 Ying Liang
Ying Liang Lingling Wang
Lingling Wang Peijun Ma
Peijun Ma Dongen Ju4*
Dongen Ju4* Minggao Zhao
Minggao Zhao Yun Shi
Yun Shi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 November 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1308264
Epigenetic mechanisms are processes that affect gene expression and cellular functions without involving changes in the DNA sequence. This abnormal or unstable expression of genes regulated by epigenetics can trigger cancer and other various diseases. The immune cells involved in anti-tumor responses and the immunogenicity of tumors may also be affected by epigenomic changes. This holds significant implications for the development and application of cancer immunotherapy, epigenetic therapy, and their combined treatments in the fight against cancer. We provide an overview of recent research literature focusing on how epigenomic changes in immune cells influence immune cell behavior and function, as well as the immunogenicity of cancer cells. And the combined utilization of epigenetic medications with immune checkpoint inhibitors that focus on immune checkpoint molecules [e.g., Programmed Death 1 (PD-1), Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA-4), T cell Immunoglobulin and Mucin Domain (TIM-3), Lymphocyte Activation Gene-3 (LAG-3)] present in immune cells and stromal cells associated with tumors. We highlight the potential of small-molecule inhibitors targeting epigenetic regulators to amplify anti-tumor immune responses. Moreover, we discuss how to leverage the intricate relationship between cancer epigenetics and cancer immunology to create treatment regimens that integrate epigenetic therapies with immunotherapies.
Epigenetic modifications are heritable and alters gene expression by changing chromatin structure and modifications, like DNA methylation, histone modifications, and non-coding RNA regulations (1). During the 1980s, DNA hypomethylation at genome-wide level was firstly discovered as a critical feature of cancer cells; meanwhile DNA hypermethylation can result to tumor suppressor genes (TSGs) silence (2). Another research found that hypermethylation of cytosine residues, as well as somatic mutations in TET2, DNMT3B, IDH1, and BRAF are exhibited in approximately 22% of tumors (3). MicroRNAs (miRNAs) are small non-coding RNAs (ncRNAs) spanning 19~22 nucleotides, which regulate gene expression by inhibiting or degrading its mRNA in a sequence-dependent way. Fabbri et al., reported the first evidence for miR-29 family involvement in directly targeting two DNA methyltransferases: DNMT3a/DNMT3b. Further, inhibiting miR-29s or DNMTs restored epigenetically silenced TSGs expression (4). Others’ findings identified Lrg1, Vegf-a, and IL-10 can be activated by histone H3K18 lactylation as reparative genes via combination of CUT&Tag and RNA-sequencing profiling, which is critical to immune homeostasis for post-myocardial infarction (5). This review will focus on epigenetic modifications during tumorigenesis and will emphasize opportunities to develop therapeutic strategies that target these modifications in cancer.
The accumulation of epigenetic changes is pivotal in cancer development (6–8). Various tumor-specific alterations create neoantigens, and epigenetic changes can contribute to the reactivation of genes that are typically restricted to immune-privileged stages, leading to their expression (9). Both tumor neoantigens and autoantigens can be immunogenic, eliciting an immune response in the body (10). Over the past few decades, significant attention has been directed towards the development of epigenetic therapies as anticancer agents, owing to their direct impact on cancer cells. recent studies have not only shed light on how epigenetic mechanisms contribute to immune evasion but have also unraveled the role of epigenetic drugs in modulating immune pathways to enhance immune recognition and promote immunogenicity (11–13). A comprehensive understanding of the role played by epigenetic regulatory mechanisms in cancer immunity is crucial to fully harnessing the potential of epigenetic drugs.
Immunotherapy is considered the fourth major technique in dealing with cancers, following surgery, radiotherapy, and chemotherapy. The fundamental objective of immunotherapy is to reinitiate and maintain the tumor-immune cycle, restoring the body’s natural anti-tumor immune response. By enhancing the immune system’s capacity to identify and focus on cancerous cells, immunotherapy aims to control and eliminate tumors. Among immunotherapies, immune checkpoint inhibitors have undergone rapid clinical development and have been widely utilized clinically. Up to now, the U.S. Food and Drug Administration (FDA) has authorized the use of a pair of anti-CTLA-4 antibodies, along with three anti-PD-1 antibodies and three anti-PD-L1 antibodies for treatment of more than a dozen different cancers (14, 15); Nonetheless, a considerable number of patients continue to encounter restricted advantages concerning their response to therapy and overall survival. In addition, epigenetic mechanisms are also involved in the development of tumors and tumor immunogenicity. Epigenetic therapies have shown remarkable clinical efficacy in hematological cancers, yet their efficacy in solid tumors has been limited (16, 17). In recent years, extensive research has revealed a significant breakthrough in tumor treatment, highlighting the effectiveness of a combined approach involving immunosuppressants and epigenetic drugs. Efforts must be directed towards the development of remedies aimed at investigating combination therapies, with the ultimate goal of improving response rates.
Epigenetic dysregulation is a common contribution to tumorigenesis. In recent years, our lab poured great energy into a systematic analysis of them to comprehensively explore the function of miRNAs (18). We analyzed the relationship between the location of human miRNA loci and the enrichment of enhancer markers Histone 3 Lysine 27 acetylation (H3K27ac) and Histone 3 Lysine 4 monomethylation (H3K4me1). Finally, we found that many miRNA precursor loci overlapped with the cognate enhancer regions enriched for H3K27ac and H3K4me1, such as miR-24-1, miR-339, and miR-3179. Interestingly, enhancer-associated miR-24-1 was also demonstrated to be able to activate its neighboring genes. Such enhancer-associated miRNAs that activate gene expression are known as nuclear activating miRNAs (NamiRNAs) (19). In the case of miR-339 and miR-24-1, in addition to exerting a positive regulatory function on neighboring gene activation via AGO2 proteins, these NamiRNAs are also involved in tumorigenesis (20, 21) (Figure 1). Therefore, NamiRNAs may be promising epigenetic targets for cancer therapy in the future.
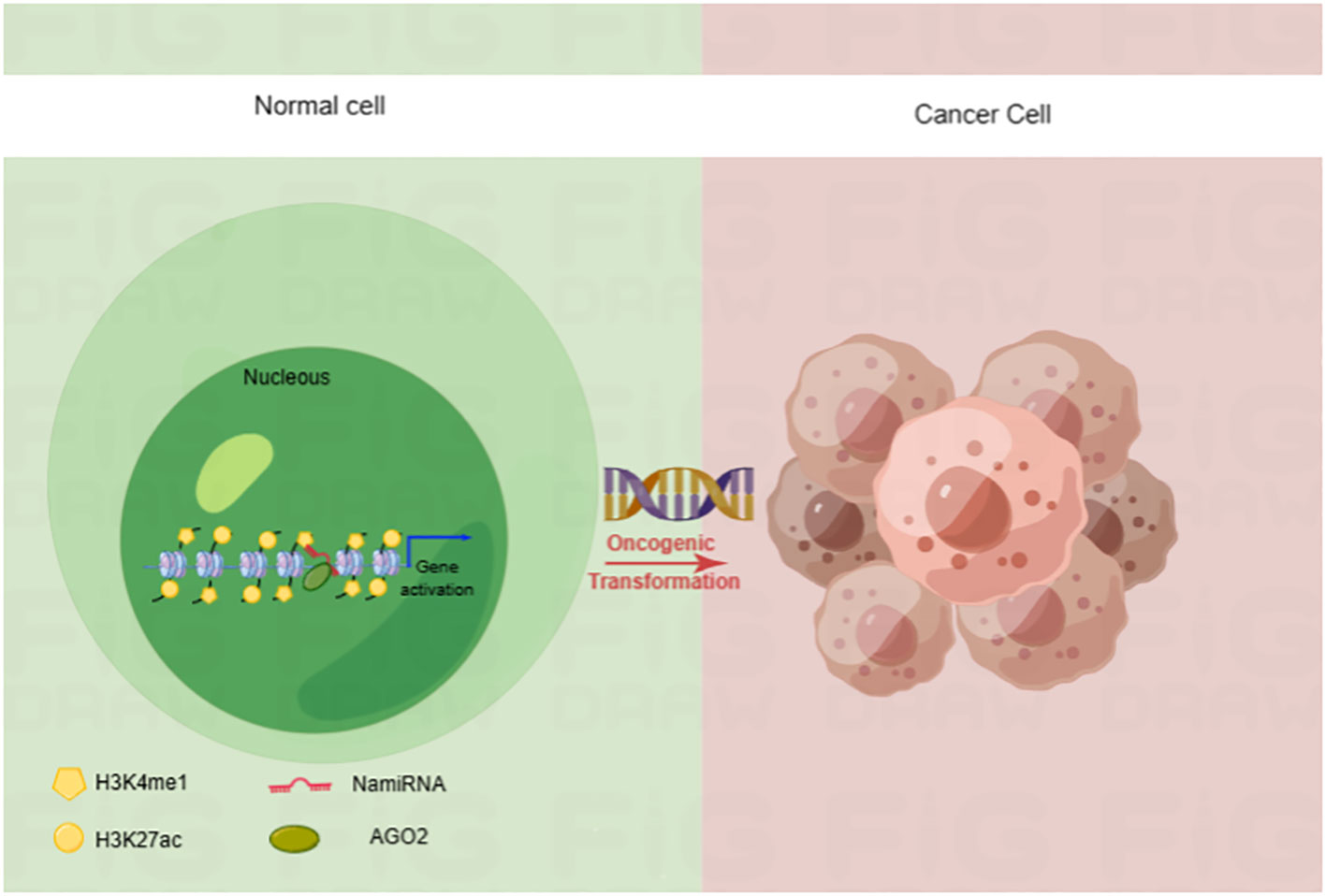
Figure 1 NamiRNA plays positive regulations on gene transcription through enhancers in nucleus. NamiRNAs exert positive roles on target gene expression to promote or block cancer progression. H3K4me1 and H3K27ac are enhancer markers.
