 Lili Yan
Lili Yan Yueran Cui
Yueran Cui Juan Feng
Juan Feng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1292022
Pellino1 (Peli1) is a highly conserved E3 Ub ligase that exerts its biological functions by mediating target protein ubiquitination. Extensive evidence has demonstrated the crucial role of Peli1 in regulating inflammation by modulating various receptor signaling pathways, including interleukin-1 receptors, Toll-like receptors, nuclear factor−κB, mitogen-activated protein kinase, and phosphoinositide 3-kinase/AKT pathways. Peli1 has been implicated in the development of several diseases by influencing inflammation, apoptosis, necrosis, pyroptosis, autophagy, DNA damage repair, and glycolysis. Peli1 is a risk factor for most cancers, including breast cancer, lung cancer, and lymphoma. Conversely, Peli1 protects against herpes simplex virus infection, systemic lupus erythematosus, esophageal cancer, and toxic epidermolysis bullosa. Therefore, Peli1 is a potential therapeutic target that warrants further investigation. This comprehensive review summarizes the target proteins of Peli1, delineates their involvement in major signaling pathways and biological processes, explores their role in diseases, and discusses the potential clinical applications of Peli1-targeted therapy, highlighting the therapeutic prospects of Peli1 in various diseases.
Ubiquitin (Ub) is an evolutionarily conserved polypeptide that plays a crucial role in post-translational modifications by binding to protein substrates, thus regulating signaling in various receptor systems (1, 2). Ubiquitination involves several reactions mediated by three classes of ubiquitin-related enzymes. First, the ubiquitin-activating enzyme (E1) activates the Ub molecules. Subsequently, the Ub-conjugating enzyme (E2) receives an activated Ub molecule from E1. Lastly, the Ub-ligase (E3) transfers the Ub molecule bound to E2 to the target protein (1).
Pellino1 (Peli1) has been extensively studied among the E3 Ub ligase family members. Initially identified in Drosophila, Peli1 has a molecular mass of 47 kDa and interacts with Pelle, a homolog of interleukin-1 receptor-associated kinase 1 (IRAK1) (3). Three members of the mammalian Peli family (Peli1, Peli2, and Peli3) have been identified, along with two selectively spliced forms of Peli3, namely Pellino3a and Pellino3b (4, 5). The N-terminal region of Peli contains a core forkhead-associated (FHA) structural domain, characterized by two inserted fragments forming a “wing” or appendage tightly bound to the FHA domain (6). Similar to the classical C3HC4 loop structure, the C-terminus of the Peli family features a RING-like domain encompassing two stable Cys-Gly-His motifs and two conserved CysPro-X-Cys motifs, enabling ubiquitination through K11, K48, and K63 linkages (7, 8). Peli1 phosphorylation is necessary for its E3 Ub- ligase activity. Substrate recognition is facilitated by the FHA structural domain, with the amino acid located at the +3 position relative to the phosphorylated threonine, a key determinant of PT peptide recognition by many FHA domains. Different Pellinos exhibit diverse phosphothreonine peptide binding properties (9). The strong binding of Peli1 to IRAK1 and receptor-interacting protein kinase 1 (RIP1) is associated with its preference for pTxxY- or pTxxS-binding motifs, whereas Pellinos no show a preference for binding to the pT141 + 3D motif (9).
Peli1 modulates immune response, cell death, autophagy, DNA damage repair, and glycolysis through its E3 Ub ligase activity in a dependent or independent manner. Peli1 exhibits remarkable versatility in immune regulation by participating in interleukin-1 receptors (IL-1R), Toll-like receptors (TLRs), nuclear factor−κB (NF-κB), mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K)/AKT pathways. These multifaceted pathways involving Peli1 regulate numerous diseases, particularly tumors, inflammatory disorders, and autoimmune conditions. E3 ligase-related targeted regulation is common; for instance, the E3 ligase SKP1-CUL1-F-box-protein (SCF) or Cullin-RING E3 ligase (CRL) has been investigated with several targeted inhibitors and agonists. For example, inhibitors such as compound A blocks substrate SKP2 binding to the SCF complex, and agonists, such as thalidomide, primarily target cereblon (CRBN), a substrate-recognizing subunit of the CUL4-DDB1 E3 Ub ligase complex (10, 11). The substrates regulated by Peli1 as an E3 ligase are also relatively specific (Table 1); therefore, selectively targeting Peli1 is essential. A few strategies to target and manipulate Peli1 for therapeutic benefits, underscoring its potential as a therapeutic target, such as the inhibitors BBT-401 and S62 (22, 48).
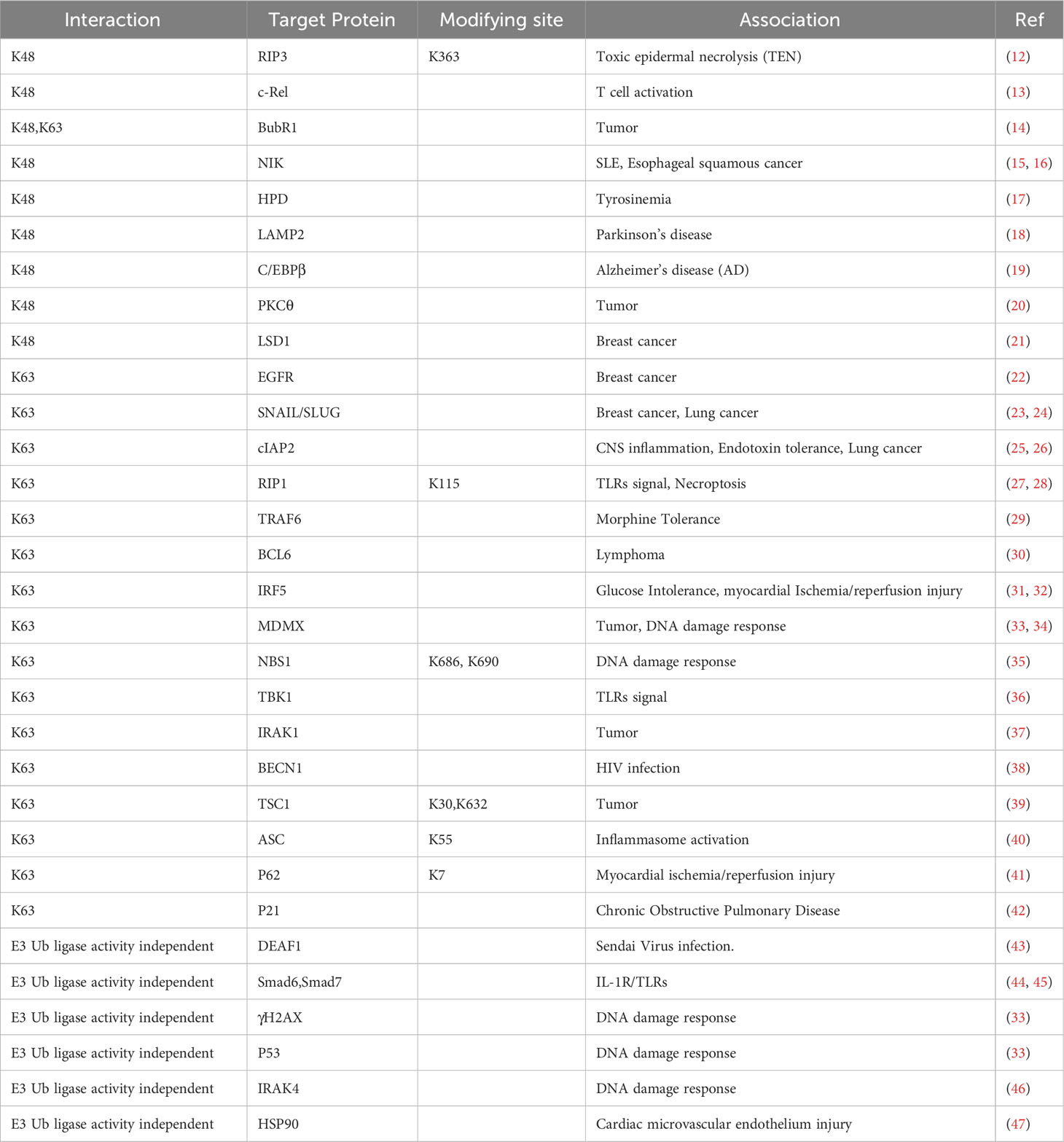
Table 1 Proteins targeted by Peli1-mediated ubiquitination or ubiquitination-independent.
This review comprehensively examines the signaling pathways involving Peli1, elucidates the target proteins under its regulation, explores their contributions to tumors and inflammation, and discusses the potential clinical applications of Peli1 as a therapeutic target. This comprehensive understanding of the multifunctionality of Peli1 in a pathological contexts provides valuable insights for future research and clinical interventions.
Peli1 exhibits robust E3 Ub ligase activity, predominantly mediating the Lys11, Lys48, and Lys63 linkages (K11, K48, and K63) of Ub (Table 1) (8). In vivo, Peli1 primarily facilitates the ubiquitination of target proteins via K48 and K63 linkages and promotes the K48-linked ubiquitination of target proteins, leading to their degradation via the Ub-proteasome system. Peli1 mediates K48-linked Ub (K48-Ub) in the HPD, resulting in its degradation and tyrosinemia (17). Peli1 mediates the K63-linked Ub (K63-Ub) of target proteins, thereby enhancing their stability or promoting subcellular localization, and mediates the K63-Ub in murine double minute X (MDMX), facilitating its nuclear export (49). Peli1 can regulate both K48-Ub and K63-Ub concurrently but differentially, where Peli1 mediates K63-Ub more substantially than the K48-Ub of BubR1 (14). Peli1-mediated K63-Ub of target proteins can signal Met1-linked Ub (M1-Ub) through K63/M1-Ub hybrids. Peli1 ubiquitinates RIP1 via K63-Ub in the TLR signaling pathway (27). Peli1 interacts with molecules and function in an E3 ligase-independent manner facilitated by its FHA structure (Table 1).
Several proteins act as transcription factors that regulate Peli1 transcription, and various molecules are involved by modulating the promoters or transcription factors (Table 2). One such transcription factor is interferon regulatory factor 3 (IRF3), which regulates Peli1 through the Toll-interleukin-1 receptor -domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway of TLR3/4, influencing Peli1 expression (36). The activation of TANK-binding kinase 1 (TBK1) and IkappaB kinase epsilon (Ikkϵ) phosphorylates and activates IRF3, leading to transcriptional Peli1 and type I interferon-beta (IFN-β) upregulation. Subsequently, IFN-β further enhances Peli1 transcription via the Janus kinase/signal transducer and activator of the transcription (JAK/STAT) signaling pathway (36). The TRIF-IRF3 signaling pathway, activated by Bid-dependent mechanisms, contributes to Peli1 transcription upon TLR3 and TLR4 activation (44, 66). The glucocorticoid receptor (GR) interacts with β-arrestin-1, maintaining GR stability and regulating the GR-sensitive region of the Peli1 promoter, thereby influencing Peli1 transcription (50).
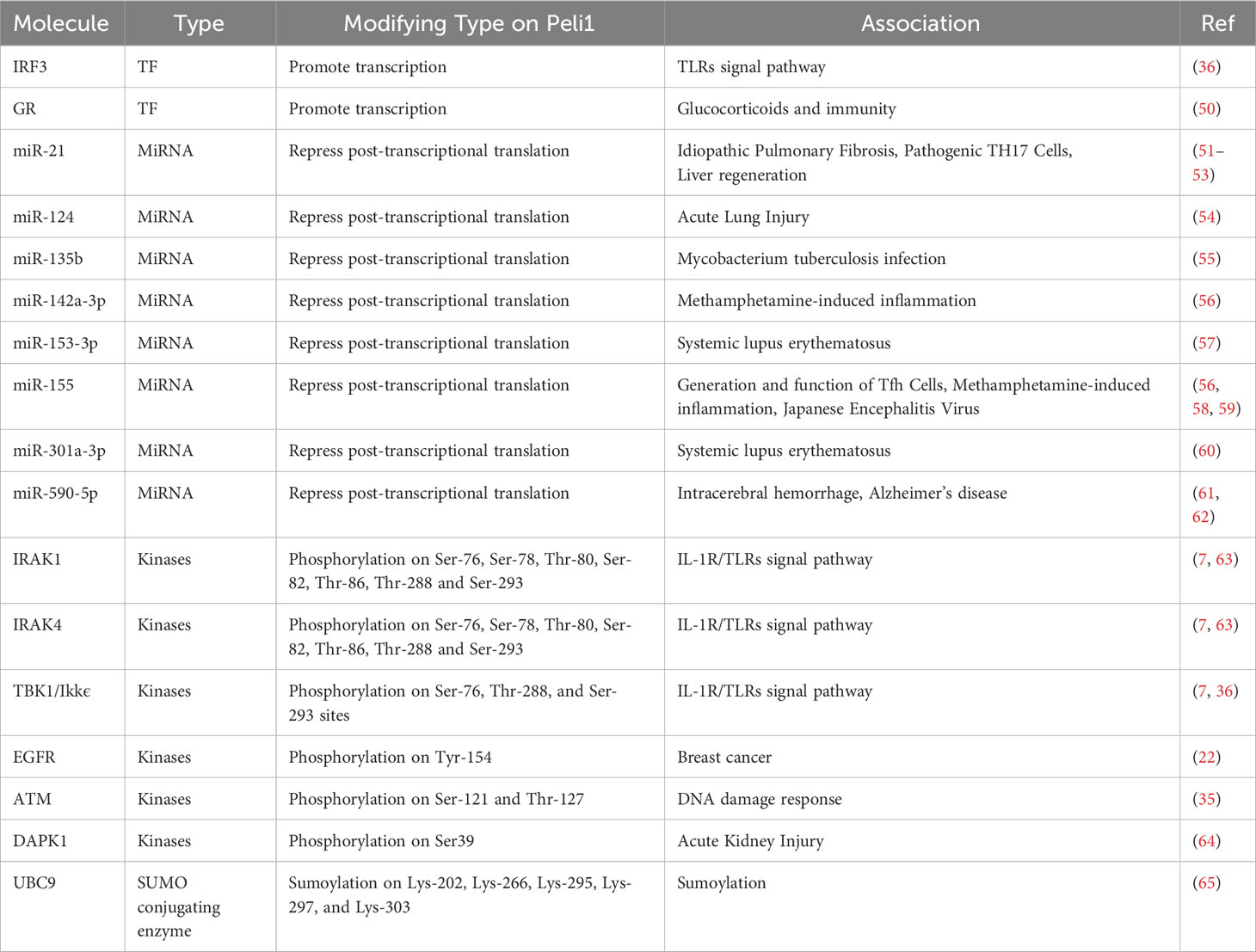
Table 2 Molecules Regulating Peli1.
Several microRNAs (miRNAs) have been identified as post-transcriptional regulators of Peli1, targeting its 3’-untranslated region (3’ UTR) and repressing its expression (Table 2). MiRNAs are small, non-coding RNA molecules that regulate gene expression at the post-transcriptional level by binding to the 3’ UTR of target genes (67). MiRNAs are closely associated with various biological functions and pathological processes, including differentiation, metabolism, aging, autophagy, cell proliferation, and apoptosis (68).
Peli1 activity is modulated by various post-translational modifications, including phosphorylation, ubiquitination, and SUMOylation (Table 2). Peli1 can be interconverted between its inactive and active forms through a reversible phosphorylation mechanism, and various kinases can phosphorylate Peli1 and enhances its E3 ligase activity (63). IRAK1/IRAK4 and TBK1/Ikkϵ are the primary kinases responsible for Peli1 phosphorylation (7, 36). IRAK1 interacts with the FHA structural domain and phosphorylates the phosphorylation sites (Ser-76, Ser-78, Ser-80, Ser-82, and Thr-86) clustered within the “wing” of Peli1 (63). TBK1/IkappaB kinase (IKK) regulates the phosphorylation at Ser-76, Thr-288, and Thr-288 of Peli1 (36). The epidermal growth factor receptor (EGFR) leads to Peli1 phosphorylation at Tyr-154 (22). In the DNA damage response context, Ser-121 and Thr-127 phosphorylation by ataxia-telangiectasia mutated (ATM) kinase activates Peli1 ubiquitination activity (35). Death-associated protein kinase 1 (DAPK1) phosphorylates Peli1 at Tyr-154, destabilizing it and releasing the TRIF-RIP1 signalosome (64).
IRAK1 promotes auto- and mutual-polyubiquitination of Peli1 in a kinase-dependent manner and induces kinase-dependent Peli1 degradation. However, the precise mechanism governing the phosphorylation-mediated auto-Ub proteasome-dependent Peli1 degradation remains unclear (7).
Peli1 can undergo SUMOylation, a process in which the small Ub-related modifier (SUMO)- conjugating enzyme UBC9 binds and modifies Peli1 (65). Five lysine residues (Lys-202, Lys-266, Lys-295, Lys-297, and Lys-303) in Peli1 serve as SUMO-1 receptor sites, partially overlapping with lysine residues involved in ubiquitination (Lys-169, Lys-202, and Lys-266), suggesting competition between SUMOylation and ubiquitination (65).
Peli1 plays a role in multiple signaling pathways upon stimulation by various factors, including IL-1R, TLRs, NF-κB, MAPK, and PI3K/AKT pathways, and participates in their crosstalk (Figure 1).
