
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 22 November 2023
Sec. NK and Innate Lymphoid Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1307398
This article is part of the Research TopicModulation of Antibody-Mediated Effector Functions in Natural Killer Cells: Protective and Detrimental Effects in Infectious DiseasesView all 8 articles
 Hillary A. Vanderven1,2,3*
Hillary A. Vanderven1,2,3* Stephen J. Kent3,4
Stephen J. Kent3,4Passive immunotherapies have been used to treat severe respiratory infections for over a century, with convalescent blood products from recovered individuals given to patients with influenza-related pneumonia as long ago as the Spanish flu pandemic. However, passive immunotherapy with convalescent plasma or hyperimmune intravenous immunoglobulin (hIVIG) has not provided unequivocal evidence of a clinical benefit for severe respiratory infections including influenza and COVID-19. Efficacy trials, primarily conducted in late-stage disease, have demonstrated inconsistent efficacy and clinical benefit for hIVIG treatment of severe respiratory infections. To date, most serological analyses of convalescent plasma and hIVIG trial samples have focused on the measurement of neutralizing antibody titres. There is, however, increasing evidence that baseline antibody levels and extra-neutralizing antibody functions influence the outcome of passive immunotherapy in humans. In this perspective, findings from convalescent plasma and hIVIG trials for severe influenza, COVID-19 and respiratory syncytial virus (RSV) will be described. Clinical trial results will be discussed in the context of the potential beneficial and deleterious roles of antibodies with Fc-mediated effector functions, with a focus on natural killer cells and antibody-dependent cellular cytotoxicity. Overall, we postulate that treating respiratory viral infections with hIVIG represents a delicate balance between protection and immunopathology.
The field of passive immunisation, or transfer of antibody to confer temporary protection, began in 1890 when Behring and Kitasato showed that a rabbit could be protected from tetanus by injecting serum from an immunized animal (1, 2). This approach is applied to protect humans against life-threatening bacterial toxins (e.g. diphtheria) and for pre- and post-exposure prophylaxis against viral infections (e.g. rabies, hepatitis A and B), but it has shown mixed results for treatment of severe viral infections. Products for passive immunotherapy include convalescent plasma (CP), intravenous immunoglobulin (IVIG), hyperimmune IVIG (hIVIG) and monoclonal antibodies (mAbs) (3). CP is pooled from blood of patients who have recovered from infection. Both IVIG and hIVIG are concentrated immunoglobulin G (IgG) prepared from pooled human plasma. IVIG is derived from plasma pooled from thousands of healthy donors, whereas hIVIG is made with plasma pooled from donors with high levels of pathogen-specific antibody (recovered or recently vaccinated). Treatment with hIVIG presents several advantages over CP including control over antibody concentration and lower infusion volume. However, CP can be produced more quickly than hIVIG, which is desirable during a pandemic (2, 4). Passive immunotherapy for viral infections introduces antibodies with direct and indirect anti-viral functions to clear virus and infected cells.
Neutralization of virus by antibody fragment antigen-binding (Fab) domains prevents infection of cells by blocking viral attachment, fusion and entry. Blood products used in passive immunotherapy are rich in IgG, the major antibody isotype found in human plasma. The IgG fragment crystallizable (Fc) domain selectively binds to activating or inhibitory Fc gamma receptors (FcγR) on immune effector cells inducing anti-viral functions such as antibody-dependent cellular cytotoxicity (ADCC) and antibody dependent phagocytosis (ADP). Fc-mediated functions may be especially important in passive immunotherapy to promote clearance of virus-infected cells and extracellular virus in an infected host (Figure 1, left box). However, antibody Fc functions can also result in antibody-dependent enhancement (ADE) of viral infection or disease.
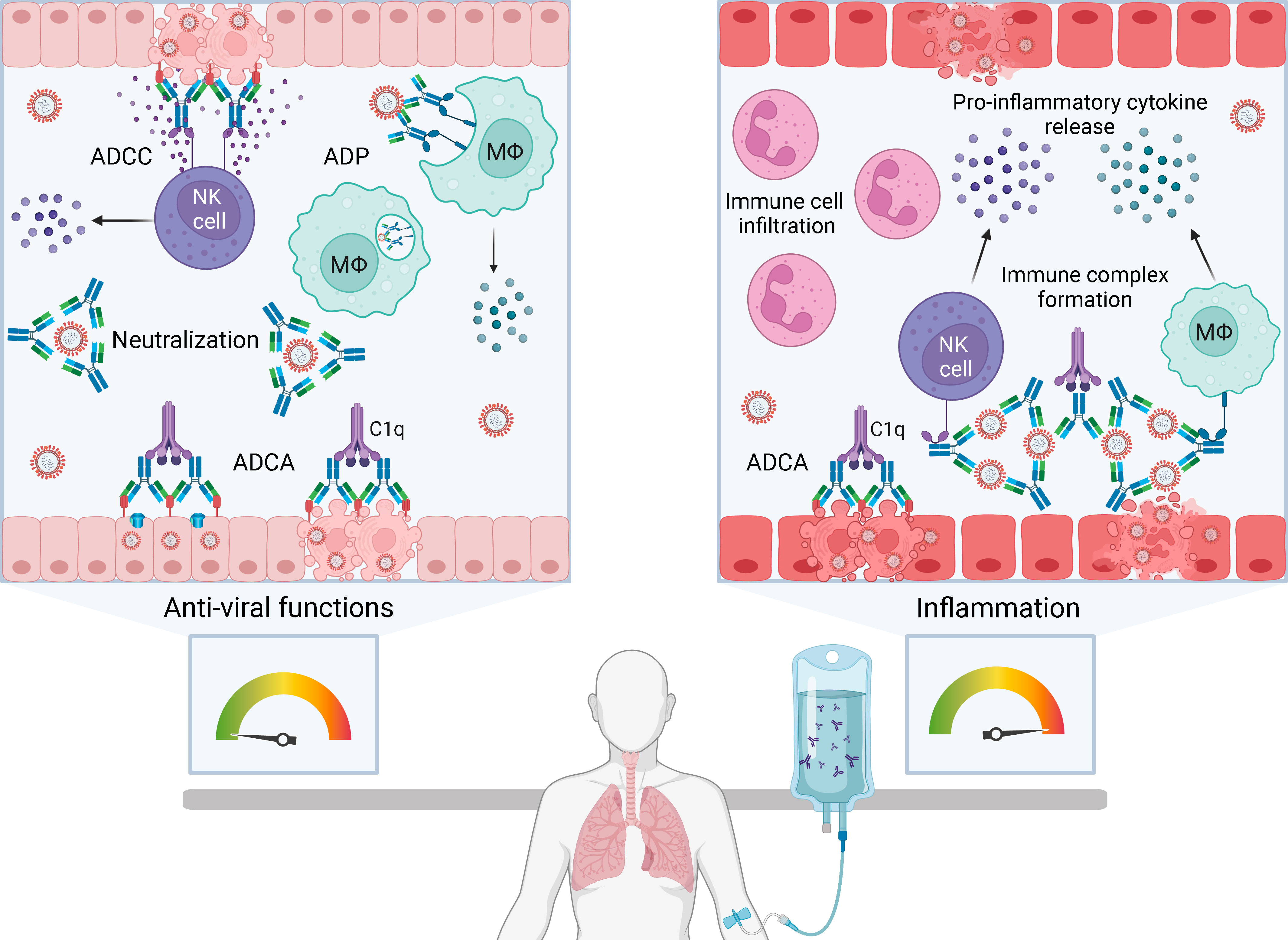
