 Shizhe Li
Shizhe Li He Zhang
He Zhang Guanning Shang1*
Guanning Shang1*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 December 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1290762
Osteosarcoma, the most common bone malignancy in children and adolescents, poses considerable challenges in terms of prognosis, especially for patients with metastatic or recurrent disease. While surgical intervention and adjuvant chemotherapy have improved survival rates, limitations such as impractical tumor removal or chemotherapy resistance hinder the treatment outcomes. Chimeric antigen receptor (CAR)-T cell therapy, an innovative immunotherapy approach that involves targeting tumor antigens and releasing immune factors, has shown significant advancements in the treatment of hematological malignancies. However, its application in solid tumors, including osteosarcoma, is constrained by factors such as low antigen specificity, limited persistence, and the complex tumor microenvironment. Research on osteosarcoma is ongoing, and some targets have shown promising results in pre-clinical studies. This review summarizes the current status of research on CAR-T cell therapy for osteosarcoma by compiling recent literature. It also proposes future research directions to enhance the treatment of osteosarcoma.
Osteosarcoma, the most common primary malignant bone tumor in children and adolescents, presents significant therapeutic challenges in terms of improving overall patient survival (1). While the combination of surgical resection and adjuvant chemotherapy has contributed to better outcomes in primary osteosarcoma, with a five-year survival rate of approximately 70% (2), recurrent or metastatic osteosarcoma poses greater difficulties. Factors such as chemotherapy resistance and the inability to surgically remove the tumor contribute to a much lower overall survival rate of approximately 25% (3). Consequently, there is an urgent need for novel treatment approaches in this context.
Chimeric antigen receptor (CAR)-T cell therapy is a novel form of immunotherapy derived from adoptive T cell transfer therapy (4). CARs are synthetic receptors composed most often of an extracellular single-chain fragment variable that recognizes tumor antigens, an intracellular signaling domain consisting of the T cell receptor and CD3ζ chain, a transmembrane structural domain, and an extracellular spacer region that adjusts the distance between CAR-T cells and the tumor (5, 6). CAR-T cells are generated by isolating T cells from the patient’s blood, genetically modifying them in vitro to express CARs, expanding and culturing them, and subsequently infusing