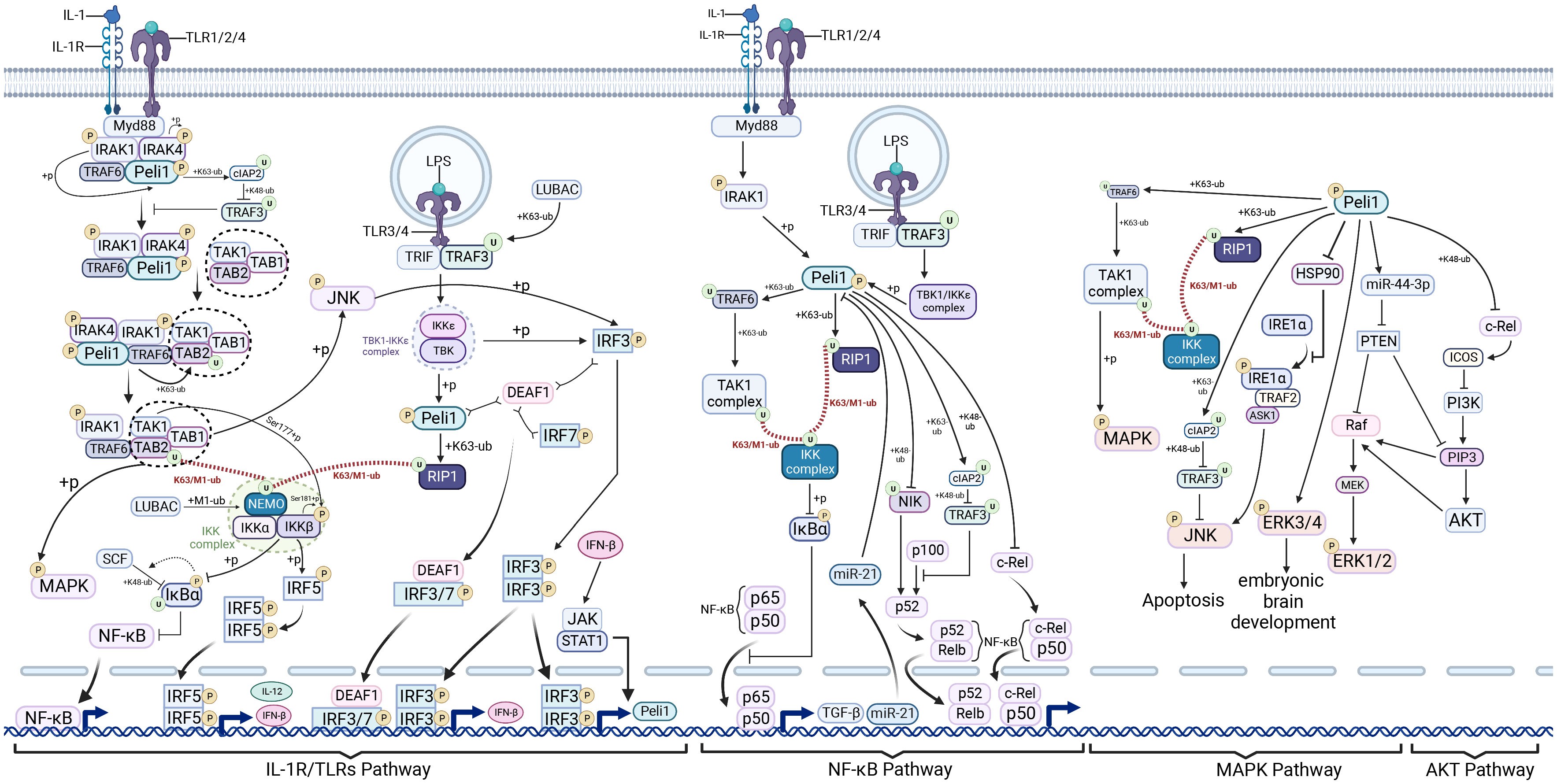
Figure 1 Pellino1 in interleukin-1 receptors/Toll-like receptors, nuclear factor−κB, mitogen-activated protein kinase, and AKT pathways. Pellino1 (Peli1), activated by interleukin-1 receptor-associated kinase (IRAK) phosphorylation, promotes the degradation of tumor necrosis factor receptor-associated factor 3 (TRAF3) by mediating cellular inhibitor of apoptosis protein 2 (cIAP2) ubiquitination in the myeloid differentiation primary response protein 88 (MyD88)-dependent interleukin-1 receptors (IL-1R)/Toll-like receptors (TLRs) signaling pathway. This process facilitates the cytoplasmic translocation of IRAK1-IRAK-TRAF6-PELI1 (an intermediate complex). The transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) complex forms complex II (IRAK1-IRAK-TRAF6-PELI1-TAK1-TAB1-TAB2) in conjunction with an intermediate complex that subsequently assembles into TRAF6-TAK1-TAB1-TAB2 (complex III). TRAF6 mediates TAK1 complex ubiquitination and recruits the IkappaB kinase (IKK) complex via the K63/M1-UB Ub chain, ultimately activating nuclear factor−κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling, and stimulating interferon regulatory factor 5 (IRF5) to induce interferon beta (IFN-β) production. In the Toll-interleukin-1 receptor-domain-containing adaptor-inducing IFN-beta (TRIF)-dependent Toll-like receptor (TLR)3/4 pathway, phosphorylated Peli1, in conjunction with the TBK1-Ikkϵ complex, mediates receptor-interacting protein kinase 1 (RIP1) ubiquitination, enabling RIP1 to recruit the IKK and TAK1 complexes through the K63/M1-UB Ub chain. This cascade activates NF-κB and MAPK signaling. The TBK1-Ikkϵ complex and Jun N-terminal kinase (JNK), activated by the TAK1 complex, can phosphorylate and activate IRF3, promoting IFN-β and Peli1 transcription. Peli1 enhances the positive feedback loop of IFN-β production. Peli1 promotes NF-κB canonical signaling by activating the IKK complex through TRAF6 and RIP1. NF-κB induces elevated miR-21 expression, inhibiting Peli1 expression, potentially forming a negative feedback loop. Peli1 mediates the ubiquitin-proteasome-dependent degradation of NIK, thus inhibiting NF-κB non-canonical signaling. However, Peli1 can also promote p100 to p52 processing and, ultimately, NF-κB non-canonical signaling by inhibiting TRAF3. Peli1 mediates ubiquitin-proteasome-dependent degradation of c-Rel. Peli1 activates the TAK1 complex via TRAF6 and RIP1, activating MAPK. Peli1 also inhibits TRAF3 via CIAP2, promoting JNK activation. Peli1 facilitates IRE1α phosphorylation by binding to HSP90, resulting in JNK activation. Peli1 inhibits phosphatase and tensin homolog (PTEN) by upregulating miR-44-3P, relieving Raf and phosphatidylinositol 3,4,5-trisphosphate (PIP3) repression, and ultimately activating extracellular signal-regulated kinase (ERK)1/2. In addition, Peli1 inhibits inducible co-stimulator (ICOS) through C-REL inhibition and activates phosphoinositide 3-kinase (PI3K), promoting PIP3 and AKT activation. Created using BioRender.com.
Peli1 plays a pro-inflammatory role in a variety of diseases through participation in the IL-1 pathway, where Peli1 was initially identified as interacting with IRAK homologous proteins (3). Peli1 participates in IL-1R signaling and to has limited modulation of IL-1 signaling (Figure 1). Upon IL-1 stimulation, the adaptor protein myeloid differentiation primary response protein 88 (MyD88) is recruited by the IL-1 receptor complex. Subsequently, the receptor complex recruits serine-threonine kinases IRAK4 and IRAK1 (69, 70). IRAK4 undergoes dimerization facilitated by MyD88, leading to IRAK4 trans-autophosphorylation (71). IRAK mediates TNF receptor-associated factor 6 (TRAF6) hyperphosphorylation, promoting its recruitment to the receptor complex (72). Peli1 interacts with IRAK4 and IRAK1 (72) and directly binds to TRAF6 and transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) (73). Ultimately, this binding results in MyD88-IRAK4-IRAK1-Peli1-TRAF6 signaling complex formation (45, 72, 74). Translocation of this complex from the membrane-bound receptor complex to the cytoplasm, necessary for MyD88-dependent signal transduction to TAK1 (75), involves Peli1-mediated TRAF3 degradation (44). Peli1 Phosphorylation by IRAK1 upon activation enhances its stability through K63-linked ubiquitination of the cellular inhibitor of apoptosis protein 2 (cIAP2). This event follows the cIAP2-mediated K48-linked Ub proteasome-dependent degradation of TRAF3, relieving cytoplasmic translocation inhibition of the MyD88-associated multiprotein complex by TRAF3 (44). Complex II (IRAK1-IRAK-TRAF6-Peli1-TAK1-TAB1-TAB2) forms the intermediate complex that transitions into complex III (TRAF6-TAK1-TAB1-TAB2) (72). The IKK complex, comprising IKKα, IKKβ, and the nuclear factor-κB essential regulator (NEMO), is a key component (76). TRAF6 mediates the ubiquitination of the K63-linked TAB2 subunit after M1-Ub chain formation from the NEMO subunit of the IKK complex catalyzed by the linear Ub chain assembly complex. The formation of K63/M1-Ub hybrids allows for the co-recruitment of both kinase complexes to the same Ub chain. TAK1 phosphorylates IKKβ at Ser-177, then autophosphorylated at Ser-181 of IKKβ, culminating in IKKβ activation (76). IKKβ has a dual role in the TLRs pathway: first, it recruits the E3 ligase SCF through IκBα phosphorylation, leading to the K48-linked IκBα ubiquitination and degradation through IκBα phosphorylation. This results in the derepression of P65 and P50, allowing their entry into the nucleus and activating of the NF-κB signaling pathway (76). Second, IKKβ phosphorylates the transcription factor interferon regulatory factor 5 (IRF5) at Ser-462, leading to its dimerization and nuclear translocation, thereby initiating the transcription of genes encoding major inflammatory cytokines such as IL-12 and IFN-β (76). TAK1 also induces MAPK phosphorylation (44). TAK1-mediated phosphorylation of Jun N-terminal kinase (JNK) activates IRF3 (77), suggesting that JNK activation facilitates crosstalk between MyD88-independent and dependent pathways associated with IRF3 activation (44).
TGF-β-BMP induces Smad6/Smad7, inhibiting the MyD88-IRAK4-IRAK1-Peli1-TRAF6 signaling complex by binding to different Peli1 regions. Consequently, the IL-1R-TLR signaling pathway is inhibited (45, 74). Moreover, the formation of the Smad6-A20 complex, involving Smad6 and the deubiquitinating enzyme A20, enhances the association of A20 with TRAF3 and TRAF6, thereby inhibiting IL-1 signaling (44).
However, Peli1 overexpression in human embryonic kidney cells (293 cells) did not affect cJNK and ELK1 expression (73). Peli1 deletion does not impact IL-1β-stimulated IKK activation, and Peli1 knockdown in primary epithelial cells does not alter the response to IL-1. Functional redundancy among Peli family proteins may account for these findings (78).
The innate immune system encompasses multiple pattern recognition receptors responsible for sensing the pathogen-associated molecular patterns of invading pathogens, thus initiating an efficient innate immune response. Peli1 has been implicated in various TLR signaling pathways (Figure 1) (79). Several ligands of MyD88-dependent TLRs, including lipopolysaccharide (LPS) (for TLR4), CpG (for TLR9), R837 (for TLR7), and Pam3CSK4 (for TLR1 and TLR2), activate pathways consistent with IL-1R-dependent MyD88 (80). Peli1 also functions in the TLR3/4-dependent TRIF signaling pathway. TLRs dimerize in endosomes upon TLR3/4 stimulation by the corresponding ligands (e.g., TLR3: poly IC, viral double-stranded RNA; TLR4: LPS) (81) and recruits TRIF and TRAF3 (82, 83). H omologous to the E6-associated protein carboxyl terminus domain containing 3 mediates the K63-linked ubiquitination of TRAF3 at K138 (84), and TRAF3 ubiquitination or auto-ubiquitination (75) is necessary for binding and activation of the TBK1/Ikkϵ complex (83). Subsequently, the TBK1-Ikkϵ complex phosphorylates Peli1, activating its E3 ligase activity (27, 36, 85). Peli1 mediates TBK1 K63-Ub, resultING in a bidirectional signaling pathway that induces TBK1/Ikkϵ-mediated Peli1 phosphorylation. Activated Peli1 ubiquitinates RIP1 at K115 via K63 linkages (27, 28). Consequently, the TAK1 and IKK complexes are recruited to the polyubiquitin chains through K63-Ub of TABs and M1-Ub of NEMO, respectively (27). The proximity between TAK1 and IKKs facilitates TAK1-mediated IKKs phosphorylation and subsequent NF-κB activation (27). TAK1-mediated JNK phosphorylation activates IRF3 (44), suggesting that crosstalk between MyD88-independent and -dependent pathways associated with IRF3 activation occurs through JNK activation (77). Thus, Peli1 serves as a critical intermediary molecule in the TRIF-dependent NF-κB activation and the inducing pro-inflammatory genes (27). The TBK1-Ikkϵ complex regulates IFN-β and Peli1 transcription by inducing IRF3 phosphorylation. IFN-β can moderately increase Peli1 transcription through the JAK/STAT1 pathway (36). Furthermore, wild-type Peli1 might negatively regulate STAT1 expression, possibly preventing JAK-STAT1/2 pathway overactivation (86). This negative feedback regulation may be part of the complex regulatory mechanisms involving Peli1. However, further investigations are required to elucidate these mechanisms fully.
Inducing IFN-β is a substantial implications of the TRIF-dependent signaling pathway of TLRs. However, the mechanisms of IFN regulation by Peli1 are yielded complex and contradictory. Virus-stimulated secretion of IFN-β relies on a positive feedback loop, reduced IRF7, IFN4, and IFN6 mRNA production, and diminished IFN secretion in mice overexpressing Peli1 lacking ligase activity. T he positive feedback loop components we are also diminished, suggesting that Peli1 positively regulates IFN-β expression through a positive feedback loop (86). The synthetic product Smaducin-6 disrupts IKKe/TBK1/Peli1 and RIR1/Peli1 complexes by binding to Peli1, reducing IFN-β1 in immune cells (87). Therefore, Peli1 is necessary for interferon production in the viral double-stranded RNA reactions.
The interaction between Peli1 and IRF3, and their association with the IFN-β promoter, facilitates inducing IFN-β expression (36). This process is influenced by DEAF1, which binds to Peli1, and Peli1 phosphorylation can potentially impede this binding (43). DEAF1 is essential for the initial phase of TLR3-dependent IFN-β production after viral stimulation. DEAF1 enters the nucleus alone or forms a heterodimer with IRF3 or IRF7, binding to the IFN-β promoter to stimulate its transcription. The interaction of DEAF1 with the IFN-β promoter requires the presence of IRF3 (43). In contrast, Peli1 negatively regulate TLR-mediated IFN-I induction in microglia by inhibiting signaling events associated with TBK1 and Ikkϵ activation. Peli1 knockdown may enhance its antiviral capacity (88). In vitro restimulation of CD4+ and CD8+ splenic T cells in Peli1-/- mice with West Nile virus (WNV)-specific peptides increased IFN-γ production compared to wild-type mice. However, WNV infection in dendritic cells of Peli1-/- mice led to reduced IFN-β levels compared to wild-type mice (89). These findings suggest that Peli1 in different cells may be dominated by different regulatory pathways, exhibiting distinct IFN regulatory directions.
Peli1 influences the classical NF-κB pathway by engaging in the IL-1R/TLR pathway (Figure 1). Peli1 triggers TAK1 activation through TRAF6 within the MyD88-dependent pathway, facilitated by K63-Ub of TRAF6 (90–92). Peli1 facilitates RIP1 recruitment to the IKK complex and TAK1 in the TRIF-dependent pathway via the K63/M1-Ub hybrid chain (91, 92). The IKK complex induces IκBα phosphorylation, leading to its degradation via SCF-mediated ubiquitination, enabling the nuclear translocation of canonical NF-κB molecules, p50/p65 (76). Peli1 participates in a negative feedback loop of NF-κB during liver regeneration, wherein NF-κB upregulates miR-21 precursor transcripts that target Peli1, suppressing the NF-κB pathway (51). Peli1 negatively regulates NF-κB in T cells by mediating the Ub proteasome-dependent degradation of the late phase NF-κB protein, c-Rel (13, 58, 93), preventing excessive NF-κB activation (13, 52).
In the non-canonical NF-κB pathway, Peli1 displays contrasting regulatory functions depending on the context. Peli1 acts as a negative regulator of esophageal squamous carcinoma’s radiotherapy sensitivity and lupus erythematosus syndrome by promoting the Ub proteasome-dependent NIK degradation, inhibiting the nuclear translocation of non-canonical NF-κB effector molecules, p52, and Rel B (15, 16). Peli1 also augments cIAP2 stability by facilitating the K63-linked cIAP2 ubiquitination (25, 94, 95), enabling cIAP2 to mediate the K48-Ub and TRAF3 degradation (26), thereby alleviating the inhibition of p100 processing into p52 by TRAF3 (59). Consequently, Peli1 promotes p52 entry into the nucleus and relieves the inhibition of the classical NF-κB molecule p65 owing to p100 accumulation (59).
