 Cheng Qian
Cheng Qian Chaoqun Liu1,2†
Chaoqun Liu1,2† Weiwei Liu
Weiwei Liu Rui Zhou
Rui Zhou Liang Zhao
Liang Zhao- 1Department of Pathology, Nanfang Hospital, Southern Medical University, Guangzhou, China
- 2Department of Pathology & Guangdong Province Key Laboratory of Molecular Tumor Pathology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China
Blood vessels are a key target for cancer therapy. Compared with the healthy vasculature, tumor blood vessels are extremely immature, highly permeable, and deficient in pericytes. The aberrantly vascularized tumor microenvironment is characterized by hypoxia, low pH, high interstitial pressure, and immunosuppression. The efficacy of chemotherapy, radiotherapy, and immunotherapy is affected by abnormal blood vessels. Some anti-angiogenic drugs show vascular normalization effects in addition to targeting angiogenesis. Reversing the abnormal state of blood vessels creates a normal microenvironment, essential for various cancer treatments, specifically immunotherapy. In addition, immune cells and molecules are involved in the regulation of angiogenesis. Therefore, combining vascular normalization with immunotherapy may increase the efficacy of immunotherapy and reduce the risk of adverse reactions. In this review, we discussed the structure, function, and formation of abnormal vessels. In addition, we elaborated on the role of the immunosuppressive microenvironment in the formation of abnormal vessels. Finally, we described the clinical challenges associated with the combination of immunotherapy with vascular normalization, and highlighted future research directions in this therapeutic area.
1 Introduction
Angiogenesis is an important hallmark of malignant tumors (1). Tumor angiogenesis refers to the process of neovascularization during tumor growth. It is induced by high concentrations of pro-angiogenic factors secreted by tumor cells to obtain an adequate supply of blood and nutrients. The concept of vascular normalization—proposed by Rakesh K. Jain in 2001 (2)—refers to the transformation of tumor blood vessels and their microenvironment from an abnormal structural and functional state to a normal one. Consequently, tumor cells are freed from hypoxia and the tumor microenvironment is reprogrammed.
Tumor blood vessels are different in structure and function from the normal tissues. The abnormal blood vessels of tumors are dilated, tortuous, disordered, immature, and impermeable and lack connections between parietal cells (3). The production of numerous abnormal blood vessels has deleterious effects that promote tumor development. Anti-angiogenic therapy deprives the tumor of oxygen and nutrients to induce necrosis. However, clinical use of anti-angiogenic drugs has led to unexpected outcomes, such as chemotherapy resistance and increased propensity to metastasis. This may be attributed to the structural changes in immature newly developed vasculature and its resulting dysfunction after the destruction of a large number of blood vessels by anti-angiogenic therapy (2, 4–6). Therefore, the focus of vascular research in oncology has shifted from understanding only anti-angiogenesis to normalizing tumor blood vessels. A window of vascular normalization can be achieved transiently during the initial days of anti-angiogenesis therapy. Normalized blood vessels can reverse drug resistance and immune suppression in patients with cancer (5). Therefore, the perfect drug dosage, exposure time, and usage range to attain the vascular normalization window are currently being explored to develop effective cancer therapeutic strategies.
A comprehensive analysis of the tumor microenvironment (TME) has revealed that tumor blood vessels cannot be understood from a single perspective. They have heterogeneous structure, function, and organization, and immune cells, cytokines, and extracellular matrix (ECM) interact with them (7, 8).
Immunotherapy has revolutionized the treatment of tumors by improving immune function in the TME. Immunotherapies, such as the chimeric antigen receptor (CAR)-T-cell therapy, enhance the function of the effector T cells. CARs are designed to create T-cell clones that can specifically recognize and clear tumors containing specific antigens or mutated proteins. Other approaches, such as immune checkpoint inhibitor therapies, relieve the immune suppression of T cells (9, 10). These inhibitors are designed to target inhibitory ligand–receptor interactions between T cells and other cells in the TME. Representative examples include monoclonal antibodies targeting cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and antibodies blocking programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1). However, all patients do not respond well to immunotherapy, and the overall clinical response rate of tumor immunotherapy needs to be improved (10). The TME is characterized by hypoxia, low pH, and high interstitial fluid pressure. Blood vessels are an important component of the TME and substantially contribute to the formation of abnormal TME, ultimately affecting the response of patients to immunotherapy (7).
Normal blood vessels help immune cells adhere to and penetrate the tumors. Recently, some authors have suggested that improving immune function can promote vascular normalization (11, 12). Therefore, combining vascular normalization with immunotherapy may be a feasible approach to improve treatment outcomes. Although several authors have evaluated the combination of vascular normalization and immunotherapy in clinical studies, the dosage and timing of anti-angiogenic drugs and immunotherapy are still controversial. Several clinically relevant biomarkers are available to quantify the extent of vascular normalization. However, the mechanism underlying the vascular normalization-mediated cancer therapies needs to be further studied.
In this review, we focused on the interplay between vascular normalization and immunotherapy from the perspective of the TME and summarized the clinical effects of combining them. In addition, we elaborated on the adverse consequences of abnormal blood vessels on tumor treatment and approaches that can normalize tumor blood vessels. Moreover, combining immunotherapy with drugs for vascular normalization, the problems caused by tumor heterogeneity, and strategies to overcome these obstacles were also discussed.
2 Normalization of tumor vasculature
Tumor blood vessels are structurally and functionally different from those in the normal tissues. They are dilated, tortuous, disordered, immature, and impermeable and lack connections between parietal cells (3). These structural abnormalities lead to hypoxia and hypoperfusion in the TME.
2.1 Abnormal structure and function of tumor blood vessels
Pericytes and endothelial cells (ECs) are proximal in the normal tissues and communicate through intercellular contacts and secreting specific molecules. This communication results in an orderly arrangement of ECs and the formation of an effective, organized, and mature vascular network (13). However, the connections among perivascular cells, basement membranes, and ECs are impaired in tumors. Tumor-associated pericytes show abnormal morphology and are low in density compared with normal tissue pericytes.The weakening of these connections renders the vessels impermeable. Consequently, blood leaks into the stroma, causing a hypoperfusion in the tumor tissues. In addition, the increased interstitial pressure due to leakage, the compression of the blood vessels by tumors, and the dysfunction of the lymphatic network aggravate the hypoperfusion in the tumor tissues.
2.2 Tumor angiogenesis
Angiogenesis refers to the formation of new blood vessel branches in an existing blood vessel network. The two phenotypes of endothelial cells, tip and stalk cells, are involved in the initial formation of blood vessels. Tip cells participate in angiogenic sprout formation, and endothelial cells contribute to lumen formation through a pinocytotic process. Finally, new vessels undergo maturation through pericyte recruitment, basement membrane formation, and stronger endothelial cell connections. Several unique neovascularization patterns exist in the tumor tissues, including vascular co-option and vascular mimicry. Vascular co-option is a non-angiogenic process in which tumor cells use pre-existing blood vessels to support tumor growth, survival, and metastasis. This process is independent of vascular endothelial growth factor (VEGF) and occurs in the absence of typical angiogenic processes. Notably, VEGF inhibitors do not the inhibit progression and invasion of such tumors. Vasculogenic mimicry is the formation of a “vessel-like” structure without endothelial cells. This structure is characterized by a fluid-conducting tube with a lumen that is capable of providing oxygen and nutrients and eliminating cellular waste (3, 14). Overall, abnormal tumor vascular function and architecture lead to malignant outcomes.
2.3 Molecular mechanisms of abnormal tumor angiogenesis
The physiologic balance between anti-angiogenic and pro-angiogenic factors is disrupted in the tumor tissues. High concentrations of pro-angiogenic factors, such as VEGF, angiopoietin 2 (ANG2), and platelet-derived growth factor (PDGF), are present in the TME (15) of various cancers, such as non-small cell lung cancer, gastric cancer, colorectal cancer, and glioma (16–20). In addition, hypoxia is responsible for tumor angiogenesis. Prolyl hydroxylase domain protein 2 is inactive under hypoxic conditions and cannot use oxygen to degrade hypoxia-inducible factor (HIF). Notably, HIF is ubiquitinated by the Von Hippel–Lindau complex and degraded by proteasomes. Hypoxic conditions inhibit HIF-α degradation, leading to the dimerization of accumulated HIF-α and HIF-β. These dimers bind to the hypoxia response elements to activate the target genes of HIF. Finally, pro-angiogenic factors, such as VEGFA, transforming growth factor (TGF)-β, and PDGF-B are released (21–23). These factors increase vascular permeability and promote endothelial cell proliferation, sprouting, migration, adhesion, and tube formation. Several authors have determined the downstream signaling of VEGF receptor (VEGFR) after the binding of VEGF. Some known downstream pathways of VEGF include the Ras/MAPK pathway regulating cell proliferation and gene expression, the FAK/paxillin pathway involved in cytoskeleton rearrangement, the PI3K/AKT pathway regulating cell survival, and the PLC-γ pathway controlling vascular permeability (19, 24). In addition, the Hippo pathway plays a role in vascular cell migration and VEGFR-induced angiogenesis. The activation of VEGFR by VEGF triggers the PI3K/MAPK signaling, which subsequently inhibits LATS (a key component of the pathway) and then activates the Hippo effectors, including Yes-associated protein and transcriptional co-activator with PDZ-binding motif (25–27).
Early growth response-1 (EGR-1) is an important upstream transcription factor that regulates cell proliferation and differentiation and plays an essential role in tumor angiogenesis (28, 29). EGR-1 is a common intermediate of VEGF and fibroblast growth factor (FGF) in regulating cell function (30). Yu, J. et al. revealed a specific mechanism by which EGR-1 leads to tumor angiogenesis. The capacity to promote angiogenesis was attenuated in the nuclear PD-L1-deficient cells in vivo and in vitro. Mechanistically, nuclear PD-L1 facilitated the binding of p-STAT3 to the EGR-1 promoter, resulting in the activation of EGR-1-mediated angiogenesis. Moreover, a new anti-angiogenic strategy has been developed to block the nuclear translocation of PD-L1 by inhibiting histone deacetylase 2 and restoring PD-L1 acetylation levels (31).
3 Consequences of abnormal tumor vasculature
Abnormal tumor vasculature can promote tumor metastasis and affect the prognosis in patients (32). In addition, abnormal tumor angiogenesis decreases the efficacy of antitumor therapies, including radiotherapy, chemotherapy, and immunotherapy (3).
3.1 Immunosuppression
Antitumor immunity is a sequential process. Chen and Mellman proposed the “cancer immune cycle” and summarized it into seven stages (33). First, dendritic cells (DCs) capture the tumor antigen and present the captured antigen to T cells, consequently initiating effector T-cell responses. Finally, activated effector T cells infiltrate the tumor bed, recognize tumor antigens, bind to tumor cells, and kill their target cells. The killed tumor cells release additional tumor-associated antigens that increase the intensity of the immune response. The abnormal formation of tumor blood vessels affects tumor immunity at various stages through several physical and biochemical mechanisms (Figure 1).
