 Kerry A. Mullan
Kerry A. Mullan Nicky de Vrij
Nicky de Vrij Sebastiaan Valkiers
Sebastiaan Valkiers Pieter Meysman
Pieter Meysman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 December 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1306169
Single-cell RNA sequencing (scRNA-seq) has become a popular technique for interrogating the diversity and dynamic nature of cellular gene expression and has numerous advantages in immunology. For example, scRNA-seq, in contrast to bulk RNA sequencing, can discern cellular subtypes within a population, which is important for heterogenous populations such as T cells. Moreover, recent advancements in the technology allow the parallel capturing of the highly diverse T-cell receptor (TCR) sequence with the gene expression. However, the field of single-cell RNA sequencing data analysis is still hampered by a lack of gold-standard cell phenotype annotation. This problem is particularly evident in the case of T cells due to the heterogeneity in both their gene expression and their TCR. While current cell phenotype annotation tools can differentiate major cell populations from each other, labelling T-cell subtypes remains problematic. In this review, we identify the common automated strategy for annotating T cells and their subpopulations, and also describe what crucial information is still missing from these tools.
The first single-cell RNA sequencing (scRNA-seq) experiments started in 2009, and the technique became commercially available in 2014 (1). Single-cell RNA sequencing has rapidly gained widespread use, as more detailed information can be acquired using it than using bulk RNA-seq. Additionally, scRNA-seq data are becoming more accessible as more companies (e.g., 10x Genomics and BD Rhapsody®) are developing and optimising the technology, leading to a higher throughput and decreasing costs. With the increasing availability of scRNA-seq data, there has been a substantial increase in our understanding of the functions of immune cells. This has led to discoveries of new immune cell subpopulations, their dynamic and heterogeneous nature, and their role in disease (2–5). A particularly useful advantage of scRNA-seq for the study of the adaptive immune system is the ability to uncover paired information on the gene expression and the immune receptor of a single cell [more extensively reviewed in (6)]. However, defining the cellular profiles for adaptive immune cells remains a complex task. For example, the T cells of the adaptive immune system are very heterogeneous and can adopt a wide variety of phenotypes. In addition to a wide variety of phenotypes, there is an increased layer of complexity due to the highly polymorphic nature of the immune cell receptors they carry, such as the T-cell receptor (TCR) for T cells. The TCR is created through somatic recombination to create a highly variable CDR3 sequence containing a variable (V), and Junction (J) for alpha (α) and gamma (γ) chains, or a V, Diversity (D) and J for beta (β) and delta (δ) chains (7). These unique TCRs can recognise a vast array of epitopes, including immunopeptides, lipids, and some small molecules [e.g., phosphoantigens and Vitamin B metabolites (8)]. The most well-studied mechanism of epitope recognition is the antigenic peptide presentation by the major histocompatibility complex (MHC) protein, encoded in humans via the human leukocyte antigen (HLA) gene loci, and then to conventional αβ T cells (7). However, there are also unconventional T cells which are thought not to interact with MHC, such as mucosal-associated invariant T cells (MAIT), natural killer (NK) T cells, and γδ T cells (9). These unconventional T cells and their cellular profiles remain poorly understood.
A crucial step in the analysis of scRNA-seq data involves annotating the cells with the correct cellular phenotype. The initial manual annotation of the cells in a scRNA-seq dataset, after (pre-)processing, is time intensive, may contain data entry errors, and requires expert knowledge of the marker genes specific to the different cellular subsets. The initial (pre-)processing is commonly done using the R Seurat package (10) or the Python Scanpy package (11). For a more comprehensive description of the different steps in the (pre-) processing of scRNA-seq data, we refer you to this excellent review by Heumos et al. (2023) (12). In brief, the manual annotation of cells in scRNA-seq data is typically approached by clustering the cells and comparing these clusters to identify the differentially expressed genes (DEGs) among them to verify if they are known marker genes that are specific to cellular populations. This is hampered by a number of factors, however, such as a high gene dropout rate, the free-floating ambient mRNA of one cell being captured in a droplet together with another cell (droplet-based methods), or the poor expression of some marker genes at the RNA level, which would be better identified at the protein level (13). More recently, this manual annotation process has been superseded by automatic methods that leverage machine learning to automate and ease the burden (12). To aid in annotating cells with their phenotypes in scRNA-seq data, several automated pipelines have been developed to infer the phenotype based on a cell’s gene expression profile. However, these tools are often focused on inferring broader cell types (i.e., annotating a cell as a T cell), and it is unknown how well these tools work for inferring the subpopulations of these broader cell types (i.e., identifying a T helper [Th] 1 cell). Thus, in this review, we describe the currently available annotation tools for identifying T-cell phenotypes from scRNA-seq datasets. We compare their annotation strategies to the literature to verify whether they are fully capturing these hard-to-delineate subpopulations. Finally, we reflect on how well some of the unconventional T-cell populations are currently being captured.
To prevent the labour-exhaustive manual annotation of new datasets, automatic annotation tools have been developed to decrease time, improve labelling accuracy, and promote consistency. Automated annotation has become part of the current gold-standard approach to single-cell RNAseq, along with manual annotation/inspection of the automated annotations by expert review (i.e., expert familiarity with the common markers of cellular populations, which enables accurate annotation) (12). Therefore, a range of tools have been developed to aid in annotation automation (Table 1). As highlighted in Table 1, the current tools fall into several subcategories, each with distinct advantages and limitations. These annotation methods can also be distinguished by the type of machine learning (ML) approach, with methods categorized into unsupervised, supervised, or semi-supervised approaches.