them back into the patient as anti-cancer therapy (7) (Figure 1A). Four generations of CAR-T cells exist; the first generation lacks co-stimulatory domains, making them less durable and effective (8). Second-generation CAR-T cells increase their proliferative capacity by adding a co-stimulatory structural domain (CD28, 4-1BB, OX40, etc.); thus, most CAR-T cell therapies currently in clinical use involve second-generation structures (9). The third generation includes additional co-stimulatory molecules compared to the second generation (10), while the fourth generation is further modified to express suicide genes or secrete cytokines (IL-12, IL-15, and IL-21) for enhanced anti-tumor efficacy (11). The concept of a “fifth-generation CAR-T” has also been proposed, involving the addition of an IL-2 receptor beta-chain fragment (IL-2Rβ) to second-generation CAR-T cells, which activates antigen specificity by regulating the JAK/STAT pathway (12, 13) (Figure 1B).
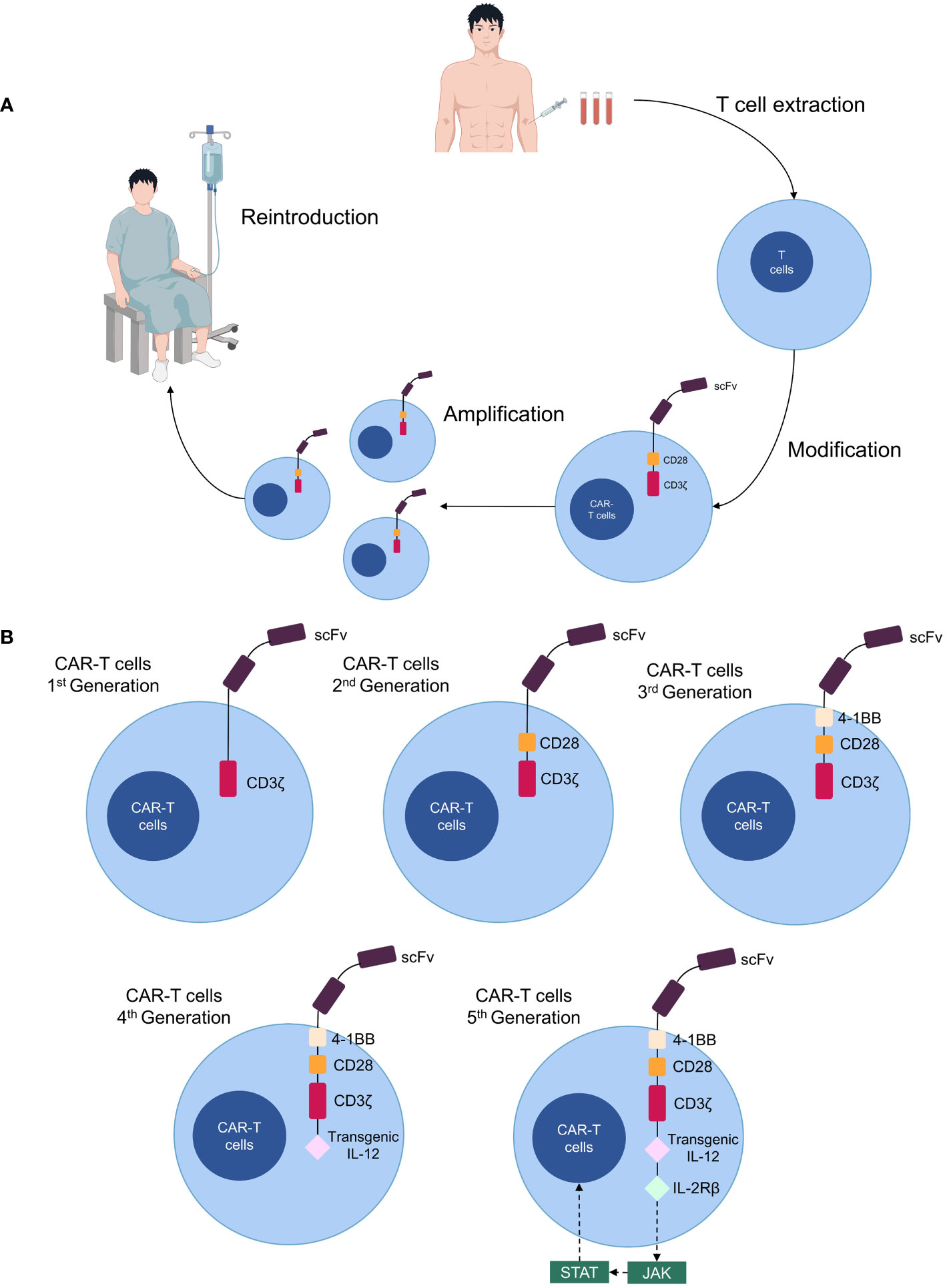
Figure 1 Production processes and structure of CAR-T cells. (A) Patient T cells are isolated from the blood and transformed into CAR-T cells in vitro, expanded, then cultured and transfused back for therapy. (B) Five generations of CAR-T cell structure.
Compared to conventional therapies, CAR-T cell therapy is more precise and durable and can secrete cytokines to improve the tumor microenvironment (TME). In recent years, significant progress has been made in the treatment of non-solid tumors (14). However, research is still ongoing for malignant solid tumors such as osteosarcoma, with some targets exhibiting satisfactory results in preclinical trials and substantial potential for clinical applications. This article reviews the potential targets of CAR-T cell therapy in osteosarcoma, summarizes the current status and future directions of CAR-T cell therapy in osteosarcoma, and provides new insights into the treatment of osteosarcoma.
The majority of potential targets for CAR-T cell therapy in osteosarcoma are tumor-associated antigens, which are expressed in both normal and tumor tissues but are more highly expressed in tumor tissues. Several current potential targets for CAR-T cell therapy in osteosarcoma include receptor tyrosine kinases (HER2, IGF1R, ROR1, and EphA2), cell surface glycoproteins (CSPG4, FRα, FRβ, and EC17), B7-H3 (CD276), disialoganglioside (GD2), natural killer group 2D (NKG2D), activated leukocyte cell adhesion molecule (ALCAM/CD166), interleukin-11 receptor alpha (IL-11Rα), and fibroblast activation protein (FAP) (Figure 2, Table 1).

Figure 2 Potential targets for CAR-T cells in the treatment of osteosarcoma.
