 Li Zhao1†
Li Zhao1† Xuezhi Sun
Xuezhi Sun Feng Gao
Feng Gao Chunlan Zheng
Chunlan Zheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 January 2024
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1305325
This article is part of the Research Topic Innate Immune Response and Interaction with Adaptive Immunity: Role in Tuberculosis Immunology/Vaccinology View all 6 articles
Tuberculosis (TB) is caused by the bacterial pathogen Mycobacterium tuberculosis (MTB) and is one of the principal reasons for mortality and morbidity worldwide. Currently, recommended anti-tuberculosis drugs include isoniazid, rifampicin, ethambutol, and pyrazinamide. TB treatment is lengthy and inflicted with severe side-effects, including reduced patient compliance with treatment and promotion of drug-resistant strains. TB is also prone to other concomitant diseases such as diabetes and HIV. These drug-resistant and complex co-morbid characteristics increase the complexity of treating MTB. Host-directed therapy (HDT), which effectively eliminates MTB and minimizes inflammatory tissue damage, primarily by targeting the immune system, is currently an attractive complementary approach. The drugs used for HDT are repositioned drugs in actual clinical practice with relative safety and efficacy assurance. HDT is a potentially effective therapeutic intervention for the treatment of MTB and diabetic MTB, and can compensate for the shortcomings of current TB therapies, including the reduction of drug resistance and modulation of immune response. Here, we summarize the state-of-the-art roles and mechanisms of HDT in immune modulation and treatment of MTB, with a special focus on the role of HDT in diabetic MTB, to emphasize the potential of HDT in controlling MTB infection.
Tuberculosis (TB), caused by infection with Mycobacterium tuberculosis (MTB), remains one of the major health problems worldwide, with millions of new cases and more than one million deaths from TB each year (1). The combination of Bacillus Calmette-Guerin (BCG) vaccination and multiple antibiotics is clinically used as a strategy to control the transmission and progression of TB. However, BCG vaccination usually fails to achieve the desired protective effect, and treatment of TB with drug combinations such as isoniazid, rifampicin, pyrazinamide, and ethambutol requires up to 6-9 months of standard treatment (2). TB treatment is lengthy and has serious side effects, including reduced patient compliance with treatment and promotion of drug-resistant strains. Treating drug-resistant strains of TB is much more difficult compared to common infections (3). Alarmingly, drug resistance to TB is becoming a knotty issue.
At the same time, TB is also prone to other concomitant diseases such as diabetes and HIV (4, 5). Epidemiologic studies have shown that as many as 25-54% of newly diagnosed TB patients also have diabetes mellitus (DM) (6, 7), and up to 80% of patients with DM reside in low and middle-income level countries, which highly overlaps with the prevalence of TB (8). These drug-resistant and complex co-morbid characteristics increase the complexity of treating TB. Therefore, there is a need to develop and apply new therapeutic approaches to TB.
Host-directed therapy (HDT) is a novel adjunctive therapeutic strategy that has emerged in recent years in the treatment of TB infections. Unlike traditional antimicrobial therapy, HDT usually acts on the immune system of the host rather than the pathogens (9). Through specific modulation of the host immune system and immune processes, HDT can achieve the goal of limiting or enhancing the inflammatory response and interfering with host defense mechanisms utilized by pathogens. These immune effects enhance the killing effect on pathogens while attenuating pathological damage to the host, which is important for combating MTB infections, especially against drug-resistant MTB (10, 11). HDT reduces lung inflammation, improves focal drug penetration, and induces antimicrobial activity of phagocytes, thereby protecting the lungs, improving long-term survival, and shortening treatment time.
Consequently, HDT helps to address the current problems of complicated TB treatment procedures and the increase of drug-resistant strains (12). In this review, we comprehensively characterize the immune features of MTB infection and focus on elucidating the mechanisms and drug candidates associated with HDT. Notably, we focus on the current state of studies on HDT in DM with MTB.
The first contact of MTB with the human body is called primary infection. Pulmonary tuberculosis is the most common consequence of MTB infection. In aerosolized form, MTB passes through the respiratory tract into the lungs and in most cases enters the distal alveolar cavities (13). MTB can infect respiratory epithelial cells or mucosal tissues and reach the alveoli with two main fates: phagocytosis by alveolar-resident immune cells such as macrophages/dendritic cells (DCs) or successful infection of alveolar epithelial cells. The latter may lead to further spread of MTB, triggering TB in other parts of the body (14, 15). In most primary infections, the immune system can either completely clear the invading MTB or control the infection by forming stable granulomas (16).
TB-specific granulomas are formed by the organized aggregation of different types of immune cells at the site of infection, in which there is a complex and dynamic interaction between MTB and the host (17). From the perspective of the host, granuloma formation effectively limits the proliferation of MTB and the progression of the disease, which is the outcome in most major infected individuals. As for MTB, granulomas, while limiting the proliferation and spread of MTB within the host, allow MTB to remain dormant without being completely cleared by the host immune system, creating a state of latent TB infection reserving the possibility of subsequent reactivation (18–20).
When the immune function of the host is impaired, the core of the granuloma undergoes caseous changes and liquefaction in response to the accumulation of foam cells transformed from macrophages and a large number of necrotic host immune cells (21, 22). This change markedly diminishes the ability of the granuloma to restrict MTB, which may lead to bacterial release and disease progression at any time (23).
Macrophages are an important component of TB pathogenesis, participating in early exposure to MTB and acting as a refuge for MTB during chronic infection. When MTB invades, alveolar macrophages rapidly recognize microbe-associated molecular patterns (MAMPs) on the surface of MTB through pattern recognition receptor (PRR), and engulf MTB into the phagosome for acidic clearance, while recruiting other types of macrophages and participating in antigen presentation (24–26). Macrophages can secrete a variety of cytokines and chemokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-23, and granulocyte-macrophage colony-stimulating factor (GM-CSF), and also fight against MTB infection through apoptosis and autophagy (27). But, accordingly, MTBs can also resist their host clearance, such as by inducing the conversion of macrophages into foam cells or inducing macrophage polarization through the production of mycolic acid, for example. Strongly toxic MTB can also prevent the fusion and acidification of phagolysosomes, thus successfully converting macrophages into cells that assist them in evading the host immune response (28, 29). Several new studies have also found that MTB can exert its pathogenic ability by modulating host cell metabolism (30–33).
DCs are involved in antigen processing and presentation of MTB and are key to bridging innate and adaptive immunity (34). With the help of DC-SIGN and toll-like receptor (TLR), DC accomplishes the recognition and self-activation of MTB (35). After activation, DC secretes IL-12 and upregulates the expression of MHC-I, MHC-II, CD 40, CD 54, CD58, and CD80, and then migrates to mediastinal lymph nodes, where it activates a Th1-type specific response marked by interferon (IFN)-γ secretion through antigen presentation (36–38). MTB can influence DC immune function by interacting with DCs and modulating the secretion of anti-inflammatory and pro-inflammatory factors (39, 40).
Neutrophils are the most abundant cell type in the respiratory secretions of patients with active TB and are the cells that can be rapidly recruited and infiltrated after MTB infection. Neutrophils appear to be a double-edged sword in MTB infection. Early in the infection, neutrophils are recruited toward the infected foci in response to various cytokines and chemokines and then exert anti-TB effects along with macrophages, by releasing non-targeted inflammatory mediators (41, 42). On the other hand, the greater number of neutrophils in the lungs correlates with increased pathological damage caused by TB (43). MTB not only induces necrosis of neutrophils, but these necrotic neutrophils phagocytosed by macrophages also promote intracellular survival of MTB (44).
NK cells are a type of innate lymphoid cells with potent cytolytic capacity. NK cells can directly lyse infected cells through cytotoxicity or secrete cytokines to exert anti-infective effects (45). NK cells can be activated by macrophage stimulation and also secrete IFN-γ and IL-22, which act on macrophages to inhibit MTB intracellular growth by enhancing phagolysosomal fusion (46, 47). Not only that, but NK cells are also involved in TB granuloma formation. During MTB infection, the function of NK cells seems to be suppressed, but the exact mechanism of action remains to be further investigated (48, 49).
After the early response of innate immunity to MTB, adaptive immunity plays a key role in the control of MTB infection in the mid- and late stages. A variety of T cells, including CD4+ effector T cells, CD4+ regulatory T cells, and CD8+ cytotoxic T cells, are involved in the anti-MTB immune process (50). During MTB infection, macrophages and DCs recruited to the lungs could phagocytose and process MTB antigens and present them to CD4+ T cells after migration to secondary lymphoid organs. CD4+ T cells are stimulated by specific antigens caused by Tuberculosis (TB). Activated CD8+ T cells can secrete a variety of cytotoxic effector molecules together with NK/NKT cells to kill intracellular bacteria by lysing infected cells (51). Humoral immunity is protective against MTB infection (52). Specific neutralizing antibodies in the mucosal barrier alone could protect against MTB invasion, whereas the bacterial load in the lungs of the B-cell-depleted MTB-infected mouse model was significantly increased (53). Theoretically, every process from the invasion of MTB into the body to its eventual removal could be a potential target for HDT (Figure 1).
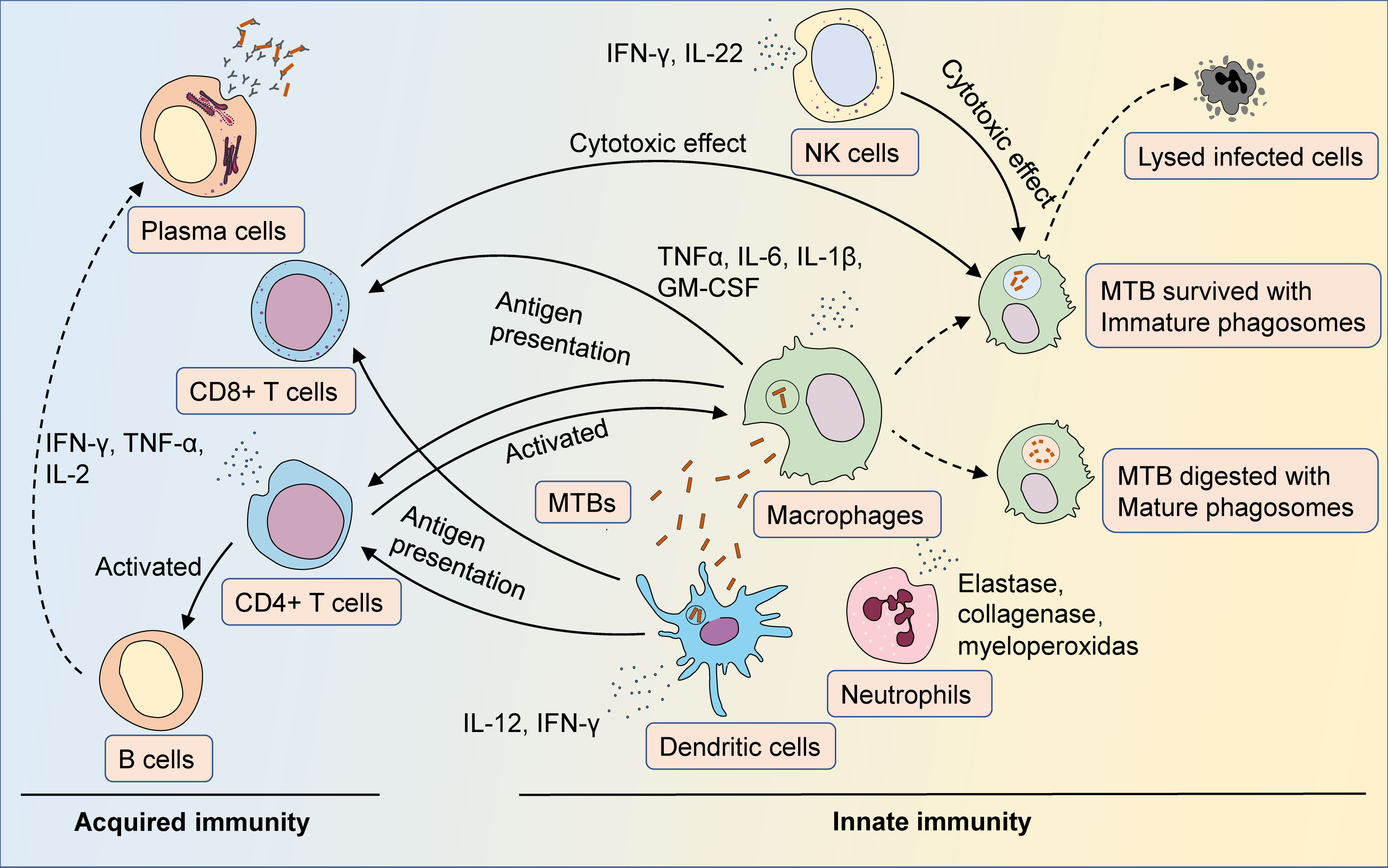
Figure 1 The immune response of MTB infection. After entering the body, MTB is first recognized by antigen-presenting cells such as macrophages and dendritic cells. Antigen-presenting cells phagocytose and process MTB, then activate T cells via antigen presentation, as well as secrete cytokines and chemokines to facilitate the initial immune response. Neutrophils could be recruited to the site of infection, fighting MTB infection by secreting cytokines and lysozyme. Macrophages are the major cells that fight MTB infection. Macrophages phagocytize MTB and eliminate bacteria by forming mature phagosomes. MTB could prevent itself from being eliminated by pathogen-host interactions that inhibit the maturation of phagosomes. Once activated, CD4+ T cells could trigger Th1- and Th17-type immune responses against MTB infection, and secrete cytokines such as IFN-γ, TNF-α, and IL-2 to enhance the antimicrobial activity of macrophages. Activated CD8+ T cells could synergize with NK/NKT cells to lyse infected cells through cytotoxic effects. Humoral immunity is also activated during MTB infection, with antibodies secreted by plasma cells assisting in countering the spread of the infection.
Cytokines are a class of biologically active small molecular peptides or glycoproteins produced by cells that possess the ability to regulate intrinsic and adaptive immunity. The main cytokines involved in MTB infection, mainly include IFN-α, IFN-γ, IL-2, IL-6, IL-10, IL-13, IL-17, TNF-α, and chemokines (54). IL-17A regulated inflammatory factors and T-cell recruitment against MTB infection. In contrast, IL-27 suppressed IL-17 A production, thereby limiting the anti-TMB immune response. Blocking IL-27R exemplified its potential as an HDT (55). During acute MTB infection, IL-4i1 deficiency led to increased M1 polarization, nitrite and IFN-γ expression, which enhanced protection against infection and was expected to be an immunomodulatory target (56). Khan et al. found that activation of CD40 and TLR-4 induced increased secretion of IL-12, IL-6, and TNF-α and enhanced autophagy by DCs, as well as enhanced adaptive immunity and bactericidal effects of anti-TB drugs. These effects led to a significant reduction of bacterial load in the lungs (57). Notably, there is a correlation between plasma levels of eicosanoids and MTB infection status in patients with MTB (58). MTB infection elicited an imbalance of eicosanoids in the host, which in turn contributed to imbalanced IL-1 and IFN-γ expression, whereas IL-1 increased host resistance to MTB by inducing arachidonic acid-like and limiting excessive I-IFN production (59).
