 Susan L. Swain
Susan L. Swain- Department of Pathology, University of Massachusetts Chan Medical School, Worcester, MA, United States
Our recent studies reveal that the persistence, location, and amount of both antigen and signals that induce pathogen recognition responses determine the number of CD4 memory cells, the subsets that develop, their location, and hence their protective efficacy. Non-replicating vaccines provide antigen that is short-lived and generate low levels of only some memory subsets that are mostly restricted to secondary lymphoid tissue. In contrast, exposure to long-lived replicating viruses and bacteria provides high levels of diverse antigens in sites of infection and induces strong pathogen recognition signals for extended periods of time, resulting in much higher levels of memory cells of diverse subsets in both lymphoid and nonlymphoid sites. These include memory subsets with highly potent functions such as T follicular helpers and cytotoxic CD4 effectors at sites of infection, where they can most effectively combat the pathogen early after re-infection. These effectors also do not develop without antigen and pathogen recognition signals at the effector stage, and both subsets must receive these signals in the tissue sites where they will become resident. We postulate that this leads to a hierarchical structure of memory, with the strongest memory induced only by replicating pathogens. This paradigm suggests a likely roadmap for markedly improving vaccine design.
1 Introduction
Our lab has identified the mechanisms that generate long-lasting, potent CD4 memory responses that can protect us from the most dangerous pathogens. We have been humbled by how evolution has selected so many complex subsets of T-cell memory with diverse protective mechanisms and correspondingly complex pathways regulating their development (1). Here we discuss recent studies that bring into focus the mechanisms that drive the development of heterogenous subsets of memory at the effector phase of the CD4 response and what implications the signals required have for designing vaccines.
2 Background and recent studies
2.1 Generation of effector subsets from naïve CD4 T cells
Early studies focused on identifying the signals that are responsible for initiating the generation of effector cells from naïve CD4 T cells. Initiation of the naïve CD4 response requires cognate interactions between naive CD4 T cells with specialized antigen-presenting cells (APC) and is mediated by T-cell receptor recognition of peptide antigen bound to MHC-II and binding of costimulatory molecules on APC and T cells (2). The APC and CD4 T cells produce cytokines (IL-4, IL-6, IL-1, IL-2, and IFN-γ), which support the development of multiple CD4 effector subsets (e.g., Th0, Th1, Th2), themselves polarized to make distinct patterns of cytokines (3, 4). Viral infections induce mostly Th1 effectors, which participate in viral control, that once antigen is cleared, contract with a cohort differentiating to memory. These memory cells persist and are powerful mediators of protection (5, 6). They are the precursors of secondary effectors that clear viruses and provide future protection (6, 7).
CD4 T-cell memory responses to influenza A virus (IAV) infection are multifunctional, with distinct subsets mediating viral clearance, helping CD8 and B-cell responses, and producing cytokines that participate in viral control (4–7). Memory cells against influenza are found in secondary lymphoid organs (SLO), mediastinal draining lymph nodes, the spleen, and in infected tissues, including the lung (4–6). The CD4 memory subsets synergize with IAV-specific CD8 T and B cells (1) to provide strong, long-lasting protection against re-challenge with lethal doses of IAV against multiple IAV strains (7).
The process of generation of the initial CD4 effector subset (Th0, Th1, Th2) is well-defined at both cellular and molecular levels (3, 4). It can be readily modeled in vitro and requires only the initial signals from the recognition of APC and cytokines from CD4 and APC (2–4). However, more specialized subsets of CD4 T cells, such as T follicular helpers (TFH) (8), require further signals and have only been uncovered in more recent studies. Shane Crotty has defined the transcription factors needed for TFH differentiation, especially Bcl6 (8). We defined key mechanisms that generate functionally distinct effector CD4 subsets from the mostly Th1 effectors present 6 days after IAV infection. We compared the generation of cytotoxic CD4 T cells (ThCTL) (9), which are found in infected lungs, and clear MHC-II+ virus-infected cells and TFH that interact with B cells and drive differentiation in germinal centers to high-affinity antibody (Ab)-secreting cells (10). In contrast to initial effector subsets, which develop by the peak of the effector response 5–7 days postinfection (dpi) (6), TFH and cytotoxic ThCTL develop only 2–3 days later. Their development requires another round of multiple signals during the effector stage, from recognition of antigens and pathogens to becoming memory cells (10, 11). Generation of both requires these signals in the local tissues, where these CD4 effectors become tissue resident and subsequently give rise to memory cells (10, 11). We propose these requirements for high levels of antigen and pathogen-derived products and create an additional checkpoint at the effector stage, which determines their fate and the make-up of late effector and memory responses (10–12).
