DNA damage and hypoxia) that control the activation of the proapoptotic BH3-only family members Bak and Bax. These two effector proteins cause mitochondrial outer membrane permeabilization, thus favoring the release of cytochrome c into the cytosol. Cytosolic cytochrome c associates with Apaf-1 to form the heptameric apoptosome, which recruits and activates caspase-9. Once activated, caspase-9 cleaves and activates downstream execution caspases (e.g., caspase-3 and caspase-7), culminating in cell apoptosis. In addition, the extrinsic and intrinsic pathways are closely linked. Caspase-8 can convert Bid into its truncated form tBid, which acts to activate Bak and Bax. TNF, tumor necrosis factor; FasL, Fas ligand; TNFR, tumor necrosis factor receptor; FADD, Fas-associated protein with death domain; BH3, B cell lymphoma-2 homology 3; Bcl-2, B cell lymphoma-2; Bcl-xL, B cell lymphoma-extra large; Bak, Bcl-2 antagonist/killer; Bax, Bcl-2-associated X protein; Bid, BH3-interacting domain death agonist; tBid, truncated BH3-interacting domain death agonist; Apaf-1, apoptotic protease activating factor-1. The figure was created by Figdraw (www.figdraw.com).
2.1.1 Extrinsic apoptotic pathway
The extrinsic apoptotic pathway, also known as the death receptor pathway, is initiated after the association of cell surface death receptors with their corresponding ligands (2) (Figure 1A). Members of the tumor necrosis factor (TNF) receptor superfamily, including TNF receptor 1 (TNFR1), CD95/Fas and two TNF-related apoptosis-inducing ligand receptors (TRAIL-R1 and TRAIL-R2) function as death receptors (27). Cytoplasmic death domains of death receptors function as a platform to recruit adaptor proteins such as Fas-associated protein with death domain (FADD) and TNF receptor-associated death domain (TRADD). Adaptor proteins then sequester pro-caspase-8 or pro-caspase-10, which results in the formation of the death-inducing signal complex (DISC) located on the cytoplasmic domain of the ligand-bound death receptor (28). Once the DISC is assembled, caspase-8 or caspase-10 is activated, which cleaves effector caspases (caspase-3 and caspase-7) to induce cell apoptosis. In addition, the FADDosome and ripoptosome, which contain caspase-8, FADD and receptor-interacting protein kinase 1 (RIPK1), are also implicated in the extrinsic pathway. Ataxia-telangiectasia and Rad3-related (ATR)-dependent caspase-10 upregulation promotes formation of the FADDosome (29). Caspase-10 and RIPK1 subsequently recruit TNFR-associated Factor 2 (TRAF2) to the FADDosome, which induces the ubiquitination and degradation of cellular FLICE-inhibitory protein (cFLIPL). This leads to nuclear factor-κB (NF-κB) activation that fosters TNF-α production, resulting in caspase-8-mediated apoptosis. In the absence of ATR, caspase-10 and TRAF2, the FADDosome cannot be formed, and caspase-8 cleaves cFLIPL to generate another apoptosis-inducing complex, the FLIPosome. The ripoptosome can change proinflammatory cytokines into death signals, leading to cell apoptosis (30).
2.1.2 Intrinsic apoptotic pathway
Alternatively, the intrinsic pathway of apoptosis, also known as the mitochondrial pathway, is activated by diverse intracellular signals, such as DNA damage, hypoxia, growth factor deprivation and oxidative stress (18) (Figure 1B). These stress signals result in mitochondrial outer membrane permeabilization (MOMP) and contribute to the extravasation of pro-apoptotic factors such as cytochrome c from the mitochondria into the cytosol. Members of the B cell lymphoma-2 (Bcl-2) family regulate MOMP and cytochrome c release into the cytoplasm (31). Bcl-2 proteins possess pro- or anti-apoptotic functions. The pro-apoptotic Bcl-2 proteins can be categorized into Bcl-2 homology 3 (BH3)-only activators (Bid, Bim and PUMA), BH3-only sensitizers (Bad, Bik, BMF, HRK and NOXA) and pore-forming effectors (Bcl-2 antagonist/killer (Bak) and Bcl-2-associated X protein (Bax)) (32, 33). Anti-apoptotic Bcl-2 proteins include Bcl-2, B cell lymphoma-extra large (Bcl-xL) and myeloid cell leukemia-1 (Mcl-1). The prevalence of pro-apoptotic Bcl-2 proteins drives apoptotic cell death, while the activation of anti-apoptotic Bcl-2 proteins contributes to the interruption of the apoptotic process (34). The equilibrium between pro- and anti-apoptotic Bcl-2 proteins is crucial for cell fate decisions. Cytosolic cytochrome c can bind to apoptotic protease activating factor-1 (Apaf-1), which leads to the assembly of the apoptosome (35). The apoptosome then recruits initiator pro-caspase-9 via the caspase activation and recruitment domain (CARD), which leads to caspase-9 activation. Caspase-9 then cleaves and activates downstream executioner caspases (e.g., caspase-3 and caspase-7), leading to cell apoptosis. It should be noted that other protein complexes are also involved in intrinsic apoptosis. The PIDDosome is a representative example and is composed of p53-induced protein with a death domain (PIDD), RIP-associated ICH-1/CED-3-homologous protein with a death domain (RAIDD) adaptor and caspase-2 (36). The PIDDosome induces caspase-2 activation in response to DNA damage and genotoxic stress, thus initiating p53-mediated apoptosis.
2.2 Overview of pyroptosis
Pyroptosis was first described in Salmonella-infected macrophages in the 1990s and was initially considered a process of apoptosis (37, 38). Later, it was realized that this bacterium-induced cell death, which mainly relies on caspase-1, was different from the more well-known process of apoptosis. The term pyroptosis was proposed in 2001 to describe this novel type of PCD (39). The major morphological characteristics of pyroptosis include chromatin condensation, DNA fragmentation, cell swelling with large bubbles, cell membrane rupture and the release of intracellular contents (40, 41). Currently, two principal (canonical and noncanonical inflammasome pathways) and several alternative pyroptosis pathways have been identified (Figure 2).
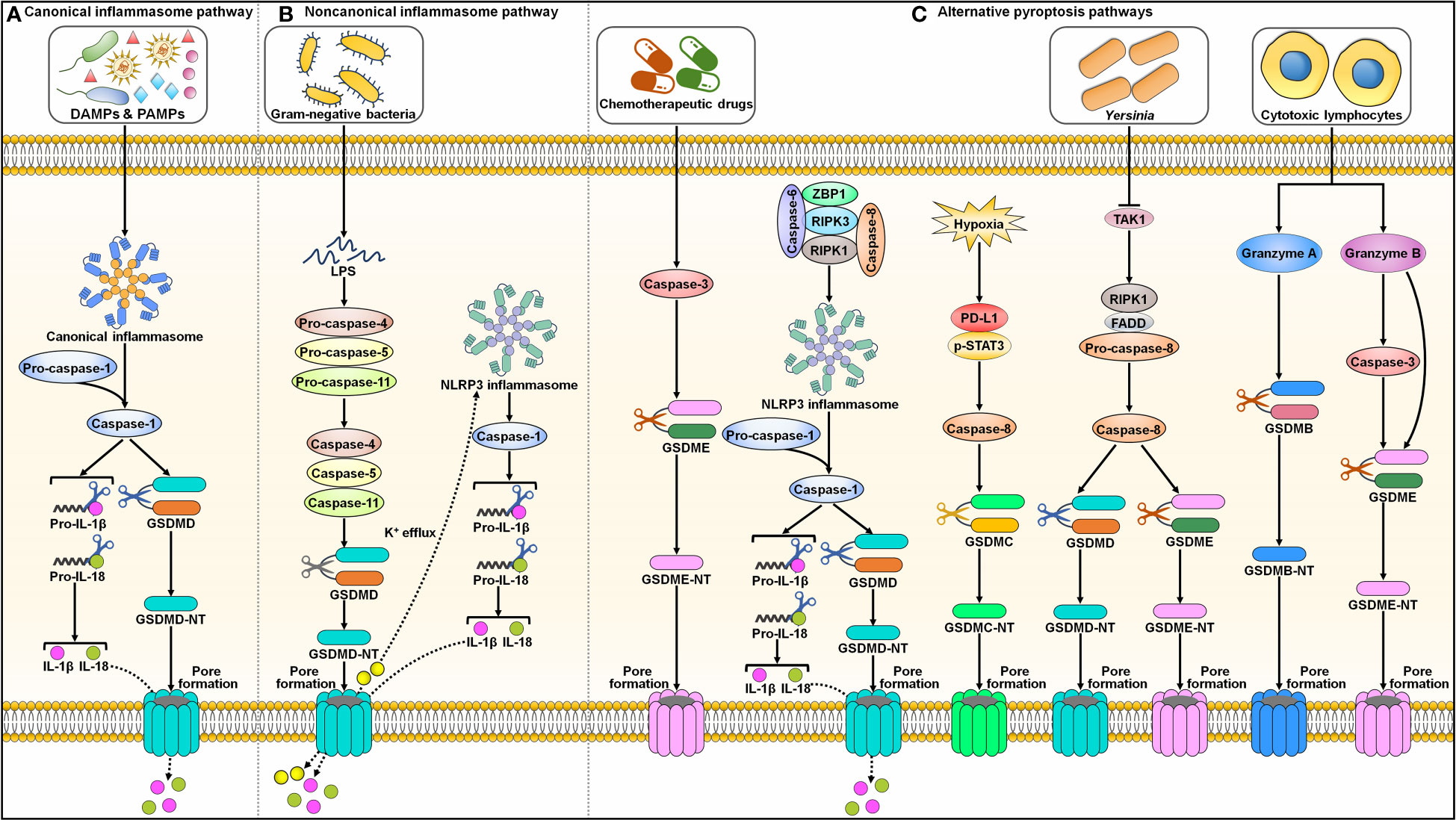
Figure 2 Graphic representation of pyroptotic signaling pathways. (A) Canonical inflammasome pathway. Canonical pyroptotic cell death is initiated through the recognition of DAMPs and PAMPs by inflammasome sensors (e.g., NLRP3 and pyrin). This stimulates the assembly of the canonical inflammasome, triggering pro-caspase-1 self-cleavage. Activated capsase-1 cleaves GSDMD to unlash its N-terminal domain (GSDMD-NT). GSDMD-NT binds to the inner leaflet of the cell membrane and oligomerizes to generate membrane pores, leading to the release of cytoplasmic contents including proinflammatory factors. Caspase-1 also processes pro-IL-1β and pro-IL-18 into their active forms (IL-1β and IL-18), which are released through the GSDMD-NT pore. (B) Noncanonical inflammasome pathway. The noncanonical pyroptosis pathway is induced by bacterial LPS-mediated activation of human caspase-4/-5 or murine caspase-11. Activated caspase-4, -5 and -11 then cleave GSDMD to facilitate pore formation on the cell membrane. The K+ efflux through the GSDMD-NT pore causes the activation of NLRP3 inflammasome and caspase-1. Caspase-1 hydrolyzes pro-IL-1β and pro-IL-18 into biologically active IL-1β and IL-18. These mature cytokines are liberated out of ruptured cells via membrane pores. (C) Alternative pyroptosis pathways. Caspase-3 activated by chemotherapeutic drugs shears GSDME to produce a functional GSDME-NT, culminating in pyroptotic cell death. Caspase-6 activates NLRP3 inflammasome to elicit pyroptosis by promoting the interaction between RIPK3 and ZBP1. Under hypoxic condition, p-STAT3 promotes PD-L1 nuclear translocation through coupling with PD-L1. This leads to increased transcription of GSDMC. Following TNF-α treatment, caspase-8 cleaves GSDMC into GSDMC-NT that penetrates the cell membrane to trigger pyroptosis. Yersinia motivates RIPK1/caspase-8 signaling cascade through TAK1 blockade. Activated caspase-8 directly processes GSDMD and GSDME, eventually contributing to pyroptosis. In addition, cytotoxic lymphocyte-derived GzmA cleaves GSDMB to actuate pyroptosis. GzmB elicits pyroptotic cell death by both indirectly activating caspase-3 to cleave GSDME and directly shearing GSDME. DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; IL-1β, interleukin-1β; IL-18, interleukin-18; GSDMD, gasdermin D; GSDMD-NT, the N-terminal domain of gasdermin D; LPS, lipopolysaccharide; NLRP3, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3; GSDME, gasdermin E; GSDME-NT, the N-terminal domain of gasdermin E; ZBP1, Z-DNA binding protein 1; RIPK3, receptor-interacting protein kinase 3; RIPK1, receptor-interacting protein kinase 1; PD-L1, programmed cell death-ligand 1; p-STAT3, phosphorylated signal transducer and activator of transcription 3; GSDMC, gasdermin C; GSDMC-NT, the N-terminal domain of gasdermin C; TAK1, transforming growth factor-β-activated kinase 1; FADD, Fas-associated protein with death domain; GSDMB, gasdermin B; GSDMB-NT, the N-terminal domain of gasdermin B.
2.2.1 Canonical inflammasome pathway
The inflammasome complex typically consists of a sensor (e.g., PRR), the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), and an effector protein (e.g., caspase-1) (42). The well-characterized PRRs include absent in melanoma 2 (AIM2), nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain-containing protein 1 (NLRP1), NLRP3, NLR family CARD-containing protein 4 (NLRC4) and pyrin (43). Inflammasomes formation provides a platform for caspase-1 activation (44) (Figure 2A). Activated capsase-1 cleaves gasdermin D (GSDMD) to unlock its N-terminal domain (GSDMD-NT). GSDMD-NT binds to the inner leaflet of the cell membrane and oligomerizes to generate pores that enhance membrane permeability, which results in water influx, cell swelling, cytolysis and extravasation of cytoplasmic contents including proinflammatory factors (45). Caspase-1 also processes the precursor forms of interleukin-1β (IL-1β) and interleukin-18 (IL-18) into their active forms, which are released through the GSDMD-NT pore (46).
2.2.2 Noncanonical inflammasome pathway
The noncanonical pyroptosis pathway is independent of inflammasomes, and instead, this pathway is initiated by bacterial lipopolysaccharide (LPS)-mediated activation of human caspase-4/-5 or murine caspase-11 (47) (Figure 2B). Activated caspase-4, -5 and -11 then cleave GSDMD to induce pore formation on the cell membrane (48). Caspase-4, -5 and -11 cannot directly trigger the maturation of pro-IL-1β and pro-IL-18. Potassium (K+) efflux through GSDMD-NT pores leads to activation of the NLRP3 inflammasome and caspase-1 (49, 50). Caspase-1 then hydrolyzes pro-IL-1β and pro-IL-18 into biologically active IL-1β and IL-18, respectively. These mature cytokines are liberated from ruptured cells via membrane pores.