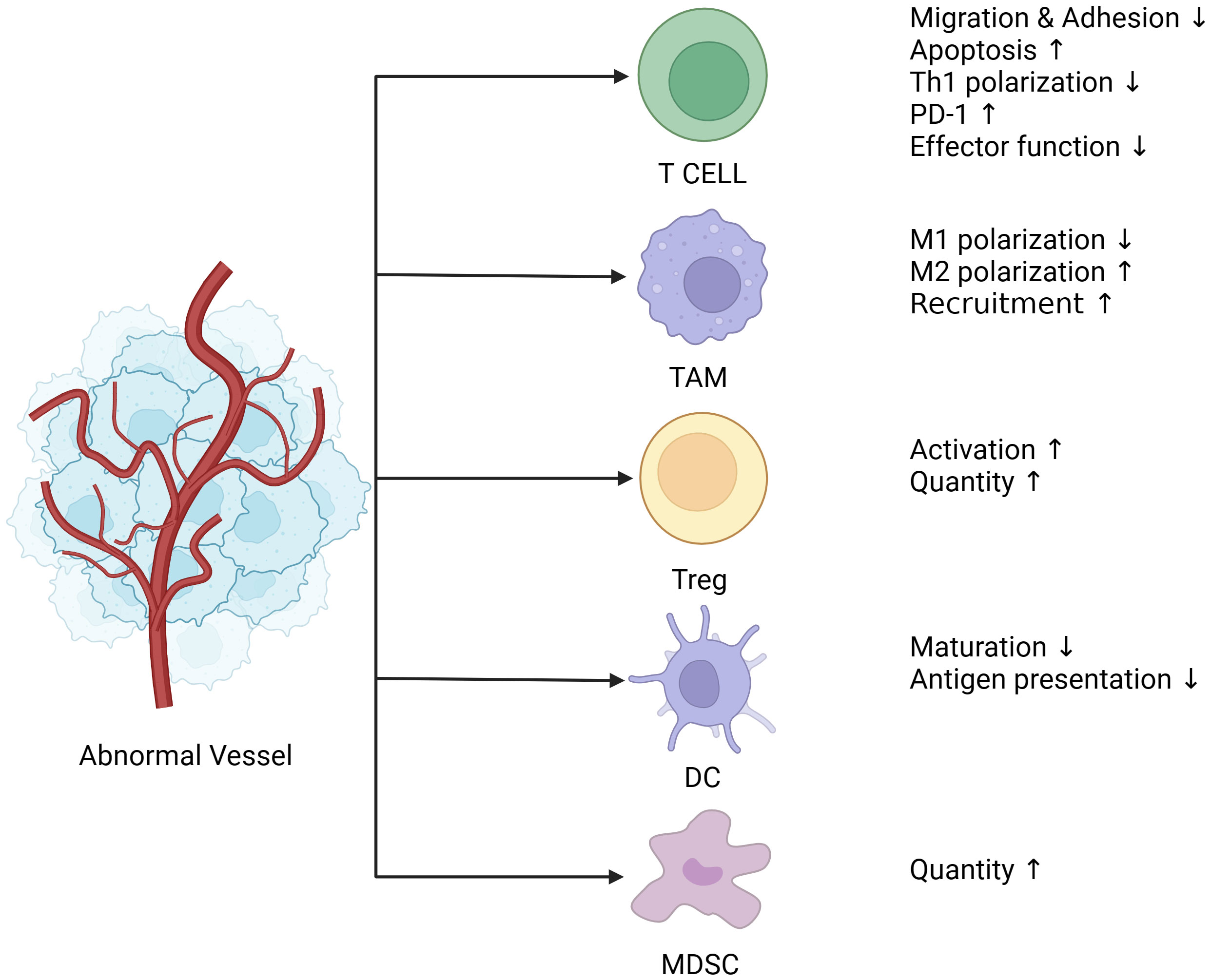
Figure 1 Abnormal blood vessels lead to immunosuppression. Tumor blood vessels are characterized by uneven distribution, tortuosity, clutter, hypertonicity, lack of pericyte coverage, and their ability to deliver oxygen and nutrients is also compromised, followed by a tumor microenvironment of hypoxia, low Ph, and high interstitial pressure. Hypoxia leads to the release of angiogenic factors such as VEGF and ANG (Angiogenin), which together with endothelial cells create an immunosuppressive microenvironment. Endothelial cells are less responsive to inflammatory stimuli, and the expression of adhesion molecules is reduced. The adhesion and migration function of immune cells such as T cells is decreased, which directly contributes to the reduction of T cell infiltration in tumors. In addition, PD-1(Programmed death-1) expression on the surface of T cells was up-regulated, and activation of T cells and polarization of CD4+T cells to Th1 cells were suppressed. Moreover, TAM is polarized to M2 type under the action of VEGF, which is an immunosuppressive phenotype and one of the major contributors to the immunosuppressive microenvironment. The release of VEGF also recruits a large number of MDSC, Treg cells, both of which are immunosuppressive cells, and Treg cells have proliferation and functional advantages under hypoxia and low Ph conditions. Finally, DCs (Dendritic cells), most of which are immature DCs infiltrated in tumors under hypoxia, have impaired presentation function. (The figure was designed in Biorender.com).
3.1.1 Effects on trafficking and infiltration of immune effector cells
The immune landscape within the TME is of three major types, namely immune infiltration, immune exclusion, and immune silent (34, 35). The “immune infiltrated” tumors are the tumors containing abundant infiltrated immune cells, such as cytotoxic T lymphoctyes (CTLs), which induce an active immune response. “Immune excluded” tumors have T cells only at their periphery, which do not infiltrate the TME. “Immune silent” tumors are the ones with little or no immune infiltration. Immune cells need to adhere and penetrate to reach the tumor tissues, and a normal vascular network is essential for this process (35). Tumor vascular tissue is structurally and functionally different from the normal vascular tissue. The heterogeneous vascular network results in hypoperfusion within tumors that have few immune cells to allocate. Moreover, immature tumor vascular tissue is prone to leakage due to insufficient connectivity between endothelial cells and the insufficient encapsulation of basement membranes and pericytes (36). Consequently, a large amount of fluid enters the interstitial space from blood vessels. Consequently, a high-pressure state is created in the interstitium, prohibits the oxygen and nutrients from entering the tumor tissues (37), resulting in a hypoxic environment. The hypoxic microenvironment attracts and sequesters tumor-associated myeloid cells and CTLs (cytotoxic T lymphocytes), resulting in the accumulation of tumor-associated myeloid cells in the hypoxic region. These cells are reprogrammed to an immunosuppressive state, limiting the efficacy of immunotherapy (38). In addition, the hypoxic environment induces the production of pro-angiogenic factors, such as VEGF, which aggravate vascular abnormality. Dorsal meningeal lymphatic vessels (MLVs) undergo extensive remodeling in mice with intracranial gliomas or metastatic melanomas. Disruption of dorsal MLVs impaired intratumor fluid drainage, disseminated brain tumor cells to deep cervical lymph nodes (dCLNs), decreased the DC trafficking from intracranial tumor tissues to dCLNs, and weakened the effect of anti-PD-1/CTLA-4 checkpoint therapy (39).
The anergy of ECs has been proposed as one of the reasons for the immunosuppression caused by abnormal blood vessels. The EC anergy results in a reduced leukocyte–vessel wall interaction by stimulating the expression of immunosuppressive molecules in abnormal tumor vessels (40). Leukocytes do not adhere to ECs without inflammatory stimulus. AP1 and NF-κB signaling activates the expression of endothelial adhesion molecules, including E-selectin, intercellular cell adhesion molecule (ICAM-1), and vascular cell adhesion molecule-1 in the presence of inflammation, which allows leukocytes to migrate across the endothelium into the extravascular space (41, 42). ECs in abnormal blood vessels, stimulated by various pro-angiogenic factors secreted by tumor tissues, exhibit a state of non-responsiveness to inflammatory stimuli. These non-responsive ECs fail to enhance the expression of endothelial adhesion molecules to stimulate the adhesion and penetration of immune cells (43, 44). Tumor ECs express immunosuppressive molecules, such as carbohydrate-binding protein galectin 1, FAS-L, TIM-3, PD-L1, and indoleamine-2,3-dioxygenase, to create an immunosuppressive TME by inducing apoptosis of activated T cells, inhibiting the activation and polarization of CD4+ Th cells to Th1 cells, promoting the activation of Treg cells, and enhancing the immunosuppressive effect of CD8+ T cells (45–48). Interestingly, tumor endothelial anergy can be overcome by sunitinib or bevacizumab treatment in renal cell carcinoma (RCC). In addition, a correlation between increased immune cell infiltration and ICAM-1 expression was observed after VEGF-targeted therapy (49).
3.1.2 Role of angiogenic factors in the TME
Angiogenic factors not only create an abnormal tumor vascular network but also contribute to the immunosuppressed TME. The angiogenic programming in tumor tissues is a multidimensional process that regulates cancer cells, tumor-associated stromal cells, and bioactive products, including cytokines and growth factors, ECM, and secreted microvesicles (50).
Abnormal tumor blood vessels often create a hypoxic TME, which affects tumor immunity by inducing the overexpression of angiogenic factors. Several major angiogenic factors play important roles in immunosuppression.
VEGF The VEGF family of proteins comprises VEGFA, VEGFB, VEGFC, VEGFD, and VEGFE (encoded by viruses) and the pro-angiogenic molecule placental growth factor (PGF/PlGF) (24). VEGF has an immunosuppressive effect. Mice exposed to pathologic VEGF concentrations showed severe thymic atrophy, characterized by a significant reduction in CD4+/CD8+ thymocytes. Specifically, VEGF, at pathologic concentrations, interfered with the development of early hematopoietic progenitor T cells to block their differentiation and migration, leading to tumor-related immunodeficiency (51). Deng, H. et al. analyzed the Kaplan–Meier Plotter database and found that high expression of VEGFA in patients with progressive hepatocellular carcinoma (HCC) is associated with a poor prognosis (52). VEGF can promote PD-1 expression on CD8+ T cells. However, both the number and function of CD8+ T cells are significantly increased after anti-VEGF therapy (53). VEGFA acts on VEGFR2 and increases the expression levels of TIM-3, CTLA-4, and Lag-3 on CD8+ T cells in a dose-dependent manner, and the expression levels of these molecules correlates with the level of T-cell exhaustion (54). Moreover, VEGF recruits numerous immunosuppressive cells, such as Tregs, myeloid-derived suppressor cells (MDSCs), and M2 macrophages, and inhibits the maturation of DCs. VEGF can promote the polarization of macrophages into M2 immunosuppressive subtype. Moreover, VEGF-induced hypoxia and a low pH microenvironment cause immunosuppression (55–57). VEGF indirectly decreases tumor necrosis factor (TNF)-α-induced lymphocyte adhesion and the expression of several inflammation-related genes, including cytokine–cytokine receptor interaction-related genes (CXCL10, CXCL11, CSF2, and FLT4) and the p38 MAPK pathway-related genes (DUSP4, IL1R1, and MEF2C) (58). In addition, exposure of ECs to pro-angiogenic factors induces a state of anergy, in which they lose the ability to respond to inflammatory cytokines and upregulate the expression of endothelial adhesion molecules (59).
TGF-β plays a dual role in tumor development. TGF-β, as a tumor suppressor, inhibits cell growth and induces apoptosis of precancerous cells. However, tumor-derived TGF-β induces tumorigenic and prometastatic responses including the formation of an immunosuppressive TME in cancer cell clones with inactivated TGF-β pathway (60). TGF-β plays a role as a pro-angiogenic factor in various diseases, including osteoarthritis, and cancer (61, 62). Leucine-rich alpha-2-glycoprotein 1 had a mitogenic effect on ECs and promoted angiogenesis in the presence of TGF-β1 (63). In addition, TGF-β stimulates the synthesis of PDGF-B by ECs and promotes the synthesis of VEGF by non-ECs during the healing phase of immune injury, suggesting that it upregulates the expression of other cytokines to promote angiogenesis (64). TGF-β is a major component of tumor-derived small extracellular vesicles in cancer patients. These vesicles stimulated macrophage chemotaxis and reprogrammed primary human macrophages to a pro-angiogenic phenotype characterized by the upregulation of pro-angiogenic factors and functions (65). TGF-β can significantly inhibit CTL activity (66) and also promote tumor growth independent of CD4+ T cells, interferon (IFN)-γ, and CTLs. Targeting the TGF-β pathway in CD4+ T cells may inhibit tumor growth by remodeling and normalizing the tumor vascular network. Further study revealed that Interleukin (IL)-4 secreted by Th2 cells plays a vital role in reprogramming the TME. Therefore, TGF-β not only inhibits the function of CTLs but also influences the type II immunity against cancer (67). Moreover, TGF-β influences the activation of macrophages. For example, phosphatidylinositol-binding protein TIPE1 can promote alternative macrophage activation and tumor progression through the PIP3/Akt/TGF-β axis (68).
ANG2 is involved in angiogenesis at the early stages and is linked to tumor immunosuppression. ANG2 was upregulated in both humans and mice in a study of liver metastases from pancreatic neuroendocrine tumors. This observation coincided with poor T-cell infiltration, suggesting an immunosuppressive TME (69). In addition, TIE2-expressing monocytes/macrophages have a tendency to polarize toward the M2 phenotype in the presence of ANG2 in a mouse model, playing an important role in the emergence of immunosuppressive microenvironment (70). ANG2 restricts the antitumor function of monocytes by inhibiting the secretion of TNF-α (71). ANG1 stimulates the binding of pericytes and vascular smooth muscle cells to ECs, thereby stabilizing newly formed blood vessels (72). However, the role of ANG1 in tumor development is controversial. ANG1 promoted colorectal tumor metastasis and growth. In addition, it upregulated carboxypeptidase A4 to promote tumor cell proliferation in triple-negative breast cancer (73, 74). Therefore, further research is required to explore the role of ANG in cancer.
PDGF-BB modulates tumor angiogenesis by inducing erythropoietin production in stromal cells (75). It can regulate various immune cells to shape the immunosuppressive TME. PDGF-BB mediated the infiltration of M2-phenotype tumor-associated macrophages (TAM) into tumor tissue by inducing pericyte- and fibroblast-derived IL-33 in a mouse tumor xenograft model (76). This growth factor enhances IL-4-induced STAT6 activation, which promotes tumor growth through the expansion of MDSCs and inhibition of cytotoxic T-cell response (77). MDSCs expressing PDGF-BB are recruited by CXCL17 in breast cancer cells and facilitate lung metastasis (78). Interestingly, the concentration of PDGF-BB can help predict the fraction of monocytic MDSCs in the peripheral blood of patients with colorectal cancer (CRC) (79). Therefore, PDGF-BB has a diagnostic value in clinical applications.
3.2 Resistance to cancer therapy
Abnormal tumor vasculature can induce resistance to various cancer treatments, which can adversely affect the prognosis of cancer patients (4). Drug resistance due to abnormal vascular networks has three main aspects. First, the low coverage rate of pericytes and the destruction of blood–tumor barrier aggravates hypoxia in the TME and leads to the accumulation of chemotherapeutic drugs in the tumor stroma (80). Pericytes promoted DNA damage repair in glioblastoma cells residing in perivascular niches, ultimately inducing temozolomide chemoresistance. The large amount of CCL5 secreted by pericytes can bind to CCR5 on glioblastoma cells to activate the DNA-dependent protein kinase catalytic subunit-mediated DNA damage repair after temozolomide treatment (81). Moreover, abnormal vascular networks with pericyte proliferation can physically affect the delivery of antitumor drugs (82). Second, tumor vasculature contributes to drug resistance. Chemotherapy-induced IL-8 secretion in tumor tissues increases the expression of ATP-binding cassette subfamily B member 1 transporter on tumor ECs, which counteracts the therapeutic effects of taxol (83). Alteration in vascular morphogenesis is a hallmark of anti-angiogenesis-resistant tumor vessels. EphB4 overexpression leads to vascular resistance by altering vascular morphogenesis and pericyte coverage in experimental SF126 glioma models (84). Tumor vascularization can occur through vascular co-option rather than angiogenesis. This type of tumor vascularization contributes to the resistance to bevacizumab therapy (85). The occurrence of vascular co-option is related to the increased expression of fibroblast activation protein-α in co-opted hepatic stellate cells in the bevacizumab-resistant CRC liver metastasis xenograft model (86). Finally, the ECM changes, such as excessive fibrosis, can reduce the perfusion of tumor tissue and the efficiency of drug delivery by constricting the tumor blood vessels. The placental growth factor (PIGF) contributes to fibrosis and tissue stiffness in HCC, leading to the development of drug resistance (87). The heterogeneous distribution of abnormal blood vessels is a common feature of solid TME and a major cause of tumor hypoxia. Radiotherapy relies on the action of reactive oxygen species to kill cancer cells, and hypoxia restricts the efficacy of radiotherapy and induces resistance, leading to poor clinical outcomes (88).