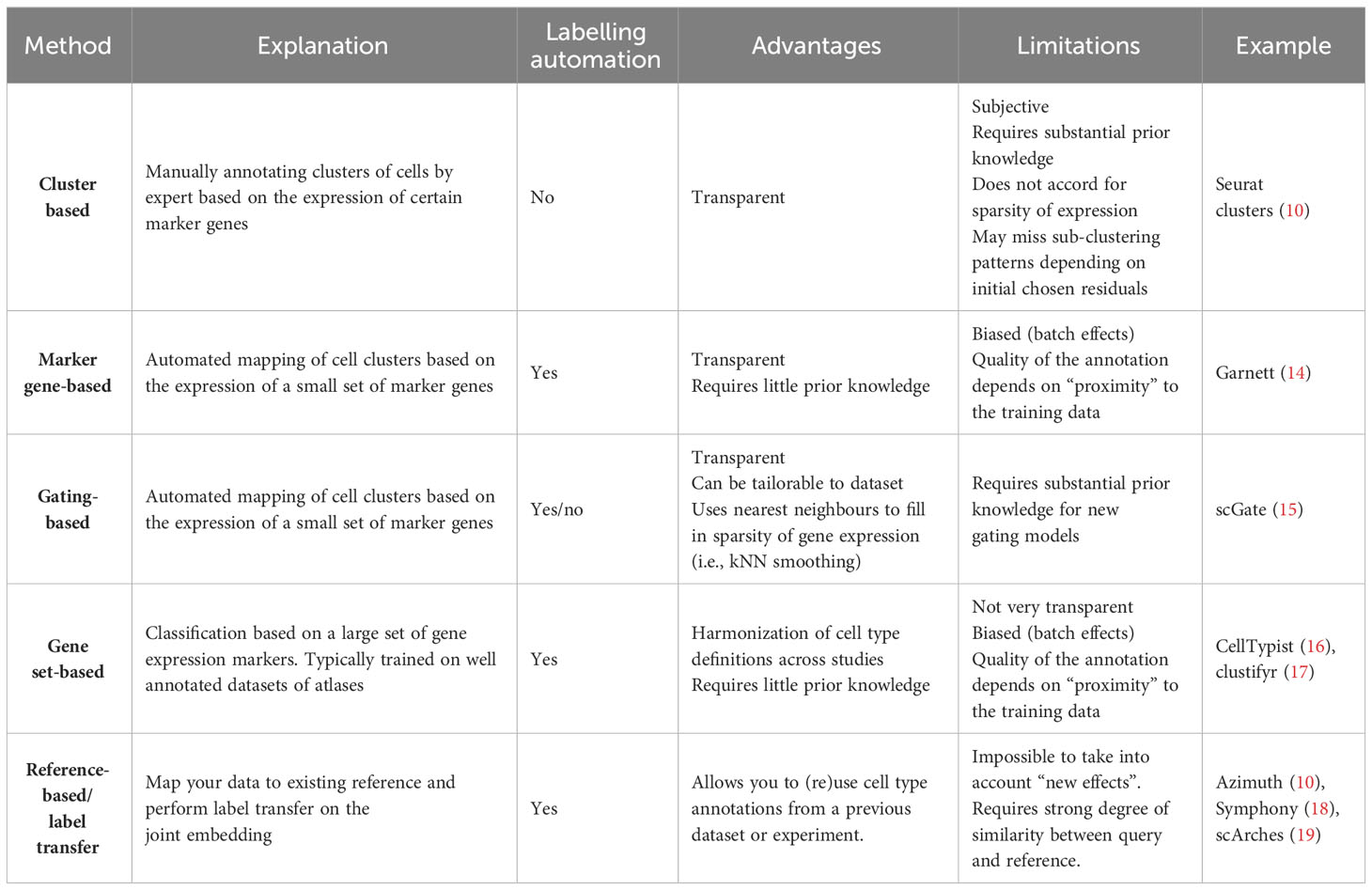
Table 1 Common strategies and programs for annotating scRNA-seq datasets.
The unsupervised approach is typically clustering-based, including, for example, k-nearest neighbours (e.g., Seurat clustering (10)), which groups together cells with similar expression profiles. Subsequently labelling the clusters then requires the manual interrogation of the distinct markers per population. Accurate annotation relies on the expert knowledge of the user for common genes expressed for each cell type.
The supervised ML classification of scRNA-seq data is available in SingleR (20), Garnett (14), and CellTypist (16). These tools enable the prediction of cell-type labels for a novel dataset based on a prediction model trained on prior datasets. The ability to annotate a new dataset with high accuracy requires the dataset to have a good overlap of genes with the prediction model. This method is more robust in handling missing marker genes in a dataset, as it relies on the entirety of a cell’s gene expression to classify a cell, rather than just a few marker genes. However, if there is too much heterogeneity between the datasets, then the prediction tools fail to identify the cells correctly. The package scTriangulate aims to overcome this limitation by using multiple annotation sources (21).
The semi-supervised annotation approach includes models such as the SCINA (22) tool, which was developed to annotate cells based on a consensus list of known markers. An alternative tool, scGate (15), follows a process similar to the gating strategy employed in flow cytometry experiments, and classifies the markers in a hierarchical structure of pure and impure cells. The latter includes prelisted markers, adding to the interpretability of the method. The scGate researchers also defined common gating strategies on common cellular markers, and this led to the development of ProjectTILs (23) to further automate the process. A particular advantage of scGate is that the user can provide their own list of markers and and is advantageous to use in instances that the dataset is dissimilar or not modelled within the pre-learned supervised models.
Therefore, the researcher will need to consider which method is most appropriate for their dataset. For instance, if their dataset is similar to a previous annotated dataset and was obtained using the same technology, then the reference-based/label transfer may be the best strategy for annotating the cells. Alternatively, if researchers have a novel cellular subset from a species that is not human or mouse, the use of reference-based, gene set-based, and marker-based tools may not be advisable, as they rely on similarity to previously curated datasets. In addition, these ML-based label transfer methods are hampered by their reliance on the quality of the annotation of the original dataset. As such, we encourage users to carefully review the latest datasets and markers that were used to define populations, if available.
Although the accuracy of these automated methods has significantly improved, a two-step annotation process is strongly recommended. This two-step process involves primary annotations of the gene expression clusters by automated algorithms, followed by expert-based manual interrogation of the cell populations. In general, a combination of both strategies will result in the most accurate definitions of cell subsets.
As highlighted above, the current annotation strategies can distinguish between populations with large phenotypic differences (e.g., B cell vs. T cells), as there are fewer overlapping transcripts. However, within each cell type there can be subspecialisations. For instance, T cells have a variety of subtypes. These subtypes are first stratified into two main lineages based on the TCR, that is, alpha-beta (αβ) and gamma-delta (γδ) T cells. Subsequently, αβ T cells, the best-described T-cell subtype, are further delineated into CD4- or CD8- expressing T cells. However, these can be further stratified based on their function and capacity for formation of immunological memory. The most well-described classical subpopulations relate to the class I (CD8+) and class II (CD4+) αβTCR cells, which are responsible for screening the peptide-loaded major histocompatibility complex for “self” and “foreign” antigens (7). Less is known about the unconventional T cells, which encompass natural killer T (NKT) cells, mucosal invariant T cells (MAIT), and γδ T cells. However, evidence that these unconventional T cells have important roles in both health and disease [reviewed in (24–26)] is emerging. Therefore, future work should consider both classical and unconventional T cells.
Given this plethora of cell subsets, how are these subpopulations currently defined by common annotation models for humans? To address this question, we looked at several tools that claim to be able to annotate for more delineated T-cell subpopulations. These annotation tools included scGate, CellTypist, and Data2Talk[online tool], as these had more extensive documentation for the T-cell subsets. Additionally, we compared these with the common protein expression panels used to identify T-cell subsets, as these are well-curated and validated panels. Last, we also included findings from the literature to fill in other annotation gaps. Tables 2–5 highlight the markers used to classify the CD4 αβ T cells (Table 2), CD8 αβ T cells (Table 3), γδ T cells (Table 4), and miscellaneous T cell markers (Table 5) that were identified by the documented annotation models or through literature searches.

Table 2 CD4+ αβ T cell markers (human).