The quick development of epidrugs over the past two decades brings new prospects to cancer therapy (22). Before the year of 2020, the range of FDA-approved epidrugs was limited, comprising solely of four pan-Histone deacetylase inhibitor (HDACi) and two DNA methyltransferase inhibitors (DNMTi). These treatments were specifically approved to target hematopoietic malignancies, including T-cell lymphoma, and myelodysplastic syndromes. Chidamide (also known as HBI-8000), an additional HDACi, received approval from the Chinese Food and Drug Administration for the treatment of peripheral T-cell lymphoma (23). Drugs like hydralazine, procaine and procainamide, originally approved by the FDA for hypertension treatment, local anesthesia and cardiac arrhythmia, respectively, have also demonstrated DNMTi activity (24). Likewise, valproic acid, an FDA-approved drug primarily used for treating seizures, also possess HDACi properties. In January 2020, a significant milestone was achieved with the approval of tazemetostat, an inhibitor targeting KMT6A (EZH2), for the treatment of epithelioid sarcoma. This marked a groundbreaking advancement as it became the first approved inhibitor of histone ‘writers’ and the first epidrug to demonstrate efficacy in treating solid tumors (25).
Understanding the influence of epigenetics on immune cells are important in tumor immunotherapy and epigenetic therapy. Epigenetic regulations play a key role in the, differentiation, development and function of various immunocyte lineages (26–28). In this section, we aim to emphasize the role of epigenetic mechanisms in governing the fate and development of immune cell subsets during anti-tumor immune responses. We specifically focus on CD4+,CD8+ T lymphocytes and natural killer (NK) cells, highlighting the impact of epigenetic regulations. Herein, we try to comprehensively review the whole picture of the crosstalk between immunity and epigenetic regulation (Figure 2), and summarize the epigenetic regulations and outcomes in CD4+ and CD8+ T cells.
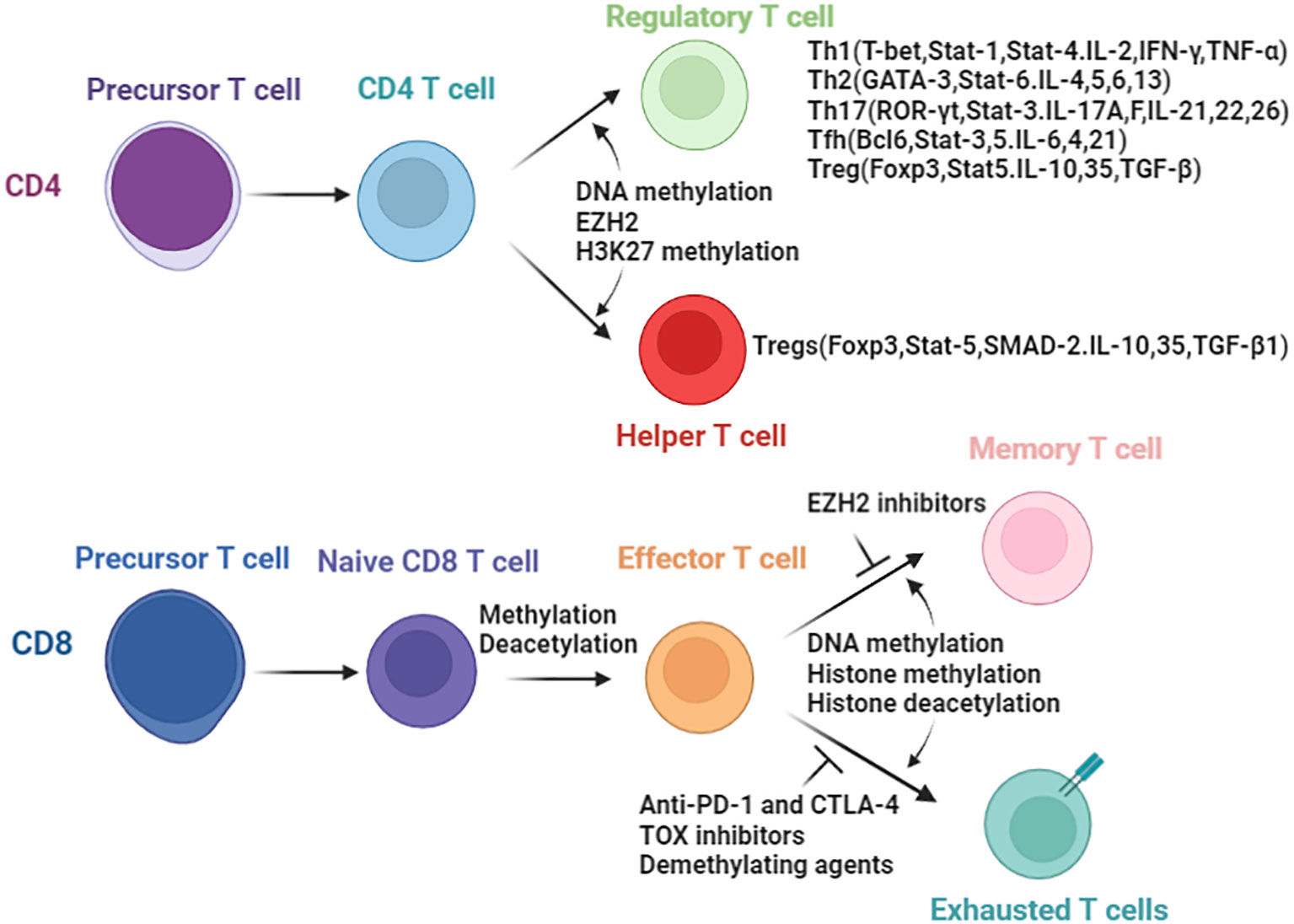
Figure 2 Epigenetic enzymes involved in the biology of T cells. Differentiation of CD4 T cells and CD8 T cells are controlled by epigenetic modifications. In parenthesis are the transcript factors and secreted cytokines resulting directly or indirectly due to epigenetic changes.
CD4+ T cells play a variety functions in the immune system and differentiate into several subsets of effector T cells. Including T helper 1 (Th1) cells, T helper 2 (Th2) cells, T helper 17 (Th17) cells, T follicular helper (Tfh) cells, and regulatory T (Treg) cells (29) (Figure 3). Th1 cells secrete gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) and are important for resistance to intracellular pathogens and cancer, whereas Th2 cells secrete cytokines interleukin-4 (IL-4), IL-5, IL-9 and IL-13, which stimulate B-cell antibody production and resist other extracellular pathogens (30–32). Th17 cells produce the cytokines IL-17A, IL-17F, IL-21 and IL-22 that direct neutrophil-mediated attack against extracellular bacteria infections (33). Treg cells secrete Inhibitory cytokines IL-10 (34) and transforming growth factor-β (TGF-β) (35–38), which suppress the activity of a variety of immune cells and play critical roles in maintaining immune homeostasis.
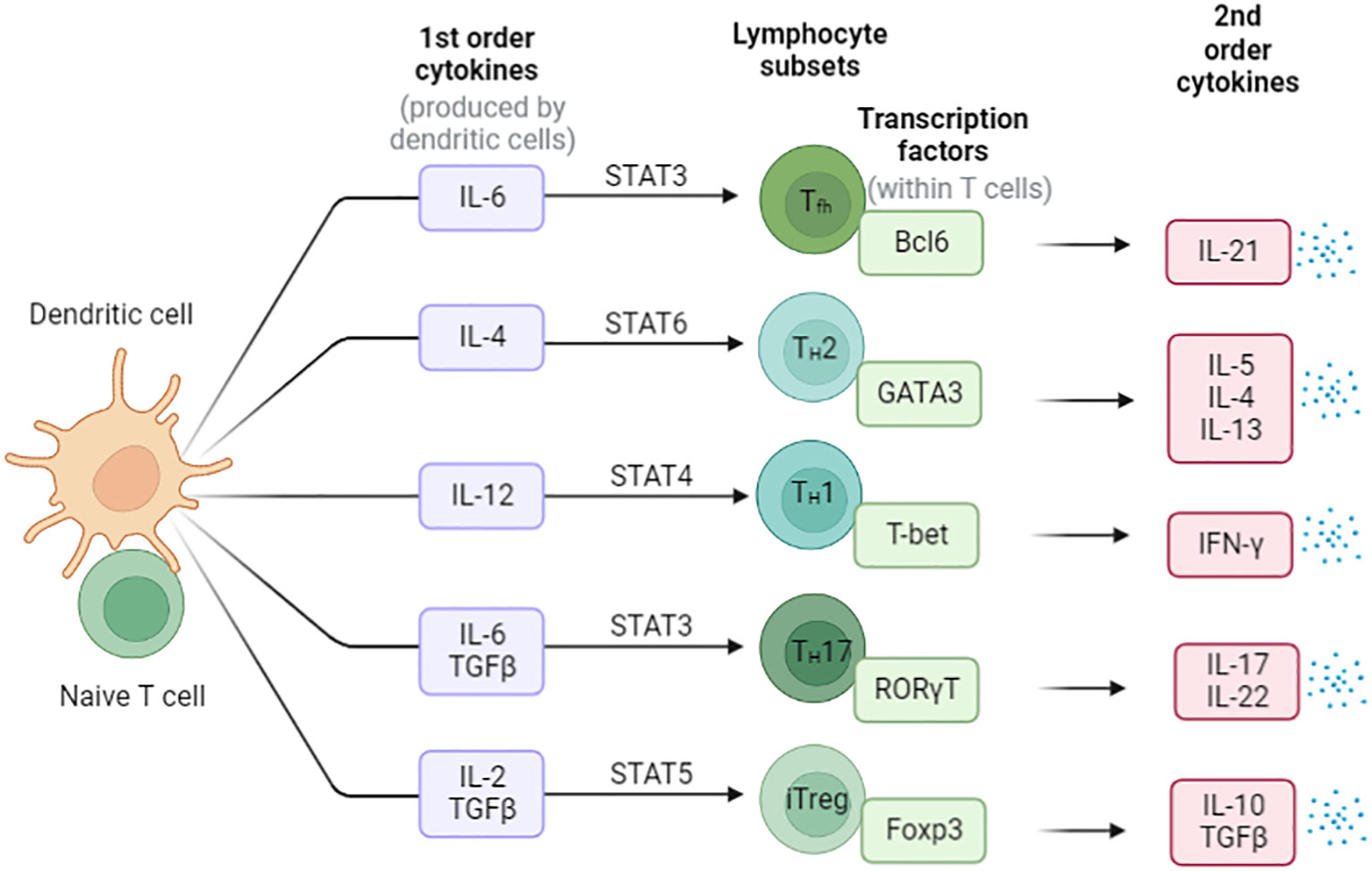
Figure 3 The differentiation of Naïve CD4+ T cells in the periphery. Cytokines direct the differentiation of these different types of helper T cells, and the major mode of helper T cell effector function is selective cytokine secretion. These T cell subsets are characterized by the expression of master transcription factors and secretion of signature cytokines.