2.2 CD4 effector cells are susceptible to programmed and activation-induced cell death
Often, vaccines that are composed of inert antigens induce short-lived effector CD4 responses with narrow specificity and modest development of immune memory against viruses (13). In contrast, previous infections with the same viruses can induce life-long protective immunity (13). We reasoned that this may be because most CD4 effectors generated by nonviable vaccines undergo wholesale default programmed cell death when survival cytokines are withdrawn (14, 15). This limits memory generation. We found that in in vitro models of CD4 effector generation from naïve CD4 T cells with antigen and activated APC, CD4 effectors undergo default programmed cell death, but they could be rescued by the addition of IL-2 and TGF-β (16). Recently, we followed CD4 effector fate in vivo after influenza infection and analyzed the signals from infection responsible for rescuing effectors from default apoptosis so that they could become memory cells (17). The infection-induced CD4 effectors required signals from autocrine IL-2 to effectively differentiate into long-lived CD4 memory (18). CD4 memory generation was drastically reduced when CD4 effectors could not make IL-2, but could be restored by IL-2-anti-IL-2 complex (IL-2C) treatment at 5–7 days postinfection (18). Without IL-2C, the IL-2 had to be autocrine and made during the cognate interaction of CD4 effectors with APC. The in vivo-generated effectors that develop in wild-type mice in response to influenza make 100- to 1,000-fold more memory than effectors from IL-2-deficient mice (18).
2.3 An effector checkpoint dictates CD4 memory development
2.3.1 Role of antigen recognition
To further analyze what drives memory generation from effectors, we developed a sequential transfer model in which 6 dpi CD4 effectors were generated from naïve CD4 T-cell receptor (TCR) transgenic cells in first hosts infected with IAV. At 6 dpi, they were isolated and transferred to a second host in which we manipulated the availability of APC and IAV infection and monitored donor effector differentiation into memory. In the second host, recovery of almost all donor-derived memory required infection with IAV expressing the peptide seen by the TCR transgenic effector CD4 T cells, but could not be induced by IAV lacking that epitope (19). However, when we introduced APC activated with pathogen-recognition (PR) signals and pulsed with the peptide antigen, memory was fully restored (19). Memory generation depended on CD4 effector (autocrine) IL-2 production for 3 days beginning as effectors were generated (20), defining a checkpoint for memory. This reduced the programmed cell death of effectors and restored robust donor CD4 memory that protected the uninfected hosts from re-challenge (18–20). We concluded that, in order to survive and become memory cells, CD4 effectors generated by infection must again receive signals from cognate recognition and costimulation, resulting in autocrine IL-2, to which they respond and become memory.
We analyzed the impact of the avidity of TCR:peptide/MHC-II interaction at the checkpoint for memory generation. We developed a new CD4 TCR transgenic mouse that recognizes a dominant influenza nucleoprotein (NP) determinant and made a series of NP peptides with a broad range of avidities for the TCR (21). We pulsed activated APC with high concentrations of each peptide to evaluate the impact of avidity on NP-specific CD4 T cells. We found that higher TCR avidity for peptide-MHC-II proportionally increased memory generation (21). Moreover, the impact of avidity on responding effectors was already apparent in their survival after only 3 dpi and was also proportional to the amount of IL-2 produced by the responding effectors, which was strictly driven by avidity and peptide dose. This supports the concept that autocrine IL-2 acts directly to support effector survival and explains how the strength of TCR signals determines the extent of CD4 memory generation (21).
2.3.2 Role of pathogen recognition pathways via activation of APC
When activated by PR pathways, APC expresses high levels of surface MHC-II that enhance antigen presentation and multiple costimulatory ligands, including CD80 and CD86, that bind to ligands on the CD4 T cells so they produce IL-6, which is needed for the strongest naïve CD4 responses (22) (Figure 1). Replicating viruses such as influenza provide high levels of both the peptide antigens (20, 23) and the mediators induced by pathogen-derived signals (11). This is in contrast to most non-replicating vaccines, such as inactivated IAV, that supply antigens and associated adjuvants that rapidly decrease with time (23). Thus, many vaccines likely provide inadequate signals for CD4 effector progression to memory at the checkpoint.