Figure 1 The balance between anti-viral functions and inflammation with passive immunotherapy for severe human respiratory viral infections. Passive immunotherapies introduce exogenous antibody that can mediate an array of protective anti-viral functions (left box) but also have the potential to cause immunopathology by enhancing pathological inflammation (right box). Antibody-based therapies can directly neutralize extracellular virus and induce Fc-mediated functions including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent phagocytosis (ADP) and antibody-dependent complement activation (ADCA). ADCC is initiated by FcγRIIIa on natural killer (NK) cells or macrophages (Mϕ) engaging Fc domains of immunoglobulin G (IgG) bound to viral antigens on infected cells. Multimeric engagement of FcγRIIIa on NK cells triggers release of cytolytic granules resulting in apoptosis of virus-infected cells and release of pro-inflammatory cytokines and chemokines, including interferon gamma (IFNγ) and macrophage inflammatory proteins (MIP1α and MIP1β). ADP is the uptake of antibody-opsonised virus or infected cells by phagocytes expressing FcγRI and FcγRIIa. ADCA is induced when C1q binds to the Fc domain of antibodies bound to virions or virus-infected cells, which can lead to lysis via membrane attack complex insertion. Formation and deposition of immune complexes can drive inflammation through FcγR-dependent activation of immune cells, such as NK cells, macrophages and neutrophils, leading to the production of high levels of pro-inflammatory cytokines. Immune complexes can also initiate ADCA resulting in anaphylatoxin release and leukocyte infiltration. Created with BioRender.com.
Two mechanisms of ADE have been described: extrinsic and intrinsic. Extrinsic ADE increases viral entry into phagocytes through FcγRIIa-dependent uptake of antibody-opsonised virus. Non-neutralizing antibody binds virus (e.g. dengue) shuttling it into macrophages that become productively infected (5–7). Intrinsic ADE involves Fc-mediated functions triggering a strong inflammatory response (8). An example of intrinsic ADE is the formation of immune complexes from non-neutralizing antibodies, leading to Fc-mediated activation of immune cells and complement resulting in anaphylatoxin release and leukocyte infiltration (Figure 1, right box) (6, 9, 10). Intrinsic ADE can promote inflammation in the respiratory tract, airway obstruction and in serious cases, acute respiratory distress syndrome (ARDS) (9–12). Whether passive immunotherapy contributes to intrinsic ADE during viral infection is unclear. IVIG has immunomodulatory and anti-inflammatory properties used to treat autoimmune and inflammatory disorders. It is postulated that CP and hIVIG may have immunomodulatory effects that blunt inflammation (13), but this is not well understood.
A systematic review and meta-analysis of the effectiveness of convalescent blood products for severe influenza and severe acute respiratory syndrome (SARS) concluded that CP may reduce mortality, but well-designed trials were necessary to establish efficacy (14). Without compelling evidence of efficacy, the high cost of treating viral infections with passive immunotherapy may present a significant limitation to its clinical use. Herein, we describe findings from CP and hIVIG trials for influenza, COVID-19 and respiratory syncytial virus (RSV) relating to potential beneficial and harmful roles for Fc-mediated antibody functions. We also highlight possible contributions of ADCC and natural killer (NK) cells to the equilibrium between protection and pathology when using passive immunotherapy to treat respiratory viruses.
Historical studies show that passive immunotherapy with convalescent blood products may be an effective treatment for severe influenza. During the Spanish flu pandemic, influenza-convalescent blood products were reported to reduce morbidity and mortality in patients with influenza-associated pneumonia and ARDS (15). A meta-analysis of 8 historical studies and 1703 patients examined the impact of transfusing influenza-convalescent whole blood, plasma or serum on mortality from Spanish influenza complicated by pneumonia (16). The overall crude case-fatality rate was lower for treated patients (16%) compared to untreated controls (37%; p < 0.001). Patients who received treatment earlier, within 4 days of pneumonia, had reduced overall crude case-fatality rates (19%) relative to patients who received later treatment (59%; p <0.001) (16). This subgroup analysis highlights that timing of treatment is critical. When administered early in the course of infection convalescent blood products reduced fatalities, but if received in later stages of disease, case-fatality rates were higher than in untreated controls. CP therapy also decreased respiratory tract viral load, serum cytokine levels and mortality (20% treated vs. 54.8% untreated; p = 0.01) in patients with severe pandemic H1N1 2009 (H1N1pdm09) influenza (17). Convalescent blood products reduced mortality from pandemic H1N1 influenza (16, 17), if given early in the course of disease, but the mechanisms underpinning this survival benefit remain largely unexplored.