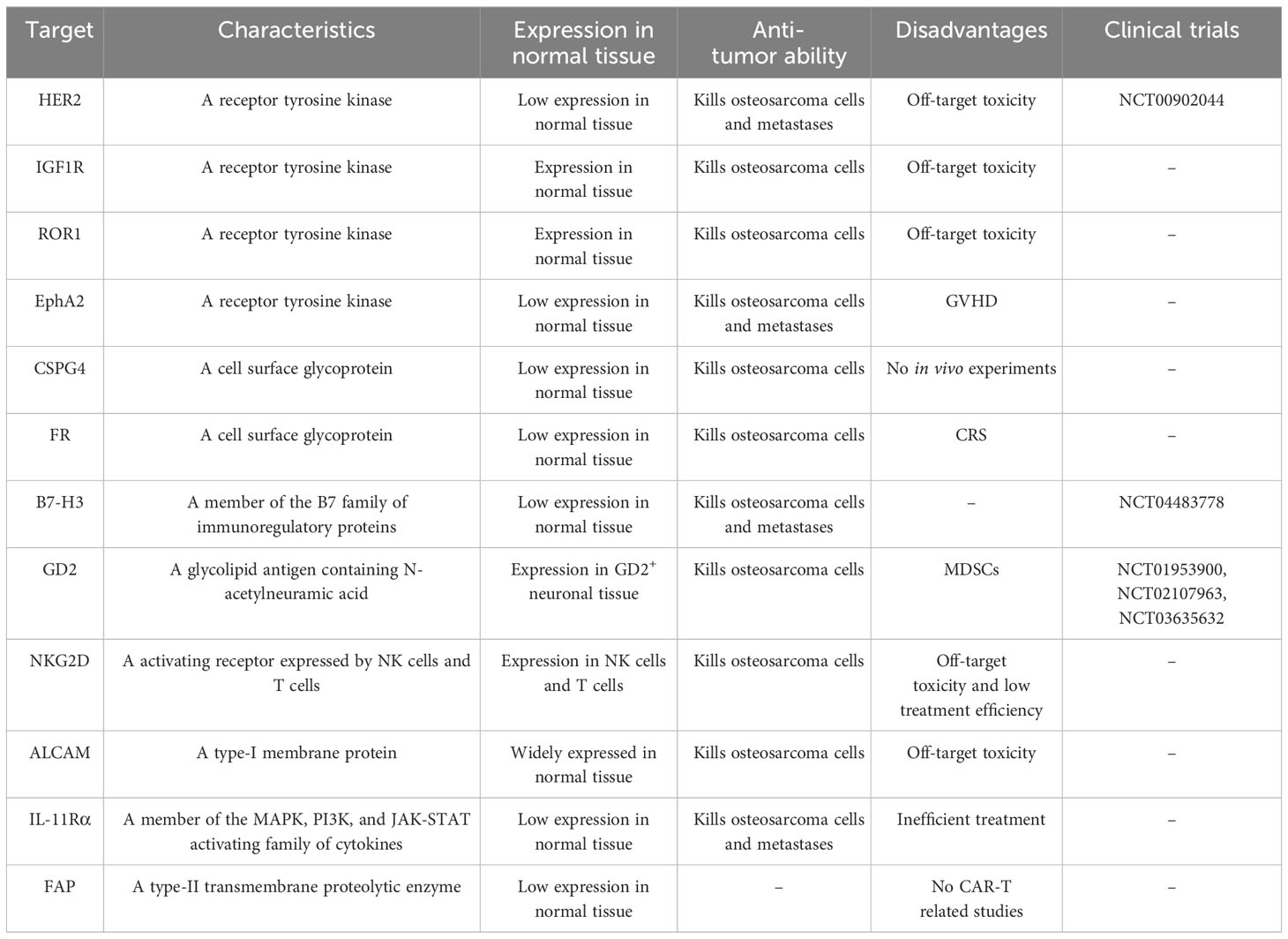
Table 1 Potential effective targets for CAR-T therapy in osteosarcoma.
Receptor tyrosine kinases play a crucial role in regulating various cellular processes such as tumor cell proliferation, invasion, migration, and differentiation through the modulation of signaling pathways such as PI3K/AKT, Ras/MEK/ERK, and JAK/STAT (15). In osteosarcoma, several receptor tyrosine kinases have the potential for CAR-T cell therapy, including HER2 and IGF1R.