Peli1 influences the MAPK pathway through multiple mechanisms (Figure 1), regulating inflammation and apoptosis and participating in brain development and angiogenesis (61, 85, 94, 96, 97). Peli1 facilitates TRAF6 and RIP1 ubiquitination in the IL-1R/TLR pathway (29, 98), leading to P38 activation via TAK1 (99). Moreover, Peli1 mediates the K63-Ub of cIAP2, activating JNK and P38 by via increasing cIAP2-mediated TRAFT3 degradation (26, 80). Peli1 induces miR-494-3p expression in cardiomyocyte exosomes, inhibiting its target, phosphatase and tensin homolog (PTEN), and activating the AKT, Smad2/3, and extracellular signal-regulated kinase (ERK) signaling pathways (100). Peli1 activates the MAPK pathway by modulating endoplasmic reticulum (ER) stress. By binding to heat shock protein 90 (HSP90), Peli1 hinders the interaction of HSP90 with IRE1α, a key protein in ER stress, promoting phosphorylation-dependent IRE1α activation (47). Activated IRE1α facilitates TRAF2 recruitment, further promoting p-JNK activation through apoptosis signal-regulating kinase 1 inducing downstream apoptotic signaling (47, 101). IRE1α promotes x-box binding protein 1 (XBP1) splicing and maturation (47), while p38MAPK phosphorylates XBP1 at Thr-48 and Ser-61, augmenting the induction of apoptotic signaling and nuclear migration of XBP1 in mice (102).
Peli1 activates the AKT pathway and is involved in various biological and pathological processes, including cancer progression (23, 103), drug resistance (104), angiogenesis (96, 100, 105), T follicular helper (Tfh) cell differentiation (93), microglia activation (98), glycolysis, and macrophage M1 polarization (31). Peli1 regulates AKT activationvia several pathways: 1) Peli1 inhibits the inducible co-stimulator (ICOS) and suppresses the PI3K/AKT pathway downstream of ICOS by mediating ubiquitination-dependent c-Rel degradation (93); 2) Peli1 upregulates miR-494-3p expression in cardiomyocyte exosomes and inhibits its target PTEN. This inhibition prevents PTEN-mediated phosphatidylinositol 3,4,5-trisphosphate (PIP3) dephosphorylation, activating downstream AKT/endothelial nitric oxide synthase/nitric oxide signaling (100).
Peli1 regulates cell death through various pathways, including apoptosis, necroptosis, and pyroptosis (Figure 2). Peli1 plays a dual regulatory role in necrosis and apoptosis by modulating the ubiquitination of RIPK1 (RIP1) and mRNA levels of cellular FLICE-like inhibitory protein (c-FLIP) (28). Upon TNF stimulation of tumor necrosis factor receptor 1 (TNFR1), RIPK1 is recruited to the TNFR1-associated death structural domain (TRADD), which recruits cIAP1 and cIAP2 via the TRAF2 and linear ubiquitin chain assembly complexes (LUBAC), culminating in complex I formation (28, 106). In complex I, RIPK1 undergoes rapid Ub chain polymerization, activating the TAK1 and IKK complexes through the K63/M1-Ub hybrids, inducing NF-κB/MAPK pathway activation. K63 and M1 ubiquitination of RIPK1 contributes to recruiting Peli1 to complex I. Without an A20-binding inhibitor of NF-κB1 (ABIN1), Peli1 further promotes K63-linked RIPK1 ubiquitination (28, 106). A20 and cylindromatosis (CYLD) mediate complex I degradation by deubiquitinating its components to form complex IIa or IIb. Complex IIa comprising the Fas-associated death domain (FADD), caspase-8, and RIPK1, triggers RIPK1-dependent apoptosis by activating caspase-8, -3, and -7 (106). TRADD in complex I recruits FADD and caspase-8 to initiate RIPK1-independent apoptosis. C-FLIP, a catalytically inactive caspase-8 homolog, interacts with procaspase-8 and impedes caspase-8 processing by counteracting the cytotoxic activity of complex IIa. Peli1 upregulates c-FLIP mRNA levels by inhibiting the repressive transcription factor c-Myc to inhibit RIPK1-dependent and RIPK1-independent apoptosis (28, 107). Upon caspase-8 inactivation, RIPK1 binds to the RIP homotypic interaction motif (RHIM) of RIPK3 to form complex IIb. This complex promotes RIPK3 oligomerization and phosphorylation, which recruits and phosphorylates Mixed lineage kinase domain-like protein (MLKL), resulting in membrane perforation and programmed necrosis (28, 106). Peli1 mediates the K63-linked ubiquitination of RIPK1 in complex II, and deleting Peli1 inhibits RIPK3, RIPK1, and FADD binding, and RIPK3 and MLKL phosphorylation, indicating the indispensable role of Peli1 in the RIPK1-RIPK3 necrosome (28). However, how Peli1 exerts its ubiquitination function requires further investigation.
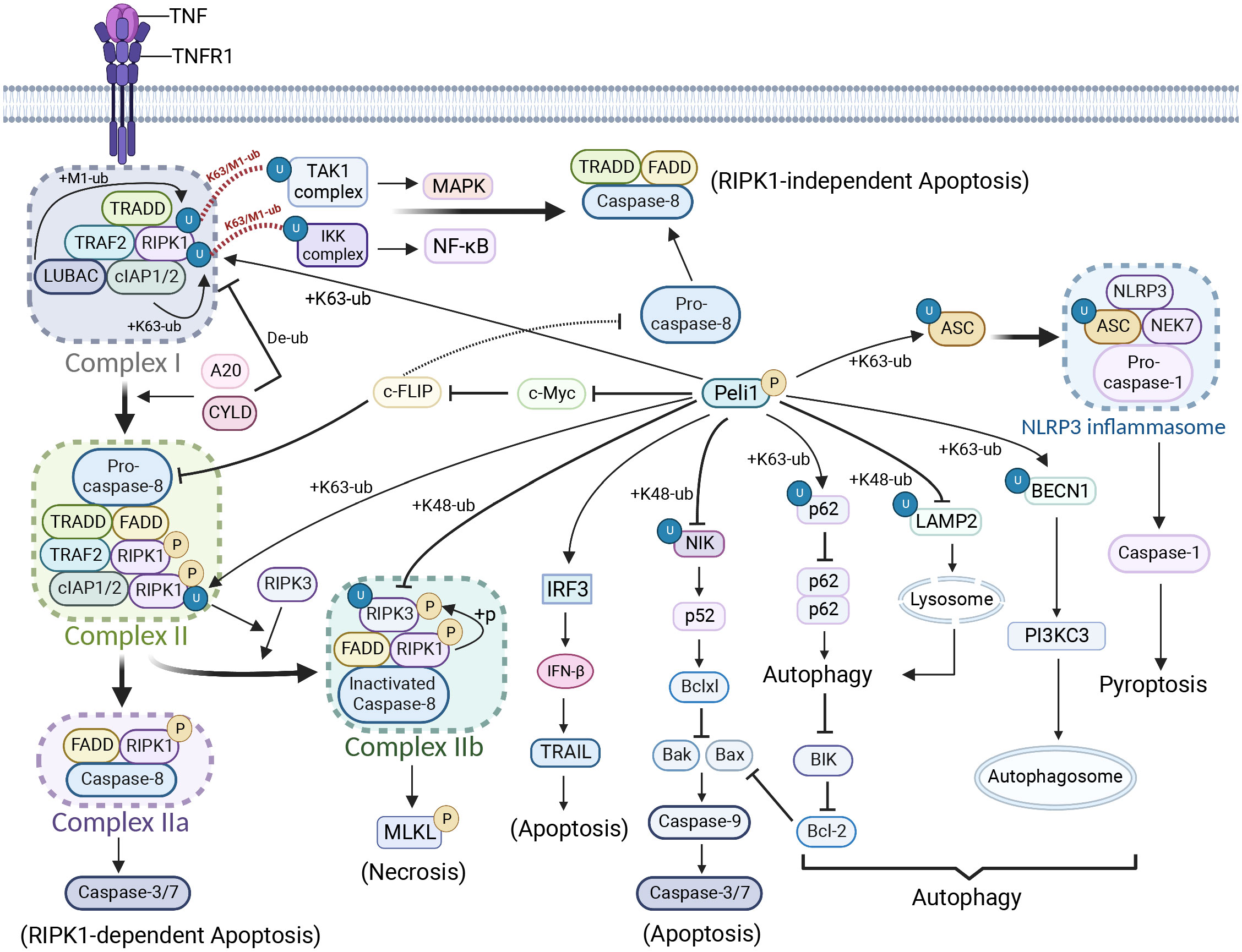
Figure 2 Peli1 regulation in cell death and autophagy. Upon tumor necrosis factor (TNF) stimulation, the TNFR1-associated death structural domain (TRADD) recruits cIAP1 and cIAP2 to RIPK1 via the TRAF2 and linear ubiquitin chain assembly complexes, forming complex I. Rapid ubiquitination of RIPK1 occurs. Complex I is deubiquitinated by A20 and cylindromatosis, forming complex II. Complex II bifurcates into complex IIa, triggering RIPK1-dependent apoptosis, and complex IIb, which induces necrosis. Complex I forms a TRADD-FADD-caspase-8 complex, leading to RIPK1-independent apoptosis. PELI1 preferentially recruits RIPK3 to form complex IIb by mediating RIPK1 ubiquitination in complex II. However, PELI1 mediates the ubiquitin-proteasome-dependent degradation of RIPK3, thereby inhibiting necrosis. PELI1 inhibits RIPK1-dependent or RIPK1-independent apoptosis by suppressing c-myc and promoting c-FLIP (a catalytically inactive caspase-8 homolog) expression. However, Peli1 can also promote apoptosis through tumor necrosis factor-related apoptosis-inducing ligand by upregulating IFN-β expression. Peli1 inhibits NF-κB non-classical signaling and downstream B-cell lymphoma-extra large via NIK, promoting apoptosis. Peli1 inhibits autophagy by mediating p62 ubiquitination, resulting in BIK accumulation, which activates downstream apoptotic signals. Peli1 inhibits lysosome formation and autophagic signaling by mediating the ubiquitin-proteasome-dependent degradation of lysosome-associated membrane protein 2. However, Peli1 can also promote autophagosome production by mediating the ubiquitination of K63-linked Beclin 1. In addition, Peli1 induces NLRP3 inflammasome production and pyroptosis through the ubiquitin-mediated modification of apoptosis-associated speck-like protein (ASC). Created using BioRender.com.
Under hypoxic LPS conditions, DAPK1 phosphorylates Ser-39 of Peli1, destabilizes it, and reduces the binding between Peli1 and RIPK1. The release of the TRIF-RIPK1 signalosome enhances RIPK1 binding to caspase-8, ultimately inducing apoptosis in renal tubular cells (64). Surprisingly, T182 phosphorylation on RIPK3 preferentially recruits Peli1, leading to the K48-linked ubiquitination-dependent degradation of kinase-active RIPK3 (12, 108), possibly functioning as a feedback mechanism. Although much progress has been made, further investigations are needed to uncover additional mechanisms by which Peli1 is involved in RIPK1-mediated necrosis and apoptosis.
In contrast, Peli1 promotes apoptosis through alternative pathways (61, 94, 109–111). Peli1 inhibits ionizing radiation-induced (IR) activation of the non-classical NF-κB pathway by promoting NIK degradation, thereby preventing IR-induced expression of B-cell lymphoma-extra large. Consequently, caspase-9 maturation and apoptotic signaling are restored (16). Peli1 mediates K63-linked ubiquitination of the autophagy-associated protein P62 at residue K7, disrupting P62 homodimer formation and autophagic degradation. Furthermore, Peli1 upregulates BIK expression by inhibiting BIK autophagic degradation, inhibiting the apoptosis suppressor protein B-cell lymphoma 2 (BCL2) and ultimately promoting apoptosis (41). Peli1 upregulates IFN-β expression by promoting IRF3/IRF5, and IFN-β1 augments the tumor necrosis factor-related apoptosis-inducing ligand in human peripheral blood T cells, inducing apoptosis in septic cells (87).
The inflammatory vesicle complex NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) comprises the ligand-sensing receptor NLRP3, adapter protein apoptosis-associated speck-like protein (ASC), pro-caspase-1, and the regulatory protein NIMA-associated kinase 7. Peli1 facilitates K63-Ub at K55 of ASC at the inflammasome junction, promoting ASC/NLRP3 interactions and ASC oligomerization for inflammasome activation and pyroptosis induction (40).
Peli1 demonstrates dual regulation of autophagy depending on specific circumstances. T in vivo in vitro the E3 ligase activity of Peli1 was significantly increased under in vivo and in vitro reoxygenation conditions (Figure 2). On the one hand, Peli1 promotes cardiomyocyte death by impairing P62 autophagic degradation and reducing autophagic flux (41). On the other hand, inhibiting BIK autophagic degradation promotes the activation of the apoptotic pathway by inhibiting BCL2 (41). T he human immunodeficiency virus (HIV) protein Tat induces increased Peli1 expression, mediating the K63-Ub of Beclin 1, leading to increased autophagy and disruption of the blood-brain barrier (BBB) through reduced tight junctional zonula occludens-1 (ZO1). In Parkinson’s disease, Peli1 is upregulated in microglia by extracellular preformed fibrils (PFF) of α-synuclein, resulting in reduced lysosomes and blocked autophagic flux due to Ub proteasome-dependent lysosome-associated membrane proteins degradation (18). Overall, Peli1 regulates autophagy through diverse pathways, necessitating further investigations to determine the underlying regulatory mechanisms depending on the specific context.
DNA double-strand breaks (DSBs) signaling and repair are crucial for maintaining genomic integrity. Peli1 is crucial in DNA damage-responsive protein accumulation and efficient homologous recombination (HR) repair, making it essential for DSB-responsive pathways (Figure 3) (33–35, 46). Peli1 participates in multiple pathways associated with the DNA damage response (35). Peli1 is recruited to DSB sites by γH2AX and activated through ATM phosphorylation.DSBs activate ATM upon laser micro-irradiation-induced DNA damage, which phosphorylates H2A histone family member X (H2AX) to its activated state, γH2AX. Peli1 binds to nibrin (NBS1), also recruited to DSBs by γH2AX. Peli1 mediates the K63-Ub of NBS1 at the K686 and K690 sites, leading to MRE11 and RAD50 recruitment to form the MRE11-RAD50-NBS1 (MRN) complex. The MRN complex phosphorylates and activates ATM, activating MDC1 and its partner RNF8 recruited by γH2AX. This activation leads to RNF8/RNF168 recruitment and establishing of a platform for downstream DNA repair proteins by mediating histone ubiquitination (112). Additionally, Peli1, recruited by γH2AX to the site of DNA damage, binds to phosphorylated P53 at the Thr-18 site through its FHA domain, facilitating the ubiquitination of the P53 repressor protein MDMX via K63 linkage. U biquitination promotes the nuclear export of MDMX and releases P53, which activates the transcription of downstream genes involved in DNA damage repair (DDR), such as P21 (33, 34). Peli1 binds to IRAK4 after IR and autophosphorylates Thr-345/Ser-346, independent of its E3 Ub ligase activity. This event recruits IRAK1 to Peli1 via IRAK4, leading to IRAK1 activation and nuclear translocation through Thr209 phosphorylation. Nuclear IRAK1 co-localizes with γH2AX and inhibits the pro-apoptotic PIDDosome complex (PIDD1-RAIDD-caspase-2), exerting an anti-apoptotic function (46).
