
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 December 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1301074
 Aoya Han1†Tingting Peng1†
Aoya Han1†Tingting Peng1† Yinyin Xie1
Yinyin Xie1 Wanwan Zhang1Wenlin Sun1Yi Xie1Yunqing Ma1Cui Wang2
Wanwan Zhang1Wenlin Sun1Yi Xie1Yunqing Ma1Cui Wang2 Nanchang Xie1*
Nanchang Xie1*Regulatory T cells (Tregs) can eliminate autoreactive lymphocytes, induce self-tolerance, and suppress the inflammatory response. Mitochondria, as the energy factories of cells, are essential for regulating the survival, differentiation, and function of Tregs. Studies have shown that patients with autoimmune diseases of the central nervous system, such as multiple sclerosis, neuromyelitis optica spectrum disorder, and autoimmune encephalitis, have aberrant Tregs and mitochondrial damage. However, the role of mitochondrial-regulated Tregs in autoimmune diseases of the central nervous system remains inconclusive. Therefore, this study reviews the mitochondrial regulation of Tregs in autoimmune diseases of the central nervous system and investigates the possible mitochondrial therapeutic targets.
Regulatory T cells (Tregs) are negative immune-regulatory cells that play a significant role in immune tolerance and the normal function of the immune system by eliminating the autoreactive lymphocytes, inducing self-tolerance, and suppressing the inflammatory response through various mechanisms (1, 2). Recent studies have also indicated the ability of Tregs to promote tissue repair or regeneration by secreting tissue-specific regenerative factors (3–5). Consistently, aberrant Tregs are a major driver of many autoimmune diseases. Reduced number and impaired function of Tregs have been reported in various autoimmune diseases, including myasthenia gravis, systemic lupus erythematosus, rheumatoid arthritis, and type 1 diabetes (6–8). Some autoimmune diseases of the central nervous system (CNS) occur due to self-tolerance defects. Self-tolerance defect in multiple sclerosis (MS) occurs mainly due to impaired Treg function, but there are also cases of decreased Treg number (6, 9, 10). However, neuromyelitis optica spectrum disorder (NMOSD) and autoimmune encephalitis are characterized by reduced number of Tregs (11, 12). The absence of immunosuppressive capacity of Tregs and decreased number of Tregs activate autoreactive cells, promote B cells to produce autoantibodies and effector T cells to secrete pro-inflammatory cytokines and chemokines, and induce the infiltration of macrophages and effector T cells into the CNS, ultimately promoting the development of autoimmune diseases of the CNS (6, 11–14). However, the specific mechanisms leading to reduced number and impaired function of Tregs in autoimmune diseases of the CNS remain to be determined.
Among numerous cellular biological processes and molecular mechanisms associated with Tregs, their unique metabolic profile has recently drawn significant interest. In physiological conditions, Tregs exhibit increased mitochondrial metabolism, characterized by high levels of mitochondrial fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) and modest glycolysis (15–19). During FAO and OXPHOS, FoxP3, a critical transcription factor of Tregs, is transcriptionally upregulated, which is essential for maintaining the immunosuppressive function and stability of Tregs and can promote Treg differentiation by inhibiting RORγt binding to DNA (15, 16, 20–23). Glycolysis is necessary for the growth and proliferation of Tregs, but it reduces the immunosuppressive ability and stability of Tregs during growth and proliferation (24–26). Glycolysis is also a key energy source for Treg migration to inflammatory tissue (27). Moreover, mitochondrial metabolite α-ketoglutarate is a substrate for ten-eleven translocase (TET)-mediated demethylation of the FoxP3 locus in Tregs, which is required for optimal expression of FoxP3 and immunosuppressive function of Tregs (28–30). Mitochondrial damage, such as damaged respiratory chain complexes and abnormal mitochondrial morphology, can markedly impair the survival, differentiation, and function of Tregs. Therefore, maintaining mitochondrial structure and function is critical for Treg homeostasis and function.
Recent studies have found abnormal mitochondrial morphology, impaired cristae organization, reduced activity and expression of respiratory chain complexes, decreased expression of cytochrome C, increased content of mitochondrial DNA (mtDNA) and mitochondrial reactive oxygen species (mtROS), and damaged mitophagy in Tregs of patients with autoimmune diseases of the CNS such as MS and NMOSD (31–33). Therefore, mitochondrial-regulated Tregs may be involved in the occurrence and progression of autoimmune diseases of the CNS (31). This paper reviews the Treg regulation by mitochondria in autoimmune diseases of the CNS and introduces the possible mitochondrial therapeutic targets.
As a signaling molecule, mtROS plays a significant role in activating signaling pathways and determining cell fate. mtROS level is strictly regulated by antioxidants such as superoxide dismutase and catalase (CAT) (34–36). However, the activities of manganese superoxide dismutase (MnSOD) and CAT are significantly decreased in Tregs of mice with experimental autoimmune encephalomyelitis (EAE), which impairs the ability of the antioxidant system to scavenge mtROS and results in mtROS accumulation (Figure 1) (31). Furthermore, aberrant mitochondrial morphology, impaired cristae organization, and reduced expression of respiratory chain complexes in MS and NMOSD can cause mitochondrial dysfunction and mtROS accumulation (Figure 1) (31–33, 37).