The epidermal growth factor receptor family comprises four transmembrane receptors: HER1, HER2, HER3, and HER4 (16). Unlike other receptors, HER2 does not require ligand binding for heterodimerization, and its overexpression leads to intracellular domain dimerization and autophosphorylation, activating downstream signaling pathways involved in tumor cell proliferation, invasion, migration, and angiogenesis. Consequently, HER2 exhibits potent oncogenic potential (17). HER2 is frequently overexpressed and associated with poor prognosis in various malignancies, including breast cancer and gastric cancer (18, 19). Recent studies have demonstrated high expression of HER2 in osteosarcoma and its correlation with poor prognosis (20, 21). While the HER2 inhibitor trastuzumab has limited efficacy in metastatic osteosarcoma, HER2 remains a potential target for immunotherapy (22). Research by Ahmed et al. showed that second-generation HER2-targeting CAR-T cells secreted immunostimulatory cytokines, including interferon IFN-γ and interleukin IL-2, upon exposure to HER2-positive osteosarcoma cells, leading to tumor growth inhibition (23). In a xenograft osteosarcoma mouse model, HER2-targeted CAR-T cells induced regression of primary lesions and lung metastases (23). Another study by Rainusso et al. demonstrated that HER2-targeted CAR-T cells reduced the myosphere-forming ability of osteosarcoma cells in vitro, suggesting the potential efficacy of HER2-targeted CAR-T cell therapy in treating metastatic osteosarcoma (24). Additionally, Park et al. found that combining HER2 CAR-T cells with PD-L1 antibodies increased T cell expression in an osteosarcoma xenotransplantation model, providing further support for the clinical application of HER2-targeted CAR-T therapy (25). Several ongoing clinical trials are evaluating the use of HER2-targeted CAR-T cells in osteosarcoma treatment. A phase I/II clinical trial (NCT00902044) involving 16 patients with HER2-positive osteosarcoma showed that escalating doses of second-generation CAR-T cells (dose range 1×104 to 1×108/m2) resulted in CAR-T cell persistence for at least six weeks. Four patients achieved stable disease for 12 weeks to 14 months, with a median progression-free survival of approximately six weeks. Only one patient experienced fever, and no other severe adverse events were reported (26). Although HER2 is a promising target for CAR-T cells in the treatment of osteosarcoma, its practical clinical applications are limited. Morgan et al. reported a case of severe respiratory failure in a patient with HER2-positive colon cancer after treatment with CAR-T cells targeting HER2 (27). They hypothesized that this may be related to CAR-T cells recognizing HER2 expressed in normal lung tissue and releasing inflammatory factors, which in turn leads to pulmonary toxicity and a cytokine storm (27). Therefore, further studies focusing on the safety aspects of CAR-T cell therapy targeting HER2 are necessary.
The insulin-like growth factor 1 receptor (IGF1R), a receptor tyrosine kinase located on the cell membrane, plays a role in the invasion and metastasis of various malignancies (28). In osteosarcoma, IGF1R is overexpressed and contributes to the early development of the disease by activating the PI3K/AKT pathway, while relying on the RAS/MAPK pathway for late lung metastasis (29, 30). Currently, clinical trials investigating IGF1R-targeted therapy for osteosarcoma are underway (31). IGF1R also holds potential for immunotherapy. Huang et al. demonstrated that third-generation CAR-T cells targeting IGF1R released cytokines such as IFN-γ, TNF-α, and IL-13, leading to anti-tumor effects on osteosarcoma cells. In a xenograft mouse model, CAR-T therapy targeting IGF1R inhibited tumor growth and prolonged survival (32). However, the expression of IGF1R in normal tissues (skeletal muscle) poses a challenge in the development of IGF1R-targeted CAR-T therapies, as off-target toxicity remains a concern (33). Further exploration and studies are necessary to address this issue and advance the development of IGF1R-targeted CAR-T therapies for osteosarcoma.
The receptor tyrosine kinase-like orphan receptor 1 (ROR1) is a type-I transmembrane glycoprotein that plays a role in cell differentiation and proliferation during embryonic and fetal development (34). In various malignancies, including osteosarcoma, ROR1 is overexpressed and contributes to the promotion of migration by regulating the Wnt signaling pathway (35, 36). A recent study demonstrated that third-generation CAR-T cells targeting ROR1 effectively inhibited osteosarcoma growth both in vitro and in vivo (32). However, similar to IGF1R, the expression of ROR1 in multiple normal tissues (parathyroid, pancreatic islets and duodenal mucosa) raises concerns regarding potential off-target toxicity (37). Lee et al. addressed this issue by utilizing the single-chain fragment variable of zilovertamab, a humanized monoclonal antibody, as the antigen recognition domain of CAR-T cells, resulting in improved safety (38). Several clinical trials have been conducted to evaluate ROR1-targeted CAR-T therapy in patients with solid tumors (NCT05274451, NCT05638828, and NCT02706392). However, these trials did not specifically include patients with osteosarcoma (39). Nevertheless, these studies provide valuable insights that can inform future investigations on the application of ROR1-targeted CAR-T therapy for osteosarcoma.