A more recent randomised phase II clinical trial (n=98) found that immune plasma [hemagglutination inhibition (HAI)≥80] was safe but did not show a clear treatment benefit for severe influenza A or B based on respiratory normalisation by day 28 (18). A larger phase III clinical trial found that high-titre immune plasma (HAI≥80) conferred no benefit over low-titre plasma (HAI ≤ 10) for severe seasonal influenza A, demonstrating that HAI titre alone does not determine clinical efficacy of CP (19). In both trials, participants had a median of 3-4 days of influenza illness before enrolment. Extra-neutralizing Fc functions, including NK cell activation and ADCC, were not measured in CP trials and their impact on efficacy is unknown.
Inconclusive results for CP led to renewed interest in hIVIG therapy for severe influenza. A small (n=35) randomised controlled trial compared hIVIG, prepared from H1N1pdm09 CP, to IVIG prepared before 2009 for hospitalised adults with influenza (4). The hIVIG had high neutralizing antibody (NAb) titres (≥640) against the H1N1pdm09 virus, and IVIG had no detectable NAbs against this virus. Respiratory viral loads were lower in the hIVIG treated group at days 5 and 7 post-infusion, but mortality and length of hospital stay were not significantly different between groups (4). A subgroup analysis of subjects who received hIVIG or IVIG within 5 days of symptom onset showed that hIVIG was the only factor that independently reduced mortality. In contrast, all 5 subjects who received hIVIG later than 5 days after symptom onset died, while all 7 subjects treated with IVIG at this later timepoint survived. This demonstrated that early administration of hIVIG conferred a survival benefit but introduced the possibility of deleterious effects if given later in the course of influenza. Investigators proposed that hIVIG had anti-viral and immunomodulatory activities including neutralization, ADCC, blocking of IL-1a-dependent leukocyte recruitment and decreased cytokine mRNA levels. However, the high mortality rate in patients who received hIVIG late in the course of disease, and possible underlying mechanisms, were not addressed.
A phase III clinical trial (n=308) used a hIVIG called Flu-IVIG to treat patients hospitalised with severe seasonal influenza A or B within 7 days of symptom onset (20). The Flu-IVIG had no clinical benefit for patients with severe influenza A, despite causing a large rise in HAI titre. However, Flu-IVIG showed a significant treatment benefit for influenza B. Flu-IVIG antibodies targeting influenza B virus had a higher affinity than antibodies against influenza A virus, leading to slower dissociation of immune complexes (20). Since immune complex functions are driven by interactions with FcγRs on effector cells, we undertook a detailed serological analysis of Flu-IVIG clinical samples to measure extra-neutralizing antibody Fc functions (21). We showed that influenza A-specific FcγRIIa- and FcγRIIIa-binding antibody, as well as antibody-dependent NK cell activation, were associated with worse outcomes in patients with severe seasonal influenza A at day 5 and 7 post-infusion. Flu-IVIG also worsened the odds of a favourable clinical outcome in influenza A patients with low pre-infusion levels of FcγR-binding antibody compared to placebo. The opposite effect was observed for severe influenza B, with a trend towards higher influenza B-specific FcγR-binding antibody being associated with better outcomes and Flu-IVIG therapy improving the odds of a favourable outcome in patients with low FcγR-binding antibody at baseline (21). Overall, Fc-mediated antibody functions appeared to be either deleterious or beneficial depending on the infecting influenza virus genotype and host immune factors including pre-existing immunity.
A growing body of work supports a potential protective role for influenza-specific ADCC (22–26), but pre-clinical and observational studies suggest that Fc-mediated functions can also cause immunopathology. Humans with more severe influenza symptoms have higher HA-specific ADCC activity (25, 27). Elevated levels of non-neutralizing antibodies and pathogenic immune complexes were identified in patients with severe and fatal infections with H1N1pdm09 virus (28, 29). Mice vaccinated with an ADCC epitope had increased perforin and inflammatory cytokine levels in the lungs, greater alveolar damage and higher mortality following H1N1pdm09 infection compared with control mice (8), suggesting that ADCC may increase pulmonary inflammation and leukocyte infiltration. A modest decrease in viral load was observed in vaccinated mice, but any protective effect was outweighed by inflammatory lung damage. Higher mortality with late administration of influenza-CP or hIVIG was independently reported across trials and meta-analyses (4, 16). Exogenous antibody introduced late in the course of influenza illness, after an endogenous antibody response has been mounted and virus replication is decreasing, may promote formation of inflammatory immune complexes. In this case, Fc-mediated functions could be detrimental by enhancing immune cell activation and inflammation without providing any significant benefit for viral clearance. Findings from the Flu-IVIG trial indicate that hIVIG may not be an effective therapy for influenza A, given the association between FcγR-binding antibody and poorer outcomes (20, 21). Conversely, hIVIG demonstrated a treatment benefit for influenza B and larger trials should be undertaken. Paradoxical hIVIG results show that a complex balance between Fc-mediated anti-viral functions and pathological inflammation may exist when treating severe influenza with passive immunotherapies.
CP therapies were tested in subjects with mild and severe COVID-19, with few showing any clinical benefit (30–37). Transfusion of CP with high titres of anti-SARS-CoV-2 IgG decreased the risk of death in COVID-19 patients hospitalised without mechanical ventilation, compared to patients who received low-titre plasma (32). Another trial reported a treatment benefit for CP in preventing progression to death and ventilation in patients hospitalised with COVID-19 at 28 days post-infusion (36). Conversely, large randomised controlled trials of COVID-19 CP showed no difference in mortality or other measures of outcome between treated and control groups (33–35, 37), with the caveat that treatment occurred late in the course of disease (median 8-9 days after symptom onset). Therapeutic mAbs were effective against pre-Omicron SARS-CoV-2 variants when administered early, but not late, in the course of disease. A recent study showed that NAb titre is a key determinant of therapeutic efficacy, but CP and hIVIG fell below the threshold for high level protection (38, 39). CP therapy failed to provide a clear clinical benefit for COVID-19 but yielded important insights into potential protective mechanisms.