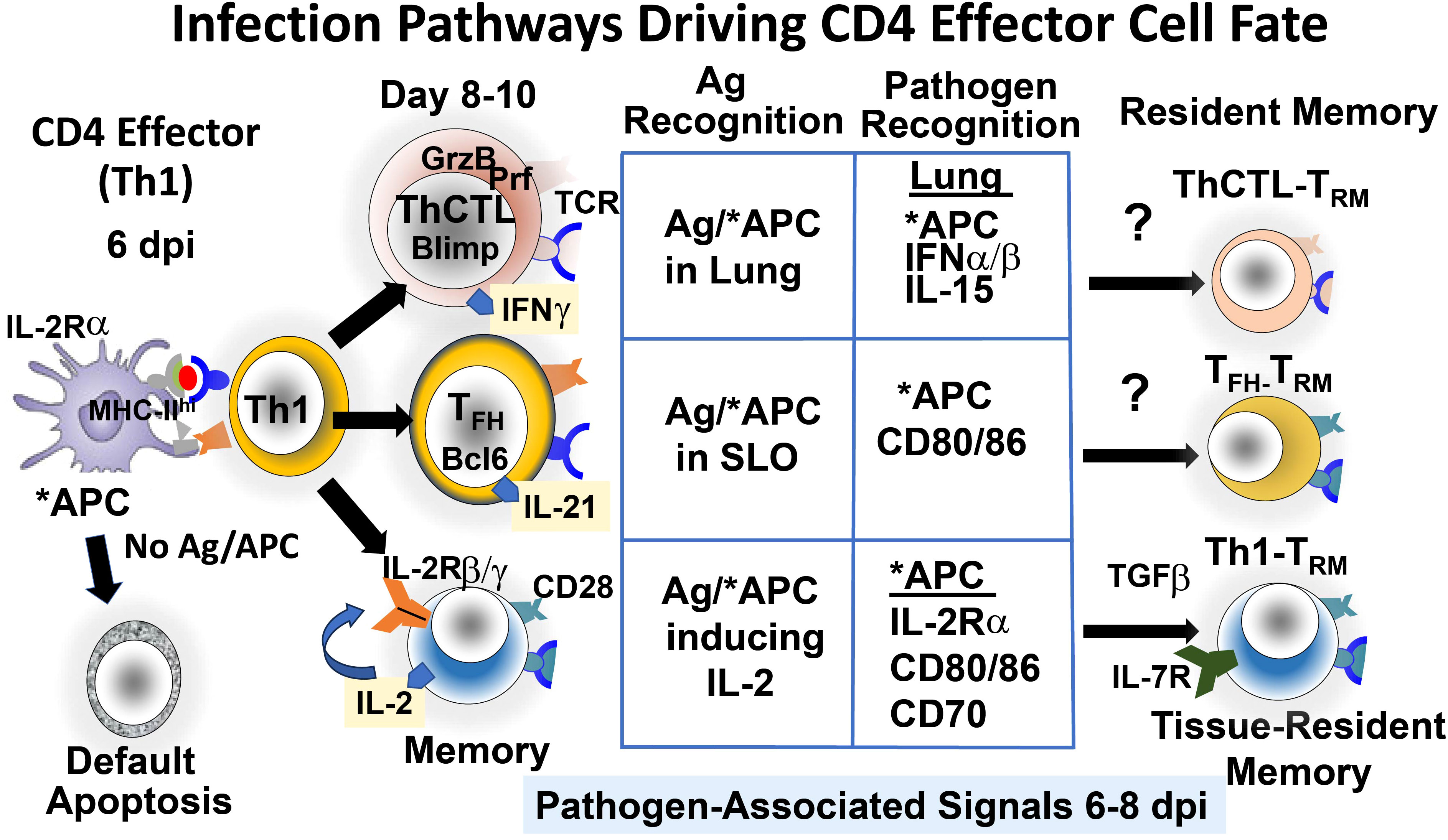
Figure 1 Pathways that drive CD4 effector cell fate at the effector checkpoint. Signals from continuing infection in the lung drive Th1 effectors to further differentiate to more specialized TFH and ThCTL and likely to memory and tissue-resident memory (TRM) subsets. Signals include abundant viral antigen (Ag) presented by antigen-presenting cells (APC), activated by pathogen recognition (PR) pathways. (*APC) and by various cytokines secreted by other cells activated by PR pathways.
We found that IAV infection induces expression of the IL-2 receptor β and γ chains on CD4 effectors, but many effectors did not express IL-2Rα (22). However, in vitro TLR agonist-activated APC and in vivo APC 6 dpi in infected mice did upregulate IL-2Rα, and APC that could not express IL-2Rα supported less memory generation than wild-type APC (21). These results suggest that IL-2 is trans-presented to the effectors by activated APC. Trans-presentation of IL-15 is a common mechanism (24), but IL-2 trans-presentation has rarely been identified (25). We suggest that the APC trans-presentation of IL-2 to the responding effectors amplifies the effective concentration of IL-2 during APC : CD4 effector cognate interaction (21). If so, the activation of IL-2Rα on APC by PR pathways is another mechanism by which infection-stimulated PR pathways contribute to CD4 memory generation. The known requirements we identified for the generation of CD4 memory from effectors are shown in Figure 1 (Memory).