Ephrin type-A receptor 2 (EphA2) is a member of the ephrin family of receptors and plays a role in normal embryonic development and kidney function. Its expression is primarily observed in highly proliferative epithelial tissues (40). EphA2 is highly expressed in various malignancies and is implicated in the epithelial-mesenchymal transition in osteosarcoma (41, 42). A study by Hsu et al. demonstrated that second-generation CAR-T cells targeting EphA2 can effectively inhibit osteosarcoma cells by releasing cytokines such as IL-2 and IFN-γ (43). In a mouse model of metastatic osteosarcoma, treatment with EphA2-targeted CAR-T cells resulted in infiltration of the lungs and liver, tumor eradication, and increased survival time (43). Unlike IGF1R and ROR1, EphA2 is expressed at lower levels in normal tissues than in highly proliferative epithelial tissues, which reduces the likelihood of off-target toxicity (44). However, Hsu et al. also reported that some models developed graft-versus-host disease (GVHD) approximately 50 days after CAR-T cell infusion. This observation may be associated with the origin of the T cells used for CAR-T therapy. The study suggested that targeting CAR-T cells with autologous-derived EphA2 may enhance treatment safety (43).
Cell surface glycoproteins are identifiable tumor antigens of CAR-T cells that can assist T cells in tumor inhibition (45). Currently, two glycoproteins (CSPG4 and FR) are potential candidates for CAR-T cell therapy in osteosarcoma.
Chondroitin sulfate proteoglycan 4 (CSPG4) is a transmembrane glycoprotein that is highly expressed in various malignancies (46). In osteosarcoma, the expression level of CSPG4 is also correlated with poor prognosis (47). The central role of CSPG4 in malignant tumor progression and metastasis, as well as its impact on the tumor microenvironment, suggests its potential as an effective target for tumor immunotherapy (48). Beard et al. found that second-generation CSPG4-targeted CAR-T cells released IFN-γ, TNF-α, and GM-CSF and exerted cytolytic effects on the MG63 cell line (49). The advantages of CSPG4-targeted CAR-T cells for the treatment of solid tumors include the inhibition of peritumoral angiogenesis and improvement of the tumor microenvironment (50). However, no in vivo trials have been conducted on CSPG4 in osteosarcoma; therefore, further studies are required to assess its utility.
Folate receptor α (FRα) and FRβ are membrane-bound proteins anchored to the extracellular surface using glycosylphosphatidylinositol and are capable of transporting folate across membranes via endocytosis (51). FRα is mainly expressed at low levels in epithelial tissues, whereas FRβ is found in placental and hematopoietic tissues (51). FRα is highly expressed in osteosarcoma, and FRβ+ tumor-associated macrophages have been detected in metastatic osteosarcoma (52, 53). Therefore, FRα and FRβ are potential therapeutic targets for the treatment of osteosarcoma. EC17, a low-molecular-weight ligand, can be stabilized on the surface of tumor cells due to its high affinity for the folate receptor and coupling with fluorescein isothiocyanate (FITC) (54). Amato et al. employed EC17 combined with folate receptors for immunotherapy in patients with metastatic renal cell carcinoma, which achieved a good effect (55). Lu et al. combined EC17 and FITC with folate receptors according to the concept of the CAR-T adaptor molecule to form second-generation FITC-specific CAR-T cells and applied these cells to the HOS osteosarcoma cell line. Both in vitro and in vivo experiments showed the strong anti-tumor activity of CAR-T cells (56). However, high doses of folate receptor CAR-T cells can lead to severe cytokine release syndrome (CRS), which can be alleviated by the intravenous administration of sodium fluorescein (56). Therefore, the safety of these targets requires further evaluation.
B7-H3 (CD276) is an immune checkpoint of the B7 family of molecules that is involved in the regulation of T cells. It may also inhibit NK cell activation and play a pro-inflammatory role (57). B7-H3 is overexpressed in a variety of malignancies and is associated with tumor proliferation, metastasis, and poor prognosis (58). Studies have shown that B7-H3 may influence tumor progression by regulating the PI3K/AKT/mTOR, JAK2/STAT3, and TLR4/NF-κB pathways (59–61). Lu et al. examined specimens from 35 patients with advanced osteosarcoma and found a positive B7-H3 expression rate of approximately 29%, suggesting the potential of B7-H3 as a target for osteosarcoma immunotherapy (62). Majzner et al. found that third-generation CAR-T cells targeting B7-H3 released IFN-γ, TNF-α, and IL-2 cytokines when co-cultured with osteosarcoma cells, showing effective anti-tumor abilities. In a mouse model of osteosarcoma, the survival time was prolonged by at least five months with CAR-T cell treatment compared to the control group (63). Talbot et al. implanted LM7 osteosarcoma cells expressing firefly luciferase (fLuc) into the tibia of a mouse model and showed that CAR-T cells targeting B7-H3 exhibited potent dose-dependent anti-lung metastatic activity, as detected by bioluminescence imaging (64). Zhang et al. subsequently constructed a third-generation CAR-T cell model targeting B7-H3, which demonstrated good anti-osteosarcoma efficacy in both in vivo and in vitro assays (65). A phase I clinical trial of second-generation B7-H3-targeted CAR-T cells for the treatment of recurrent osteosarcoma in children and adolescents is currently underway (NCT04483778). Compared to other immunotherapeutic targets, B7-H3 is expressed at lower levels in normal tissues than in malignant tissues and has less potential for off-target toxicity (63). Additionally, B7-H3 not only affects innate and adaptive immunity but also regulates tumor progression through various non-immune signaling pathways (58–61). Therefore, B7-H3 may be an ideal target for CAR-T cell therapy in osteosarcoma.