2.2.3 Alternative pyroptosis pathways
In addition to caspase-1, other caspases can also induce pyroptosis (Figure 2C). Reportedly, caspase-3 activated by chemotherapeutic drugs shears gasdermin E (GSDME) to produce a functional pore-forming fragment (GSDME-NT), culminating in pyroptotic cell death (51, 52). Caspase-6 activates the NLRP3 inflammasome to elicit pyroptosis by promoting the interaction between RIPK3 and Z-DNA binding protein 1 (ZBP1) (53). Programmed cell death-ligand 1 (PD-L1) transforms TNF-α-induced apoptosis into pyroptosis in cancer cells (54). Under hypoxic conditions, phosphorylated signal transducer and activator of transcription 3 (p-STAT3) promotes PD-L1 nuclear translocation through coupling with PD-L1. This leads to increased transcription of gasdermin C (GSDMC). Following TNF-α treatment, caspase-8 cleaves GSDMC into GSDMC-NT, which penetrates the cell membrane to trigger pyroptosis. Yersinia activates the RIPK1/caspase-8 signaling cascade through blockade of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) (55–57). Activated caspase-8 directly processes GSDMD and GSDME, eventually contributing to pyroptosis.
In addition, granzymes (Gzms) are found to participate in GSDM-dependent pyroptosis. Cytotoxic lymphocyte-derived GzmA cleaves gasdermin B (GSDMB) in target cells, which leads to pyroptosis (58). GzmB elicits pyroptotic cell death in cancer cells by both indirectly activating caspase-3 to cleave GSDME and by directly shearing GSDME (59). The molecular mechanisms of pyroptosis are still poorly understood and warrant in-depth investigation.
2.3 Overview of ferroptosis
Ferroptosis is a recently discovered mode of PCD that is mainly characterized by excessive iron accumulation, lipid peroxidation, and cell membrane rupture (60, 61). Ferroptosis inducers were discovered prior to when this process was formally termed. In 2003, Dolma et al. (62) revealed a novel form of nonapoptotic PCD caused by the small molecule erastin. Later, in 2008, synthetic compounds RAS-selective lethal 3 (RSL3) and RSL5 were found to trigger this pattern of cell death (63, 64). It was not until 2012 that such PCD was formally named ferroptosis (65). Since then, a great deal of effort has been devoted to elucidating the characteristics and regulatory mechanisms of ferroptosis. Ferroptosis can be induced through either the extrinsic or intrinsic pathway (66, 67). The extrinsic pathway, also known as the transporter-dependent pathway, is triggered through decreased cystine uptake or increased iron uptake (Figure 3). The intrinsic pathway, also called the enzyme-regulated pathway, is primarily induced by the inhibition of intracellular antioxidant enzymes (e.g., glutathione peroxidase 4 (GPX4)).
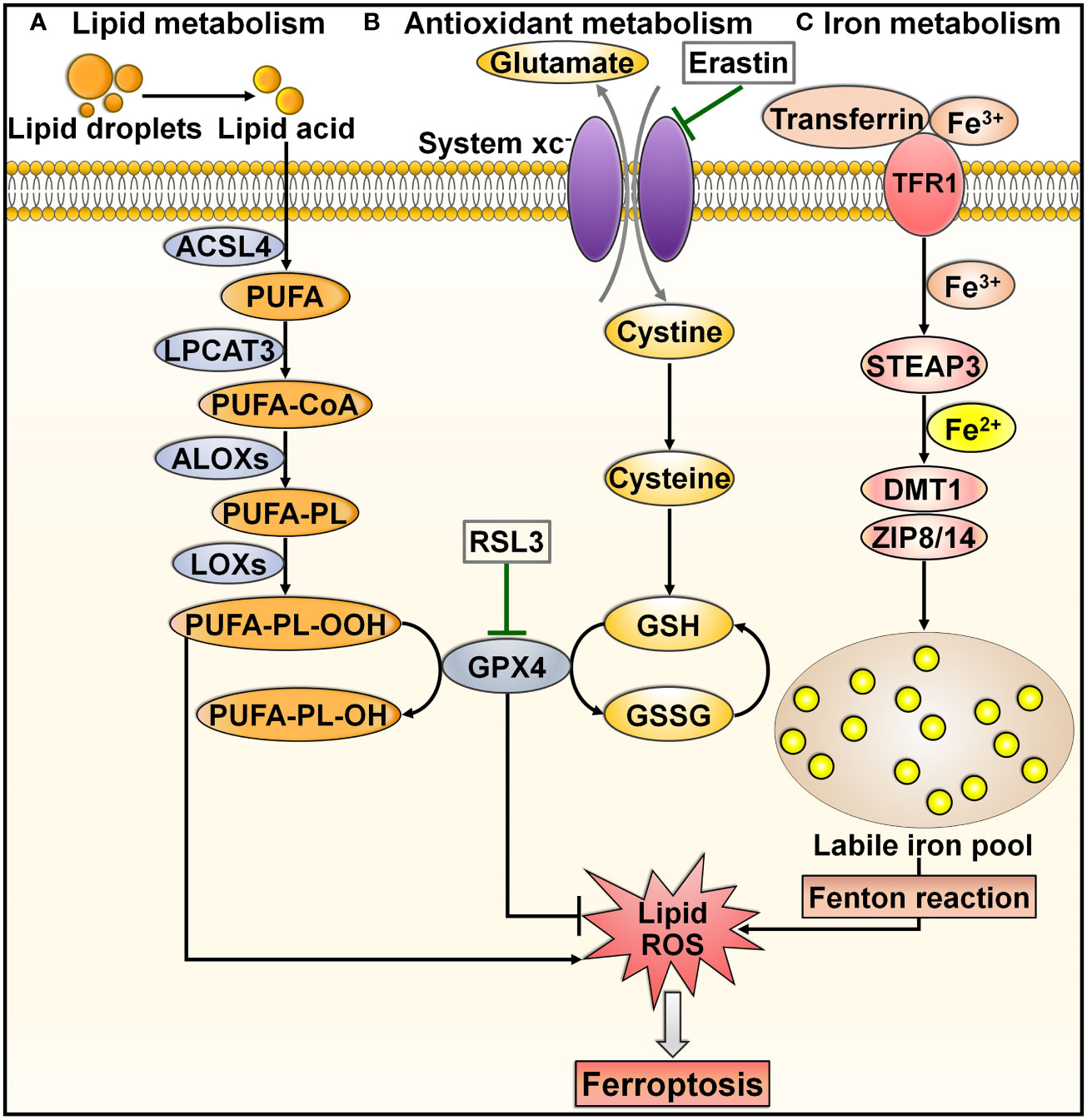
Figure 3 Overview of molecular mechanisms regulating ferroptosis. Ferroptosis is can be induced through either transporter-dependent pathway or enzyme-regulated pathway. (A) Lipid metabolism. Under energetic or oxidative stress, free PUFA is catalyzed by ACSL4, LPCAT3 and ALOX to produce PUFA-PL. PUFA-PL is then converted into toxic PUFA-PL-OOH under the assistance of LOXs and eventually induces ferroptotic cell death. (B) Antioxidant metabolism. Extracellular cystine is exchanged with glutamate to enter the cell through system xc-, and it converts into cysteine to promote GSH synthesis. GPX4 transforms GSH into GSSG and simultaneously reduces toxic lipid hydroperoxides (LOOHs) to nontoxic lipid alcohols (L-OHs), thus protecting cells from ferroptosis. Blockade of system xc- by erastin, or GPX4 inhibitor (e.g., RSL3) can trigger ferroptosis by promoting lipid-ROS production. (C) Iron metabolism. Extracellular Fe3+ enters the cell through transferrin and TFR1. In cells, Fe3+ is released from transferrin and reduced to Fe2+ by STEAP3. Fe2+ is then transferred into a labile iron pool, which is mediated by DMT1 or ZIP8/14. Excess Fe2+ contributes to the formation of massive ROS and consequent oxidative stress through the Fenton reaction. These events drive the peroxidation of lipids, nucleic acids and proteins, culminating in cell ferroptosis. ACSL4, acyl-CoA synthetase long-chain family member 4; PUFA, polyunsaturated fatty acid; LPCAT3, lysophosphatidylcholine acyltransferase 3; PUFA-CoA, coenzyme A-activated polyunsaturated fatty acid; ALOXs, arachidonate lipoxygenases; PUFA-PL, PUFA-containing phospholipid; LOXs, lipoxygenases; PUFA-PL-OOH, PUFA phospholipid hydroperoxide; PUFA-PL-OH, PUFA phospholipid alcohol; RSL3, RAS-selective lethal 3; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; ROS, reactive oxygen species; TFR1, transferrin receptor 1; STEAP3, six-transmembrane epithelial antigen of prostate 3; DMT1, divalent metal transporter 1; ZIP8/14, zinc-iron regulatory protein family 8/14.
Lipid peroxidation is a key initiator of ferroptosis (68). Under energetic or oxidative stress, free polyunsaturated fatty acids (PUFAs) are catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3) and arachidonate lipoxygenases (ALOXs) to produce PUFA-containing phospholipids (PUFA-PLs) (69) (Figure 3A). Membrane electron transfer proteins are involved in reactive oxygen species (ROS) production during lipid peroxidation. PUFA-PL is then converted into toxic lipid peroxides (PUFA phospholipid hydroperoxides (PUFA-PL-OOH)) with the assistance of lipoxygenases (LOXs) and eventually induces ferroptotic cell death (70).
System xc-, a cystine/glutamate transporter, is composed of solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) (71). System xc- acts to sustain the intracellular content of glutathione (GSH) through regulation of cystine uptake (72). Extracellular cystine is exchanged with glutamate to enter the cell through system xc- and is then converted into cysteine (73) (Figure 3B). Cysteine promotes the synthesis of GSH. GPX4 can transform GSH into oxidized glutathione (GSSG) and simultaneously reduce toxic lipid hydroperoxide (LOOHs) to nontoxic lipid alcohols (L-OHs), thus inhibiting the generation of lipid-based ROS and protecting cells from ferroptosis (60). Blockade of system xc- by erastin, GSH deficiency, or a GPX4 inhibitor (e.g., RSL3) induces ferroptotic cell death by promoting lipid-ROS production (74).
Accumulated intracellular iron is another contributor to ferroptosis (75). In iron metabolism, extracellular ferric iron (Fe3+) enters the cell through transferrin and transferrin receptor 1 (TFR1) (76) (Figure 3C). In cells, Fe3+ is released from transferrin and is reduced to ferrous iron (Fe2+) by the iron reductase six-transmembrane epithelial antigen of prostate 3 (STEAP3). Fe2+ is transferred into a labile iron pool under the action of divalent metal transporter 1 (DMT1) or zinc-iron regulatory protein (ZIP) family 8/14 (77, 78). Labile iron is generally stored in the iron-storage protein ferritin, thereby restricting the high reactivity of iron and inhibiting ROS generation (79). The imbalance in iron metabolism can lead to ferroptosis (80). In practice, nuclear receptor coactivator 4 (NCOA4) binds to ferritin and transports it to the lysosome for degradation, which results in the release of free Fe2+ (81). Excess Fe2+ contributes to ROS formation and consequent oxidative stress through the Fenton reaction (65). These events drive the peroxidation of lipids, nucleic acids and proteins, culminating in cell ferroptosis (82). In addition to facilitating lipid peroxidation through nonenzymatic mechanisms, excessive intracellular iron can induce ferroptosis by activating ALOX or EGLN prolyl hydroxylase (PHD), which participate in lipid peroxidation and oxygen homeostasis (83, 84).
2.4 Overview of necroptosis
Necroptosis, also referred to as programmed necrosis, is a necrosis-like form of cell death driven by RIPK1, RIPK3 and mixed lineage kinase domain-like protein (MLKL) (85, 86). Necroptosis has been closely associated with diverse pathologies, including cancer and cardiovascular diseases (87, 88). It shares similar morphological features with necrosis, such as a translucent cytoplasm, swelling of organelles, plasma membrane rupture, mitochondrial dysfunction, increased cell volume, cell lysis, extravasation of intracellular contents and chromatin condensation (89). Unlike apoptosis and pyroptosis, necroptosis is initiated by caspase-independent signaling pathways (90) (Figure 4). Necroptosis can be activated by a plethora of internal and external stimuli, including the TNF superfamily members (e.g., Fas ligand (FasL), TNF-α and TNF-related apoptosis-inducing ligand (TRAIL)), interferon-γ (IFN-γ), bacteria, DAMPs, endoplasmic reticulum (ER) stress, hypoxia, LPS, metabolic and genotoxic stresses, ROS, viral infection and chemotherapeutic agents (91, 92). Necroptosis occurs after necroptotic activators are detected by cytosolic nucleic acid sensors, death receptors, IFN receptors, PRRs, T cell receptors (TCRs) and Toll-like receptors (TLRs) (88, 93). The most well-characterized necroptosis mechanism is triggered by ligation of the death receptor TNFR1 to its ligand TNF-α (92). TNFR1 then undergoes conformational changes that allow it to recruit a series of proteins, such as cellular inhibitor of apoptosis protein 1 (cIAP1), cIAP2, linear ubiquitin chain assembly complex (LUBAC), RIPK1, TRAF2, TRAF5 and TRADD (94). This short-lived membrane signaling complex is known as complex I. LUBAC and cIAPs induce RIPK1 ubiquitination, which creates stable complex I and triggers an alternative pathway that facilitates cell survival via mitogen-activated protein kinase (MAPK) and NF-κB signalings (95). When cIAPs are degraded or inhibited, ubiquitination of RIPK1 is blocked, and A20 and cylindromatosis (CYLD) mediate RIPK1 deubiquitination (96, 97). This promotes RIPK1 dissociation from the cell membrane and leads to activation of cell death pathways (e.g., apoptosis and necroptosis) (98). RIPK1 deubiquitination encourages the development of a cytosolic apoptotic complex (complex II) that is composed of caspase-8, FADD, RIPK1 and TRADD (99). Caspase-8 triggers apoptosis and impedes necroptosis by cleaving RIPK1 and RIPK3 (100). In conditions where caspase-8 is inhibited or RIPK3 is overexpressed, complex II does not initiate the apoptosis pathway, but rather, it induces necroptotic cell death (101). In the absence of caspase-8 activity, RIPK1 recruits and phosphorylates RIPK3, leading to assembly of the necrosome (102). Following this, the RIPK1/RIPK3 complex facilitates phosphorylation and oligomerization of MLKL (103). With the ability to interact with lipids, oligomerized MLKL translocates toward the cell membrane, where it creates large pores and causes uncontrollable release of cellular contents (e.g., DAMPs and inflammatory cytokines) (104). The MLKL oligomer combines with ion channels to disrupt cellular ion homeostasis, which then drives osmotic cell membrane rupture and ultimately induces necroptosis (89). In addition to the TNF-mediated necroptosis mechanism, other pathways (nonclassical necroptotic pathways) that elicit necroptosis have been reported (98). For instance, Z-DNA-binding protein 1 (ZBP1) and Toll/IL-1 receptor (TIR) domain-containing adapter-inducing IFN-β (TRIF) directly bind to RIPK3, leading to RIPK1-independent activation of necroptosis (105, 106).