2.4 Checkpoint signals regulate the generation of highly differentiated TFH and ThCTL effectors
IAV infection generates a population of CD4 effectors, which are mostly Th1 cytokine-polarized cells with a few Th17, and this population gives rise 2–3 days later to TFH and ThCTL (10, 11). These more differentiated effectors play critical roles in viral clearance and protection: TFH support the germinal center response and selection of high-affinity B cells, which provide optimum B-cell Ab responses (26), and ThCTL kill infected cells expressing MHC-II (9), thus counteracting viral evasion mechanisms that downregulate MHC-I. We postulate that each effector then gives rise to distinct memory CD4 cells of related function, including TRM memory, in tissue sites where the effectors reside (10, 11). We asked if the development of TFH and ThCTL effectors from the 6 dpi Th1 effectors also requires antigen recognition and/or other signals from infection. When 6 dpi effectors were transferred to second hosts without antigen or infection, few TFH or ThCTL developed. Even the transfer of peptide-pulsed APC could not restore TFH (10) or ThCTL (11) generation, though they restored memory production (19). Thus, virus infection, in addition to antigen recognition, was required for both these subsets (Figure 1). TFH and ThCTL development is programmed by gene pathways on the opposing Bcl6-Blimp1 axes (9, 26). CD28 costimulation is required during this interaction for TFH development but not for ThCTL generation, and only ThCTL is required for infection-induced 1L-15 (11), supporting the dichotomy between TFH and ThCTL effector stage differentiation.
Both TFH and ThCTL effectors express a gene program associated with tissue residence (loss of S1PR1, Klf2, CCR7) (10, 11, 26). Since TFH are found almost exclusively in secondary lymphoid sites vs. ThCTL in the lung (the site of IAV infection), we asked if local antigen presentation is needed. We restricted presentation to the lung and draining lymph node (dLN) by introducing the APC intranasally and restricted presentation to the spleen with intrasplenic injection (10, 11). The usual intravenous route resulted in presentation mostly in the spleen. TFH developed in dLN with i.v. introduction and in the spleen with i.s. or i.v., while lung ThCTL in the lung required i.n. inoculation (10). We suggest that local antigen presentation contributes to inducing residency and drives the final differentiation to TFH and ThCTL (10, 11). Additional PR-induced pathways independent of how infection impacts APC, are also required for the development of TFH and ThCTL (IL-15 and IFN type I) (10, 11). Unlike memory responses, the development of TFH and ThCTL effectors from earlier effectors does not require IL-2 (10, 11), distinguishing the pathways to memory (18) from those of more differentiated effectors. We are currently investigating if the tissue-resident effectors can become resident memory CD4 T cells (CD4 TRM).
Both ThCTL and TFH exert potent effector functions that, while critical for protection, have also been implicated in autoimmune diseases and disease pathologies (27, 28). Thus, it is likely beneficial that their development at this late effector checkpoint is tightly regulated by multiple concurrent signals of pathogen infection: antigen presentation, costimulation, and other signals generated by infection, as these requirements ensure TFH and ThCTL only develop when infection continues through the effector checkpoint, indicating a more potent immune response is needed to effectively mediate viral clearance and induce strong memory.
2.5 Hypothesis: the generation of distinct CD4 effectors and additional memory subsets by infection results in greater protection and a hierarchy of memory
CD4 T-cell immunity is important because T cells often target internal conserved proteins shared among heterosubtypic virus strains, presented by MHC-II, which is not readily downregulated by viral mechanisms, and thus they respond to emerging viral variants that frequently mutate their surface proteins that are targeted by Ab (8). The strict requirements at the CD4 effector stage described here to generate memory directly, and the TFH and ThCTL effectors and memory they become, are readily supplied by replicating viruses but not by benign foreign or self-antigens. Infection drives more memory cells, memory cells with more potent functions, and in sites of potential re-infection, providing greater protection against replicating pathogens (Figure 2). Thus, we postulate the checkpoint requirements we have defined, create a striking hierarchy of CD4 memory. Those memory cells generated by homeostatic exposure to self-antigen (29) induce memory in the central sites (TCM) of the secondary lymphoid organs (SLO) that are Th1-like (30). Exposure to foreign, but nonreplicating, antigens, even if of high affinity and associated with some adjuvant, gives short-lasting signals and induces memory mostly directly from initial CD4 effectors (Th0, Th1, Th2, etc.) and mostly in central sites (TCM). In contrast, infection, producing high levels of antigen and PR recognition signals through the effector checkpoint, induces the greatest functional diversity, additional more potent CD4 memory subsets, and memory that is resident in the tissues of infection as well as SLO (Figure 2).