Anti-SARS-CoV-2 spike protein (S) IgG and NAb titre were the main serological parameters measured in COVID-19 CP trials, but ADCC and antibody-dependent NK cell activation were explored. The CP for COVID-19 Respiratory Illness (CONCOR-1) trial found that antibody content of CP modified its therapeutic effect. High ADCC activity in CP was independently associated with lower risk of intubation and death 30 days post-infusion (35), suggesting that enhanced ADCC may provide a therapeutic benefit. This is consistent with findings in animal models and severe COVID-19 cohorts that showed a protective role for Fc-mediated functions in SARS-CoV-2 infection (40–45). A small (n=80) study showed that CP blunted inflammatory S-specific antibody features, like C1q- and FcγRIIa-binding, while enhancing nucleocapsid (N)-specific antibody responses (13). Patients who received the greatest benefit from COVID-19 CP therapy had the lowest baseline Fc-functionality, including low ADP and antibody-dependent NK cell macrophage inflammatory protein 1β (MIP1β) production. Interestingly, N-specific antibody-dependent NK cell activation and MIP1β production were the features most strongly associated with better clinical outcomes, despite not being enriched in the treatment group. Ullah et al. showed that robust Fc functions in CP protected mice from lethal SARS-CoV-2 infection (46). Together, these findings suggest that CP efficacy against COVID-19 may partially depend on Fc-mediated functions like NK cell activation and ADCC.
Standardised hIVIG preparations with higher concentrations of anti-SARS-CoV-2 antibody may overcome CP variability and improve therapeutic efficacy. A small trial reported that COVID-19 hIVIG was safe, increased survival and reduced the risk of disease progression in subjects severely or critically ill with ARDS (47). A phase III clinical trial (n=593) of hIVIG showed no efficacy in patients hospitalised with COVID-19 without end-stage organ failure. The hIVIG was administered late in the course of infection, a median of 8 days after symptom onset (48). Interestingly, hIVIG safety was altered by the presence of endogenous SARS-CoV-2 antibody at randomisation. In the hIVIG group, there was an increased risk of adverse events at day 7 post-infusion for subjects who were NAb-positive at baseline, compared to those who were NAb-negative. The underlying cause of adverse events in hIVIG-treated NAb-positive subjects remains to be elucidated, but hIVIG may be harmful in certain subgroups of hospitalised COVID-19 patients and beneficial in others (48), as we observed for influenza (21). This highlights the need to analyse FcγR-binding and ADCC in these clinical samples to uncover associations with outcome.
Antibody-dependent NK cell activation and ADCC have been linked to SARS-CoV-2 protection in CP trials and cohort studies (35, 44, 45), but Fc-mediated functions may drive inflammation in severe COVID-19. Antibody features, such as S-specific C1q- and FcγRIIa-binding, were associated with more severe disease (13). Antibody-dependent complement activation is enhanced in patients with severe COVID-19 and may increase inflammatory lung injury (49, 50). FcγR-dependent uptake of SARS-CoV-2 by macrophages/monocytes results in inflammatory cell death and systemic inflammation (51), and FcγR-mediated activation of monocytes by anti-SARS-CoV-2 IgG promotes tumer necrosis factor alpha (TNFα) release (49). Severe COVID-19 is associated with an afucosylated anti-S antibody signature (52, 53). Afucosylated IgG has a high affinity for FcγRIIIa and is a potent mediator of ADCC, indicating that antibody-dependent NK cell activation and ADCC may be exacerbating COVID-19 disease. Patients with worse COVID-19 symptoms have elevated serum ADCC activity, and the high affinity variant of FcγRIIIa (158-V/V) is overrepresented in hospitalised and deceased COVID-19 patients (54, 55). There is no unequivocal evidence of ADE of human SARS-CoV-2 infection, but immune complexes formed with inflammatory anti-S antibodies may over-activate complement and immune cells by binding to C1q and FcγRs, contributing to COVID-19 pathogenesis in some individuals (56).
Collectively, CP and hIVIG have not demonstrated significant impact on COVID-19 mortality or clinical outcome. However, these trials revealed key information about protective and pathological features of SARS-CoV-2 antibodies. Further insights can be gained into the balance of harmful and beneficial antibody profiles in COVID-19 by measuring antibody Fc-functionality in hIVIG trials.
A hIVIG (RSV-IVIG) and mAbs against the RSV fusion (F) protein (palivizumab and nirsevimab) have been approved for prophylactic use against RSV in high-risk infants and children (57–59). RSV-IVIG was marketed from 1996-2003 for RSV prevention in children younger than 2 years of age with a history of prematurity or chronic lung disease (60). Combination therapy with RSV-IVIG and ribavirin improved outcomes in bone marrow transplant recipients and reduced lung viral load in RSV-infected cotton rats compared to ribavirin alone (61, 62). Prophylactic administration of RSV-IVIG to children and infants with bronchopulmonary dysplasia and/or prematurity reduced the incidence of lower respiratory tract infections and hospitalisations (63, 64), but there is no conclusive evidence to support a role for RSV-IVIG in treatment of severe RSV (65, 66). RSV-IVIG was prone to unpredictable efficacy profiles and was replaced by palivizumab (58), which neutralizes RSV with a potency ~50 times greater than RSV-IVIG (60, 67). Palivizumab and nirsevimab are administered intramuscularly and indicated for prophylaxis only (58, 59).