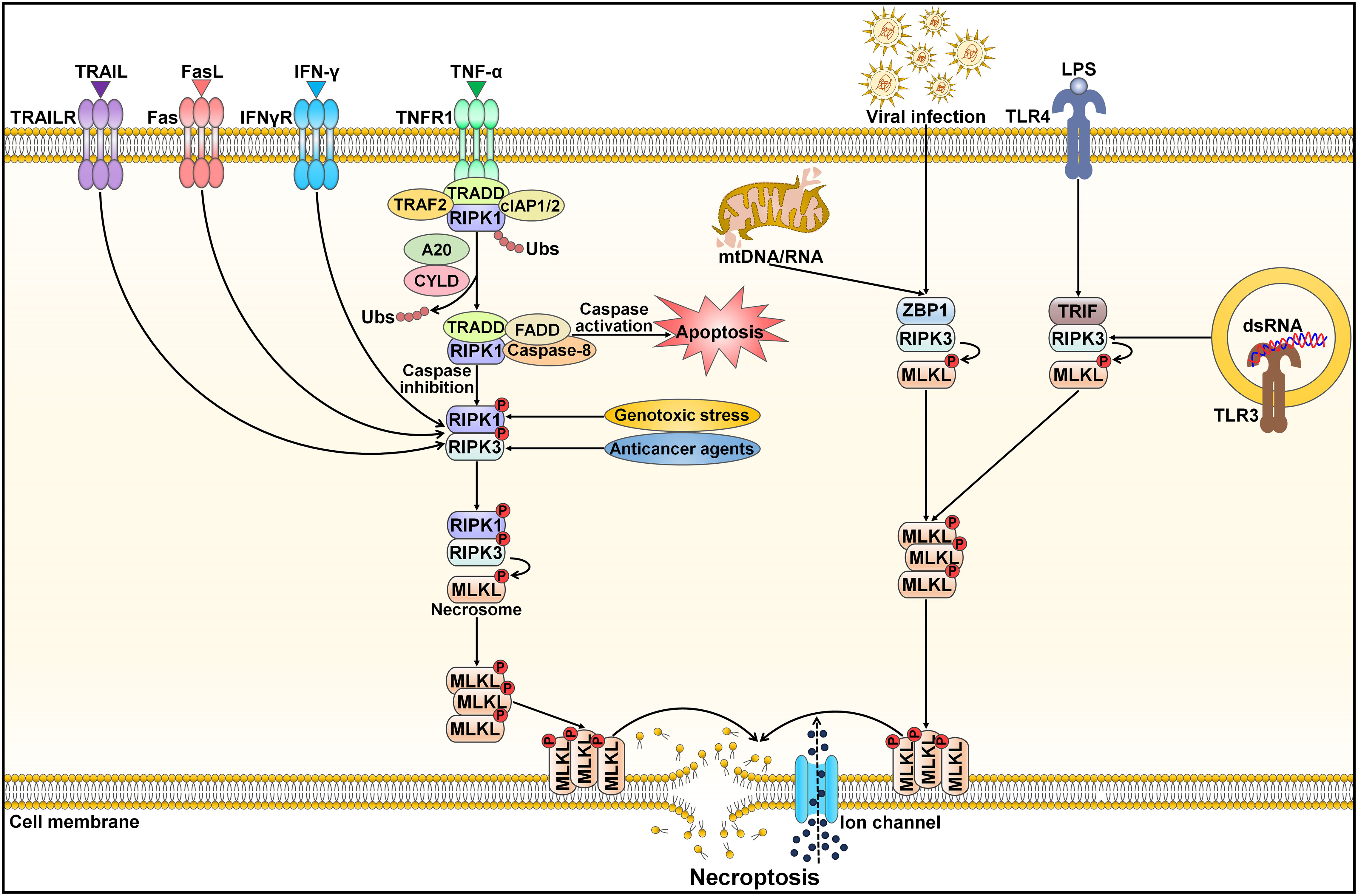
Figure 4 Schematic representation of necroptotic cell death. Necroptosis can be activated by ligand binding to TNF receptor family proteins (e.g., TNFR1, IFNγR, Fas and TRAILR). The most extensively studied subtype of regulated necrosis is TNF-α-induced necroptosis pathway, which relies on the necrosome composed of RIPK1, RIPK3 and MLKL. TNF-α interacts with TNFR1 leading to the structural alteration of TNFR1. A signaling complex (complex I) is assembled at the cytoplasmic domains of TNFR1 and comprises TRAF2, TRADD, RIPK1 and cIAP1/2. Several factors, especially the ubiquitination status of RIPK1 and the activation state of caspase-8, influence whether or not the cell adopts a necroptotic fate. Polyubiquitinated RIPK1 contributes to the stabilization of complex I and triggers an alternative pathway that facilitates cell survival via MAPK and NF-κB signals. When cIAPs are degraded or inhibited, ubiquitination of RIPK1 is blocked, and A20 and CYLD induce RIPK1 deubiquitination. This promotes RIPK1 dissociation from the cell membrane and induces the activation of cell death pathways (apoptosis and necroptosis). RIPK1 deubiquitination facilitates the generation of a cytosolic apoptotic complex (complex II) that includes TRADD, FADD, RIPK1 and caspase-8. Caspase-8 triggers apoptosis and prevents necroptosis by cleaving RIPK1 and RIPK3. In the absence of caspase-8, RIPK1 recruits and phosphorylates RIPK3, following which activated RIPK3 phosphorylates MLKL and results in its oligomerization. The MLKL oligomer migrates to the cell membrane and generates large pores that contribute to necroptotic cell death by allowing ion influx, and cell swelling and lysis. Moreover, necroptosis can be initiated by other inducers, including genotoxic stress, anticancer agents, nucleic acids (DNAs and RNAs) released from damaged mitochondria, viral infection, LPS and dsRNA. After sensing cytosolic mtDNA/mtRNA or virus-derived RNA, ZBP1 induces necroptosis via the RIPK3/MLKL axis. TLR3 and TLR4 are activated by dsRNA and LPS, respectively. Thereafter, TLR stimulation triggers RIPK1-independent necroptosis via TRIF. TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRAILR, tumor necrosis factor-related apoptosis-inducing ligand receptor; FasL, Fas ligand; IFN-γ, interferon-γ; IFNγR, interferon-γ receptor; TNF-α, tumor necrosis factor-α; TNFR1, tumor necrosis factor-α receptor 1; TRAF2, tumor necrosis factor receptor-associated factor 2; TRADD, tumor necrosis factor receptor-associated death domain; cIAP1/2, cellular inhibitor of apoptosis protein 1/2; RIPK1, receptor-interacting protein kinase 1; Ubs, ubiquitins; CYLD, cylindromatosis; FADD, Fas-associated protein with death domain; RIPK3, receptor-interacting protein kinase 3; MLKL, mixed lineage kinase domain-like protein; mtDNA/RNA, mitochondrial DNA/RNA; ZBP1, Z-DNA binding protein 1; LPS, lipopolysaccharide; TLR4, Toll-like receptor 4; TRIF, Toll/interleukin-1 receptor domain-containing adapter-inducing interferon-β; dsRNA, double-stranded RNA; TLR3, Toll-like receptor 3.
2.5 Overview of autophagy-dependent cell death
Autophagy (self-eating) is an evolutionarily conserved mechanism for the degradation and recycling of cytoplasmic materials (e.g., organelles, lipids and proteins) via lysosomal digestion (107). Autophagy is engaged in many biological processes, such as development and differentiation (108). There are three main types of autophagy in mammals: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) (109) (Figure 5). Macroautophagy, the most well-known form of autophagy, is a nonselective cellular process. This autophagic process starts with the nucleation of a double-membraned structure termed phagophore (110) (Figure 5A). The phagophore is elongated to enclose cytoplasmic contents, which leads to the formation of a double-membrane vesicle called autophagosome. The autophagosome subsequently fuses with the lysosome to form an autolysosome, where degradation of cargo contents occurs. Microautophagy involves the direct incorporation of cytoplasmic components into the lysosome or late endosome for degradation (111) (Figure 5B). Endosomal sorting complexes required for transport (ESCRT) machinery is needed for the membrane fission step (112). In the CMA pathway, the cytosolic chaperone heat shock cognate protein 70 (HSC70) recognizes CMA-targeted proteins harboring a pentapeptide motif (113) (Figure 5C). The HSC70-target protein complex is translocated to the lysosome through binding to the lysosome-associated membrane protein 2A (LAMP2A) receptor on the lysosomal membrane. CMA-targeted proteins undergo degradation within the lysosome. In all types of autophagy, the resultant products of the degradation process are recycled into the cytosol and can be reutilized in a variety of biological processes, such as adenosine triphosphate (ATP) generation, and nucleotide, protein and lipid biosynthesis (114).
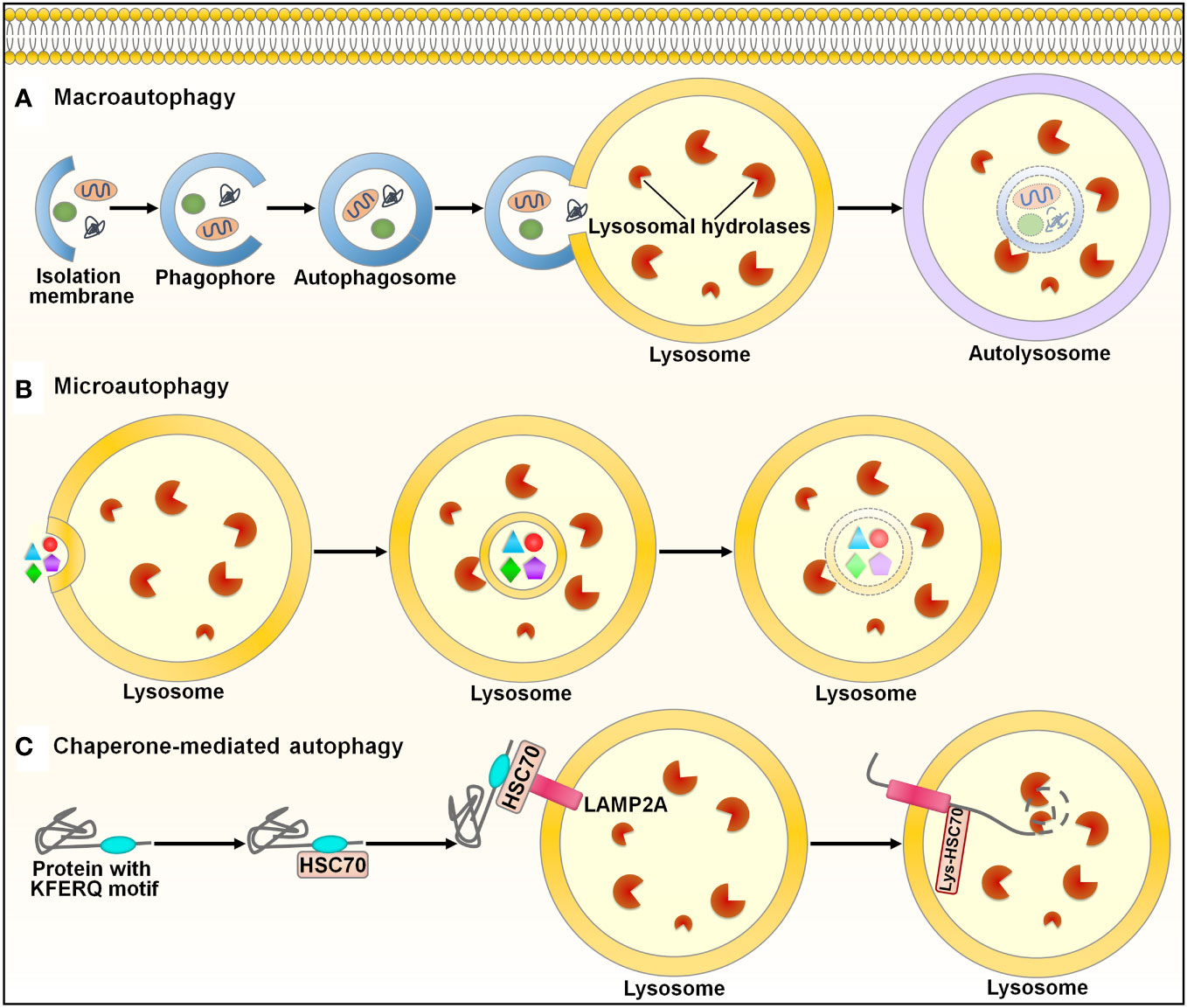
Figure 5 Schematic illustration of the autophagy pathways. Autophagy is categorized into three main types: macroautophagy, microautophay and chaperone-mediated autophagy. (A) Macroautophagy. This pathway is initiated with the nucleation of a double-membraned structure, named phagophore. The phagophore is elongated to enclose cytoplasmic contents, leading to the formation of a double-membrane vesicle called autophagosome. The autophagosome subsequently fuses with the lysosome to form an autolysosome, where degradation of cargo contents occurs. (B) Microautophagy. Microautophagy involves direct incorporation of cytoplasmic components into the lysosome for degradation. (C) Chaperone-mediated autophagy. In this pathway, the cytosolic chaperone HSC70 recognizes proteins containing the KFERQ-like motif. The HSC70-target protein complex is translocated to the lysosome through binding to the LAMP2A receptor on the lysosomal membrane. CMA-targeted proteins undergo degradation within the lysosome. HSC70, heat shock cognate protein 70; LAMP2A, lysosome-associated membrane protein 2A.