Epigenetic mechanisms play important roles in the differentiation and functions of T cells (39, 40). Enhancer of zeste homology 2 (EZH2) is a histone H3K27 methyltransferase. Deficiency of EZH2 was found to specifically reduce the differentiation and plasticity of Th1 and Th2 cell depend on loss of H3K27me3 at genes encoding T-bet and Gata3 for Th1 and Th2 cells, respectively (41). Up to 10% of the mouse genome is made up of endogenous retrovirus (ERV) sequences (42). Véronique Adoue et al. showed that the histone methyltransferase SET domain bifurcated 1 (SETDB1) deposits the repressive H3K9me3 mark at ERVs in Th2 cells. The level of chromatin by the SETDB1-dependent deposition of H3K9me3 at ERVs control Th2 cell stability (43). For Th2 cells, the acquisition and maintenance of the Th2 cell identity relies on epigenetic modifications that establish a stable gene expression program that supports Th2-specific cytokine production and effector responses (44).
As for Th17 cells, Epigenetic modifications have emerged as important mechanisms for regulating the production of IL-17 during Th17 cell differentiation (45, 46). Reseachers uncovered that the enrichment of permissive histone modifications such as H3K4me3 and DNA demethylation in the promoters of cytokine and genes such as IL17a, IL17f, IL21, IL23r, and Rorc in TH17 cells. CXXC finger protein 1 (Cxxc1) is an epigenetic regulator, Feng Lin et al. showed Cxxc1 is critical for T-cell intrathymic development and through regulating H3K4 trimethylation, Cxxc1 deficiency decreased the generation of TH17 cells and prevented their differentiation into Treg cells (47). Cxxc1-dependent H3K4me3 plays a critical role in thymocyte development, phagocytosis, and the bactericidal activity of macrophages (48, 49).
CD8+ T cells play important role in anti-tumor immunotherapies and pathogen clearance and resist re-infection through long-lived immune memory T cells (50). Similar to the differentiation of naïve CD4+ cells, naive CD8+ T cells differentiate into a variety of memory and effector cell types under stimulation.The epigenetic regulation of CD8+ T cells has been extensively investigated (51).
T cell metabolism is influenced by environmental nutrient availability, which impacting T cell function. Ketone bodies (KBs) include β-hydroxybutyrate (βOHB) and acetoacetate (AcAc). Katarzyna et al. showed that CD8+ Teff cells prefer to use KBs rather than glucose to fuel the tricarboxylic acid (TCA) cycle both in vitro and in vivo. βOHB increased CD8+ Teff cells cytokine production and cytolytic activity responses through effects on histone acetylation at effector gene loci (52). The activation process of CD8+ T cell involves dynamic changes in DNA and histone modifications. Eomesodermin (EOMES) is a key transcription factor in regulating the differentiation of naïve CD8+ T cells into effector and memory CD8+ T cells (53). Perforin (PRF1), granzyme B (GZMB), and Gamma interferon (IFNG) are the target genes of EOMES, and promotes effector function in CD8+ T cells (54–56). Yasuto Araki et al. showed that compared with naive CD8+ T cells, the levels of H3K9Ac were significant higher in the EOMES, PRF1, and GZMB loci in memory CD8+ T cells.
Treg cells inhibit inflammatory effector T cells and play an opposite effect in anti-tumor immunity. The generation, maintenance, and function of CD4+ CD25+ Treg cells is critically dependent on transcription factor forkhead box P3 (Foxp3) (36, 57, 58), and epigenetic regulation is important for Foxp3 activation (59–61). nTreg exhibit a specific CpG hypomethylation pattern which is independent of Foxp3 expression, what’s more, Foxp3 induction and CpG hypomethylation are both required in the development of Treg cell (46). Furthermore,Treg exhibit specific DNA methylation signature that help distinguish Treg from Tconv cells (62), especially in the Foxp3 gene region.There are at least three conserved non-coding sequence (CNS1–CNS3) cis-elements at the foxp3 gene locus (Figure 4), the methylation status of which determine the expression and stability of Foxp3 (63, 64). (Conserved non-coding sequence 22) CNS2 is a CpG-rich Foxp3 intronic cis-element specifically demethylated in mature Treg, helps prevents autoimmunity and maintain immune homeostasis, thus CNS2 play an important role in the stability and function of Tregs (65, 66). The demethylation of the CNS2 is dependent on the activity of Ten-eleven translocation (TET) family members and induction of TET activity has been recognized as a mechanism for promoting the function of Treg cells in autoimmune diseases (67, 68). Mice conditionally deficient for Tet2/Tet3 in Tregs develop inflammatory disease, and the Treg cells gene signatures are altered in these mice, indicating that Tet2/3 are master regulators that stabilize Treg cells identity and immune homeostasis (69).
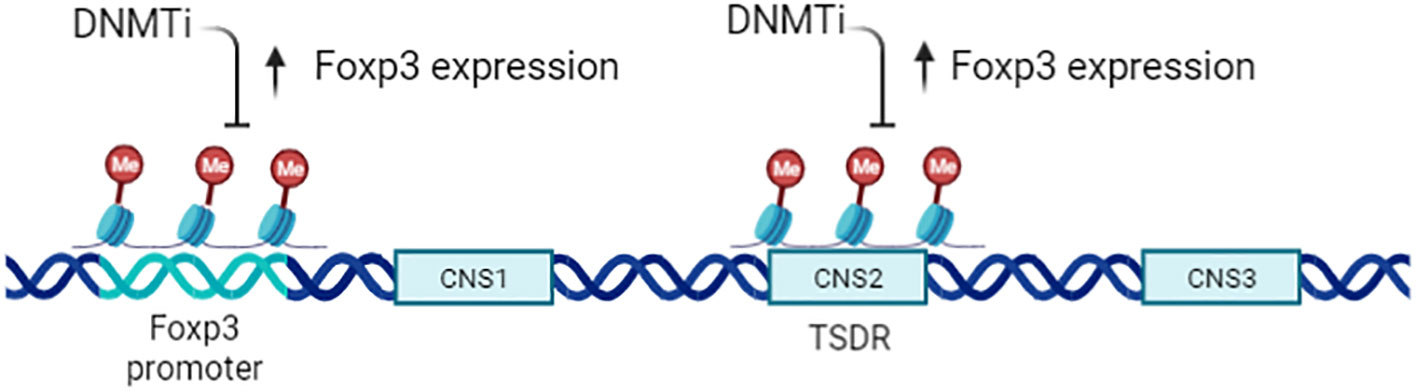
Figure 4 Epigenetic regulation of CD4+CD25+ Tregs. Expression of Foxp3 is tightly controlled by the methylation status of the gene, particularly the Treg -specific demethylation region (TSDR;CNS2). A DNMTi can enhance Foxp3 expression but does not increase Treg function, owing to the altered epigenetic state of Foxp3 target genes.
Natural killer (NK) cells have been classified as a type of lymphocyte, NK cells are generally considered components of the innate system. These cells have a high cytolytic capacity, unlike cytotoxic T cells, can induce tumor cell death directly and are imperative for tumor immunotherapy (70).
The tumor immune escape of NK cells has multiple molecular mechanisms. These include some upregulation of inhibitory receptors on NK cells such as HLA-E and HLA-G (71, 72), the secretion of chemokines and cytokines like IL-10, transforming growth factor β (TGF-β), CXCL9 and prostaglandin E2 (73, 74), as well as the expression of inhibitory receptors including PD-1,CTLA-4,TIM-3,and LAG3 (75–77). DNA and histone methylation have been implicated in the downregulation of NK inhibitory receptor CD94/NK group 2 member A (NKG2A). What’s more, the methylation of H3K27, mediated by EZH2, leads to the suppress NK cells via ligation of the natural killer group 2D (NKG2D) receptor. Moreover, H3K27 methylation also induce silencing of IL-15, CD122 NKG2D receptor proteins, which consequently suppress NK cell expansion and decreases their cytotoxic targeting of tumor cells (78–80). The cytotoxicity of NK cells can also be suppressed by H3K9 and H3K4 methylation, leading to the downregulation of the NKG2D receptor. However, when the methyltransferases responsible for these methylation processes were inhibited, the expression level of the ligands increases, resulting in enhanced anti-tumor cytotoxicity of NK cells (81–84).