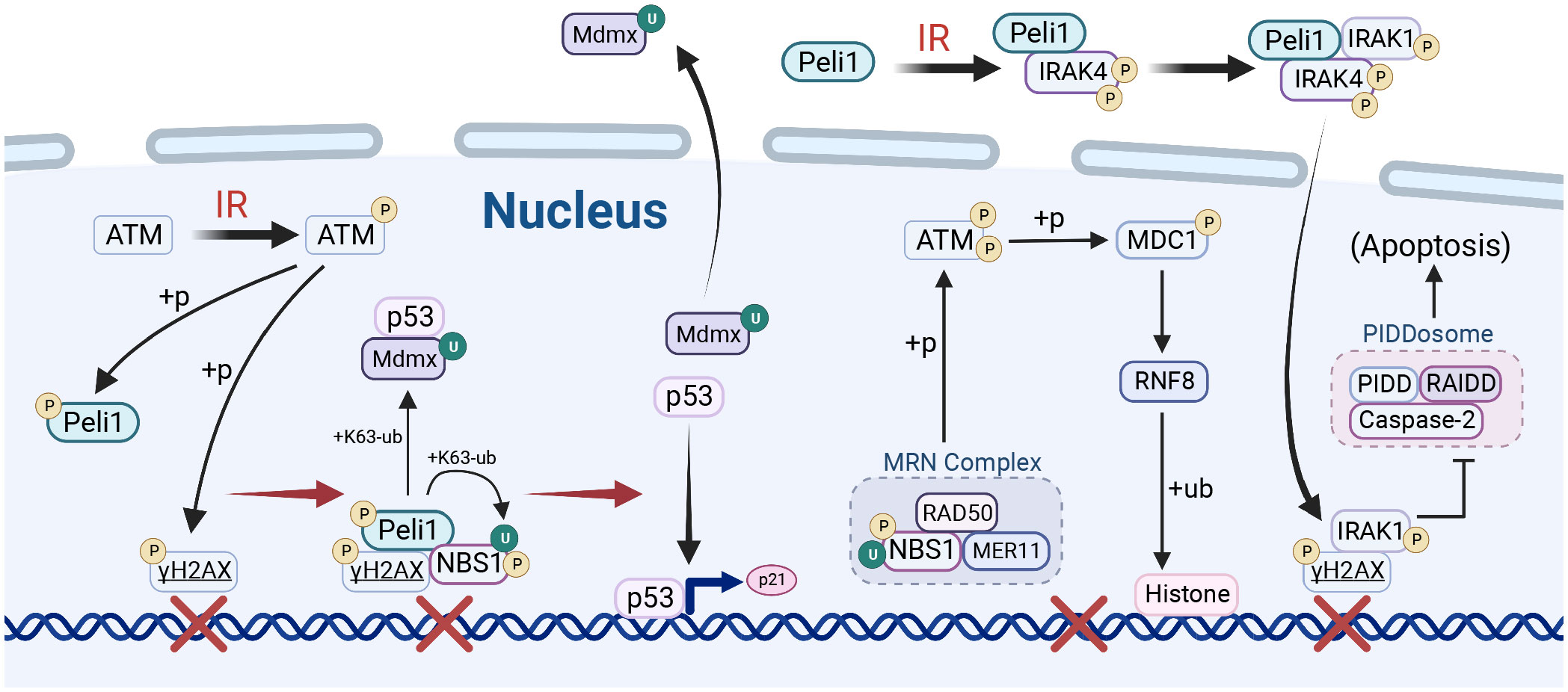
Figure 3 Peli1 in DNA damage repair. Upon ionizing radiation (IR) stimulation, PELI1 mediates nibrin (NBS1) ubiquitination and facilitates MRE11-RAD50-NBS1 (MRN) complex assembly, which is recruited to the DNA double-strand break (DSB) site by γH2AX and activated by ataxia-telangiectasia mutated (ATM) phosphorylation. The MRN complex enhances ATM phosphorylation, which mediates the phosphorylation of MDC1, thereby promoting the ubiquitination of histones by RNF8/RNF18 and establishing a platform for DNA damage repair. Peli1, recruited to the DSB site by γH2AX, promotes the nuclear export of murine double minute X (MDMX) by mediating MDMX ubiquitination, thus liberating P53 to activate the transcription of downstream genes in response to DNA damage repair. Peli1 binds to IRAK4 to promote IRAK1 recruitment and phosphorylation, which then translocates to the nucleus and co-localizes with γH2A, inhibiting the pro-apoptotic PIDDosome complex. Created using BioRender.com.
Peli1 exerts a complex and context-dependent regulation of glycolysis through various mechanisms. Peli1 is reportedly a negative regulator of glycolysis (37, 39, 52); however, other studies have shown that Peli1 promotes glycolysis (31). Peli1 enhances the stability of the tuberous sclerosis 1 (TSC1)/TSC2 complex in tumor-infiltrating CD8+ T cells by mediating K63-linked TSC1 ubiquitination (39). Peli1 inhibits TSC2 phosphorylation and inactivation by TCR/CD28 via the AKT pathway. Consequently, the stabilized complex hampers mammalian target of rapamycin complex 1 (mTORC1) activation by inactivating Rheb. Peli1 suppresses the phosphorylation of mTORC1 target proteins S6K and S6, accompanied by downregulating the downstream glycolytic genes, such as GLUT1, HK2, PGK1, Eno1, Pkm, Hif1a, and Myc. In pathogenic Th17 cells, Peli1 downregulates c-Myc by mediating the K48-Ub and ubiquitination-dependent degradation of c-Rel, a potent activator of genes involved in glycolysis and mitochondrial respiratory pathways (113), thereby inhibiting glycolysis (52). However, contradictory findings have been reported regarding glycolysis regulation by Peli1 in macrophages.
Peli1 promotes K63-linked ubiquitination of IRF5 in response to LPS/IFN-γ stimulation, enhancing glycolysis and M1 polarization by increasing the nuclear translocation and transcription factor activity of IRF5 (31). Conversely, Peli1-mediated K63-linked ubiquitination of IRAK1 and STAT1 activation inhibits IL-10-induced polarization of M2c macrophages and IL-10 production, thereby inhibiting tumor growth. Peli1-deficient bone marrow-derived macrophages exhibit defective mitochondrial respiration but enhanced glycolysis during M2c polarization (37). However, IL-10 inhibits LPS-induced glucose uptake and macrophage glycolysis (114). Therefore, the regulation of glycolysis by Peli1 in the tumor microenvironment and different cell types may involve additional pathways that warrant further in-depth investigation.
Peli1 plays a pivotal role in tumors, with complex roles across different cancer types, impacting tumor behavior, therapeutic response, and immune modulation (Figure 4). Understanding Peli1 functions holds promise for developing targeted cancer therapies and improving clinical management.
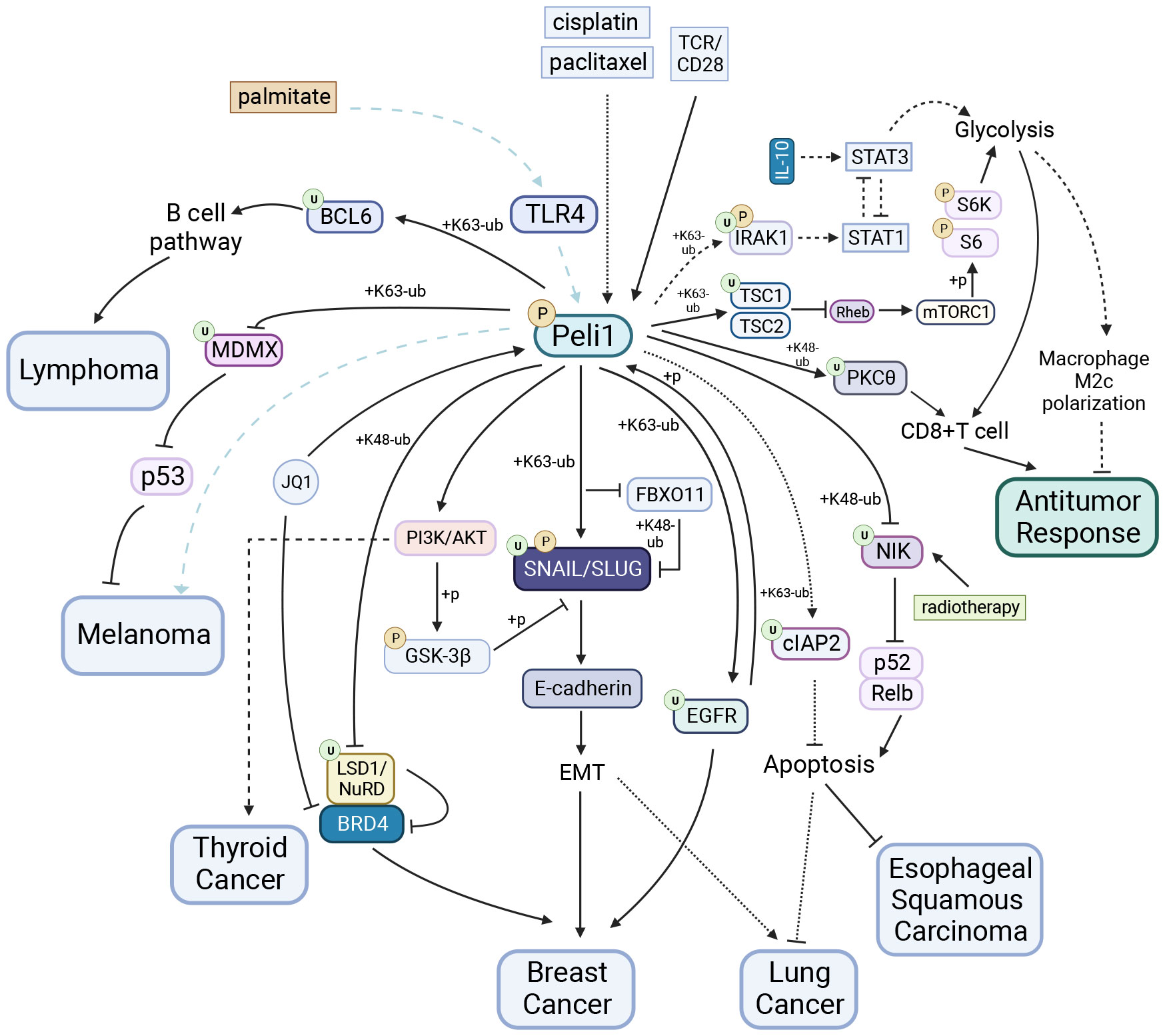
Figure 4 Peli1 in Tumors. Peli1 stabilizes SNAIL/SLUG via ubiquitination and the PI3K/AKT/GSK-3βpathway, contributing to breast and lung cancers EMT. Peli1 is detrimental to JQ1 drug tolerance in breast cancer through LSD1 degradation and the BRD4/LSD1/NuRD complex dissociation. Peli1 interacts with EGFR to promote breast cancer metastasis. Peli1 overexpression upregulates the expression of the apoptosis-inhibitory protein cIAP2, conferring resistance to cisplatin- and paclitaxel-induced apoptosis in lung cancers. Peli1 expression is upregulated in papillary thyroid carcinoma and promotes cancer cell proliferation and migration by activating the PI3K/AKT pathway. Elevated Peli1 expression induces lymphoma development by facilitating BCL6 ubiquitination and promoting the constitutive activation of the post-BCL6 B-cell signaling pathway. Peli1 is involved in palmitate-induced, TLR4-dependent lung metastasis in melanoma, whereas Peli1 inhibits melanoma progression through MDMX ubiquitination and cytoplasmic localization. Peli1 enhances the sensitivity of esophageal squamous carcinoma to radiotherapy by inhibiting the IR-induced activation of the non-canonical NF-κB pathway. Peli1 mediates PKCθubiquitination and inhibits glycolysis via TSC1 ubiquitination, negatively regulating antitumor activity in CD8+ T cells. In contrast, Peli1 exhibits positive antitumor capacity in macrophages by inhibiting glycolysis through IRAK1 TSC1 ubiquitination.Created using BioRender.com.
Triple-negative breast cancer (TNBC), which is aggressive and heterogeneous, is the second most common malignancy among women. Peli1 is upregulated in breast cancer tumor tissues, and higher Peli1 expression levels correlate with reduced survival rates. Peli1 is a positive regulator of tumor metastasis that significantly contributes to breast cancer mortality (24). Silencing Peli1 reduces tumor migration, invasion, and tumor-sphere formation. Loss of E-cadherin expression, a characteristic feature of epithelial-mesenchymal transition (EMT), is associated with the upregulation of E-cadherin transcriptional repressors (e.g., SNAIL, SLUG, TWIST, ZEB1, and ZEB2) (115–119). Peli1 stabilizes SNAIL/SLUG by mediating K63-linked polyubiquitination while inhibiting FBXO11 binding to SNAIL/SLUG and subsequent K48-linked Ub proteasome-dependent degradation (24). Peli1 inhibits glycogen synthase kinase-3 beta activity by activating AKT-mediated phosphorylation, disrupting the SCF-mediated ubiquitination degradation of SNAIL through SNAIL phosphorylation (105, 120). Peli1 interacts with EGFR to promote breast cancer metastasis. Activated EGFR phosphorylates and activates Peli1, enabling it to mediate the K63-linked EGFR ubiquitination and protect it from degradation. Understanding this process provides insights for refining therapies targeting EGFR (22).
Prognostic markers and biological therapies of Peli1 hold promise for improved disease management due to resistance to conventional therapies. Peli1 is detrimental to JQ1 drug tolerance by targeting bromodomain-containing protein 4 (BRD4). BRD4 is known for its role in the super-enhancer organization and the transcriptional activation of major oncogenes, including c-MYC and BCL2 (121, 122). BRD4 inhibitors, such as JQ1, have emerged as promising therapeutic agents for cancer (123). BRD4 unexpectedly interacts with the LSD1/NuRD complex and co-domains with this inhibitory complex at the super-enhancer, similar to the BRD4/LSD1/NuRD complex. However, long-term treatment with JQ1 leads to elevated Peli1 expression, resulting in the dissociation of the BRD4/LSD1/NuRD complex, LSD1 degradation via Ub-proteasome mechanisms, and JQ1 resistance development (21). Peli1 is associated with resistance to combination chemotherapy in breast cancer (21). Therefore, Peli1 plays a pathological role in breast cancer and contributes to chemotherapy tolerance, underscoring the importance of comprehensive investigations to enhance the clinical management of breast cancer.
Peli1 overexpression, implicated in lymphoma progression, is significantly elevated in various lymphomas. Peli1 expression may be a valuable prognostic indicator for patients with lymphomas, especially those with diffuse large B cell lymphoma (DLBCL). High Peli1 expression in lymphoma is associated with frequent bone marrow involvement and shorter relapse-free survival (124). Peli1 expression may be an independent prognostic indicator of DLBCL (30). Elevated Peli1 expression induces lymphoma development by facilitating B-cell lymphoma 6 (BCL6) ubiquitination through K63 linkage and promoting the constitutive activation of the post-BCL6 B-cell signaling pathway. This activation increases mature B cells and B220+ lymphocytes infiltration into the tumor (30). Peli1 expression positively correlates with the MYC, BCL6, BCL2, and MUM1 expression in lymphomas (30, 124). A study focusing on the Bcl-6-dependent risk stratification of DLBCL based on Peli1 nuclear expression highlighted the potential role of Peli1 and Bcl-6 in assessing DLBCL risk (125) Peli1 expression is highly elevated in high-grade lymphomas but significantly reduced in low-grade lymphomas (124). A completely different role for Peli1 in cHL exists, where miR-21-5p acts as an abundantly expressed oncogene that protects cHL from apoptosis by targeting Peli1 (109). This paradoxical role of Peli1 in different lymphomas may partly explain the differential expression of Peli1 in distinct grades of lymphomas and provide insights into developing t of personalized medical treatments for lymphomas.
Elevated Peli1 expression in lung cancer plays a crucial role in the development and progression and drug resistance (23, 25, 126). Peli1 expression is an essential prognostic indicator of survival in patient with lung cancer (126). First, Peli1 enhances the stability of SNAIL/SLUG by mediating SNAIL/SLUG polyubiquitination through K63 linkage, consequently inhibiting E-cadherin expression. This process promotes various malignant characteristics of lung cancer cells, including proliferation, survival, colony formation, invasion, and migration, by inducing EMT (23). Second, Peli1 overexpression upregulates the expression of the apoptosis-inhibitory proteins cIAP1 and cIAP2, conferring resistance to cisplatin- and paclitaxel-induced apoptosis in tumor cells. Peli1 achieves this effect by directly interacting with cIAP2 and stabilizing it through its E3 ligase activity, involving K63-linked polyubiquitination (25). The combination of low Peli1 expression and high necrosis factor RIPK3 expression along with DDR factor p53 is a significant predictor of survival in patients with stage I non-small cell lung cancer squamous cell carcinoma subtype (126). Therefore, Peli1 plays a pathological role in lung cancer and represents a novel therapeutic target to treat for this disease.
The role of Peli1 varies across cancer types. Peli1 promotes tumor progression and sensitivity to radiotherapy in papillary thyroid and esophageal squamous carcinomas (16, 103). Conversely, its functions are contradictory in melanoma, suggesting a complex role in tumor biology (34, 127).
Peli1 expression is increased in papillary thyroid carcinoma and promotes cancer cell proliferation and migration by activating the PI3K/AKT pathway, possibly associated with the deletion of miR-30c-5P, which targets Peli1 (103).
However, Peli1 plays contradictory roles in melanoma progression. Peli1 mediates MDMX ubiquitination by binding to its RING structural domain, leading to MDMX cytoplasmic localization. This activation promotes P53-mediated tumor progression inhibition, with higher Peli1 expression associated with improved survival patients with melanoma (34). In contrast Peli1 is involved in palmitate-induced, TLR4-dependent lung metastasis in melanoma (127). The diverse roles of Peli1 in the different phases of melanoma progression warrant further investigation.
Peli1 also plays a protective role in esophageal squamous carcinoma; Peli1 enhances the sensitivity of esophageal squamous carcinoma to radiotherapy by inhibiting the IR-induced activation of the non-canonical NF-κB pathway. This effect is achieved through the Peli1-mediated NIK ubiquitination and degradation, leading to increased tumor cell apoptosis (16). Consistent with this, a comprehensive analysis of transcriptome and methylation data from the Gene Expression Omnibus database revealed that high Peli1 expression in in patients with esophageal cancer is associated with longer overall survival (128).