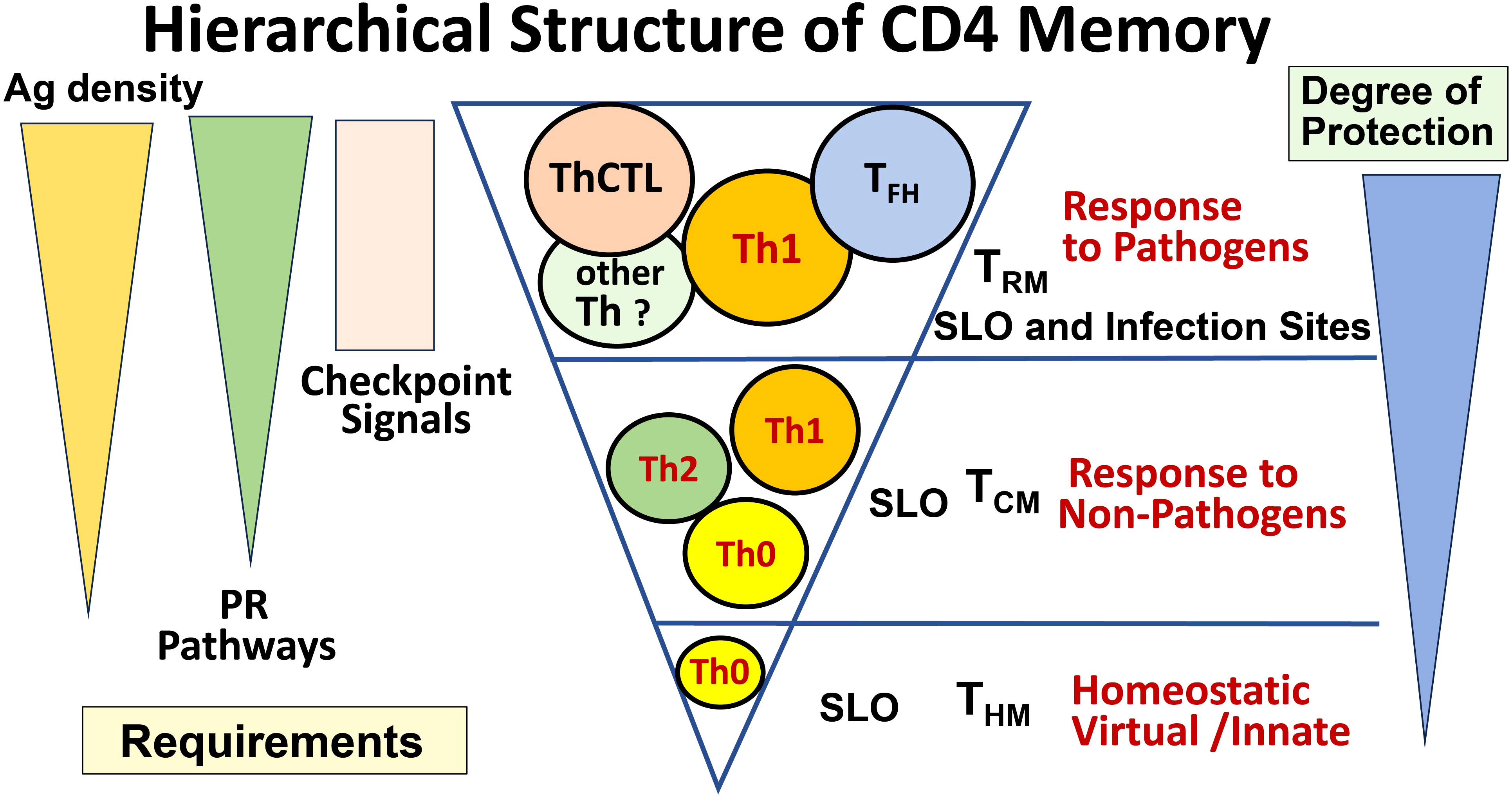
Figure 2 Hierarchical structure of CD4 memory. The signals driving the generation of CD4 effector and memory subsets result in a very different spectrum of memory subsets depending on the nature of the stimulus. When no foreign Ag is present, only a small number of homeostatic (aka innate, virtual) memory cells develop (THM bottom). When noninfectious Ag is present at sufficient levels, especially if accompanied by a source of PR signals, cytokine-polarized effectors are generated, and they can generate some central memory (TCM middle). When a pathogen is present that persists into the effector phase, such that high levels of Ag and PR signals persist in infected tissues, high numbers of more specialized effectors (TFH, ThCTL) can be generated, and we postulate that these give rise to tissue-resident memory (TRM). We predict that the highest level of long-lasting protection requires these specialized effectors and memory from them.