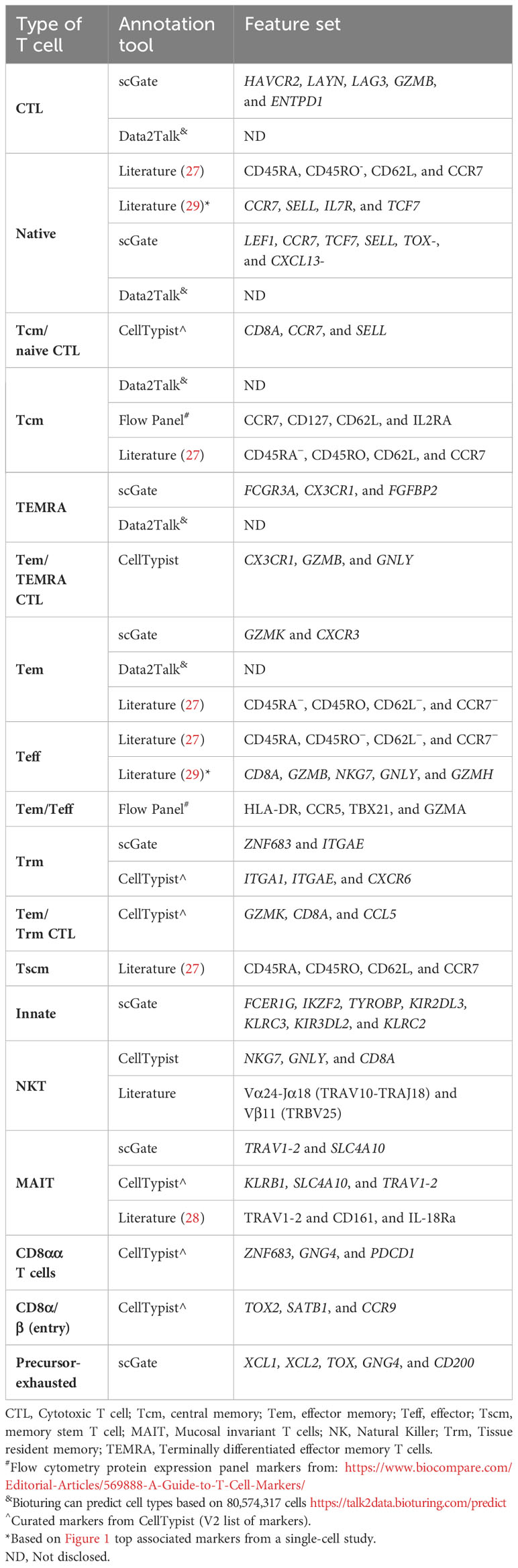
Table 3 CD8+ Markers (human).
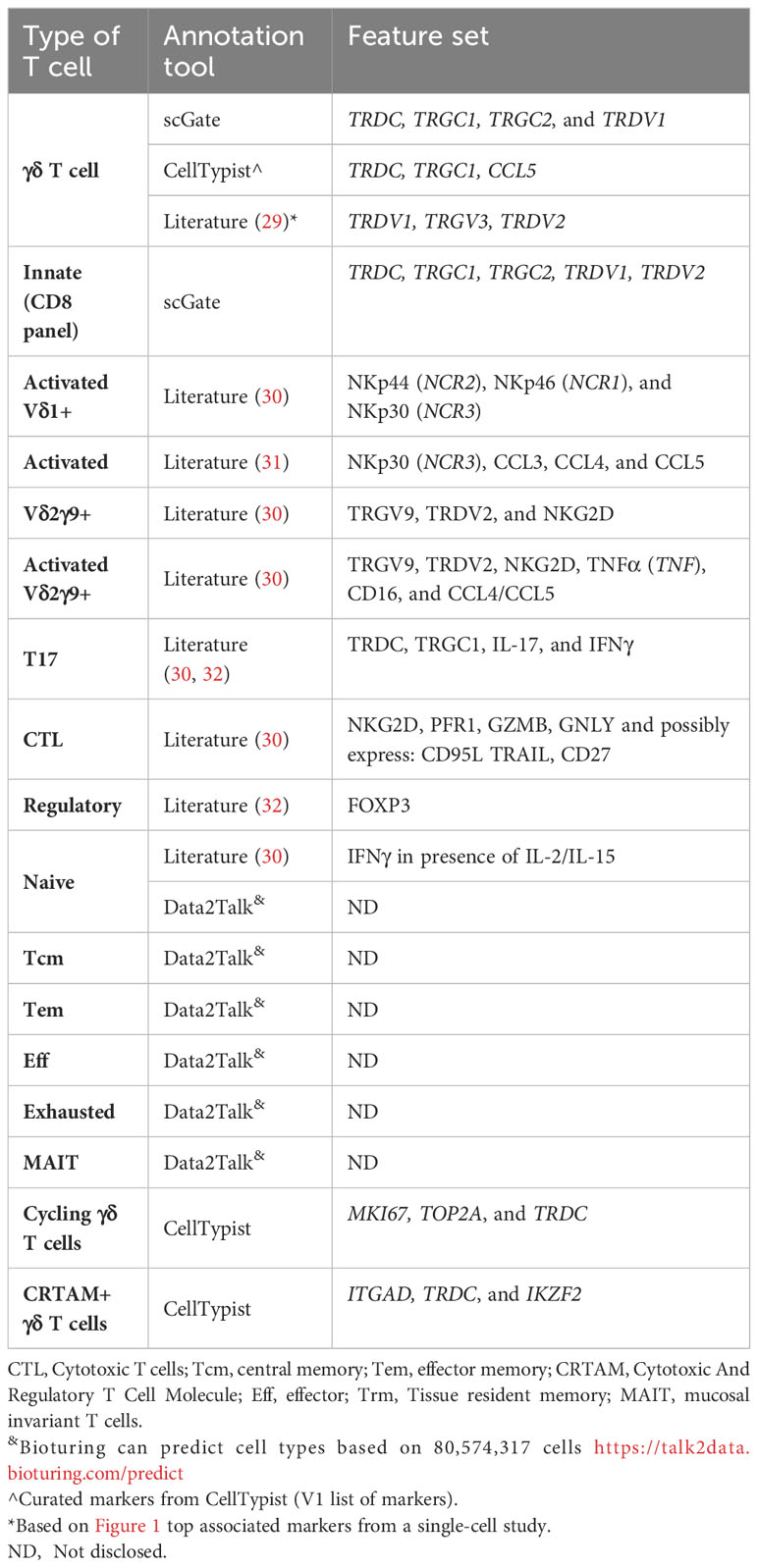
Table 4 γδ T cell Markers (human).