3.3 Tumor metastasis
Metastasis is a major issue in current cancer treatment, and the majority of cancer patients die from metastatic disease than from primary tumors. Tumor cells penetrate the basement membrane and invade deeper tissue layers during metastasis. This is followed by intravasation into proximal vessels or lymphatics and ultimately extravasation to distant organs by transepithelial migration and capillary rupture. Finally, tumor cells migrate along neurons or directly diffuse into adjacent spaces (89). Unlike those in normal tissue, tumor blood vessels—characterized by immaturity, lack of connection between parietal cells, and high permeability—cannot prevent the dissemination of tumor cells. Matrix metalloproteinases (MMPs) are the important regulators of the TME. They play a unique role in tumor angiogenesis and regulate vascular stability and permeability. MMPs can hydrolyze type IV collagen to increase the permeability of blood vessels and promote tumor invasion. In addition, MMPs can induce the secretion of VEGF by tumor cells to increase the permeability of blood vessels and remodel the ECM (90–93). However, the role of MMPs in cancer is complex. MMP-2 secreted by ovarian tumor cells can regulate their attachment to the peritoneum for metastasis (94). This complexity may be one of the reasons for the failure of multiple clinical trials involving small-molecule MMP inhibitors for cancer treatment (91). In addition, vasculogenic mimicry is one of the manifestations of an abnormal tumor vascular network in which highly aggressive tumor cells replace endothelial cells. This abnormal network not only complements the traditional angiogenic pattern but also plays an important role in tumor metastasis. Vasculogenic mimicry induced by the miR-124/Foxq1/EGFR axis promotes metastasis in nasopharyngeal carcinoma. Anti-EGFR treatment can inhibit the formation of this abnormal vascular network and consequently tumor growth and metastasis (95). Therefore, targeting EGFR signaling can improve the prognosis of metastatic CRC (96).
3.4 Ferroptosis
Ferroptosis is a unique mode of cell death driven by iron-dependent phospholipid peroxidation, and resistance to ferroptosis is related to tumorigenesis (97). In addition, ferroptosis is related to the formation of abnormal blood vessels in tumors. The hypoxia in the TME caused by abnormal vascular structure and function is associated with ferroptosis resistance, resulting in tumor development. A hypoxia-induced lncRNA, cystathionine beta-synthase mRNA-transgenic lncRNA, protects gastric cancer cells from ferroptosis, leading to chemoresistance (98). Aberrant expression of various angiogenic factors and receptors caused by abnormal blood vessels inhibits ferroptosis. For example, upregulation of FGFR4 confers anti-HER2 resistance by attenuating ferroptosis in breast cancer. The ANGPTL4 protein, a decisive regulator of angiogenesis, can inhibit ferroptosis to induce resistance against radiation therapy (99, 100). Angiogenic drugs, such as apatinib and sorafenib, sensitize tumor cells to ferroptosis (101, 102). However, ferroptosis has a two-sided effect on tumor progression, and the induction of ferroptosis is sufficient to convert non-suppressive polymorphonuclear-MDSCs (PMN-MDSCs) into immunosuppressive ones. Moreover, the immunosuppressive molecules released by PMN-MDSCs undergoing ferroptosis have an inhibitory effect on T cells (103). Therefore, ferroptosis can lead to tumor immunosuppression. Mouse models with conditional deletion of ferroptosis suppressor genes, such as GPX4 and SLC7A11, accelerated pancreatitis and pancreatic tumorigenesis. The products of oxidative DNA damage during ferroptosis can increase the infiltration of macrophages into pancreatic ductal adenocarcinoma (PDAC), and ferroptotic PDAC cells can release the KRASG12D protein, which promotes macrophage polarization into the M2 phenotype. Notably, inflammation-related immunosuppression due to cell death has the potential to promote tumorigenesis (104).
4 TME leads to abnormal vasculature
The TME includes tumor cells, surrounding non-tumor cells, the secretory products of corresponding cells, and other cellular and non-cellular components of the ECM, such as stromal cells, fibroblasts, and immune cells (105). The progression of the tumor indicates that the TME has the characteristics of immunosuppression. This feature is closely related to the dysfunction of the adaptive immune system, the diversity and plasticity of myeloid cells, and the remodeling of cancer-associated fibroblasts (CAFs) and ECM (106, 107). CD4+-deficient mice lack the coverage of pericytes, suggesting that the immunosuppressive TME may have a significant effect on the formation of abnormal tumor vessels (Figure 2) (108). Several researchers have proposed the concept of “immunoediting.” Cancer immunoediting has three stages, namely elimination, equilibrium, and escape. The immune system recognizes and kills tumor cells in the elimination phase. Only a few tumor subclones progress to the equilibrium phase, where tumor growth can also be limited or even eliminated by a normal immune system. However, “immunoedited” tumor cell subclones that can evade immune recognition and destruction may enter the escape phase, in which their growth is not restricted and immunosuppressive properties are apparent. The vascular abnormalities are exacerbated due to the appearance of immunosuppressive TME in the immune escape phase (109).
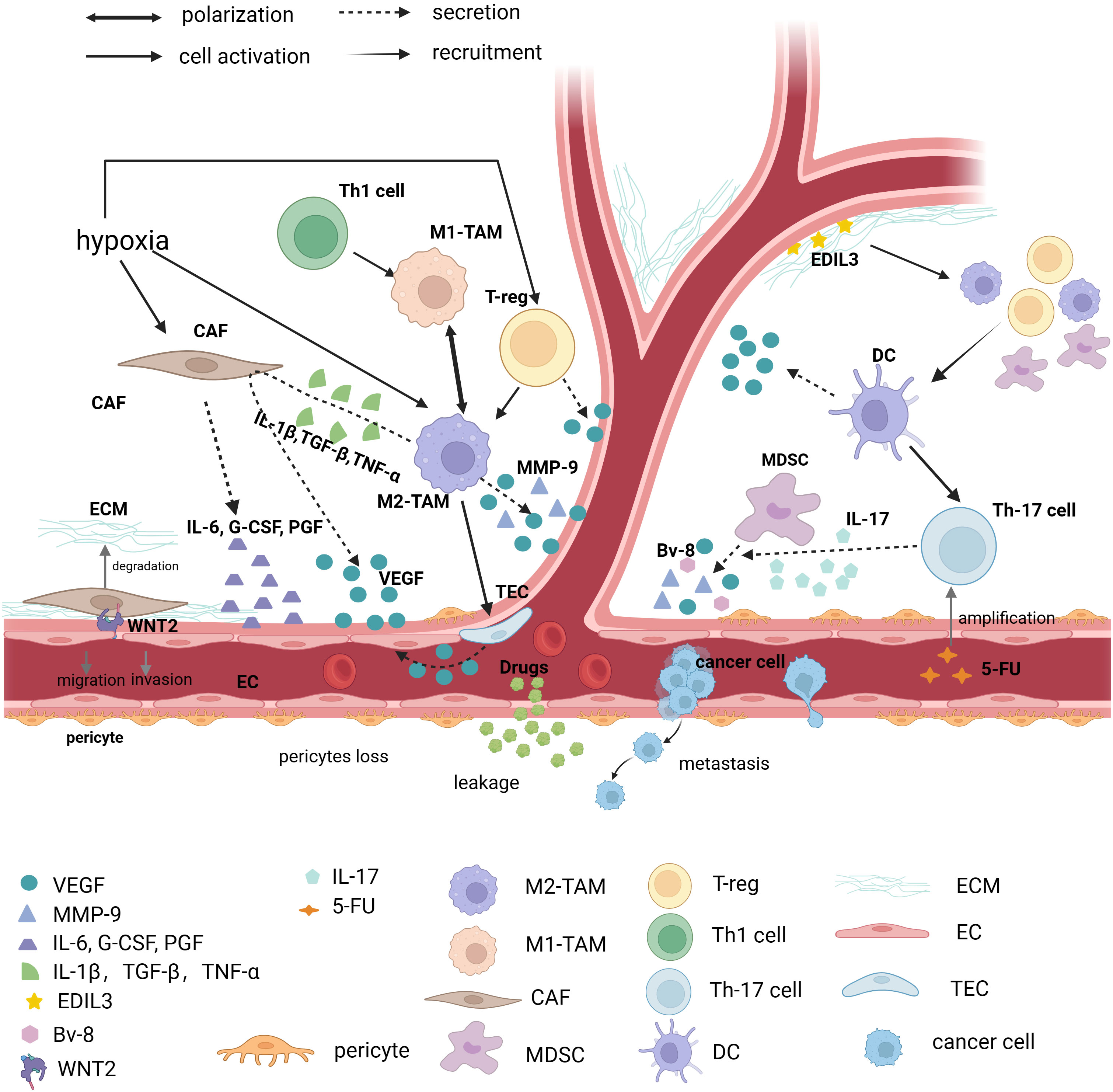
Figure 2 Immunosuppressed TME causes abnormal vessels. Tumor vessels are in an abnormal state of TME (Tumor microenvironment), and various factors that cause immunosuppression such as immune cells, CAF (Cancer associated fibroblasts), ECM (Extracellular matrix), and various immune factors contribute to tumor vessel abnormalities. TAM (Tumor-associated macrophage), and in particular TAM of the M2 phenotype, promotes angiogenesis in at least three ways. First, it directly secretes factors that cause vascular abnormalities, such as: VEGF-A (Vascular endothelial growth factor-A), EGF (Epidermal growth factor), MMP-9 (Matrix metalloproteinase-9), etc., secondly activate tumor endothelial cells to secrete VEGF for angiogenesis, and finally M2-type TAM stimulates CAF to secrete VEGF through IL-1β (Interleukin-1β), TGF-β (Transforming growth factor-β) and TNF-α (Tumor necrosis factor-α). CAF, one of the focuses of TME research, regulates angiogenesis by secreting IL-6, G-CSF (Granulocyte colony-stimulating factor) and PGF (Placental growth factor), and is involved in ECM degradation. Recent studies have found that direct contact of CAF with endothelial cells via WNT2 leads to endothelial cell migration. Other immune cells such as MDSC (Myeloid-derived suppressor cell) secrete VEGF, BV-8, and MMP-9. It is worth noting that Th17 (T helper cell 17) cells can promote MDSC progenitor function through IL-17. Moreover, it has been found that 5-FU can induce immunosuppressive microenvironment by promoting Th17 proliferation via caspase-1. The vascular regulatory function of a T cell depends on its molecular type. Th1 cells can polarize Tams towards the M1 phenotype, but Treg cells drive Tams towards the M2 phenotype and secrete VEGF to promote angiogenesis. DCs (Dendritic cells) are able to secrete VEGF directly, but it can also recruit Treg, MDSC, and M2 macrophages, which exacerbate the immunosuppressive effects of TME. In addition, as one of the important components of TME, ECM can secrete EDIL3 to promote angiogenesis. (The figure was designed in Biorender.com).
4.1 Tumor-associated macrophages
Macrophages are an important component of innate immunity. These cells not only promote inflammation and destroy pathogens but also play a significant role in tissue repair and neovascularization (110, 111). TAMs are strongly associated with an immunosuppressive TME (106). TAMs promote tumor cell proliferation and play a role in cancer progression in sustained cell cultures (112). M0 macrophages differentiate from monocytes under the influence of macrophage colony-stimulating factors. Lipopolysaccharides and Th1-derived cytokines promote the conversion of M0 to M1 macrophages, and IL-4, IL-10, IL-13, IL-33, and TGF-β induce the conversion of M0 to M2 macrophages (113). Although this simplistic classification does not describe the different states of macrophages within tumors, it is commonly assumed that M1 and M2 macrophages can inhibit and promote tumor growth, respectively (114). Macrophages secrete growth factors, such as epidermal growth factor and VEGF; therefore, abnormal blood vessels formed by macrophage activity may be a vital reason for tumor progression in breast cancer (115–117).
M2 macrophages are involved in tumor angiogenesis and invasion (118). Ultrasound-targeted microbubble destruction (UTMD) inhibits the growth and metastasis of pancreatic cancer by regulating vascular normalization. UTMD-induced vascular normalization depends on the polarization of macrophages from the M2 to M1 phenotype (119), indicating that the polarization of TAMs is critical to vascular normalization. Macrophages in injured tissues secrete cytokines, such as insulin-like growth factor-1, VEGFA, and TGF-β, which directly or indirectly promote angiogenesis (120, 121). Similarly, hypoxic TME-induced TAMs can secrete VEGFA and MMP-9 to support angiogenesis (122). In addition, TAMs can promote VEGFA expression in endothelial cells in vitro (123). Therefore, the secretion of pro-angiogenic factors in the TME is associated with TAMs. The M2 polarization of TAMs in phosphatase and tensin homolog-silenced esophageal cancer enhanced the malignant behavior of tumor-associated vascular ECs to form abnormal blood vessels through the PI3K/ATK-dependent pathway (124). TNF-α, secreted by TAMs, upregulated the expression of MALR, an lncRNA, which stabilized the HIF-1α mRNA in esophageal squamous cell carcinoma to promote angiogenesis (125).