The role of autophagy in initiating cell death is highly contextual. It is still unclear how autophagy-dependent processes cause cell death. Autosis, a specific form of autophagy-dependent cell death, is critically dependent on plasma membrane Na+/K+-ATPase (115). Moreover, cell death is promoted when feedback mechanisms to suppress autophagy are interrupted (116). Cells undergoing autophagy-dependent cell death are characterized by the generation of abundant autophagic vacuoles/vesicles. Thus, hyperactivation of autophagy may induce cell death. Likewise, degraded cytoplasmic components, intracellular membranes and organelles are usually detected in cells undergoing autophagy, which suggests that deprivation of cytoplasmic materials upon persistent autophagy contributes to cell death. It is still unknown whether the degradation of specific organelles is the primary cause of autophagy-dependent cell death or whether excessive degradation of cellular components by autophagy triggers cell death. Since autophagy mainly relies on lysosome function, lysosome-mediated bulk degradation of cytoplasmic contents can induce cellular demise (117). The involvement of the lysosome in autophagy-dependent cell death therefore requires further investigation. Ubiquitination is important for the engulfment of intracellular cargos in selective autophagy (118). Cargo receptors combine with specific ubiquitinated substrates to guide the selective packaging of cargos into autophagosomes (119). The ubiquitin system may play a role in autophagy-dependent cell death, but further investigation is required to substantiate and expand this assumption. The autophagic machinery may selectively degrade prosurvival factors to elicit cell death. Reportedly, autophagy promoted the degradation of the anti-apoptotic dBruce to drive caspase-dependent apoptosis in nurse cells during late Drosophila melanogaster oogenesis (120). The molecular machinery necessary for autophagy-dependent cell death may be different from that favoring cell survival. The rate of autophagic flux, the duration of activated autophagy and the fate of engulfed components may bring about different requirements for autophagy machinery components. Altogether, autophagy-dependent cell death seems to be coordinated differently from prosurvival autophagy. In addition, various signaling pathways, such as the phosphoinositide-3-kinase (PI3K) pathway and MAPK signaling pathway, may regulate autophagy-dependent cell death (121, 122). The detailed mechanisms regulating autophagy-dependent cell death have not yet been fully defined. Future research efforts are necessary to better understand the modulatory mechanisms of autophagy-dependent cell death.
3 Crosstalk between programmed cell death and tumor microenvironment
Increasing evidence has shown that various types of PCD including apoptosis, autophagy-dependent cell death, pyroptosis and ferroptosis can enhance antitumor immunity via stimulation of immune-promoting cells or release of proinflammatory mediators. In some cases, PCD also limits antitumor immune responses through elimination of antitumor immune cells or activation of immunosuppressive cells.
3.1 Apoptosis in tumor immunity
Several studies have demonstrated that apoptosis can stimulate antitumor immune cells (Table 1). High expression of STAT3 was associated with chemotherapy resistance and immune evasion of hepatocellular carcinoma (HCC) cells (123). STAT3 absence aggravated therapeutically induced-ER stress-mediated apoptosis in HCC. The DNA derived from apoptotic HCC cells stimulated the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)/stimulator of interferon genes (STING) signaling cascade in CD103+ DCs and triggered type I IFN (IFN-I) production, which eventually augmented the antitumor function of CD8+ T and NK cells. Accordingly, STAT3 knockdown sensitized HCC cells to the cytotoxic agent sorafenib. Insulin-like growth factor-2 mRNA-binding protein 1 (IGF2BP1) recognized m6A target transcripts, including c-MYC and PTEN (155). IGF2BP1 acted as an oncogenic driver in HCC (124). IGF2BP1 dysfunction efficiently prevented its attachment to m6A mRNA targets and led to enhanced apoptosis of HCC cells. These events promoted the activation and intratumoral infiltration of various immune cells (e.g., macrophages, NK cells, CD4+ T cells and CD8+ T cells) and restricted PD-L1 expression to block immunosuppression.
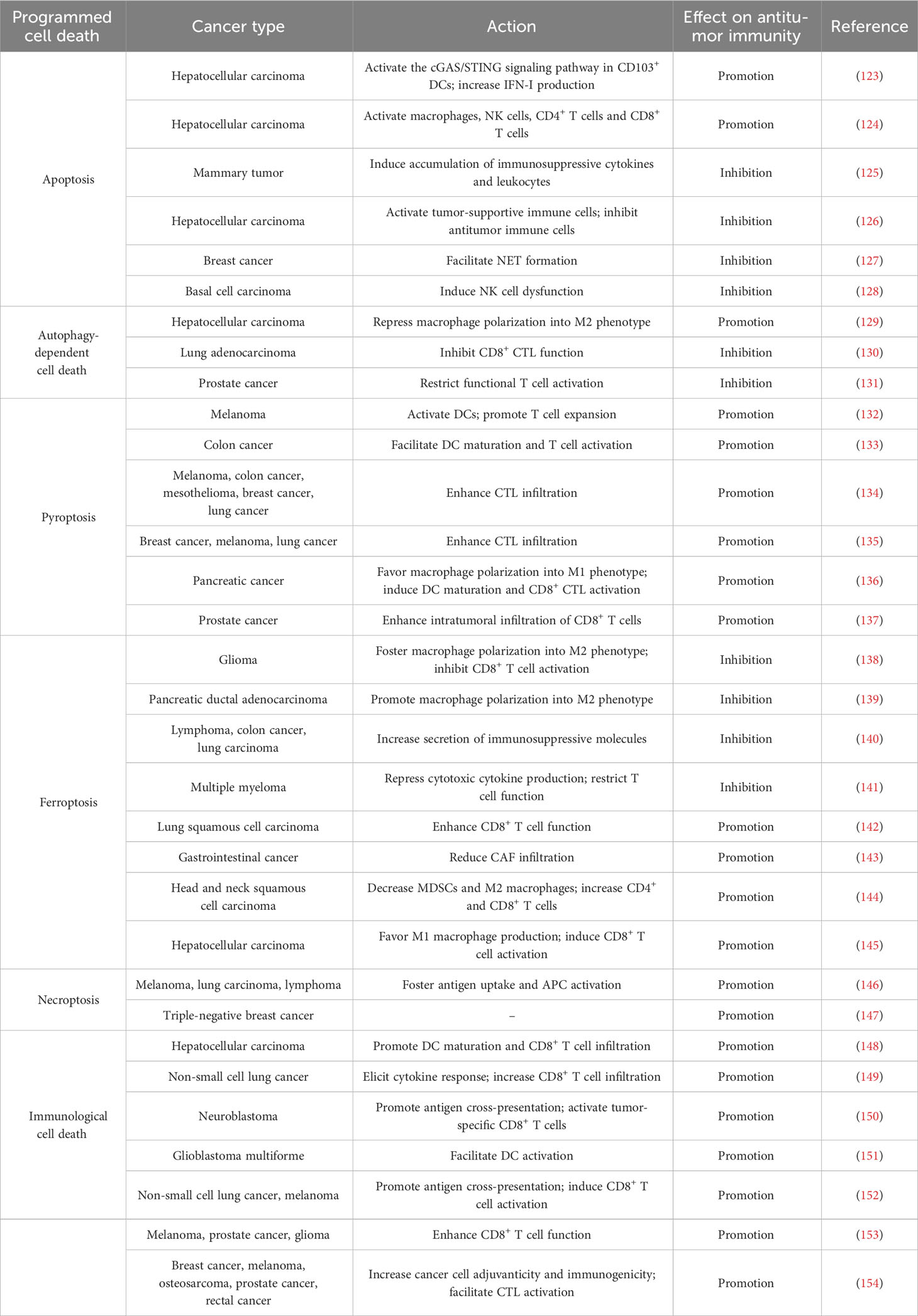
Table 1 Overview of the effects of programmed cell death on tumor immunity.
Apoptosis contributes to the reshaping of an immunosuppressive TME. Apoptotic cells are commonly cleared from tissues via efferocytosis (156). The clearance of apoptotic cells by efferocytosis prevents these cells from undergoing secondary necrosis and secreting proinflammatory factors. Efferocytosis expeditiously eradicated lapatinib-induced apoptotic cancer cells from mammary tumors in vivo (125). This process upregulated immunosuppressive cytokines (e.g., IL-10, IL-13 and TGF-β1) and recruited immunosuppressive leukocytes (e.g., myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs)) to the tumors. Thus, cancer cell efferocytosis following chemotherapy imparted tolerance to cancer cells and enabled them to escape treatment-induced apoptosis. Inhibition of efferocytosis triggered secondary necrosis of apoptotic cells but failed to prevent the aforementioned immunosuppressive alternations in response to cancer cell death. Mechanistically, necrosis secondary to impaired efferocytosis stimulated IFN-γ-inducible expression of indoleamine 2,3-dioxygenase 1 (IDO1), culminating in immunosuppression and cancer development. Combined suppression of efferocytosis and IDO1 dampened cancer cell death-induced immunosuppression and restricted mammary tumor progression in murine models. Altogether, apoptotic and necrotic cell death pathways affect cancer progression through different mechanisms. Additional work is warranted to gain a fundamental understanding of the intricate communication between diverse cell death pathways and the host immune system. A recent study showed that apoptosis, ferroptosis and pyroptosis were common types of cell death processes in the TME of HCC (126). These cell death pathways were cooperatively involved in establishing an immunosuppressive TME, given their close association with increased intratumoral infiltration of tumor-supportive immune cells (e.g., activated mast cells and Tregs) as well as decreased infiltration of antitumor immune cells (e.g., γδ T cells, monocytes and neutrophils). The Treg-like Vδ1+ γδ T cell population was dominant over the cytotoxic Vδ2+ population and comprised the major γδ T cell subset in HCC. Adoptive transfer of allogeneic Vδ2+ γδ T cells may represent an effective immunotherapeutic approach for HCC.
Apoptosis can facilitate cancer immune evasion by repressing antitumor immunity. Intrinsic apoptosis in cancer cells affects the ability of the TME to escape antitumor immunity. Tumor-associated neutrophil-released neutrophil extracellular traps (NETs) repressed the function of effector immune cells (157). Reportedly, apoptotic breast cancer cells secreted spermidine to foster NET formation in a pannexin 1 (Panx1) channel-dependent manner, which attenuated antitumor immunity (127). Suppression of spermidine synthesis inhibited breast cancer growth in vivo. Thus, cancer cells remodel the tumor immune microenvironment to evade immunosurveillance through the release of metabolites from intrinsic apoptotic cells. The effects of other metabolites derived from apoptotic cells on the tumor immune microenvironment deserve further study. Basal cell carcinoma (BCC)-expressed CD200 was released into the TME as soluble CD200 (sCD200) (128). In the TME, sCD200 bound the respective receptor on NK cells to block the MAPK signaling pathway, which promoted peroxisome proliferator-activated receptor γ (PPARγ)-mediated gene transcription of Fas death receptor family members (FasL, Fas and FADD). This in turn contributed to autoregulatory activation-induced NK cell apoptosis. Blocking sCD200-mediated suppression of the MAPK or PPARγ signaling cascade promoted the survival and tumor-killing activity of NK cells. Therefore, BCC cells can remodel the TME via sCD200 release, which leads to NK cell exhaustion and cancer immune evasion. It is intriguing whether sCD200-mediated NK cell dysfunction also constitutes an immune escape mechanism in other cancer types.
3.2 Autophagy-dependent cell death in tumor immunity
Autophagy-dependent cell death plays contradictory roles in the regulation of the tumor immune microenvironment. Autophagy-dependent cell death is involved in macrophage polarization. For example, HCC cells polarized cocultured macrophages into the M2-like phenotype (129). Blockade of autophagy-dependent cell death in macrophages inhibited the NF-κB pathway by promoting ubiquitination-mediated degradation of TGF-β-activated kinase-binding protein 3 (TAB3), which facilitated M2-like macrophage polarization. These effects could be impeded by activation of autophagy-dependent cell death. Interference with M2 type macrophage polarization may be a promising treatment strategy alone or in combination with cancer immunotherapy.
Autophagy-dependent cell death can affect the response to immunotherapy through the coordination of adaptive immune cells. Loss of AT-rich interaction domain containing protein 1A (ARID1A) suppressed autophagy-dependent cell death by inducing the epidermal growth factor receptor (EGFR)/PI3K/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway (130). This caused increased production of IFN-I and enhanced infiltration of CD8+ cytotoxic T lymphocytes (CTLs), which resulted in an improved response to anti-programmed cell death protein-1 (PD-1)/anti-PD-L1 in patients with EGFR-mutant lung adenocarcinoma (LUAD). The multityrosine kinase inhibitor (MTKI) ESK981 exhibited an autophagy-inhibitory property, as it downregulated the lipid kinase PIKfyve (131). ESK981-mediated inhibition of autophagy-dependent cell death promoted C-X-C motif chemokine (CXCL10) secretion via the IFN-γ pathway and enhanced functional T cell infiltration, which conferred increased sensitivity to anti-PD-1 immunotherapy in preclinical models of prostate cancer. Collectively, inhibition of autophagy-dependent cell death can turn tumors from an immunologically “cold” state to an inflamed “hot” state, contributing to priming of the tumor immune microenvironment.