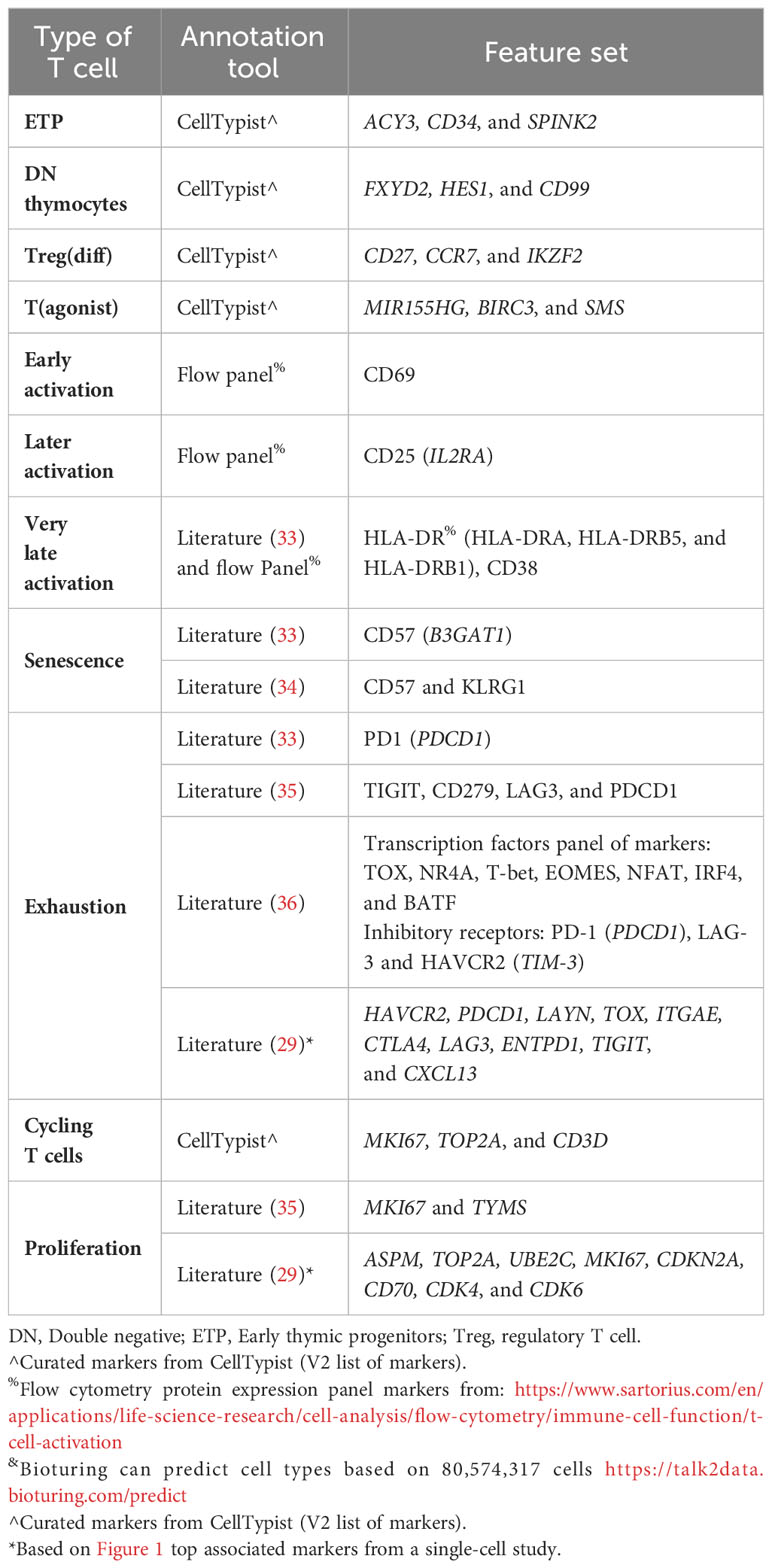
Table 5 Miscellaneous T cell markers (human).
The T-cell annotation models include most of the well-defined effector CD4+ populations, including T helper 1, Th17, follicular Th (Tfh) and regulatory T cells (Tregs) (Table 2). CellTypist was the only annotation model to included memory markers for the CD4+ T-cell population, while Data2Talk included Th2 cells, but the markers were not disclosed. The CD8+ T cells were classified into cytotoxic T cells (CTL; granzymes [GZMB, GZMK, etc.], perforin [PFR1], granulysin [GNLY]), NKT cells (KLR gene family, CD160, etc.), and MAIT cells (Table 3). These CD8+ T-cell subsets were also broken down into memory features, naive, effector, effector memory (Tem), terminal memory (TEMRA), resident memory (Trm), and central memory (Tcm) (Table 3) cells. The three annotation models cover many of the common classical CD8+ and CD4+ T-cell populations, except for Th2, Th9, and Th22 cells. The identification of these populations has relied on cytokine expression. However, the current technology inadequately captures the transcription factors and cytokines (e.g., interleukins) (37). In addition, these populations may also be missed due to a bias in the experimental choices, that is, no focused Th2 specific single-cell experiments. Therefore, we need to identify appropriate markers for the transcriptional level before we can add them to the label transfer models.
T cells can also be defined by their functional states, which are not necessarily restricted to T-cell lineage (e.g., γδ TCR vs. αβ TCR), or a specific subtype (e.g., CD4, CD8, or DN). These functional features include activation (e.g., CD69 [early], CD25 [late] and CD38/HLA-DR [very late]), exhaustion (PD-1, TIGIT, LAG3, and TIM3) (36), senescent (CD57 and KLRG1) (34), and cell cycling/proliferation markers (Table 5). However, the current automated annotation includes only the cell cycling markers. Given that these functional features are important in determining if a T cell is functioning properly, they need to be included in annotation models to identify the most biologically relevant T cell clones. It should be mentioned that when a cell expresses a marker associated with activation, senescence, or exhaustion, it does not mean a cell is activated, senescent, or exhausted. For instance, exhaustion is a functional state characterized by multiple features, including not only the expression of a combination of inhibitory genes such as PD-1, TIGIT, LAG3, and TIM3, and others, but also a lack of effector capacity, that is, a lack of cytokine production or cytotoxic activity (38). Defining these states is even further complicated by the fact that certain genes are associated with multiple states. For example, sole PD-1 expression can indicate an activated state, but it can also indicate a differentiation state to exhaustion, or be a marker of exhaustion if expressed together with other immune checkpoint genes (39). Similarly, when KLRG1 is expressed together with CD57, this can point to T-cell senescence; however, KLRG1 can also be a defining feature of antigen-experienced memory T cells when expressed by itself (40). Therefore, to accurately annotate the exhausted and senescent cellular states, identification of the expression (or lack thereof) of multiple markers is needed. Researchers need to carefully design their annotation panels and be transparent about what markers were used to identify the subpopulations.
Currently, γδ T cells have limited representation in the annotation models (Table 4). The γδ T-cell population is subdivided into innate (Vδ2γ9+) and adaptive-like (e.g., Vδ1+, Vδ2+Vγ9-, Vδ3+) γδ T cells. The most studied γδ T-cell subpopulation is that of the invariant innate Vδ2γ9+ T cells that respond to (E)-4-Hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP). However, the models fail to adequately differentiate between these innate and adaptive-like γδ T-cell subpopulations. For instance, the scGate general annotation model classifies γδ T cells into the innate T-cell population along with NKT cells. CellTypist classifies γδ T cells as γδ TCR or CRTAM+ γδ T cells. Only Talk2Data includes γδ T cell sub-populations, but the markers used for the classifications are unknown (Table 4). Therefore, at this point in time, fully capturing the diversity of γδ T-cell subsets in scRNAseq data analysis requires expert knowledge of marker genes.