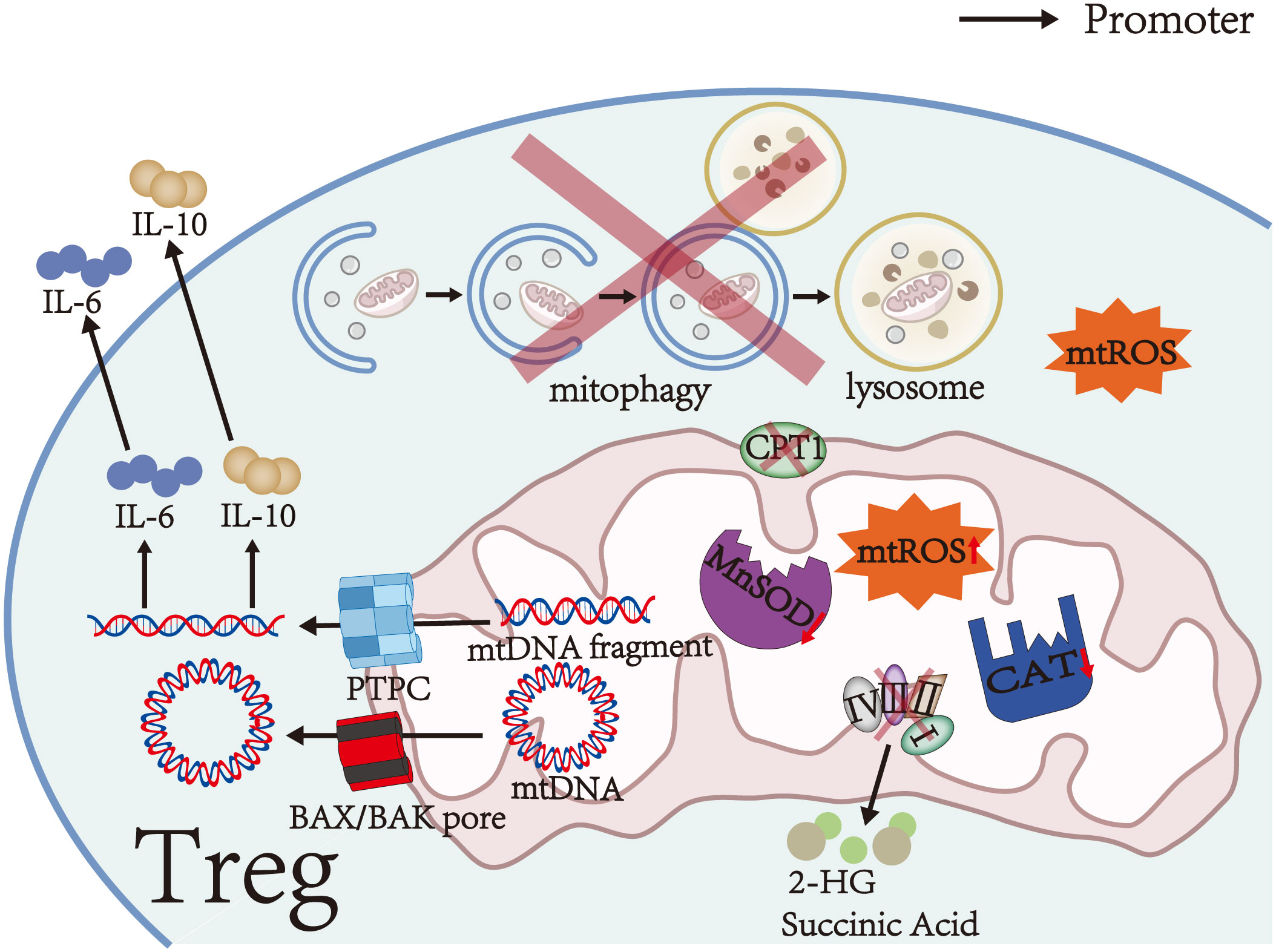
Figure 1 Mitochondrial damage in Tregs. Aberrant mitochondrial morphology, impaired cristae organization, reduced expression of respiratory chain complexes, and significantly decreased activities of MnSOD and CAT in Tregs cause mitochondrial dysfunction and mtROS accumulation. Furthermore, respiratory chain complex III deficiency leads to the accumulation of succinate and 2-HG in Tregs. During mitochondrial dysfunction, mtDNA and mtDNA fragments are released into the cytosol via the PTPC or BAX/BAK pores. Cytosolic mtDNA and mtDNA fragments can promote the secretion of IL-6 and IL-10. CPT1 on the outer mitochondrial membrane is a rate-limiting enzyme in FAO, which is the main energy source of Tregs. Therefore, reduced or defective CPT1 activity causes FAO impairment. Moreover, impaired initiation of mitophagy, incorrect autophagosome formation, or aberrant lysosomal degradation impairs mitophagy and leads to damaged mitochondria accumulation in Tregs.
mtROS upregulates the transcription of hypoxia-inducible factor 1α (HIF-1α) subunits through NF-κB activation and stabilizes HIF-1α subunits by inhibiting prolyl hydroxylase and asparaginyl hydroxylase (38–40). Stabilized HIF-1α subunits dimerize with HIF-1β subunits (also known as aryl hydrocarbon receptor nuclear translocator) to form HIF-1 (41). Subsequently, HIF-1 translocates into the nucleus and induces the transcription of genes encoding glycolytic enzymes and glucose transporters, leading to a metabolic shift from OXPHOS to glycolysis in the early stages of T cell differentiation (42, 43). Data from several animal studies suggest that glycolysis reduces the expression of FoxP3, CD25, PD-1, CTLA-4, and ICOS (Table 1), disrupts their stability, and inhibits induced Treg (iTreg) differentiation, thereby impairing the immunosuppressive function of iTregs and decreasing the number of iTregs (15, 25, 43, 49). Moreover, excessive activation of glycolysis reduces the stability of thymus-derived Tregs (tTregs) by downregulating the expression of FoxP3 and CD25 and converts tTregs into pathogenic cells with effector or memory T cell phenotype (25, 50–52). These cells contribute to autoimmune diseases of the CNS by producing pro-inflammatory cytokines, such as IFN-γ and IL-17 (51, 53–55). However, recent studies have shown that glycolysis is essential for Treg differentiation and function. For example, some in vitro studies have shown that glycolysis can induce the differentiation of naive T cells into iTreg and upregulate the expression of CTLA-4, PD-1, CD39, and ICOS (Table 1) to maintain the immunosuppressive function of iTregs (24, 56). These processes are achieved by regulating the expression of FoxP3 exon 2 splicing variants via glycolytic enzyme enolase-1 (24, 56). In addition, glycolysis is required for the optimal expression of the inhibitory molecules CTLA-4 and ICOS and is essential for the immunosuppressive function of tTregs (24, 57). The controversial roles of glycolysis in Treg differentiation and function may be due to differences in the origin of Tregs (thymic or peripheral T cells, human or animal), external and internal environments, metabolic requirements, and cytokines environments. Therefore, the effect of glycolysis on Tregs still needs to be explored. Interestingly, due to the dimerization of HIF-1α subunits and HIF-1β subunits and aryl hydrocarbon receptor degradation by HIF-1α subunits via the ubiquitin-proteasome pathway, the binding rate of aryl hydrocarbon receptor to HIF-1β subunits decreases, causing reduced transcriptional activity of genes encoding ectoenzymes and IL-10 during Treg differentiation and eventually inhibiting Treg differentiation and reducing the number of Tregs (58, 59). Furthermore, HIF-1 binds to the transcription factor FoxP3 in T cell cytoplasm to degrade the latter via the ubiquitin-proteasome pathway, thereby downregulating FoxP3 levels and inhibiting Treg differentiation (Figure 2) (60, 61). However, the exact role of HIF-1 in Tregs remains controversial. Clambey et al. (62) and Flück et al. (63) found that HIF-1 can promote the proliferation of Tregs by inducing FoxP3 transcription, thus inhibiting T cell-mediated colitis. The contradictory roles may be due to the tissue heterogeneity of Tregs, suggesting that HIF-1 expression is essential for Tregs in specific tissues. In addition to inhibiting Treg differentiation by stabilizing HIF-1, mtROS can induce DNA breaks. Subsequently, DNA breaks induce Treg apoptosis by initiating a DNA damage response, ultimately decreasing Treg number (Figure 2) (31, 64).