The Fc-mediated functions of RSV-IVIG have not been assessed, but detection of RSV-specific ADCC in the respiratory tract of infants during natural infection was first reported over 40 years ago (68). RSV-specific ADCC activity was independent of symptom severity and waned rapidly compared to neutralization (68, 69). Antibodies targeting RSV attachment (G) protein are more efficient inducers of ADCC compared to anti-RSV F protein antibodies (70, 71). Evidence of a protective role for ADCC in animal models of RSV is limited. Fab fragments derived from anti-RSV G protein mAbs did not reduce viral load in RSV-infected mice, but corresponding mAbs with intact Fc domains conferred protection (72).
In the 1960s, a formalin-inactivated RSV vaccine caused vaccine-induced ADE in children (73, 74). Children were exposed to RSV infection shortly after vaccination and developed enhanced respiratory disease (ERD), which was fatal in two cases (73, 75). Follow-up studies revealed that non-neutralizing antibodies formed immune complexes that deposited in the airway activating complement and pro-inflammatory cytokine release (9, 10). Serum antibody and anti-RSV mAbs showed ADE of RSV infection in FcγR-expressing monocytic cell lines (76–78), and binding to FcγRs appeared to reduce neutralizing activity of anti-RSV mAbs except palivizumab (79). RSV-IVIG did not lead to reported cases of ERD, but a history of RSV vaccine-induced ADE is damaging. Future antibody therapies or vaccines for RSV must be evaluated for harmful Fc-mediated effects and characterised to identify protective and inflammatory antibody features.
Prophylactic use of NAbs is efficacious for targeted protection of at-risk individuals, but antibody-based therapies are being widely tested for treatment of existing viral infections. Passive immunotherapy for severe respiratory virus infection is most effective if administered early in the course of disease, within 4-5 days of symptom onset. At early stages of infection, infused antibodies may enhance anti-viral activity, including neutralization and Fc functions, to clear virus and infected cells. In late-stage disease, Fc-mediated functions may contribute to inflammation outweighing any beneficial effects from viral clearance. High NAb titre correlates with therapeutic efficacy, but the level of Fc functions required for protection with CP or hIVIG remains uncharacterised. Moving forward, in-depth serological analyses, along with other measures of immune function including T cell-mediated immunity, are required to dissect the utility or harm of Fc-mediated antibody functions when treatment is received relatively late in the course of disease.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
HV: Conceptualization, Writing – original draft. SK: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Australian National Health and Medical Research Council (NHMRC) and James Cook University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kaufmann SHE. Remembering Emil von Behring: from tetanus treatment to antibody cooperation with phagocytes. mBio (2017) 8(1):e00117-17. doi: 10.1128/mbio.00117-17
2. Subbarao K, Mordant F, Rudraraju R. Convalescent plasma treatment for COVID-19: tempering expectations with the influenza experience. Eur J Immunol (2020) 50(10):1447–53. doi: 10.1002/eji.202048723
3. Beigel JH. Polyclonal and monoclonal antibodies for the treatment of influenza. Curr Opin Infect Dis (2018) 31(6):527–34. doi: 10.1097/qco.0000000000000499
4. Hung IFN, To KKW, Lee C-K, Lee K-L, Yan W-W, Chan K, et al. Hyperimmune iv immunoglobulin treatment: A multicenter double-blind randomized controlled trial for patients with severe 2009 influenza a(H1n1) infection. Chest (2013) 144(2):464–73. doi: 10.1378/chest.12-2907
5. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol (2020) 5(10):1185–91. doi: 10.1038/s41564-020-00789-5
6. Thomas S, Smatti MK, Ouhtit A, Cyprian FS, Almaslamani MA, Thani AA, et al. Antibody-dependent enhancement (Ade) and the role of complement system in disease pathogenesis. Mol Immunol (2022) 152:172–82. doi: 10.1016/j.molimm.2022.11.010
7. Halstead S, O’Rourke E. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med (1977) 146(1):201–17. doi: 10.1084/jem.146.1.201
8. Ye Z-W, Yuan S, Poon K-M, Wen L, Yang D, Sun Z, et al. Antibody-dependent cell-mediated cytotoxicity epitopes on the hemagglutinin head region of pandemic H1n1 influenza virus play detrimental roles in H1n1-infected mice. Front Immunol (2017) 8:317. doi: 10.3389/fimmu.2017.00317
9. Polack FP, Hoffman SJ, Crujeiras G, Griffin DE. A role for nonprotective complement-fixing antibodies with low avidity for measles virus in atypical measles. Nat Med (2003) 9(9):1209–13. doi: 10.1038/nm918
10. Polack FP, Teng MN, L.Collins P, Prince GA, Exner M, Regele H, et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med (2002) 196(6):859–65. doi: 10.1084/jem.20020781
11. Nader PR, Horwitz MS, Rousseau J. Atypical exanthem following exposure to natural measles: eleven cases in children previously inoculated with killed vaccine. J Pediatr (1968) 72(1):22–8. doi: 10.1016/S0022-3476(68)80396-8
12. Graham BS. Vaccines against respiratory syncytial virus: the time has finally come. Vaccine (2016) 34(30):3535–41. doi: 10.1016/j.vaccine.2016.04.083
13. Herman JD, Wang C, Burke JS, Zur Y, Compere H, Kang J, et al. Nucleocapsid-specific antibody function is associated with therapeutic benefits from COVID-19 convalescent plasma therapy. Cell Rep Med (2022) 3(11):100811. doi: 10.1016/j.xcrm.2022.100811
14. Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw F-M, Lim WS, et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: A systematic review and exploratory meta-analysis. J Infect Dis (2014) 211(1):80–90. doi: 10.1093/infdis/jiu396