With extensive research on tumor immunogenicity and immune cells, cancer treatment has witnessed a notable advancement with the emergence of tumor immunotherapy, specifically immune checkpoint inhibitors (ICIs). It has revolutionized the field of oncology and shown remarkable clinical efficacy in certain types of cancer, especially in treating solid tumors. Presently, ICIs targeting three distinct molecules, namely CTLA-4, PD-1, and PD-L1, have gained widespread usage. CTLA-4 exerts inhibitory actions on T cells through multiple mechanisms. Firstly, it initiates receptor signaling that recruits phosphatases, subsequently suppressing the activity of essential transcription factors crucial for T cell function, like NF-κB, AP-1, and NFAT. Moreover, CTLA-4’s competition with CD28 for ligand binding diminishes the co-stimulatory signals accessible to T cells, resulting in a continued suppression of their activation (85). Additionally, research has demonstrated that CTLA-4 also contributes to the augmentation of Tregs’ function and activity. It promotes the suppressive capacity of Tregs, thereby exerting an additional regulatory effect on immune responses (14, 86). The effectiveness of inhibiting CTLA-4 has been extensively validated in a diverse range of tumors, both in preclinical studies and clinical trials. Notably, it obtained FDA approval in March 2011 for treating advanced melanoma. Additionally, ipilimumab, an anti-CTLA-4 antibody, is presently being studied as a possible therapeutic option for a range of cancer types, including prostate cancer, non-small-cell lung carcinoma (NSCLC), renal cell carcinoma (RCC), and other malignancies (87). PD-1 is predominantly expressed during the later phases of T cell activation. It can also be found on the surface of B cells and macrophages. Contrastingly, CTLA-4 primarily regulates the extent of early activation in both naïve and memory T cells. These distinct expression patterns and functions highlight the complementary roles played by PD-1 and CTLA-4 in modulating immune responses at different stages. The interplay of PD-1 with its counterpart PD-L1 initiates a signaling cascade that can induce T cell apoptosis, leading to a state of T cell exhaustion. This exhaustion impairs the functionality and responsiveness of T cells, ultimately aiding in tumor cells’ evasion of the immune response (88). In the context of immunotherapy, PD-1/PD-L1 antibodies function by interrupting the PD-1 and PD-L1 interaction. This disruption enables T cells to undergo reactivation, proliferation and enhances their capacity to identify and eradicate tumor cells. PD-1 blockade primarily operates by attenuating the proximal T cell receptor (TCR) signaling pathway, thereby restoring the depleted CD8+ effector T cell population. The reinvigoration of T cell function plays a vital role in bolstering the anti-tumor immune response, effectively combating tumor growth. In 2014, the FDA granted approval for two anti-PD-1 antibodies, namely pembrolizumab and nivolumab, to be used in treating advanced melanoma. Furthermore, over the recent years, the FDA has approved three anti-PD-L1 antibodies—namely atezolizumab, avelumab and durvalumab—for treating various types of cancer. In addition, an expanding array of checkpoint receptors and ligands are being targeted in clinical settings, adding to the list of potential therapeutic options. These include TIM3, B7H3, LAG3, CD39, TIGIT, IDO-1, CD73, adenosine A2A receptor, among others (Figure 5). The exploration and development of therapies targeting these checkpoints highlight the expanding landscape of immune checkpoint inhibition and the potential for new treatment options in various cancers (75, 89–93). The efficacy of various combinations-immune checkpoint inhibitors is being investigated currently in clinical trials.
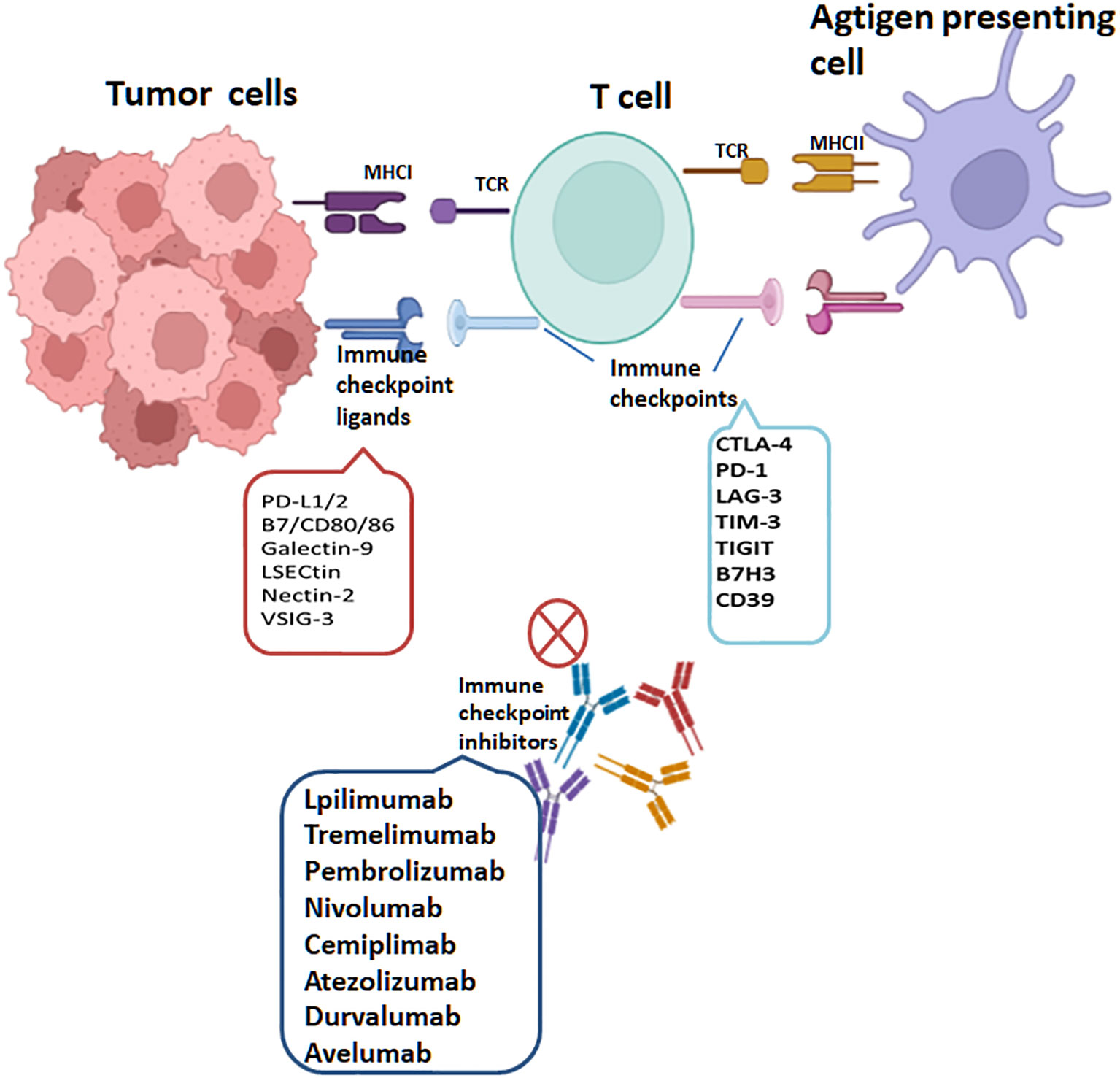
Figure 5 T cells are activated through MHC class I expressed on tumor cells or MHC class II molecules expressed on antigen-presenting cells, recognized by T-Cell Receptor (TCR). PD-1, PD-L1, CTLA-4, LAG-3, TIM-3, TIGIT, CD39 and B7H3 are targets of immune checkpoints (ICs). Interactions of ICs with their ligands on tumor cells lead to T cell inactivation. The immune checkpoint inhibitors(ICIs) currently approved through the FDA include lpilimumab and tremelimumab targeting CTLA-4, pembrolizumab, cemiplimab and nivolumab targeting PD-1 and atezolizumab, avelumab and durvalumab targeting PD-L1.
While ICIs have shown remarkable efficacy in treating certain cancers, a subset of patients may experience primary or acquired resistance to these therapies. Additionally, the use of ICIs can give rise to immune-related adverse events (IRAEs) impacting multiple organs (94). As a result, in many tumor indications the objective response rate (ORR) to ICIs can be less than 40% (95). The primary resistance mechanism to ICIs mainly includes: 1) a diminished expression of PD-L1 on tumor cells; 2) diminished response to ICIs due to tumor-intrinsic factors, such as genetic alterations within tumor cells that impact antigen presentation, interferon signaling, or downstream signaling pathways; 3) the loss of the IFN-γ signaling pathway; and 4) other factors that influence the enteric microbiome, effector T-cell infiltration in the tumor microenvironment (TME) and T-cell exhaustion. Another crucial mechanism that promotes resistance to ICIs involves epigenetic modulation, primarily encompassing changes in histone marks and chromatin structure, alterations in DNA methylation and dysregulation of regulatory RNA expression levels (96–98). For instance, HDACi can increase the levels of tumor antigens and reactivate proapoptotic genes, thereby sensitizing tumor cells to immune-mediated destruction (99). Histone methyltransferases (HMT) such as EZH2 have been identified as playing a role in the regulation of immune checkpoint molecules. HMTs have the capacity to inhibit the expression of PD-L1 by increasing H3K27me3 levels on the promoters of CD274 (PD-L1 gene), potentially reducing the immune evasion capabilities of tumor cells (100). In certain human melanoma cell lines, DNA hypomethylation has been associated with constant elevation of cytokines like IL-6 and VEGF. This dysregulated cytokine expression may contribute to resistance against immunotherapy (28). MiR-214, miR-568 and miR-126 contribute to T-cell exhaustion by influencing the development and function of Tregs. These microRNAs are also involved in downregulating cytotoxic T lymphocyte (CTL) activity (101).
Epigenetic targeting agents as a novel class of immune modulators have shown significant efficacy in hematological malignancies (102). However, their efficacy in solid tumors is constrained. Given that both epigenetic therapy and immune checkpoint blockade target the tumor microenvironment, combining ICIs with epigenetic modulators shows great promise in overcoming various cases of ICI resistance. This combinatory approach has the potential not only to generate synergistic effects but also mitigate adverse effects and deter the emergence of drug resistance. Some clinical trials and structures of the chemical compounds[ (103–105)] are underway or completed are listed below (Table 1).