From the literature we know that there are difficulties obtaining data from the adaptive γδ T cells. γδ T cells are challenging to study as no antigen-specific culturing methods (41) exist for them, they have a minority fraction in the blood (comprising up to 10% of all T cells) (40), and have a high prevalence in mucosal membranes (e.g., skin, liver, and intestines) (42). Nevertheless, we are slowly defining adaptive γδ T cells that have overlapping phenotypes with the αβ T cells. For instance, functional information derived from mouse models has been used to identify several phenotypes, including T17+ (IL-17 and Th17-like) and T1 (IFNγ and Th1-like) cells (43). Intriguingly, on average ~30% of γδ T cells express the CD8 marker (40). Importantly, recent studies show CD8+ γδ T cells exhibit peptide restriction, similar to classical αβ T cells (44, 45). Consequently, γδ T cells express the same cytotoxic T-cell markers as CD8+ αβ T cells (30). Therefore, CD8+ γδ T cells may be functionally indistinguishable from CD8+ αβ T cells. We also note that γδ T cells can interact with CD1 and MR1, but their molecular signature is not well defined (46). Overall, the innate γδ T cells can be readily identified by their TCR arrangement in single-cell experiments. To bridge the adaptive γδ T cells annotation gap, therefore, we need to use TCR information along with what is known about classical and unconventional αβ T cells.
In addition to the issues with γδ T-cell classification, there are also issues with annotating other unconventional populations, such as MAIT and NKT cells. MAIT cells exhibit MR1 restriction and the semi-invariant TCR arrangement of TRAV1-2 with TRAJ33, TRAJ12, or TRAJ23, often paired with either the TRBV6 or TRBV20 gene families. In flow cytometry experiments, TRAV1-2 and CD161 (KLRB1), IL-18Ra or CD26 are commonly used to identify the MAIT population; however, there can be individual variability (28). MAIT cells can also exhibit the expression of Th17 markers (RORγt and IL-17), in addition to Th1-like features (T-bet, IFNγ) (28). Moreover, the semi-invariant type I NKT cells are identified by an invariant pairing of Vα24Jα18 (TRAV10-TRAJ18) with Vβ11 (TRBV25) and exhibit CD1d restriction, while the type II NKT cells have highly variable TCR combinations, and little is known about what their lipid-restriction is (9). It appears that there may be many subtypes of NKT cells, including Th17-like, Th2-like (GATA3), and Th1 (T-bet) cells (9). Therefore, both MAIT cells and NKT cells cannot be distinguished from other T-cell subpopulations based on gene expression alone. The easiest way to identify MAIT cells and Type I NKT cells will be to utilise the scTCR-seq data layer in combination with the gene expression layer. However, the type II NKT cells cannot be accurately identified until we can identify if they have specific gene(s) that are distinct from the other T-cell subsets.
Other considerations for annotation at the single-cell transcriptome level concern the gene sparsity, low abundance of transcripts captured, and poor correlation of mRNA expression to protein expression for several markers. To illustrate these problems, we highlight a common issue with the identification of the CD4+ T-cell population due to the abundance and sparsity of CD4 cells being lower than those of CD8A and CD8B cells (Figure 1). For this, we used the publicly available dataset GSE145370 (47), which was derived from CD45+ sorted cells from oesophageal tumour and adjacent tissue. The 14 available samples (~108,000 single cells) were then processed through the STEGO.R pipeline (48). The low abundance and high sparsity of CD4 cells makes it difficult to distinguish the double negative (CD4−CD8−) T-cell population from the true CD4+ population (Figure 1, left column). As an illustration of missing populations, we looked at a common marker associated with CD4+ Tregs, FOXP3. The semi-automated method may miss many of the CD4+ T regulatory cells if CD4 is used in the annotation model. Therefore, other common CD4-specific markers, in the absence of CD8, may need to be used as a surrogate for correctly annotating CD4 subpopulations. Alternatively, the sorting of pure CD4+ T-cell subpopulations (i.e., Th1 and Th2), followed by bulk RNA-seq and differential expression analysis, may be required to identify new population specific transcriptional markers. This would aid in finding alternative transcriptional markers to identify CD4 subpopulations without the need to use the CD4 transcript for annotation purposes.
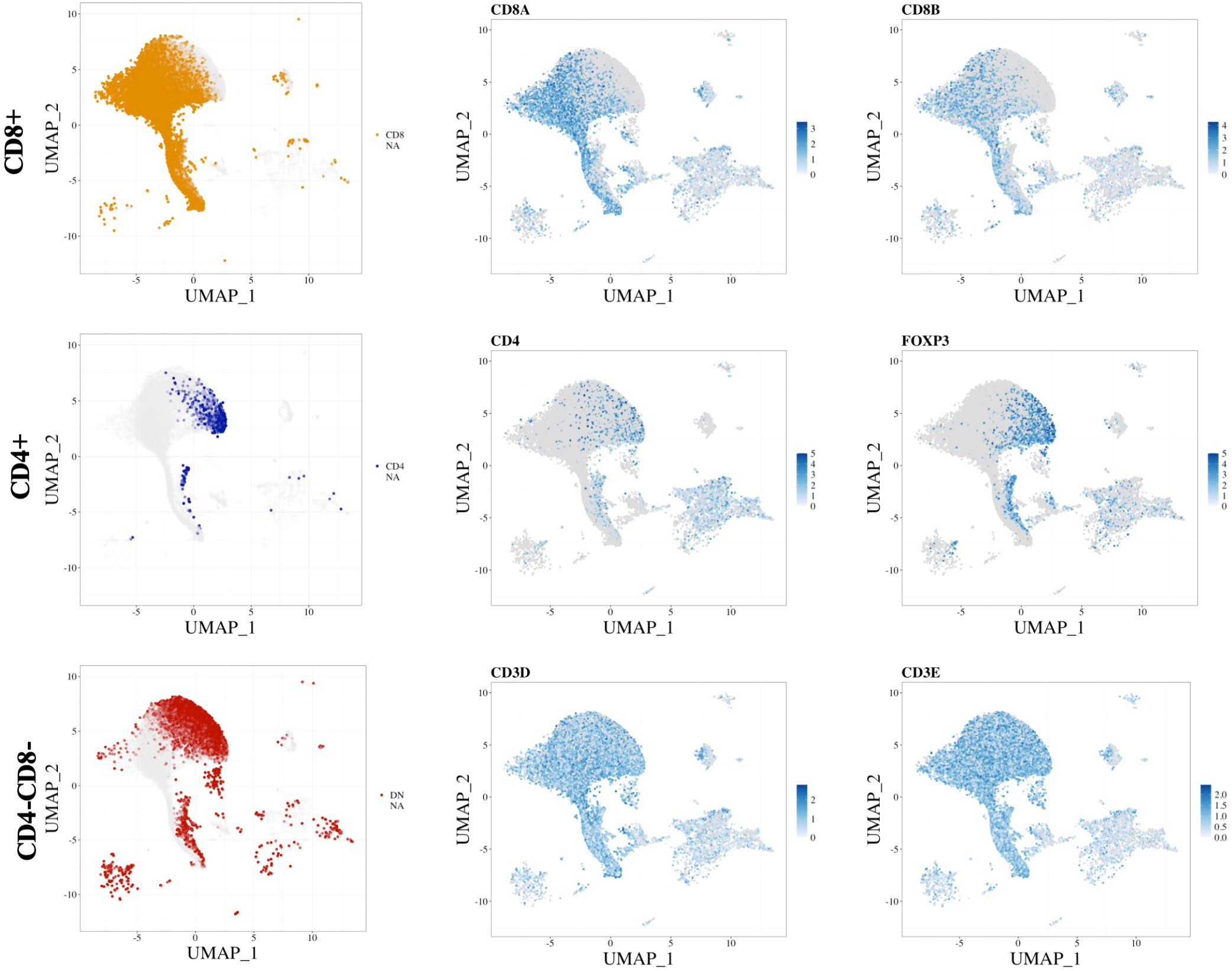
Figure 1 Marker sparsity of common T cell markers. represent the (top row) CD8+ T cells, (middle row) CD4+ T cells and (bottom row) double negative. The (left column) represents the scGate annotations, while the (middle and right columns) show the scaled expression of the markers of individual transcripts with the name listed above. The data was derived from an esophageal cancer set: GSE145370 (47), with the data processed and figure made with the aid of STEGO.R (48).