3 Discussion
The above paradigm for posteffector CD4 effector differentiation leads to a hierarchical scheme of CD4 memory with ascending functional diversity, tissue residence, and potency in clearing infection. They each correlate with multiple, synergizing signals they receive at the effector stage checkpoint: the level, avidity, and duration of antigen presentation, the level of activation of antigen-presenting cells, and the level of signals from other pathogen recognition pathways. This hierarchy should restrict the development of the most potent, and hence risky, CD4 immune memory to infection with the most dangerous pathogens.
3.1 Unanswered questions and limitations
The paradigm that CD4 effectors require re-exposure to high levels of Ag and pathogen recognition signals for memory and TFH and ThCTL development has not yet been definitively verified in humans or nonhuman primate models. Also, long-lived humans accumulate a higher proportion of memory cells as they age, and thus more responses may be derived from CD4 memory and fewer from naïve CD4 T cells. However, our studies in mice suggest that memory CD4 T cells have equivalent requirements for these signals to make secondary memory cells (21). We do not know if other CD4 effector subsets may also require checkpoint signals. CD8 T-cell memory benefits from prolonged antigen exposure (31, 32), and CD8 TRM requires recognition at the site of residency (33, 34, 35). Whether a comparable set of requirements applies to CD8 effectors and creates a comparable hierarchy is not established, but it seems likely, considering they follow a parallel scheme of development.
Our studies define only the role of Ag and PR signals at the checkpoint where effectors have formed and their proximal fate, whether apoptosis or further differentiation to TFH, ThCTL, or memory, is determined (Figure 1). In our transfer studies and in acute infections that are effectively cleared, both signals soon disappear, although Ag may persist in some specialized APCs. In situations of chronic infections or continuing exposure to Ag, exhaustion of CD4 effectors can occur. An advantage of vaccine strategies is that they can be designed to provide signals through the effector phase but not much longer thereafter.
3.2 Implications for vaccine designs
We suggest a major weakness of many current non-replicating vaccines is their failure to provide the prolonged antigen and pathogen signals needed to support high levels of CD4 memory and TFH and ThCTL subsets (10, 11, 17, 19, 23). Indeed, inactivated influenza presents CD4 antigens for only a few days, producing mostly Th0 and Th1 effectors in the SLO with few TFH, limited generation of B-cell Ab. The addition of live, attenuated influenza at the effector peak enhances TFH, effectors in the lung, and Ab production (23). Protein vaccines present only a fraction of viral epitopes. CD4 T cells’ memory generation depends on high affinity and peptide density on APC during the cognate interaction of CD4 effectors at the checkpoint, which determines the level of IL-2 and hence the extent of memory generation. However, functional avidity depends on peptide interaction with MHC-II and that will vary with individual alleles. This argues that the extent of CD4 T-cell memory generation in a human population will increase with the breadth of antigens included in the vaccine. Inducing CD4 memory to more epitopes will also provide less chance viral variants have of escaping CD4 immunity.
We need vaccines to generate strong multifunctional CD4 memory against potentially deadly pathogens such as influenza and SARS-CoV2 that rapidly mutate and evade Ab-mediated protection. Current vaccine immunity is short-lived, and both these infections require frequently updated vaccines to the new target selected mutants.
We predict that the pathways we have defined in influenza will be found to be equally applicable to many other viral infections. Thus, we suggest that providing missing signals to current vaccine platforms could make them much more effective. In mouse models, we have provided: in vivo peptide antigen on TLR-activated APC (18, 19, 23), IL-2:anti-IL-2 Ab complexes (18), infection surrogates (10, 19, 23), and others that have supplied PR signals in humans (36) and all enhance CD4 memory and protection. Thus, there is support for the concept that providing the missing signals, at the effector stage, to a basic vaccine platform should improve CD4 immunity.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee University of Massachusetts Chan Medical School. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The main research contributions on which this perspective was based were supported by NIH R21AI153120, R21AI146532, R01AI118820, R01AI076534, R01AI076534, and P01AI046530. P01AI046539, R37AG025805, and P01AG021600 to Susan Swain and U19AI09858 to Raymond Welsh. Graduate students received support from T32 AI007349 and T32 AI132152; T32 AI007349 to UMMS.