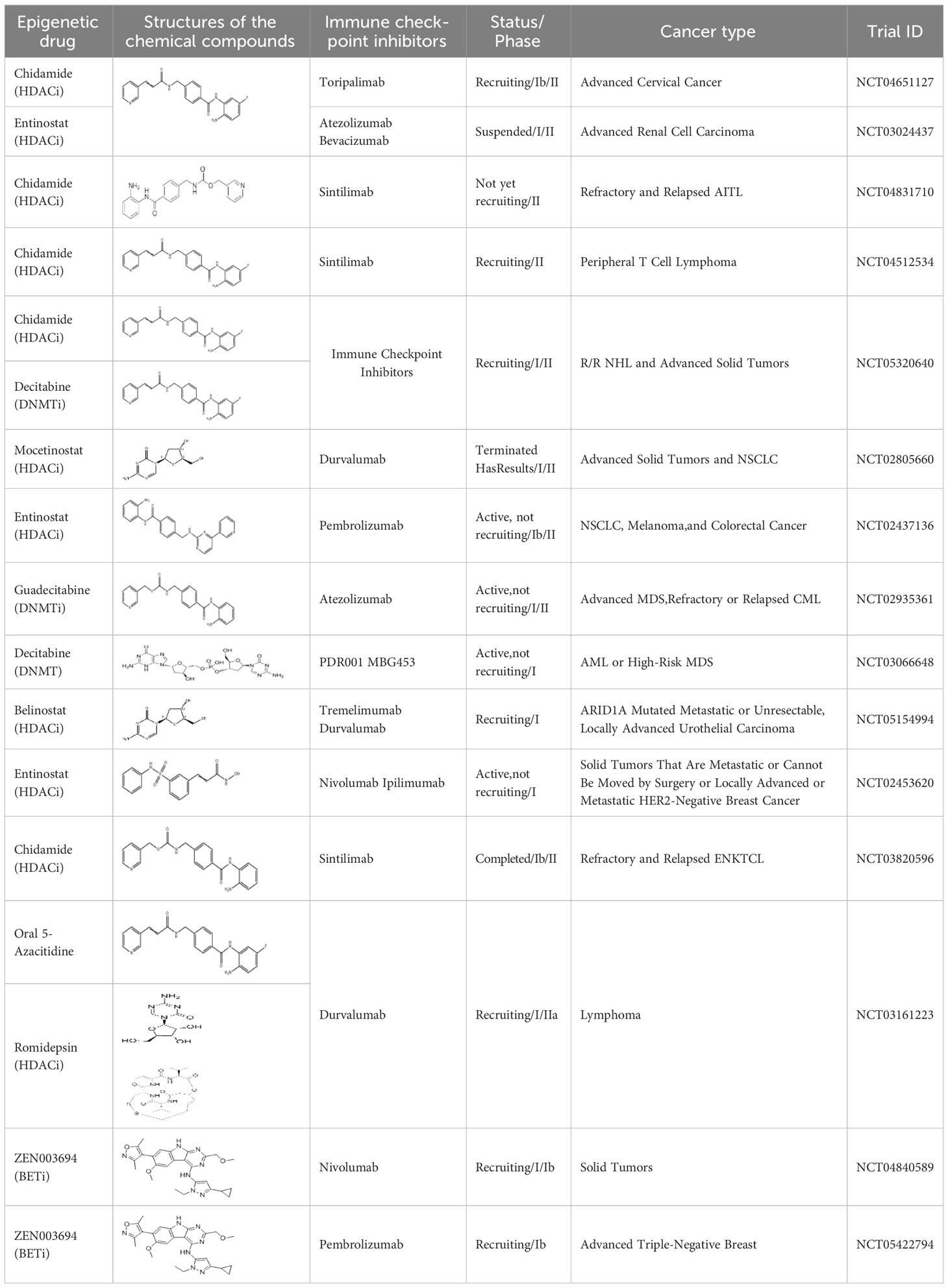
Table 1 Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies.
DNMTi holds a pivotal position during the outset of the immune cycle, particularly by augmenting the immunogenicity of tumors and facilitating immune recognition through the re-expression of tumor-associated antigens. Additionally, the upregulation of MHC class I and II molecules by DNMTi has been observed in numerous cancer types (106). In individuals with melanoma, global DNA hypermethylation is often linked to decreased expression levels of PD-L1. Research has shown that DNA hypomethylation can enhance the expression of CTLA-4, PD-L1, PD-1, and TIGIT in human colorectal cancer (107). In a murine ovarian cancer model, the administration of decitabine in combination with anti-CTLA-4 therapy led to increased efficacy and prolonged CD8+ tumor-infiltrating lymphocytes (TILs) response (108). Peng et al. (109) further demonstrated that inhibitors targeting DNMT1 and EZH2 have the ability to reawaken the production of T-helper 1 (Th1) chemokines, specifically CXCL9 and CXCL10, in human ovarian cancer. This reactivation resulted in an augmented infiltration of effector T cells into the tumor, subsequently decelerating tumor progression and enhancing the therapeutic effectiveness of anti-PD-L1 treatment. It is worth noting that, the DNA methylation status of LAG-3 is correlated with the expression levels of this immune checkpoint molecule in both tumor cells and immune cells in clear cell renal cell carcinoma (ccRCC) (110); this phenomenon significantly influences the destiny of immune cell infiltration within the tumor microenvironment.
The key histone modifications implicated in the cancer process primarily involve HDACs, HMTs like EZH2, and the Histone Reader Proteins of the BET (Bromodomain and Extra-Terminal) family. HDAC inhibitors have shown encouraging outcomes in overcoming ICI resistance in diverse cancer cell lines (110, 111), and in patients with metastatic non-small cell lung cancer (NSCLC), the use of HDAC inhibitor entinostat increased the PD-1 blockade immunotherapy response and facilitated heightened T cell infiltration (112). In a separate study, entinostat was found to enhance the therapeutic effectiveness of anti-PD-1 treatment by inhibiting myeloid-derived suppressor cells (MDSCs) in lung and kidney cancer mouse models (113). These findings underscore the potential of HDAC inhibitors in modulating the immune microenvironment and enhancing the efficacy of immunotherapies in certain cancer types. Multiple arguments support the association between EZH2 expression and tumor immunogenicity, indicating that interfering with EZH2 expression may have an impact on the response to ICIs. Indeed, another study has reported that inactivating EZH2 can reverse resistance to anti-CTLA-4 and IL-2 immunotherapies and inhibit the growth of melanoma (114). In the context of hepatocellular carcinoma, EZH2-mediated upregulation of H3K27me3 levels on the CD274 promoter, which encodes PD-L1, leads to a reduction in the expression of PD-L1 (100). Targeting EZH2 has been proposed as a potential strategy to enhance tumor immunogenicity. Additional investigation is warranted to comprehensively grasp the intricate interplay between EZH2, tumor immunogenicity, and the response to ICIs. However, the evidence thus far suggests that targeting EZH2 may represent a promising approach to enhance the effectiveness of immune-based cancer therapies. Indeed, the BET family are known to bind to acetylated histones and modulate immune function-related gene transcription. Inhibition of BET family members has demonstrated a decrease in PD-L1 expression, cytokine production, and nuclear factor-κB (NF-kB) activity in cancer cells (97, 101, 115). JQ1, a potent BET inhibitor, effectively boosts the body’s anti-tumor immune response by enhancing the cytotoxicity of CTLs while simultaneously reducing the infiltration of Tregs. JQ1 can not only coordinate the anti-tumor effect of PD-1 but also regulate PD-L1 and promote the expression of MHC-I molecules in colorectal cancer (CRC) cells (116). However, the exact mechanism underlying this regulation is not yet fully understood and needs further study. The modulation of BET proteins provides a potential therapeutic avenue for targeting the immune response in cancer and other immune-related diseases (117, 118). Inhibitors targeting the BET family are currently under investigation for their potential to augment the effectiveness of immunotherapies and surmount resistance to immune checkpoint blockade across diverse cancer types.
MiRNAs are also a critical component of epigenetic modifications. MiRNAs, both directly and indirectly, regulate the expression of PD-L1 and several other immune checkpoints. These miRNAs exert their regulatory influence by either binding directly to the PD-L1 mRNA or targeting other molecules involved in the signaling pathways that control PD-L1 expression. At least two independent studies noted that miRNAs are associated with the expression of PD-L1 or PD-1. In various tumor models, a majority of the identified miRNAs have demonstrated the ability to down-regulate the expression of PD-L1. For instance, in non-small cell lung cancer miR-140 (119), miR-142, and miR-197 have been observed to down-regulate PD-L1 expression, potentially attenuating its inhibitory effects on immune responses (120, 121). On the other hand, it is interesting to note that miR-3127-5p, in contrast, has been found to up-regulate PD-L1 expression (97). Some studies demonstrate the diverse roles of different miRNAs in regulating PD-L1 expression in colorectal cancer, with some miRNAs acting as suppressors and others as enhancers of PD-L1 expression through distinct molecular mechanisms. In colorectal cancer the miR-200 family, encompassing miR-200a, miR-200b, miR-429 and miR-200c act as inhibitors of PD-L1 expression. On the contrary, miR-20b, miR-21 and miR-130b have been reported to elevate PD-L1 expression in colorectal cancer by inhibiting the expression of PTEN (phosphatase and tensin homolog) (122, 123). Other miRNA (e.g., miR-138-5p) are also involved in the regulation of CTLA-4 (109). More miRNAs are yet to be discovered in relation to PD-1/PD-L1 and the exact mechanisms need to be further investigated. It is encouraging that our previous research found that NamiRNAs can regulate kidney cancer development by targeting enhancers (21). As to whether NamiRNAs play positive functions on their target genes by binding to the corresponding enhancers involved in immune therapy resistance for ccRCC remains to be eclucidated. Interestingly, our recent work suggests that miR-93-5p upregulates its target gene MCM7 in ccRCC to evade an immune response (Figure 6).