15. Luke TC, Casadevall A, Watowich SJ, Hoffman SL, Beigel JH, Burgess TH. Hark back: passive immunotherapy for influenza and other serious infections. Crit Care Med (2010) 38:e66–73. doi: 10.1097/CCM.0b013e3181d44c1e
16. Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for spanish influenza pneumonia: A future H5n1 treatment? Ann Intern Med (2006) 145(8):599–609. doi: 10.7326/0003-4819-145-8-200610170-00139
17. Hung IF, To KK, Lee CK, Lee KL, Chan K, Yan WW, et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza a (H1n1) 2009 virus infection. Clin Infect Dis (2011) 52(4):447–56. doi: 10.1093/cid/ciq106
18. Beigel JH, Tebas P, Elie-Turenne M-C, Bajwa E, Bell TE, Cairns CB, et al. Immune plasma for the treatment of severe influenza: an open-label, multicentre, phase 2 randomised study. Lancet Respir Med (2017) 5(6):500–11. doi: 10.1016/S2213-2600(17)30174-1
19. Beigel JH, Aga E, Elie-Turenne M-C, Cho J, Tebas P, Clark CL, et al. Anti-influenza immune plasma for the treatment of patients with severe influenza A: A randomised, double-blind, phase 3 trial. Lancet Respir Med (2019) 7(11):941–50. doi: 10.1016/S2213-2600(19)30199-7
20. Davey RT Jr., Fernández-Cruz E, Markowitz N, Pett S, Babiker AG, Wentworth D, et al. Anti-influenza hyperimmune intravenous immunoglobulin for adults with influenza a or B infection (Flu-IVIG): A double-blind, randomised, placebo-controlled trial. Lancet Respir Med (2019) 7(11):951–63. doi: 10.1016/S2213-2600(19)30253-X
21. Vanderven HA, Wentworth DN, Han WM, Peck H, Barr IG, Davey RT Jr., et al. Understanding the treatment benefit of hyperimmune anti-influenza intravenous immunoglobulin (Flu-IVIG) for severe human influenza. JCI Insight (2023) 8(14):e167464. doi: 10.1172/jci.insight.167464
22. DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk–specific antibodies require fcγr interactions for protection against influenza virus in vivo. Nat Med (2014) 20(2):143–51. doi: 10.1038/nm.3443
23. DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require fc receptor engagement for in vivo protection. J Clin Invest (2016) 126(2):605–10. doi: 10.1172/jci84428
24. Henry Dunand Carole J, Leon Paul E, Huang M, Choi A, Chromikova V, Ho Irvin Y, et al. Both neutralizing and non-neutralizing human H7n9 influenza vaccine-induced monoclonal antibodies confer protection. Cell Host Microbe (2016) 19(6):800–13. doi: 10.1016/j.chom.2016.05.014
25. Jegaskanda S, Luke C, Hickman HD, Sangster MY, Wieland-Alter WF, McBride JM, et al. Generation and protective ability of influenza virus–specific antibody-dependent cellular cytotoxicity in humans elicited by vaccination, natural infection, and experimental challenge. J Infect Dis (2016) 214(6):945–52. doi: 10.1093/infdis/jiw262
26. Liu Y, Tan HX, Koutsakos M, Jegaskanda S, Esterbauer R, Tilmanis D, et al. Cross-lineage protection by human antibodies binding the influenza B hemagglutinin. Nat Commun (2019) 10(1):324. doi: 10.1038/s41467-018-08165-y
27. Vanderven HA, Ana-Sosa-Batiz F, Jegaskanda S, Rockman S, Laurie K, Barr I, et al. What lies beneath: antibody dependent natural killer cell activation by antibodies to internal influenza virus proteins. EBioMedicine (2016) 8:277–90. doi: 10.1016/j.ebiom.2016.04.029
28. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, et al. Severe pandemic 2009 H1n1 influenza disease due to pathogenic immune complexes. Nat Med (2011) 17(2):195–9. doi: 10.1038/nm.2262
29. To KKW, Zhang AJX, Hung IFN, Xu T, Ip WCT, Wong RTY, et al. High titer and avidity of nonneutralizing antibodies against influenza vaccine antigen are associated with severe influenza. Clin Vaccine Immunol (2012) 19(7):1012–8. doi: 10.1128/CVI.00081-12
30. Libster R, Pérez Marc G, Wappner D, Coviello S, Bianchi A, Braem V, et al. Early high-titer plasma therapy to prevent severe COVID-19 in older adults. N Engl J Med (2021) 384(7):610–8. doi: 10.1056/NEJMoa2033700
31. Korley FK, Durkalski-Mauldin V, Yeatts SD, Schulman K, Davenport RD, Dumont LJ, et al. Early convalescent plasma for high-risk outpatients with COVID-19. N Engl J Med (2021) 385(21):1951–60. doi: 10.1056/NEJMoa2103784
32. Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, et al. Convalescent plasma antibody levels and the risk of death from COVID-19. N Engl J Med (2021) 384(11):1015–27. doi: 10.1056/NEJMoa2031893
33. Simonovich VA, Burgos Pratx LD, Scibona P, Beruto MV, Vallone MG, Vázquez C, et al. A randomized trial of convalescent plasma in COVID-19 severe pneumonia. N Engl J Med (2020) 384(7):619–29. doi: 10.1056/NEJMoa2031304
34. Abani O, Abbas A, Abbas F, Abbas M, Abbasi S, Abbass H, et al. Convalescent plasma in patients admitted to hospital with COVID-19 (Recovery): A randomised controlled, open-label, platform trial. Lancet (2021) 397(10289):2049–59. doi: 10.1016/S0140-6736(21)00897-7
35. Bégin P, Callum J, Jamula E, Cook R, Heddle NM, Tinmouth A, et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat Med (2021) 27(11):2012–24. doi: 10.1038/s41591-021-01488-2
36. Avendaño-Solá C, Ramos-Martínez A, Muñez-Rubio E, Ruiz-Antorán B, Malo de Molina R, Torres F, et al. A multicenter randomized open-label clinical trial for convalescent plasma in patients hospitalized with COVID-19 pneumonia. J Clin Invest (2021) 131(20):e152740. doi: 10.1172/JCI152740
37. Janiaud P, Axfors C, Schmitt AM, Gloy V, Ebrahimi F, Hepprich M, et al. Association of convalescent plasma treatment with clinical outcomes in patients with COVID-19: A systematic review and meta-analysis. JAMA (2021) 325(12):1185–95. doi: 10.1001/jama.2021.2747