Acknowledgments
Thanks to all my coauthors referenced here, whose work led to the body of knowledge and the hypotheses described here. Special thanks to Priyahardshini Devarajan, K. Karl McKinstry, Tara M. Strutt, Bianca Bautista, and Michael C. Jones, who made the key discoveries defining the requirements of CD4 effectors for further differentiation.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Swain SL, McKinstry K,K, Strutt T,M. Expanding roles for CD4 memory T cells in protection against viruses. Nat Rev Immunol (2012) 12:136–48. doi: 10.1038/nri3152
2. Swain SL, Croft M, Dubey C, Haynes L, Rogers MP, Zhang X, et al. From naive to memory T cells. Immunol Rev (1996) 150:143–67. doi: 10.1111/j.1600-065X.1996.tb00700.x
3. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol/ Rev (2013) 252:12–23. doi: 10.1111/imr.12032
4. Swain SL, Bradley LM, Croft M, Tonkonogy S, Atkins G, Weinberg AD, et al. Helper T cell subsets: Phenotype, function and the role of lymphokines in regulating their development. Immunol Rev (1991) 123:115–44. doi: 10.1111/j.1600-065X.1991.tb00608.x
5. Strutt TM, McKinstry KK, Marshall NB, Vong AM, Dutton RW, Swain SL. Multipronged CD4 T cell effector and memory responses cooperate to provide potent immunity against respiratory virus. Immunol Rev (2013) 255:149–64. doi: 10.1111/imr.12088
6. McKinstry KK, Strutt TS, Kuang Y, Brown DB, Sell S, Dutton RW, et al. Memory CD4+ T cells protect against influenza through multiple, synergizing mechanisms. J Clin Invest (2012) 122:2847–56. doi: 10.1172/JCI63689
7. Strutt TM, McKinstry KK, Kuang Y, Bradley LM, Swain SL. Memory CD4+ T-cell-mediated protection depends on secondary effectors that are distinct from and superior to primary effectors. Proc Natl Acad Sci U.S.A. (2012) 109:2551–60. doi: 10.1073/pnas.1205894109
8. Powell TJ, Strutt T, Reome J, Hollenbaugh JA, Roberts AD, Woodland DL, et al. Priming with cold adapted influenza A does not prevent infection but elicits long lived protection against supralethal challenge with heterosubtypic virus. J Immunol (2006) 178:1030–8. doi: 10.4049/jimmunol.178.2.1030
9. Marshall NB, Vong AM, Devarajan P, Brauner MD, Kuang Y, Nayar R, et al. NKG2C/E marks the unique cytotoxic CD4 T cell subset, ThCTL, generated by influenza infection. J Immunol (2017) 198:1142–55. doi: 10.4049/jimmunol.1601297
10. Devarajan P, Vong AM, Castonguay CH, Kugler-Umana O, Bautista BL, Jones MC, et al. Strong influenza-induced TFH generation requires CD4 effectors to recognize antigen locally and receive signals from continuing infection. Proc Natl Acad Sci U.S.A. (2022) 119:119–31. doi: 10.1073/pnas.2111064119
11. Devarajan P, Vong AM, Castonguay CH, Kugler-Umana O, Bautista BL, Jones MC, et al. Cytotoxic CD4 development requires CD4 effectors to concurrently recognize local antigen and encounter Type-I-IFN-induced IL-15. Cell Reports. (2023). doi: 10.1016/j.celrep.2023.113429
12. Devarajan P, Jones MC, Umana OK, Vong AM, Xia J, Swain SL. Pathogen recognition by CD4 effectors drives further protective effector and memory cell generation against respiratory viruses. Front Immunol (2018) 6:596(9). doi: 10.3389/fimmu.2018.00596
13. Krammer F. The human antibody response to influenza A virus infection and vaccination. Nat Rev Immunol (2019) 19:383–97. doi: 10.1038/s41577-019-0143-6
14. Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. (1991) 353:858–61. doi: 10.1038/353858a0
15. Zheng L, Li J, Lenardo M. Restimulation-induced cell death: new medical and research perspectives. Immunol Rev (2017) 277:44–60. doi: 10.1111/imr.12535
16. Zhang X, Giangreco L, Broome HE, Dargan CM, Swain SL. Control of CD4 effector fate: transforming growth factor beta 1 and interleukin 2 synergize to prevent apoptosis and promote effector expansion. J Exp Med (1995) 1182:699–709. doi: 10.1084/jem.182.3.699
17. McKinstry KK, Strutt TM, Swain SL. Regulation of CD4 T cell contraction during pathogen challenge. Immunol Rev (2010) 236:110–24. doi: 10.1111/j.1600-065X.2010.00921.x
18. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, et al. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Comm (2014) 5:5377. doi: 10.1038/ncomms6377
19. Bautista BL, Devarajan P, McKinstry KK, Strutt TM, Vong AM, Jones MC, et al. Short-lived antigen recognition but not viral infection at a defined checkpoint programs effector CD4 T cells to become protective memory. J Immunol (2016) 197:3936–49. doi: 10.4049/jimmunol.1600838
20. Swain SL, Jones MC, Devarajan P, Xia J, Dutton RW, Strutt TM, et al. Durable CD4 T cell memory generation depends on persistence of high levels of infection at an effector checkpoint that determines multiple fates. Cold Spring Harbor Perspect (2021). doi: 10.1101/cshperspect.a038182
21. Jones MC, Castonguay C, Nanaware PP, Weaver GC, Stadinski B, Kugler-Umana OA, et al. CD4 effector TCR avidity for peptide on APC determines the level of memory generated. J Immunol (2023) 210:1950–61. doi: 10.4049/jimmunol.2200337
22. Brahmakshatriya V, Kuang Y, Deverajan P, Xia J, Zhang W, Vong AM, et al. IL-6 production by TLR-activated APC broadly enhances aged cognate CD4 helper and B cell antibody responses in vivo. J Immunol (2017) 198:2819–33. doi: 10.4049/jimmunol.1601119
23. Xia J, Kuang Y, Liang. J, Jones M, Swain SL. Influenza vaccine-induced CD4 effectors require antigen recognition at an effector checkpoint to generate CD4 lung memory and antibody Production. J Immunol (2020) 205:2077–90. doi: 10.4049/jimmunol.2000597
24. Waldmann TA, Waldmann. R, Lin JX, Leonard WJ. The implications of IL-15 trans-presentation on the immune response. Adv Immunol (2022) 156:103–32. doi: 10.1016/bs.ai.2022.09.002
25. Wuest SC, Edwan JH, Martin JF, Han S, Perry. JS, Cartagena CM, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med (2011) 17:604–9. doi: 10.1038/nm.2365
26. Crotty S. T follicular helper cell biology: A decade of discovery and diseases. Immunity. (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
27. Juno JA, van Bockel D, Kent SJ, Kelleher AD, Zaunders JJ, Munier CM. Cytotoxic CD4 T cells-friend or foe during viral infection? Front Immunol (2017) 8:19.mmunol. doi: 10.3389/fimmu.2017.00019.mmunol
28. Gensous. N, Charrier. M, Duluc. D, Contin-Bordes C, Truchete ME, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Front Immunol (2018) 9:1637. doi: 10.3389/fimmu.2018.01637
29. Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naïve cells to memory phenotype cells. Nat Immunol (2011) 12:478–84. doi: 10.1038/ni.2018
30. Kawabe T, Jankovic D, Kawabe S, Huang Y, Lee PH, Yamane H, et al. Memory-phenotype CD4+ T cells spontaneously generated under steady-state conditions exert innate TH1-like effector function. Sci Immunol (2017) 2, 12:eaam9304. doi: 10.1126/sciimmunol.aam9304
31. Obst R, van Santen H-M, Mathis D, Benoist. C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med (2005) 201:1555–65. doi: 10.1084/jem.20042521
32. León. B, Ballesteros-Tato A, Randall TD, Lund FE. Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J Exp Med (2014) 211:1637–55. doi: 10.1084/jem.20131692
33. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med (2016) 213:951–66. doi: 10.1084/jem.20151855
34. McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol (2018) 11:1071–8. doi: 10.1038/s41385-018-0003-x
35. Abdelbary M, Hobbs SJ, Gibbs JS, Yewdell JW, Nolz JC. T cell receptor signaling strength establishes the chemotactic properties of effector CD8+ T cells that control tissue-residency. Nat Commun (2023) 14:3928. doi: 10.1038/s41467-023-39592-1
Keywords: immune memory, pathogen, antigen, CD4, effector
Citation: Swain SL (2023) CD4 memory has a hierarchical structure created by requirements for infection-derived signals at an effector checkpoint. Front. Immunol. 14:1306433. doi: 10.3389/fimmu.2023.1306433
Received: 03 October 2023; Accepted: 28 November 2023;
Published: 12 December 2023.
Edited by:
Vandana Kalia, University of Washington, United StatesReviewed by:
Emma Teixeiro, University of Missouri, United StatesCopyright © 2023 Swain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan L. Swain, c3VzYW4uc3dhaW5AdW1hc3NtZWQuZWR1