Furthermore, Peli1 exhibits opposing effects on different immune cells during antitumor responses. In CD8+ T cells, Peli1 negatively regulates antitumor activity. First, Peli1 mediates the ubiquitination degradation of PKCθ via K48 linkage (20), inhibiting T cell receptor signaling and impairing CD8+ T cell function. Second, Peli1 increases the stability of the TSC1/TSC2 complex by mediating the K63-Ub of TSC1 (39). This stabilization inhibited TSC2 phosphorylation and inactivation by TCR/CD28 signaling through the AKT pathway. Consequently, the stable complex suppressed mTORC1 activation and phosphorylation S6K and S6 by inactivating Rheb. Ultimately, these mechanisms lead to a reduced antitumor response in T cells. In contrast, Peli1 exhibits positive antitumor capacity in macrophages by inhibiting glycolysis. This is achieved by mediating the K63-linked IRAK1 ubiquitination, leading to STAT1 activation and the inhibition of M2c macrophage polarization induced by IL-10 (37). As a result, tumor growth was inhibited in vivo. Furthermore, Peli1 inactivates the mitotic spindle checkpoints by mediating K48-Ub and Ub degradation of BubR1, inducing extensive chromosomal aneuploidy and tumorigenesis (14).
Peli1 is mainly pathogenic in infarction, primarily through mechanisms involving inflammation, autophagy, and apoptosis. Targeting Peli1 could rescue infarction and improve cardiac function (95–97, 105).
Inflammation is a crucial pathway through which Peli1 is involved in infarction. Peli1 silencing reduces inflammatory infiltration and ultimately improves cardiac dysfunction by regulating the TLR/IL1R pathway (92), inhibiting RIP1 and TRAF6 ubiquitination, and inhibiting NF-κB activity. Peli1 promotes the nuclear translocation of IRF5 by mediating the K63-Ub of IRF5, promoting M1 polarization of macrophages andcardiomyocyte death (32). Peli1 also regulates infarction via autophagy and apoptosis. Reoxygenation, in vivo and in vitro, significantly increases the E3 ligase activity of Peli1 (41). Furthermore, Peli1 promotes the ubiquitination of the autophagic protein P62 at residue K7 through K63 linkage. Disrupting P62 homodimer formation due to ubiquitination prevents P62 autophagic degradation, consequently reducing autophagic flux and promoting cardiomyocyte death. Inhibiting BIK autophagic degradation by Peli1 promotes apoptotic pathway activation by inhibiting BCL2, which increases myocardial infarction and contributes to cardiac dysfunction.
In an LPS-induced myocarditis model, Peli1 was activated and promoted pro-inflammatory genes expression (129). Si-Peli1 Treatment alleviated or even reversed LPS-induced cellular injury by modulating cardiomyocyte energy metabolism and altering the expression of key genes (Cs, Cpt2, and Acadm) and metabolites (3-oxoocotanoyl-CoA, hydroxypyruvic acid, lauroyl-CoA, and nicotinamide adenine dinucleotide phosphate) (129). In the context of diabetes-induced cardiovascular response, Peli1 elevates its interaction with HSP90, competitively inhibiting IRE1α binding to HSP90 and promoting IRE1α phosphorylation and ER stress. Peli1 also enhances TRAF2 recruitment to IRE1α by promoting XBP1 splicing and maturation, activating the MAPK pathway and mediating apoptosis of cardiac microvascular endothelial cells (47).
Peli1 has been proposed as a potential predictive biomarker for coronary artery disease (130). Peli1 plays a protective role in atherosclerosis as its deficiency increases pathogenic immune cell subsets (Th1 cells and Tfh cells)and decreases protective subsets (Treg and Breg cells), promoting systemic inflammation, immune cell infiltration, and foam cell formation in vascular smooth muscle cells, thereby exacerbating atherosclerosis (131). Peli1 is also downregulated in giant cell arteritis (132).
In summary, Peli1 is involved in cardiovascular disease progression by regulating inflammation autophagy, and apoptosis, thus providing novel insights and potential therapeutic targets for individualized treatment.
Peli1 plays distinct pathogenic and protective roles in response to various viral infections. Peli1 exerts pathogenic effects on central nervous system (CNS) antiviral infections (38, 88, 89). Peli1 negatively regulates TLR-mediated IFN-I induction by inhibiting TBK1/Ikkϵ activation-related signaling events. Peli1-deficient mice and microglia infected with vesicular stomatitis virus (VSV) exhibited significantly enhanced IFN-α/IFN-β expression and increased antiviral responses (88). Furthermore, Peli1 acts as a pathogenic factor during WNV infection by promoting cell attachment, entry, replication, and neuroinflammation via microglial activation (89). Peli1 is also involved in HIV invasion of the BBB, where Tat induces elevated Peli1 expression. Peli1 mediates the K63-linked ubiquitination of beclin 1, resulting in increased autophagy and disruption of the BBB through the disassembly of tightly linked ZO1 (38). The Japanese encephalitis virus (JEV) suppresses Peli1 in microglia by upregulating microRNA-155, increasing TRAF3 expression. This mechanism facilitates immune escape from JEV by inhibiting the non- canonical NF-κB pathway through NF-κBp100 accumulation (59). Peli1 promotes Zika virus (ZIKV) infection and placental inflammation and is involved in multiple stages of ZIKV infection, including cellular attachment, entry, replication, and translation (133).
However, Peli1 also plays a protective role against several viral infections. Peli1 participates in the positive feedback loop of IFN-β secretion by promoting IRF3 binding to the IFN promoter, which is crucial for IFN production during viral double-stranded RNA exposure (86). Peli1 also restricts herpes simplex virus type 1 (HSV-1) skin infection by suppressing HSV-1 replication and local dissemination. Peli1 enhances T cell recruitment to the infection by increasing Gpr156 expression (134). Peli1 positively regulates the antiviral response of isolated epithelial cells and the systemic response activated by TLR3. However, Peli1 plays an opposing role in the antiviral response in lung cells (78, 135). For example, Peli1 acts as a pro-inflammatory molecule during viral infection, and its knockdown reduces rhinovirus (RV)-induced CXCL8 and IL-6 production (78) without affecting RV replication (78, 135).
The role of Peli1 in infection depens on the infection type, which is common in studies on the role of other E3 ligases in infectious diseases. For example, TRIM29 exerts a pathogenic role by inhibiting I-IFN production during infections with Epstein–Barr and RNA viruses such as influenza and eutherian viruses (136–138). Peli1 positively regulates Helicobacter pylori and non-typeable Haemophilus influenzae (NTHi) infections (139, 140). Peli1 enhances TLR2-mediated NF-κB activity and chemokine-induced responses to H. pylori LPS (140). Inhibiting Peli1 improves bacterial clearance during NTHi infection (139). Thus, in infectious diseases, the differential targeting of Peli1 according to differences in its regulatory functions provides new pathways for treating infections.
Peli1 is pathogenic in several respiratory diseases, notably Chronic obstructive pulmonary disease (COPD), asthma, acute lung injury, and persistent bacterial bronchitis (PBB). Peli1 primarily functions as a pro-inflammatory agent, influencing disease progression and severity. Peli1-mediated K63-Ub of P21 prevents P21 degradation, leading to an increased senescence-associated secretory phenotype and promotion of COPD and inflammation (42). Peli1 regulates the pro-inflammatory response of airway epithelial cells in patients with asthma, and Peli1 knockdown significantly reduces CXCL8 expression in the airway epithelium of these patients (78). E levation of IL-1 signaling factors, including Peli1, contributes to in childhood asthma. Elevaed Peli1 expression is associated with severity and relapse of asthma in patients (141).
Peli1 is also upregulated in acute lung injury and promotes disease progression (54, 90). TGF-β1 upregulates Peli1 expression in acute lung injury by suppressing microRNA-124 through DNA-methyltransferase 1 upregulation. This promotes M1 alveolar macrophage polarization via IRF5 nuclear translocation (54). Peli1 increases TRAF6 expression, activates the NF-κB pathway, and exerts pro-inflammatory effects (90). Furthermore, Peli1 expression is higher in children with persistent bacterial bronchitis (PBB), particularly in recurrent cases (142). In summary, Peli1 predominantly plays a pro-inflammatory role in COPD, asthma, acute lung injury, and PBB.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by β-amyloid (Aβ) accumulation in the brain (143, 144). Peli1 is a genetic risk factor for AD, and Peli1 expression is upregulated in the brain tissues of patients with AD and plays a pathogenic role (19, 61, 145). In AD, Peli1 mediates C/EBPβ ubiquitination degradation, which inhibits the CD36 expression, impairing Aβ phagocytosis by microglia (19). Peli1 has been implicated in AD pathogenesis through the TRAF3/MAPK and the BCL2 apoptotic pathways, thereby reducing microglial and neuronal cell viability. Peli1 downregulation through overexpression or mimicking MIR-590-5P, which is downregulated in AD, can attenuate neuronal damage caused by Aβ (61).
Stroke, one of the leading causes of death and disability, can benefit from accurate and rapid etiological classificationto determine treatment options and reduce the risk of recurrence. Peli1 is a diagnostic risk marker for Cardiogenic (CE) strokes (146–148). Elevated Peli1 levels are associated with a high risk of stroke in patients with atrial fibrillation, suggesting its potential use as a marker for stroke prediction, prevention, and treatment in this population (147).
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating, and neurodegenerative disease of the central nervous system that affects young adults (149, 150). The role of Peli1 in Experimental autoimmune encephalomyelitis (EAE) pathogenesis is subject to contrasting viewpoints. Some studies have proposed that Peli1 positively regulates EAE progression by promoting microglial activation, whereas others suggest a protective role of Peli1 in EAE by inhibiting T-cell activation and pathogenicity. Peli1 promotes K48-Ub and degradation of TRAF3 by C-IAP (80), leading to MAPK activation, AP-1 activation, and inflammatory factors transcription, thus specifically activating microglia and promoting neuroinflammation. Conversely, Peli1 inhibits T-cell glycolysis, TH17 cell activation, and pathogenicity by mediating c-Rel ubiquitination degradation (13). Peli1-deficient EAE models have exhibited decreased inflammatory factors and EAE scores but elevated levels of peripheral autoimmunity and increased antigen-presenting proteins (151). Therefore, further investigations are required to comprehensively elucidate the specific mechanisms underlying the role of Peli1 in EAE, considering different cell types and their spatial and temporal contexts.
Peli1 is also involved in Methamphetamine (meth) abuse development, which leads to neurological symptoms, including memory impairment, altered cognitive function, and attention deficits (152). Peli1 plays a pro-inflammatory role in the effects of meth, as meth treatment results in elevated Peli1 expression owing to decreased levels of miR-142a-3p and miR-155-5p levels (56). Meth also upregulates Peli1 expression via the TRIF signaling pathway (99). Peli1 induces neuroinflammation by activating the NF-κB and MAPK pathways and RIPK1 (99, 153).
Peli1 is a potential therapeutic target for the treatment of neurological hemorrhagic diseases. Following subarachnoid hemorrhage (SAH), Peli1 upregulation induces MAPK activation and inflammatory factor production, activating microglia (94). Decreased miR-590-5p levels after intracranial hemorrhage lead to elevated Peli1 expression, inducing neuroinflammation and cerebral edema (62). Peli1 controls the survival of dopaminergic neurons by regulating microglia-mediated neuroinflammation and the production of neurotoxic factors through the NF-κB/MAPK pathway (154).
In summary, Peli1 is involved in the progression of various diseases within the nervous system via inflammation modulation, especially in the microglia. However, further research is required to elucidate the precise mechanisms and therapeutic implications of these effects.
Peli1acts as a pro-inflammatory pathogenic factor under various obstetric conditions. During ZIKV infection, Peli1 promotes ZIKV infection and placental inflammation, exacerbating congenital abnormalities (133). Peli1 is a novel regulator of TNF and TLR signaling in human myometrial and amniotic cells and upregulates the expression of pro-inflammatory factors, adhesion factors, and contractile proteinsPeli1 upregulation in the amniotic membranes of patients with preterm histological chorioamnionitis suggests its potential as a therapeutic target for reducing preterm delivery caused by inflammation and infection (155). I in utero, vitamin D intervention downregulate Peli1, contributing to immune protection against uterine inflammation (156). Peli1 positively correlates with miR-21 in patients with autoimmune premature ovarian insufficiency (POI). However, the underlying mechanisms and significance of Peli1 in POI pathogenesis require further investigation (157).
Peli1 regulates multiple autoimmune diseases with protective and pathogenic roles. Peli1 plays a pathogenic role in psoriasis (48, 158) and acts as an inflammatory modulatior in EAE, primarily promotes inflammation but inhibiting excessive inflammation (13, 80, 85). Systemic lupus erythematosus (SLE) is a complex, multisystem autoimmune disease characterized by genetic and environmental factors. In SLE, Peli1 is downregulated due to targeted inhibition by elevated miR-153-3p and miR-301a-3p levels (57, 60). Peli1 exerts its protective effects against SLE through multiple mechanisms. First, Peli1 downregulates the expression of inducible T cell co-stimulatory ICOS in CD4+ T cells by inhibiting c-Rel, inhibiting PI3K-AKT signaling. Upregulating downstream KLF2 inhibits Tfh and Th17 cell differentiation, ultimately attenuating autoimmunity in SLE (93). Second, Peli1 mediates the NIK ubiquitination and degradation, inhibits the nuclear translocation of Relb/P52, and prevents antibody production by B cells, thereby suppressing SLE (15). Peli1 inhibition of the non-classical NF-κB pathway in response to Poly IC treatment also attenuated SLE autoimmunity (159). Therefore, Peli1 is a promising protective factor against SLE and a potential therapeutic target.
Glucocorticoids are used extensively to treat inflammatory diseases. Glucocorticoid-induced GR downregulation is a well-known response that occurs in most cells and is necessary to limit the duration of glucocorticoid action (160). β-arrestin-1 binds to and remains stable at the GR (50). Silencing β-arrestin-1 results in the release of GR, leading to increased GR binding to the Peli1 promoter region. Subsequently, Peli1 transcription and expression are enhanced, and Peli1 mediates K48-linked ubiquitination of GR, contributing to GR turnover by shortening its half-life (50).
Sepsis, characterized by a systemic inflammatory response to infection, remains the leading cause of death in intensive care units (161). Pathological changes in sepsis are associated with the initial acute phase of hyperinflammation triggered by the innate immune system (162). Peli1 plays a prominent pro-inflammatory role in sepsis as an inflammatory factor. Peli1 promotes inflammation through TRAF6/NF-κB signaling and TRAF3/MAPK signaling (26, 90, 163). Peli1 couples the K63 Ub chain to ASC at the inflammatory vesicle junctions and promotes ASC/NLRP3 interactions and ASC oligomerization, leading to inflammatory vesicle activation (40). Peli1 induces TLR3- and TLR4-driven co-stimulatory gene expression, proliferation, and B cells survival (27). Peli1 deficiency enhances resistance to LPS endotoxic shock (40) and attenuates the induction of pro-inflammatory factors by TLR3/TLR4 ligands (27). In septic acute kidney injury (AKI) (64), DAPK1 Peli1 phosphorylation promotes RIP1 binding to caspase-8 and ultimately induces tubular apoptosis. In mice, synergistic DAPK1 inactivation or ablation and MyD88 inhibitors prevent septic AKI. In conclusion, Peli1 is implicated in sepsis and its progression in various organs and tissues through multiple pathways. Therefore, targeting Peli1 has a significant therapeutic potential for managing sepsis.
The targeted regulation of Peli1 has emerged as an intriguing avenue for therapeutic intervention, given its diverse roles in disease pathogenesis. Several drugs have been identified as Peli1 inhibitors. For example, resistin inhibits Peli1 activity and suppresses invasive breast cancer metastasis. This inhibition is achieved through hydrogen bonding with specific Peli1 residues, inhibiting Peli1 enhanced SNAIL/SLUG activity via the K63 linkage (24) and slowing TNBC progression. BBT-401, a pharmacologically targeted Peli1 inhibitor developed by Bridge Biotherapeutics, is undergoing phase II clinical trials to treat ulcerative colitis. BBT-401-1S inhibits Th17 cell effector responses in psoriasis models by decreasing p65 phosphorylation and producing IL-17A. Peli1 inhibitors have also demonstrated a dose-dependent inhibition of IL-17A and IL-22 production (48).
Given that Peli1 often functions through protein interactions, the regulation of Peli1 binding to target proteins and co-targeting strategies have been explored. Peli1 and EGFR promote metastasis in breast cancer. Compound S62, which does not individually bind to Peli1 or EGFR, can block both linkages and shows promise for treating breast cancer with combined targeting of Peli1 and EGFR (22). Furthermore, Peli1 has been implicated in modulating the tolerance to JQ1 targeting BRD4 in breast cancer. Combining BRD4 and Peli1 targeting is necessary for effective breast cancer treatment (21). Peli1 expression is increased in breast cancer samples from patients receiving multiple chemotherapeutic agents, and its expression level is positively correlated with the number of agents used by patients (21). This finding indicates that Peli1 is involved in resistance to various agents, warranting further investigation (21). A novel membrane-bound palmitate-coupled Smad6-derived peptide called Smaducin-6 has been developed to disrupt IRAK1-, RIP1-, and Ikkϵ-mediated TLR4 signaling complex formation by interacting with membrane-bound Peli1 and to restore neutrophil recruitment by reducing GRK2 expression in neutrophils through CXCR2 re-expression is sepsis (87).