A specific classification of TAM surface markers will help understand their role in the TME. The F4/80+CD115+C3AR+CD88+ TAMs show a high heme oxygenase-1 (a stress-responsive enzyme) activity, which plays a critical role in shaping a prometastatic TME to favor immunosuppression, angiogenesis, and epithelial-to-mesenchymal transition (126). A subset of TAMs that expressed podoplanin showed high collagenolytic and gelatinase activity, and these TAMs mediated the destruction of the vascular basement membrane to increase vascular permeability. These macrophages directly acted on blood vessels to affect EC junctions, leading to intravasation of tumor cells into blood vessels (127). Tie2-expressing macrophages functioned as a key factor in vascular remodeling after chemotherapy. However, Tie2 expression on macrophages is not effective for tumor angiogenesis. Therefore, additional research is needed to target this phenotype of macrophages for tumor treatment (128, 129).
4.2 MDSCs
MDSCs show immunosuppressive activity, and their three recognized subsets are granulocytic/PMN, monocytic, and immature/early MDSCs (130, 131). These cells mainly promote and maintain tumor angiogenesis by secreting MMPs. Mechanistically, MMP-9 promotes angiogenesis and stimulate tumor neovascularization by increasing the bioavailability of VEGF (132). Moreover, VEGF can trigger the recruitment of MDSCs. BV8, highly expressed in MDSCs, promotes angiogenesis and enhances MDSC mobilization in tumors (133). The use of JAK2/STAT3 inhibitors inhibited angiogenesis and decreased the MDSC population in the TME of head and neck carcinoma by inhibiting VEGFA and CK2 (134, 135). Notably, MDSCs isolated 24 h after lung cancer surgery were more potent in promoting angiogenesis and tumor growth than pre-surgery MDSCs, and the number of pulmonary metastatic tumors and MDSCs were positively related to the extent of surgical manipulation (136).
4.3 DCs
DCs are antigen-presenting cells that play a crucial role in bridging innate and adaptive immune responses. DCs act as professional antigen-presenting cells to process an antigen and present it to naive T lymphocytes (137). DCs are classified as conventional or classical, plasmacytoid, and monocyte-derived DCs (138). However, antigen presentation is disrupted by the large amount of immature DCs in the TME. Tumor-associated DCs produce IL-23, which can induce IL-17 secretion by Th17 cells to indirectly induce angiogenesis. The newly formed abnormal vessels promote the accumulation of MDSCs and further inhibit the maturity of DCs (139). The classical DCs produce bioactive pro-angiogenic factors, such as VEGFA, FGF2, and ET-1. In addition, DCs can express several chemokines, including CXCL8 and CCL2, which can induce angiogenesis by directly acting on ECs. In addition, CXCL1, CXCL2, CXCL3, and CXCL5 exert an indirect pro-angiogenic effect by recruiting other pro-angiogenic bone marrow cells, including neutrophils (140–142).
4.4 T lymphocytes
T lymphocytes are one of the most critical players in adaptive immunity. The effect of T cells on angiogenesis depends on their subtypes and their cytokine profiles. CD4+ T lymphocyte deficiency leads to transcriptomic alterations in tumor-associated vascular ECs, resulting in modifications in the pathways or genes known to regulate vascular normalization. Examples of these modifications include the increased expression of VEGFA, decreased expression of Angpt1/Angpt2, and the downregulation of adhesion and ECM molecules (108). CTLs and CD4+ Th1 cells can secrete IFN-γ, which restricts the proliferation of ECs and induces their apoptosis, resulting in the restriction of blood flow in the tumor (143). Interestingly, CD8+ T lymphocytes exude from blood vessels by regulating the CXCR3/CXCL10 axis, migrate to the retinal tissue, and secrete several factors, such as TNF, IFN-γ, perforin, and granzyme A/B, to promote angiogenesis (144). Moreover, T cells can also affect blood vessels by regulating TAMs in the TME. The IL-8-producing CD4+ T lymphocytes in glioma can recruit TAMs and induce their M2 polarization. Notably, blocking IL-8 transforms the M2 TAMs into anti-angiogenic macrophages (145–147). CD4+ T lymphocytes, such as Th17 cells and γδT cells, secrete IL-22, which can promote angiogenesis through the STAT3 and MAPK pathways (148, 149).
IL-17-secreting CD4+ Th17 cells directly promote angiogenesis and modulate MDSCs to promote angiogenesis independent of VEGF (150). 5-Fluorouracil, an extensively used chemotherapeutic agent, is toxic to MDSCs in the TME. However, 5-fluorouracil activates caspase-1, which could expand the Th17 population. Th17 cells can counter the effects of anti-angiogenic therapy by stimulating neovascularization (151). Hypoxic TME can induce the expression of the chemokine CCL28, leading to the accumulation of Tregs, which limit tumor immunity and promote angiogenesis. Mechanistically, miRNA21 can induce the ICOS expression on Tregs, and the crosstalk of ICOS ligands with Tregs can activate ECs to induce abnormal angiogenesis (152–154).
4.5 Cancer-associated fibroblasts
CAFs play a key role in the TME and affect the malignant progression of tumors through multiple processes, including remodeling of ECM, increasing the production of growth factors, and promoting angiogenesis (155). Bioinformatic analysis has demonstrated that CAFs can be considered prognostic markers in certain tumors. Higher stromal/CAF scores were associated with poor overall survival in patients with ovarian cancer and poor immune response to treatment (156). CAF-derived wingless-type MMTV integration site family member 2 (WNT2) increased tumor angiogenesis in colon cancer, and its overexpression increased blood vessel density and tumor volume in CRC xenografts. Unterleuthner, D. et al. analyzed publicly available datasets for human CRC and found decreased survival rates in patients diagnosed with tumors having high WNT2 expression (157). Furthermore, IL-6 mediates the crosstalk between tumor cells and CAFs by supporting tumor growth and promoting fibroblast activation. Athymic nude mice with head and neck squamous cell carcinoma patient-derived xenografts (PDX) and gastric adenocarcinoma PDX were treated with tocilizumab, an anti-IL6Rα antibody that inhibits tumor growth in vivo in part by inhibiting the STAT3 and MEK/ERK signaling. The treatment induced tumor growth arrest and reduced STAT3 and ERK1/2 signaling (158). The analysis of phosphorylated STAT3 expression in CAFs of human tissue microarrays demonstrated a negative correlation between its increased stromal expression and the survival of patients with CRC. Mechanically, STAT3 activation plays an essential role in the development of CAF-mediated CRC. Therefore, STAT3 activation increases the expression of several angiogenic signals, thereby promoting CRC progression (159). CAFs can mediate the tumor vascular abnormalities caused by hypoxia. They increase the secretion of the hypoxia-induced angiogenesis regulator NCBP2-AS2 (the uncharacterized protein renamed from hypoxia-induced angiogenesis regulator), thereby enhancing the VEGF signaling to promote endothelial sprouting (160).
4.6 Extracellular matrix
ECM is a complex and dynamic structure composed of macromolecular substances secreted by cells into the extracellular space comprising interstitial matrix and basement membrane. In addition, ECM contains various secreted proteins, including cytokines, chemokines, and growth factors, involved in immune cell regulation. Collagen is one of the main components of the ECM, and high-density collagen can guide macrophages to acquire an immunosuppressive phenotype (161). Therefore, ECM is a highly dynamic partner of the immune system (162, 163). Epidermal growth factor-like repeats and discoidin I-like domains 3 (EDIL3), an ECM protein highly expressed in HCC, contributes to angiogenesis. Moreover, autocrine EDIL3 can support tumorigenesis by promoting resistance to anoikis in HCC cells (164). Notably, VEGF induces high EDIL3 expression in malignant cells, suggesting that high VEGF in an immunosuppressive microenvironment can indirectly affect tumor blood vessels by changing the composition of ECM (165, 166).
5 Vascular normalization and immunotherapy
Solid tumors require blood vessels to grow, and many new cancer therapies target tumor blood vessels. Traditional anti-angiogenic “vessel blocking” strategies attempt to inhibit new blood vessel formation and destroy existing blood vessels to cause nutrient starvation in tumors. However, their success is limited by inadequate efficacy or the development of drug resistance. In recent years, the concept of “vascular normalization” has been applied to cancer treatment. Some anti-angiogenic agents can temporarily “normalize” the abnormal structure and function of the tumor vasculature, making it more efficient for oxygen and drug delivery. The appropriate use of drugs that induce vascular normalization can alleviate hypoxia and improve the efficacy of conventional therapies (167, 168).
The inhibition and reprogramming of the immune system play a key role in the occurrence and development of tumors. Immunotherapy aims to reactivate antitumor immune cells and overcome the immune escape mechanism in the tumors (109). The discovery of IL-2, T-cell dual signal activation pathway, CTLA-4, PD-1, and several other immune molecules led to the concept of “immune checkpoint blockade” in tumor treatment. However, all patients do not respond to these therapies, indicating the complexity of tumor-induced immune alterations. Therefore, finding a synergistic method to activate anti-tumor T cell responses and target inhibitory TME has been a major research focus in tumor immunotherapy (169). The crosstalk between vascular abnormalities and immunosuppression suggests that a combination of the two approaches would have a greater therapeutic effect.
5.1 Vascular normalization strategies
5.1.1 Targeting the VEGF/VEGFR pathway
VEGFA, VEGFB, and PIGF are primarily involved in the regulation of angiogenesis and vascular permeability. These molecules bind to VEGFR-1, which is primarily located on the surface of vascular endothelial cells. However, the primary function of VEGFA is accomplished by binding to VEGFR-2. Neuropilins-1 and -2 act as co-receptors for the VEGF ligands (53, 170). VEGFA induces the VEGFR-2 dimerization and triggers its autophosphorylation of VEGFR-2 to activate the downstream signal transduction pathways, including the PI3K, PLC-γ, Akt, Ras, and MAPK pathways, to promote cell proliferation, survival, migration, permeability, and differentiation and regulate cell adhesion molecules in addition to other functions (171, 172). VEGFs regulate EC proliferation, increase vascular permeability, and aggravate immunosuppression. Drugs targeting the VEGF/VEGFR pathway can achieve vascular normalization by inhibiting these functions. Bevacizumab (a recombinant humanized monoclonal antibody) was the first clinically approved drug, which increased the function of other cytotoxic agents through vascular normalization. Notably, vessel diameter, density, permeability, and interstitial fluid pressure decreased after the treatment (173).
Tyrosine kinase inhibitors (TKIs), such as apatinib, cedianib, and anlotinib, are being used to induce vascular normalization in tumors. TKIs inhibit the catalytic domain of receptor tyrosine kinases in a competitive or allosteric manner. Specifically, apatinib targets VEGFR-2, anlotinib targets VEGFR, PDGFR, and FGFR, and desinib targets VEGFR-1/2/3, PDGFR-α, CSF-1R, and Flt3. Notably, TKIs have more targets than macromolecular monoclonal antibodies. The combination of TKIs and cytotoxic drugs is one of the active research areas in cancer therapeutics. Wang, T. et al. conducted a randomized clinical trial based on the effect of apatinib plus pegylated liposomal doxorubicin (PLD) versus PLD alone in patients with platinum-resistant recurrent ovarian cancer. The combination treatment showed a promising efficacy with manageable toxic effects. Apatinib plus PLD group has improved median progression-free survival (5.8 months) compared with the PLD group (3.3 months). The median overall survival was 23.0 months in the apatinib plus PLD group compared with 14.4 months in the PLD group. The disease control rate was 81.5% in the apatinib plus PLD group and 53.1% in the PLD group (174). However, patients receiving combination therapy with cedeinib and cytotoxic drugs for glioblastoma showed improvements in OS only in the subgroup with increased tumor perfusion and oxygenation. The investigators suggested that this phenomenon was related to vascular normalization after combination therapy. In addition, this finding explains the failure of sildenib and bevacizumab therapy to prolong overall survival in clinical studies (175, 176). Therefore, further research is required to select appropriate TKIs for cancer treatment.
Moreover, anti-VEGF therapy could improve the delivery and efficacy of CAR-T therapy. Combination treatment with an anti-mouse VEGF antibody improved the CAR-T cell infiltration and distribution throughout the TME, delayed tumor growth, and improved the ability of CAR-T cells to penetrate and distribute throughout the TME compared with CAR-T-cell therapy alone. In addition, the combination therapy prolonged the survival of tumor-bearing mice (177). Other therapies, such as bispecific antibodies, have shown promising antitumor activity in experimental studies. Combined blockade of ANGPT2 and VEGFA with a bispecific antibody (A2V) showed superior therapeutic efficacy compared with single agents in genetically engineered and transplanted tumor models, including metastatic breast cancer, pancreatic neuroendocrine tumors, and melanoma models. Notably, this therapeutic effect is related to immune function (178).