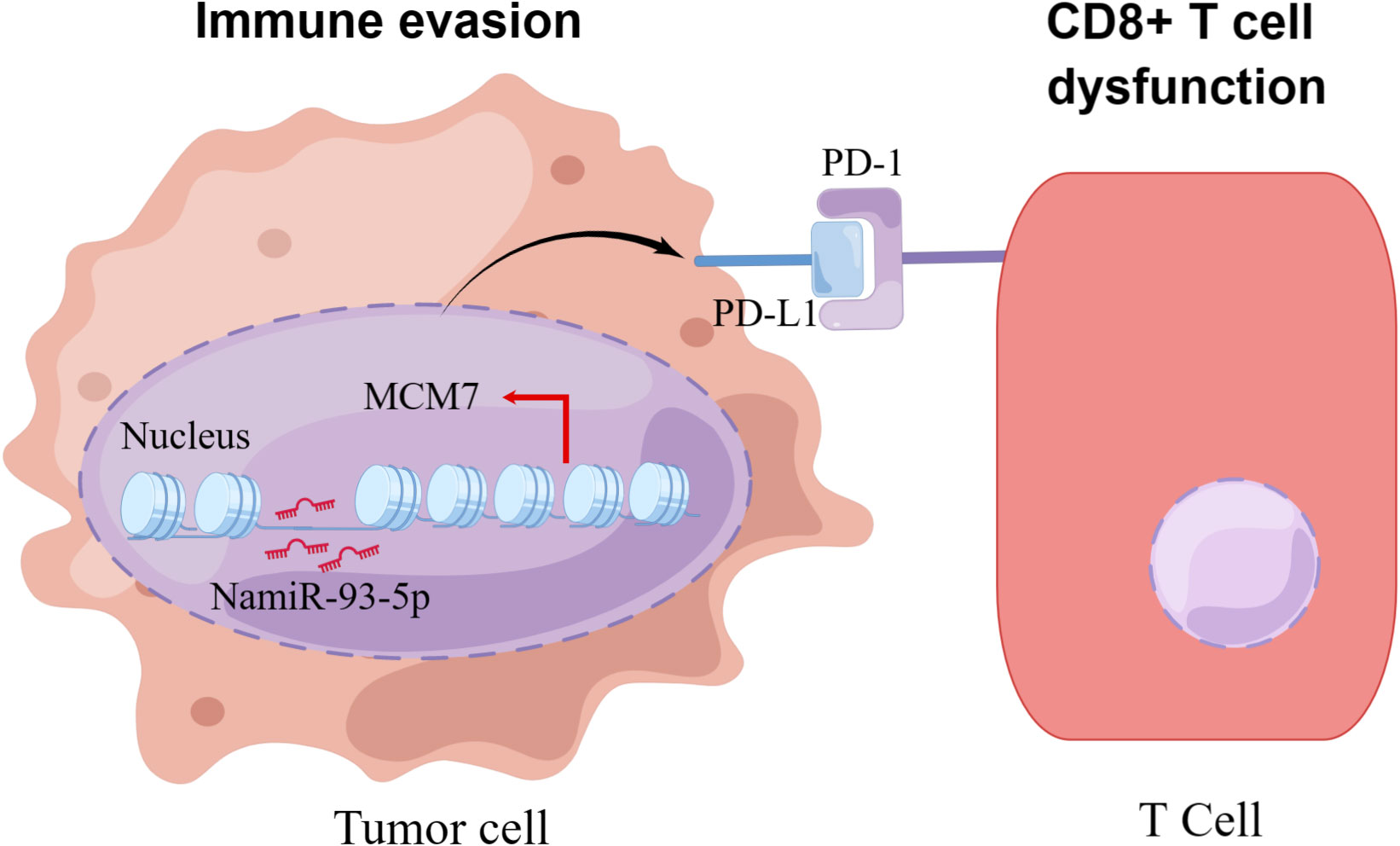
Figure 6 The schematic diagram of how NamiRNA-93-5p activates MCM7 to promote immune escape. In kidney cancer cells, NamiRNA-93-5p binds to the enhancer of MCM7 to activate its expression in the nucleus, thus inducing activation of PD-1/PD-L1 pathway and dysfunction of CD8+ T cells, and finally contributes to immune escape.
Initially, tumorigenesis was believed to be primarily promoted via genetic alterations. However, it is now widely recognized that epigenetic factors play a critical role in the initiation and progression of cancers. Epigenetic features, including DNA methylations and histone modifications, should lead to altered gene expression patterns and contribute to cancer progression. Targeting these epigenetic aberrations appears to be a potential method in cancer therapy due to several reasons (124). First, cancers frequently exhibit dysregulation of epigenetic characteristics and recurrent mutations of epigenetic modulators. Second, unlike hereditary changes, epigenetic modifications can often be reversed. Third, it is possible to target enzymes or chromatin-binding proteins that control epigenetic traits. As a result, oncogenic epigenetic regulators can be suppressed, and normal epigenetic features can be restored to create epigenetic medications that can treat cancer. Although many epigenetic agents have been approved and have demonstrated some benefits over conventional chemotherapy drugs, there are still many problems that need to be resolved (125). For instance, epigenetic agents mostly target blood malignancies, and further research is still needed to better treat solid tumors and other challenging and complex disorders. Additionally, the majority of epigenetic changes are loss of function mutations, which are challenging to treat (126). Thus, target specificity of epigenetic agents remain to be resolved. In the future, one of the most important approaches for applying such medications may be to create pharmaceuticals that can interfere with the specific target.
Immune cells play a crucial role in anti-tumor therapy, and epigenetics regulate the differentiation, development, and function of various immune cells. More and more epigenetic drugs targeting immune cell surface receptors have been developed. Therefore, studying the regulation of epigenetics on immune cells is a key factor in the treatment of tumors. Moreover, we described how epigenetics regulates the differentiation and plasticity of CD4+, CD8+, Tregs and other immune cells. Although there are many related studies on the regulatory molecular mechanisms of epigenetics in regulating immune cell function, there are still many issues that are not very clear. For example, how do epigenetic molecular mechanisms regulate T cell differentiation into other subsets of T cell, and how do they control T cell differentiation and function? How do these epigenetic modifications play a role in anti-tumor immunity?
As an emerging class of immune modulators, epigenetic regulation plays a critical role in both facilitating the anti-cancer immune response and enabling tumor cells to evade immunosurveillance. Understanding and targeting these epigenetic mechanisms hold great promise for developing effective immunotherapies and overcoming immune evasion strategies employed by cancer cells. Epigenetic drugs not only have potential as standalone therapies, but may also be used in combination with ICIs to synergistically enhance the immune response by modulating gene expression and reversing immune suppression within the tumor microenvironment. This combined strategy has demonstrated potential in both preclinical research and clinical trials, offering new avenues for improving treatment outcomes. Here, we described how ICIs have achieved very encouraging results in cancer treatment in recent years. However, long-term clinical effectiveness of ICIs has been compromised by the development of primary and acquired drug resistance, and the adverse events associated with ICIs have not been extensively studied. Epigenetic drugs in contrast play an important role in the immune tumor microenvironment and may also reduce resistance to ICIs, thereby improving remission rates. Epigenetic therapy also faces challenges and limitations in the treatment of cancer, and it may exhibit varying specificity towards different tumor cell subtypes. For example, a particular epigenetic therapy might be effective in non-small cell lung cancer (NSCLC) but have limited effectiveness in small cell lung cancer (SCLC) (127). Future research needs to encompass a broader exploration of epigenetic changes within different subtypes to develop more specific treatment approaches. Overall, the combination of these two drugs represents a promising strategy that takes advantage of their synergistic effects to improve treatment outcomes in both solid tumors and hematological tumors. And, identifying the most effective biologic dosing strategies may hold the key to the success of these combined therapeutic approaches. Nevertheless, the specific cellular and molecular mechanisms that account for the superior effectiveness observed in combination therapy compared to monotherapy-driven anti-tumor effects are yet to be fully understood. Defining biomarkers that help identify patients suitable for treatment and monitor treatment responses is also a future challenge. Numerous therapeutic trials combining these two treatments are currently underway; however, the majority of them are in the initial phases of development (112, 128, 129), Moreover, other epigenetic mechanisms, such as long non-coding RNA (lncRNA) or miRNA, are not yet clinically targetable at present. The next significant advancements are expected to occur through investment in Phase 3 clinical trials. These trials are pivotal in evaluating the safety and effectiveness of prospective treatments, and their successful completion can pave the way for regulatory approval and widespread implementation.
YL: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. LW: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. PM: Software, Resources. DJ: Data curation, Methodology, Supervision, Writing – review & editing. MZ: Supervision, Writing – review & editing. YS: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by “Phoenix Introduction Plan” Talent Startover Project of Tangdu Hospital, Fourth Military Medical University (grant number 2022YFJH001), Project of Shanghai Mental Health Center (grant number 2020-YJ05) and the National Natural Science Foundation of China (grant number 82203445).