A combination of gene and protein expression layers could be used to resolve several of these annotation problems. This inclusion of a protein expression layer has been made possible by the cellular indexing of transcriptomes and epitopes through sequencing (CITE-seq), allowing the use of protein-specific antibodies within scRNA-seq. For instance, the issue of identifying CD4+ T cells that stems from low mRNA abundance could be resolved by the addition of CD4-specific antibodies to capture CD4 protein expression. Another such problem that could be resolved by CITE-seq is the identification of memory subsets within T-cell populations. CITE-seq resolves this by capturing the expression of two CD45 protein isoforms that originate from alternative splicing, CD45RO and CD45RA, to differentiate between naive T cells (CD45RA+/CD45RO−) and memory T cells (CD45RA−/CD45RO+) (10). However, while the inclusion of protein antibodies is easily able to resolve isoform expression, this task is not as simple as relying on RNA expression alone. The sequencing of isoforms typically requires full-length transcripts, and the read lengths required to cover these transcripts are not obtained by the commonly used short-read methods for scRNA-seq (49, 50). To illustrate this limitation, using CellTypist to annotate cells in a scRNA-seq experiment, we are currently unable to differentiate between a naive T cell and central memory T cell (Table 3). However, scRNA-seq with the CITE-seq has been able to identify the memory populations (51). Alternatively, long-read sequencing, for example by Oxford Nanopore Technologies (ONT) or PacBio, can be used for scRNA-seq, and can readily resolve splicing/isoform information. For instance, ONT-based single-cell RNA sequencing led to the detection of multiple CD45 isoforms, consistent with CITE-seq data (52). Thus, to properly annotate memory T-cell populations or other T-cell populations that are defined by protein markers with poor mRNA expression correlation, we will either need to include protein expression (e.g., CITE-seq), or sequence isoforms using techniques that capture the full length of a transcript, such as long-read sequencing.
An additional inconsistency between the protein expression and transcriptional profiling pertains to the degree of expression. With flow cytometry, protein expression can readily capture dose, including low, moderate, and high, based on arbitrary cut-offs. However, due to fewer transcripts being captured, there is limited capacity to have these grades of expression in scRNA-seq, and they can mostly only be differentiated by binary (e.g., present or absent) thresholds. For instance, CD127low protein expression is a marker for Tregs; however, this would be an inappropriate transcriptional marker (Table 2). Therefore, when designing a panel of transcriptional phenotyping markers, the expert will need to consider this technological limitation.
Overall, the above analysis identified inconsistencies with marker choice (Tables 2–5), which represents a concerning issue regarding the reproducibility of these T-cell studies. Additionally, there was a plethora of missing annotations (e.g., for Th2 cells, γδ T cell phenotypes, and functional features). Consequently, if these missing annotations are essential to identifying the T cell associated with a particular disease(s)/pathology (e.g., infection, cancer, autoimmune disease, and transplantation), using the automated models will lead to the T-cell subset of interest being missed. Therefore, filling in the missing annotations will need to be done manually or by way of a semi-automated process using custom gene sets.
T cells remain a challenging subset of immune cells to interrogate due to their complex and variable subspecialisations, together with the diversity of the TCR repertoire. There has been some progress made in the development of T cell-specific annotation strategies and in TCR repertoire interrogation [reviewed in (6)]. Technology has progressed to now include simultaneous scRNA-seq and scTCR-seq, which can capture both the αβTCR and γδTCR genes (e.g., 10x Genomics and BD Rhapsody). Both these layers of data are likely needed to identify the role individual T-cell clones are performing at a given time point. For example, scTCR-seq can capture the paired αβTCR or γδTCR sequence and identify if the clone was expanded. Clonal expansion may indicate whether or not a particular TCR has responded to an epitope/antigen. The functional state will also further indicate if it is worth undertaking further analysis of the T cell and enable bystander clones to be ruled out. This information is needed for functional validation so that sorting based on phenotype-specific biomarkers and TCR genes can be done, which in turn can eventually be used as immunotherapies (e.g., CAR-T or TCR-T) (53). Having access to both layers in the initial discovery single-cell experiment will decrease the time needed to identify the most biologically relevant T-cell clones.
A deep dive into the current annotation strategies identified that inconsistences exist in the subclassification of T cells, along with missing T-cell subsets. To rectify these phenotyping inconsistencies, we will need a central resource of well-curated classifications so we can estimate the robustness of the markers for any given T-cell subpopulation. We may need to consider not segregating the classification based on γδTCR vs. αβTCR, as new understanding is showcasing overlapping, if not identical, markers (Tables 2–4). To achieve this database, the T-cell community requires the development of a public repository for protein markers, bulk RNA-seq derived markers, and, if possible, scRNA-seq with scTCR-seq and protein antibody information. Once this is built, we can determine the most robust markers per T-cell subset. We believe this literature review provides a useful reference and may serve as a foundation in the realization of this effort.
Once a consistent gene-set list of markers is established, we need to tackle the remaining problems regarding how to efficiently interrogate scRNA with paired scTCR-seq data. To achieve this, expert T-cell functional knowledge and computational expertise will be needed. This could help in determining which T cells should be functionally tested, and may lead to groundbreaking discoveries that lead to novel T cell-based therapeutics or help guide patient management in current immunotherapy protocols.