Gene therapy targeting Peli1 has also shown promise in improving perfusion and cardiac function in ischemic infarction models (95, 105). In an LPS-induced myocarditis model, Peli1 activatio was associated with promoting pro-inflammatory genes, and si-Peli1 treatment alleviated or reversed LPS-induced cellular injury by altering cardiomyocyte energy metabolism (129). MiRNAs have been explored as regulators of Peli1 and human umbilical cord mesenchymal stem cell-derived extracellular vesicles modified with miR-30c-5p effectively suppress Peli1 expression and inhibit papillary thyroid carcinoma progression in vitro and in vivo (103).
Targeted protein degradation (TPD) and targeted covalent inhibitors (TCIs) may provide insights into the targeting Peli1 (164, 165). TPD primarily comprises molecular glue and proteolysis targeting chimera (PROTAC), which mediate the binding of E3 Ub ligases to target proteins, leading to target proteins degradation. PROTACs are heterobifunctional molecules comprising two specific ligands and a chemical linker that enables them to bind to E3 Ub ligases and the target protein (166). For example, von Hippel-Lindau (VHL) and cereblon (CRBN) are still the most widely used E3 ligases, and many of the developed PROTACs are based on their efficacy (167–169). Molecular glues are low- molecular- weight inducers or protein-protein interaction stabilizers.Upon binding to a protein, the small molecule induces a conformational change and causes the small molecule-protein complex to become a “new substrate” for the E3 ligase and thus undergoes ubiquitination. For example, thalidomide and its derivatives are effective cancer therapeutic agents and are among the best understood molecular glue degraders (11). These drugs selectively reprogram the E3 ligase cereblon (CRBN) to allow the Ub-proteasome system to degrade target proteins. In addition to promoting Peli1 degradation as a target protein through TPD, inhibiting Peli1 function through covalent inhibitors is also a potent and effective strategy. Covalent inhibitors are a class of small molecule compounds that can covalently bind to specific target proteins, inhibiting their biological functions, similar to the study of BBT-401 inhibiting Peli1 (48). However, Peli1 agonists are not being developed, which is detrimental to studies targeting the protective role of Peli1 in diseases such as SLE. Recently, with advances in computational tools, structure-targeted, high-throughput virtual screening and molecular docking have become effective methods for inverse drug discovery. TCI and agonist by high-throughput virtual screening of Peli1 may be helpful in targeted therapy.
Targeting Peli1 and therapy via TPD-linked Peli- targeting of downstream substrates may have side effects. Due to the diversity of Peli1 substrates and the wide range of biological functions in which Peli1 is involved, we also noted that Peli1 is primarily pathogenic in some diseases such as cancer, myocardial infarction, and AD. In contrast, Peli1 plays a protective role in other diseases such as atherosclerosis, some infectious diseases, and SLE. Complex roles make targeting Peli1 challenging, and Peli1 inhibition or promotion should be discussed according to the disease context, especially when patients simultaneously have different diseases in which Peli1 is involved.
Peli1 is critical in the development and progression of various diseases, particularly immune-related disorders and cancers. Further research on Peli1 as a diagnostic and therapeutic target, aided by cutting-edge therapeutic tools, may provide novel treatment options for various of human diseases. T he side effects of therapeutic interventions on Peli1 in different contexts in response to the complex regulation of Peli1 are also worth studying.
As a significant member of the E3 ligase family Peli, Peli1 exerts its regulatory function by binding to various proteins and mediating post-translational ubiquitination modifications (7, 8). Peli1 also exhibits multiple functions independent of its E3-Ub ligase activity, expanding its regulatory repertoire (35, 43, 47). The reversible phosphorylation mechanism enables the interconversion of Peli1 between inactive and active forms, with various kinases (such as IRAK1, IRAK4, TBK1/Ikkϵ) phosphorylating Peli1 to enhance its E3 ligase activity (36, 63). Peli1 possesses numerous phosphorylation sites, rendering it prone to activation through kinase phosphorylation and less susceptible to dephosphorylation-mediated inactivation (22, 85). Peli1 production and degradation may represent crucial pathways for its negative regulation. Peli1 transcription is regulated by IRF3 and the GR (50). Several miRNAs have been identified as Peli1 regulators via direct targeting (53–55, 60). Peli1 can also promote miR-21-mediated negative regulation of its expression through NF-κB (51), suggesting a potential negative feedback loop. IRAK-1 facilitates the Ub proteasome-dependent Peli1 degradation in a kinase-dependent manner (7); however, the specific mechanism of Peli1 degradation remains unknown.
Peli1 participates in the signaling through receptor systems such as IL-1R and TLRs (27, 72, 80). Peli1 exerts a multifunctional regulatory role in signaling pathways, including NF-κB, MAPK, and AKT (90, 93, 97), which are involved in pro-inflammatory responses and immune progression. Paradoxically, Peli1 also inhibits T cell activation by suppressing excessive NF-κB activation (13). Peli1 is involved in cell death, autophagy, DNA repair, glycolysis, and immune cell activation. The reasons underlying the paradoxical role of Peli1 in inflammatory regulation remain unknown, and a deeper understanding of its biology will contribute to our understanding of innate and adaptive immunity.
Peli1 has distinct roles in the progression of various diseases within different disease contexts. Peli1 acts as a pathogenic agent in most tumors (23, 24, 103), where it promotes tumor cell migration and proliferation and negatively affects antitumor immunity. However, in some tumor backgrounds, Peli1 is a beneficial factor with a protective function in antitumor immunity (37), and promotes radiation therapy sensitivity in esophageal cancer (16). Peli1 is a crucial regulatory molecule of the immune system in the cardiovascular system, exerting pro-inflammatory and pro-apoptotic effects (92). Given its specific expression in microglial, Peli1 functions in progression of various CNS diseases (e.g., AD, cardiogenic stroke, and MS) by regulating microglia (19, 80, 146). Peli1 promotes neuroinflammation after treatment with central nervous system drugs such as meth and morphine. Peli1 also differentially regulates several autoimmune diseases. Although its pathogenic role in MS and psoriasis is well- recognized, Peli1 plays a protective role against SLE. During viral infections, Peli1 plays a pro-inflammatory role (38, 88, 89, 133) and contributes to the pathogenesis of viral infections such as VSV, WNV, HIV, and ZIKV by promoting viral replication. However, Peli1 protects against HSV infection (134) by limiting viral invasion and spread. In contrast, Peli1 downregulation in JEV infection promotes immune evasion (59).
Thus, the significance of targeting Peli1 and its downstream target genes is evident. BBT-401 (48), a Peli1 target, is undergoing phase II clinical trials to treat ulcerative colitis. Several other drugs have shown promise in targeting Peli1, including the Smad6-derived peptide Smaducin-6, compound S62, and resistin (22, 24, 133). Blocking Peli1 can confer protective effects in numerous diseases; however, exploring Peli1 agonists remains an intriguing area of investigation, as they could potentially enhance the protective effects of Peli1 in diseases such as SLE. TPDs and TCIs have been used as cutting-edge therapeutic strategies based on several E3 ligases, providing new directions for developing of targeted therapies for Peli1. However, side effects of targeting Peli1 are inevitable, considering the involvement of Peli1 in multiple diseases and biological functions. Therefore, novel drug development and individualized therapeutic strategies are needed to target Peli1 within reasonable limits and avoid adverse events as much as possible.
Mounting evidence supports the predictive and prognostic value of Peli1. Therefore, a comprehensive understanding of the biological properties and molecular functions of Peli1 will facilitate the development of novel clinical therapeutic strategies. This review will advance future research on the role of Peli1 in immune diseases, cancers, and other conditions. This review provides information on the molecular mechanisms and directions for the clinical diagnosis and treatment of diseases, offering fundamental insights and evidence for potential future research areas, particularly in inflammation and cancer.
LY: Visualization, Writing – original draft, Writing – review & editing. YC: Investigation, Resources, Writing – review & editing. JF: Writing – review & editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Outstanding Scientific Fund Q18 of the Shengjing Hospital (to JF) and the National Natural Science Foundation of China (No. 82271275 to JF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
2. Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol (2011) 12(5):295–307. doi: 10.1038/nrm3099
3. Grosshans J, Schnorrer F, Nüsslein-Volhard C. Oligomerisation of tube and pelle leads to nuclear localisation of dorsal. Mech Dev (1999) 81(1-2):127–38. doi: 10.1016/s0925-4773(98)00236-6
4. Resch K, Jockusch H, Schmitt-John T. Assignment of homologous genes, peli1/peli1 and peli2/peli2, for the pelle adaptor protein pellino to mouse chromosomes 11 and 14 and human chromosomes 2p13.3 and 14q21, respectively, by physical and radiation hybrid mapping. Cytogenet Cell Genet (2001) 92(1-2):172–4. doi: 10.1159/000056895
5. Jensen LE, Whitehead AS. Pellino3, a novel member of the pellino protein family, promotes activation of C-jun and elk-1 and may act as a scaffolding protein. J Immunol (2003) 171(3):1500–6. doi: 10.4049/jimmunol.171.3.1500
6. Lin CC, Huoh YS, Schmitz KR, Jensen LE, Ferguson KM. Pellino proteins contain a cryptic fha domain that mediates interaction with phosphorylated irak1. Structure (2008) 16(12):1806–16. doi: 10.1016/j.str.2008.09.011
7. Butler MP, Hanly JA, Moynagh PN. Kinase-active interleukin-1 receptor-associated kinases promote polyubiquitination and degradation of the pellino family: direct evidence for pellino proteins being ubiquitin-protein isopeptide ligases. J Biol Chem (2007) 282(41):29729–37. doi: 10.1074/jbc.M704558200
8. Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, et al. The irak-catalysed activation of the E3 ligase function of pellino isoforms induces the lys63-linked polyubiquitination of irak1. Biochem J (2008) 409(1):43–52. doi: 10.1042/bj20071365
9. Huoh YS, Ferguson KM. The pellino E3 ubiquitin ligases recognize specific phosphothreonine motifs and have distinct substrate specificities. Biochemistry (2014) 53(30):4946–55. doi: 10.1021/bi5005156
10. Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the P27 E3 ligase scf(Skp2) results in P27- and skp2-mediated cell-cycle arrest and activation of autophagy. Blood (2008) 111(9):4690–9. doi: 10.1182/blood-2007-09-112904
11. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science (2010) 327(5971):1345–50. doi: 10.1126/science.1177319
12. Choi SW, Park HH, Kim S, Chung JM, Noh HJ, Kim SK, et al. Peli1 selectively targets kinase-active rip3 for ubiquitylation-dependent proteasomal degradation. Mol Cell (2018) 70(5):920–35.e7. doi: 10.1016/j.molcel.2018.05.016
13. Chang M, Jin W, Chang JH, Xiao Y, Brittain GC, Yu J, et al. The ubiquitin ligase peli1 negatively regulates T cell activation and prevents autoimmunity. Nat Immunol (2011) 12(10):1002–9. doi: 10.1038/ni.2090
14. Park J, Park HY, Kim S, Kim HS, Park JY, Go H, et al. Pellino 1 inactivates mitotic spindle checkpoint by targeting bubr1 for ubiquitinational degradation. Oncotarget (2017) 8(19):32055–67. doi: 10.18632/oncotarget.16762
15. Liu J, Huang X, Hao S, Wang Y, Liu M, Xu J, et al. Peli1 negatively regulates noncanonical nf-κb signaling to restrain systemic lupus erythematosus. Nat Commun (2018) 9(1):1136. doi: 10.1038/s41467-018-03530-3
16. Dai D, Zhou H, Yin L, Ye F, Yuan X, You T, et al. Peli1 promotes radiotherapy sensitivity by inhibiting noncanonical nf-κb in esophageal squamous cancer. Mol Oncol (2022) 16(6):1384–401. doi: 10.1002/1878-0261.13134
17. Xie Y, Lv X, Ni D, Liu J, Hu Y, Liu Y, et al. Hpd degradation regulated by the ttc36-stk33-peli1 signaling axis induces tyrosinemia and neurological damage. Nat Commun (2019) 10(1):4266. doi: 10.1038/s41467-019-12011-0
18. Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial exosomes facilitate α-synuclein transmission in parkinson’s disease. Brain (2020) 143(5):1476–97. doi: 10.1093/brain/awaa090
19. Xu J, Yu T, Pietronigro EC, Yuan J, Arioli J, Pei Y, et al. Peli1 impairs microglial Aβ Phagocytosis through promoting C/ebpβ Degradation. PloS Biol (2020) 18(10):e3000837. doi: 10.1371/journal.pbio.3000837
20. Park J, Lee SY, Jeon Y, Kim KM, Lee JK, Ko J, et al. The pellino1-pkcθ Signaling axis is an essential target for improving antitumor cd8+ T-lymphocyte function. Cancer Immunol Res (2022) 10(3):327–42. doi: 10.1158/2326-6066.Cir-21-0419
21. Liu B, Liu X, Han L, Chen X, Wu X, Wu J, et al. Brd4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc Natl Acad Sci USA (2022) 119(6):e2109133119. doi: 10.1073/pnas.2109133119
22. Qi J, Xu G, Wu X, Lu C, Shen Y, Zhao B. Peli1 and egfr cooperate to promote breast cancer metastasis. Oncogenesis (2023) 12(1):9. doi: 10.1038/s41389-023-00457-3
23. Jeon YK, Kim CK, Hwang KR, Park HY, Koh J, Chung DH, et al. Pellino-1 promotes lung carcinogenesis via the stabilization of slug and snail through K63-mediated polyubiquitination. Cell Death Differ (2017) 24(3):469–80. doi: 10.1038/cdd.2016.143
24. Liu SS, Qi J, Teng ZD, Tian FT, Lv XX, Li K, et al. Resistomycin attenuates triple-negative breast cancer progression by inhibiting E3 ligase pellino-1 and inducing snail/slug degradation. Signal Transduct Target Ther (2020) 5(1):133. doi: 10.1038/s41392-020-00255-y
25. Jeon YK, Kim CK, Koh J, Chung DH, Ha GH. Pellino-1 confers chemoresistance in lung cancer cells by upregulating ciap2 through lys63-mediated polyubiquitination. Oncotarget (2016) 7(27):41811–24. doi: 10.18632/oncotarget.9619
26. Li P, Liu H, Zhang Y, Liao R, He K, Ruan X, et al. Endotoxin tolerance inhibits degradation of tumor necrosis factor receptor-associated factor 3 by suppressing pellino 1 expression and the K48 ubiquitin ligase activity of cellular inhibitor of apoptosis protein 2. J Infect Dis (2016) 214(6):906–15. doi: 10.1093/infdis/jiw279
27. Chang M, Jin W, Sun SC. Peli1 facilitates trif-dependent toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol (2009) 10(10):1089–95. doi: 10.1038/ni.1777
28. Wang H, Meng H, Li X, Zhu K, Dong K, Mookhtiar AK, et al. Peli1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of ripk1 and mrna levels of C-flip. Proc Natl Acad Sci USA (2017) 114(45):11944–9. doi: 10.1073/pnas.1715742114
29. Wang L, Yin C, Xu X, Liu T, Wang B, Abdul M, et al. Pellino1 contributes to morphine tolerance by microglia activation via mapk signaling in the spinal cord of mice. Cell Mol Neurobiol (2020) 40(7):1117–31. doi: 10.1007/s10571-020-00797-3