Disialoganglioside (GD2) is a tumor-associated surface antigen expressed in the fetus and is also present in neural stem cells with multiple biological functions, including signal transduction and cell adhesion (66). As a tumor antigen, GD2 expression is highly specific to tumors and enhances the interaction between tumor cells and extracellular matrix proteins. In fact, GD2 is highly expressed in various malignancies (67). Roth et al. conducted immunohistochemistry on 44 osteosarcoma specimens and revealed high expression of GD2 in osteosarcoma, with higher staining intensity in recurrent osteosarcoma specimens (68). This suggests that GD2 may play a key role in the progression of osteosarcoma and has potential value in targeted therapy. Long et al. demonstrated that third-generation GD2-targeted CAR-T cells significantly inhibited the growth of osteosarcoma cells in vitro. However, in a xenograft model, an increase in myeloid-derived suppressor cells (MDSCs) hindered the efficacy of CAR-T cells in vivo (69). They also suggested that MDSCs may inhibit T cell responses by affecting the CD28-containing co-stimulatory domains in CAR-T cells (69). All-trans retinoic acid (ATRA) can be used to inhibit MDSC activity. In xenografted mouse models, the use of ATRA enhanced CAR-T cell efficacy and improved the survival rate of mice (69). Furthermore, Long et al. found that ATRA may enhance efficacy by increasing the expression of glutathione synthase in MDSCs and mediating the neutralization of reactive oxygen species (69, 70). In addition, the combination of GD2-targeted CAR-T cells and PD-L1 antibodies can improve T cell infiltration in osteosarcoma and enhance its efficacy (25). Chulanetra et al. found that the combination of fourth-generation GD2-targeted CAR-T cells and doxorubicin enhanced the efficacy of doxorubicin against osteosarcoma cells (71). Moreover, Tanaka et al. suggested that live attenuated varicella-zoster virus vaccination enhances the therapeutic efficacy of third-generation GD2-targeted CAR-T cells against malignant solid tumors by affecting the TME (NCT01953900) (72). These results indicate that GD2-targeted CAR-T cell therapy may have potential in multiple drug combinations for osteosarcoma, with broad application prospects. However, Chulanetra et al. showed that an increased ratio of tumor cells to CAR-T cells could reduce the durability of CAR-T cell therapy (71). CAR-T cells acting on osteosarcoma cells upregulate PD-L1 and PD-1 expression, which may diminish the effect of GD2-targeted CAR-T cells (71). Therefore, the efficiency of GD2-targeted CAR-T cells must be further explored. Several clinical trials on GD2-targeted CAR-T therapy for relapsed or refractory osteosarcoma are currently underway (NCT01953900, NCT02107963, and NCT03635632).
Natural killer group 2D (NKG2D) is an activated immune receptor expressed on NK and T cells, playing a role in the regulation of innate and secondary immune responses (73). NKG2D is normally expressed in NK cells, CD8 T cells, γδ T cells, and some CD4 T cells. In humans, NKG2D primarily recognizes ligands such as MHC I chain-related molecules A and B (MICA and MICB) and the UL16 binding protein family (ULBP1-6) (74). These ligands are often overexpressed in malignant tumors, suggesting an association of NKG2D with tumor progression (75). Fernandez et al. found that activated and amplified NK cells could target osteosarcoma cells in vivo and in vitro through interactions between NKG2D and its ligands, indicating the potential of NKG2D and its ligands as immunotherapeutic targets for osteosarcoma (76). Subsequently, Fernandez et al. demonstrated that third-generation CAR-T cells targeting NKG2D inhibited osteosarcoma cells in both in vitro and in vivo assays, prolonging the survival time of xenograft mouse models without associated toxic effects (77). Additionally, Parihar et al. suggested that cytotoxic-chain fusion of NKG2D with the T cell receptor could alleviate MDSCs caused by CAR-T treatment, indicating potential therapeutic options for NKG2D in combination with other CAR-T targets (78). Although NKG2D is minimally expressed on normal cell surfaces compared with NK and T cells, off-target toxicity cannot be ruled out. For example, Van Seggelen et al. reported lethal toxicity in NKG2D-targeted CAR-T cells in a mouse model (79). Moreover, Sentman et al. found that the acute toxic response in a mouse model resembled the symptoms of cytokine release syndrome (CRS) observed in patients treated with CAR-T cells, which was associated with two effectors of CAR-T therapy: perforin and GM-CSF (80). However, no adverse reactions associated with CAR-T cells were observed in a phase I clinical trial of NKG2D-targeted CAR-T cells in patients with refractory multiple myeloma (81). Therefore, further investigation is needed to assess the safety of NKG2D as a therapeutic target.