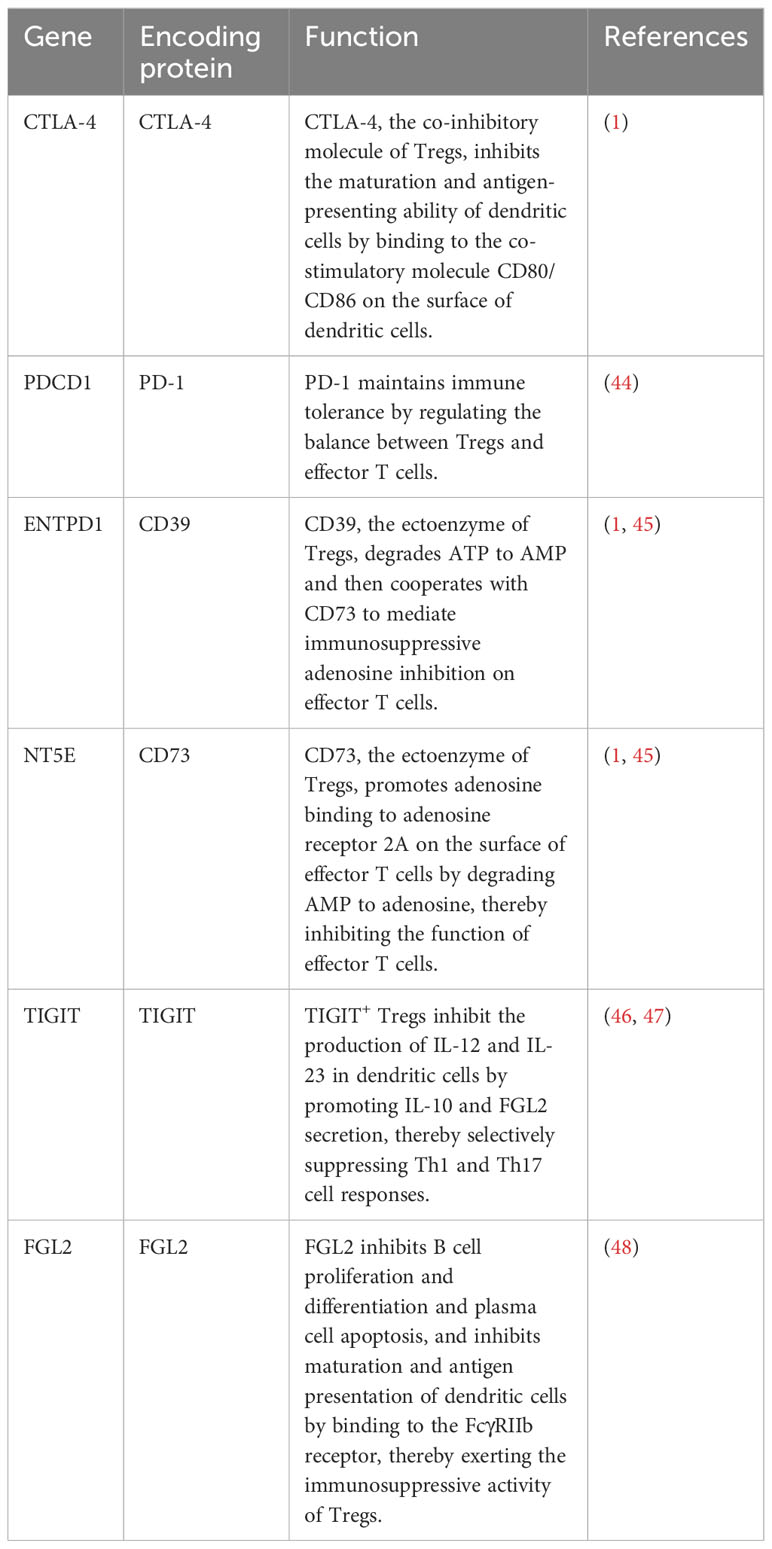
Table 1 Genes and molecules associated with Treg function.
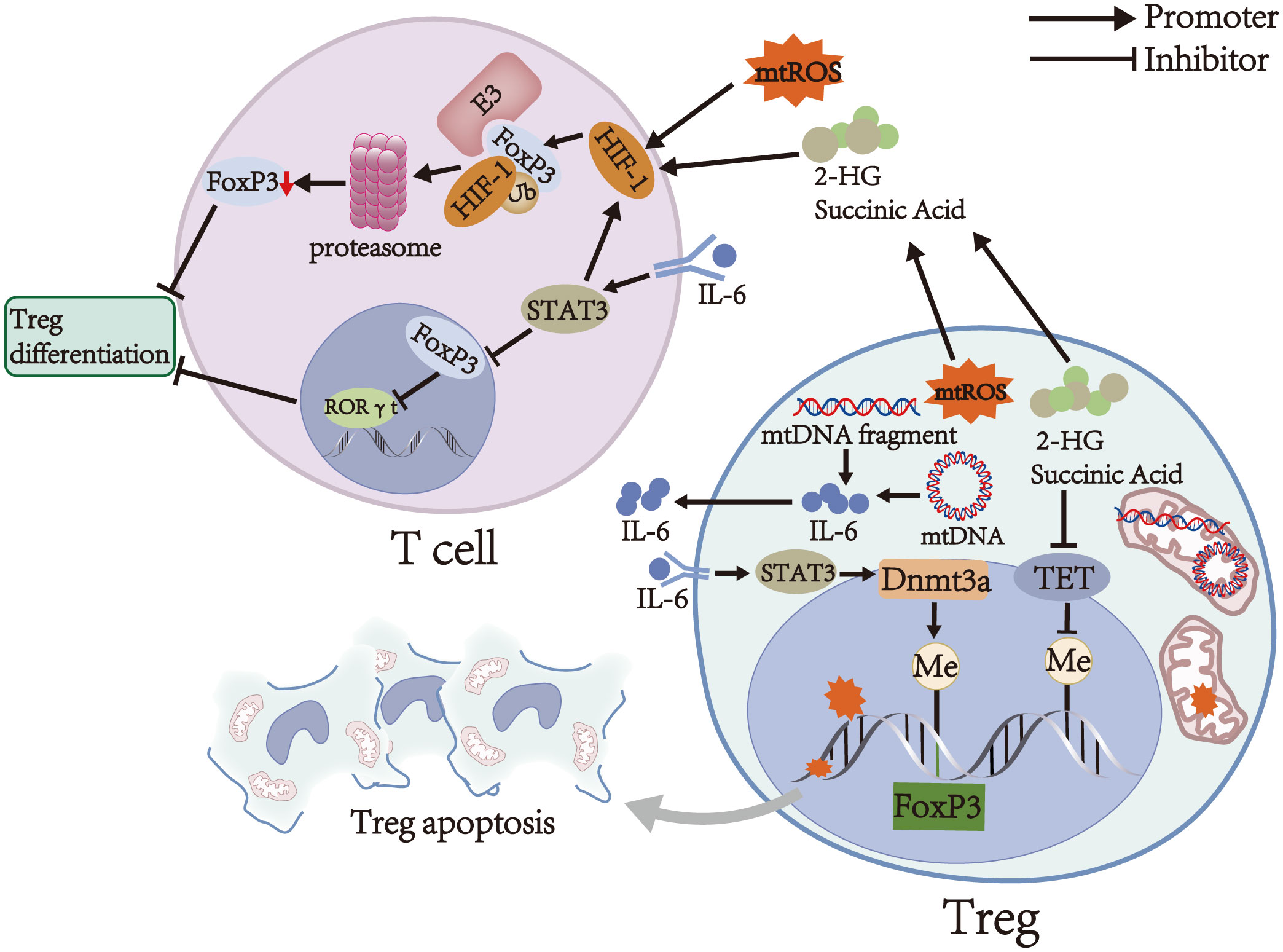
Figure 2 Main mechanisms of Treg regulation by mitochondria. mtROS induces DNA breaks, and the latter can induce Treg apoptosis. Moreover, mtROS upregulates and stabilizes HIF-1, which can downregulate FoxP3 levels and inhibit Treg differentiation by binding to FoxP3 in T cells to degrade the latter via the ubiquitin-proteasome pathway. mtDNA promotes the secretion of IL-6, which activates STAT3 and promotes Foxp3 locus methylation in Tregs in a Dnmt3a-dependent manner, thereby decreasing the expression of FoxP3 and weakening the immunosuppressive function of Tregs. On the other hand, IL-6 activates STAT3 in T cells to attenuate RORγt inhibition by FoxP3 and promote the transcription of HIF-1, thereby inhibiting Treg differentiation. Succinate and 2-HG inhibit TET, thereby causing DNA hypermethylation in specific regulatory regions of Tregs and impairing the immunosuppressive function of Tregs. Furthermore, succinate and 2-HG stabilize HIF-1 and inhibit Treg differentiation.
During mitochondrial dysfunction, mtDNA is cleaved into small fragments by endonucleases. The fragments are released into the cytosol through the permeability transition pore complex (PTPC, including mitochondrial permeability transition pores/MPTP and voltage-dependent anion channels/VDAC) (Figure 1) (65). Moreover, BAX and BAK oligomerize in the outer mitochondrial membrane and form BAX/BAK pores, which allow the inner mitochondrial membrane to herniate into the cytosol and release mtDNA (Figure 1) (66). Cytosolic mtDNA activates the inflammasome NLRP3, which in turn increases the release of mtDNA through a positive feedback mechanism (65, 67, 68). In the EAE model, the increase of mtDNA fluorescent particles in the cytoplasm confirmed the release of mtDNA fragments (31).
As an upstream effector, cytosolic mtDNA promotes the secretion of IL-6 and IL-10 by activating several signal pathways, including cGAS-STING and TLR9-MyD88 signals, finally affecting the number and function of Tregs (Figure 1) (68–70). Among them, IL-10 is an immunosuppressive cytokine that suppresses the activation of autoreactive T cells and downregulates pro-inflammatory cytokines to enhance the immunosuppressive function of Tregs in stress conditions (1, 71). In contrast, IL-6 activates STAT3 and promotes Foxp3 locus methylation in Tregs in a DNA methyltransferase 3a (Dnmt3a)-dependent manner, thereby decreasing the expression of FoxP3 (Figure 2) (55, 72). Low FoxP3 expression can downregulate co-inhibitory molecules and ectoenzymes on the surfaces of Tregs and turn Tregs into pathogenic cells with effector or memory T cell phenotype (20, 53, 55). On the other hand, IL-6 activates STAT3 in T cells by binding to IL-6 receptor and gp130, which can attenuate RORγt inhibition by FoxP3 and promote the transcription of HIF-1α subunits (Figure 2) (21, 60, 73). These alterations finally inhibit Treg differentiation and decrease Treg number (43, 61). Thus, the influence of mtDNA on the number and function of Tregs may depend on the balance between the mechanisms mentioned above.