3.3 Pyroptosis in tumor immunity
Recently, the role of the pyroptotic pathway in antitumor immunity has been revealed (Table 1). The combination of the B-raf proto-oncogene, serine/threonine kinase (BRAF) inhibitors and mitogen-activated extracellular signal-regulated kinase (MEK) inhibitors (BRAFi + MEKi) impeded the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling cascade in melanoma cells, which induced the activation of caspase-3 and resulted in GSDME-executed pyroptosis as well as extravasation of DAMPs (132). Pyroptotic cancer cell-derived DAMPs activated DCs to promote T cell expansion, which led to tumor regression. Conversely, GSDME-knockdown melanoma displayed defective DAMP release and reduced intratumoral T cell infiltration during BRAFi + MEKi treatment. ERK1/2 inhibition produces intense antitumor immune responses through the induction of GSDME-mediated pyroptosis. BRAFi + MEKi-resistant melanoma cells did not undergo pyroptosis but were susceptible to pyroptosis-inducing chemotherapy. Pharmacological reinduction of pyroptosis retarded the growth of BRAFi + MEKi-resistant melanoma cells and might represent an effective salvage option for targeted therapy-resistant melanoma. Listeria monocytogenes (Lmo)-based immunotherapy (Lmo@RBC) induced GSDMC-dependent pyroptosis in colon cancer cells (133). Proinflammatory factors released by pyroptotic cancer cells promoted DC maturation and T cell activation, which culminated in efficient inhibition of primary and metastatic tumors. These findings provide evidence for the potential use of this live bacterial vaccine in cancer immunotherapy. Oncolytic parapoxvirus ovis (ORFV) promoted intratumoral infiltration of CTLs and exhibited tumor killing effects in vivo by inducing GSDME-dependent pyroptosis in cancer cells (134). Moreover, ORFV also sensitized immune-cold tumors to immune checkpoint blockade (ICB) therapy. Blocking GSDME-mediated pyroptosis abolished these effects. Similarly, oncolytic vesicular stomatitis virus (VSV) induced cancer cell pyroptosis by activating GSDME (135). VSV treatment suppressed tumor growth by attracting CTLs to the tumor site. Knockdown of GSDME dampened VSV-mediated tumor-antagonizing effects and reduced the ability of VSV to stimulate antitumor immune responses. Furthermore, VSV therapy increased the sensitivity of immunologically “cold” tumors to anti-PD-1 treatment. The combination of oncolytic virus (OV)-based approaches and immunotherapy might be a powerful treatment paradigm against cancer. Membrane anchoring photosensitizer-driven pyroptosis of pancreatic cancer cells reshaped the TME by fostering M1 macrophage polarization, DC maturation and CD8+ CTL activation (136). Pyroptosis-mediated reprogramming of the tumor immune microenvironment turned poorly immunogenic tumors into T cell-inflamed and -tumoricidal microenvironments, which led to the inhibition of primary and metastatic tumors. These results highlighted the critical role of pyroptosis in light-controlled antitumor immunity.
The E3 ubiquitin ligase cell division cycle 20 homolog (CDC20) has been recognized as an oncogenic driver (158). A recent study revealed that CDC20 exerted an inhibitory effect on antitumor immune responses and facilitated prostate cancer pathogenesis (137). Mechanistically, CDC20 enhanced ubiquitination-mediated GSDME degradation to prevent pyroptosis in prostate cancer cells, hence reducing intratumoral infiltration of CD8+ T cells. Consequently, CDC20 inhibition improved the response to anti-PD-1 in murine models of prostate cancer. Activated immune cells can in turn coordinate cancer cell pyroptosis. It was reported that blockade of cytotoxic T lymphocyte-associated protein-4 (CTLA-4) stimulated CD8+ T cells and upregulated the expression of IFN-γ and TNF-α in the TME, which induced the STAT1/interferon regulatory factor 1 (IRF1) pathway to trigger pyroptosis in head and neck squamous cell carcinoma (HNSCC) (159). The impact of the tumor immune microenvironment on cancer cell pyroptosis warrants further study.
3.4 Ferroptosis in tumor immunity
Ferroptosis serves as a key mechanism that regulates the crosstalk between cancer cells and the TME. Reportedly, ferroptosis was the most enriched PCD pathway in glioma (138). Increased ferroptosis was linked to exacerbated immunosuppression and poor patient outcomes. Ferroptosis fostered the recruitment and polarization of tumor-associated macrophages (TAMs) into an immunosuppressive M2-like phenotype and reduced the cytotoxic activity of CD8+ T cells. Pharmacological inhibition of ferroptosis in combination with ICB produced a synergistic therapeutic outcome in glioma-bearing mice. Autophagy-dependent ferroptosis induced by oxidative stress promoted the transfer of KRASG12D from pancreatic ductal adenocarcinoma (PDAC) to macrophages through exosomes (139). KRASG12D then drove macrophage polarization into the M2-like pro-tumor phenotype by inducing fatty acid oxidation. Inhibition of KRASG12D release and uptake prevented macrophage-mediated PDAC progression in vivo. Polymorphonuclear (PMN)-MDSCs in the TME underwent spontaneous ferroptosis, which increased the liberation of immunosuppressive molecules (e.g., peroxidized lipids) from ferroptotic cells, leading to compromised CD8+ T cell-mediated immune responses and cancer progression (140). The secretion of immunosuppressive factors preceded PMN-MDSC cell death during ferroptosis. Blockade of ferroptosis prevented PMN-MDSC death and the release of immunosuppressive molecules, which shifted PMN-MDSCs into classical nonsuppressive PMNs. Furthermore, pharmacological inhibition of ferroptosis magnified the anticancer activity of anti-PD-1 in experimental animal models. CD36, a scavenger receptor involved in lipid metabolism, promoted ferroptosis in CD8+ T cells by facilitating the uptake of fatty acids to induce lipid peroxidation (141). CD36-mediated ferroptosis inhibited cytotoxic cytokine production and dampened T cell function in multiple myeloma (141). Blocking ferroptosis in CD8+ T cells effectively restored their tumor-killing effects and augmented the anticancer efficacy of anti-PD-1 therapy.
In contrast, ferroptosis functions to induce antitumor immune responses. For instance, the activation of ferroptosis mediated the tumor-suppressive role of resveratrol in lung squamous cell carcinoma (LUSC) by enhancing the cytotoxic effect of CD8+ T cells (142). Anoctamin 1 (ANO1) promoted TGF-β secretion by gastrointestinal (GI) cancer cells, which attracted cancer-associated fibroblasts (CAFs) into the TME by blocking ferroptosis in a PI3K/Akt signaling-dependent manner (143). CAFs impaired CD8+ T cell-mediated immunity via the CAF-related secretome, thus enhancing resistance to anti-PD-1 immunotherapy and accelerating GI cancer progression. ANO1-mediated ferroptosis inhibition represents a vital mechanism underlying TME reprogramming and immunotherapy resistance in GI cancer. Moreover, ferroptosis can reverse the immunosuppressive microenvironment. RSL3-induced ferroptosis in HNSCC decreased the number of MDSCs and M2 macrophages and increased the number of tumor-infiltrating CD4+ and CD8+ T cells in the TME (144). Ferroptosis-inducing agents may be an attractive therapeutic option for HNSCC. Apolipoprotein C1 (APOC1) was found to be involved in cancer pathogenesis (160). Overexpression of APOC1 in TAMs in the HCC microenvironment repressed ROS production to inhibit ferroptosis through the regulation of iron and lipid metabolism, which facilitated the transformation of TAMs into the tumor-supportive M2 phenotype and the establishment of a proinflammatory TME (145). Loss of APOC1 promoted the conversion of M2 macrophages into the M1 type and induced the activation of CD8+ T cells. In vivo experiments demonstrated that APOC1 depletion resulted in immune activation and improved HCC sensitivity to anti-PD-1 treatment. Ferroptosis may act in an opposite manner in cancer progression through a myriad of mechanisms, such as promotion of cancer cell death, mobilization of cytotoxic T cells and induction of an immunosuppressive phenotype. Induction of ferroptosis significantly inhibits cancer development. However, in some cancers, the immunosuppressive effect of ferroptosis occupies a dominant position, which may be an important explanation for the limited therapeutic potential of ferroptosis inducers in preclinical models. The effect of ferroptosis on carcinogenesis and tumor immunity may differ depending on the type of cells in which this process occurs. The therapeutic benefit of ferroptosis-regulating agents must therefore be carefully evaluated. Continual studies will be required to maximize the efficacy of ferroptosis modulation by curbing the inhibition of cancer cell death and surmounting the immunosuppressive phenotype. Cell-specific delivery of ferroptosis-inducing or -suppressing agents may be a clinically relevant therapeutic approach. Undoubtedly, elucidating the detailed mechanisms of ferroptosis in cancer-TME interaction networks will offer valuable opportunities to develop innovative therapeutic approaches for cancer intervention.
In addition, immune cells can regulate ferroptosis in cancer cells. ICB (e.g., anti-CTLA-4 and anti-PD-L1)-activated CD8+ T cells reduced the expression of two subunits (SLC3A2 and SLC7A11) of the glutamate-cystine antiporter system xc- and limited cystine uptake by cancer cells via IFN-γ release (161). As a result, CD8+ T cells facilitated lipid peroxidation and ferroptosis in cancer cells. Increased ferroptosis further enhanced the anticancer efficacy of immunotherapy. Moreover, cysteine or cysteine deprivation synergized with ICB to actuate T cell-mediated antitumor immunity and induce cancer cell ferroptosis. The expression of system xc- was shown to be negatively correlated with the CD8+ T cell signature, IFN-γ level and clinical outcomes in cancer patients. Accordingly, T cell-induced cancer cell ferroptosis constitutes an anticancer mechanism.
3.5 Other cell death pathways in tumor immunity
Ectopic activation of RIPK1/RIPK3 induced necroptosis of fibroblasts within the TME, which contributed to enhanced production of NF-κB-dependent cytokines (146). These events promoted antigen uptake and activation of antigen-presenting cells (APCs) to strengthen tumor-specific CD8+ T cell immune responses. RIPK1/RIPK3 activators synergized with ICB to foster systemic tumor control in preclinical animal models. Oncolytic alphavirus M1 induced necroptosis in triple-negative breast cancer (TNBC) (147). This necroptotic virotherapy acted synergistically with doxorubicin (DOX) to inhibit TNBC growth in vivo.
ICD, a type of PCD induced by many stressors, involves diverse mechanisms of cell death including apoptosis, ferroptosis, necroptosis and pyroptosis (162). ICD encompasses the emission of DAMPs from dying cancer cells that stimulate antigen cross-presentation and initiate antitumor adaptive immunity (2). Thus, harnessing ICD may represent a promising treatment modality for cancer therapy (163). ICD is emerging as a critical component of chemotherapy-induced antitumor immunity. For instance, oxaliplatin was found to induce immunogenic apoptosis in HCC cells, as evidenced by increased secretion of ICD biomarkers (ATP and high-mobility group box 1 (HMGB1)) from dying cells (148). As expected, oxaliplatin elicited DC maturation and CD8+ T cell infiltration. Likewise, sequential treatment with pemetrexed and cisplatin induced ICD in non-small cell lung cancer (NSCLC) cells by activating the STING signaling pathway (149). This treatment contributed to robust cytokine responses and enhanced infiltration of CD8+ T cells. Similarly, STING-activating nanoparticles (STING-NPs) effectively activated the STING signaling pathway and induced ICD in neuroblastoma (150). STING-NP-mediated ICD promoted cancer cell phagocytosis and DC cross-presentation of cancer-derived antigens, which eventually triggered tumor-specific CD8+ T cell immune responses. Furthermore, STING-NPs retarded cancer growth and induced immunological memory that protected against tumor rechallenge. STING-NPs acted synergistically with PD-L1 blockade in a murine model of neuroblastoma. STING-NPs may have utility as an adjuvant therapy for enhancing responses to immunotherapy in neuroblastoma. Polyglycerol-functionalized nanodiamonds bearing DOX (Nano-DOX) induced autophagy-dependent cell death in glioblastoma multiforme (GBM) and promoted the secretion of cancer antigens and DAMPs by GBM cells, which contributed to DC activation (151). Autophagy-dependent cell death serves as a major mechanism through which Nano-DOX subverts the GBM-associated immunosuppressive microenvironment. These pharmaceuticals combined with ICB may effectively stimulate the adaptive immune system to produce intense tumor-killing effects. OVs are emerging as innovative immunotherapeutic agents and can activate adaptive antitumor immunity through the induction of ICD, such as immunogenic apoptosis, autophagy-dependent cell death, pyroptosis and necroptosis (152). OVs have been engineered to facilitate ICD through mechanisms involving insertion or deletion of death pathway-regulating genes. Genetically modified OVs act to skew infected cancer cells toward specific death pathways for increased immunogenicity. OV-based immunotherapy in combination with standard therapeutic regimens may be effective in potentiating antitumor immunity and improving therapeutic benefits. For instance, the oncolytic virus M1 was shown to be capable of eliciting ICD in cancer (153). M1 promoted DC cell activation to actuate antitumor CD8+ T cell responses and contributed to long-term antitumor immune memory in vivo. M1 treatment also increased the efficacy of anti-PD-L1 therapy. In addition, many types of radiation (e.g., nonionizing (ultraviolet) and ionizing radiation) can initiate ICD, thus enhancing the adjuvanticity and immunogenicity of dying cancer cells and activating CTL function (154). Radiation therapy cooperated with conventional immunotherapies (e.g., ICB) to induce systemic antitumor immunity and cancer regression.
4 Potential clinical application of targeted therapies against programmed cell death in cancer
Considering the association between PCD and tumor immunity, targeted therapies against PCD have the potential to reinforce the clinical benefit of conventional immunotherapy. Targeting PCD has recently gained substantial attention in the field of cancer immunotherapy. Several clinically used drugs can coordinate tumor immunity through the regulation of PCD pathways in cancer. Paclitaxel, a representative chemotherapeutic agent for lung cancer, induced pyroptosis in lung cancer cells by activating caspase-3 and GSDME (164). Paclitaxel increased the immunogenicity of cancer cells, facilitated antigen presentation, and augmented antitumor immunity (165). As a result, paclitaxel enhanced the efficacy of immunotherapy. BRAFi + MEKi, which are FDA-approved for the treatment of BRAFV600E/K-mutant melanoma, activated DCs and tumor-specific T cells in melanoma by triggering GSDME-executed pyroptosis and promoting DAMP release (132). BRAFi + MEKi may improve patient response to immunotherapy, which merits further verification. Sorafenib, a clinically approved anticancer agent, repressed system xc- function and elicited ER stress, which contributed to ferroptotic cell death (166). Sorafenib combined with PD-L1 inhibition induced antitumor T cell immunity and effectively suppressed HCC growth (167). DOX triggered immunogenic cell death (autophagy-dependent cell death and pyroptosis), activated innate/adaptive immune cells, and induced systemic antitumor immunity (168, 169). Cisplatin acted as an inducer of apoptosis and ferroptosis in cancer cells and promoted intratumoral infiltration of CTLs (170, 171). Taken together, the evidence shows that combination treatment with antineoplastic agents and immunotherapy may be superior to each component alone due to adjunctive or synergistic effects.