Activated leukocyte cell adhesion molecule (ALCAM), also known as CD166, is a type-I transmembrane immunoglobulin that plays a role in mediating adhesion between homogeneous and heterogeneous cells in the body (82). ALCAM is highly expressed in various tumors and is involved in processes such as tumor angiogenesis and distant metastasis (83, 84). Immunohistochemical staining conducted by Federman et al. revealed positive expression of ALCAM in the cell membrane and cytoplasm of osteosarcoma cells (85). Furthermore, Cardenes et al. demonstrated that ALCAM facilitates the binding and uptake of tumor-derived extracellular vesicles in colorectal and ovarian cancers, thereby promoting the migration of tumor cells (86). These findings suggest that ALCAM plays a role in regulating osteosarcoma metastasis. In the context of immunotherapy, Wang et al. developed second-generation CD166-BBζ-targeted CAR-T cells that exhibited significant cytotoxicity against osteosarcoma cells in vitro, with the cytotoxic effect strongly correlated with the expression of CD166 (87). Additionally, in vivo studies showed that CAR-T cells targeting CD166 effectively inhibited osteosarcoma growth without causing off-target toxicity (87). However, it should be noted that CD166 is also expressed in normal tissues, such as epithelial cells (88), raising the possibility of off-target toxicity.
Interleukin-11 (IL-11) is a member of the IL-6 family of cytokines and plays a role in various biological processes such as hematopoiesis, bone development, and tissue repair (89). IL-11 exerts its effects through the IL-11 receptor, which consists of interleukin-11 receptor alpha (IL-11Rα) and GP130 receptors, and activates the JAK signaling pathway. IL-11Rα is believed to be involved in tumor proliferation, invasion, and angiogenesis (90). Lewis et al. found that IL-11 and IL-11Rα were specifically upregulated in primary osteosarcoma cells and lung metastases but minimally expressed in normal bone tissue, and that IL-11Rα expression was associated with poor prognosis (91, 92). Huang et al. applied third-generation IL-11Rα-targeting CAR-T cells to osteosarcoma cells and found that CAR-T cells effectively killed tumor cells and inhibited lung metastases, and that the expression level of IL-11Rα was positively correlated with the killing effect (93). In addition to CAR-T cell therapy, Jiang et al. utilized the high expression of IL-11Rα in osteosarcoma to develop a drug delivery system called IL11-PDox. This system involved functionalizing REDOX-sensitive polymersomes with IL-11 ligand and encapsulating the chemotherapy drug Dox. Their findings showed that IL11-PDox enhanced the efficacy of chemotherapy in mouse models of recurrent and metastatic osteosarcoma, suggesting a potential combination strategy of IL-11Rα-targeted therapy with chemotherapy for advanced osteosarcoma (94). Although no toxic reactions were observed in CAR-T trials targeting IL-11Rα, concerns have been raised about the efficiency of IL-11Rα-targeted therapy because of its underexpression in osteosarcoma (93).
The high number of cancer-associated fibroblasts (CAFs) in malignant solid tumors provides an ideal microenvironment for the development of tumor cells (95). CAFs play a crucial role in tumors by secreting various cytokines and metabolites that inhibit immune cells and promote tumor invasion and metastasis. One specific protein expressed by CAFs is fibroblast activation protein (FAP), which belongs to the prolyloligopeptidase family and is minimally expressed in normal human tissues (96). It is now recognized as an effective target for CAFs, and an increasing number of studies have shown that targeting FAP can inhibit tumor progression by modulating the TME (97, 98). Wang et al. demonstrated that CAR-T cells targeting FAP could effectively inhibit tumor growth by targeting tumor stromal cells both in vitro and in vivo, without significant toxicity. This suggests that FAP-targeted CAR-T therapy may be an effective approach for treating malignant solid tumors (99). Additionally, Zhang et al. found that FAP is highly expressed in osteosarcoma and its expression level correlates with poor prognosis (100). Although no studies have investigated FAP-targeted CAR-T therapy in osteosarcoma, FAP remains a potential target for CAR-T therapy in this disease due to its impact on the TME and its high expression level in osteosarcoma.