Recent studies have shown that Tregs mainly rely on high levels of FAO and modest glycolysis to meet their energy requirements in steady-state conditions (15, 16). FAO is also necessary for T cell differentiation to Tregs (74, 75). During FAO-driven OXPHOS, fatty acids increase the stability and immunosuppressive activity of Tregs by upregulating FoxP3 transcription and inducing CD25 and STAT5 expression (23, 76). At the same time, FoxP3 increases the transcription of FAO and OXPHOS-related genes and inhibits glycolysis by binding to the Myc promoter and downregulating the expression of Myc in Tregs, establishing a positive feedback loop to maximize the immunosuppressive function of Tregs (22, 25, 77). Carnitine palmitoyltransferase 1 (CPT1) on the outer mitochondrial membrane is a rate-limiting enzyme in FAO (Figure 1). Previous studies have demonstrated that CPT1 inhibitors can prevent Treg differentiation and significantly reduce the expression of granzymes, ectoenzymes, and co-inhibitory molecules in Tregs by suppressing FAO, thereby reducing the number and immunosuppressive activity of Tregs (15, 78, 79). Therefore, FAO impairment due to reduced or defective CPT1 activity reduces the number and immunosuppressive activity of Tregs. In addition, fatty acid concentrations in patients with MS are lower than those in healthy individuals, thereby decreasing FAO in Tregs and weakening the inhibitory function of Tregs (23).
OXPHOS is a critical metabolic pathway for the differentiation and immunosuppressive function of Tregs (15–17). Therefore, decreased activity and expression of respiratory chain complexes in autoimmune diseases of the CNS, such as MS and NMOSD, can contribute to the abnormal number and function of Tregs (32, 33, 77, 80, 81). Weinberg et al. (81) found that accumulation of succinate and 2-hydroxyglutarate (2-HG) can inhibit TET by competing with α-ketoglutarate in respiratory chain complex III-deficient Tregs (Figure 1), thereby causing DNA hypermethylation in specific regulatory regions of Tregs and reducing the expression of PDCD1 (encoding PD-1), NT5E (encoding CD73), TIGIT, and FGL2 genes associated with the immunosuppressive function of Tregs (Figure 2) (82, 83). These alterations finally downregulate co-inhibitory molecules and ectoenzymes, such as PD-1, CD73, TIGIT, and FGL2, and impair the immunosuppressive function of Tregs (Table 1) (44–46, 48, 81). On the other hand, succinate and 2-HG stabilize HIF-1α subunits by inhibiting prolyl hydroxylases, thereby inhibiting Treg differentiation and decreasing Treg number (Figure 2) (83–85). Similar to respiratory chain complex III inhibition, respiratory chain complex I inhibition can downregulate the expression of FoxP3 and decrease the number of Tregs (16). In addition, Angelin et al. (77) found that Tregs with mitochondrial ND6 gene mutations have reduced immunosuppressive function due to the inability of respiratory chain complex I to oxidize NADH to NAD.