We thank Dr. Chathurani S. Jayasena for critically reviewing the manuscript. Figures are created with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell (2007) 128(4):635–8. doi: 10.1016/j.cell.2007.02.006
2. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science (2001) 293(5532):1068–70. doi: 10.1126/science.1063852
3. Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjostrom M, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet (2020) 52(8):778–89. doi: 10.1038/s41588-020-0648-8
4. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U.S.A. (2007) 104(40):15805–10. doi: 10.1073/pnas.0707628104
5. Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res (2022) 131(11):893–908. doi: 10.1161/CIRCRESAHA.122.320488
6. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
7. Herceg Z, Hainaut P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol Oncol (2007) 1(1):26–41. doi: 10.1016/j.molonc.2007.01.004
8. Garcia-Martinez L, Zhang Y, Nakata Y, Chan HL, Morey L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat Commun (2021) 12(1):1786. doi: 10.1038/s41467-021-22024-3
9. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
10. Kalejs M, Erenpreisa J. Cancer/testis antigens and gametogenesis: a review and “brain-storming” session. Cancer Cell Int (2005) 5(1):4. doi: 10.1186/1475-2867-5-4
11. Sigalotti L, Fratta E, Coral S, Maio M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol Ther (2014) 142(3):339–50. doi: 10.1016/j.pharmthera.2013.12.015
12. Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol (2015) 6:29. doi: 10.3389/fimmu.2015.00029
13. Terranova-Barberio M, Thomas S, Munster PN. Epigenetic modifiers in immunotherapy: a focus on checkpoint inhibitors. Immunotherapy (2016) 8(6):705–19. doi: 10.2217/imt-2016-0014
14. Perrier A, Didelot A, Laurent-Puig P, Blons H, Garinet S. Epigenetic mechanisms of resistance to immune checkpoint inhibitors. Biomolecules (2020) 10(7). doi: 10.3390/biom10071061
15. Wu Q, Qian W, Sun X, Jiang S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J Hematol Oncol (2022) 15(1). doi: 10.1186/s13045-022-01362-9
16. Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol (2019) 17(2):75–90. doi: 10.1038/s41571-019-0266-5
17. Oleksiewicz U, Machnik M. Causes, effects, and clinical implications of perturbed patterns within the cancer epigenome. Semin Cancer Biol (2022) 83:15–35. doi: 10.1016/j.semcancer.2020.12.014
18. Liang Y, Xu P, Zou QP, Luo HB, Yu WQ. An epigenetic perspective on tumorigenesis: Loss of cell identity, enhancer switching, and NamiRNA network. Semin Cancer Biol (2022) 83:596–604. doi: 10.1016/j.semcancer.2018.09.004
19. Xiao M, Li J, Li W, Wang Y, Wu FZ, Xi YP, et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol (2017) 14(10):1326–34. doi: 10.1080/15476286.2015.1112487
20. Liang Y, Lu Q, Li W, Zhang DP, Zhang FL, Zou QP, et al. Reactivation of tumour suppressor in breast cancer by enhancer switching through NamiRNA network. Nucleic Acids Res (2021) 49(15):8556–72. doi: 10.1093/nar/gkab626
21. Ju D, Liang Y, Hou G, Zheng W, Zhang G, Dun X, et al. FBP1/miR-24-1/enhancer axis activation blocks renal cell carcinoma progression via Warburg effect. Front Oncol (2022) 12:928373. doi: 10.3389/fonc.2022.928373
22. Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol (2020) 17(2):91–107. doi: 10.1038/s41571-019-0267-4
23. Borcoman E, Kamal M, Marret G, Dupain C, Castel-Ajgal Z, Le Tourneau C. HDAC inhibition to prime immune checkpoint inhibitors. Cancers (Basel) (2021) 14(1). doi: 10.3390/cancers14010066
24. Singh V, Sharma P, Capalash N. DNA methyltransferase-1 inhibitors as epigenetic therapy for cancer. Curr Cancer Drug Targets (2013) 13(4):379–99. doi: 10.2174/15680096113139990077
25. Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol (2020) 21(11):1433–42. doi: 10.1016/S1470-2045(20)30441-1
26. Yang L, Jing Y, Kang D, Jiang P, Li N, Zhou X, et al. Ubiquitin-specific peptidase 18 regulates the differentiation and function of Treg cells. Genes Dis (2021) 8(3):344–52. doi: 10.1016/j.gendis.2020.03.004
27. Wilson CB, Makar KW, Perez-Melgosa M. Epigenetic regulation of T cell fate and function. J Infect Dis (2002) 185 Suppl 1:S37–45. doi: 10.1086/338001
28. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery (2020) 19(11):776–800. doi: 10.1038/s41573-020-0077-5
29. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238(1):247–62. doi: 10.1111/j.1600-065X.2010.00951.x
30. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature (1996) 383(6603):787–93. doi: 10.1038/383787a0
31. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
32. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-gamma in tumor progression and regression: a review. Biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
33. Valeri M, Raffatellu M. Cytokines IL-17 and IL-22 in the host response to infection. Pathog Dis (2016) 74(9):1–15. doi: 10.1093/femspd/ftw111
34. Komai T, Inoue M, Okamura T, Morita K, Iwasaki Y, Sumitomo S, et al. Transforming growth factor-beta and interleukin-10 synergistically regulate humoral immunity via modulating metabolic signals. Front Immunol (2018) 9:1364. doi: 10.3389/fimmu.2018.01364
35. Bauché D, Campbell S, Marie JC. Integrin αvβ8-mediated TGF-β Activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity (2015) 42(5):903–15. doi: 10.1016/j.immuni.2015.04.012
36. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1158/1557-3125.HIPPO19-B11
37. Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol (2022) 15(1):1–33. doi: 10.1186/s13045-022-01282-8
38. Chen W. TGF-beta regulation of T cells. Annu Rev Immunol (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
39. Dutta A, Venkataganesh H, Love PE. New insights into epigenetic regulation of T cell differentiation. Cells (2021) 10(12):3459. doi: 10.3390/cells10123459
40. Frias AB, Boi SK, Lan X, Youngblood B. Epigenetic regulation of T cell adaptive immunity. Immunol Rev (2021) 300(1):9–21. doi: 10.1111/imr.12943
41. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39(5):819–32. doi: 10.1016/j.immuni.2013.09.012
42. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature (2002) 420(6915):520–62. doi: 10.1038/nature01262
43. Adoue V, Binet B, Malbec A, Fourquet J, Romagnoli P, Meerwijk JP, et al. The histone methyltransferase SETDB1 controls T helper cell lineage integrity by repressing endogenous retroviruses. Immunity (2019) 50(3):629–644 e8. doi: 10.1016/j.immuni.2019.01.003
44. Onodera A, Kokubo K, Nakayama T. Epigenetic and transcriptional regulation in the induction, maintenance, heterogeneity, and recall-response of effector and memory Th2 cells. Front Immunol (2018) 9:2929. doi: 10.3389/fimmu.2018.02929
45. Philpott M, Oppermann U. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc Natl Acad Sci United States America (2020) 117(11):6056–66. doi: 10.1073/pnas.1919893117
46. Friedman MJ, Lee H, Lee J, Oh S. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc Natl Acad Sci United States America (2020) 117(11):6056–66. doi: 10.1073/pnas.1919893117
47. Lin F, Meng X, Guo Y, Cao W, Liu W, Xia Q, et al. Epigenetic initiation of the Th17 differentiation programme is promoted by Cxxc finger protein 1. Eur J Immunol (2019) 49:407–7. doi: 10.1126/sciadv.aax1608
48. Cao W, Guo J, Wen X, Miao L, Lin F, Xu G, et al. CXXC finger protein 1 is critical for T-cell intrathymic development through regulating H3K4 trimethylation. Nat Commun (2016) 7:1–11. doi: 10.1038/ncomms11687
49. Hui Z, Zhou L, Xue Z, Zhou L, Luo Y, Lin F, et al. Cxxc finger protein 1 positively regulates GM-CSF-derived macrophage phagocytosis through csf2rα-mediated signaling. Front Immunol (2018) 9:1885. doi: 10.3389/fimmu.2018.01885
50. Philip M, Schietinger A. CD8 T cell differentiation and dysfunction in cancer. Nat Rev Immunol (2022) 22(4):209–23. doi: 10.1038/s41577-021-00574-3
51. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
52. Luda KM, Longo J, Kitchen-Goosen SM, Duimstra LR, Ma EH, Watson MJ, et al. Ketolysis drives CD8(+) T cell effector function through effects on histone acetylation. Immunity (2023) 56(9):2021–2035.e8. doi: 10.1016/j.immuni.2023.07.002
53. Llaó-Cid L, Roessner PM, Chapaprieta V, Öztürk S, Roider T, Bordas M, et al. EOMES is essential for antitumor activity of CD8(+) T cells in chronic lymphocytic leukemia. Leukemia (2021) 35(11):3152–62. doi: 10.1038/s41375-021-01198-1
54. van der Leun AM, Thommen DS, Schumacher TN. CD8 T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20(4):218–32. doi: 10.1038/s41568-019-0235-4
55. Araki Y, Fann M, Wersto R, Weng N. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J Immunol (2008) 180(12):8102–8. doi: 10.4049/jimmunol.180.12.8102
56. Wong P, Foltz JA, Chang L, Neal CC, Yao T, Cubitt CC, et al. T-BET and EOMES sustain mature human NK cell identity and antitumor function. J Clin Invest (2023) 133(13):1–18. doi: 10.1172/JCI162530
57. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
58. Hariyanto AD, Permata TBM, Gondhowiardjo SA. Role of CD4(+)CD25(+)FOXP3(+) T(Reg) cells on tumor immunity. Immunol Med (2022) 45(2):94–107. doi: 10.1080/25785826.2021.1975228
59. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity (2013) 38(3):414–23. doi: 10.1016/j.immuni.2013.03.002
60. Joudi AM, Reyes Flores CP, Singer BD. Epigenetic control of regulatory T cell stability and function: implications for translation. Front Immunol (2022) 13:861607. doi: 10.3389/fimmu.2022.861607
61. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 30(6):465–74. doi: 10.1038/s41422-020-0324-7
62. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Träger U, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol (2017) 18(10):1160–72. doi: 10.1038/ni.3799
63. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol (2016) 37(11):803–11. doi: 10.1016/j.it.2016.08.012
64. Kitagawa Y, Ohkura N, Sakaguchi S. Epigenetic control of thymic Treg-cell development. Eur J Immunol (2015) 45(1):11–6. doi: 10.1002/eji.201444577
65. Bai L, Hao X, Keith J, Feng Y. DNA methylation in regulatory T cell differentiation and function: challenges and opportunities. Biomolecules (2022) 12(9):1282. doi: 10.3390/biom12091282
66. Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, et al. Molecular mechanisms controlling foxp3 expression in health and autoimmunity: from epigenetic to post-translational regulation. Front Immunol (2019) 10:3136. doi: 10.3389/fimmu.2019.03136
67. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med (2016) 213(3):377–97. doi: 10.1084/jem.20151438
68. Gerecke C, Rodrigues CE, Homann T, Kleuser B. The role of ten-eleven translocation proteins in inflammation. Front Immunol (2022) 13:861351. doi: 10.3389/fimmu.2022.861351
69. Yue XJ, Lio CW, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat Commun (2019) 10(1):2011. doi: 10.1038/s41467-019-09541-y
70. Rahman M, Bordoni B. Histology, natural killer cells. In: StatPearls. Treasure Island (FL): StatPearls Publishing (2023). http://www.ncbi.nlm.nih.gov/books/NBK565844/.