Th1 cells in the TME play a crucial role in vascular normalization. The disruption of vascular normalization reduced the T-lymphocyte infiltration and depletion, or inactivation of CD4+ T lymphocytes reduced vascular normalization, suggesting a reciprocal regulatory cycle (108). Anti-angiogenic immune-modulating therapy affects the lymphotoxin/lymphotoxin beta-receptor (LT/LTβR) signaling pathway through CD8+ T and NK cell signaling and eventually induces the differentiation of postcapillary venules into inflamed high endothelial venules (HEVs). Tumor HEVs enhance the proliferation of TCF-1+PD-1+ lymphocytes and the production of cytotoxic PD-1+TIM-3+ lymphocytes by changing the perivascular microenvironment to promote antitumor immunity (179). Therefore, the combination of anti-VEGF/VEGFR and immunotherapy can enhance tumor immunity by multiple ways, such as relieving the inhibition of antigen presentation and inhibiting the recruitment of immunosuppressive cells.
5.1.2 Blocking other alternative targets
Angiopoietins include ANG1, ANG2, and ANG4, and their receptors include Tie1 and Tie2. ANG1 can tightly bind Tie2 at nanomolar affinity. This binding can reduce angiogenesis and vascular permeability, promote endothelial cell maturation, favor vascular stabilization, and modulate vascular normalization during anti-angiogenic therapy. ANG2 is a partially competitive antagonist of ANG1 for binding to Tie2. ANG2 prevents the phosphorylation of Tie2 by ANG1. However, when ANG1 is absent, ANG2 can replace ANG1 and binds to Tie2. The level of ANG2 increases under inflammatory and hypoxic conditions, which reduces vascular stability and enhances EC activation, angiogenesis, and remodeling (180). Therefore, neutralizing ANG2 by preventing its binding to Tie2 is the main research direction for developing drugs targeting the ANG–Tie pathway. However, multiple clinical trials of monoclonal antibodies against ANGs have been unsuccessful. A phase I clinical study on MEDI3617, a monoclonal antibody against ANG2, showed long-term grade 3 edema-related adverse events in patients, and the drug development was terminated due to its limited clinical activity. Nesvacumab was also discontinued because no significant benefit was found with reference to the comparison arm of the experimental therapy (181, 182). Moreover, resistance to classical anti-VEGF agents inevitably emerges, and the most common mechanism involves increased tumor hypoxia levels induced by anti-angiogenic therapy (183). A hypoxic environment induces the upregulation of other angiogenic factors, such as ANG2, FGFR, and EGFR (184). Ectopic expression of ANG2 reduces the beneficial effects of VEGFR-2 blockade by inhibiting vascular normalization. Therefore, simultaneous blocking of these angiogenic pathways (7, 185) will increase the efficacy of the treatment (178, 186).
FGFs are angiogenic factors that stimulate vascular cell proliferation, migration, and differentiation (187). Upregulation of FGF has been associated with tumor resistance to anti-VEGF therapy (188), which illustrates that combining FGF/FGFR inhibition with anti-VEGF therapy can lead to higher antitumor efficacy. Infigratinib, a pan-FGFR inhibitor, targets FGFR to inhibit cell proliferation, angiogenesis rescue programs, hypoxia, invasion, and metastasis (189). FGF401, another agent targeted on FGFR4, selectively improves the chemotherapy outcomes in mice bearing high FGF19-expressing HCC tumors (190). Combining anti-VEGF agents with the inhibition of the receptor kinase of VEGF, FGF, and PDGF can synergistically inhibit tumor growth and enhance response to radiation therapy (191).
PDGF/PDGFR is one of the important targets, and PDGF-B and PDGFR-β are being actively explored. However, the effect of tumor PDGF-B on vascular maturity has been controversial (192). PDGFR-β is required for the recruitment of pericytes to tumor blood vessels, and EC-derived PDGF-B is essential for the proper integration of pericytes in the vessel wall (193). PDGF-B can stabilize blood vessels; however, its high expression in tumors is inconsistent with the occurrence of numerous immature blood vessels in tumors (194, 195). The above-threshold level of PDGF-BB, such as high PDGF-B expression in metastatic breast cancer can induce pericyte loss and promote vascular leakage. On the contrary, low PDGF-B expression was found in metformin-induced vascular normalization. The blockade of PDGF-B/PDGFR-β significantly enhances vascular maturity in tumors with high PDGF-B expression, whereas an opposite effect is observed in tumors with low PDGF-B expression (194, 196). Therefore, further studies are required to define the optimal conditions and the amount of PDGF-B inhibitors for vascular normalization.
5.2 Immunotherapy interacts with vascular normalization
5.2.1 Mechanism of reciprocal inhibition
A fundamental mechanism of vascular normalization in synergy with immunotherapy is to increase the effective immune response by increasing the number of immunostimulatory cells or decreasing the immunosuppressive cells. Combined anti-VEGF and immune checkpoint blockade treatment increased M1-TAM subpopulations in HCC. The infiltration and activation of cytotoxic T cells increased, whereas the infiltration of Tregs and CCR2+ monocytes decreased after combination therapy (197). Compared with nintedanib monotherapy for lung cancer, combined therapy with αPD-L1 was more effective in promoting the infiltration of activated T cells and DCs and eliminating the immunosuppressive environment by reducing the proportion of MDSCs and TAMs (198). Combination therapy can improve the efficacy of immunotherapy by relieving high interstitial pressure, increasing the infiltration of immune cells, enhancing the response to IFN-γ, and upregulating the MHC-I expression in tumor cells (198). In addition, the endothelial ICAM-1-induced transendothelial migration of leukocytes increased the infiltration of immune cells after vascular normalization. Antigenic compounds induce the expression of endothelial ICAM-1, implying that VEGF-targeted therapy can counteract tumor endothelial cell anergy and promote the formation of inflammatory infiltrates in tumors (49). Moreover, Rac1 and its effector molecules, such as Trio, Tiam, and Rhog, may contribute to the increased trans-endothelial migration of T cells. Rac1 and Rhog are the members of the rho-GTPase family. Tiam and Trio are the guanine nucleotide exchange factors, which stimulate the release of GDP and promote GTP binding. Rac1, widely expressed in tissues, is a regulator associated with cell motility and invasion (199). Myct1, a direct target gene of ETV2, is involved in regulating the angiogenic function of ECs, and more CD8+ T cells can migrate through the Myct1-deficient EC barrier. The expression of Rac1 was increased in Myct1-deficient ECs in tumors, and inhibition of Rac1 abolished the increase in T-cell migratory phenotype (200). Finally, the formation of HEVs (the blood vessels usually found in secondary lymphoid tissues) promotes the infiltration of lymphocytes. HEV promotes the infiltration of CD8+ CTLs in solid tumors, and their presence is associated with slower tumor growth and better prognosis in patients (201). Combined anti-VEGFR2 therapy with anti-PD-L1 antibodies induced HEV in breast cancer and pancreatic neuroendocrine tumors (202).
Moreover, VEGFR-2 blockade alone increased the expression of PD-1 in tumor-infiltrating CD4+ cells in an endothelial IFN-γ dependent manner (197). This suggests that the combined therapy with PD-1 can reduce the negative effects of the anti-VEGF monotherapy. In addition, combination therapy increases the density of DCs and promotes their activation (198). This change indirectly restores the function of suppressed T cells, a goal that cannot be achieved with either drug alone. In addition, targeting peroxisome proliferator-activated receptor-gamma, a factor that activates VEGFA transcription, can avoid T-cell dysfunction caused by T-cell exhaustion (203).
Compared with anti-VEGFR-2 therapy alone, dual anti-PD-1/VEGFR-2 therapy can promote CD4+ T cell-induced vascular normalization, indicated by the increased density of microvessels covered by pericytes and the protection against increased hypoxia in HCC (197). The blocking of TGF-β1 signaling favors the proliferation and expression of adhesion molecules, such as E-selectin in ECs, leading to the densification and normalization of the vasculature within the tumors. Moreover, the co-blockade of TGF-β1/PD-1 increased the density of blood vessels covered by pericytes (204). CD4+ and CD8+ T cells effectively mediate vascular normalization in breast cancer models (108, 205). Therefore, the concepts of vascular normalization and immunotherapy are combined to avoid excessive vascular pruning caused by the use of anti-angiogenic drugs and normalize the tumor vascular network. The normalized blood vessels can stimulate the infiltration of immune cells and enhance the efficacy of immunotherapy. Simultaneously, an immune-stimulated TME can promote tumor vascular normalization; therefore, a virtuous circle is formed between vascular normalization and immunotherapy.
5.2.2 Regimens of combined therapy
Anti-angiogenic agents synergize with immune checkpoint blockade; however, the sequence, dose, and timing of administration remain to be studied. Huang, Y. et al. suggested that a normalization “window” exists, depending on the timing and dose of anti-angiogenic therapy (206). Li, Q. et al. showed that both standard and low-dose anti-angiogenic drugs had vascular normalization effects in breast cancer. However, low-dose anti-angiogenic drugs were more effective in promoting the activated immune cell infiltration and PD-1 expression compared with standard dose drugs. Clinical studies on patients with advanced triple-negative breast cancer revealed that the combination of low-dose apatinib and immune checkpoint blockade shows better efficacy and tolerance (207). Given the reciprocal mechanisms, the sequence of anti-angiogenic and immunotherapy treatments is critical. The ideal effect can be achieved by using anti-angiogenic drugs to mediate vascular normalization before immune checkpoint blockade treatment. The efficacy and mechanism of immunotherapy followed by anti-angiogenic therapy have been explored. Administration of sorafenib, an inhibitor of the pan-vascular endothelial growth factor receptor, improved outcomes in patients with HCC after initial treatment with anti-PD-1 antibody. In addition, the infiltration of immune cells and the degree of vascular normalization were improved (208). This study specified the order of drug use but failed to compare the tested order with other orders of drug use; therefore, a further expansion of this concept is needed.
5.3 Clinical trials of combination therapy
Combined therapy includes anti-angiogenic drugs, such as large-molecule monoclonal antibodies and small-molecule TKIs, for vascular normalization and immune checkpoint inhibitors for immunotherapy in clinical trials. In addition, bispecific antibodies, such as PD-L1 and VEGF dual fusion antibodies, have also been used for combination therapy. Several combination regimens have already been approved by the FDA, and a large number of therapies are being tested (Table 1).
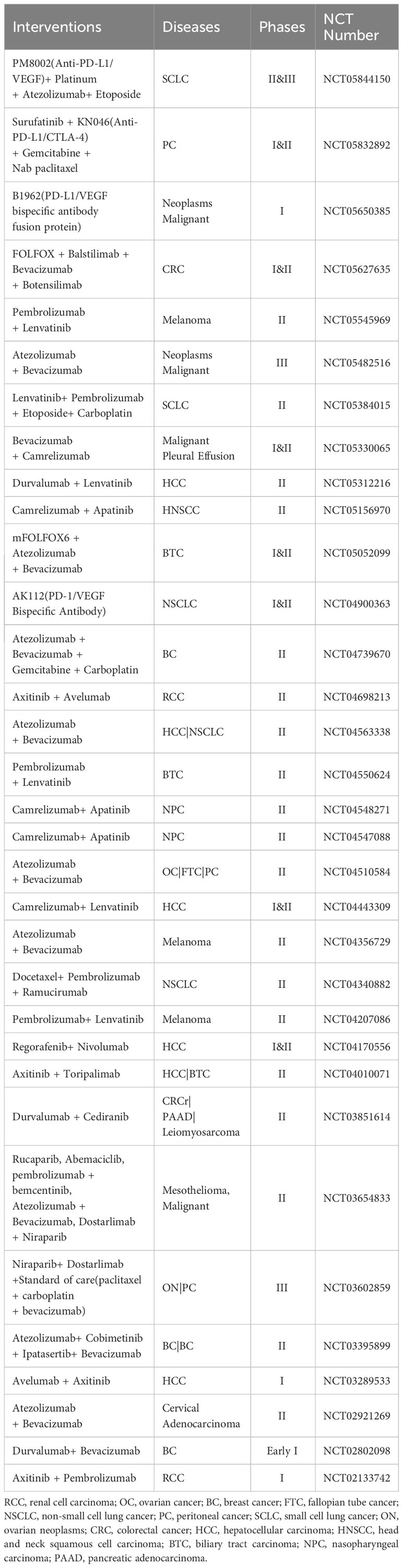
Table 1 Clinical trials based on vascular normalization and immunotherapy.
Vascular normalization has proven to be a successful therapeutic strategy in RCC. The response to chemotherapy is poor, and targeted therapy against VEGFR is used as the first-line treatment in advanced RCC (overall survival: 8–9 months) (209). Several immune checkpoint inhibitors combined with anti-angiogenic drugs have been approved for the treatment of RCC, including atixinib/pembrolizumab, axitinib/avelumab, cabozantinib/nivolumab, and lenvatinib/pembrolizumab (170). The combination of lenvatinib and pembrolizumab leads to a longer overall and progression-free survival compared with sunitinib monotherapy. Moreover, this combination shows certain advantages in progression-free survival over lenvatinib plus everolimus treatment (210). In addition, the addition of atezolizumab to bevacizumab plus chemotherapy significantly improved overall and progression-free survival in patients with metastatic nonsquamous non-small cell lung cancer, irrespective of PD-L1 expression and EGFR or ALK genetic alteration status (211). This treatment was subsequently approved for clinical use. Overall, the concept of combining vascular normalization and immunotherapy is gradually being validated in clinical practice.