Mitophagy, a specific form of autophagy, is essential for clearing damaged mitochondria and maintaining cell homeostasis (86). Mitochondrial depolarization induces the ubiquitination of mitochondrial outer membrane proteins and promotes the recruitment of mitophagy receptors, followed by autophagosome formation to degrade damaged mitochondria (86). Therefore, impaired initiation of mitophagy, incorrect autophagosome formation, or aberrant lysosomal degradation in autoimmune diseases of the CNS can impair mitophagy in Tregs (Figure 1) (31, 87).
Crosstalk between mitochondria and lysosomes has been demonstrated. Lysosomal dysfunction induces mitochondrial defect and vice versa (88, 89). Mitochondrial dysfunction in Tregs of EAE mice can decrease the activity of several hydrolases in lysosomes and downregulate the expression of Rab7, which regulates the fusion of autophagosomes with lysosomes, thus preventing lysosomal degradation of damaged mitochondria (31, 88, 90). In addition, downregulation of the AMPK-PIKFYVE-PtdIns (3, 5) P2-MCOLN1 pathway in the presence of damaged respiratory chain complexes leads to lysosomal calcium accumulation and impairs lysosomal hydrolysis, thereby impairing mitophagy (91, 92). In addition to lysosomal dysfunction, significantly reduced expression of autophagy protein LC3-II in Tregs in patients with myasthenia gravis can impair autophagosome formation and reduce the number of autophagosomes, eventually impairing mitophagy (87). Thanks to the damaged mitophagy of Tregs, the accumulation of damaged mitochondria increases mtROS production and exacerbates mitochondrial oxidative stress, thereby forming a vicious circle (31). Eventually, mtROS accumulation induces DNA breaks, and DNA damage induces Treg apoptosis by initiating a DNA damage response (31, 64).
Accumulation of mtROS decreases the number of Tregs; thus, a better understanding of the role of mitochondria-specific antioxidants in reducing mtROS may provide a new modality for immunotherapy of autoimmune diseases of the CNS. Animal studies applying mitochondrial antioxidants have reported promising results. Mito-TEMPO, a mitochondria-targeting antioxidant mimicking superoxide dismutase, can restore lysosome function and inhibit Treg apoptosis in the EAE mice model by reducing mtROS levels and mitigating mtROS damage, thereby inhibiting effector T cell infiltration into the spinal cord and increasing Treg infiltration to alleviate the symptoms (Table 2) (31, 93). The ability of superoxide dismutase mimics to delay the progression of EAE suggests that these novel antioxidants can be applied to autoimmune diseases of the CNS, such as NMOSD and autoimmune encephalitis, in the future. Furthermore, Cyclosporin A (CsA), an MPTP inhibitor, can reduce the production of mtROS and attenuate mitochondrial dysfunction by inhibiting MPTP opening through the blockade of the interaction of cyclophilin D with adenine nucleotide translocator (Table 2) (94, 101). Studies have found that CsA can induce Treg proliferation and prevent T cell proliferation by inhibiting calcineurin, thus ameliorating the symptoms of EAE, MS, and NMOSD (95, 96, 102). However, some immunosuppressive effects of CsA may be caused by MPTP inhibition, but this possibility has not been investigated and remains to be explored in the future. In addition, genetic or pharmacological downregulation of mtROS lowers HIF-1α levels, and HIF-1α-deficient mice with an increased number of Tregs are resistant to EAE (61, 103, 104). However, these results do not demonstrate a direct link between reduced mtROS levels, lowered HIF-1α levels, and an increased number of Tregs. In the future, it is necessary to explore whether mitochondrial antioxidants inhibit HIF-1α subunits and whether this inhibition affects Tregs in autoimmune diseases of the CNS.
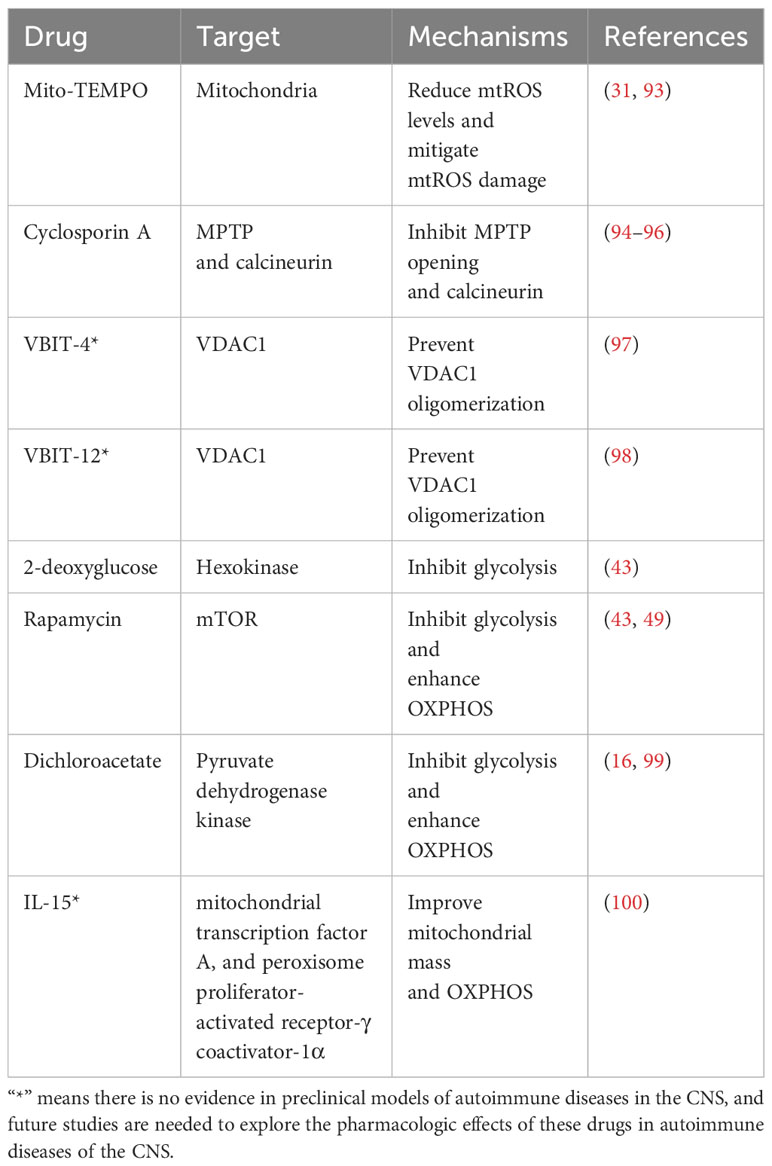
Table 2 Experimental therapeutic drugs targeting mitochondria of Tregs for autoimmune diseases of the CNS.