Histone deacetylase (HDAC), an important class of enzymes in histone modification, is involved in epigenetic regulatory processes (60). MTB promotes self-survival by affecting HDAC transcript levels, and inhibition of HDAC activity enhances macrophage MTB resistance (61). HDAC3 inhibitor RGFP966 could inhibit MTB growth through anti-microbial activity and modulation of macrophage pro-inflammatory factor secretion (62). SIRT1 and SIRT7 expression was down-regulated in MTB-infected macrophages. Cheng et al. found that SIRT1-induced phagocytosis of lysosomal-lysosomal fusion and autophagy was activated and inhibited the inflammatory response through RelA/p65 deacetylation, reducing the intracellular growth of MTB and MTB-resistant strains (63). Zhang et al. found that silencing SIRT7 expression could limit NO production in macrophages, reduce their frequency of early apoptosis, and promote intracellular growth of MTB, suggesting a candidate molecule for HDT.
Ferroptosis is a regulated necrosis triggered by a combination of iron toxicity, lipid peroxidation, and plasma membrane damage involved multiple infection-immunomodulatory processes (40, 64). Dai et al. found that MTB infection enhanced intracellular MTB iron utilization and promoted its proliferation through TRIM21- and NCOA4-dependent ferritinophagy. This confirmed the effectiveness of ferritin metabolism as an anti-MTB target (65). Amaral et al. found that infection of GPX4-deficient mice with MTB produced higher levels of lung necrosis and bacterial load. GPX4-deficient macrophages infected with MTB exhibited necrosis, suggesting the involvement of ferroptosis in MTB infection and the possibility of glutathione (GSH)/GPX4 as an HDT target for MTB (66).
In macrophages, galectin-8 recognized damaged MTB-containing phagosomes and then targeted MTBs for selective autophagy (67). Galectin-8 appeared to be required for targeting and controlling MTB in macrophages. Alarmin S100A8/A9 mediated neutrophil accumulation by upregulating CD11b expression during progression to chronic TB, leading to worse disease outcomes (68). SP110b is an IFN-induced nuclear protein that governs host innate immunity to MTB infection. SP110b regulated the activity of NF-κB to inhibit IFN-γ-mediated monocyte and macrophage death, effectively controlling the growth of MTB while reducing inflammatory lesions in the lungs (69). MTB promoted intracellular survival in macrophages by elevating triacylglycerol (TAG) storage in macrophages via the WNT6/ACC2 pathway and could be considered as an HDT target (70). Pires et al. demonstrated that silencing Cystatin C significantly increased intracellular killing of MTBs by macrophages, with a concomitant promotion of HLA-II expression and IFN-γ secretion, and CD4+ T cell proliferation in macrophages (71). The use of siRNA Cystatin F produced a similar effect (72). Smyth et al. found that MTB infection induced up-regulation of protein kinase R (PKR) expression and activation in macrophages and inhibited intracellular MTB survival through activation of IFN-γ-mediated autophagy, confirming the validity of PKR as an HDT target (73). Inhibition of transglutaminase 2 (TG2) could limit MTB growth within macrophages (74). TG2 inhibitors can inhibit MTB growth by affecting macrophage autophagy and inhibit granuloma growth (75). In addition, inhibition of PARP is a potential strategy to effectively alter macrophage chemokine secretion and inhibit MTB and multidrug-resistant (MDR)-MTB levels, which could be used to develop HDT therapies targeting intracellular MTBs (76).
Non-coding RNAs (ncRNAs) account for 98% of total RNAs and are the major RNA transcripts in organisms. These transcribed sequences mainly include lncRNAs, miRNAs, and circRNAs, which generally do not encode proteins but play regulatory roles at the transcriptional or post-transcriptional level (77). NcRNAs interact with each other in a complex network of roles. Currently, specific ncRNA species and expression levels abnormally occur in MTB infection, and these ncRNAs may be involved in the regulation of host-MTB interactions, and thus are expected to be potential candidates for HDT.
MTB could promote its survival by affecting the activity of lysosomal tissue proteases in host macrophages (78). Notably, miR-106b-5p, specifically up-regulated by MTB, was involved in the process of MTB on lysosomal tissue protease activity, which was achieved by binding to CTSS mRNA. Knockdown of miR-106b-5p in infected cells significantly increased the expression and activity of CTSS, as well as the level of T-cell activation and intracellular killing of MTB by macrophages. Additionally, the expression levels of miR-143 and miR-365 were significantly increased in MTB-infected macrophages (79). Knockdown of the expression of both could reduce the production of IL-6 and CCL-5, inhibit the proliferation and growth of MTB in macrophages, and induce apoptosis in infected macrophages. Further, BACH-1 and C-Maf were direct targets of miR-143, whereas miR-365 directly targeted cMaf, Bach1, and Elmo-1. Fu et al. demonstrated that the miR-342-3p/SOCS6 axis could affect the expression of inflammatory cytokines and chemokines and regulate the transition between MTB-induced apoptosis and necrosis by mediating K48-linked ubiquitination and RIPK3 degradation via A20 (80). Overexpression of MiR-342-3p improved resistance to MTB in mice. MiR-27a was abundantly expressed in MTB-infected animal, human, and macrophage cells (81). MiR-27a inhibited Ca2+ signaling by targeting the ER-located Ca2+ transporter protein CACNA2D3. Down-regulation of miR-27a levels increased resistance to MTB infection in mice. Targeting these miRNAs may be able to serve as the candidates for HDT.
LncRNAs are a category of ncRNAs with lengths greater than 200 nt, characterized by tissue-specific and spatiotemporal specificity, and low sequence conservation (82). In MTB infection, the expression signature of lncRNAs associated with immunoregulatory pathways is dysregulated (83). Gcanga et al. revealed that the expression of lincRNA-MIR99AHG correlated with the polarization status of macrophages (84). Lower levels of lincRNA-MIR99AHG were present in macrophages infected with MTB and in peripheral blood monocyte-derived macrophages from patients with active TB, and its expression was induced by stimulation of macrophages with IL-4/13. Knockdown of lincRNA-MIR99AHG significantly reduced intracellular MTB growth and proinflammatory cytokine production in mouse/human macrophages and reduced MTB load in mouse lungs and spleens.
Ibrutinib is a classical anti-chronic lymphocytic leukemia agent. Hu et al. found that ibrutinib induced macrophage autophagy by inhibiting the BTK/Akt/mTOR pathway, inhibited intracellular MTB growth in macrophages, and significantly attenuated MTB loading in mice (85). Boland et al. proved that the antitumor agent tamoxifen, although it lacked a direct antimicrobial effect, could produce an anti-TB effect by inducing autophagy rather than targeting its classical estrogen receptor pathway (86). This finding was further validated in a zebrafish model of TB infection, confirming the HDT potential of tamoxifen.
Linezolid is used in anti-tuberculosis therapy but carries some host toxicity. Winchell et al. demonstrated that co-administration of IL-1 blockers and linezolid attenuated side effects resulting from linezolid treatment and enhanced the therapeutic effect of linezolid in MTB-infected rhesus monkeys (87). Pires et al. found that lysosomal proteases were down-regulated after MTB infection of macrophages, thereby promoting macrophage survival. Treatment with saquinavir induced an increase in lysosomal protease activity and promoted intracellular MTB killing, as well as improved the expression of HLA class II antigens and the secretion of IFN-γ, while promoting the activation and proliferation of T lymphocytes (88). They further found that liposomal saquinavir could improve the transformation efficiency and reduce the drug cytotoxicity, and significantly enhance the killing effect of macrophages against MDR and XDR strains (89). There have been no clinical trials of saquinavir as an HDT drug for the treatment of tuberculosis. However, Ribera et al. found that the combination of saquinavir, ritonavir, and rifampicin, may result in lower blood levels of saquinavir and ritonavir in patients (90). Considering the wide use of rifampicin in the treatment of TB, more studies are needed on the methodology and efficacy of the use of saquinavir as an HDT drug for TB.
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase with a molecular weight of 289 kDa, which has been universally used for the treatment of cancer and against organ transplant rejection (91). By analyzing the gene expression of MTB-infected DCs, Etna et al. found that rapamycin could regulate the transcriptional expression of IFN and downstream products in a GSK-3β-dependent manner, as well as regulate the balance of pro- and anti-inflammatory factors (92). But another mTOR inhibitor everolimus showed unsatisfactory results. A relapse of tuberculosis was found in the renal transplant patient and the renal cell carcinoma patient treated with everolimus (93, 94). Additionally, the patient with a neuroendocrine pancreatic tumor developed two tuberculosis infections within a year and a half while being treated with everolimus (95). Studies of strains isolated from his body showed that although everolimus stimulated autophagy and the production of reactive oxygen species, it did not exhibit direct or indirect antimicrobial activity against MTB. Although everolimus promoted the recovery of lung function in a phase II clinical trial evaluating the effectiveness of everolimus as HDT in the treatment of TB (96), further preclinical and clinical studies are needed to determine the safety and scope of its use as HDT for TB.
Appropriate levels of prostaglandin E2 (PGE2) are necessary to induce an optimal innate immune response early in MTB infection. Inhibition of cyclooxygenase-2 (COX-2) with celecoxib significantly reduced PGE2, enhanced IFN-γ production and NO release, and increased phagocytosis of MTB by macrophages (97). However, Mortensen et al. showed in different mouse models that although COX inhibitor treatment was commonly used to alleviate TB symptoms, it actually impaired the Th1-type immune response after MTB infection, reduced Th1 differentiation and IFN-γ expression, and decreased the protective capacity of CD4+ T cells against overt transfer (98). Mo et al. evaluated the effects of 4 calcium modulators on MTB in macrophages and found that flunarizine treatment could enhance macrophage bactericidal capacity by promoting maturation and acidification of phagolysosomes by increasing CaM and pCaMKII levels (99) (Figure 2).
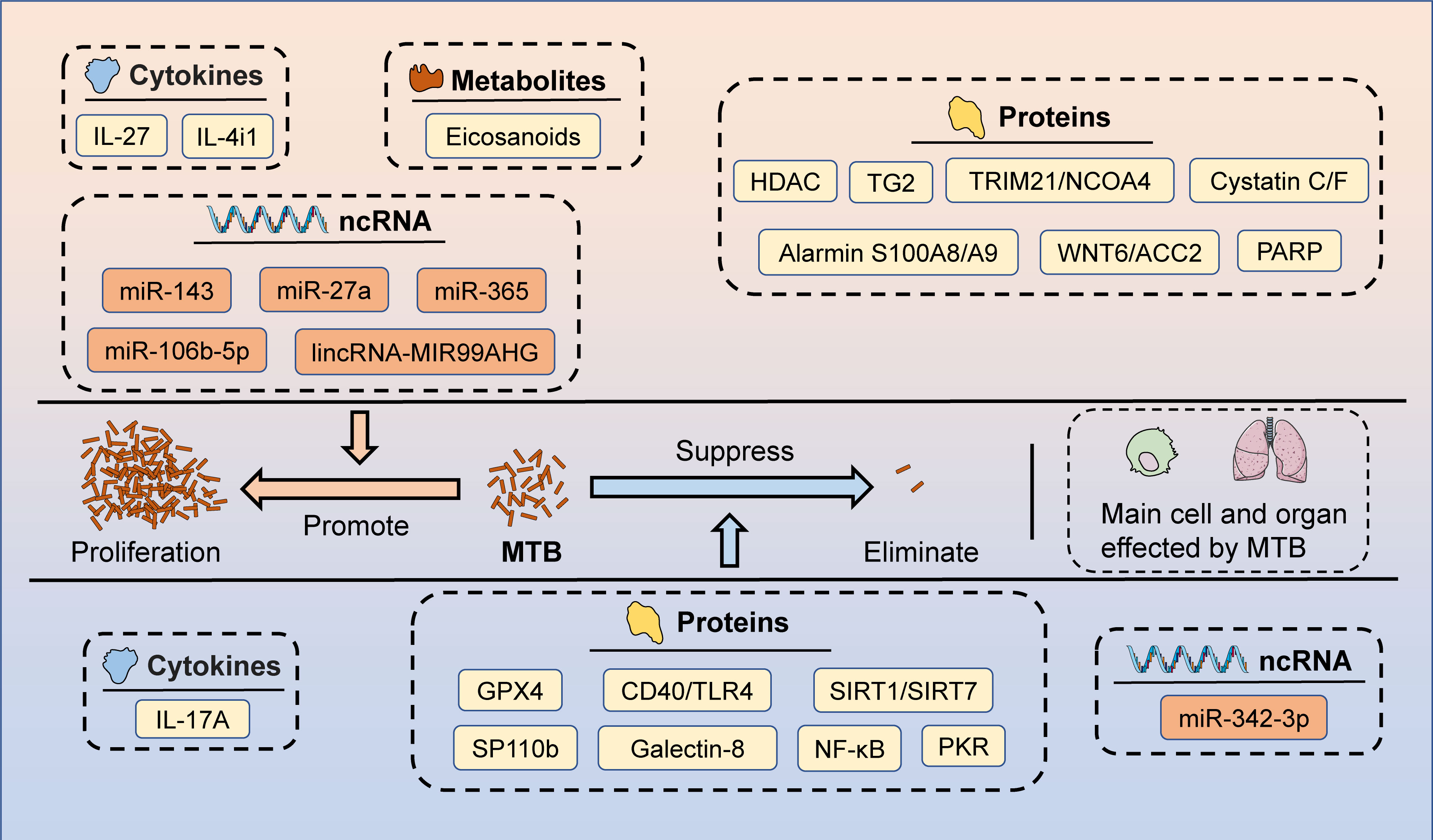
Figure 2 The potential targets of HDT for MTB infection. Macrophages and lungs are the primary cells and organs affected by MTB after the invasion of the body. Several candidate targets have been identified that could potentially be used for HDT, including cytokines (promote MTB proliferation: IL-27 and IL-4i1; inhibit MTB proliferation: IL-17A), metabolites (promote MTB proliferation: Eicosanoids), proteins (promote MTB proliferation: HDAC, TG2, TRIM21/NCOA4, Cystatin C/F, Alarmin S100A8/A9, WNT6/ACC2, and PARP; inhibit MTB proliferation: GPX4, CD40/TLR4, SIRT1/SIRT7, SP110b, Galectin-8, NF-κB, and PKR), and ncRNAs (promote MTB proliferation: miR-143, miR-27a, miR-365, miR-106b-5p, and lincRNA-MIR99AHG; inhibit MTB proliferation: miR-342-3p).