6 Discussion
All the current drugs available for vascular normalization target angiogenic factors and their upstream/downstream pathways, reflecting the importance of TME in tumor therapy. Moreover, the concept of immune response in the TME has also been extended from the role of immune cells to that of other components, such as blood vessels, ECM, and CAFs. In addition, the relationship between tumor vascular normalization and tumor immunity has also been extended to the entire TME. Here, we summarized the role of CAF, ECM, and cytokines in the cycle of tumor vascular normalization and tumor immunity. The combination of anti-angiogenic drugs and immunotherapy focuses on vascular normalization and provides a new direction for cancer treatment by increasing the efficacy through reciprocal mechanisms and decreasing side effects by reducing the dosage of each drug (7). However, the availability of combination strategies is limited, and numerous combination regimens are only available for highly angiogenic tumors such as RCC. Clinical trials for combination therapy in poorly vascularized tumors, such as PDAC, after the failure of anti-VEGF and immune checkpoint blockade monotherapy are lacking (212–214). It is difficult to achieve the same efficacy for the same therapy for different types of tumors or even the same type of tumors, which denotes the heterogeneity in tumor blood vessels and tumor immunity. Circulating markers, such as serum ANG2, and the ECM molecule EMILIN-2 (a pro-angiogenic molecule) are associated with the therapeutic effect of immune checkpoint blockade (215, 216), indicating that they may be considered new markers for predicting the efficacy of immunotherapy. Changes in vascular function can be directly visualized with imaging studies, such as Doppler ultrasonography, perfusion scanning, or dynamic contrast-enhanced magnetic resonance imaging (217, 218). In conclusion, the close relationship between vascular normalization and tumor immunity determines their potential applications in cancer treatment.
Author contributions
CQ: Conceptualization, Validation, Writing – original draft. CL: Conceptualization, Validation, Writing – original draft. WL: Data curation, Visualization, Writing – original draft. RZ: Data curation, Visualization, Writing – review & editing. LZ: Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81972813, 81902946 and 82173172), the Natural Science Foundation of Guangdong Province (2023A1515011991, 2022A151511088, 2021B1515120001, 2021A1515111190, 2020A1515011389), Science and Technology Program of Guangzhou (201904010074, 202102080560, 202102080542) and Medical Scientific Research Foundation of Guangdong Province of China (B2021018) and the Beijing Xisike Clinical Oncology Research Foundation (Y-Roche2019/2-0025).
Acknowledgments
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Huang M, Lin Y, Wang C, Deng L, Chen M, Assaraf YG, et al. New insights into antiangiogenic therapy resistance in cancer: Mechanisms and therapeutic aspects. Drug Resist Update (2022) 64:100849. doi: 10.1016/j.drup.2022.100849
2. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med (2001) 7:987–9. doi: 10.1038/nm0901-987
3. Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis (2017) 20:409–26. doi: 10.1007/s10456-017-9562-9
4. Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
5. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT, et al. Angiogenesis in brain tumours. Nat Rev Neurosci (2007) 8:610–22. doi: 10.1038/nrn2175
6. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev (2011) 91:1071–121. doi: 10.1152/physrev.00038.2010
7. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol (2018) 15:325–40. doi: 10.1038/nrclinonc.2018.29
8. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol (2018) 18:195–203. doi: 10.1038/nri.2017.145
9. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin (2020) 70(2):86–104. doi: 10.3322/caac.21596
10. Dagher OK, Schwab RD, Brookens SK, Posey AD. Advances in cancer immunotherapies. Cell (2023) 186(8):1814–1814.e1. doi: 10.1016/j.cell.2023.02.039
11. Yan W, Qiu L, Yang M, Xu A, Ma M, Yuan Q, et al. CXCL10 mediates CD8+ T cells to facilitate vessel normalization and improve the efficacy of cetuximab combined with PD-1 checkpoint inhibitors in colorectal cancer. Cancer Lett (2023) 567:216263. doi: 10.1016/j.canlet.2023.216263
12. Zhang N, Yin R, Zhou P, Liu X, Fan P, Qian L, et al. DLL1 orchestrates CD8+ T cells to induce long-term vascular normalization and tumor regression. Proc Natl Acad Sci U.S.A. (2021) 118(22):e2020057118. doi: 10.1073/pnas.2020057118
13. Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol (2005) 7:452–64. doi: 10.1215/S1152851705000232
14. Rosińska S, Gavard J. Tumor vessels fuel the fire in glioblastoma. Int J Mol Sci (2021) 22(12):6514. doi: 10.3390/ijms22126514
15. Zheng W, Qian C, Tang Y, Yang C, Zhou Y, Shen P, et al. Manipulation of the crosstalk between tumor angiogenesis and immunosuppression in the tumor microenvironment: Insight into the combination therapy of anti-angiogenesis and immune checkpoint blockade. Front Immunol (2022) 13:1035323. doi: 10.3389/fimmu.2022.1035323
16. Manzat Saplacan RM, Balacescu L, Gherman C, Chira RI, Craiu A, Mircea PA, et al. The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflammation (2017) 2017:4708076. doi: 10.1155/2017/4708076
17. Akiyama T, Yasuda T, Uchihara T, Yasuda-Yoshihara N, Tan BJY, Yonemura A, et al. Stromal reprogramming through dual PDGFRα/β Blockade boosts the efficacy of anti-PD-1 immunotherapy in fibrotic tumors. Cancer Res (2023) 83:753–70. doi: 10.1158/0008-5472.CAN-22-1890
18. Zou X, Tang XY, Qu ZY, Sun ZW, Ji CF, Li YJ, et al. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int J Biol Macromol (2022) 202:539–57. doi: 10.1016/j.ijbiomac.2022.01.113
19. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer (2013) 13:871–82. doi: 10.1038/nrc3627
20. Liu N, Liu M, Fu S, Wang J, Tang H, Isah AD, et al. Ang2-targeted combination therapy for cancer treatment. Front Immunol (2022) 13:949553. doi: 10.3389/fimmu.2022.949553
21. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci (2021) 22(11):5703. doi: 10.3390/ijms22115703
22. McGettrick AF, O'Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
23. Tirpe AA, Gulei D, Ciortea SM, Crivii C, Berindan-Neagoe I. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci (2019) 20(24):6140. doi: 10.3390/ijms20246140
24. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
25. Azad T, Janse van Rensburg HJ, Lightbody ED, Neveu B, Champagne A, Ghaffari A, et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat Commun (2018) 9:1061. doi: 10.1038/s41467-018-03278-w
26. Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc Natl Acad Sci U.S.A. (2017) 114:10918–23. doi: 10.1073/pnas.1704030114
27. Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L, et al. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev Cell (2017) 42(5):462–478.e7. doi: 10.1016/j.devcel.2017.08.002
28. Fahmy RG, Dass CR, Sun L-Q, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med (2003) 9:1026–32. doi: 10.1038/nm905
29. Santiago FS, Li Y, Khachigian LM. Serine 26 in early growth response-1 is critical for endothelial proliferation, migration, and network formation. J Am Heart Assoc (2021) 10:e020521. doi: 10.1161/JAHA.120.020521
30. Punetha M, Chouhan VS, Sonwane A, Singh G, Bag S, Green JA, et al. Early growth response gene mediates in VEGF and FGF signaling as dissected by CRISPR in corpus luteum of water buffalo. Sci Rep (2020) 10:6849. doi: 10.1038/s41598-020-63804-z
31. Yu J, Zhuang A, Gu X, Hua Y, Yang L, Ge S, et al. Nuclear PD-L1 promotes EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell Discovery (2023) 9:33. doi: 10.1038/s41421-023-00521-7
32. Miranda-Galvis M, Teng Y. Targeting hypoxia-driven metabolic reprogramming to constrain tumor progression and metastasis. Int J Mol Sci (2020) 21(15):5487. doi: 10.3390/ijms21155487
33. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
34. Mellman I, Chen DS, Powles T, Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity (2023) 56:2188–205. doi: 10.1016/j.immuni.2023.09.011
35. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
36. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
37. Zanotelli MR, Reinhart-King CA. Mechanical forces in tumor angiogenesis. Adv Exp Med Biol (2018) 1092:91–112. doi: 10.1007/978-3-319-95294-9_6
38. Sattiraju A, Kang S, Giotti B, Chen Z, Marallano VJ, Brusco C, et al. Hypoxic niches attract and sequester tumor-associated macrophages and cytotoxic T cells and reprogram them for immunosuppression. Immunity (2023) 56(8):1825–1843.e6. doi: 10.1016/j.immuni.2023.06.017
39. Hu X, Deng Q, Ma L, Li Q, Chen Y, Liao Y, et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res (2020) 30:229–43. doi: 10.1038/s41422-020-0287-8
40. Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol (2021) 18:527–40. doi: 10.1038/s41571-021-00496-y
41. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi: 10.1038/nri2171
42. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15:692–704. doi: 10.1038/nri3908
43. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 88:667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
44. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human Malignancies: the role of angiogenic factors. Cancer Res (1996) 56:1111–7.
45. Anderson AC, Joller N, Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
46. Bohana-Kashtan O, Civin CI. Fas ligand as a tool for immunosuppression and generation of immune tolerance. Stem Cells (2004) 22:908–24. doi: 10.1634/stemcells.22-6-908
47. Cagnoni AJ, Giribaldi ML, Blidner AG, Cutine AM, Gatto SG, Morales RM, et al. Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8+ regulatory T cells. Proc Natl Acad Sci U.S.A. (2021) 118(21):e2102950118. doi: 10.1073/pnas.2102950118
48. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
49. Nowak-Sliwinska P, van Beijnum JR, Griffioen CJ, Huinen ZR, Sopesens NG, Schulz R, et al. Proinflammatory activity of VEGF-targeted treatment through reversal of tumor endothelial cell anergy. Angiogenesis (2022) 26(2):279–93. doi: 10.1007/s10456-022-09863-4
50. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
51. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
52. Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L, et al. Dual vascular endothelial growth factor receptor and fibroblast growth factor receptor inhibition elicits antitumor immunity and enhances programmed cell death-1 checkpoint blockade in hepatocellular carcinoma. Liver Cancer (2020) 9:338–57. doi: 10.1159/000505695
53. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol (2018) 52:117–24. doi: 10.1016/j.semcancer.2017.12.002
54. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med (2015) 212:139–48. doi: 10.1084/jem.20140559
55. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):1–12. doi: 10.1038/s41423-019-0306-1
56. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
57. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2:1096–103. doi: 10.1038/nm1096-1096
58. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J (2015) 29:227–38. doi: 10.1096/fj.14-250985
59. Huijbers EJM, Khan KA, Kerbel RS, Griffioen AW. Tumors resurrect an embryonic vascular program to escape immunity. Sci Immunol (2022) 7:eabm6388. doi: 10.1126/sciimmunol.abm6388
60. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
61. Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med (2013) 19:704–12. doi: 10.1038/nm.3143
62. Liu S, Ren J, Ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther (2021) 6:8. doi: 10.1038/s41392-020-00436-9
63. Wang X, Abraham S, McKenzie JAG, Jeffs N, Swire M, Tripathi VB, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature (2013) 499:306–11. doi: 10.1038/nature12345
64. Pintavorn P, Ballermann BJ. TGF-beta and the endothelium during immune injury. Kidney Int (1997) 51:1401–12. doi: 10.1038/ki.1997.192
65. Ludwig N, Yerneni SS, Azambuja JH, Pietrowska M, Widłak P, Hinck CS, et al. TGFβ+ small extracellular vesicles from head and neck squamous cell carcinoma cells reprogram macrophages towards a pro-angiogenic phenotype. J Extracell Vesicles (2022) 11:e12294. doi: 10.1002/jev2.12294
66. Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-β1 cytokine. Immunity (2011) 35:123–34. doi: 10.1016/j.immuni.2011.04.019
67. Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, et al. TGF-β suppresses type 2 immunity to cancer. Nature (2020) 587:115–20. doi: 10.1038/s41586-020-2836-1
68. Cheng Y, Bai F, Ren X, Sun R, Guo X, Liu W, et al. Phosphoinositide-binding protein TIPE1 promotes alternative activation of macrophages and tumor progression via PIP3/akt/TGFβ Axis. Cancer Res (2022) 82:1603–16. doi: 10.1158/0008-5472.CAN-21-0003
69. Lee E, O'Keefe S, Leong A, Park HR, Varadarajan J, Chowdhury S, et al. Angiopoietin-2 blockade suppresses growth of liver metastases from pancreatic neuroendocrine tumors by promoting T cell recruitment. J Clin Invest (2023) 133(20):e167994. doi: 10.1172/JCI167994
70. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
71. Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol (2007) 178:7405–11. doi: 10.4049/jimmunol.178.11.7405
72. Metheny-Barlow LJ, Li LY. The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res (2003) 13:309–17. doi: 10.1038/sj.cr.7290176
73. Liu X, Liang H, Fang H, Xiao J, Yang C, Zhou Z, et al. Angiopoietin-1 promotes triple-negative breast cancer cell proliferation by upregulating carboxypeptidase A4. Acta Biochim Biophys Sin (Shanghai) (2023) 55:1487–95. doi: 10.3724/abbs.2023082
74. Ibrahim NS, Lazaris A, Rada M, Petrillo SK, Huck L, Hussain S, et al. Angiopoietin1 deficiency in hepatocytes affects the growth of colorectal cancer liver metastases (CRCLM). Cancers (Basel) (2019) 12(1):35. doi: 10.3390/cancers12010035
75. Xue Y, Lim S, Yang Y, Wang Z, Jensen LD, Hedlund EM, et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med (2011) 18:100–10. doi: 10.1038/nm.2575
76. Yang Y, Andersson P, Hosaka K, Zhang Y, Cao R, Iwamoto H, et al. The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat Commun (2016) 7:11385. doi: 10.1038/ncomms11385
77. Jayakumar A, Bothwell ALM. Stat6 promotes intestinal tumorigenesis in a mouse model of adenomatous polyposis by expansion of MDSCs and inhibition of cytotoxic CD8 response. Neoplasia (2017) 19:595–605. doi: 10.1016/j.neo.2017.04.006
78. Hsu Y-L, Yen MC, Chang WA, Tsai PH, Pan YC, Liao SH, et al. CXCL17-derived CD11b+Gr-1+ myeloid-derived suppressor cells contribute to lung metastasis of breast cancer through platelet-derived growth factor-BB. Breast Cancer Res (2019) 21:23. doi: 10.1186/s13058-019-1114-3
79. Shirasuna K, Ito M, Matsuda T, Enomoto T, Ohara Y, Yamamoto M, et al. Correlation analysis of the proportion of monocytic myeloid-derived suppressor cells in colorectal cancer patients. PloS One (2020) 15:e0243643. doi: 10.1371/journal.pone.0243643
80. Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X, et al. Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell (2017) 21(5):591–603.e4. doi: 10.1016/j.stem.2017.10.002
81. Zhang X-N, Yang KD, Chen C, He ZC, Wang QH, Feng H, et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res (2021) 31:1072–87. doi: 10.1038/s41422-021-00528-3
82. Park J-S, Kim IK, Han S, Park I, Kim C, Bae J, et al. Normalization of tumor vessels by tie2 activation and ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell (2016) 30:953–67. doi: 10.1016/j.ccell.2016.10.018
83. Kikuchi H, Maishi N, Annan DA, Alam MT, Dawood RIH, Sato M, et al. Chemotherapy-induced IL8 upregulates MDR1/ABCB1 in tumor blood vessels and results in unfavorable outcome. Cancer Res (2020) 80:2996–3008. doi: 10.1158/0008-5472.CAN-19-3791
84. Uhl C, Markel M, Broggini T, Nieminen M, Kremenetskaia I, Vajkoczy P, et al. EphB4 mediates resistance to antiangiogenic therapy in experimental glioma. Angiogenesis (2018) 21:873–81. doi: 10.1007/s10456-018-9633-6
85. Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, et al. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med (2013) 2:427–36. doi: 10.1002/cam4.105
86. Qi M, Fan S, Huang M, Pan J, Li Y, Miao Q, et al. Targeting FAPα-expressing hepatic stellate cells overcomes resistance to antiangiogenics in colorectal cancer liver metastasis models. J Clin Invest (2022) 132(19):e157399. doi: 10.1172/JCI157399
87. Aoki S, Inoue K, Klein S, Halvorsen S, Chen J, Matsui A, et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut (2022) 71:185–93. doi: 10.1136/gutjnl-2020-322493
88. Sørensen BS, Horsman MR. Tumor hypoxia: impact on radiation therapy and molecular pathways. Front Oncol (2020) 10:562. doi: 10.3389/fonc.2020.00562
89. Gerstberger S, Jiang Q, Ganesh K. Metastasis. Cell (2023) 186:1564–79. doi: 10.1016/j.cell.2023.03.003
90. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
91. Shay G, Lynch CC, Fingleton B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol (2015) 44-46:200–6. doi: 10.1016/j.matbio.2015.01.019
92. Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol (2000) 10:415–33. doi: 10.1006/scbi.2000.0379
93. Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, et al. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res (2003) 63:5224–9.