mtDNA release affects the number and function of Tregs and induces the inflammatory response in autoimmune diseases of the CNS. Therefore, targeting mtDNA release may be another novel therapeutic approach for treating autoimmune diseases of CNS. VBIT-4 and VBIT-12, two VDAC1 oligomerization inhibitors, prevent VDAC1 oligomerization by directly interacting with VDAC1, thereby inhibiting apoptosis, reducing mtDNA release, inhibiting inflammatory cell infiltration and inflammasome NLRP3 activation, protecting against mitochondrial dysfunction, and reducing inflammatory response and disease severity (Table 2) (97, 98). This VDAC1-based treatment strategy has been effective in some animal models of autoimmune diseases, such as inflammatory bowel disease, systemic lupus erythematosus, and type 2 diabetes (97, 98, 105). Future studies on autoimmune diseases of the CNS are needed to determine the efficacy of VDAC1-based treatment for these diseases.
Increased mitochondrial metabolism promotes Treg differentiation, while inhibition of mitochondrial metabolism or increased glycolysis inhibits Treg differentiation. Therefore, shifting cellular energy metabolism to OXPHOS may be a treatment strategy for autoimmune diseases of the CNS. Animal studies of cellular metabolic reprogramming have yielded promising results in autoimmune diseases of the CNS. For example, inhibition of glycolysis by HIF-1α gene knockout or application of 2-deoxyglucose or rapamycin can promote Treg differentiation, increase the number of Tregs, and reduce spinal cord inflammation in EAE mice (Table 2) (43, 49). Similarly, decreasing pyruvate dehydrogenase kinase activity genetically (gene knockout) or pharmacologically (by dichloroacetate) can enhance OXPHOS levels, promote Treg differentiation, increase the number of Tregs, and protect mice against EAE (Table 2) (16, 99). The development of this therapeutic strategy requires further research. In the future, we should explore whether these drugs affecting cellular metabolic reprogramming can modulate the activity of Tregs in autoimmune diseases of the human CNS. Furthermore, IL-15 has been shown to improve mitochondrial mass and OXPHOS in Tregs from HIV-infected immune non-responders by inducing the expression of mitochondrial transcription factor A and peroxisome proliferator-activated receptor-γ coactivator-1α (Table 2) (100). Currently, IL-15 and some of its derivatives, such as IL-15 super-agonists, are in clinical trials for cancer and AIDS. Future studies are needed to investigate the immunologic role of IL-15 in autoimmune diseases of the CNS.
Recent studies have investigated the role of mitochondrial regulation on Treg number and function. However, the details of the mitochondrial regulation process remain to be elucidated. For example, the type of E3 ligase responsible for HIF-1-mediated ubiquitination of FoxP3 is still unclear. Similarly, the effects of glycolysis on Tregs, the mechanisms by which mtDNA affects Tregs and the mechanisms by which respiratory chain complex I damage causes Treg dysfunction need more studies. Moreover, whether drugs targeting mitochondria can improve human autoimmune diseases of the CNS by selectively modulating Treg activity remains to be explored.
In conclusion, understanding the mechanisms by which mitochondrial dysfunction affects the number and function of Tregs in autoimmune diseases may pave the way for developing new therapeutic approaches. Future in-depth studies in this field will be a significant entry point for exploring the molecular mechanisms and therapeutic targets in autoimmune diseases of the CNS.
AH: Writing – original draft, Writing – review & editing. TP: Writing – review & editing. YYX: Writing – review & editing. WZ: Writing – review & editing. WS: Writing – review & editing. YX: Writing – review & editing. YM: Writing – review & editing. CW: Writing – review & editing. NX: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Outstanding Young Talent Cultivation Project of Henan Science and Technology Innovation Talents (Grant number YXKC2022037) and the National Natural Science Foundation of China (Grant number 81971214).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Treg, regulatory T cell; CNS, central nervous system; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; ten-eleven translocase, TET; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species; catalase, CAT; manganese superoxide dismutase, MnSOD; EAE, experimental autoimmune encephalomyelitis; HIF-1, hypoxia-inducible factor 1; induced Treg, iTreg; thymus-derived Treg, tTreg; permeability transition pore complex, PTPC; MPTP, mitochondrial permeability transition pore; VDAC, voltage-dependent anion channel; DNA methyltransferase 3a, Dnmt3a; CPT1, carnitine palmitoyltransferase 1; 2-hydroxyglutarate, 2-HG; Cyclosporin A, CsA.
1. Grover P, Goel PN, Greene MI. Regulatory T cells: regulation of identity and function. Front Immunol (2021) 12:750542. doi: 10.3389/fimmu.2021.750542
2. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
3. Kikuchi K. New function of zebrafish regulatory T cells in organ regeneration. Curr Opin Immunol (2020) 63:7–13. doi: 10.1016/j.coi.2019.10.001
4. Dombrowski Y, O'Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci (2017) 20(5):674–80. doi: 10.1038/nn.4528
5. Arpaia N, Green Jesse A, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162(5):1078–89. doi: 10.1016/j.cell.2015.08.021
6. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflamm (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
7. Goschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to Treg-based therapies. Semin Immunopathol (2019) 41(3):301–14. doi: 10.1007/s00281-019-00741-8
8. Long SA, Buckner JH. CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J Immunol (2011) 187(5):2061–6. doi: 10.4049/jimmunol.1003224
9. Kouchaki E, Salehi M, Reza Sharif M, Nikoueinejad H, Akbari H. Numerical status of CD4(+)CD25(+)FoxP3(+) and CD8(+)CD28(-) regulatory T cells in multiple sclerosis. Iranian J Basic Med Sci (2014) 17(4):250–5.
10. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199(7):971–9. doi: 10.1084/jem.20031579
11. Platt MP, Bolding KA, Wayne CR, Chaudhry S, Cutforth T, Franks KM, et al. Th17 lymphocytes drive vascular and neuronal deficits in a mouse model of postinfectious autoimmune encephalitis. Proc Natl Acad Sci USA (2020) 117(12):6708–16. doi: 10.1073/pnas.1911097117
12. Ma X, Qin C, Chen M, Yu HH, Chu YH, Chen TJ, et al. Regulatory T cells protect against brain damage by alleviating inflammatory response in neuromyelitis optica spectrum disorder. J Neuroinflamm (2021) 18(1):201. doi: 10.1186/s12974-021-02266-0
13. Liba Z, Kayserova J, Elisak M, Marusic P, Nohejlova H, Hanzalova J, et al. Anti-N-methyl-D-aspartate receptor encephalitis: the clinical course in light of the chemokine and cytokine levels in cerebrospinal fluid. J Neuroinflamm (2016) 13(1):55. doi: 10.1186/s12974-016-0507-9
14. Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev (2014) 259(1):231–44. doi: 10.1111/imr.12169
15. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
16. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/JCI76012
17. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J (2015) 29(6):2315–26. doi: 10.1096/fj.14-268409
18. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential regulation of human treg and Th17 cells by fatty acid synthesis and glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
19. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17(6):618–25. doi: 10.1038/ni.3466
20. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol (2007) 8(3):277–84. doi: 10.1038/ni1437
21. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453(7192):236–40. doi: 10.1038/nature06878
22. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
23. Pompura SL, Wagner A, Kitz A, LaPerche J, Yosef N, Dominguez-Villar M, et al. Oleic acid restores suppressive defects in tissue-resident FOXP3 Tregs from patients with multiple sclerosis. J Clin Invest (2021) 131(2):e138519. doi: 10.1172/JCI138519
24. Tanimine N, Germana SK, Fan M, Hippen K, Blazar BR, Markmann JF, et al. Differential effects of 2-deoxy-D-glucose on in vitro expanded human regulatory T cell subsets. PloS One (2019) 14(6):e0217761. doi: 10.1371/journal.pone.0217761
25. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
26. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity (2016) 44(2):406–21. doi: 10.1016/j.immuni.2016.01.028
27. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity (2017) 47(5):875–89.e10. doi: 10.1016/j.immuni.2017.10.017
28. Kaelin William G, McKnight Steven L. Influence of metabolism on epigenetics and disease. Cell (2013) 153(1):56–69. doi: 10.1016/j.cell.2013.03.004
29. Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med (2016) 213(3):377–97. doi: 10.1084/jem.20151438
30. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol (2007) 5(2):e38. doi: 10.1371/journal.pbio.0050038
31. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab (2020) 32(4):591–604 e7. doi: 10.1016/j.cmet.2020.07.001
32. Foolad F, Khodagholi F, Nabavi SM, Javan M. Changes in mitochondrial function in patients with neuromyelitis optica; correlations with motor and cognitive disabilities. PloS One (2020) 15(3):e0230691. doi: 10.1371/journal.pone.0230691
33. Gonzalo H, Nogueras L, Gil-Sanchez A, Hervas JV, Valcheva P, Gonzalez-Mingot C, et al. Impairment of mitochondrial redox status in peripheral lymphocytes of multiple sclerosis patients. Front Neurosci (2019) 13:938. doi: 10.3389/fnins.2019.00938
34. Wang Y, Branicky R, Noe A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol (2018) 217(6):1915–28. doi: 10.1083/jcb.201708007
35. Galasso M, Gambino S, Romanelli MG, Donadelli M, Scupoli MT. Browsing the oldest antioxidant enzyme: catalase and its multiple regulation in cancer. Free Radic Biol Med (2021) 172:264–72. doi: 10.1016/j.freeradbiomed.2021.06.010
36. Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci (2011) 12(10):7114–62. doi: 10.3390/ijms12107114
37. Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science (2015) 350(6265):1204–7. doi: 10.1126/science.aac4357
38. Lee G, Won HS, Lee YM, Choi JW, Oh TI, Jang JH, et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1alpha Activation. Sci Rep (2016) 6:18928. doi: 10.1038/srep18928
39. Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep (2012) 13(3):251–7. doi: 10.1038/embor.2012.9
40. Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol (2007) 27(4):755–61. doi: 10.1161/01.ATV.0000258979.92828.bc
41. Liu YV, Semenza GL. RACK1 vs. HSP90: Competition for HIF-1α Degradation vs. Stabilization. Cell Cycle (2014) 6(6):656–9. doi: 10.4161/cc.6.6.3981
42. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol (2020) 599(1):23–37. doi: 10.1113/jp280572
43. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
44. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA (2016) 113(30):8490–5. doi: 10.1073/pnas.1608873113
45. Sauer AV, Brigida I, Carriglio N, Hernandez RJ, Scaramuzza S, Clavenna D, et al. Alterations in the adenosine metabolism and CD39/CD73 adenosinergic machinery cause loss of Treg cell function and autoimmunity in ADA-deficient SCID. Blood (2012) 119(6):1428–39. doi: 10.1182/blood-2011-07-366781
46. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
47. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
48. Shalev I, Liu H, Koscik C, Bartczak A, Javadi M, Wong KM, et al. Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J Immunol (2008) 180(1):249–60. doi: 10.4049/jimmunol.180.1.249
49. Chen X, Li S, Long D, Shan J, Li Y. Rapamycin facilitates differentiation of regulatory T cells via enhancement of oxidative phosphorylation. Cell Immunol (2021) 365:104378. doi: 10.1016/j.cellimm.2021.104378
50. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol (2016) 17(3):277–85. doi: 10.1038/ni.3365
51. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39(5):949–62. doi: 10.1016/j.immuni.2013.10.016
52. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16(2):188–96. doi: 10.1038/ni.3077
53. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10(9):1000–7. doi: 10.1038/ni.1774
54. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17(6):673–5. doi: 10.1038/nm.2389
55. Garg G, Muschaweckh A, Moreno H, Vasanthakumar A, Floess S, Lepennetier G, et al. Blimp1 prevents methylation of foxp3 and loss of regulatory T cell identity at sites of inflammation. Cell Rep (2019) 26(7):1854–68 e5. doi: 10.1016/j.celrep.2019.01.070
56. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
57. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature (2013) 499(7459):485–90. doi: 10.1038/nature12297
58. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med (2015) 21(6):638–46. doi: 10.1038/nm.3868
59. Quintana FJ, Sherr DH, Insel PA. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev (2013) 65(4):1148–61. doi: 10.1124/pr.113.007823
60. Barbi J, Pardoll DM, Pan F. Ubiquitin-dependent regulation of Foxp3 and Treg function. Immunol Rev (2015) 266(1):27–45. doi: 10.1111/imr.12312
61. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
62. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci (2012) 109(41):E2784–93. doi: 10.1073/pnas.1202366109