Furthermore, some clinical trials have shown the therapeutic efficacy of targeted treatments against PCD combined with immunotherapy (172) (Table 2). A phase 1 trial showed that NK cell therapy combined with trastuzumab had good tolerance in patients with treatment-refractory HER2-positive solid tumors (173). This combined therapy promoted cancer cell apoptosis and increased NK cell expansion and lymphocytic infiltrates. Phase 2 trials are recommended to further assess the safety and efficacy of NK cell therapy in combination with trastuzumab. Cytokine-induced killer (CIK) cell immunotherapy combined with sintilimab plus chemotherapeutic agents (carboplatin and pemetrexed or paclitaxel) was well tolerated and exhibited antitumor activity in NSCLC patients (174). Tiragolumab plus atezolizumab (31.3%) improved the objective response rate compared with placebo plus atezolizumab (16.2%) in NSCLC patients (175). The tiragolumab plus atezolizumab group (5.4 months) exhibited better median progression-free survival than the placebo plus atezolizumab group (3.6 months). Tiragolumab plus atezolizumab had a manageable safety profile and represented a prospective combination immunotherapy for NSCLC intervention. In a phase 3 trial involving 800 NSCLC patients, durvalumab plus chemotherapy prolonged progression-free survival and overall survival (176). This combined treatment had a tolerable safety profile. NSCLC patients who received pembrolizumab in combination with chemotherapy (gemcitabine, docetaxel or pemetrexed) exhibited higher progression-free survival than patients treated with chemotherapy alone (177). In another study, sitravatinib plus tislelizumab controlled disease progression in the majority (88.5%) of NSCLC patients (178). Nanoparticle albumin-bound paclitaxel (nab-paclitaxel) improved the objective response rate (55.2%) in NSCLC patients following PD-(L)1 inhibitor treatment (179). Lenalidomide is an immune modulator that has been approved for the treatment of relapsed/refractory follicular lymphoma (183). The DNA methyltransferase inhibitor azacytidine activated apoptosis- and immune-related pathways (180). In a phase 1b study, azacytidine in combination with lenalidomide and dexamethasone contributed to an overall response rate of 22% and a clinical benefit response rate of 32% in patients with relapsed or refractory multiple myeloma (180). Azacytidine might enhance sensitivity to lenalidomide and dexamethasone in patients with relapsed and/or refractory multiple myeloma. The efficacy of combined nivolumab and ibrutinib was previously assessed in patients with chronic lymphocytic leukemia (CLL) in a phase 2 clinical trial (181). Of 24 patients, 10 responded to this combined treatment, which corresponded to an overall response rate of 42%. Recently, fifty patients with advanced acral melanoma were enrolled in a phase 2 nonrandomized clinical study (182). Camrelizumab plus apatinib and temozolomide treatment contributed to a disease control rate of 88.0% in these patients. In addition, extensive clinical trials are planned or are currently underway to expand therapeutic avenues and improve patient outcomes, relevant examples of which are summarized in Table 3. Despite the encouraging clinical results, continual research efforts are warranted to reveal the mechanism by which PCD-targeted pharmaceuticals affect immunotherapy response in cancer patients.
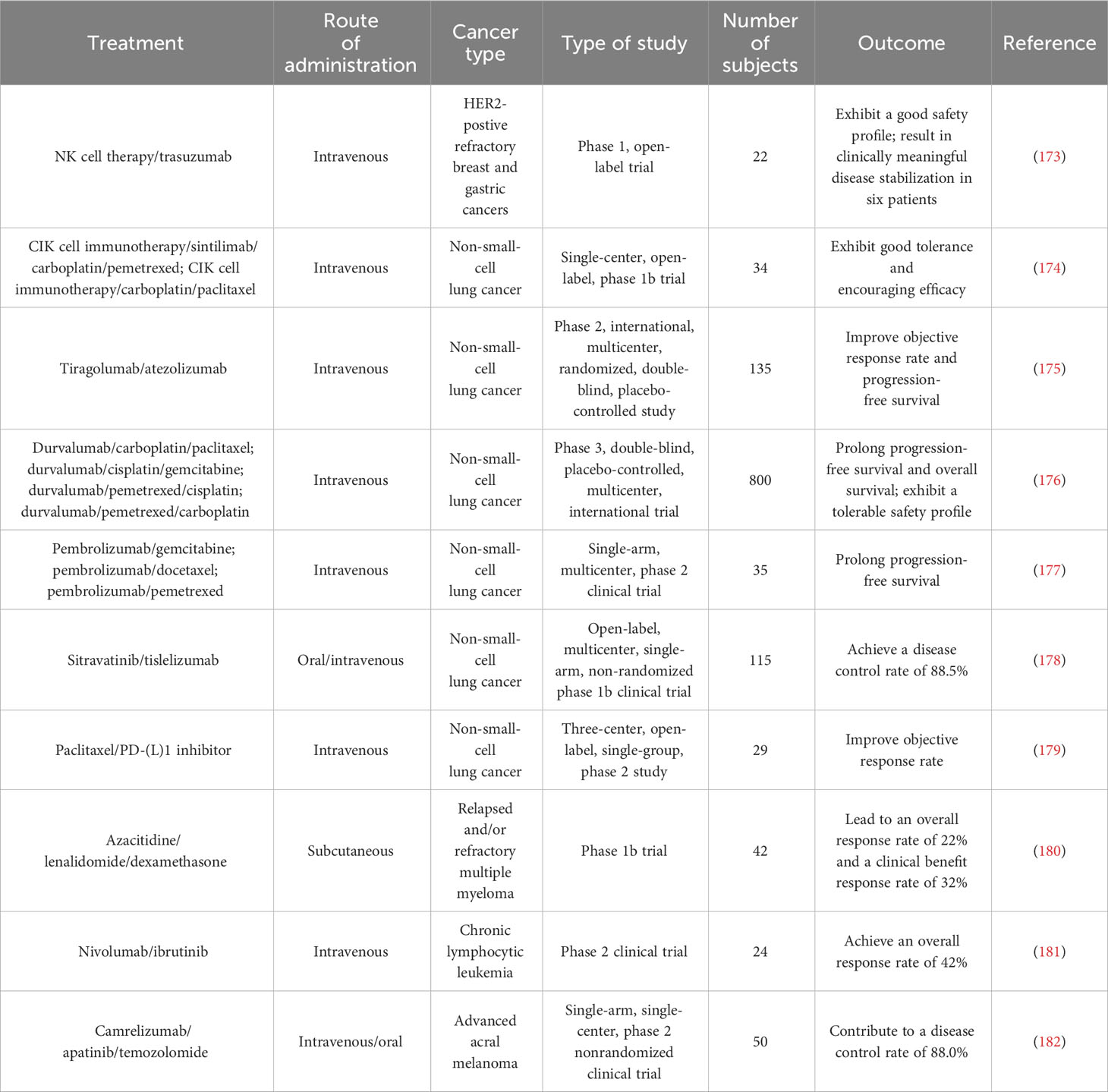
Table 2 Summary of clinical trials testing combined programmed cell death-regulating drugs and immunotherapy in diverse cancer types.
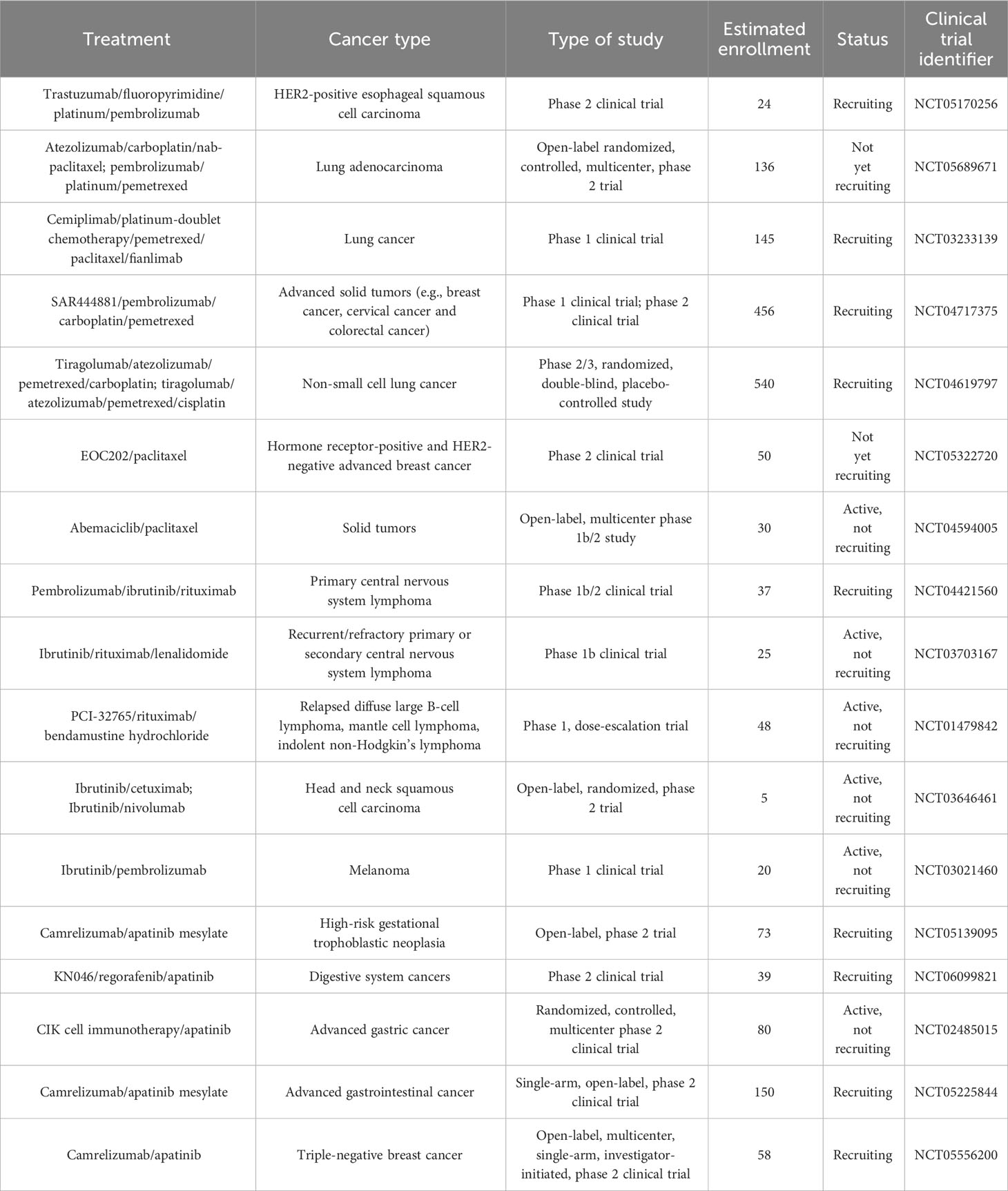
Table 3 List of ongoing clinical trials testing combined programmed cell death-regulating drugs and immunotherapy in diverse cancer types.
5 Conclusions and future perspectives
It is increasingly acknowledged that multiple PCD pathways act under both physiological and pathological conditions. Due to its important role in cancer biology, PCD has become a hot spot in cancer research. Remarkably, various forms of PCD, such as apoptosis, autophagy-dependent cell death, pyroptosis and ferroptosis, are immunogenic and can therefore affect the immune system. Importantly, these PCD pathways exert a regulatory action on immune cells within the TME. The study of PCD in tumor immunity is a rapidly developing field. Accumulating evidence shows that PCD acts as a double-edged sword in cancer pathogenesis. PCD synergizes with host antitumor immune responses while contributing to cancer immunosuppression and immune evasion. The role of PCD in remodeling the TME is still poorly understood, and it was not until recently that the regulation and mechanisms of pyroptosis and ferroptosis were gradually recognized. Therefore, the impact of these newly characterized forms of PCD on tumor immunity deserves special attention. The traditional view holds that apoptotic cell death is an immunologically “silent” process and cannot trigger inflammation. However, emerging evidence suggests that certain stimuli can elicit an immunogenic subtype of apoptosis in cancer (184). Investigations of ICD, especially immunogenic apoptosis, have just begun. Accordingly, considerable research efforts are needed to gain a thorough comprehension of the broad communication between PCD and antitumor immunity. This interaction pattern may even vary in distinct tumor types or in different contexts. It will be equally important to determine how cancer cells and diverse cells within the TME interact with each other to activate or block tumor immunity. An in-depth investigation of the complex TME may help to elucidate the mechanisms underlying PCD-mediated immune regulation. Cancer cell populations can simultaneously undergo various types of PCD. Different forms of PCD may act in synergy or in opposition at the same tumor site. Certain cell death pathways can induce or restrict other death mechanisms. It is thus necessary to adequately decipher the sophisticated regulatory network behind distinct forms of PCD in the TME and define which PCD type plays a dominant role in different contexts. Moreover, factors that determine the ultimate effect of various types of PCD on cancer progression and tumor immunity must be carefully identified.
According to the available evidence from both preclinical and clinical studies, PCD can affect the efficacy of immunotherapy through crosstalk with immune cells infiltrating the TME. Manipulation of cell death pathways seems to be an attractive strategy to enhance the treatment advantages of cancer immunotherapy. A variety of antitumor agents can affect the immunotherapy response by initiating cell death programs in cancer. The exact mechanisms responsible for the synergistic actions of chemotherapeutic agents and ICB require further investigation. Noncancerous cells, including normal cells and immune cells, may also die upon recognition of signals (e.g., DAMPs) released from dying cancer cells. It remains uncertain whether chemotherapy-induced PCD is beneficial for cancer patients in the long term. The side effects of chemotherapy in combination with immunotherapy must be completely examined. The screening and discovery of tumoricidal drugs that specifically act on cancer cells with minimal adverse effects on normal cells are urgently needed. Tumor-targeted delivery systems, such as exosomes and polymeric nanoparticles, seem to offer an effective way to deliver therapeutic agents to cancer cells (185). The development of safe and efficient delivery vehicles may help to balance therapeutic goals and potential side effects. The administration sequence and timing of various drugs in combination therapies need to be determined. In addition, further clinical studies are needed to investigate the efficacy, safety and mechanisms of antineoplastic drugs combined with immunotherapy in large cohorts of cancer patients. Taken together, gaining better insight into PCD involvement in carcinogenesis and tumor immunity will finally accelerate the translation of scientific discoveries into new ways to treat cancer.