94. Kenny HA, Kaur S, Coussens LM, Lengyel E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest (2008) 118:1367–79. doi: 10.1172/JCI33775
95. Luo Y, Wang J, Wang F, Liu X, Lu J, Yu X, et al. Foxq1 promotes metastasis of nasopharyngeal carcinoma by inducing vasculogenic mimicry via the EGFR signaling pathway. Cell Death Dis (2021) 12:411. doi: 10.1038/s41419-021-03674-z
96. Xie Y-H, Chen Y-X, Fang J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther (2020) 5:22. doi: 10.1038/s41392-020-0116-z
97. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
98. Yang H, Hu Y, Weng M, Liu X, Wan P, Hu Y, et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J Adv Res (2022) 37:91–106. doi: 10.1016/j.jare.2021.10.001
99. Zhang Y, Liu X, Zeng L, Zhao X, Chen Q, Pan Y, et al. Exosomal protein angiopoietin-like 4 mediated radioresistance of lung cancer by inhibiting ferroptosis under hypoxic microenvironment. Br J Cancer (2022) 127:1760–72. doi: 10.1038/s41416-022-01956-7
100. Zou Y, Zheng S, Xie X, Ye F, Hu X, Tian Z, et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat Commun (2022) 13:2672. doi: 10.1038/s41467-022-30217-7
101. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer (2013) 133:1732–42. doi: 10.1002/ijc.28159
102. Xia L, Gong M, Zou Y, Wang Z, Wu B, Zhang S, et al. Apatinib induces ferroptosis of glioma cells through modulation of the VEGFR2/nrf2 pathway. Oxid Med Cell Longev (2022) 2022:9925919. doi: 10.1155/2022/9925919
103. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature (2022) 612:338–46. doi: 10.1038/s41586-022-05443-0
104. Chen X, Kang R, Kroemer G, Tang D. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer (2021) 7:891–901. doi: 10.1016/j.trecan.2021.04.005
105. Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer (2019) 18:14. doi: 10.1186/s12943-018-0927-5
106. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
107. Hernández-Camarero P, López-Ruiz E, Marchal JA, Perán M. Cancer: a mirrored room between tumor bulk and tumor microenvironment. J Exp Clin Cancer Res (2021) 40:217. doi: 10.1186/s13046-021-02022-5
108. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544:250–4. doi: 10.1038/nature21724
109. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
110. Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol (2012) 167:195–205. doi: 10.1111/j.1365-2249.2011.04515.x
111. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
112. Rohan TE, Xue X, Lin HM, D'Alfonso TM, Ginter PS, Oktay MH, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J Natl Cancer Inst (2014) 106(8):dju136. doi: 10.1093/jnci/dju136
113. Gharib SA, McMahan RS, Eddy WE, Long ME, Parks WC, Aitken ML, et al. Transcriptional and functional diversity of human macrophage repolarization. J Allergy Clin Immunol (2019) 143:1536–48. doi: 10.1016/j.jaci.2018.10.046
114. Cassetta L. & Pollard, J. W. A timeline of tumour-associated macrophage biology. Nat Rev Cancer (2023) 23:238–57. doi: 10.1038/s41568-022-00547-1
115. Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol (2000) 192:150–8. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G
116. Lewis CE, Leek R, Harris A, McGee JO. Cytokine regulation of angiogenesis in breast cancer: the role of tumor-associated macrophages. J Leukoc Biol (1995) 57:747–51. doi: 10.1002/jlb.57.5.747
117. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia (2002) 7:177–89. doi: 10.1023/A:1020304003704
118. Chen P, Bonaldo P. Role of macrophage polarization in tumor angiogenesis and vessel normalization: implications for new anticancer therapies. Int Rev Cell Mol Biol (2013) 301:1–35. doi: 10.1016/B978-0-12-407704-1.00001-4
119. Lin L, Du Y, Hao J, Wu R, Du L. UTMD inhibits pancreatic cancer growth and metastasis by inducing macrophage polarization and vessel normalization. BioMed Pharmacother (2023) 160:114322. doi: 10.1016/j.biopha.2023.114322
120. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
121. Lin S, Zhang Q, Shao X, Zhang T, Xue C, Shi S, et al. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif (2017) 50(6):e12390. doi: 10.1111/cpr.12390
122. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol (2014) 5:75. doi: 10.3389/fphys.2014.00075
123. Qiao T, Yang W, He X, Song P, Chen X, Liu R, et al. Dynamic differentiation of F4/80+ tumor-associated macrophage and its role in tumor vascularization in a syngeneic mouse model of colorectal liver metastasis. Cell Death Dis (2023) 14:117. doi: 10.1038/s41419-023-05626-1
124. Yang C, Chen C, Xiao Q, Wang X, Shou Y, Tian X, et al. Relationship between PTEN and angiogenesis of esophageal squamous cell carcinoma and the underlying mechanism. Front Oncol (2021) 11:739297. doi: 10.3389/fonc.2021.739297
125. Liu J, Liu ZX, Li JJ, Zeng ZL, Wang JH, Luo XJ, et al. The macrophage-associated lncRNA MALR facilitates ILF3 liquid-liquid phase separation to promote HIF1α signaling in esophageal cancer. Cancer Res (2022) 83(9):1476–89. doi: 10.1158/0008-5472.CAN-22-1922
126. Consonni FM, Bleve A, Totaro MG, Storto M, Kunderfranco P, Termanini A, et al. Heme catabolism by tumor-associated macrophages controls metastasis formation. Nat Immunol (2021) 22:595–606. doi: 10.1038/s41590-021-00921-5
127. Bieniasz-Krzywiec P, Martín-Pérez R, Ehling M, García-Caballero M, Pinioti S, Pretto S, et al. Podoplanin-expressing macrophages promote lymphangiogenesis and lymphoinvasion in breast cancer. Cell Metab (2019) 30(5):917–936.e10. doi: 10.1016/j.cmet.2019.07.015
128. Chen L, Li J, Wang F, Dai C, Wu F, Liu X, et al. Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res (2016) 76:6828–38. doi: 10.1158/0008-5472.CAN-16-1114
129. Jakab M, Rostalski T, Lee KH, Mogler C, Augustin HG. Tie2 receptor in tumor-infiltrating macrophages is dispensable for tumor angiogenesis and tumor relapse after chemotherapy. Cancer Res (2022) 82:1353–64. doi: 10.1158/0008-5472.CAN-21-3181
130. Cole K, Al-Kadhimi Z, Talmadge JE. Role of myeloid-derived suppressor cells in tumor recurrence. Cancer Metastasis Rev (2023) 42:113–42. doi: 10.1007/s10555-023-10079-1
131. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
132. Jacob A, Prekeris R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol (2015) 3:4. doi: 10.3389/fcell.2015.00004
133. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature (2007) 450:825–31. doi: 10.1038/nature06348
134. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
135. Liu J-F, Deng WW, Chen L, Li YC, Wu L, Ma SR, et al. Inhibition of JAK2/STAT3 reduces tumor-induced angiogenesis and myeloid-derived suppressor cells in head and neck cancer. Mol Carcinog (2018) 57:429–39. doi: 10.1002/mc.22767
136. Wang J, Su X, Yang L, Qiao F, Fang Y, Yu L, et al. The influence of myeloid-derived suppressor cells on angiogenesis and tumor growth after cancer surgery. Int J Cancer (2016) 138:2688–99. doi: 10.1002/ijc.29998
137. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol (2012) 30:1–22. doi: 10.1146/annurev-immunol-100311-102839
138. Del Prete A, Salvi V, Soriani A, Laffranchi M, Sozio F, Bosisio D, et al. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell Mol Immunol (2023) 20(5):432–47. doi: 10.1038/s41423-023-00990-6
139. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
140. Bosisio D, Salvi V, Gagliostro V, Sozzani S. Angiogenic and antiangiogenic chemokines. Chem Immunol Allergy (2014) 99:89–104. doi: 10.1159/000353317
141. Riboldi E, Musso T, Moroni E, Urbinati C, Bernasconi S, Rusnati M, et al. Cutting edge: proangiogenic properties of alternatively activated dendritic cells. J Immunol (2005) 175:2788–92. doi: 10.4049/jimmunol.175.5.2788
142. Sozzani S, Rusnati M, Riboldi E, Mitola S, Presta M. Dendritic cell-endothelial cell cross-talk in angiogenesis. Trends Immunol (2007) 28:385–92. doi: 10.1016/j.it.2007.07.006
143. Gocher AM, Workman CJ, Vignali DAA. Interferon-γ: teammate or opponent in the tumour microenvironment? Nat Rev Immunol (2022) 22:158–72. doi: 10.1038/s41577-021-00566-3
144. Deliyanti D, Figgett WA, Gebhardt T, Trapani JA, Mackay F, Wilkinson-Berka JL, et al. CD8+ T cells promote pathological angiogenesis in ocular neovascular disease. Arterioscler Thromb Vasc Biol (2023) 43:522–36. doi: 10.1161/ATVBAHA.122.318079
145. Liu H, Zhao Q, Tan L, Wu X, Huang R, Zuo Y, et al. Neutralizing IL-8 potentiates immune checkpoint blockade efficacy for glioma. Cancer Cell (2023) 41(4):693–710.e8. doi: 10.1016/j.ccell.2023.03.004
146. Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and IL8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res (2021) 81:5876–88. doi: 10.1158/0008-5472.CAN-21-1456
147. Zhang M, Huang L, Ding G, Huang H, Cao G, Sun X, et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J Immunother Cancer (2020) 8(1):e000308. doi: 10.1136/jitc-2019-000308
148. Fleming C, Morrissey S, Cai Y, Yan J. γδ T cells: unexpected regulators of cancer development and progression. Trends Cancer (2017) 3:561–70. doi: 10.1016/j.trecan.2017.06.003
149. Khosravi N, Caetano MS, Cumpian AM, Unver N, De la Garza Ramos C, Noble O, et al. IL22 promotes kras-mutant lung cancer by induction of a protumor immune response and protection of stemness properties. Cancer Immunol Res (2018) 6:788–97. doi: 10.1158/2326-6066.CIR-17-0655
150. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med (2013) 19:1114–23. doi: 10.1038/nm.3291
151. Pilot T, Fratti A, Thinselin C, Perrichet A, Demontoux L, Limagne E, et al. Heat shock and HSP70 regulate 5-FU-mediated caspase-1 activation in myeloid-derived suppressor cells and tumor growth in mice. J Immunother Cancer (2020) 8(1):e000478. doi: 10.1136/jitc-2019-000478
152. Facciabene A, Motz GT. & Coukos, G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res (2012) 72:2162–71. doi: 10.1158/0008-5472.CAN-11-3687
153. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475:226–30. doi: 10.1038/nature10169
154. Zheng Z, Xu PP, Wang L, Zhao HJ, Weng XQ, Zhong HJ, et al. MiR21 sensitized B-lymphoma cells to ABT-199 via ICOS/ICOSL-mediated interaction of Treg cells with endothelial cells. J Exp Clin Cancer Res (2017) 36:82. doi: 10.1186/s13046-017-0551-z
155. De P, Aske J, Dey N. Cancer-associated fibroblast functions as a road-block in cancer therapy. Cancers (Basel) (2021) 13(1):e000478. doi: 10.3390/cancers13205246
156. Feng S, Xu Y, Dai Z, Yin H, Zhang K, Shen Y, et al. Integrative analysis from multicenter studies identifies a WGCNA-derived cancer-associated fibroblast signature for ovarian cancer. Front Immunol (2022) 13:951582. doi: 10.3389/fimmu.2022.951582
157. Unterleuthner D, Neuhold P, Schwarz K, Janker L, Neuditschko B, Nivarthi H, et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis (2020) 23:159–77. doi: 10.1007/s10456-019-09688-8
158. Karakasheva TA, Lin EW, Tang Q, Qiao E, Waldron TJ, Soni M, et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res (2018) 78:4957–70. doi: 10.1158/0008-5472.CAN-17-2268
159. Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut (2020) 69:1269–82. doi: 10.1136/gutjnl-2019-319200
160. Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal (2019) 12(567):eaan8247. doi: 10.1126/scisignal.aan8247
161. Larsen AMH, Kuczek DE, Kalvisa A, Siersbæk MS, Thorseth ML, Johansen AZ, et al. Collagen density modulates the immunosuppressive functions of macrophages. J Immunol (2020) 205:1461–72. doi: 10.4049/jimmunol.1900789
162. He Y, Liu T, Dai S, Xu Z, Wang L, Luo F, et al. Tumor-associated extracellular matrix: how to be a potential aide to anti-tumor immunotherapy? Front Cell Dev Biol (2021) 9:739161. doi: 10.3389/fcell.2021.739161
163. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: A mutually dependent relationship. Science (2023) 379:eabp8964. doi: 10.1126/science.abp8964
164. Feng M-X, Ma MZ, Fu Y, Li J, Wang T, Xue F, et al. Elevated autocrine EDIL3 protects hepatocellular carcinoma from anoikis through RGD-mediated integrin activation. Mol Cancer (2014) 13:226. doi: 10.1186/1476-4598-13-226
165. Aoki M, Kanamori M, Ohmori K, Takaishi M, Huh NH, Nogami S, et al. Expression of developmentally regulated endothelial cell locus 1 was induced by tumor-derived factors including VEGF. Biochem Biophys Res Commun (2005) 333:990–5. doi: 10.1016/j.bbrc.2005.06.009
166. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H, et al. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol (2019) 37:2. doi: 10.1007/s12032-019-1329-2
167. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discovery (2011) 10:417–27. doi: 10.1038/nrd3455
168. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307:58–62. doi: 10.1126/science.1104819
169. Wang D-R, Wu X-L, Sun Y-L. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct Target Ther (2022) 7:331. doi: 10.1038/s41392-022-01136-2
170. Patel SA, Nilsson MB, Le X, Cascone T, Jain RK, Heymach JV, et al. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin Cancer Res (2023) 29:30–9. doi: 10.1158/1078-0432.CCR-22-1366
171. Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE 2001 re21 (2001) 2001(112):re21. doi: 10.1126/stke.2001.112.re21
172. Melder RJ, Koenig GC, Witwer BP, Safabakhsh N, Munn LL, Jain RK, et al. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med (1996) 2:992–7. doi: 10.1038/nm0996-992
173. Gerber H-P, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res (2005) 65:671–80. doi: 10.1158/0008-5472.671.65.3
174. Wang T, Tang J, Yang H, Yin R, Zhang J, Zhou Q, et al. Effect of apatinib plus pegylated liposomal doxorubicin vs pegylated liposomal doxorubicin alone on platinum-resistant recurrent ovarian cancer: the APPROVE randomized clinical trial. JAMA Oncol (2022) 8:1169–76. doi: 10.1001/jamaoncol.2022.2253
175. Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci U.S.A. (2013) 110:19059–64. doi: 10.1073/pnas.1318022110
176. Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol (2013) 31:3212–8. doi: 10.1200/JCO.2012.47.2464
177. Dong X, Ren J, Amoozgar Z, Lee S, Datta M, Roberge S, et al. Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. J Immunother Cancer (2023) 11(3):e005583. doi: 10.1136/jitc-2022-005583
178. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
179. Hua Y, Vella G, Rambow F, Allen E, Antoranz Martinez A, Duhamel M, et al. Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1+ T lymphocyte niches through a feed-forward loop. Cancer Cell (2022) 40(12):1600–1618.e10. doi: 10.1016/j.ccell.2022.11.002
180. Wang R, Yang M, Jiang L, Huang M. Role of Angiopoietin-Tie axis in vascular and lymphatic systems and therapeutic interventions. Pharmacol Res (2022) 182:106331. doi: 10.1016/j.phrs.2022.106331
181. Brown DM, Boyer DS, Csaky K, Vitti R, Perlee L, Chu KW, et al. INTRAVITREAL NESVACUMAB (ANTIANGIOPOIETIN 2) PLUS AFLIBERCEPT IN DIABETIC MACULAR EDEMA: phase 2 RUBY randomized trial. Retina (2022) 42:1111–20. doi: 10.1097/IAE.0000000000003441
182. Hyman DM, Rizvi N, Natale R, Armstrong DK, Birrer M, Recht L, et al. Phase I study of MEDI3617, a selective angiopoietin-2 inhibitor alone and combined with carboplatin/paclitaxel, paclitaxel, or bevacizumab for advanced solid tumors. Clin Cancer Res (2018) 24:2749–57. doi: 10.1158/1078-0432.CCR-17-1775
183. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8:592–603. doi: 10.1038/nrc2442
184. Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest (2011) 121:1313–28. doi: 10.1172/JCI42405
185. Chae S-S, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, et al. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin Cancer Res (2010) 16:3618–27. doi: 10.1158/1078-0432.CCR-09-3073
186. Koh YJ, Kim HZ, Hwang SI, Lee JE, Oh N, Jung K, et al. Double antiangiogenic protein, DAAP, targeting VEGF-A and angiopoietins in tumor angiogenesis, metastasis, and vascular leakage. Cancer Cell (2010) 18:171–84. doi: 10.1016/j.ccr.2010.07.001
187. Javerzat S, Auguste P, Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med (2002) 8:483–9. doi: 10.1016/S1471-4914(02)02394-8
188. Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med (2018) 10(432):eaag0945. doi: 10.1126/scitranslmed.aag0945
189. Huynh H, Lee LY, Goh KY, Ong R, Hao HX, Huang A, et al. Infigratinib mediates vascular normalization, impairs metastasis, and improves chemotherapy in hepatocellular carcinoma. Hepatology (2019) 69:943–58. doi: 10.1002/hep.30481
190. Huynh H, Prawira A, Le TBU, Vu TC, Hao HX, Huang A, et al. FGF401 and vinorelbine synergistically mediate antitumor activity and vascular normalization in FGF19-dependent hepatocellular carcinoma. Exp Mol Med (2020) 52:1857–68. doi: 10.1038/s12276-020-00524-4
191. Griffin RJ, Williams BW, Wild R, Cherrington JM, Park H, Song CW, et al. Simultaneous inhibition of the receptor kinase activity of vascular endothelial, fibroblast, and platelet-derived growth factors suppresses tumor growth and enhances tumor radiation response. Cancer Res (2002) 62:1702–6.
192. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med (2013) 19:460–73. doi: 10.1016/j.molmed.2013.05.002
193. Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest (2003) 112:1142–51. doi: 10.1172/JCI200318549
194. Cao R, Björndahl MA, Religa P, Clasper S, Garvin S, Galter D, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell (2004) 6:333–45. doi: 10.1016/j.ccr.2004.08.034
195. Hosaka K, Yang Y, Seki T, Nakamura M, Andersson P, Rouhi P, et al. Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat Commun (2013) 4:2129. doi: 10.1038/ncomms3129
196. Wang J-C, Li GY, Wang B, Han SX, Sun X, Jiang YN, et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J Exp Clin Cancer Res (2019) 38:235. doi: 10.1186/s13046-019-1211-2
197. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (2020) 71:1247–61. doi: 10.1002/hep.30889
198. Tu J, Xu H, Ma L, Li C, Qin W, Chen X, et al. Nintedanib enhances the efficacy of PD-L1 blockade by upregulating MHC-I and PD-L1 expression in tumor cells. Theranostics (2022) 12:747–66. doi: 10.7150/thno.65828
199. Liang J, Oyang L, Rao S, Han Y, Luo X, Yi P, et al. Rac1, A potential target for tumor therapy. Front Oncol (2021) 11:674426. doi: 10.3389/fonc.2021.674426
200. Kabir AU, Subramanian M, Lee DH, Wang X, Krchma K, Wu J, et al. Dual role of endothelial Myct1 in tumor angiogenesis and tumor immunity. Sci Transl Med (2021) 13(583):eabb6731. doi: 10.1126/scitranslmed.abb6731
201. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71:5678–87. doi: 10.1158/0008-5472.CAN-11-0431
202. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med (2017) 9(385):eaak9679. doi: 10.1126/scitranslmed.aak9679
203. Xiong Z, Chan SL, Zhou J, Vong JSL, Kwong TT, Zeng X, et al. Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut (2023) 72(9):1758–73. doi: 10.1136/gutjnl-2022-328364
204. Bertrand C, Van Meerbeeck P, de Streel G, Vaherto-Bleeckx N, Benhaddi F, Rouaud L, et al. Combined blockade of GARP:TGF-β1 and PD-1 increases infiltration of T cells and density of pericyte-covered GARP+ Blood vessels in mouse MC38 tumors. Front Immunol (2021) 12:704050. doi: 10.3389/fimmu.2021.704050
205. Zheng X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest (2018) 128:2104–15. doi: 10.1172/JCI96582
206. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res (2013) 73:2943–8. doi: 10.1158/0008-5472.CAN-12-4354
207. Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin Cancer Res (2020) 26:1712–24. doi: 10.1158/1078-0432.CCR-19-2179
208. Kikuchi H, Matsui A, Morita S, Amoozgar Z, Inoue K, Ruan Z, et al. Increased CD8+ T-cell infiltration and efficacy for multikinase inhibitors after PD-1 blockade in hepatocellular carcinoma. J Natl Cancer Inst (2022) 114:1301–5. doi: 10.1093/jnci/djac051
209. Jin J, Xie Y, Zhang JS, Wang JQ, Dai SJ, He WF, et al. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug Resist Update (2023) 67:100929. doi: 10.1016/j.drup.2023.100929
210. Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med (2021) 384:1289–300. doi: 10.1056/NEJMoa2035716
211. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
212. Di Maggio F, Arumugam P, Delvecchio FR, Batista S, Lechertier T, Hodivala-Dilke K, et al. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology (2016) 16(6):995–1004. doi: 10.1016/j.pan.2016.05.393
213. Kindler HL, Ioka T, Richel DJ, Bennouna J, Létourneau R, Okusaka T, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol (2011) 12:256–62. doi: 10.1016/S1470-2045(11)70004-3
214. Park W, Chawla A, O'Reilly EM. Pancreatic cancer: A review. JAMA (2021) 326:851–62. doi: 10.1001/jama.2021.13027
215. Fejza A, Polano M, Camicia L, Poletto E, Carobolante G, Toffoli G, et al. The efficacy of anti-PD-L1 treatment in melanoma is associated with the expression of the ECM molecule EMILIN2. Int J Mol Sci 22 (2021) 22(14):7511. doi: 10.3390/ijms22147511
216. Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res (2017) 5:17–28. doi: 10.1158/2326-6066.CIR-16-0206
217. Padhani AR, Miles KA. Multiparametric imaging of tumor response to therapy. Radiology (2010) 256:348–64. doi: 10.1148/radiol.10091760
Keywords: vascular normalization, cancer immunotherapy, TME (tumor microenvironment), angiogenic, anti-angiogenic
Citation: Qian C, Liu C, Liu W, Zhou R and Zhao L (2023) Targeting vascular normalization: a promising strategy to improve immune–vascular crosstalk in cancer immunotherapy. Front. Immunol. 14:1291530. doi: 10.3389/fimmu.2023.1291530
Received: 09 September 2023; Accepted: 01 December 2023;
Published: 15 December 2023.
Edited by:
Aparna Jayachandran, Fiona Elsey Cancer Research Institute, AustraliaReviewed by:
Ilkka Pietilä, Uppsala University, SwedenRodolfo Chavez-Dominguez, National Autonomous University of Mexico, Mexico
Copyright © 2023 Qian, Liu, Liu, Zhou and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Zhao, liangsmu@foxmail.com; Rui Zhou, yaruisunny@sina.com
†These authors have contributed equally to this work