63. Flück K, Breves G, Fandrey J, Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol (2016) 9(2):379–90. doi: 10.1038/mi.2015.67
64. Corrado M, Campello S. Autophagy inhibition and mitochondrial remodeling join forces to amplify apoptosis in activation-induced cell death. Autophagy (2016) 12(12):2496–7. doi: 10.1080/15548627.2016.1226738
65. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity (2022) 55(8):1370–85 e8. doi: 10.1016/j.immuni.2022.06.007
66. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science (2018) 359(6378):eaao6047. doi: 10.1126/science.aao6047
67. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
68. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol (2022) 23(3):159–73. doi: 10.1038/s41577-022-00760-x
69. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for Treg suppressive function. Cell Metab (2020) 31(2):422–37.e5. doi: 10.1016/j.cmet.2019.11.021
70. Yang D, Sun H, Hu Z, Tang J. Mitochondrial DNA-toll-like receptor 9 pathway mediated inflammatory responses. Med Recapitulate (2019) 25(21):4201–6. doi: 10.3969/j.issn.1006-2084.2019.21.008
71. Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J Clin Invest (2013) 123(11):4859–74. doi: 10.1172/JCI65180
72. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol (2009) 182(1):259–73. doi: 10.4049/jimmunol.182.1.259
73. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29(1):44–56. doi: 10.1016/j.immuni.2008.05.007
74. Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J (2016) 30(11):3800–9. doi: 10.1096/fj.201600522R
75. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
76. Goverman JM. Regulatory T cells in multiple sclerosis. N Engl J Med (2021) 384(6):578–80. doi: 10.1056/NEJMcibr2033544
77. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93 e7. doi: 10.1016/j.cmet.2016.12.018
78. Zhang Q, Fang Y, Lv C, Zhu Y, Xia Y, Wei Z, et al. Norisoboldine induces the development of Treg cells by promoting fatty acid oxidation-mediated H3K27 acetylation of Foxp3. FASEB J (2022) 36(4):e22230. doi: 10.1096/fj.202101643R
79. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep (2019) 27(1):226–37.e4. doi: 10.1016/j.celrep.2019.03.029
80. Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol (2006) 59(3):478–89. doi: 10.1002/ana.20736
81. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
82. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38. doi: 10.1101/gad.191056.112
83. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19(1):17–30. doi: 10.1016/j.ccr.2010.12.014
84. Steinert EM, Vasan K, Chandel NS. Mitochondrial metabolism regulation of T cell-mediated immunity. Annu Rev Immunol (2021) 39:395–416. doi: 10.1146/annurev-immunol-101819-082015
85. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene (2006) 25(34):4675–82. doi: 10.1038/sj.onc.1209594
86. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy (2020) 16(1):3–17. doi: 10.1080/15548627.2019.1603547
87. Wang N, Yuan J, Karim MR, Zhong P, Sun YP, Zhang HY, et al. Effects of mitophagy on regulatory T cell function in patients with myasthenia gravis. Front Neurol (2020) 11:238. doi: 10.3389/fneur.2020.00238
88. Demers-Lamarche J, Guillebaud G, Tlili M, Todkar K, Belanger N, Grondin M, et al. Loss of mitochondrial function impairs lysosomes. J Biol Chem (2016) 291(19):10263–76. doi: 10.1074/jbc.M115.695825
89. Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis (2012) 45(3):962–72. doi: 10.1016/j.nbd.2011.12.015
90. Wang T, Ming Z, Xiaochun W, Hong W. Rab7: role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal (2011) 23(3):516–21. doi: 10.1016/j.cellsig.2010.09.012
91. Fernandez-Mosquera L, Yambire KF, Couto R, Pereyra L, Pabis K, Ponsford AH, et al. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy (2019) 15(9):1572–91. doi: 10.1080/15548627.2019.1586256
92. Baixauli F, Acin-Perez R, Villarroya-Beltri C, Mazzeo C, Nunez-Andrade N, Gabande-Rodriguez E, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab (2015) 22(3):485–98. doi: 10.1016/j.cmet.2015.07.020
93. Trnka J, Blaikie FH, Smith RAJ, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radical Biol Med (2008) 44(7):1406–19. doi: 10.1016/j.freeradbiomed.2007.12.036
94. Gan X, Zhang L, Liu B, Zhu Z, He Y, Chen J, et al. CypD-mPTP axis regulates mitochondrial functions contributing to osteogenic dysfunction of MC3T3-E1 cells in inflammation. J Physiol Biochem (2018) 74(3):395–402. doi: 10.1007/s13105-018-0627-z
95. Satake A, Schmidt AM, Archambault A, Leichner TM, Wu GF, Kambayashi T. Differential targeting of IL-2 and T cell receptor signaling pathways selectively expands regulatory T cells while inhibiting conventional T cells. J Autoimmun (2013) 44:13–20. doi: 10.1016/j.jaut.2013.06.009
96. Swanson SK, Born T, Zydowsky LD, Cho H, Chang HY, Walsh CT, et al. Cyclosporin-mediated inhibition of bovine calcineurin by cyclophilins A and B. Proc Natl Acad Sci USA (1992) 89(9):3741–5. doi: 10.1073/pnas.89.9.3741
97. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science (2019) 366(6472):1531–6. doi: 10.1126/science.aav4011
98. Verma A, Pittala S, Alhozeel B, Shteinfer-Kuzmine A, Ohana E, Gupta R, et al. The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: a potential therapeutic target. Mol Ther (2022) 30(2):726–44. doi: 10.1016/j.ymthe.2021.06.024
99. Makita N, Ishiguro J, Suzuki K, Nara F. Dichloroacetate induces regulatory T-cell differentiation and suppresses Th17-cell differentiation by pyruvate dehydrogenase kinase-independent mechanism. J Pharm Pharmacol (2017) 69(1):43–51. doi: 10.1111/jphp.12655
100. Younes SA, Talla A, Pereira Ribeiro S, Saidakova EV, Korolevskaya LB, Shmagel KV, et al. Cycling CD4+ T cells in HIV-infected immune nonresponders have mitochondrial dysfunction. J Clin Invest (2018) 128(11):5083–94. doi: 10.1172/JCI120245
101. Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol (2022) 23(4):266–85. doi: 10.1038/s41580-021-00433-y
102. Kageyama T, Komori M, Miyamoto K, Ozaki A, Suenaga T, Takahashi R, et al. Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol (2012) 260(2):627–34. doi: 10.1007/s00415-012-6692-2
103. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab (2005) 1(6):401–8. doi: 10.1016/j.cmet.2005.05.001
104. Lin X, David CA, Donnelly JB, Michaelides M, Chandel NS, Huang X, et al. A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc Natl Acad Sci USA (2008) 105(1):174–9. doi: 10.1073/pnas.0706585104
Keywords: mitochondria, regulatory T cell, Foxp3, self-tolerance, central nervous system, autoimmune diseases
Citation: Han A, Peng T, Xie Y, Zhang W, Sun W, Xie Y, Ma Y, Wang C and Xie N (2023) Mitochondrial-regulated Tregs: potential therapeutic targets for autoimmune diseases of the central nervous system. Front. Immunol. 14:1301074. doi: 10.3389/fimmu.2023.1301074
Received: 24 September 2023; Accepted: 30 November 2023;
Published: 12 December 2023.
Edited by:
Bieke Broux, University of Hasselt, BelgiumReviewed by:
Alessandra Colamatteo, University of Naples Federico II, ItalyCopyright © 2023 Han, Peng, Xie, Zhang, Sun, Xie, Ma, Wang and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nanchang Xie, eGllbmFuY2hhbmcyMDAxQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.