71. de Kruijf EM, Sajet AGH, van Nes J, Natanov R, Putter HTHBM, Smit V, et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol (2010) 185(12):7452–9. doi: 10.4049/jimmunol.1002629
72. Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J Immunother Cancer (2021) 9(8):e002899. doi: 10.1136/jitc-2021-002899
73. Chiu J, Ernst DM, Keating A. Acquired natural killer cell dysfunction in the tumor microenvironment of classic hodgkin lymphoma. Front Immunol (2018) 9:267. doi: 10.3389/fimmu.2018.00267
74. Yang Z, Qi Y, Lai N, Zhang J, Chen Z, Liu M, et al. Notch1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-beta1. J Exp Clin Cancer Res (2018) 37(1):1. doi: 10.1186/s13046-017-0664-4
75. Anderson AC, Joller N, Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
76. Shi X, Li C, Tan L, Wen S, Liao T, Zhang Y, et al. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: A large cohort study. J Clin Endocrinol Metab (2021) 106(1):120–32. doi: 10.1210/clinem/dgaa701
77. Takamatsu K, Tanaka N, Hakozaki K, Takahashi R, Teranishi Y, Murakami T, et al. Profiling the inhibitory receptors LAG-3, TIM-3, and TIGIT in renal cell carcinoma reveals Malignancy. Nat Commun (2021) 12(1):5547. doi: 10.1038/s41467-021-25865-0
78. Sun WY, Lv S, Li H, Cui W, Wang L. Enhancing the anticancer efficacy of immunotherapy through combination with histone modification inhibitors. Genes (2018) 9(12):633. doi: 10.3390/genes9120633
79. Shreeve N, Depierreux D, Hawkes D, Traherne JA, Sovio U, Huhn O, et al. The CD94/NKG2A inhibitory receptor educates uterine NK cells to optimize pregnancy outcomes in humans and mice. Immunity (2021) 54(6):1231–1244 e4. doi: 10.1016/j.immuni.2021.03.021
80. Bugide S, Gupta R, Green MR, Wajapeyee N. EZH2 inhibits NK cell-mediated antitumor immunity by suppressing CXCL10 expression in an HDAC10-dependent manner. Proc Natl Acad Sci United States America (2021) 118(30):e2102718118. doi: 10.1073/pnas.2102718118
81. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U.S.A. (2017) 114(51):E10981–90. doi: 10.1073/pnas.1712514114
82. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci USA (2018) 115(15):E3509–18. doi: 10.1073/pnas.1802691115
83. Xia MR, Wang B, Wang Z, Zhang X, Wang X. Epigenetic regulation of NK cell-mediated antitumor immunity. Front Immunol (2021) 12:672328. doi: 10.3389/fimmu.2021.672328
84. Rechberger JS, Toll SA, Vanbilloen J FW, Daniels DJ, Khatua S. Exploring the molecular complexity of medulloblastoma: implications for diagnosis and treatment. Diagnostics (2023) 13(14):2398. doi: 10.3390/diagnostics13142398
85. Sreya Bagchi RY, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
86. Zhang A, Ren Z, Tseng KF, Liu X, Li H, Lu C, et al. Dual targeting of CTLA-4 and CD47 on Treg cells promotes immunity against solid tumors. Sci Trans Med (2021) 13(605):eabg8693. doi: 10.1126/scitranslmed.abg8693
87. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
88. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
89. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
90. Yi KH, Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev (2009) 229(1):145–51. doi: 10.1111/j.1600-065X.2009.00768.x
91. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519
92. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
93. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discovery (2020) 10(1):40–53. doi: 10.1158/2159-8290.CD-19-0980
94. Khan S, Gerber DE. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin Cancer Biol (2020) 64:93–101. doi: 10.1016/j.semcancer.2019.06.012
95. Karimian Z, Mavoungou S, Salem JE, Tubach F, Dechartres A. The quality of reporting general safety parameters and immune-related adverse events in clinical trials of FDA-approved immune checkpoint inhibitors. BMC Cancer (2020) 20(1):1128. doi: 10.1186/s12885-020-07518-5
96. Zhang P, Zhang M. Epigenetic alterations and advancement of treatment in peripheral T-cell lymphoma. Clin Epigenet (2020) 12(1):169. doi: 10.1186/s13148-020-00962-x
97. Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer (2021) 20(1):171. doi: 10.1186/s12943-021-01464-x
98. Avella Patino DM, Radhakrishnan V, Suvilesh KN, Manjunath Y, Li G, Kimchi ET, et al. Epigenetic regulation of cancer immune cells. Semin Cancer Biol (2022) 83:377–83. doi: 10.1016/j.semcancer.2021.06.022
99. Sborov DW, Canella A, Hade EM, Mo X, Khountham S, Wang J, et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk Lymphoma (2017) 58(10):2310–8. doi: 10.1080/10428194.2017.1298751
100. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer (2019) 7(1):300. doi: 10.1186/s40425-019-0784-9
101. Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B (2020) 10(5):723–33. doi: 10.1016/j.apsb.2019.09.006
102. Zhao A, Zhou H, Yang J, Li M, Niu T. Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic Malignancies. Signal Transduction Targeted Ther (2023) 8(1). doi: 10.1038/s41392-023-01342-6
103. Loharch S, Bhutani I, Jain K, Gupta P, Sahoo DK, Parkesh R. EpiDBase: a manually curated database for small molecule modulators of epigenetic landscape. Database (2015) 2015. doi: 10.1093/database/bav013
104. Aguirre-Portoles C, Feliu J, Reglero G, Ramirez de Molina A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol Oncol (2018) 12(10):1735–52. doi: 10.1002/1878-0261.12367
105. Kalantar-Zadeh K, Schwartz GG, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, et al. Effect of apabetalone on cardiovascular events in diabetes, CKD, and recent acute coronary syndrome: results from the BETonMACE randomized controlled trial. Clin J Am Soc Nephrol (2021) 16(5):705–16. doi: 10.2215/CJN.16751020
106. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol (2020) 65:99–113. doi: 10.1016/j.semcancer.2019.12.019
107. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin Epigenet (2018) 10(1):104. doi: 10.1186/s13148-018-0539-3
108. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res (2015) 3(9):1030–41. doi: 10.1158/2326-6066.CIR-15-0073
109. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
110. Wang L, Li H, Ren Y, Zou S, Fang W, Jiang X, et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis (2016) 7(1):e2063. doi: 10.1038/cddis.2015.328
111. Gray JE, Saltos A, Tanvetyanon T, Haura EB, Creelan B, Antonia SJ, et al. Phase I/ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin Cancer Res (2019) 25(22):6623–32. doi: 10.1158/1078-0432.CCR-19-1305
112. Hellmann MD, Janne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin Cancer Res (2021) 27(4):1019–28. doi: 10.1158/1078-0432.CCR-20-3305
113. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res (2017) 23(17):5187–201. doi: 10.1158/1078-0432.CCR-17-0741
114. Goswami S AI, Zhang J, Skepner J, Anandhan S, Zhang X, Xiong L, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest (2018) 128(9):3813–18. doi: 10.1172/JCI99760
115. Zou Z, Huang B, Wu X, Zhang H, Qi J, Bradner J, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene (2014) 33(18):2395–404. doi: 10.1038/onc.2013.179
116. Wang H, Liu G, Jin X, Song S, Chen S, Zhou P, et al. BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC. J Cancer (2022) 13(7):2126–37. doi: 10.7150/jca.69375
117. Mills RJ, Humphrey SJ, Fortuna PRJ, Lor M, Foster SR, Quaife-Ryan GA, et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell (2021) 184(8):2167–2182 e22. doi: 10.1016/j.cell.2021.03.026
118. Mohammed SA, Albiero M, Ambrosini S, Gorica E, Karsai G, Caravaggi CM, et al. The BET protein inhibitor apabetalone rescues diabetes-induced impairment of angiogenic response by epigenetic regulation of thrombospondin-1. Antioxid Redox Signal (2022) 36(10-12):667–84. doi: 10.1089/ars.2021.0127
119. Xie WB, Liang LH, Wu KG, Wang LX, He X, Song C, et al. MiR-140 expression regulates cell proliferation and targets PD-L1 in NSCLC. Cell Physiol Biochem (2018) 46(2):654–63. doi: 10.1159/000488634
120. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther (2015) 23(4):717–27. doi: 10.1038/mt.2015.10
121. Wan J, Ling X, Peng B, Ding G. miR-142-5p regulates CD4+ T cells in human non-small cell lung cancer through PD-L1 expression via the PTEN pathway. Oncol Rep (2018) 40(1):272–82. doi: 10.3892/or.2018.6439
122. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
123. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
124. Salarinia R, Sahebkar A, Peyvandi M, Mirzaei HR, Jaafari MR, Riahi MM, et al. Epi-drugs and epi-miRs: moving beyond current cancer therapies. Curr Cancer Drug Targets (2016) 16(9):773–88. doi: 10.2174/1568009616666151207110143
125. Rath S, Chakraborty D, Pradhan J, Imran Khan M, Dandapat J. Epigenomic interplay in tumor heterogeneity: Potential of epidrugs as adjunct therapy. Cytokine (2022) 157:155967. doi: 10.1016/j.cyto.2022.155967
126. Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C, et al. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics (2019) 14(12):1164–76. doi: 10.1080/15592294.2019.1640546
127. Zhang C, Wang H. Accurate treatment of small cell lung cancer: Current progress, new challenges and expectations. Biochim Biophys Acta Rev Cancer (2022) 1877(5):188798. doi: 10.1016/j.bbcan.2022.188798
128. Palomba ML, Cartron G, Popplewell L, Ribrag V, Westin J, Huw LY, et al. Combination of atezolizumab and tazemetostat in patients with relapsed/refractory diffuse large B-cell lymphoma: results from a phase ib study. Clin Lymphoma Myeloma Leuk (2022) 22(7):504–12. doi: 10.1016/j.clml.2021.12.014
129. Gatti-Mays ME, Gameiro SR, Ozawa Y, Knudson KM, Hicks KC, Palena C, et al. Improving the odds in advanced breast cancer with combination immunotherapy: stepwise addition of vaccine, immune checkpoint inhibitor, chemotherapy, and HDAC inhibitor in advanced stage breast cancer. Front Oncol (2021) 10. doi: 10.3389/fonc.2020.581801
Keywords: epigenetic regulations, immune cells, T cells, anti-tumor immunity, immune checkpoint inhibitors
Citation: Liang Y, Wang L, Ma P, Ju D, Zhao M and Shi Y (2023) Enhancing anti-tumor immune responses through combination therapies: epigenetic drugs and immune checkpoint inhibitors. Front. Immunol. 14:1308264. doi: 10.3389/fimmu.2023.1308264
Received: 06 October 2023; Accepted: 06 November 2023;
Published: 23 November 2023.
Edited by:
Mohd Wajid Ali Khan, University of Hail, Saudi ArabiaReviewed by:
Shafeeq Ahmed Mohammed, University Hospital Zürich, SwitzerlandCopyright © 2023 Liang, Wang, Ma, Ju, Zhao and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Shi, eXVuc2hpQGNvaC5vcmc=; Minggao Zhao, bWluZ2dhb0BmbW11LmVkdS5jbg==; Dongen Ju, ZG4xOTk1MTExOUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.