Adjuvant HDT is expected to be a potentially innovative alternative to improve the efficacy of conventional antimicrobial therapy for MTB. Some immunomodulators or compounds can effectively improve the concentration and effect of therapeutic drugs such as rifampicin, functioning as sensitizers with therapeutic safety for MTB therapy. For example, Subbian et al. prepared a rabbit model infected with virulent MTB tuberculosis using aerosol exposure and administered a combination regimen of phosphodiesterase-4 inhibitor (PDE4i) CC-11050 and isoniazid (100). INH + CC-11050 treatment was more efficient than isoniazid alone in terms of increased MTB clearance efficiency, and the inflammatory state was suppressed, as evidenced by the downregulation of inflammatory factors such as TNF-α. Previously, they had demonstrated the effectiveness of another PDE4i, CC-3052, in combination with isoniazid for the treatment of rabbit tuberculosis (101, 102). Similarly, in a mouse model, they also confirmed the effectiveness of PDE4-i CC11050/3052 in controlling the inflammation in the lungs of MTB-infected mice (103, 104). These demonstrated that PDE4-i adjuvant therapy significantly improved the response to isoniazid treatment in model with experimental tuberculosis and could be a candidate for HDT. In a Phase II clinical trial, CC11050 was shown to have a favorable safety and tolerability profile in the adjuvant treatment of tuberculosis, along with facilitating the recovery of lung function (96). Enhancement of NOD-2 and TLR-4 signaling contributed to the enhancement of host immunity in a mouse model of MTB, and reduced the dose and duration of rifampicin and isoniazid treatment (105).
Natural products are substances naturally produced by organisms that are pharmacologically or biologically active. Natural products have been used since ancient times to treat human diseases. Recent advances in modern science and technology have made it possible to purify and identify the active ingredients in natural products, thus further improving the efficiency of their utilization (106, 107). Several studies have focused on the utilization of active ingredients in natural products for HDT, and some progress has been made.
Andrographolide inhibited the Notch1/Akt/NF-κB signaling pathway, contributing to the inhibition of NLRP3 inflammasome activation and subsequent IL-1β production, which probably facilitated the improvement of macrophage resistance to MTB infection (108). Ursolic acid boosted macrophage autophagy and enhanced intracellular killing of MTB by synergistically inhibiting Akt/mTOR and TNF-α/TNFR1 (109). Therefore, ursolic acid may be a potential adjuvant drug for anti-tuberculosis HDT by virtue of its macrophage-inhibiting, inflammation-suppressing function. Baicalin was capable of inducing autophagy activation in MTB-infected macrophages by inhibiting the PI3K/Akt/mTOR pathway. This further limited the generation of inflammation-associated NLRP3 inflammasome and IL-1β. Thus, baicalin exhibited anti-inflammatory and anti-MTB functions with HDT potency (110). Baicalein promoted autophagy by inhibiting the Akt/mTOR pathway, downregulating AIM2 and NLRP3 inflammasomes, and inhibiting pyroptosis in MTB-infected macrophages (111). The immune function and antimicrobial capacity of baicalein made it a candidate for HDT in treating MTB.
The marine natural product Clionamines stimulated macrophage autophagy by down-regulating PI4KB and inhibited MTB survival in macrophages (112). Guttiferone K isolated from Yunnan Baiyao was able to inhibit the downstream NF-κB and Akt/mTOR signaling pathways via the TLR/IRAK-1 pathway, thereby significantly inducing autophagy and suppressing MTB-induced inflammatory responses in macrophages (113). Berbamine was shown to activate macrophage autophagy by modulating the ROS/Ca2+ axis, and could efficiently inhibit MTB, demonstrating the advantage of improving the efficacy or shortening the course of treatment in drug-resistant TB (114). The antigout drug colchicine enhanced macrophage resistance to MTB by modulating NLRP3-dependent IL-1β signaling and COX-2-mediated PGE2 production in macrophages (115).
Negi et al. investigated that the β-glucan polysaccharide, curdlan, activated STAT-1 to induce inducible nitric oxide synthase (iNOS) and NO expression, thereby enhancing the anti-MTB activity of macrophages (116). In a mouse model infected with MTB, curdlan activated protective Th1 and Th17 immunity and significantly reduced MTB load in the spleen and lung of mice. Polysaccharide-rich extract (PRE) isolated from Tinospora cordifolia treatment upregulated pro-inflammatory factor expression and activation of p38, ERK, JNK, and MAPK, and inhibited the survival of drug-resistant MTB in macrophages by inducing NO expression (117).
Curcumin, a component of traditional medicine derived from turmeric, is a lipophilic polyphenol. Curcumin has demonstrated impressive therapeutic efficacy in inflammatory, tumor, and infectious diseases, accompanied by low cost and excellent tolerability (118). Curcumin treatment induced autophagy and apoptosis in infected/uninfected MTB macrophages, with the conversion of LC3-I to LC3-II, the degradation of p62, and upregulation of the pro-apoptotic proteins Bax, Casp3, and PARP, as well as the reduced expression of the anti-apoptotic protein Bcl (119). CMN encapsulation in PLGA to form nanoparticles (ISCurNP) improved CMN bioavailability and more effectively assisted in killing MTBs in RAW 264.7 macrophages.
Nebulized inhalation is a direct drug delivery method using the respiratory tract and lungs as target organs, with the advantages of rapid onset of action, high local concentration, easy application, and fewer systemic adverse effects (120). The use of inhaled drugs for MTB treatment is a potentially efficient therapeutic tool that is gradually receiving attention. For instance, the utilization of STAT3 inhibitor ST3-H2A2 or IL-10Ra inhibitor IL10R1-7, when administered in aerosol form, effectively reduced MTB load in mouse lungs and was accompanied by an increase in the anti-bactericidal activities of NOS, NADPH oxidase and lysozyme (121). Targeting the host lung IL-10-STAT3 pathway in aerosol form, potentiated host bactericidal capacity and modulated apoptosis and autophagy pathways, serving as a novel HDT option.
All-trans retinoic acid (ATRA), the most common metabolite of vitamin A, promotes transcriptional activation of differentiation-associated genes and plays an important role in the treatment of cancer, dermatological disorders, acute promyelocytic leukemia (APL), and MTB infections (122). In the treatment of MTB, ATRA can modulate the immune robustness of the host body by regulating the expression of specific genes, monocyte activation, and induction of autophagy, which are promising candidates for HDT. The application of ATRA may result in varying degrees of side-effects due to off-target effects, and therefore the development of different ATRA-loaded drug delivery systems is highly prospective. O’Connor et al. developed an ATRA-loaded microparticle (ATRA-MP) that could be inhaled via the respiratory tract and was better targeted for delivery to alveolar macrophages than ATRA solutions (123). In the MTB-infected mouse model, three doses of ATRA-MP administered intratracheally resulted in a significant reduction in lung MTB load and improved lung pathology. Furthermore, Bahlool et al. also constructed ATRA-loaded PLGA nanoparticles (ATRA-PLGA NPs) in HDT against Tuberculosis (124). ATRA-PLGA NPs could target and be delivered to THP-1 cells in large quantities, leading to inhibition of H37Ra MTB proliferation in macrophages in a dose-dependent manner. ATRA-PLGA NPs also reached high concentrations locally in the lungs and were ultimately uptaken and therapeutically acted upon by MTB-infected cells. These studies highlighted the targeting, efficacy, and safety of inhalable, controlled-release ATRA formulations in HDT, providing a novel therapeutic tool for MTB. Singh et al. designed liposomes targeting CD44 using thioaptamers (CD44TA-LIP) that modulated antigen presentation and inflammatory factor expression in macrophages, enhancing the ability to enhance the killing effect on MTB (125) (Table 1).
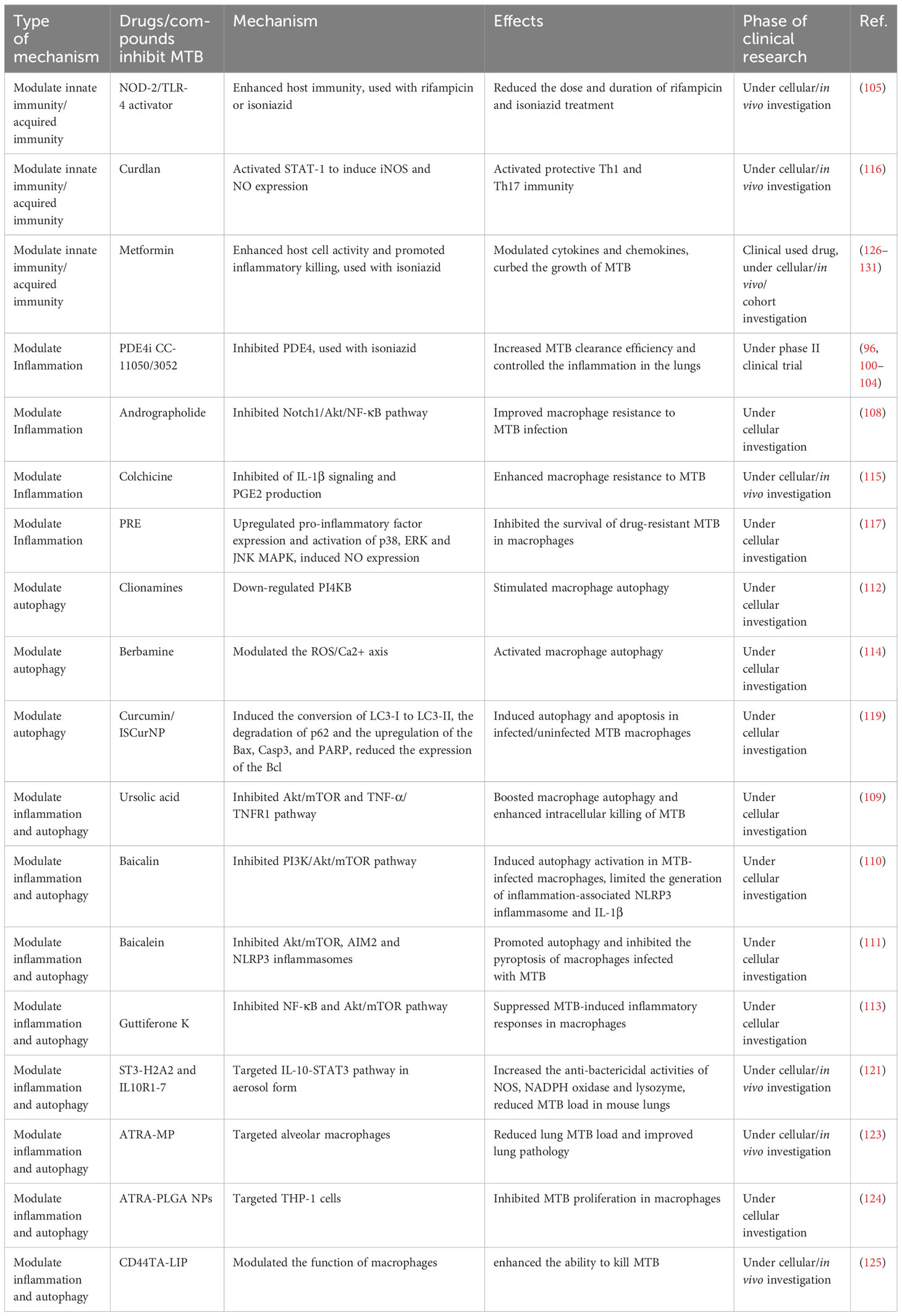
Table 1 Potential HDT drugs for TB therapy.
Diabetes is a complex metabolic disorder that impacts the functioning of several systems of the body. The link between DM and TB has been recognized for centuries (132). Several studies have confirmed that diabetic patients have a higher risk of contracting tuberculosis than non-diabetics, approximately two to eight fold (133). In 2021, about 400,000 people have already suffered from both tuberculosis and diabetes (134).
Type 1 diabetes mellitus is a typical autoimmune disease, in which specific activation of pancreatic β-cell-reactive T-cells is the key factor contributing to the pathologic changes. In the pathogenesis of type 2 diabetes, aberrant immune cell activation and the accompanying chronic inflammatory milieu are consequences of diabetes, which in turn lead to pathologic damage and disease progression. Thus, aberrant immunity plays an important role in disease progression in both type 1 and type 2 diabetes.
The characteristic hyperglycemia of diabetes cause a variety of immune response dysfunctions. The abundance of DCs in diabetic patients is significantly reduced, and their activation is also impaired (135, 136). Macrophages in a hyperglycemic environment could lead to higher levels of IL-6 and lower levels of IL-1β, as well as dysfunctional activation of the NLRP3 inflammasome (137). The conversion of macrophages from a pro-inflammatory to an anti-inflammatory phenotype is also impeded, leading to abnormal chronic inflammation in various organs, such as skin tissue wounds and pancreatic islets, resulting in their tissue damage and dysfunction (138, 139). Similarly, there is abnormal activation of neutrophils in diabetic patients, which damages tissues and organs by yielding increased amounts of ROS and pro-inflammatory cytokines (140, 141). Chronic hyperglycemia also causes NK cell dysfunction (142). In adaptive immunity, hyperglycemia could reduce the number of lymphocytes in the germinal centers and glycosylate immunoglobulins, thereby affecting their normal functions (143, 144). Upon antigenic stimulation, adaptive immunity in diabetic patients produces lower levels of immune responses, including reduced cell activation and proliferation as well as reduced cytokines (145, 146). These innate and adaptive immune deficiencies make diabetics more susceptible to infection, while some microorganisms are more nutrient-rich in a hyperglycemic environment and are more prone to uncontrollable infections (147). Thus, TB-diabetes complications are detrimental to the treatment of TB (148). Therefore, higher rates of treatment failure and higher mortality can be observed in patients with TB-DM than in patients with TB alone (149, 150). Meanwhile, patients with TB-DM are also more likely to develop resistance to MTB and a greater risk of transmission (151, 152).
On the one hand, MTB infection also influences the progression of diabetes, and it is well established that MTB infection leads to impaired glucose tolerance in the host, which is a key component in the pathogenesis of diabetes (153). Impaired glucose tolerance can be normalized in the host after successful treatment, but even latently infected MTB increases the risk of future diabetes, possibly by affecting insulin resistance in the host (154–156). A systematic review also concluded that TB promoted the onset and progression of diabetes (156). So, the state of co-morbidity of MTB and DM negatively affects the disease progression and outcome of both, which reinforces the difficulty and importance of their treatment.
Imbalanced glycemic control impairs immunity to MTB infection. In MTB-infected diabetic patients, MTB antigen-induced levels of IFN-γ, TNFα, and IL-6 mRNA, were significantly higher, leading to an increased inflammatory response and worsened disease progression at the site of MTB infection.
The high-glycemic environment directly impacts the process of antigen processing and presentation, which in turn affects the clearance of MTB (157). Furthermore, T2DM altered the basal phenotype of human macrophages and diminished their capacity to respond, antigen expression, internalise, cytokines (IL-6, IL-1β, IL-10, and IL-12), and chemokines (MCP-1, MIG, and RANTES) secretion, and controlled MTB (158). T2DM significantly increased mortality, bacterial load in organs, and inflammatory lesions in a mouse model of MTB infection (159). Sinha et al. found that glucose tolerance-impaired mice were associated with more significant pulmonary pathologic changes, lower TNF-α, IFN-γ, IFN-β, and IL-10 levels, and a tendency toward higher bacterial loads after MTB infection. IFN-γ levels were restored after restoration of glucose tolerance following a normal diet. Prediabetes increased tuberculosis severity, but high body fat with unimpaired glucose tolerance was protective (160).