In this study, we presented a comprehensive review of the tools used to annotate T cells from scRNA-seq datasets and also analysed the single-cell derived TCR repertoire. There are a multitude of automated strategies used to annotate T cells. However, the biggest shortcomings are a lack of consistency among tools concerning the markers used to annotate the T cell subsets, leading to severe issues with reproducibility. To overcome this challenge, collation of the currently available T cell-based data should be stored in a single repository, and development of new tools that make use of this harmonised framework is needed. Without this progress, there will continue to be issues with reproducibility, which will hamper progress in the development of T cell-based therapies.
KAM: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. NdV: Data curation, Writing – review & editing. SV: Data curation, Writing – review & editing. PM: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work has been made possible by grant number 2022-249472 from the Chan Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation. In addition, this work was supported by the Research Foundation Flanders [1S71721N to NdV, 1S40321N to SV].
The authors thank Professor Benson Ogunjimi for his feedback on the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wu X, Yang B, Udo-Inyang I, Ji S, Ozog D, Zhou L, et al. Research techniques made simple: single-cell RNA sequencing and its applications in dermatology. J Invest Dermatol (2018) 138(5):1004–9. doi: 10.1016/j.jid.2018.01.026
2. Liu J, Chang HW, Huang ZM, Nakamura M, Sekhon S, Ahn R, et al. Single-cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8(+) T cells in autoimmunity and cancer. J Allergy Clin Immunol (2021) 147(6):2370–80. doi: 10.1016/j.jaci.2020.11.028
3. Argyriou A, Wadsworth MH 2nd, Lendvai A, Christensen SM, Hensvold AH, Gerstner C, et al. Single cell sequencing identifies clonally expanded synovial CD4(+) T(PH) cells expressing GPR56 in rheumatoid arthritis. Nat Commun (2022) 13(1):4046. doi: 10.1038/s41467-022-31519-6
4. Penkava F, Velasco-Herrera MDC, Young MD, Yager N, Nwosu LN, Pratt AG, et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat Commun (2020) 11(1):4767. doi: 10.1038/s41467-020-18513-6
5. Moon JS, Younis S, Ramadoss NS, Iyer R, Sheth K, Sharpe O, et al. Cytotoxic CD8(+) T cells target citrullinated antigens in rheumatoid arthritis. Nat Commun (2023) 14(1):319. doi: 10.1038/s41467-022-35264-8
6. Valkiers S, Vrij N, Gielis S, Verbandt S, Ogunjimi B, Laukens K, et al. Recent advances in T-cell receptor repertoire analysis: Bridging the gap with multimodal single-cell RNA sequencing. ImmunoInformatics (2022) 5. doi: 10.1016/j.immuno.2022.100009
7. Attaf M, Huseby E, Sewell AK. alphabeta T cell receptors as predictors of health and disease. Cell Mol Immunol (2015) 12(4):391–9. doi: 10.1038/cmi.2014.134
8. Gully BS, Rossjohn J, Davey MS. Our evolving understanding of the role of the gammadelta T cell receptor in gammadelta T cell mediated immunity. Biochem Soc Trans (2021) 49(5):1985–95. doi: 10.1042/BST20200890
9. Pellicci DG, Koay HF, Berzins SP. Thymic development of unconventional T cells: how NKT cells, MAIT cells and gammadelta T cells emerge. Nat Rev Immunol (2020) 20(12):756–70. doi: 10.1038/s41577-020-0345-y
10. Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell (2021) 184(13):3573–3587 e29. doi: 10.1016/j.cell.2021.04.048
11. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol (2018) 19(1):15. doi: 10.1186/s13059-017-1382-0
12. Heumos L, Schaar AC, Lance C, Litinetskaya A, Drost F, Zappia L, et al. Best practices for single-cell analysis across modalities. Nat Rev Genet (2023) 24(8):550–72. doi: 10.1038/s41576-023-00586-w
13. Reimegard J, Tarbier M, Danielsson M, Schuster J, Baskaran S, Panagiotou S, et al. A combined approach for single-cell mRNA and intracellular protein expression analysis. Commun Biol (2021) 4(1):624. doi: 10.1038/s42003-021-02142-w
14. Pliner HA, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods (2019) 16(10):983–6. doi: 10.1038/s41592-019-0535-3
15. Andreatta M, Berenstein AJ, Carmona SJ. scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets. Bioinformatics (2022) 38(9):2642–4. doi: 10.1093/bioinformatics/btac141
16. Dominguez Conde C, Xu C, Jarvis LB, Rainbow DB, Wells SB, Gomes T, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science (2022) 376(6594):eabl5197. doi: 10.1126/science.abl5197
17. Fu R, Gillen AE, Sheridan RM, Tian C, Daya M, Hao Y, et al. clustifyr: an R package for automated single-cell RNA sequencing cluster classification. F1000Res (2020) 9:223. doi: 10.12688/f1000research.22969.2
18. Kang JB, Nathan A, Weinand K, Zhang F, Millard N, Rumker L, et al. Efficient and precise single-cell reference atlas mapping with Symphony. Nat Commun (2021) 12(1):5890. doi: 10.1038/s41467-021-25957-x
19. Lotfollahi M, Naghipourfar M, Luecken MD, Khajavi M, Buttner M, Wagenstetter M, et al. Mapping single-cell data to reference atlases by transfer learning. Nat Biotechnol (2022) 40(1):121–30. doi: 10.1038/s41587-021-01001-7
20. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol (2019) 20(2):163–72. doi: 10.1038/s41590-018-0276-y
21. Li G, Song B, Singh H, Prasath Surya VB, Grimes Leighton H, Salomonis N. Decision level integration of unimodal and multimodal single cell data with scTriangulate. Nat Commun (2023) 14(1):406. doi: 10.1038/s41467-023-36016-y
22. Zhang Z, Luo D, Zhong X, Choi JH, Ma Y, Wang S, et al. SCINA: A semi-supervised subtyping algorithm of single cells and bulk samples. Genes (Basel) (2019) 10(7). doi: 10.3390/genes10070531
23. Andreatta M, Corria-Osorio J, Muller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun (2021) 12(1):2965. doi: 10.1038/s41467-021-23324-4
24. Provine NM, Klenerman P. MAIT cells in health and disease. Annu Rev Immunol (2020) 38:203–28. doi: 10.1146/annurev-immunol-080719-015428
25. Dhodapkar MV, Kumar V. Type II NKT cells and their emerging role in health and disease. J Immunol (2017) 198(3):1015–21. doi: 10.4049/jimmunol.1601399