Author contributions
MW: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – original draft. FY: Investigation, Resources, Writing – review & editing. YZ: Writing – review & editing, Resources. PL: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the grant from the Natural Science Foundation of Shandong Province, China (No. ZR2021MH018).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gong L, Huang D, Shi Y, Liang Z, Bu H. Regulated cell death in cancer: from pathogenesis to treatment. Chin Med J (2023) 136(6):653–65. doi: 10.1097/CM9.0000000000002239
2. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death differentiation (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
3. Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death differentiation (2021) 28(7):2029–44. doi: 10.1038/s41418-021-00814-y
4. Liu Y, Ding W, Wang J, Ao X, Xue J. Non-coding RNA-mediated modulation of ferroptosis in cardiovascular diseases. Biomedicine pharmacotherapy (2023) 164:114993. doi: 10.1016/j.biopha.2023.114993
5. Ao X, Ding W, Li X, Xu Q, Chen X, Zhou X, et al. Non-coding RNAs regulating mitochondrial function in cardiovascular diseases. J Mol Med (2023) 101(5):501–26. doi: 10.1007/s00109-023-02305-8
6. Ashkenazi A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discovery (2008) 7(12):1001–12. doi: 10.1038/nrd2637
7. Liu Y, Wang Y, Li X, Jia Y, Wang J, Ao X. FOXO3a in cancer drug resistance. Cancer Lett (2022) 540:215724. doi: 10.1016/j.canlet.2022.215724
8. Zhou X, Ao X, Jia Z, Li Y, Kuang S, Du C, et al. Non-coding RNA in cancer drug resistance: underlying mechanisms and clinical applications. Front Oncol (2022) 12:951864. doi: 10.3389/fonc.2022.951864
9. Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol (2022) 13:847345. doi: 10.3389/fimmu.2022.847345
10. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol (2009) 9(5):353–63. doi: 10.1038/nri2545
11. Li X, Dai H, Wang H, Han W. Exploring innate immunity in cancer immunotherapy: opportunities and challenges. Cell Mol Immunol (2021) 18(6):1607–9. doi: 10.1038/s41423-021-00679-8
12. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol (2008) 9(3):231–41. doi: 10.1038/nrm2312
13. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol (2017) 17(4):262–75. doi: 10.1038/nri.2017.9
14. Alvarez-Meythaler JG, Garcia-Mayea Y, Mir C, Kondoh H, ME LL. Autophagy takes center stage as a possible cancer hallmark. Front Oncol (2020) 10:586069. doi: 10.3389/fonc.2020.586069
15. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
16. Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol (2018) 20(10):1110–7. doi: 10.1038/s41556-018-0201-5
17. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
18. Green DR, Llambi F. Cell death signaling. Cold Spring Harbor Perspect Biol (2015) 7(12):a006080. doi: 10.1101/cshperspect.a006080
19. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202(12):1691–701. doi: 10.1084/jem.20050915
20. Park W, Wei S, Kim BS, Kim B, Bae SJ, Chae YC, et al. Diversity and complexity of cell death: A historical review. Exp Mol Med (2023) 55(8):1573–94. doi: 10.1038/s12276-023-01078-x
21. Zhang R, Kang R, Tang D. The STING1 network regulates autophagy and cell death. Signal transduction targeted Ther (2021) 6(1):208. doi: 10.1038/s41392-021-00613-4
22. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death differentiation (2014) 21(1):39–49. doi: 10.1038/cdd.2013.84
23. Decraene B, Yang Y, De Smet F, Garg AD, Agostinis P, De Vleeschouwer S. Immunogenic cell death and its therapeutic or prognostic potential in high-grade glioma. Genes Immun (2022) 23(1):1–11. doi: 10.1038/s41435-021-00161-5
24. Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26(4):239–57. doi: 10.1038/bjc.1972.33
25. Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res (2000) 45(3):528–37. doi: 10.1016/s0008-6363(99)00384-3
26. Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene (1998) 17(25):3237–45. doi: 10.1038/sj.onc.1202581
27. D'Arcy MS. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int (2019) 43(6):582–92. doi: 10.1002/cbin.11137
28. Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in fas/APO-1- and TNF receptor-induced cell death. Cell (1996) 85(6):803–15. doi: 10.1016/s0092-8674(00)81265-9
29. Mohr A, Deedigan L, Jencz S, Mehrabadi Y, Houlden L, Albarenque SM, et al. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death differentiation (2018) 25(2):340–52. doi: 10.1038/cdd.2017.164
30. Feoktistova M, Geserick P, Panayotova-Dimitrova D, Leverkus M. Pick your poison: the ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle (2012) 11(3):460–7. doi: 10.4161/cc.11.3.19060
31. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxidants Redox Signaling (2013) 19(6):546–58. doi: 10.1089/ars.2012.4905
32. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death differentiation (2018) 25(1):65–80. doi: 10.1038/cdd.2017.186
33. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol (2019) 20(3):175–93. doi: 10.1038/s41580-018-0089-8
34. Li MX, Dewson G. Mitochondria and apoptosis: emerging concepts. F1000prime Rep (2015) 7:42. doi: 10.12703/P7-42
35. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (2016) 8(4):603–19. doi: 10.18632/aging.100934
36. Tinel A, Tschopp J. The piddosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science (2004) 304(5672):843–6. doi: 10.1126/science.1095432
37. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358(6382):167–9. doi: 10.1038/358167a0
38. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci United States America (1996) 93(18):9833–8. doi: 10.1073/pnas.93.18.9833
39. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9(3):113–4. doi: 10.1016/s0966-842x(00)01936-3
40. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
41. Fink SL, Cookson BT. Pyroptosis and host cell death responses during salmonella infection. Cell Microbiol (2007) 9(11):2562–70. doi: 10.1111/j.1462-5822.2007.01036.x
42. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6:36. doi: 10.1038/s41421-020-0167-x
43. Kanneganti TD. Intracellular innate immune receptors: life inside the cell. Immunol Rev (2020) 297(1):5–12. doi: 10.1111/imr.12912
44. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol (2009) 10(3):241–7. doi: 10.1038/ni.1703
45. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β Secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
46. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature (2021) 593(7860):607–11. doi: 10.1038/s41586-021-03478-3
47. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
48. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
49. Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ Efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
50. Li Z, Ji S, Jiang ML, Xu Y, Zhang CJ. The regulation and modification of GSDMD signaling in diseases. Front Immunol (2022) 13:893912. doi: 10.3389/fimmu.2022.893912
51. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
52. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
53. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell (2020) 181(3):674–87.e13. doi: 10.1016/j.cell.2020.03.040
54. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22(10):1264–75. doi: 10.1038/s41556-020-0575-z
55. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362(6418):1064–9. doi: 10.1126/science.aau2818
56. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during yersinia infection. Proc Natl Acad Sci United States America (2018) 115(46):E10888–97. doi: 10.1073/pnas.1809548115
57. Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med (2020) 217(3):jem.20191644. doi: 10.1084/jem.20191644
58. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
59. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
60. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
61. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep (2018) 8(1):5155. doi: 10.1038/s41598-018-23408-0
62. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3(3):285–96. doi: 10.1016/s1535-6108(03)00050-3
63. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
64. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447(7146):864–8. doi: 10.1038/nature05859
65. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
67. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
68. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biology (2021) 10(3):184. doi: 10.3390/biology10030184
69. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
70. Jia M, Zhang H, Qin Q, Hou Y, Zhang X, Chen D, et al. Ferroptosis as a new therapeutic opportunity for nonviral liver disease. Eur J Pharmacol (2021) 908:174319. doi: 10.1016/j.ejphar.2021.174319
71. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (2018) 38(1):12. doi: 10.1186/s40880-018-0288-x
72. Parker JL, Deme JC, Kolokouris D, Kuteyi G, Biggin PC, Lea SM, et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc. Nat Commun (2021) 12(1):7147. doi: 10.1038/s41467-021-27414-1
73. Jyotsana N, Ta KT, DelGiorno KE. The role of cystine/glutamate antiporter SLC7A11/xCT in the pathophysiology of cancer. Front Oncol (2022) 12:858462. doi: 10.3389/fonc.2022.858462
74. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discovery (2021) 7(1):193. doi: 10.1038/s41420-021-00579-w
75. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
76. Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics (2017) 9(10):1367–75. doi: 10.1039/c7mt00143f
77. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol (2019) 12(1):34. doi: 10.1186/s13045-019-0720-y
78. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
79. Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PL, Levy D, et al. Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci (2020) 21(22):8765. doi: 10.3390/ijms21228765
80. Tang M, Chen Z, Wu D, Chen L. Ferritinophagy/ferroptosis: iron-related newcomers in human diseases. J Cell Physiol (2018) 233(12):9179–90. doi: 10.1002/jcp.26954
81. Chen J, Li X, Ge C, Min J, Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death differentiation (2022) 29(3):467–80. doi: 10.1038/s41418-022-00941-0
82. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci (2016) 73(11-12):2195–209. doi: 10.1007/s00018-016-2194-1
83. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci United States America (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
84. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol (2020) 8:590226. doi: 10.3389/fcell.2020.590226
85. Roberts JZ, Crawford N, Longley DB. The role of ubiquitination in apoptosis and necroptosis. Cell Death differentiation (2022) 29(2):272–84. doi: 10.1038/s41418-021-00922-9
86. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and tumor progression. Trends Cancer (2022) 8(1):21–7. doi: 10.1016/j.trecan.2021.09.003
87. Najafov A, Chen H, Yuan J. Necroptosis and cancer. Trends Cancer (2017) 3(4):294–301. doi: 10.1016/j.trecan.2017.03.002
88. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer (2019) 18(1):100. doi: 10.1186/s12943-019-1029-8
89. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight (2019) 4(15):e128834. doi: 10.1172/jci.insight.128834
90. Liu X, Xie X, Ren Y, Shao Z, Zhang N, Li L, et al. The role of necroptosis in disease and treatment. MedComm (2021) 2(4):730–55. doi: 10.1002/mco2.108
91. Chen J, Kos R, Garssen J, Redegeld F. Molecular insights into the mechanism of necroptosis: the necrosome as a potential therapeutic target. Cells (2019) 8(12):1486. doi: 10.3390/cells8121486
92. Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death differentiation (2017) 24(7):1184–95. doi: 10.1038/cdd.2017.65
93. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
94. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
95. Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (2014) 343(6177):1357–60. doi: 10.1126/science.1249361
96. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature (2004) 430(7000):694–9. doi: 10.1038/nature02794
97. Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PloS One (2013) 8(10):e76841. doi: 10.1371/journal.pone.0076841
98. Seo J, Nam YW, Kim S, Oh DB, Song J. Necroptosis molecular mechanisms: recent findings regarding novel necroptosis regulators. Exp Mol Med (2021) 53(6):1007–17. doi: 10.1038/s12276-021-00634-7
99. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell (2011) 43(3):432–48. doi: 10.1016/j.molcel.2011.06.006
100. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature (2011) 471(7338):363–7. doi: 10.1038/nature09852
101. Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol (2020) 190(2):272–85. doi: 10.1016/j.ajpath.2019.10.012
102. Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell (2009) 138(2):229–32. doi: 10.1016/j.cell.2009.07.006
103. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54(1):133–46. doi: 10.1016/j.molcel.2014.03.003
104. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res (2018) 28(1):9–21. doi: 10.1038/cr.2017.133
105. Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J (2017) 36(17):2529–43. doi: 10.15252/embj.201796476
106. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
107. Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of autophagy in oxidative stress. Int J Mol Sci (2020) 21(9):3289. doi: 10.3390/ijms21093289
108. Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development (2018) 145(4):dev146506. doi: 10.1242/dev.146506
109. Jacob JA, Salmani JMM, Jiang Z, Feng L, Song J, Jia X, et al. Autophagy: an overview and its roles in cancer and obesity. Clinica chimica Acta (2017) 468:85–9. doi: 10.1016/j.cca.2017.01.028
110. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi: 10.1038/cr.2013.168
111. Yamamoto H, Zhang S, Mizushima N. Autophagy genes in biology and disease. Nat Rev Genet (2023) 24(6):382–400. doi: 10.1038/s41576-022-00562-w
112. Schuck S. Microautophagy - distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci (2020) 133(17):jcs246322. doi: 10.1242/jcs.246322
113. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol (2012) 22(8):407–17. doi: 10.1016/j.tcb.2012.05.006
114. Schaaf MB, Houbaert D, Mece O, Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death differentiation (2019) 26(4):665–79. doi: 10.1038/s41418-019-0287-8
115. Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, et al. Autosis is a na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci United States America (2013) 110(51):20364–71. doi: 10.1073/pnas.1319661110
116. Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, et al. The histone H4 lysine 16 acetyltransferase HMOF regulates the outcome of autophagy. Nature (2013) 500(7463):468–71. doi: 10.1038/nature12313