38. Stadler E, Burgess MT, Schlub TE, Khan SR, Chai KL, McQuilten ZK, et al. Monoclonal antibody levels and protection from COVID-19. Nat Commun (2023) 14(1):4545. doi: 10.1038/s41467-023-40204-1
39. Stadler E, Chai KL, Schlub TE, Cromer D, Polizzotto MN, Kent SJ, et al. Determinants of passive antibody effectiveness in SARS-CoV-2 infection. medRxiv (2022). doi: 10.1101/2022.03.21.22272672
40. Winkler ES, Gilchuk P, Yu J, Bailey AL, Chen RE, Chong Z, et al. Human neutralizing antibodies against SARS-CoV-2 require intact fc effector functions for optimal therapeutic protection. Cell (2021) 184(7):1804–20.e16. doi: 10.1016/j.cell.2021.02.026
41. Suryadevara N, Shrihari S, Gilchuk P, VanBlargan LA, Binshtein E, Zost SJ, et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell (2021) 184(9):2316–31.e15. doi: 10.1016/j.cell.2021.03.029
42. Ullah I, Prévost J, Ladinsky MS, Stone H, Lu M, Anand SP, et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require fc function for optimal efficacy. Immunity (2021) 54(9):2143–58.e15. doi: 10.1016/j.immuni.2021.08.015
43. Schäfer A, Muecksch F, Lorenzi JCC, Leist SR, Cipolla M, Bournazos S, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J Exp Med (2020) 218(3):e20201993. doi: 10.1084/jem.20201993
44. Brunet-Ratnasingham E, Anand SP, Gantner P, Dyachenko A, Moquin-Beaudry G, Brassard N, et al. Integrated immunovirological profiling validates plasma SARS-CoV-2 rna as an early predictor of COVID-19 mortality. Sci Adv (2021) 7(48):eabj5629. doi: 10.1126/sciadv.abj5629
45. Zohar T, Loos C, Fischinger S, Atyeo C, Wang C, Slein MD, et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell (2020) 183(6):1508–19.e12. doi: 10.1016/j.cell.2020.10.052
46. Ullah I, Beaudoin-Bussières G, Symmes K, Cloutier M, Ducas E, Tauzin A, et al. The fc-effector function of COVID-19 convalescent plasma contributes to SARS-CoV-2 treatment efficacy in mice. Cell Rep Med (2023) 4(1):100893. doi: 10.1016/j.xcrm.2022.100893
47. Ali S, Uddin SM, Shalim E, Sayeed MA, Anjum F, Saleem F, et al. Hyperimmune anti-COVID-19 IVIG (C-IVIG) treatment in severe and critical COVID-19 patients: A phase I/ii randomized control trial. eClinicalMedicine (2021) 36:100926. doi: 10.1016/j.eclinm.2021.100926
48. Polizzotto MN, Nordwall J, Babiker AG, Phillips A, Vock DM, Eriobu N, et al. Hyperimmune immunoglobulin for hospitalised patients with COVID-19 (Itac): A double-blind, placebo-controlled, phase 3, randomised trial. Lancet (2022) 399(10324):530–40. doi: 10.1016/S0140-6736(22)00101-5
49. Jarlhelt I, Nielsen SK, Jahn CXH, Hansen CB, Pérez-Alós L, Rosbjerg A, et al. SARS-CoV-2 antibodies mediate complement and cellular driven inflammation. Front Immunol (2021) 12:767981. doi: 10.3389/fimmu.2021.767981
50. Ma L, Sahu SK, Cano M, Kuppuswamy V, Bajwa J, McPhatter JN, et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci Immunol (2021) 6(59):eabh2259. doi: 10.1126/sciimmunol.abh2259
51. Junqueira C, Crespo Â, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, et al. Fcγr-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature (2022) 606(7914):576–84. doi: 10.1038/s41586-022-04702-4
52. Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated igg characterizes enveloped viral responses and correlates with COVID-19 severity. Science (2021) 371(6532):eabc8378. doi: 10.1126/science.abc8378
53. Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory igg fc structures in patients with severe COVID-19. Nat Immunol (2021) 22(1):67–73. doi: 10.1038/s41590-020-00828-7
54. Yu Y, Wang M, Zhang X, Li S, Lu Q, Zeng H, et al. Antibody-dependent cellular cytotoxicity response to SARS-CoV-2 in COVID-19 patients. Signal Transduct Target Ther (2021) 6(1):346. doi: 10.1038/s41392-021-00759-1
55. Vietzen H, Danklmaier V, Zoufaly A, Puchhammer-Stöckl E. High-Affinity Fcγriiia Genetic Variants and Potent Nk cell-Mediated Antibody-Dependent Cellular Cytotoxicity (Adcc) Responses Contributing to Severe COVID-19. Genet Med (2022) 24(7):1449–58. doi: 10.1016/j.gim.2022.04.005
56. Li D, Edwards RJ, Manne K, Martinez DR, Schäfer A, Alam SM, et al. In vitro and In vivo Functions of SARS-CoV-2 Infection-Enhancing and Neutralizing Antibodies. Cell (2021) 184(16):4203–19.e32. doi: 10.1016/j.cell.2021.06.021
57. Robinson RF, Nahata MC. Respiratory syncytial virus (Rsv) immune globulin and palivizumab for prevention of rsv infection. Am J Health Syst Pharm (2000) 57(3):259–64. doi: 10.1093/ajhp/57.3.259
58. Diethelm-Varela B, Soto JA, Riedel CA, Bueno SM, Kalergis AM. New developments and challenges in antibody-based therapies for the respiratory syncytial virus. Infect Drug Resist (2023) 16:2061–74. doi: 10.2147/idr.S379660
59. Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, et al. Nirsevimab for prevention of rsv in healthy late-preterm and term infants. N Engl J Med (2022) 386(9):837–46. doi: 10.1056/NEJMoa2110275
60. Wasserman RL, Greener BN, Mond J. Ri-002, an intravenous immunoglobulin containing high titer neutralizing antibody to rsv and other respiratory viruses for use in primary immunodeficiency disease and other immune compromised populations. Expert Rev Clin Immunol (2017) 13(12):1107–19. doi: 10.1080/1744666x.2017.1389647
61. Gruber WC, Wilson SZ, Throop BJ, Wyde PR. Immunoglobulin administration and ribavirin therapy: efficacy in respiratory syncytial virus infection of the cotton rat. Pediatr Res (1987) 21(3):270–4. doi: 10.1203/00006450-198703000-00013
62. Whimbey E, Champlin RE, Englund JA, Mirza NQ, Piedra PA, Goodrich JM, et al. Combination therapy with aerosolized ribavirin and intravenous immunoglobulin for respiratory syncytial virus disease in adult bone marrow transplant recipients. Bone Marrow Transplant (1995) 16(3):393–9.