30. Park HY, Go H, Song HR, Kim S, Ha GH, Jeon YK, et al. Pellino 1 promotes lymphomagenesis by deregulating bcl6 polyubiquitination. J Clin Invest (2014) 124(11):4976–88. doi: 10.1172/jci75667
31. Kim D, Lee H, Koh J, Ko JS, Yoon BR, Jeon YK, et al. Cytosolic pellino-1-mediated K63-linked ubiquitination of irf5 in M1 macrophages regulates glucose intolerance in obesity. Cell Rep (2017) 20(4):832–45. doi: 10.1016/j.celrep.2017.06.088
32. Chen H, Hou Y, Zhai Y, Yang J, Que L, Liu J, et al. Peli1 deletion in macrophages attenuates myocardial ischemia/reperfusion injury by suppressing M1 polarization. J Leukoc Biol (2023) 113(2):95–108. doi: 10.1093/jleuko/qiac012
33. Dai L, Lin J, Said AB, Yau YH, Shochat SG, Ruiz-Carrillo D, et al. Pellino1 specifically binds to phospho-thr18 of P53 and is recruited to sites of DNA damage. Biochem Biophys Res Commun (2019) 513(3):714–20. doi: 10.1016/j.bbrc.2019.03.095
34. Li D, Tavana O, Sun SC, Gu W. Peli1 modulates the subcellular localization and activity of mdmx. Cancer Res (2018) 78(11):2897–910. doi: 10.1158/0008-5472.Can-17-3531
35. Ha GH, Ji JH, Chae S, Park J, Kim S, Lee JK, et al. Pellino1 regulates reversible atm activation via nbs1 ubiquitination at DNA double-strand breaks. Nat Commun (2019) 10(1):1577. doi: 10.1038/s41467-019-09641-9
36. Smith H, Liu XY, Dai L, Goh ET, Chan AT, Xi J, et al. The role of tbk1 and ikkϵ in the expression and activation of pellino 1. Biochem J (2011) 434(3):537–48. doi: 10.1042/bj20101421
37. Kim D, Koh J, Ko JS, Kim HY, Lee H, Chung DH. Ubiquitin E3 ligase pellino-1 inhibits il-10-mediated M2c polarization of macrophages, thereby suppressing tumor growth. Immune Netw (2019) 19(5):e32. doi: 10.4110/in.2019.19.e32
38. Liao K, Niu F, Hu G, Guo ML, Sil S, Buch S. Hiv tat-mediated induction of autophagy regulates the disruption of zo-1 in brain endothelial cells. Tissue Barriers (2020) 8(2):1748983. doi: 10.1080/21688370.2020.1748983
39. Ko CJ, Zhang L, Jie Z, Zhu L, Zhou X, Xie X, et al. The E3 ubiquitin ligase peli1 regulates the metabolic actions of mtorc1 to suppress antitumor T cell responses. EMBO J (2021) 40(2):e104532. doi: 10.15252/embj.2020104532
40. Zhang L, Ko CJ, Li Y, Jie Z, Zhu L, Zhou X, et al. Peli1 facilitates nlrp3 inflammasome activation by mediating asc ubiquitination. Cell Rep (2021) 37(4):109904. doi: 10.1016/j.celrep.2021.109904
41. Yang J, Tong T, Zhu C, Zhou M, Jiang Y, Chen H, et al. Peli1 contributes to myocardial ischemia/reperfusion injury by impairing autophagy flux via its E3 ligase mediated ubiquitination of P62. J Mol Cell Cardiol (2022) 173:30–46. doi: 10.1016/j.yjmcc.2022.09.004
42. Ma JH, Zhang YT, Wang LP, Sun QY, Zhang H, Li JJ, et al. K63 ubiquitination of P21 can facilitate pellino-1 in the context of chronic obstructive pulmonary disease and lung cellular senescence. Cells (2022) 11(19):3115. doi: 10.3390/cells11193115
43. Ordureau A, Enesa K, Nanda S, Le Francois B, Peggie M, Prescott A, et al. Deaf1 is a pellino1-interacting protein required for interferon production by sendai virus and double-stranded rna. J Biol Chem (2013) 288(34):24569–80. doi: 10.1074/jbc.M113.479550
44. Kinsella S, Fichtner M, Watters O, König HG, Prehn JHM. Increased A20-E3 ubiquitin ligase interactions in bid-deficient glia attenuate tlr3- and tlr4-induced inflammation. J Neuroinflamm (2018) 15(1):130. doi: 10.1186/s12974-018-1143-3
45. Lee YS, Kim JH, Kim ST, Kwon JY, Hong S, Kim SJ, et al. Smad7 and smad6 bind to discrete regions of pellino-1 via their mh2 domains to mediate tgf-beta1-induced negative regulation of il-1r/tlr signaling. Biochem Biophys Res Commun (2010) 393(4):836–43. doi: 10.1016/j.bbrc.2010.02.094
46. Li Y, Shah RB, Sarti S, Belcher AL, Lee BJ, Gorbatenko A, et al. A non-canonical irak signaling pathway triggered by DNA damage. bioRxiv (2023). doi: 10.1101/2023.02.08.527716
47. Zhao Q, Yang J, Chen H, Li J, Que L, Zhu G, et al. Peli1 induction impairs cardiac microvascular endothelium through hsp90 dissociation from ire1α. Biochim Biophys Acta Mol Basis Dis (2019) 1865(10):2606–17. doi: 10.1016/j.bbadis.2019.06.017
48. Kim SH, Oh J, Roh WS, Park J, Chung KB, Lee GH, et al. Pellino-1 promotes intrinsic activation of skin-resident il-17a-producing T cells in psoriasis. J Allergy Clin Immunol (2023) 151(5):1317–28. doi: 10.1016/j.jaci.2022.12.823
49. Cheng X, Chen R, Zhou T, Zhang B, Li Z, Gao M, et al. Leveraging the multivalent P53 peptide-mdmx interaction to guide the improvement of small molecule inhibitors. Nat Commun (2022) 13(1):1087. doi: 10.1038/s41467-022-28721-x
50. Petrillo MG, Oakley RH, Cidlowski JA. β-arrestin-1 inhibits glucocorticoid receptor turnover and alters glucocorticoid signaling. J Biol Chem (2019) 294(29):11225–39. doi: 10.1074/jbc.RA118.007150
51. Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP. Microrna-21 is upregulated during the proliferative phase of liver regeneration, targets pellino-1, and inhibits nf-kappab signaling. Am J Physiol Gastrointest Liver Physiol (2010) 298(4):G535–41. doi: 10.1152/ajpgi.00338.2009
52. Qiu R, Yu X, Wang L, Han Z, Yao C, Cui Y, et al. Inhibition of glycolysis in pathogenic T(H)17 cells through targeting a mir -21-peli1-C-rel pathway prevents autoimmunity. J Immunol (2020) 204(12):3160–70. doi: 10.4049/jimmunol.2000060
53. Díaz-Piña G, Ordoñez-Razo RM, Montes E, Páramo I, Becerril C, Salgado A, et al. The role of adar1 and adar2 in the regulation of mirna-21 in idiopathic pulmonary fibrosis. Lung (2018) 196(4):393–400. doi: 10.1007/s00408-018-0115-9
54. Wang Y, Wang X, Zhang H, Han B, Ye Y, Zhang M, et al. Transforming growth factor-β1 promotes M1 alveolar macrophage polarization in acute lung injury by up-regulating dnmt1 to mediate the microrna-124/peli1/irf5 axis. Front Cell Infect Microbiol (2021) 11:693981. doi: 10.3389/fcimb.2021.693981
55. Malardo T, Gardinassi LG, Moreira BP, Padilha É, Lorenzi JC, Soares LS, et al. Microrna expression signatures in lungs of mice infected with mycobacterium tuberculosis. Tuberculosis (Edinb) (2016) 101:151–9. doi: 10.1016/j.tube.2016.09.003
56. Yu G, Song Y, Xie C, Tao L, Wan F, Jiang L, et al. Mir-142a-3p and mir-155-5p reduce methamphetamine-induced inflammation: role of the target protein peli1. Toxicol Appl Pharmacol (2019) 370:145–53. doi: 10.1016/j.taap.2019.03.019
57. Li D, Li X, Duan M, Dou Y, Feng Y, Nan N, et al. Mir-153-3p induces immune dysregulation by inhibiting peli1 expression in umbilical cord-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Autoimmunity (2020) 53(4):201–9. doi: 10.1080/08916934.2020.1750011
58. Liu WH, Kang SG, Huang Z, Wu CJ, Jin HY, Maine CJ, et al. A mir-155-peli1-C-rel pathway controls the generation and function of T follicular helper cells. J Exp Med (2016) 213(9):1901–19. doi: 10.1084/jem.20160204
59. Rastogi M, Singh SK. Japanese encephalitis virus exploits microrna-155 to suppress the non-canonical nf-κb pathway in human microglial cells. Biochim Biophys Acta Gene Regul Mech (2020) 1863(11):194639. doi: 10.1016/j.bbagrm.2020.194639
60. Luo S, Wu R, Li Q, Zhang G. Mir-301a-3p advances irak1-mediated differentiation of th17 cells to promote the progression of systemic lupus erythematosus via targeting peli1. J Healthc Eng (2021) 2021:2982924. doi: 10.1155/2021/2982924
61. Shang L, Peng T, Chen X, Yan Z, Wang J, Gao X, et al. Mir-590-5p overexpression alleviates β-amyloid-induced neuron damage via targeting pellino-1. Anal Cell Pathol (Amst) (2022) 2022:7657995. doi: 10.1155/2022/7657995
62. Guo Q, Su H, He JB, Li HQ, Sha JJ. Mir-590-5p alleviates intracerebral hemorrhage-induced brain injury through targeting peli1 gene expression. Biochem Biophys Res Commun (2018) 504(1):61–7. doi: 10.1016/j.bbrc.2018.08.121
63. Smith H, Peggie M, Campbell DG, Vandermoere F, Carrick E, Cohen P. Identification of the phosphorylation sites on the E3 ubiquitin ligase pellino that are critical for activation by irak1 and irak4. Proc Natl Acad Sci USA (2009) 106(12):4584–90. doi: 10.1073/pnas.0900774106
64. Hu BC, Wu GH, Shao ZQ, Zheng Y, Liu JQ, Zhang R, et al. Redox dapk1 destabilizes pellino1 to govern inflammation-coupling tubular damage during septic aki. Theranostics (2020) 10(25):11479–96. doi: 10.7150/thno.49870
65. Kim JH, Sung KS, Jung SM, Lee YS, Kwon JY, Choi CY, et al. Pellino-1, an adaptor protein of interleukin-1 receptor/toll-like receptor signaling, is sumoylated by ubc9. Mol Cells (2011) 31(1):85–9. doi: 10.1007/s10059-011-0006-x
66. Kinsella S, König HG, Prehn JH. Bid promotes K63-linked polyubiquitination of tumor necrosis factor receptor associated factor 6 (Traf6) and sensitizes to mutant sod1-induced proinflammatory signaling in microglia. eNeuro (2016) 3(2):0099-15. doi: 10.1523/eneuro.0099-15.2016
67. Fabian MR, Sonenberg N. The mechanics of mirna-mediated gene silencing: A look under the hood of mirisc. Nat Struct Mol Biol (2012) 19(6):586–93. doi: 10.1038/nsmb.2296
68. Liu B, Li J, Cairns MJ. Identifying mirnas, targets and functions. Brief Bioinform (2014) 15(1):1–19. doi: 10.1093/bib/bbs075
69. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking irak-4. Nature (2002) 416(6882):750–6. doi: 10.1038/nature736
70. Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. Interleukin-1 (Il-1) receptor-associated kinase-dependent il-1-induced signaling complexes phosphorylate tak1 and tab2 at the plasma membrane and activate tak1 in the cytosol. Mol Cell Biol (2002) 22(20):7158–67. doi: 10.1128/mcb.22.20.7158-7167.2002
71. Vollmer S, Strickson S, Zhang T, Gray N, Lee KL, Rao VR, et al. The mechanism of activation of irak1 and irak4 by interleukin-1 and toll-like receptor agonists. Biochem J (2017) 474(12):2027–38. doi: 10.1042/bcj20170097
72. Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. Pellino 1 is required for interleukin-1 (Il-1)-mediated signaling through its interaction with the il-1 receptor-associated kinase 4 (Irak4)-irak-tumor necrosis factor receptor-associated factor 6 (Traf6) complex. J Biol Chem (2003) 278(13):10952–6. doi: 10.1074/jbc.M212112200
73. Jensen LE, Whitehead AS. Pellino2 activates the mitogen activated protein kinase pathway. FEBS Lett (2003) 545(2-3):199–202. doi: 10.1016/s0014-5793(03)00533-7
74. Choi KC, Lee YS, Lim S, Choi HK, Lee CH, Lee EK, et al. Smad6 negatively regulates interleukin 1-receptor-toll-like receptor signaling through direct interaction with the adaptor pellino-1. Nat Immunol (2006) 7(10):1057–65. doi: 10.1038/ni1383
75. Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor traf3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol (2010) 11(1):70–5. doi: 10.1038/ni.1819
76. Cohen P, Strickson S. The role of hybrid ubiquitin chains in the myd88 and other innate immune signalling pathways. Cell Death Differ (2017) 24(7):1153–9. doi: 10.1038/cdd.2017.17
77. Zhang B, Li M, Chen L, Yang K, Shan Y, Zhu L, et al. The tak1-jnk cascade is required for irf3 function in the innate immune response. Cell Res (2009) 19(4):412–28. doi: 10.1038/cr.2009.8
78. Bennett JA, Prince LR, Parker LC, Stokes CA, de Bruin HG, van den Berge M, et al. Pellino-1 selectively regulates epithelial cell responses to rhinovirus. J Virol (2012) 86(12):6595–604. doi: 10.1128/jvi.06755-11
79. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
80. Xiao Y, Jin J, Chang M, Chang JH, Hu H, Zhou X, et al. Peli1 promotes microglia-mediated cns inflammation by regulating traf3 degradation. Nat Med (2013) 19(5):595–602. doi: 10.1038/nm.3111
81. Vobořil M, Brabec T, Dobeš J, Šplíchalová I, Březina J, Čepková A, et al. Toll-like receptor signaling in thymic epithelium controls monocyte-derived dendritic cell recruitment and treg generation. Nat Commun (2020) 11(1):2361. doi: 10.1038/s41467-020-16081-3
82. Lin YC, Huang DY, Chu CL, Lin YL, Lin WW. The tyrosine kinase syk differentially regulates toll-like receptor signaling downstream of the adaptor molecules traf6 and traf3. Sci Signal (2013) 6(289):ra71. doi: 10.1126/scisignal.2003973
83. Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, et al. Specificity in toll-like receptor signalling through distinct effector functions of traf3 and traf6. Nature (2006) 439(7073):204–7. doi: 10.1038/nature04369
84. Li F, Li Y, Liang H, Xu T, Kong Y, Huang M, et al. Hectd3 mediates traf3 polyubiquitination and type I interferon induction during bacterial infection. J Clin Invest (2018) 128(9):4148–62. doi: 10.1172/jci120406
85. Goh ET, Arthur JS, Cheung PC, Akira S, Toth R, Cohen P. Identification of the protein kinases that activate the E3 ubiquitin ligase pellino 1 in the innate immune system. Biochem J (2012) 441(1):339–46. doi: 10.1042/bj20111415
86. Enesa K, Ordureau A, Smith H, Barford D, Cheung PC, Patterson-Kane J, et al. Pellino1 is required for interferon production by viral double-stranded rna. J Biol Chem (2012) 287(41):34825–35. doi: 10.1074/jbc.M112.367557
87. Lee YS, Park JS, Jung SM, Kim SD, Kim JH, Lee JY, et al. Inhibition of lethal inflammatory responses through the targeting of membrane-associated toll-like receptor 4 signaling complexes with a smad6-derived peptide. EMBO Mol Med (2015) 7(5):577–92. doi: 10.15252/emmm.201404653
88. Xiao Y, Jin J, Zou Q, Hu H, Cheng X, Sun SC. Peli1 negatively regulates type I interferon induction and antiviral immunity in the cns. Cell Biosci (2015) 5:34. doi: 10.1186/s13578-015-0024-z
89. Luo H, Winkelmann ER, Zhu S, Ru W, Mays E, Silvas JA, et al. Peli1 facilitates virus replication and promotes neuroinflammation during west nile virus infection. J Clin Invest (2018) 128(11):4980–91. doi: 10.1172/jci99902
90. Liu X, Lin Z, Xu Y. Pellino1 promoted inflammation in lung injury model of sepsis by traf6/nf-κb signal pathway. J Inflamm (Lond) (2021) 18(1):11. doi: 10.1186/s12950-021-00276-6
91. Song J, Zhu Y, Li J, Liu J, Gao Y, Ha T, et al. Pellino1-mediated tgf-β1 synthesis contributes to mechanical stress induced cardiac fibroblast activation. J Mol Cell Cardiol (2015) 79:145–56. doi: 10.1016/j.yjmcc.2014.11.006
92. Wu W, Hu Y, Li J, Zhu W, Ha T, Que L, et al. Silencing of pellino1 improves post-infarct cardiac dysfunction and attenuates left ventricular remodelling in mice. Cardiovasc Res (2014) 102(1):46–55. doi: 10.1093/cvr/cvu007
93. Huang X, Hao S, Liu J, Huang Y, Liu M, Xiao C, et al. The ubiquitin ligase peli1 inhibits icos and thereby tfh-mediated immunity. Cell Mol Immunol (2021) 18(4):969–78. doi: 10.1038/s41423-021-00660-5
94. Huang XP, Peng JH, Pang JW, Tian XC, Li XS, Wu Y, et al. Peli1 contributions in microglial activation, neuroinflammatory responses and neurological deficits following experimental subarachnoid hemorrhage. Front Mol Neurosci (2017) 10:398. doi: 10.3389/fnmol.2017.00398
95. Selvaraju V, Thirunavukkarasu M, Joshi M, Oriowo B, Shaikh IA, Rishi MT, et al. Deletion of newly described pro-survival molecule pellino-1 increases oxidative stress, downregulates ciap2/nf-κb cell survival pathway, reduces angiogenic response, and thereby aggravates tissue function in mouse ischemic models. Basic Res Cardiol (2020) 115(4):45. doi: 10.1007/s00395-020-0804-4
96. Rednam CK, Wilson RL, Selvaraju V, Rishi MT, Thirunavukkarasu M, Coca-Soliz V, et al. Increased survivability of ischemic skin flap tissue in flk-1(+/-) mice by pellino-1 intervention. Microcirculation (2017) 24(6). doi: 10.1111/micc.12362
97. Thirunavukkarasu M, Pradeep SR, Ukani G, Abunnaja S, Youssef M, Accorsi D, et al. Gene therapy with pellino-1 improves perfusion and decreases tissue loss in flk-1 heterozygous mice but fails in mapkap kinase-2 knockout murine hind limb ischemia model. Microvasc Res (2022) 141:104311. doi: 10.1016/j.mvr.2022.104311