117. Denton D, Xu T, Dayan S, Nicolson S, Kumar S. Dpp regulates autophagy-dependent midgut removal and signals to block ecdysone production. Cell Death differentiation (2019) 26(4):763–78. doi: 10.1038/s41418-018-0154-z
118. Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol (2016) 26(1):6–16. doi: 10.1016/j.tcb.2015.08.010
119. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death differentiation (2019) 26(4):605–16. doi: 10.1038/s41418-018-0252-y
120. Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late drosophila melanogaster oogenesis. J Cell Biol (2010) 190(4):523–31. doi: 10.1083/jcb.201002035
121. Saxton RA, Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell (2017) 169(2):361–71. doi: 10.1016/j.cell.2017.03.035
122. Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol (2010) 22(2):157–68. doi: 10.1016/j.ceb.2009.11.005
123. Wang X, Hu R, Song Z, Zhao H, Pan Z, Feng Y, et al. Sorafenib combined with STAT3 knockdown triggers ER stress-induced HCC apoptosis and cGAS-STING-mediated anti-tumor immunity. Cancer Lett (2022) 547:215880. doi: 10.1016/j.canlet.2022.215880
124. Liu Y, Guo Q, Yang H, Zhang XW, Feng N, Wang JK, et al. Allosteric regulation of IGF2BP1 as a novel strategy for the activation of tumor immune microenvironment. ACS Cent Sci (2022) 8(8):1102–15. doi: 10.1021/acscentsci.2c00107
125. Werfel TA, Elion DL, Rahman B, Hicks DJ, Sanchez V, Gonzales-Ericsson PI, et al. Treatment-induced tumor cell apoptosis and secondary necrosis drive tumor progression in the residual tumor microenvironment through merTK and IDO1. Cancer Res (2019) 79(1):171–82. doi: 10.1158/0008-5472.CAN-18-1106
126. Hu Y, Chen D, Hong M, Liu J, Li Y, Hao J, et al. Apoptosis, pyroptosis, and ferroptosis conspiringly induce immunosuppressive hepatocellular carcinoma microenvironment and γδ T-cell imbalance. Front Immunol (2022) 13:845974. doi: 10.3389/fimmu.2022.845974
127. Mukai H, Miki N, Yamada H, Goto H, Kawakami T, Suzuki A, et al. Pannexin1 channel-dependent secretome from apoptotic tumor cells shapes immune-escape microenvironment. Biochem Biophys Res Commun (2022) 628:116–22. doi: 10.1016/j.bbrc.2022.08.062
128. Morgan HJ, Rees E, Lanfredini S, Powell KA, Gore J, Gibbs A, et al. CD200 ectodomain shedding into the tumor microenvironment leads to NK cell dysfunction and apoptosis. J Clin Invest (2022) 132(21):e150750. doi: 10.1172/JCI150750
129. Gao Z, Li XG, Feng SR, Chen JF, Song K, Shi YH, et al. Autophagy suppression facilitates macrophage M2 polarization via increased instability of NF-κB pathway in hepatocellular carcinoma. Int Immunopharmacol (2023) 123:110685. doi: 10.1016/j.intimp.2023.110685
130. Sun D, Qian H, Wang J, Xie T, Teng F, Li J, et al. ARID1A deficiency reverses the response to anti-PD(L)1 therapy in EGFR-mutant lung adenocarcinoma by enhancing autophagy-inhibited type I interferon production. Cell communication Signaling CCS (2022) 20(1):156. doi: 10.1186/s12964-022-00958-5
131. Qiao Y, Choi JE, Tien JC, Simko SA, Rajendiran T, Vo JN, et al. Autophagy inhibition by targeting PIKfyve potentiates response to immune checkpoint blockade in prostate cancer. Nat Cancer (2021) 2:978–93. doi: 10.1038/s43018-021-00237-1
132. Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discovery (2020) 10(2):254–69. doi: 10.1158/2159-8290.CD-19-0672
133. Liu Y, Lu Y, Ning B, Su X, Yang B, Dong H, et al. Intravenous delivery of living listeria monocytogenes elicits gasdmermin-dependent tumor pyroptosis and motivates anti-tumor immune response. ACS nano (2022) 16(3):4102–15. doi: 10.1021/acsnano.1c09818
134. Lin J, Sun S, Zhao K, Gao F, Wang R, Li Q, et al. Oncolytic parapoxvirus induces gasdermin E-mediated pyroptosis and activates antitumor immunity. Nat Commun (2023) 14(1):224. doi: 10.1038/s41467-023-35917-2
135. Lin J, Liu F, Gao F, Chen Y, Wang R, Wang X, et al. Vesicular stomatitis virus sensitizes immunologically cold tumors to checkpoint blockade by inducing pyroptosis. Biochim Biophys Acta Mol basis Dis (2022) 1868(12):166538. doi: 10.1016/j.bbadis.2022.166538
136. Wang M, Wu M, Liu X, Shao S, Huang J, Liu B, et al. Pyroptosis remodeling tumor microenvironment to enhance pancreatic cancer immunotherapy driven by membrane anchoring photosensitizer. Advanced Sci (2022) 9(29):e2202914. doi: 10.1002/advs.202202914
137. Wu F, Wang M, Zhong T, Xiao C, Chen X, Huang Y, et al. Inhibition of CDC20 potentiates anti-tumor immunity through facilitating GSDME-mediated pyroptosis in prostate cancer. Exp Hematol Oncol (2023) 12(1):67. doi: 10.1186/s40164-023-00428-9
138. Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro-oncology (2022) 24(7):1113–25. doi: 10.1093/neuonc/noac033
139. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 16(11):2069–83. doi: 10.1080/15548627.2020.1714209
140. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature (2022) 612(7939):338–46. doi: 10.1038/s41586-022-05443-0
141. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–12.e5. doi: 10.1016/j.cmet.2021.02.015
142. Shan G, Minchao K, Jizhao W, Rui Z, Guangjian Z, Jin Z, et al. Resveratrol improves the cytotoxic effect of CD8 +T cells in the tumor microenvironment by regulating HMMR/ferroptosis in lung squamous cell carcinoma. J Pharm Biomed Anal (2023) 229:115346. doi: 10.1016/j.jpba.2023.115346
143. Jiang F, Jia K, Chen Y, Ji C, Chong X, Li Z, et al. ANO1-mediated inhibition of cancer ferroptosis confers immunotherapeutic resistance through recruiting cancer-associated fibroblasts. Advanced Sci (2023) 10(24):e2300881. doi: 10.1002/advs.202300881
144. Zhao YY, Lian JX, Lan Z, Zou KL, Wang WM, Yu GT. Ferroptosis promotes anti-tumor immune response by inducing immunogenic exposure in HNSCC. Oral Dis (2023) 29(3):933–41. doi: 10.1111/odi.14077
145. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol (2022) 56:102463. doi: 10.1016/j.redox.2022.102463
146. Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol (2019) 4(36):eaaw2004. doi: 10.1126/sciimmunol.aaw2004
147. Zhang J, Liu Y, Tan J, Zhang Y, Wong CW, Lin Z, et al. Necroptotic virotherapy of oncolytic alphavirus M1 cooperated with doxorubicin displays promising therapeutic efficacy in TNBC. Oncogene (2021) 40(29):4783–95. doi: 10.1038/s41388-021-01869-4
148. Zhu H, Shan Y, Ge K, Lu J, Kong W, Jia C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell Oncol (2020) 43(6):1203–14. doi: 10.1007/s13402-020-00552-2
149. Yu J, Zhang Q, Li J, Si Z, Guo Y, Xu X, et al. Sequential administration of pemetrexed and cisplatin reprograms tumor immune microenvironment and potentiates PD-1/PD-L1 treatment in a lung cancer model. J Invest Med (2022) 70(3):792–9. doi: 10.1136/jim-2021-002159
150. Wang-Bishop L, Wehbe M, Shae D, James J, Hacker BC, Garland K, et al. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J immunotherapy Cancer (2020) 8(1):e000282. doi: 10.1136/jitc-2019-000282
151. Li TF, Xu YH, Li K, Wang C, Liu X, Yue Y, et al. Doxorubicin-polyglycerol-nanodiamond composites stimulate glioblastoma cell immunogenicity through activation of autophagy. Acta biomaterialia (2019) 86:381–94. doi: 10.1016/j.actbio.2019.01.020
152. Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol (2014) 4:74. doi: 10.3389/fonc.2014.00074
153. Liu Y, Cai J, Liu W, Lin Y, Guo L, Liu X, et al. Intravenous injection of the oncolytic virus M1 awakens antitumor T cells and overcomes resistance to checkpoint blockade. Cell Death Dis (2020) 11(12):1062. doi: 10.1038/s41419-020-03285-0
154. Galassi C, Klapp V, Yamazaki T, Galluzzi L. Molecular determinants of immunogenic cell death elicited by radiation therapy. Immunol Rev (2023). doi: 10.1111/imr.13271
155. Samuels TJ, Jarvelin AI, Ish-Horowicz D, Davis I. Imp/IGF2BP Levels Modulate Individual Neural Stem Cell Growth and Division through myc mRNA Stability. eLife (2020) 9:e51529. doi: 10.7554/eLife.51529
156. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: A critical assessment. Front Immunol (2018) 9:127. doi: 10.3389/fimmu.2018.00127
157. Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity (2020) 52(5):856–71.e8. doi: 10.1016/j.immuni.2020.03.001
158. Wang L, Zhang J, Wan L, Zhou X, Wang Z, Wei W. Targeting CDC20 as a novel cancer therapeutic strategy. Pharmacol Ther (2015) 151:141–51. doi: 10.1016/j.pharmthera.2015.04.002
159. Wang S, Wu ZZ, Zhu SW, Wan SC, Zhang MJ, Zhang BX, et al. CTLA-4 blockade induces tumor pyroptosis via CD8+ T cells in head and neck squamous cell carcinoma. Mol Ther (2023) 31(7):2154–68. doi: 10.1016/j.ymthe.2023.02.023
160. Zhang H, Wang Y, Liu C, Li W, Zhou F, Wang X, et al. The apolipoprotein C1 is involved in breast cancer progression via EMT and MAPK/JNK pathway. Pathology Res Pract (2022) 229:153746. doi: 10.1016/j.prp.2021.153746
161. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
162. Catanzaro E, Feron O, Skirtach AG, Krysko DV. Immunogenic cell death and role of nanomaterials serving as therapeutic vaccine for personalized cancer immunotherapy. Front Immunol (2022) 13:925290. doi: 10.3389/fimmu.2022.925290
163. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
164. Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis (2019) 24(3-4):312–25. doi: 10.1007/s10495-019-01515-1
165. Zhu L, Chen L. Progress in research on paclitaxel and tumor immunotherapy. Cell Mol Biol Lett (2019) 24:40. doi: 10.1186/s11658-019-0164-y
166. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife (2014) 3:e02523. doi: 10.7554/eLife.02523
167. Li Z, Bu J, Zhu X, Zhou H, Ren K, Chu PK, et al. Anti-tumor immunity and ferroptosis of hepatocellular carcinoma are enhanced by combined therapy of sorafenib and delivering modified GO-based PD-L1 siRNAs. Biomaterials Adv (2022) 136:212761. doi: 10.1016/j.bioadv.2022.212761
168. Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta pharmacologica Sin (2019) 40(9):1237–44. doi: 10.1038/s41401-019-0222-z
169. Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun (2018) 9(1):1074. doi: 10.1038/s41467-018-03473-9
170. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res Treat (2018) 50(2):445–60. doi: 10.4143/crt.2016.572
171. Ursic K, Kos S, Kamensek U, Cemazar M, Scancar J, Bucek S, et al. Comparable effectiveness and immunomodulatory actions of oxaliplatin and cisplatin in electrochemotherapy of murine melanoma. Bioelectrochemistry (2018) 119:161–71. doi: 10.1016/j.bioelechem.2017.09.009
172. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal transduction targeted Ther (2022) 7(1):196. doi: 10.1038/s41392-022-01046-3
173. Lee SC, Shimasaki N, Lim JSJ, Wong A, Yadav K, Yong WP, et al. Phase I trial of expanded, activated autologous NK-cell infusions with trastuzumab in patients with HER2-positive cancers. Clin Cancer Res (2020) 26(17):4494–502. doi: 10.1158/1078-0432.CCR-20-0768
174. Zhou L, Xiong Y, Wang Y, Meng Y, Zhang W, Shen M, et al. A phase ib trial of autologous cytokine-induced killer cells in combination with sintilimab, monoclonal antibody against programmed cell death-1, plus chemotherapy in patients with advanced non-small-cell lung cancer. Clin Lung Cancer (2022) 23(8):709–19. doi: 10.1016/j.cllc.2022.07.009
175. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (Cityscape): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol (2022) 23(6):781–92. doi: 10.1016/S1470-2045(22)00226-1
176. Heymach JV, Mitsudomi T, Harpole D, Aperghis M, Jones S, Mann H, et al. Design and rationale for a phase III, double-blind, placebo-controlled study of neoadjuvant durvalumab + Chemotherapy followed by adjuvant durvalumab for the treatment of patients with resectable stages II and III non-small-cell lung cancer: the AEGEAN trial. Clin Lung Cancer (2022) 23(3):e247–51. doi: 10.1016/j.cllc.2021.09.010
177. Salous T, Shukla NA, Althouse SK, Perkins SM, Furqan M, Leal T, et al. A phase 2 trial of chemotherapy plus pembrolizumab in patients with advanced non-small cell lung cancer previously treated with a PD-1 or PD-L1 inhibitor: big ten cancer research consortium BTCRC-LUN15-029. Cancer (2023) 129(2):264–71. doi: 10.1002/cncr.34565
178. Zhao J, Yu X, Huang D, Ma Z, Gao B, Cui J, et al. Saffron-103: A phase 1b study of the safety and efficacy of sitravatinib combined with tislelizumab in patients with locally advanced or metastatic non-small cell lung cancer. J immunotherapy Cancer (2023) 11(2):e006055. doi: 10.1136/jitc-2022-006055
179. Sonoda T, Umeda Y, Demura Y, Tada T, Nakashima K, Anzai M, et al. Efficacy and safety of nanoparticle albumin-bound paclitaxel monotherapy after immune checkpoint inhibitor administration for advanced non-small cell lung cancer: A multicenter phase 2 clinical trial. Cancer Med (2023) 12(12):13041–53. doi: 10.1002/cam4.5978
180. Khouri J, Faiman BM, Grabowski D, Mahfouz RZ, Khan SN, Wei W, et al. DNA methylation inhibition in myeloma: experience from a phase 1b study of low-dose continuous azacitidine in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. Semin Hematol (2021) 58(1):45–55. doi: 10.1053/j.seminhematol.2020.12.004
181. Jain N, Senapati J, Thakral B, Ferrajoli A, Thompson P, Burger J, et al. A phase 2 study of nivolumab combined with ibrutinib in patients with diffuse large B-cell richter transformation of CLL. Blood Adv (2023) 7(10):1958–66. doi: 10.1182/bloodadvances.2022008790
182. Mao L, Lian B, Li C, Bai X, Zhou L, Cui C, et al. Camrelizumab plus apatinib and temozolomide as first-line treatment in patients with advanced acral melanoma: the CAP 03 phase 2 nonrandomized clinical trial. JAMA Oncol (2023) 9(8):1099–107. doi: 10.1001/jamaoncol.2023.1363
183. Flowers CR, Leonard JP, Fowler NH. Lenalidomide in follicular lymphoma. Blood (2020) 135(24):2133–6. doi: 10.1182/blood.2019001751
184. Pitt JM, Kroemer G, Zitvogel L. Immunogenic and non-immunogenic cell death in the tumor microenvironment. Adv Exp Med Biol (2017) 1036:65–79. doi: 10.1007/978-3-319-67577-0_5
Keywords: programmed cell death, cancer development, tumor immunity, immunosuppression, immunotherapy, anticancer therapeutics
Citation: Wang M, Yu F, Zhang Y and Li P (2024) Programmed cell death in tumor immunity: mechanistic insights and clinical implications. Front. Immunol. 14:1309635. doi: 10.3389/fimmu.2023.1309635
Received: 08 October 2023; Accepted: 28 December 2023;
Published: 12 January 2024.
Edited by:
Hongwei Liang, China Pharmaceutical University, ChinaReviewed by:
Zong Sheng Guo, University at Buffalo, United StatesJi-Xin Tang, Guangdong Medical University, China
Copyright © 2024 Wang, Yu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Wang, d2FuZ21hbkBxZHUuZWR1LmNu; Peifeng Li, cGVpZmxpQHFkdS5lZHUuY24=