63. Groothuis JR, Simoes E, Levin MJ, Hall CB, Long CE, Rodriguez WJ, et al. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. N Engl J Med (1993) 329(21):1524–30. doi: 10.1056/nejm199311183292102
64. The PREVENT Study Group. Reduction of respiratory syncytial virus hospitalization among premature infants and infants with bronchopulmonary dysplasia using respiratory syncytial virus immune globulin prophylaxis. Pediatrics (1997) 99(1):93–9. doi: 10.1542/peds.99.1.93
65. Fuller H, Del Mar C. Immunoglobulin treatment for respiratory syncytial virus infection. Cochrane Database Syst Rev (2006) 4:Cd004883. doi: 10.1002/14651858.CD004883.pub2
66. Sanders SL, Agwan S, Hassan M, van Driel ML, Del Mar CB. Immunoglobulin treatment for hospitalised infants and young children with respiratory syncytial virus infection. Cochrane Database Syst Rev (2019) 8:CD009417. doi: 10.1002/14651858.CD009417.pub2
67. Eiland LS. Respiratory syncytial virus: diagnosis, treatment and prevention. J Pediatr Pharmacol Ther (2009) 14(2):75–85. doi: 10.5863/1551-6776-14.2.75
68. Kaul TN, Welliver RC, Ogra PL. Development of antibody-dependent cell-mediated cytotoxicity in the respiratory tract after natural infection with respiratory syncytial virus. Infect Immun (1982) 37(2):492–8. doi: 10.1128/iai.37.2.492-498.1982
69. Meguro H, Kervina M, Wright PF. Antibody-dependent cell-mediated cytotoxicity against cells infected with respiratory syncytial virus: characterization of in vitro and in vivo properties1. J Immunol (1979) 122(6):2521–6. doi: 10.4049/jimmunol.122.6.2521
70. Cortjens B, Yasuda E, Yu X, Wagner K, Claassen YB, Bakker AQ, et al. Broadly reactive anti-respiratory syncytial virus G antibodies from exposed individuals effectively inhibit infection of primary airway epithelial cells. J Virol (2017) 91(10):e02357-16. doi: 10.1128/jvi.02357-16
71. Gupta N, LeGoff J, Chamat S, Mercier-Delarue S, Touzelet O, Power UF, et al. Affinity-purified respiratory syncytial virus antibodies from intravenous immunoglobulin exert potent antibody-dependent cellular cytotoxicity. PloS One (2013) 8(7):e69390. doi: 10.1371/journal.pone.0069390
72. Miao C, Radu GU, Caidi H, Tripp RA, Anderson LJ, Haynes LM. Treatment with respiratory syncytial virus G glycoprotein monoclonal antibody or F(Ab′)2 components mediates reduced pulmonary inflammation in mice. J Gen Virol (2009) 90(5):1119–23. doi: 10.1099/vir.0.009308-0
73. Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine12. Am J Epidemiol (1969) 89(4):422–34. doi: 10.1093/oxfordjournals.aje.a120955
74. van Erp EA, Luytjes W, Ferwerda G, van Kasteren PB. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front Immunol (2019) 10:548. doi: 10.3389/fimmu.2019.00548
75. Polack FP, Alvarez-Paggi D, Libster R, Caballero MT, Blair RV, Hijano DR, et al. Fatal enhanced respiratory syncytial virus disease in toddlers. Sci Transl Med (2021) 13(616):eabj7843. doi: 10.1126/scitranslmed.abj7843
76. Gimenez HB, Keir HM, Cash P. In vitro enhancement of respiratory syncytial virus infection of U937 cells by human sera. J Gen Virol (1989) 70(1):89–96. doi: 10.1099/0022-1317-70-1-89
77. Krilov LR, Anderson LJ, Marcoux L, Bonagura VR, Wedgwood JF. Antibody-mediated enhancement of respiratory syncytial virus infection in two monocyte/macrophage cell lines. J Infect Dis (1989) 160(5):777–82. doi: 10.1093/infdis/160.5.777
78. Gimenez HB, Chisholm S, Dornan J, Cash P. Neutralizing and enhancing activities of human respiratory syncytial virus-specific antibodies. Clin Diagn Lab Immunol (1996) 3(3):280–6. doi: 10.1128/cdli.3.3.280-286.1996
Keywords: antibody-based therapy, passive immunotherapy, influenza virus, SARS-CoV-2, respiratory syncytial virus (RSV), antibody-dependent cellular cytotoxicity, natural killer cells
Citation: Vanderven HA and Kent SJ (2023) Fc-mediated functions and the treatment of severe respiratory viral infections with passive immunotherapy – a balancing act. Front. Immunol. 14:1307398. doi: 10.3389/fimmu.2023.1307398
Received: 04 October 2023; Accepted: 06 November 2023;
Published: 22 November 2023.
Edited by:
Ricciarda Galandrini, Sapienza University of Rome, ItalyReviewed by:
Veronica Ueckermann, University of Pretoria, South AfricaCopyright © 2023 Vanderven and Kent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hillary A. Vanderven, aGlsbGFyeS52YW5kZXJ2ZW5AamN1LmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.