98. Wang L, Yin C, Liu T, Abdul M, Zhou Y, Cao JL, et al. Pellino1 regulates neuropathic pain as well as microglial activation through the regulation of mapk/nf-κb signaling in the spinal cord. J Neuroinflamm (2020) 17(1):83. doi: 10.1186/s12974-020-01754-z
99. Yang T, Zang S, Wang Y, Zhu Y, Jiang L, Chen X, et al. Methamphetamine induced neuroinflammation in mouse brain and microglial cell line bv2: roles of the tlr4/trif/peli1 signaling axis. Toxicol Lett (2020) 333:150–8. doi: 10.1016/j.toxlet.2020.07.028
100. Tang C, Hou YX, Shi PX, Zhu CH, Lu X, Wang XL, et al. Cardiomyocyte-specific peli1 contributes to the pressure overload-induced cardiac fibrosis through mir-494-3p-dependent exosomal communication. FASEB J (2023) 37(1):e22699. doi: 10.1096/fj.202200597R
101. Deldicque L. Endoplasmic reticulum stress in human skeletal muscle: any contribution to sarcopenia? Front Physiol (2013) 4:236. doi: 10.3389/fphys.2013.00236
102. Lee J, Sun C, Zhou Y, Lee J, Gokalp D, Herrema H, et al. P38 mapk-mediated regulation of xbp1s is crucial for glucose homeostasis. Nat Med (2011) 17(10):1251–60. doi: 10.1038/nm.2449
103. Zheng T, Zhou Y, Xu X, Qi X, Liu J, Pu Y, et al. Mir-30c-5p loss-induced peli1 accumulation regulates cell proliferation and migration via activating pi3k/akt pathway in papillary thyroid carcinoma. J Transl Med (2022) 20(1):20. doi: 10.1186/s12967-021-03226-1
104. Liu Y, Tang LL, Liang C, Wu MM, Zhang ZR. Insulin resistance and pellino-1 mediated decrease in the activities of vasodilator signaling contributes to sunitinib-induced hypertension. Front Pharmacol (2021) 12:617165. doi: 10.3389/fphar.2021.617165
105. Thirunavukkarasu M, Selvaraju V, Joshi M, Coca-Soliz V, Tapias L, Saad I, et al. Disruption of vegf mediated flk-1 signaling leads to a gradual loss of vessel health and cardiac function during myocardial infarction: potential therapy with pellino-1. J Am Heart Assoc (2018) 7(18):e007601. doi: 10.1161/jaha.117.007601
106. Wang Q, Fan D, Xia Y, Ye Q, Xi X, Zhang G, et al. The latest information on the ripk1 post-translational modifications and functions. BioMed Pharmacother (2021) 142:112082. doi: 10.1016/j.biopha.2021.112082
107. Ju E, Park KA, Shen HM, Hur GM. The resurrection of rip kinase 1 as an early cell death checkpoint regulator-a potential target for therapy in the necroptosis era. Exp Mol Med (2022) 54(9):1401–11. doi: 10.1038/s12276-022-00847-4
108. Park HH, Morgan MJ, Kang HC, Kim YS. Regulation of rip3 protein stability by peli1-mediated proteasome-dependent degradation. BMB Rep (2018) 51(10):484–5. doi: 10.5483/BMBRep.2018.51.10.217
109. Yuan Y, Niu F, Nolte IM, Koerts J, de Jong D, Rutgers B, et al. Microrna high throughput loss-of-function screening reveals an oncogenic role for mir-21-5p in hodgkin lymphoma. Cell Physiol Biochem (2018) 49(1):144–59. doi: 10.1159/000492850
110. Mo H, Wang Z, He Z, Wan J, Lu R, Wang C, et al. Decreased peli1 expression attenuates osteoarthritis by protecting chondrocytes and inhibiting M1-polarization of macrophages. Bone Joint Res (2023) 12(2):121–32. doi: 10.1302/2046-3758.122.Bjr-2022-0214.R1
111. Tian J, Gu L, Adams A, Wang X, Huang R. Pellino-1 protects periodontal ligament stem cells against H(2)O(2)-induced apoptosis via activation of nf-κb signaling. Mol Biotechnol (2018) 60(8):533–8. doi: 10.1007/s12033-018-0067-6
112. Eliezer Y, Argaman L, Kornowski M, Roniger M, Goldberg M. Interplay between the DNA damage proteins mdc1 and atm in the regulation of the spindle assembly checkpoint. J Biol Chem (2014) 289(12):8182–93. doi: 10.1074/jbc.M113.532739
113. Li X, Luo G, Li T, Sun H, Wang W, Eiler E, et al. The C-rel-C-myc axis controls metabolism and proliferation of human T leukemia cells. Mol Immunol (2020) 125:115–22. doi: 10.1016/j.molimm.2020.06.029
114. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of il-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356(6337):513–9. doi: 10.1126/science.aal3535
115. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol (2000) 2(2):76–83. doi: 10.1038/35000025
116. Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, et al. The transcriptional repressor snail promotes mammary tumor recurrence. Cancer Cell (2005) 8(3):197–209. doi: 10.1016/j.ccr.2005.07.009
117. Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Invest (2012) 122(4):1469–86. doi: 10.1172/jci57349
118. Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of zeb1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res (2008) 68(7):2479–88. doi: 10.1158/0008-5472.Can-07-2559
119. Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, et al. Sip1/zeb2 induces emt by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res (2005) 33(20):6566–78. doi: 10.1093/nar/gki965
120. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of snail by gsk-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol (2004) 6(10):931–40. doi: 10.1038/ncb1173
121. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. Bet bromodomain inhibition as a therapeutic strategy to target C-myc. Cell (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
122. Peirs S, Frismantas V, Matthijssens F, Van Loocke W, Pieters T, Vandamme N, et al. Targeting bet proteins improves the therapeutic efficacy of bcl-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia (2017) 31(10):2037–47. doi: 10.1038/leu.2017.10
123. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of bet bromodomains. Nature (2010) 468(7327):1067–73. doi: 10.1038/nature09504
124. Choe JY, Park M, Yun JY, Na HY, Go H, Kim HJ, et al. Peli1 expression is correlated with myc and bcl6 expression and associated with poor prognosis in diffuse large B-cell lymphoma. Mod Pathol (2016) 29(11):1313–23. doi: 10.1038/modpathol.2016.128
125. Lee KR, Lee JO, Lee JS, Paik JH. Bcl-6-dependent risk stratification by nuclear expression of peli1 in diffuse large B-cell lymphoma. J Cancer (2022) 13(15):3598–605. doi: 10.7150/jca.67569
126. Lim JH, Oh S, Kim L, Suh YJ, Ha YJ, Kim JS, et al. Low-level expression of necroptosis factors indicates a poor prognosis of the squamous cell carcinoma subtype of non-small-cell lung cancer. Transl Lung Cancer Res (2021) 10(3):1221–30. doi: 10.21037/tlcr-20-1027
127. Zhang X, Li X, Xiong G, Yun F, Feng Y, Ni Q, et al. Palmitic acid promotes lung metastasis of melanomas via the tlr4/trif-peli1-pnf-κb pathway. Metabolites (2022) 12(11):1132. doi: 10.3390/metabo12111132
128. Wang J, Luo J, Sun Z, Sun F, Kong Z, Yu J. Identification of mthfd2 as a novel prognosis biomarker in esophageal carcinoma patients based on transcriptomic data and methylation profiling. Med (Baltimore) (2020) 99(37):e22194. doi: 10.1097/md.0000000000022194
129. Yang C, Zhao K, Chen X, Jiang L, Li P, Huang P. Pellino1 deficiency reprograms cardiomyocytes energy metabolism in lipopolysaccharide-induced myocardial dysfunction. Amino Acids (2021) 53(5):713–37. doi: 10.1007/s00726-021-02978-w
130. Huang KK, Zheng HL, Li S, Zeng ZY. Identification of hub genes and their correlation with immune infiltration in coronary artery disease through bioinformatics and machine learning methods. J Thorac Dis (2022) 14(7):2621–34. doi: 10.21037/jtd-22-632
131. Burger F, Baptista D, Roth A, Brandt KJ, Miteva K. The E3 ubiquitin ligase peli1 deficiency promotes atherosclerosis progression. Cells (2022) 11(13):2014. doi: 10.3390/cells11132014
132. Kuret T, Lakota K, Čučnik S, Jurčič V, Distler O, Rotar Ž, et al. Dysregulated expression of arterial micrornas and their target gene networks in temporal arteries of treatment-naïve patients with giant cell arteritis. Int J Mol Sci (2021) 22(12):6520. doi: 10.3390/ijms22126520
133. Luo H, Li G, Wang B, Tian B, Gao J, Zou J, et al. Peli1 signaling blockade attenuates congenital zika syndrome. PloS Pathog (2020) 16(6):e1008538. doi: 10.1371/journal.ppat.1008538
134. Cai KQ, Shellhamer C, Akiyama T, Jensen LE. Pellino1 restricts herpes simplex virus infections in the epidermis and dissemination to sebaceous glands. J Invest Dermatol (2022) 143(4):639–47.E2. doi: 10.1016/j.jid.2022.09.007
135. Marsh EK, Prestwich EC, Williams L, Hart AR, Muir CF, Parker LC, et al. Pellino-1 regulates the responses of the airway to viral infection. Front Cell Infect Microbiol (2020) 10:456. doi: 10.3389/fcimb.2020.00456
136. Xing J, Zhang A, Minze LJ, Li XC, Zhang Z. Trim29 negatively regulates the type I ifn production in response to rna virus. J Immunol (2018) 201(1):183–92. doi: 10.4049/jimmunol.1701569
137. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. Trim29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun (2017) 8(1):945. doi: 10.1038/s41467-017-00101-w
138. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for trim29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi: 10.1038/ni.3580
139. Hughes BM, Burton CS, Reese A, Jabeen MF, Wright C, Willis J, et al. Pellino-1 regulates immune responses to haemophilus influenzae in models of inflammatory lung disease. Front Immunol (2019) 10:1721. doi: 10.3389/fimmu.2019.01721
140. Smith SM, Freeley M, Moynagh PN, Kelleher DP. Differential modulation of helicobacter pylori lipopolysaccharide-mediated tlr2 signaling by individual pellino proteins. Helicobacter (2017) 22(1):2014. doi: 10.1111/hel.12325
141. Wei W, Huang J, Ma Y, Ma X, Fang L, Fang W, et al. Il-1 signaling pathway molecules as key markers in childhood asthma. Pediatr Allergy Immunol (2021) 32(2):305–13. doi: 10.1111/pai.13388
142. Baines KJ, Upham JW, Yerkovich ST, Chang AB, Marchant JM, Carroll M, et al. Mediators of neutrophil function in children with protracted bacterial bronchitis. Chest (2014) 146(4):1013–20. doi: 10.1378/chest.14-0131
143. Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med (2010) 362(4):329–44. doi: 10.1056/NEJMra0909142
144. Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell (2012) 148(6):1204–22. doi: 10.1016/j.cell.2012.02.040
145. Santiago JA, Quinn JP, Potashkin JA. Co-expression network analysis identifies molecular determinants of loneliness associated with neuropsychiatric and neurodegenerative diseases. Int J Mol Sci (2023) 24(6):5909. doi: 10.3390/ijms24065909
146. García-Berrocoso T, Palà E, Consegal M, Piccardi B, Negro A, Gill N, et al. Cardioembolic ischemic stroke gene expression fingerprint in blood: A systematic review and verification analysis. Transl Stroke Res (2020) 11(3):326–36. doi: 10.1007/s12975-019-00730-x
147. Li Y, Tan W, Ye F, Wen S, Hu R, Cai X, et al. Inflammation as a risk factor for stroke in atrial fibrillation: data from a microarray data analysis. J Int Med Res (2020) 48(5):300060520921671. doi: 10.1177/0300060520921671
148. Ye X, Kong D, Wang J, Ishrat T, Shi H, Ding X, et al. Myd88 contributes to neuroinflammatory responses induced by cerebral ischemia/reperfusion in mice. Biochem Biophys Res Commun (2016) 480(1):69–74. doi: 10.1016/j.bbrc.2016.10.007
149. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Primers (2018) 4(1):43. doi: 10.1038/s41572-018-0041-4
150. Laaker C, Hsu M, Fabry Z, Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc (2021) 1(12):e300. doi: 10.1002/cpz1.300
151. Lereim RR, Oveland E, Xiao Y, Torkildsen Ø, Wergeland S, Myhr KM, et al. The brain proteome of the ubiquitin ligase peli1 knock-out mouse during experimental autoimmune encephalomyelitis. J Proteomics Bioinform (2016) 9(9):209–19. doi: 10.4172/jpb.1000408
152. Dang J, Tiwari SK, Agrawal K, Hui H, Qin Y, Rana TM. Glial cell diversity and methamphetamine-induced neuroinflammation in human cerebral organoids. Mol Psychiatry (2021) 26(4):1194–207. doi: 10.1038/s41380-020-0676-x
153. Xu W, Yang T, Lou X, Chen J, Wang X, Hu M, et al. Role of the peli1-ripk1 signaling axis in methamphetamine-induced neuroinflammation. ACS Chem Neurosci (2023) 14(5):864–74. doi: 10.1021/acschemneuro.2c00623
154. Dai D, Yuan J, Wang Y, Xu J, Mao C, Xiao Y. Peli1 controls the survival of dopaminergic neurons through modulating microglia-mediated neuroinflammation. Sci Rep (2019) 9(1):8034. doi: 10.1038/s41598-019-44573-w
155. Lim R, Barker G, Lappas M. Pellino 1 is a novel regulator of tnf and tlr signalling in human myometrial and amnion cells. J Reprod Immunol (2018) 127:24–35. doi: 10.1016/j.jri.2018.04.003
156. Zhang Q, Zhao J, Ni M, Shen Q, Zhou W, Liu Z. Vitamin D(3) reverses the transcriptional profile of offspring cd4(+) T lymphocytes exposed to intrauterine inflammation. J Steroid Biochem Mol Biol (2022) 221:106120. doi: 10.1016/j.jsbmb.2022.106120
157. Li X, Xie J, Wang Q, Cai H, Xie C, Fu X. Mir-21 and pellino-1 expression profiling in autoimmune premature ovarian insufficiency. J Immunol Res (2020) 2020:3582648. doi: 10.1155/2020/3582648
158. Kim S, Lee SY, Bae S, Lee JK, Hwang K, Go H, et al. Pellino1 promotes chronic inflammatory skin disease via keratinocyte hyperproliferation and induction of the T helper 17 response. Exp Mol Med (2020) 52(9):1537–49. doi: 10.1038/s12276-020-00489-4
159. Wang Y, Yuan J, Dai D, Liu J, Xu J, Miao X, et al. Poly ic pretreatment suppresses B cell-mediated lupus-like autoimmunity through induction of peli1. Acta Biochim Biophys Sin (Shanghai) (2018) 50(9):862–8. doi: 10.1093/abbs/gmy082
160. Ramamoorthy S, Cidlowski JA. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev (2013) 24:41–56. doi: 10.1159/000342502
161. Yan J, Li S, Li S. The role of the liver in sepsis. Int Rev Immunol (2014) 33(6):498–510. doi: 10.3109/08830185.2014.889129
162. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest (2016) 126(1):23–31. doi: 10.1172/jci82224
163. Murphy M, Xiong Y, Pattabiraman G, Qiu F, Medvedev AE. Pellino-1 positively regulates toll-like receptor (Tlr) 2 and tlr4 signaling and is suppressed upon induction of endotoxin tolerance. J Biol Chem (2015) 290(31):19218–32. doi: 10.1074/jbc.M115.640128
164. Békés M, Langley DR, Crews CM. Protac targeted protein degraders: the past is prologue. Nat Rev Drug Discov (2022) 21(3):181–200. doi: 10.1038/s41573-021-00371-6
165. Bianco G, Goodsell DS, Forli S. Selective and effective: current progress in computational structure-based drug discovery of targeted covalent inhibitors. Trends Pharmacol Sci (2020) 41(12):1038–49. doi: 10.1016/j.tips.2020.10.005
166. Rana S, Mallareddy JR, Singh S, Boghean L, Natarajan A. Inhibitors, protacs and molecular glues as diverse therapeutic modalities to target cyclin-dependent kinase. Cancers (Basel) (2021) 13(21):5506. doi: 10.3390/cancers13215506
167. Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S, et al. Homo-protacs: bivalent small-molecule dimerizers of the vhl E3 ubiquitin ligase to induce self-degradation. Nat Commun (2017) 8(1):830. doi: 10.1038/s41467-017-00954-1
168. Ren C, Sun N, Kong Y, Qu X, Liu H, Zhong H, et al. Structure-based discovery of siais001 as an oral bioavailability alk degrader constructed from alectinib. Eur J Med Chem (2021) 217:113335. doi: 10.1016/j.ejmech.2021.113335
Keywords: Peli1, ubiquitination, IL-1R/TLRs, NF-κB, inflammation, cancer, immune
Citation: Yan L, Cui Y and Feng J (2023) Biology of Pellino1: a potential therapeutic target for inflammation in diseases and cancers. Front. Immunol. 14:1292022. doi: 10.3389/fimmu.2023.1292022
Received: 11 September 2023; Accepted: 04 December 2023;
Published: 18 December 2023.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Ran Cheng, Baylor College of Medicine, United StatesCopyright © 2023 Yan, Cui and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Feng, anVhbmZlbmdAY211LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.