LTBi is associated with an increased risk of developing diabetes (161, 162). At the patient level, Masood et al. found that peripheral blood mononuclear cells from diabetic patients with latent TB (LTBi) could elicit a dysfunctional immune response upon stimulation with PPD. The peripheral blood mononuclear cells secreted higher levels of IFN-γ, IL-6, IL-2, TNF-α, and GM-CSF, accompanied by reduced SOCS3 mRNA expression. This suggested that LTBi was strongly associated with exacerbating immune dysregulation in diabetic patients (163). Kathamuthu et al. found that in MAIT cells of diabetic patients with latent infection of MTB, the expression of Th1 and Th 17-type cytokines was increased, while the expression of cytotoxicity markers PFN, GZE B, and GNLSN was downregulated (164). In LTB PDM/DM combined, there was a decrease in γδ T cells expressing cytokines, cytotoxicity, and other immune markers as well as elevated levels of cytokines in supernatants (165).
There could be an interaction between MTB infection and the development of DM, and MTB can also promote the progression of diabetes. Oswal et al. found that adipose tissue/cells could modulate lung pathology and bacterial loads in MTB-infected mice (166). MTB infection could induce adipocyte dysfunction, leading to increased lipid accumulation in the lungs and altering cellular energy metabolism through inhibition of signaling pathways such as insulin, AMPK, and mTOR (167). Podell et al. demonstrated using a guinea pig model that MTB infection alone induced insulin resistance and chronic hyperglycemia and resulted in the accumulation of AGEs in tissues and serum (168).
T2DM mice exhibit increased body weight, blood glucose levels, glucose tolerance, insulin resistance, and increased susceptibility to MTB infection. Cheekatla et al. found that CD11c+ was the major source of IL-6 in MTB-infected T2DM mice, while NK-CD11c+ cell interactions increased IL-6 production. Anti-IL-6 treatment and anti-NK1.1 treatment significantly increased the survival rate of MTB-infected DM mice (169). The level of IL-22 was significantly lower in MTB-T2DM mice than in MTB mice (170). ILC3 was an important source of IL-22, and recombinant IL-22 treatment or overtransfer of ILC3 significantly improved the survival of MTB-T2DM-infected mice. The IL-22 pathway may be a novel target for therapeutic intervention in T2DM patients with active tuberculosis.
The level of macrophages response to MTB is also altered in metabolically altered groups. Macrophages in chronic diabetic patients show lower MTB killing capacity. Rapamycin activates macrophages to enhance anti-MTB activity independent of tuberculosis status, but this effect is poor in diabetic patients (171). Vrieling et al. found that plasma oxidized low-density lipoprotein (oxLDL) levels were significantly elevated in diabetic patients. In vitro, oxLDL could affect macrophage control of MTB by inducing lysosomal and autophagic dysfunction (172). Butyrate affected a variety of cellular functions, and its levels were altered in diabetic individuals. Lachmandas et al. found that physiological doses of butyrate were able to inhibit MTB-induced immune response in vitro, and blocking IL-10 reversed the effect of butyrate (173).
Oxysterol receptor GPR183 expression is reduced in diabetic patients infected with MTB and correlates with disease severity (174). Further studies in mice infected with MTB in the lung showed that macrophage targeting and recruitment to the site of infection was associated with activation of the GPR183/oxysterol axis. The lack of GPR183 in diabetic patients may be one of the important factors contributing to the difficulty in controlling their infections (175). Wang et al. constructed a T2DM-pulmonary tuberculosis (PTB) mouse model by injecting streptozotocin, nicotinamide, and MTB, and the expression of pSTAT3 was increased in T2DM-associated PTB mice. This confirmed that pSTAT3 could promote NFAT5 expression by inhibiting miR-19b/1281 transcription, which led to exacerbated lung injury in MTB-infected T2DM mice (176). Zhao et al. demonstrated that SP1 could activate Akt by inhibiting PTEN transcription, which exacerbated MTB-infected in diabetic mice infected with MTB (177).
Glibenclamide, a widely used sulfonylurea hypoglycemic agent, is an insulin secretion enhancer. Kewcharoenwong et al. found that pretreatment of monocytes with glibenclamide significantly decreased the levels of M1 surface markers (HLA-DR+ and CD 86+) and TNF-α production, and increased M2 (CD 163+ and CD 206+) surface markers and IL-10 expression. This resulted in impaired bactericidal capacity and potential susceptibility of primary human monocytes from T2DM patients to MTB (178). MTBs are able to invade host cells or survive within host cells by utilizing cholesterol, hence, some lipid-controlling drugs may have potential anti-TB effect (179, 180). Statins are one of the most widely used lipid-lowering drugs worldwide, which are extensively applied to reduce serum cholesterol levels and cardiovascular risk. Kang et al. investigated the effect of statins as adjunctive therapy for tuberculosis in a retrospective cohort study and found no significant association between statin use and the risk of developing tuberculosis in diabetic patients (181). Results from another cohort study by Lee et al. also showed that statin use was weakly associated with tuberculosis risk in diabetic patients > 65 years of age (182). However, in a study by Kim et al., it was found that statin users had a significantly lower risk of developing TB than non-statin users, although this protective effect would be attenuated by diabetes mellitus (183). This protective effect was confirmed by Pan et al., who also found that the higher the statin dose, the stronger the protective effect (184). The contradictions between the results of these large cohort studies may be due to some flaws in experimental design and data selection, and the presence of confounding factors may also be responsible. A series of preclinical studies and a number of clinical trials have demonstrated the potential therapeutic role of statins in non-diabetic tuberculosis patients (185–189). Accordingly, well-designed prospective studies are necessary to confirm the protective effect of statins in diabetic comorbidities with tuberculosis. Ezetimibe is an orally potent lipid-lowering drug that inhibits cholesterol absorption. Tsai et al. demonstrated that Ezetimibe reduced intracellular lipid content, and its treatment was associated with a reduced prevalence of latent TB and a significant inhibition of intracellular growth of MTB (190). Vitamin D can control MTB growth by inducing some anti-microbial peptides (AMP). Lopez-Lopez et al. found that vitamin D receptor (VDR) and AMP expression levels were lower in patients with T2DM, and supplementation with vitamin D was effective in improving their ability to clear MTB (191) (Figure 3).
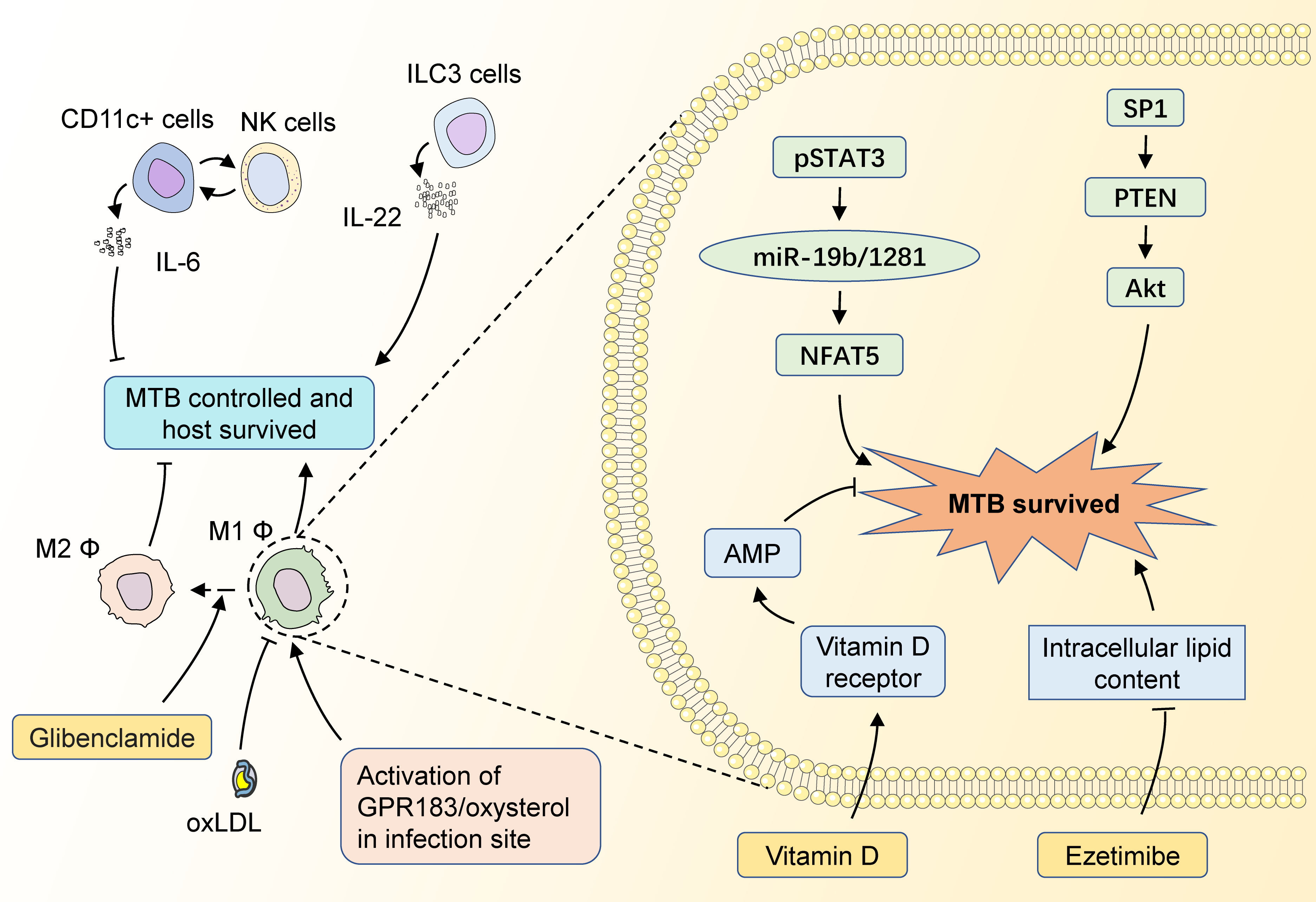
Figure 3 The potential targets of HDT for MTB infection with diabetic mellitus. CD11c+ cells could interact with NK cells to impair MTB control in the host by increasing the level of IL-6 secretion, while ILC3 cells could enhance the restriction of MTB infection by secreting IL-22. M1 macrophages could fight against MTB infection by secreting multiple inflammatory factors, and M2 macrophages have a significantly weakened ability to limit MTB. GPR183/oxysterol activated at the site of infection is necessary for macrophage recruitment, which facilitates the control of infection in the early stages. The diabetes-control drug Glibenclamide induces a shift from M1 to M2 macrophages, which adversely affects the control of MTB. OxLDL in the blood also impairs the antimicrobial capacity of M1 macrophages. pSTAT3 could promote NFAT5 expression by inhibiting miR-19b/1281 transcription, thus promoting the intracellular survival of MTBs. SP1 activates Akt by inhibiting PTEN transcription, which also impairs the MTB control of the host. MTB survival depends on the intracellular lipids, while Ezetimibe could inhibit MTB survival by decreasing the intracellular lipid contents. Meanwhile, Vitamin D could restrain MTB survival by inducing AMP expression.
It is worth noting that metformin is a first-line therapy for the management of diabetes mellitus type 2 (T2DM) with potent glucose-lowering properties, well-characterized biosafety, and relatively low cost (192). Metformin has been reported as an adjunctive therapy against MTB. Naicker et al. demonstrated that metformin modulated cellular interactions between MTB and macrophages by enhancing host cell activity and promoting inflammatory killing (126). The combination of metformin and isoniazid possessed a more potent ability to modulate cytokines and chemokines than isoniazid alone, effectively curbing the growth of MTB. Several clinical trials testing metformin as an adjunctive therapy for TB have been conducted. Numerous clinical studies have demonstrated that the use of metformin significantly reduced the incidence of active TB in diabetic patients, with a superior protective effect of high-dose metformin compared to low-dose metformin (127–129). Singhal et al. observed in two independent cohorts that MET-adjuvant therapy augmented the efficacy of conventional anti-TB drugs, thereby improving the severity of MTB infection and providing a better prognosis (130). Metformin reduced mortality in diabetic patients infected with tuberculosis during tuberculosis treatment, and the improved mortality rate was similar to that of non-diabetic tuberculosis patients (131).
GSH is an antioxidant, which is essential for the maintenance of redox homeostasis in vivo. It has been shown that GSH exhibits both direct antimycobacterial activity and in-direct antimycobacterial activity by enhancing the function of NK cells and T cells (47, 193, 194). In diabetic individuals, GSH levels are decreased (195). Liposomal glutathione (L-GSH) contains reduced GSH (rGSH) that could be readily absorbed and directly utilized by immune cells, resulting in rapid restoration of intracellular rGSH levels. Kachour et al. verified the therapeutic effects of L-GSH supplementation in a mouse model of tuberculosis, as manifested by a reduction in MTB burden and oxidative stress levels in the lungs, as well as an increase in lung levels of IL-12, IFN-γ, IL-2, IL-17, and TNF-α, and a decrease in IL-6, IL-10, and TGF-β production (196). Sasaninia et al. confirmed the favorable controlling effect of L-GSH supplementation in extrapulmonary tuberculosis in mice as well (197). (L-GSH supplementation has been shown to be an effective adjuvant therapy in both pulmonary TB and extrapulmonary TB). Beever et al. utilized a TB-diabetic mouse model to assess the therapeutic effects of combining L-GSH and rifampicin in TB (198). The results showed that L-GSH alone significantly reduced MTB burden and increased IFN-γ, TNF-α, IL-17, IL-10 and TGF-β1 levels. The combination of L-GSH and rifampicin provided better control of MTB infection and oxidative stress levels compared to rifampicin alone. In human-derived in vitro models, L-GSH also exhibited promising capacity in controlling MTB infection (195, 199). To et al. conducted a three month duration randomized controlled trial to evaluate the therapeutic efficacy of L-GSH in TB-DM, in which a total of 11 T2DM-positive subjects were enrolled in the L-GSH supplementation group and a total of 7 T2DM-positive subjects were enrolled in the placebo group (200). The use of L-GSH decreased the level of oxidative stress in the blood components of the subjects, with increased levels of Th 1-related cytokines IFN-γ, TNF-α, and IL-2, and decreased levels of IL-6 and IL-10. Despite the small sample size of this clinical trial, this results, as well as those of other clinical and preclinical trials, suggest that L-GSH supplementation has the potential to be an effective adjunctive therapy for the treatment of TB-DM (Table 2).