26. Wo J, Zhang F, Li Z, Sun C, Zhang W, Sun G. The role of gamma-delta T cells in diseases of the central nervous system. Front Immunol (2020) 11:580304. doi: 10.3389/fimmu.2020.580304
27. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel) (2016) 8(3). doi: 10.3390/cancers8030036
28. Gherardin NA, Souter MN, Koay HF, Mangas KM, Seemann T, Stinear TP, et al. Human blood MAIT cell subsets defined using MR1 tetramers. Immunol Cell Biol (2018) 96(5):507–25. doi: 10.1111/imcb.12021
29. Cheng D, Qiu K, Rao Y, Mao M, Li L, Wang Y, et al. Proliferative exhausted CD8(+) T cells exacerbate long-lasting anti-tumor effects in human papillomavirus-positive head and neck squamous cell carcinoma. Elife (2023) 12. doi: 10.7554/eLife.82705.sa2
30. Lawand M, Dechanet-Merville J, Dieu-Nosjean MC. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol (2017) 8:761. doi: 10.3389/fimmu.2017.00761
31. Hudspeth K, Fogli M, Correia DV, Mikulak J, Roberto A, Bella Della S, et al. Engagement of NKp30 on Vdelta1 T cells induces the production of CCL3, CCL4, and CCL5 and suppresses HIV-1 replication. Blood (2012) 119(17):4013–6. doi: 10.1182/blood-2011-11-390153
32. Zhao Y, Niu C, Cui J. Gamma-delta (gammadelta) T cells: friend or foe in cancer development? J Transl Med (2018) 16(1):3. doi: 10.1186/s12967-017-1378-2
33. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun (2020) 11(1):3434. doi: 10.1038/s41467-020-17292-4
34. Shive CL, Freeman ML, Younes SA, Kowal CM, Canaday DH, Rodriguez B, et al. Markers of T cell exhaustion and senescence and their relationship to plasma TGF-beta levels in treated HIV+ Immune non-responders. Front Immunol (2021) 12:638010. doi: 10.3389/fimmu.2021.638010
35. Su Y, Chen D, Yuan D, Lausted C, Choi J, Dai CL, et al. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell (2020) 183(6):1479–1495 e20. doi: 10.1016/j.cell.2020.10.037
36. Jenkins E, Whitehead T, Fellermeyer M, Davis SJ, Sharma S. The current state and future of T-cell exhaustion research. Oxf Open Immunol (2023) 4(1):iqad006. doi: 10.1093/oxfimm/iqad006
37. Hughes TK, Wadsworth MH 2nd, Gierahn TM, Do T, Weiss D, Andrade PR, et al. Second-strand synthesis-based massively parallel scRNA-seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity (2020) 53(4):878–894 e7. doi: 10.1016/j.immuni.2020.09.015
38. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol (2019) 19(11):665–74. doi: 10.1038/s41577-019-0221-9
39. Sauce D, Almeida JR, Larsen M, Haro L, Autran B, Freeman GJ, et al. PD-1 expression on human CD8 T cells depends on both state of differentiation and activation status. Aids (2007) 21(15):2005–13. doi: 10.1097/QAD.0b013e3282eee548
40. Garcillan B, Marin AV, Jimenez-Reinoso A, Briones AC, Munoz-Ruiz M, Garcia-Leon MJ, et al. gammadelta T lymphocytes in the diagnosis of human T cell receptor immunodeficiencies. Front Immunol (2015) 6:20. doi: 10.3389/fimmu.2015.00020
41. Gao J, Zhao L, Wan YY, Zhu B. Mechanism of action of IL-7 and its potential applications and limitations in cancer immunotherapy. Int J Mol Sci (2015) 16(5):10267–80. doi: 10.3390/ijms160510267
42. Qi C, Wang Y, Li P, Zhao J. Gamma delta T cells and their pathogenic role in psoriasis. Front Immunol (2021) 12:627139. doi: 10.3389/fimmu.2021.627139
43. Huber SA, Graveline D, Newell MK, Born WK, O'Brien RL. V gamma 1+ T cells suppress and V gamma 4+ T cells promote susceptibility to coxsackievirus B3-induced myocarditis in mice. J Immunol (2000) 165(8):4174–81. doi: 10.4049/jimmunol.165.8.4174
44. Benveniste PM, Roy S, Nakatsugawa M, Chen ELY, Nguyen L, Millar DG, et al. Generation and molecular recognition of melanoma-associated antigen-specific human gammadelta T cells. Sci Immunol (2018) 3(30):eaav4036. doi: 10.1126/sciimmunol.aav4036
45. Kierkels GJJ, Scheper W, Meringa AD, Johanna I, Beringer DX, Janssen A, et al. Identification of a tumor-specific allo-HLA-restricted gammadeltaTCR. Blood Adv (2019) 3(19):2870–82. doi: 10.1182/bloodadvances.2019032409
46. Van Rhijn I, Le Nours J. CD1 and MR1 recognition by human gammadelta T cells. Mol Immunol (2021) 133:95–100. doi: 10.1016/j.molimm.2020.12.008
47. Zheng Y, Chen Z, Han Y, Han L, Zou X, Zhou B, et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat Commun (2020) 11(1):6268. doi: 10.1038/s41467-020-20019-0
48. Mullan KA, Ha M, Valkiers S, Ogunjimi B, Laukens K, Meysman P. STEGO. R: an application to aid in scRNA-seq and scTCR-seq processing and analysis. bioRxiv (2023). doi: 10.1101/2023.09.27.559702
49. Arzalluz-Luque A, Conesa A. Single-cell RNAseq for the study of isoforms-how is that possible? Genome Biol (2018) 19(1):110. doi: 10.1186/s13059-018-1496-z
50. Ray TA, Cochran K, Kozlowski C, Wang J, Alexander G, Cady MA, et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat Commun (2020) 11(1):3328. doi: 10.1038/s41467-020-17009-7
51. Lakkis J, Schroeder A, Su K, Lee MYY, Bashore AC, Reilly MP, et al. A multi-use deep learning method for CITE-seq and single-cell RNA-seq data integration with cell surface protein prediction and imputation. Nat Mach Intell (2022) 4(11):940–52. doi: 10.1038/s42256-022-00545-w
52. Tian L, Jabbari JS, Thijssen R, Gouil Q, Amarasinghe SL, Voogd O, et al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol (2021) 22(1):310. doi: 10.1186/s13059-021-02525-6
Keywords: T cells, single cell, RNA-seq, annotation, bioinformatics, adaptive immunity, T-cell receptor
Citation: Mullan KA, de Vrij N, Valkiers S and Meysman P (2023) Current annotation strategies for T cell phenotyping of single-cell RNA-seq data. Front. Immunol. 14:1306169. doi: 10.3389/fimmu.2023.1306169
Received: 03 October 2023; Accepted: 27 November 2023;
Published: 21 December 2023.
Edited by:
Qi-Jing Li, Institute of Molecular and Cell Biology (A*STAR), SingaporeReviewed by:
Arundhoti Das, National Institutes of Health (NIH), United StatesCopyright © 2023 Mullan, de Vrij, Valkiers and Meysman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pieter Meysman, cGlldGVyLm1leXNtYW5AdWFudHdlcnBlbi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.