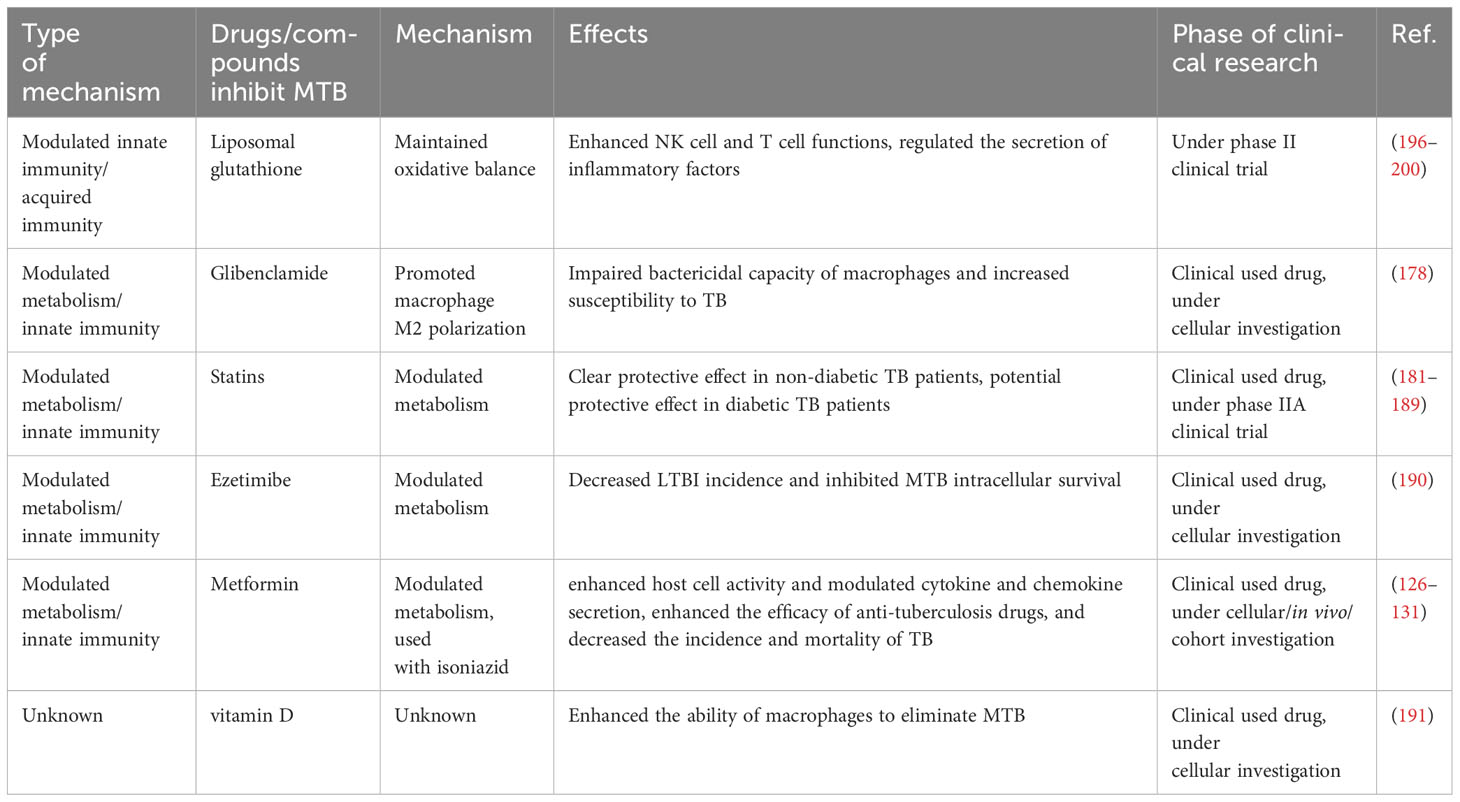
Table 2 Potential HDT drugs for TB-DM therapy.
Systematically, this paper provides a meticulous elucidation of HDT treatment for MTB, including the immune mechanisms of MTB infection, potential therapeutic agents, and the immunological characteristics and potential HDT of diabetic MTB. Specifically, HDT can kill MTB by directly modulating immune response; second, HDT can modulate immune cells and factors in a multifaceted manner and does not involve pathogen-specific targets, limiting MTB to develop resistance and tolerance to HDT in the short term; third, HDT reduces the dosage and duration of antibiotics currently in use, thus improving patient adherence to treatment, and reduces the probability of drug-resistant strains. At present, HDT still holds several challenges that are worth attention.
First, there may be conflicting results between certain HDT treatment clinical trial data, possibly due to a variety of additional factors such as age, dose selection, subject lifestyle changes, race, co-infections, and genetic patterns. Therefore, more rigorously designed clinical trials are needed to validate the efficacy of HDT drugs. More importantly, it is difficult for mouse models to accurately simulate the immune response in humans infected with MTB. It is necessary to conduct a variety of other animal models of MTB infection to be able to validate the efficacy of HDT. These more accurate preclinical study data confirm the effectiveness of HDT and may provide a basis for clinical trials.
The activation of the immune system is a very finely tuned process. HDT involves the regulation of the immune system and needs to be applied with caution. Some immune targets have a narrow therapeutic window and require a high degree of accuracy in the dosage of the drug used. Enhancing the immune response may lead to aggravated histopathologic damage, while mitigating the immune response may lead to the spread and recurrence of infection. The development of novel HDT targets remains a necessity as there is still a lack of clinically available HDT drugs. The process of new target development needs to focus on the balance between destructive and protective immune responses, as well as the protection and reversal of organ tissues that have already produced pathological damage. Consideration has to be given to saving as much organ function as possible, the possibility of combining or avoiding interfering therapeutic effects with existing therapeutic modalities, as well as potential toxicity and safety issues. Especially in combination with other diseases, such as the MTB diabetic state, the organism may face a more complex immune environment. How to precisely regulate the immune response to achieve a balance between the therapeutic needs of different diseases is one of the difficult issues to overcome. Precise modulation of the immune response to achieve a balanced therapeutic need for different co-morbidities is the challenge to be overcome.
Finally, HDT may exhibit off-target effects. How to enhance the targeting ability of HDT drugs and avoid off-target effects on other biological processes in the body are areas of concern for future studies. This means that reliable biomarkers need to be developed for efficacy monitoring. Since individuals have different strengths of immune response to MTB infection and different cycles of treatment initiation, the selection of HDT type, dose, duration of use, mode of action, and combination drugs requires precise individualization, which is another issue that needs to be focused on in preclinical and clinical trials.
Overall, the drugs for HDT are repositioned drugs that have been approved and used in actual clinical practice, with a certain degree of relative safety and efficacy guaranteed. HDT is a potentially effective therapeutic tool for the treatment of MTB and diabetic MTB, and can compensate for the shortcomings of other TB therapies, including the reduction of drug resistance and modulation of immune response. In the context of co-morbidities, such as DM, HDT provides an individualized treatment tailored to individualized therapy. The application of HDT for the treatment of MTB and diabetic MTB warrants in-depth basic and clinical research.
LZ: Investigation, Visualization, Writing – original draft. KF: Investigation, Visualization, Writing – original draft. XS: Writing – review & editing. WL: Writing – review & editing. FQ: Writing – review & editing. LS: Writing – review & editing. FG: Writing – review & editing. CZ: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; AMP, anti-microbial peptides; ATRA-MP, ATRA-loaded microparticle; ATRA-PLGA NPs, ATRA-loaded PLGA nanoparticles; BCG, Bacillus Calmette-Guerin; COX-2, cyclooxygenase-2; DCs, dendritic cells; DM, diabetes mellitus; T2DM, diabetes mellitus type 2; GSH, glutathione; GM-CSF, granulocyte-macrophage colony stimulating factor; HDAC, histone deacetylase; HDT, host-directed therapy; iNOS, inducible nitric oxide synthase; IFN, interferon; IL, interlukin; CD44TA-LIP, liposomes targeting CD44 using thioaptamers; L-GSH, Liposomal glutathione; mTOR, mammalian target of rapamycin; MAMPs, microbe-associated molecular patterns; MDR, multidrug-resistant; MTB, mycobacterium tuberculosis; NK, natural killer; NcRNAs, non-coding RNAs; oxLDL, oxidized low density lipoprotein; PRR, pattern recognition receptor; PDE4i, phosphodiesterase-4 inhibitor; PRE, polysaccharide-rich extract; PGE2, prostaglandin E2; PKR, protein kinase R; PTB, pulmonary tuberculosis; TLR, toll-like receptor; TG2, transglutaminase 2; TAG, triacylglycerol; TB, tuberculosis; TB-DM, TB-diabetes mellitus; TNF, tumor necrosis factor; VDR, vitamin D receptor.
1. Chakaya J, Khan M, Ntoumi F, Aklillu E, Fatima R, Mwaba P, et al. Global Tuberculosis Report 2020 - Reflections on the Global TB burden, treatment and prevention efforts. Int J Infect Dis (2021) 113 Suppl 1(Suppl 1):S7–12. doi: 10.1016/j.ijid.2021.02.107
2. Singh R, Dwivedi SP, Gaharwar US, Meena R, Rajamani P, Prasad T. Recent updates on drug resistance in Mycobacterium tuberculosis. J Appl Microbiol (2020) 128(6):1547–67. doi: 10.1111/jam.14478
3. Sulis G, Pai M. Isoniazid-resistant tuberculosis: A problem we can no longer ignore. PloS Med (2020) 17(1):e1003023. doi: 10.1371/journal.pmed.1003023
4. Azevedo-Pereira JM, Pires D, Calado M, Mandal M, Santos-Costa Q, Anes E. HIV/Mtb co-infection: from the amplification of disease pathogenesis to an “Emerging syndemic”. Microorganisms (2023) 11(4):853. doi: 10.3390/microorganisms11040853
5. Bisht MK, Dahiya P, Ghosh S, Mukhopadhyay S. The cause-effect relation of tuberculosis on incidence of diabetes mellitus. Front Cell Infect Microbiol (2023) 13. doi: 10.3389/fcimb.2023.1134036
6. Viney K, Cavanaugh J, Kienene T, Harley D, Kelly PM, Sleigh A, et al. Tuberculosis and diabetes mellitus in the Republic of Kiribati: a case-control study. Trop Med Int Health (2015) 20(5):650–7. doi: 10.1111/tmi.12462
7. Abdelbary BE, Garcia-Viveros M, Ramirez-Oropesa H, Rahbar MH, Restrepo BI. Tuberculosis-diabetes epidemiology in the border and non-border regions of Tamaulipas, Mexico. Tuberculosis (Edinb) (2016) 101S:S124–34. doi: 10.1016/j.tube.2016.09.024
8. Wu Q, Liu Y, Ma YB, Liu K, Chen SH. Incidence and prevalence of pulmonary tuberculosis among patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Ann Med (2022) 54(1):1657–66. doi: 10.1080/07853890.2022.2085318
9. Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discovery (2018) 17(1):35–56. doi: 10.1038/nrd.2017.162
10. Guler R, Brombacher F. Host-directed drug therapy for tuberculosis. Nat Chem Biol (2015) 11(10):748–51. doi: 10.1038/nchembio.1917
11. Hawn TR, Shah JA, Kalman D. New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunol Rev (2015) 264(1):344–62. doi: 10.1111/imr.12255
12. Jeong EK, Lee HJ, Jung YJ. Host-directed therapies for tuberculosis. Pathogens (2022) 11(11):1291. doi: 10.3390/pathogens11111291
13. Sia JK, Rengarajan J. Immunology of mycobacterium tuberculosis infections. Microbiol Spectr (2019) 7(4). doi: 10.1128/microbiolspec.GPP3-0022-2018
14. Danelishvili L, McGarvey J, Li YJ, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol (2003) 5(9):649–60. doi: 10.1046/j.1462-5822.2003.00312.x
15. Hernández-Pando R, Jeyanathan M, Mengistu G, Aguilar D, Orozco H, Harboe M, et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet (2000) 356(9248):2133–8. doi: 10.1016/S0140-6736(00)03493-0
16. Rahlwes KC, Dias BRS, Campos PC, Alvarez-Arguedas S, Shiloh MU. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence (2023) 14(1):e1004917. doi: 10.1080/21505594.2022.2150449
17. Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol (2017) 17(11):691–702. doi: 10.1038/nri.2017.69
18. Luies L, du Preez I. The echo of pulmonary tuberculosis: mechanisms of clinical symptoms and other disease-induced systemic complications. Clin Microbiol Rev (2020) 33(4):1–19. doi: 10.1128/CMR.00036-20
19. Cohen SB, Gern BH, Urdahl KB. The tuberculous granuloma and preexisting immunity. Annu Rev Immunol (2022) 40:589–614. doi: 10.1146/annurev-immunol-093019-125148
20. Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol (2003) 48(3):833–43. doi: 10.1046/j.1365-2958.2003.03474.x
21. Martin CJ, Carey AF, Fortune SM. A bug’s life in the granuloma. Semin Immunopathol. (2016) 38(2):213–20. doi: 10.1007/s00281-015-0533-1
22. Ong CWM, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PloS Pathog (2015) 11(5):e1004650. doi: 10.1371/journal.ppat.1004917
23. Grosset J. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob Agents Chemother (2003) 47(3):833–6. doi: 10.1128/AAC.47.3.833-836.2003
24. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
25. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature (2014) 506(7489):503–6. doi: 10.1038/nature12902
26. Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol (2017) 14(12):963–75. doi: 10.1038/cmi.2017.88
27. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, et al. Cutting edge: mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol (2016) 196(6):2444–9. doi: 10.4049/jimmunol.1501612
28. Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol (2012) 12(8):581–91. doi: 10.1038/nri3259
29. Simeone R, Sayes F, Song O, Gröschel MI, Brodin P, Brosch R, et al. Cytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PloS Pathog (2015) 11(2):e1004265. doi: 10.1371/journal.ppat.1004650
30. Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, et al. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PloS Pathog (2014) 10(7):2150449. doi: 10.1371/journal.ppat.1004265
31. Ó Maoldomhnaigh C, Cox DJ, Phelan JJ, Mitermite M, Murphy DM, Leisching G, et al. Lactate alters metabolism in human macrophages and improves their ability to kill mycobacterium tuberculosis. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.663695
32. Borah K, Mendum TA, Hawkins ND, Ward JL, Beale MH, Larrouy-Maumus G, et al. Metabolic fluxes for nutritional flexibility of Mycobacterium tuberculosis. Mol Syst Biol (2021) 17(5):e10280. doi: 10.15252/msb.202110280
33. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med (2018) 215(4):1135–52. doi: 10.1084/jem.20172020
34. Prendergast KA, Kirman JR. Dendritic cell subsets in mycobacterial infection: control of bacterial growth and T cell responses. Tuberculosis (Edinb) (2013) 93(2):115–22. doi: 10.1016/j.tube.2012.10.008
35. Mihret A. The role of dendritic cells in Mycobacterium tuberculosis infection. Virulence (2012) 3(7):654–9. doi: 10.4161/viru.22586
36. Mihret A, Mamo G, Tafesse M, Hailu A, Parida S. Dendritic Cells Activate and Mature after Infection with Mycobacterium tuberculosis. BMC Res Notes (2011) 4:247. doi: 10.1186/1756-0500-4-247
37. Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med (2005) 202(12):1715–24. doi: 10.1084/jem.20051782
38. Tallieux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med (2003) 197(1):121–7. doi: 10.1084/jem.20021468
39. Nigou J, Zelle-Rieser C, Gilleron M, Thurnher M, Puzo G. Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J Immunol (2001) 166(12):7477–85. doi: 10.4049/jimmunol.166.12.7477
40. Chadha A, Mehto S, Selvakumar A, Vashishta M, Kamble SS, Popli S, et al. Suppressive role of neddylation in dendritic cells during Mycobacterium tuberculosis infection. Tuberculosis (Edinb) (2015) 95(5):599–607. doi: 10.1016/j.tube.2015.05.014
41. Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Faé KC, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest (2014) 124(3):1268–82. doi: 10.1172/JCI72030
42. Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe (2012) 12(3):301–12. doi: 10.1016/j.chom.2012.07.009
43. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol (2017) 2:17072. doi: 10.1038/nmicrobiol.2017.72
44. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe (2017) 22(4):519–530.e3. doi: 10.1016/j.chom.2017.09.003
45. Zhang Y, Huang B. The development and diversity of ILCs, NK cells and their relevance in health and diseases. Adv Exp Med Biol (2017) 1024:225–44. doi: 10.1007/978-981-10-5987-2_11
46. Esin S, Counoupas C, Aulicino A, Brancatisano FL, Maisetta G, Bottai D, et al. Interaction of Mycobacterium tuberculosis cell wall components with the human natural killer cell receptors NKp44 and Toll-like receptor 2. Scand J Immunol (2013) 77(6):460–9. doi: 10.1111/sji.12052
47. Allen M, Bailey C, Cahatol I, Dodge L, Yim J, Kassissa C, et al. Mechanisms of control of mycobacterium tuberculosis by NK cells: role of glutathione. Front Immunol (2015) 6(OCT). doi: 10.3389/fimmu.2015.00508
48. Lu CC, Wu TS, Hsu YJ, Chang CJ, Lin CS, Chia JH, et al. NK cells kill mycobacteria directly by releasing perforin and granulysin. J Leukoc Biol (2014) 96(6):1119–29. doi: 10.1189/jlb.4A0713-363RR
49. Bozzano F, Costa P, Passalacqua G, Dodi F, Ravera S, Pagano G, et al. Functionally relevant decreases in activatory receptor expression on NK cells are associated with pulmonary tuberculosis in vivo and persist after successful treatment. Int Immunol (2009) 21(7):779–91. doi: 10.1093/intimm/dxp046
50. Robinson RT, Orme IM, Cooper AM. The onset of adaptive immunity in the mouse model of tuberculosis and the factors that compromise its expression. Immunol Rev (2015) 264(1):46–59. doi: 10.1111/imr.12259
51. Stegelmann F, Bastian M, Swoboda K, Bhat R, Kiessler V, Krensky AM, et al. Coordinate expression of CC chemokine ligand 5, granulysin, and perforin in CD8+ T cells provides a host defense mechanism against Mycobacterium tuberculosis. J Immunol (2005) 175(11):7474–83. doi: 10.4049/jimmunol.175.11.7474
52. Williams A, Reljic R, Naylor I, Clark SO, Falero-Diaz G, Singh M, et al. Passive protection with immunoglobulin A antibodies against tuberculous early infection of the lungs. Immunology (2004) 111(3):328–33. doi: 10.1111/j.1365-2567.2004.01809.x
53. Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol (2007) 178(11):7222–34. doi: 10.4049/jimmunol.178.11.7222
54. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in mycobacterium tuberculosis infection. Microbiol Spectr (2016) 4(5). doi: 10.1128/microbiolspec.TBTB2-0018-2016
55. Erdmann H, Behrends J, Ritter K, Hölscher A, Volz J, Rosenkrands I, et al. The increased protection and pathology in Mycobacterium tuberculosis-infected IL-27R-alpha-deficient mice is supported by IL-17A and is associated with the IL-17A-induced expansion of multifunctional T cells. Mucosal Immunol (2018) 11(4):1168–80. doi: 10.1038/s41385-018-0026-3
56. Harding JS, Herbath M, Chen Y, Rayasam A, Ritter A, Csoka B, et al. VEGF-A from granuloma macrophages regulates granulomatous inflammation by a non-angiogenic pathway during mycobacterial infection. Cell Rep (2019) 27(7):2119–2131.e6. doi: 10.1016/j.celrep.2019.04.072
57. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation through CD40 and TLR-4 Is an Effective Host Directed Therapy against Mycobacterium tuberculosis. Front Immunol (2016) 7(SEP). doi: 10.3389/fimmu.2016.00386
58. Nore KG, Jørgensen MJ, Dyrhol-Riise AM, Jenum S, Tonby K. Elevated levels of anti-inflammatory eicosanoids and monocyte heterogeneity in mycobacterium tuberculosis infection and disease. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.579849
59. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi: 10.1038/nature13489
60. Biersack B, Polat S, Höpfner M. Anticancer properties of chimeric HDAC and kinase inhibitors. Semin Cancer Biol (2022) 83:472–86. doi: 10.1016/j.semcancer.2020.11.005
61. Moreira JD, Koch BEV, van Veen S, Walburg KV, Vrieling F, Mara Pinto Dabés Guimarães T, et al. Functional Inhibition of Host Histone Deacetylases (HDACs) Enhances in vitro and in vivo Anti-mycobacterial Activity in Human Macrophages and in Zebrafish. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00036
62. Campo M, Heater S, Peterson GJ, Simmons JD, Skerrett SJ, Mayanja-Kizza H, et al. HDAC3 inhibitor RGFP966 controls bacterial growth and modulates macrophage signaling during Mycobacterium tuberculosis infection. Tuberculosis (Edinb) (2021) 127:102062. doi: 10.1016/j.tube.2021.102062
63. Cheng CY, Gutierrez NM, Marzuki MB, Lu X, Foreman TW, Paleja B, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol (2017) 2(9):eaaj1789. doi: 10.1126/sciimmunol.aaj1789
64. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
65. Dai Y, Zhu C, Xiao W, Huang K, Wang X, Shi C, et al. Mycobacterium tuberculosis hijacks host TRIM21- and NCOA4-dependent ferritinophagy to enhance intracellular growth. J Clin Invest (2023) 133(8):e159941. doi: 10.1172/JCI159941
66. Amaral EP, Foreman TW, Namasivayam S, Hilligan KL, Kauffman KD, Bomfim CCB, et al. GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. J Exp Med (2022) 219(11):e20220504. doi: 10.1084/jem.20220504
67. Bell SL, Lopez KL, Cox JS, Patrick KL, Watson RO. Galectin-8 senses phagosomal damage and recruits selective autophagy adapter TAX1BP1 to control mycobacterium tuberculosis infection in macrophages. mBio (2021) 12(4):e0187120. doi: 10.1128/mBio.01871-20
68. Scott NR, Swanson RV, Al-Hammadi N, Domingo-Gonzalez R, Rangel-Moreno J, Kriel BA, et al. S100A8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J Clin Invest (2020) 130(6):3098–112. doi: 10.1172/JCI130546
69. Leu JS, Chen ML, Chang SY, Yu SL, Lin CW, Wang H, et al. SP110b controls host immunity and susceptibility to tuberculosis. Am J Respir Crit Care Med (2017) 195(3):369–82. doi: 10.1164/rccm.201601-0103OC
70. Brandenburg J, Marwitz S, Tazoll SC, Waldow F, Kalsdorf B, Vierbuchen T, et al. WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J Clin Invest (2021) 131(16):e141833. doi: 10.1172/JCI141833
71. Pires D, Calado M, Velez T, Mandal M, Catalão MJ, Neyrolles O, et al. Modulation of cystatin C in human macrophages improves anti-mycobacterial immune responses to mycobacterium tuberculosis infection and coinfection with HIV. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.742822
72. Mandal M, Pires D, Catalão MJ, Azevedo-Pereira JM, Anes E. Modulation of cystatin F in human macrophages impacts cathepsin-driven killing of multidrug-resistant mycobacterium tuberculosis. Microorganisms (2023) 11(7):1861. doi: 10.3390/microorganisms11071861
73. Smyth R, Berton S, Rajabalee N, Chan T, Sun J. Protein kinase R restricts the intracellular survival of mycobacterium tuberculosis by promoting selective autophagy. Front Microbiol (2021) 11. doi: 10.3389/fmicb.2020.613963
74. Palucci I, Matic I, Falasca L, Minerva M, Maulucci G, De Spirito M, et al. Transglutaminase type 2 plays a key role in the pathogenesis of Mycobacterium tuberculosis infection. J Intern Med (2018) 283(3):303–13. doi: 10.1111/joim.12714
75. Palucci I, Maulucci G, De Maio F, Sali M, Romagnoli A, Petrone L, et al. Inhibition of transglutaminase 2 as a potential host-directed therapy against mycobacterium tuberculosis. Front Immunol (2020) 10. doi: 10.3389/fimmu.2019.03042
76. van Doorn CLR, Steenbergen SAM, Walburg KV, Ottenhoff THM. Pharmacological poly (ADP-ribose) polymerase inhibitors decrease mycobacterium tuberculosis survival in human macrophages. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.712021
77. Yan H, Bu P. Non-coding RNA in cancer. Essays Biochem (2021) 65(4):625–39. doi: 10.1042/EBC20200032
78. Pires D, Bernard EM, Pombo JP, Carmo N, Fialho C, Gutierrez MG, et al. Mycobacterium tuberculosis Modulates miR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front Immunol (2017) 8(DEC). doi: 10.3389/fimmu.2017.01819
79. Tamgue O, Gcanga L, Ozturk M, Whitehead L, Pillay S, Jacobs R, et al. Differential Targeting of c-Maf, Bach-1, and Elmo-1 by microRNA-143 and microRNA-365 Promotes the Intracellular Growth of Mycobacterium tuberculosis in Alternatively IL-4/IL-13 Activated Macrophages. Front Immunol (2019) 10(MAR). doi: 10.3389/fimmu.2019.00421
80. Fu B, Lin X, Tan S, Zhang R, Xue W, Zhang H, et al. MiR-342 controls Mycobacterium tuberculosis susceptibility by modulating inflammation and cell death. EMBO Rep (2021) 22(9):e52252. doi: 10.15252/embr.202052252
81. Liu F, Chen J, Wang P, Li H, Zhou Y, Liu H, et al. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat Commun (2018) 9(1):4295. doi: 10.1038/s41467-018-06836-4
82. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol (2021) 220(2):e202009045. doi: 10.1083/jcb.202009045
83. Zhang X, Chen C, Xu Y. Long non-coding RNAs in tuberculosis: from immunity to biomarkers. Front Microbiol (2022) 13. doi: 10.3389/fmicb.2022.883513
84. Gcanga L, Tamgue O, Ozturk M, Pillay S, Jacobs R, Chia JE, et al. Host-directed targeting of lincRNA-MIR99AHG suppresses intracellular growth of mycobacterium tuberculosis. Nucleic Acid Ther (2022) 32(5):421–37. doi: 10.1089/nat.2022.0009
85. Hu Y, Wen Z, Liu S, Cai Y, Guo J, Xu Y, et al. Ibrutinib suppresses intracellular mycobacterium tuberculosis growth by inducing macrophage autophagy. J Infect (2020) 80(6):e19–26. doi: 10.1016/j.jinf.2020.03.003
86. Boland R, Heemskerk MT, Forn-Cuní G, Korbee CJ, Walburg KV, Esselink JJ, et al. Repurposing tamoxifen as potential host-directed therapeutic for tuberculosis. mBio (2023) 14(1):e0302422. doi: 10.1128/mbio.03024-22
87. Winchell CG, Mishra BB, Phuah JY, Saqib M, Nelson SJ, Maiello P, et al. Evaluation of IL-1 blockade as an adjunct to linezolid therapy for tuberculosis in mice and macaques. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00891
88. Pires D, Valente S, Calado M, Mandal M, Azevedo-Pereira JM, Anes E. Repurposing saquinavir for host-directed therapy to control mycobacterium tuberculosis infection. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.647728
89. Pires D, Mandal M, Pinho J, Catalão MJ, Almeida AJ, Azevedo-Pereira JM, et al. Liposomal delivery of saquinavir to macrophages overcomes cathepsin blockade by mycobacterium tuberculosis and helps control the phagosomal replicative niches. Int J Mol Sci (2023) 24(2):1142. doi: 10.3390/ijms24021142
90. Ribera E, Azuaje C, Lopez RM, Domingo P, Curran A, Feijoo M, et al. Pharmacokinetic interaction between rifampicin and the once-daily combination of saquinavir and low-dose ritonavir in HIV-infected patients with tuberculosis. J Antimicrob Chemother (2007) 59(4):690–7. doi: 10.1093/jac/dkl552
91. Zhang Y, Zhang J, Wang S. The role of rapamycin in healthspan extension via the delay of organ aging. Ageing Res Rev (2021) 70:101376. doi: 10.1016/j.arr.2021.101376
92. Etna MP, Severa M, Licursi V, Pardini M, Cruciani M, Rizzo F, et al. Genome-Wide Gene Expression Analysis of Mtb-Infected DC Highlights the Rapamycin-Driven Modulation of Regulatory Cytokines via the mTOR/GSK-3β Axis. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.649475
93. Jeon SY, Yhim HY, Lee NR, Song EK, Kwak JY, Yim CY. Everolimus-induced activation of latent Mycobacterium tuberculosis infection in a patient with metastatic renal cell carcinoma. Korean J Intern Med (2017) 32(2):365–8. doi: 10.3904/kjim.2015.121
94. FijaŁkowska-Morawska JB, Jagodzińska M, Nowicki M. Pulmonary embolism and reactivation of tuberculosis during everolimus therapy in a kidney transplant recipient. Ann Transplant (2011) 16(4):107–10. doi: 10.12659/AOT.882226
95. Bianco DM, De Maio F, Santarelli G, Palucci I, Salustri A, Bianchetti G, et al. Evaluation of Everolimus Activity against Mycobacterium tuberculosis Using In Vitro Models of Infection. Antibiot (Basel) (2023) 12(1):171. doi: 10.3390/antibiotics12010171
96. Wallis RS, Ginindza S, Beattie T, Arjun N, Sebe M, Likoti M, et al. Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med (2021) 9(8):897–908. doi: 10.1016/S2213-2600(20)30448-3
97. Sorgi CA, Soares EM, Rosada RS, Bitencourt CS, Zoccal KF, Pereira PAT, et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim Biophys Acta Mol Basis Dis (2020) 1866(3):165574. doi: 10.1016/j.bbadis.2019.165574
98. Mortensen R, Clemmensen HS, Woodworth JS, Therkelsen ML, Mustafa T, Tonby K, et al. Cyclooxygenase inhibitors impair CD4 T cell immunity and exacerbate Mycobacterium tuberculosis infection in aerosol-challenged mice. Commun Biol (2019) 2(1):288. doi: 10.1038/s42003-019-0530-3
99. Mo S, Liu X, Zhang K, Wang W, Cai Y, Ouyang Q, et al. Flunarizine suppresses Mycobacterium tuberculosis growth via calmodulin-dependent phagosome maturation. J Leukoc Biol (2022) 111(5):1021–9. doi: 10.1002/JLB.4A0221-119RR
100. Subbian S, Tsenova L, Holloway J, Peixoto B, O’Brien P, Dartois V, et al. Adjunctive phosphodiesterase-4 inhibitor therapy improves antibiotic response to pulmonary tuberculosis in a rabbit model. EBioMedicine (2016) 4:104–14. doi: 10.1016/j.ebiom.2016.01.015
101. Subbian S, Tsenova L, O’Brien P, Yang G, Koo MS, Peixoto B, et al. Phosphodiesterase-4 inhibition combined with isoniazid treatment of rabbits with pulmonary tuberculosis reduces macrophage activation and lung pathology. Am J Pathol (2011) 179(1):289–301. doi: 10.1016/j.ajpath.2011.03.039
102. Subbian S, Tsenova L, O’Brien P, Yang G, Koo MS, Peixoto B, et al. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PloS Pathog (2011) 7(9):e1002262. doi: 10.1371/journal.ppat.1002262
103. Subbian S, Koo MS, Tsenova L, Khetani V, Zeldis JB, Fallows D, et al. Pharmacologic inhibition of host phosphodiesterase-4 improves isoniazid-mediated clearance of mycobacterium tuberculosis. Front Immunol (2016) 7(JUN). doi: 10.3389/fimmu.2016.00238
104. Koo MS, Manca C, Yang G, O’Brien P, Sung N, Tsenova L, et al. Phosphodiesterase 4 inhibition reduces innate immunity and improves isoniazid clearance of Mycobacterium tuberculosis in the lungs of infected mice. PloS One (2011) 6(2):e17091. doi: 10.1371/journal.pone.0017091
105. Aqdas M, Maurya SK, Pahari S, Singh S, Khan N, Sethi K, et al. Immunotherapeutic role of NOD-2 and TLR-4 signaling as an adjunct to antituberculosis chemotherapy. ACS Infect Dis (2021) 7(11):2999–3008. doi: 10.1021/acsinfecdis.1c00136
106. Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem (2016) 8(6):531–41. doi: 10.1038/nchem.2479
107. Petrocelli G, Marrazzo P, Bonsi L, Facchin F, Alviano F, Canaider S. Plumbagin, a natural compound with several biological effects and anti-inflammatory properties. Life (Basel) (2023) 13(6):1303. doi: 10.3390/life13061303
108. He W, Sun J, Zhang Q, Li Y, Fu Y, Zheng Y, et al. Andrographolide exerts anti-inflammatory effects in Mycobacterium tuberculosis-infected macrophages by regulating the Notch1/Akt/NF-κB axis. J Leukoc Biol (2020) 108(6):1747–64. doi: 10.1002/JLB.3MA1119-584RRR
109. Shen J, Fu Y, Liu F, Ning B, Jiang X. Ursolic acid promotes autophagy by inhibiting akt/mTOR and TNF-α/TNFR1 signaling pathways to alleviate pyroptosis and necroptosis in mycobacterium tuberculosis-infected macrophages. Inflammation (2023) 46(5):1749–63. doi: 10.21203/rs.3.rs-2738766/v1
110. Zhang Q, Sun J, Wang Y, He W, Wang L, Zheng Y, et al. Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium tuberculosis. Front Microbiol (2017) 8(NOV). doi: 10.3389/fmicb.2017.02142
111. Ning B, Shen J, Liu F, Zhang H, Jiang X. Baicalein Suppresses NLRP3 and AIM2 Inflammasome-Mediated Pyroptosis in Macrophages Infected by Mycobacterium tuberculosis via Induced Autophagy. Microbiol Spectr (2023) 11(3). doi: 10.1128/spectrum.04711-22
112. Persaud R, Li SC, Chao JD, Forestieri R, Donohue E, Balgi AD, et al. Clionamines stimulate autophagy, inhibit Mycobacterium tuberculosis survival in macrophages, and target Pik1. Cell Chem Biol (2022) 29(5):870–882.e11. doi: 10.1016/j.chembiol.2021.07.017
113. Zhang Q, Sun J, Fu Y, He W, Li Y, Tan H, et al. Guttiferone K exerts the anti-inflammatory effect on mycobacterium tuberculosis- (H37Ra-) infected macrophages by targeting the TLR/IRAK-1 mediated akt and NF- κ B pathway. Mediators Inflammation (2020) 2020:8528901. doi: 10.1155/2020/8528901
114. Zhang S, Zhou X, Ou M, Fu X, Lin Q, Tao X, et al. Berbamine promotes macrophage autophagy to clear Mycobacterium tuberculosis by regulating the ROS/Ca2+ axis. mBio (2023) 14(4):e0027223. doi: 10.1128/mbio.00272-23
115. Kwon KW, Kim LH, Kang SM, Lee JM, Choi E, Park J, et al. Host-directed anti-mycobacterial activity of colchicine, an anti-gout drug, via strengthened host innate resistance reinforced by the IL-1β/PGE2 axis. Br J Pharmacol (2022) 179(15):3951–69. doi: 10.1111/bph.15838
116. Negi S, Pahari S, Das DK, Khan N, Agrewala JN. Curdlan limits mycobacterium tuberculosis survival through STAT-1 regulated nitric oxide production. Front Microbiol (2019) 10(MAY). doi: 10.3389/fmicb.2019.01173
117. Gupta PK, Kulkarni S. Polysaccharide rich extract (PRE) from Tinospora cordifolia inhibits the intracellular survival of drug resistant strains of Mycobacterium tuberculosis in macrophages by nitric oxide induction. Tuberculosis (Edinb) (2018) 113:81–90. doi: 10.1016/j.tube.2018.09.005
118. Sadeghi M, Dehnavi S, Asadirad A, Xu S, Majeed M, Jamialahmadi T, et al. Curcumin and chemokines: mechanism of action and therapeutic potential in inflammatory diseases. Inflammopharmacology (2023) 31(3):1069–93. doi: 10.1007/s10787-023-01136-w
119. Gupta PK, Jahagirdar P, Tripathi D, Devarajan PV, Kulkarni S. Macrophage targeted polymeric curcumin nanoparticles limit intracellular survival of Mycobacterium tuberculosis through induction of autophagy and augment anti-TB activity of isoniazid in RAW 264.7 macrophages. Front Immunol (2023) 14:1233630. doi: 10.3389/fimmu.2023.1233630
120. Braunstein M, Hickey AJ, Ekins S. Why wait? The case for treating tuberculosis with inhaled drugs. Pharm Res (2019) 36(12):166. doi: 10.1007/s11095-019-2704-6
121. Upadhyay R, Sanchez-Hidalgo A, Wilusz CJ, Lenaerts AJ, Arab J, Yeh J, et al. Host directed therapy for chronic tuberculosis via intrapulmonary delivery of aerosolized peptide inhibitors targeting the IL-10-STAT3 pathway. Sci Rep (2018) 8(1):16610. doi: 10.1038/s41598-018-35023-0
122. Liang C, Qiao G, Liu Y, Tian L, Hui N, Li J, et al. Overview of all-trans-retinoic acid (ATRA) and its analogues: Structures, activities, and mechanisms in acute promyelocytic leukaemia. Eur J Med Chem (2021) 220:113451. doi: 10.1016/j.ejmech.2021.113451
123. O’Connor G, Krishnan N, Fagan-Murphy A, Cassidy J, O’Leary S, Robertson BD, et al. Inhalable poly(lactic-co-glycolic acid) (PLGA) microparticles encapsulating all-trans-Retinoic acid (ATRA) as a host-directed, adjunctive treatment for Mycobacterium tuberculosis infection. Eur J Pharm Biopharm (2019) 134:153–65. doi: 10.1016/j.ejpb.2018.10.020
124. Bahlool AZ, Fattah S, O’Sullivan A, Cavanagh B, MacLoughlin R, Keane J, et al. Development of inhalable ATRA-loaded PLGA nanoparticles as host-directed immunotherapy against tuberculosis. Pharmaceutics (2022) 14(8):1745. doi: 10.3390/pharmaceutics14081745
125. Singh VK, Chau E, Mishra A, DeAnda A, Hegde VL, Sastry JK, et al. CD44 receptor targeted nanoparticles augment immunity against tuberculosis in mice. J Control Release (2022) 349:796–811. doi: 10.1016/j.jconrel.2022.07.040
126. Naicker N, Rodel H, Perumal R, Ganga Y, Bernstein M, Benede N, et al. Metformin increases cell viability and regulates pro-inflammatory response to mtb. Infect Drug Resist (2023) 16:3629–38. doi: 10.2147/IDR.S401403
127. Lee MC, Chiang CY, Lee CH, Ho CM, Chang CH, Wang JY, et al. Metformin use is associated with a low risk of tuberculosis among newly diagnosed diabetes mellitus patients with normal renal function: A nationwide cohort study with validated diagnostic criteria. PloS One (2018) 13(10):e0205807. doi: 10.1371/journal.pone.0205807
128. Fu CP, Lee CL, Li YH, Lin SY. Metformin as a potential protective therapy against tuberculosis in patients with diabetes mellitus: A retrospective cohort study in a single teaching hospital. J Diabetes Investig (2021) 12(9):1603–9. doi: 10.1111/jdi.13523
129. Heo E, Kim E, Jang EJ, Lee CH. The cumulative dose-dependent effects of metformin on the development of tuberculosis in patients newly diagnosed with type 2 diabetes mellitus. BMC Pulm Med (2021) 21(1):303. doi: 10.1186/s12890-021-01667-4
130. Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med (2014) 6(263):263ra159. doi: 10.1126/scitranslmed.3009885
131. Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin use reverses the increased mortality associated with diabetes mellitus during tuberculosis treatment. Clin Infect Dis (2018) 66(2):198–205. doi: 10.1093/cid/cix819
132. Restrepo BI, Schlesinger LS. Impact of diabetes on the natural history of tuberculosis. Diabetes Res Clin Pract (2014) 106(2):191–9. doi: 10.1016/j.diabres.2014.06.011
133. Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis (2009) 9(12):737–46. doi: 10.1016/S1473-3099(09)70282-8
134. Global tuberculosis report 2022 . Available at: https://iris.who.int/handle/10665/363752.
135. Hinkmann C, Knerr I, Hahn EG, Lohmann T, Seifarth CC. Reduced frequency of peripheral plasmacytoid dendritic cells in type 1 diabetes. Horm Metab Res (2008) 40(11):767–71. doi: 10.1055/s-2008-1080896
136. Gilardini Montani MS, Granato M, Cuomo L, Valia S, Di Renzo L, D’Orazi G, et al. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/β-catenin and p38 MAPK. Biochim Biophys Acta (2016) 1862(4):805–13. doi: 10.1016/j.bbadis.2016.01.001
137. Kousathana F, Georgitsi M, Lambadiari V, Giamarellos-Bourboulis EJ, Dimitriadis G, Mouktaroudi M. Defective production of interleukin-1 beta in patients with type 2 diabetes mellitus: Restoration by proper glycemic control. Cytokine (2017) 90:177–84:2017. doi: 10.1016/j.cyto.2016.11.009
138. Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PloS One (2010) 5(3):e9539. doi: 10.1371/journal.pone.0009539
139. Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes (2014) 63(5):1698–711. doi: 10.2337/db13-0863
140. Ridzuan N, John CM, Sandrasaigaran P, Maqbool M, Liew LC, Lim J, et al. Preliminary study on overproduction of reactive oxygen species by neutrophils in diabetes mellitus. World J Diabetes (2016) 7(13):271. doi: 10.4239/wjd.v7.i13.271
141. Wang H, Meng QH, Gordon JR, Khandwala H, Wu L. Proinflammatory and proapoptotic effects of methylglyoxal on neutrophils from patients with type 2 diabetes mellitus. Clin Biochem (2007) 40(16–17):1232–9. doi: 10.1016/j.clinbiochem.2007.07.016
142. Berrou J, Fougeray S, Venot M, Chardiny V, Gautier JF, Dulphy N, et al. Natural killer cell function, an important target for infection and tumor protection, is impaired in type 2 diabetes. PloS One (2013) 8(4):e62418. doi: 10.1371/journal.pone.0062418
143. Farnsworth CW, Schott EM, Benvie A, Kates SL, Schwarz EM, Gill SR, et al. Exacerbated staphylococcus aureus foot infections in obese/diabetic mice are associated with impaired germinal center reactions, ig class switching, and humoral immunity. J Immunol (2018) 201(2):560–72. doi: 10.4049/jimmunol.1800253
144. Danze PM, Tarjoman A, Rousseaux J, Fossati P, Dautrevaux M. Evidence for an increased glycation of IgG in diabetic patients. Clin Chim Acta (1987) 166(2–3):143–53. doi: 10.1016/0009-8981(87)90416-5
145. Martinez PJ, Mathews C, Actor JK, Hwang SA, Brown EL, De Santiago HK, et al. Impaired CD4+ and T-helper 17 cell memory response to Streptococcus pneumoniae is associated with elevated glucose and percent glycated hemoglobin A1c in Mexican Americans with type 2 diabetes mellitus. Transl Res (2014) 163(1):53–63. doi: 10.1016/j.trsl.2013.07.005
146. Richard C, Wadowski M, Goruk S, Cameron L, Sharma AM, Field CJ. Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res Care (2017) 5(1):e000379. doi: 10.1136/bmjdrc-2016-000379
147. Gan YH. Host susceptibility factors to bacterial infections in type 2 diabetes. PloS Pathog (2013) 9(12):1–3. doi: 10.1371/journal.ppat.1003794
148. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev (2020) 16(5):442–9. doi: 10.2174/1573399815666191024085838
149. Viswanathan V, Vigneswari A, Selvan K, Satyavani K, Rajeswari R, Kapur A. Effect of diabetes on treatment outcome of smear-positive pulmonary tuberculosis–a report from South India. J Diabetes Complications (2014) 28(2):162–5. doi: 10.1016/j.jdiacomp.2013.12.003
150. Evangelista M do SN, Maia R, Toledo JP, de Abreu RG, Barreira D. Tuberculosis associated with diabetes mellitus by age group in Brazil: a retrospective cohort study, 2007-2014. Braz J Infect Dis (2020) 24(2):130–6. doi: 10.1016/j.bjid.2020.03.005
151. Tegegne BS, Mengesha MM, Teferra AA, Awoke MA, Habtewold TD. Association between diabetes mellitus and multi-drug-resistant tuberculosis: evidence from a systematic review and meta-analysis. Syst Rev (2018) 7(1):161. doi: 10.1186/s13643-018-0828-0
152. Behr MA, Warren SA, Salamon H, Hopewell PC, Ponce De Leon A, Daley CL, et al. Transmission of Mycobacterium tuberculosis from patients smear-negative for acid-fast bacilli. Lancet (1999) 353(9151):444–9. doi: 10.1016/S0140-6736(98)03406-0
153. Jeon CY, Harries AD, Baker MA, Hart JE, Kapur A, Lönnroth K, et al. Bi-directional screening for tuberculosis and diabetes: a systematic review. Trop Med Int Health (2010) 15(11):1300–14. doi: 10.1111/j.1365-3156.2010.02632.x
154. Krishnappa D, Sharma SK, Singh AD, Sinha S, Ammini AC, Soneja M. Impact of tuberculosis on glycaemic status: A neglected association. Indian J Med Res (2019) 149(3):384–8. doi: 10.4103/ijmr.IJMR_1927_17
155. Menon S, Rossi R, Dusabimana A, Zdraveska N, Bhattacharyya S, Francis J. The epidemiology of tuberculosis-associated hyperglycemia in individuals newly screened for type 2 diabetes mellitus: systematic review and meta-analysis. BMC Infect Dis (2020) 20(1):937. doi: 10.1186/s12879-020-05512-7
156. Workneh MH, Bjune GA, Yimer SA. Prevalence and associated factors of tuberculosis and diabetes mellitus comorbidity: A systematic review. PloS One (2017) 12(4):e0175925. doi: 10.1371/journal.pone.0175925
157. Monroy-Mérida G, Guzmán-Beltrán S, Hernández F, Santos-Mendoza T, Bobadilla K. High glucose concentrations impair the processing and presentation of mycobacterium tuberculosis antigens in vitro. Biomolecules (2021) 11(12):1763. doi: 10.3390/biom11121763
158. Lopez-Lopez N, Martinez AGR, Garcia-Hernandez MH, Hernandez-Pando R, Castañeda-Delgado JE, Lugo-Villarino G, et al. Type-2 diabetes alters the basal phenotype of human macrophages and diminishes their capacity to respond, internalise, and control Mycobacterium tuberculosis. Mem Inst Oswaldo Cruz (2018) 113(4):e170326. doi: 10.1590/0074-02760170326
159. Alim MA, Kupz A, Sikder S, Rush C, Govan B, Ketheesan N. Increased susceptibility to Mycobacterium tuberculosis infection in a diet-induced murine model of type 2 diabetes. Microbes Infect (2020) 22(8):303–11. doi: 10.1016/j.micinf.2020.03.004
160. Sinha R, Ngo MD, Bartlett S, Bielefeldt-Ohmann H, Keshvari S, Hasnain SZ, et al. Pre-diabetes increases tuberculosis disease severity, while high body fat without impaired glucose tolerance is protective. Front Cell Infect Microbiol (2021) 11. doi: 10.3389/fcimb.2021.691823
161. Magee MJ, Khakharia A, Gandhi NR, Day CL, Kornfeld H, Rhee MK, et al. Increased risk of incident diabetes among individuals with latent tuberculosis infection. Diabetes Care (2022) 45(4):880–7. doi: 10.2337/dc21-1687
162. Tepekule B, Kusejko K, Zeeb M, Tarr PE, Calmy A, Battegay M, et al. Impact of latent tuberculosis on diabetes. J Infect Dis (2022) 225(12):2229–34. doi: 10.1093/infdis/jiac054
163. Masood KI, Irfan M, Masood Q, Yameen M, Jamil B, Ram N, et al. Latent M. tuberculosis infection is associated with increased inflammatory cytokine and decreased suppressor of cytokine signalling (SOCS)-3 in the diabetic host. Scand J Immunol (2022) 95(4):e13134. doi: 10.1111/sji.13134
164. Kathamuthu GR, Pavan Kumar N, Moideen K, Dolla C, Kumaran P, Babu S. Multi-dimensionality immunophenotyping analyses of MAIT cells expressing th1/th17 cytokines and cytotoxic markers in latent tuberculosis diabetes comorbidity. Pathogens (2022) 11(1):87. doi: 10.3390/pathogens11010087
165. Kathamuthu GR, Kumar NP, Moideen K, Menon PA, Babu S. Decreased frequencies of gamma/delta T cells expressing Th1/Th17 cytokine, cytotoxic, and immune markers in latent tuberculosis-diabetes/pre-diabetes comorbidity. Front Cell Infect Microbiol (2021) 11:756854. doi: 10.3389/fcimb.2021.756854
166. Ayyappan JP, Ganapathi U, Lizardo K, Vinnard C, Subbian S, Perlin D, et al. Adipose tissue regulates pulmonary pathology during TB infection. mBio (2019) 10(2):e02771-18. doi: 10.1128/mBio.02771-18
167. Oswal N, Lizardo K, Dhanyalayam D, Ayyappan JP, Thangavel H, Heysell SK, et al. Host metabolic changes during mycobacterium tuberculosis infection cause insulin resistance in adult mice. J Clin Med (2022) 11(6):1646. doi: 10.3390/jcm11061646
168. Podell BK, Ackart DF, Kirk NM, Eck SP, Bell C, Basaraba RJ. Non-diabetic hyperglycemia exacerbates disease severity in Mycobacterium tuberculosis infected Guinea pigs. PloS One (2012) 7(10):e46824. doi: 10.1371/journal.pone.0046824
169. Cheekatla SS, Tripathi D, Venkatasubramanian S, Nathella PK, Paidipally P, Ishibashi M, et al. NK-CD11c+ Cell Crosstalk in Diabetes Enhances IL-6-Mediated Inflammation during Mycobacterium tuberculosis Infection. PloS Pathog (2016) 12(10):e1005972. doi: 10.1371/journal.ppat.1005972
170. Tripathi D, Radhakrishnan RK, Thandi RS, Paidipally P, Devalraju KP, Neela VSK, et al. IL-22 produced by type 3 innate lymphoid cells (ILC3s) reduces the mortality of type 2 diabetes mellitus (T2DM) mice infected with Mycobacterium tuberculosis. PloS Pathog (2019) 15(12):e1008140. doi: 10.1371/journal.ppat.1008140
171. Restrepo BI, Khan A, Singh VK, de-Leon E, Aguillón-Durán GP, Ledezma-Campos E, et al. Human monocyte-derived macrophage responses to M. tuberculosis differ by the host’s tuberculosis, diabetes or obesity status, and are enhanced by rapamycin. Tuberculosis (Edinb) (2021) 126:102047. doi: 10.1016/j.tube.2020.102047
172. Vrieling F, Wilson L, Rensen PCN, Walzl G, Ottenhoff THM, Joosten SA. Oxidized low-density lipoprotein (oxLDL) supports Mycobacterium tuberculosis survival in macrophages by inducing lysosomal dysfunction. PloS Pathog (2019) 15(4):e1007724. doi: 10.1371/journal.ppat.1007724
173. Lachmandas E, Van Den Heuvel CNAM, Damen MSMA, Cleophas MCP, Netea MG, Van Crevel R. Diabetes mellitus and increased tuberculosis susceptibility: the role of short-chain fatty acids. J Diabetes Res (2016) 2016:6014631. doi: 10.1155/2016/6014631
174. Bartlett S, Gemiarto AT, Ngo MD, Sajiir H, Hailu S, Sinha R, et al. GPR183 regulates interferons, autophagy, and bacterial growth during mycobacterium tuberculosis infection and is associated with TB disease severity. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.601534
175. Ngo MD, Bartlett S, Bielefeldt-Ohmann H, Foo CX, Sinha R, Arachchige BJ, et al. A blunted GPR183/oxysterol axis during dysglycemia results in delayed recruitment of macrophages to the lung during mycobacterium tuberculosis infection. J Infect Dis (2022) 225(12):2219–28. doi: 10.1093/infdis/jiac102
176. Wang X, Lin Y, Liang Y, Ye Y, Wang D, Tai A, et al. Phosphorylated STAT3 suppresses microRNA-19b/1281 to aggravate lung injury in mice with type 2 diabetes mellitus-associated pulmonary tuberculosis. J Cell Mol Med (2020) 24(23):13763–74. doi: 10.1111/jcmm.15954
177. Zhao H, Shi L, Wang X, Yu X, Wang D. Sp1 transcription factor represses transcription of phosphatase and tensin homolog to aggravate lung injury in mice with type 2 diabetes mellitus-pulmonary tuberculosis. Bioengineered (2022) 13(4):9928–44. doi: 10.1080/21655979.2022.2062196
178. Kewcharoenwong C, Prabowo SA, Bancroft GJ, Fletcher HA, Lertmemongkolchai G. Glibenclamide reduces primary human monocyte functions against tuberculosis infection by enhancing M2 polarization. Front Immunol (2018) 9(SEP):2109. doi: 10.3389/fimmu.2018.02109
179. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U.S.A. (2008) 105(11):4376–80. doi: 10.1073/pnas.0711159105
180. Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science (2000) 288(5471):1647–50. doi: 10.1126/science.288.5471.1647
181. Kang YA, Choi NK, Seong JM, Heo EY, Koo BK, Hwang SS, et al. The effects of statin use on the development of tuberculosis among patients with diabetes mellitus. Int J Tuberc Lung Dis (2014) 18(6):717–24. doi: 10.5588/ijtld.13.0854
182. Lee MY, Lin KD, Hsu WH, Chang HL, Yang YH, Hsiao PJ, et al. Statin, calcium channel blocker and Beta blocker therapy may decrease the incidence of tuberculosis infection in elderly Taiwanese patients with type 2 diabetes. Int J Mol Sci (2015) 16(5):11369–84. doi: 10.3390/ijms160511369
183. Kim MC, Yun SC, Lee SO, Choi SH, Kim YS, Woo JH, et al. Association between tuberculosis, statin use, and diabetes: A propensity score-matched analysis. Am J Trop Med Hyg (2019) 101(2):350–6. doi: 10.4269/ajtmh.18-0983
184. Pan SW, Yen YF, Feng JY, Chuang PH, Su VYF, Kou YR, et al. Opposite effects of statins on the risk of tuberculosis and herpes zoster in patients with diabetes: A population-based cohort study. Br J Clin Pharmacol (2020) 86(3):569–79. doi: 10.1111/bcp.14142
185. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother (2016) 71(6):1570–7. doi: 10.1093/jac/dkw014
186. Lobato LS, Rosa PS, Da Silva Ferreira J, Da Silva Neumann A, Da Silva MG, Nascimento DC, et al. Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother (2014) 58(10):5766–74. doi: 10.1128/AAC.01826-13
187. Lai CC, Lee MTG, Lee SH, Hsu WT, Chang SS, Chen SC, et al. Statin treatment is associated with a decreased risk of active tuberculosis: an analysis of a nationally representative cohort. Thorax (2016) 71(7):646–51. doi: 10.1136/thoraxjnl-2015-207052
188. Adewole OO, Omotoso BA, Ogunsina M, Aminu A, Odeyemi AO, Awopeju OF, et al. Atorvastatin accelerates Mycobacterium tuberculosis clearance in pulmonary TB: a randomised phase IIA trial. Int J Tuberc Lung Dis (2023) 27(3):226–8. doi: 10.5588/ijtld.22.0548
189. Su VYF, Su WJ, Yen YF, Pan SW, Chuang PH, Feng JY, et al. Statin use is associated with a lower risk of TB. Chest (2017) 152(3):598–606. doi: 10.1016/j.chest.2017.04.170
190. Tsai IF, Kuo CP, Lin AB, Chien MN, Ho HT, Wei TY, et al. Potential effect of ezetimibe against Mycobacterium tuberculosis infection in type II diabetes. Respirology (2017) 22(3):559–66. doi: 10.1111/resp.12948
191. Lopez-Lopez N, Gonzalez-Curiel I, Castañeda-Delgado J, Montoya-Rosales A, Gandara-Jasso B, Enciso-Moreno JA, et al. Vitamin D supplementation promotes macrophages’ anti-mycobacterial activity in type 2 diabetes mellitus patients with low vitamin D receptor expression. Microbes Infect (2014) 16(9):755–61. doi: 10.1016/j.micinf.2014.06.010
192. Foretz M, Guigas B, Viollet B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinol (2023) 19(8):460–476. doi: 10.1038/s41574-023-00833-4
193. Millman AC, Salman M, Dayaram YK, Connell ND, Venketaraman V. Natural killer cells, glutathione, cytokines, and innate immunity against Mycobacterium tuberculosis. J Interferon Cytokine Res (2008) 28(3):153–65. doi: 10.1089/jir.2007.0095
194. Guerra C, Morris D, Sipin A, Kung S, Franklin M, Gray D, et al. Glutathione and adaptive immune responses against Mycobacterium tuberculosis infection in healthy and HIV infected individuals. PloS One (2011) 6(12):e28378. doi: 10.1371/journal.pone.0028378
195. Islamoglu H, Cao R, Teskey G, Gyurjian K, Lucar S, Fraix MP, et al. Effects of ReadiSorb L-GSH in Altering Granulomatous Responses against Mycobacterium tuberculosis Infection. J Clin Med (2018) 7(3):40. doi: 10.3390/jcm7030040
196. Kachour N, Beever A, Owens J, Cao R, Kolloli A, Kumar R, et al. Liposomal glutathione helps to mitigate mycobacterium tuberculosis infection in the lungs. Antioxid (Basel) (2022) 11(4):673. doi: 10.3390/antiox11040673
197. Sasaninia K, Kelley M, Abnousian A, Owens J, Yoon SY, Beever A, et al. Liposomal glutathione supplementation mitigates extrapulmonary tuberculosis in the liver and spleen. Front Biosci (Elite Ed) (2023) 15(3):15. doi: 10.31083/j.fbe1503015
198. Beever A, Kachour N, Owens J, Sasaninia K, Kolloli A, Kumar R, et al. L-GSH supplementation in conjunction with rifampicin augments the treatment response to mycobacterium tuberculosis in a diabetic mouse model. Front Pharmacol (2022) 13:879729. doi: 10.3389/fphar.2022.879729
199. Abrahem R, Cao R, Robinson B, Munjal S, Cho T, To K, et al. Elucidating the efficacy of the bacille calmette-guérin vaccination in conjunction with first line antibiotics and liposomal glutathione. J Clin Med (2019) 8(10):1556. doi: 10.3390/jcm8101556
200. To K, Cao R, Yegiazaryan A, Owens J, Nguyen T, Sasaninia K, et al. Effects of Oral Liposomal Glutathione in Altering the Immune Responses Against Mycobacterium tuberculosis and the Mycobacterium bovis BCG Strain in Individuals With Type 2 Diabetes. Front Cell Infect Microbiol (2021) 11:657775. doi: 10.3389/fcimb.2021.657775
Keywords: mycobacterium tuberculosis, diabetes mellitus, host-directed therapy, drug-resistance, immune
Citation: Zhao L, Fan K, Sun X, Li W, Qin F, Shi L, Gao F and Zheng C (2024) Host-directed therapy against mycobacterium tuberculosis infections with diabetes mellitus. Front. Immunol. 14:1305325. doi: 10.3389/fimmu.2023.1305325
Received: 01 October 2023; Accepted: 11 December 2023;
Published: 08 January 2024.
Edited by:
Subash Babu, International Centers for Excellence in Research (ICER), IndiaReviewed by:
Afsal Kolloli, Rutgers University, Newark, United StatesCopyright © 2024 Zhao, Fan, Sun, Li, Qin, Shi, Gao and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunlan Zheng, MTUwMDcxODU5MjBAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.