Research on malignant solid tumors, including osteosarcoma, has been prompted by the excellent performance of CAR-T cells in the treatment of hematological malignancies. Currently, potential effective targets for CAR-T cell therapy in osteosarcoma include HER2, IGF1R, ROR1, EphA2, CSPG4, folate receptor with EC17, B7-H3, GD2, NKG2D, ALCAM/CD166, IL-11Rα, and FAP. Among these targets, HER2, EphA2, B7-H3, and IL-11Rα CAR-T cells warrant further investigation owing to their effectiveness in metastatic osteosarcoma (24, 43, 64, 93). HER2, B7-H3, and GD2 are currently undergoing clinical trials, so they will likely be the first targets used for the clinical treatment of osteosarcoma. Although CAR-T therapy is a promising treatment for osteosarcoma, low antigen specificity, poor durability, numerous side effects, and the complexity of the tumor microenvironment (TME) still limit its widespread application (101). Therefore, scholars have recently proposed counterstrategies to overcome the various limitations of CAR-T therapy (Table 2).

Table 2 Current limitations and potential counterstrategies of CAR-T cell therapy in osteosarcoma.
Low antigen specificity is a major limitation of CAR-T cell therapy for solid tumors. The ideal targets for CAR-T cells are tumor-specific antigens (TSAs), which are expressed at high levels in tumor tissue but virtually absent in normal tissue (9). In contrast, the majority of CAR-T targets in osteosarcoma are tumor-associated antigens, and differences in expression between tumor-associated antigens and TSAs in normal tissues may lead to severe off-target toxicity and insufficient efficacy (102). Current responses to this limitation include the search for additional TSA targets and the development of novel CAR-T cells. However, TSA targets in osteosarcoma are rare and difficult to identify, making the development of novel CAR-T cells a more feasible solution to address low specificity (101). For example, Park et al. constructed bispecific CAR-T cells targeting both HER2 and GD2 to act on osteosarcoma cells and improve efficacy (25). Bielamowicz et al. developed multi-specific CAR-T cells targeting HER2, IL13Rα2, and EphA2 to enhance antigen specificity in glioblastoma, which could serve as a reference for their application in osteosarcoma (103). Furthermore, the concept of “logic gating” has been proposed, wherein T cells can improve their specificity by expressing both antigens (A and B) or only antigens A or B (104). Srivastava et al. designed a logically gated ROR1-targeted CAR-T cell that reduced off-target toxicity and selectively targeted tumor cells (105). Similarly, Lee et al. suggested that combining CAR-T cells with multiple bispecific junctions (e.g. FITC) could help target multiple antigens and enhance efficacy in heterogeneous solid tumors (106).
In addition to specificity, the durability of CAR-T cells impacts their treatment efficacy, as insufficient durability can affect the long-term therapeutic effects of CAR-T cells in vivo (107). Antigen heterogeneity or antigen loss is the main cause of antigen-dependent drug resistance in CAR-T cells (108). Hegde et al. found that when CAR-T cells act on HER2-positive U373 cells (glioblastoma), HER2-negative tumor cells are produced secondarily, which leads to drug resistance in CAR-T cells (109). In addition to the development of multi-specific CAR-T cells mentioned earlier, it is currently believed that genetic manipulation interventions and drug combinations can enhance the durability of CAR-T cells (110). CAR-T cells can be categorized as autologous or allogeneic based on their source. DiNofia et al. discovered that autologous CAR-T cells exhibited significantly greater durability compared to allogeneic CAR-T cells (111). However, the widespread availability of autologous CAR-T cell preparation is limited due to variations in patient conditions. Therefore, allogeneic CAR-T cells may still be a suitable source, considering their short production cycle and lower cost. Furthermore, Dimitri et al. suggested that the use of CRISPR-Cas9 technology could genetically regulate allogeneic CAR-T cells and improve their persistence (112). Van der Stegen et al. found that induced pluripotent stem cells derived from T cells could contribute to the large-scale development of potent allogeneic CAR-T cells (113). Ex vivo culture time also impacts durability in the preparation of CAR-T cells. Ghassemi et al. found that shortening the ex vivo culture time of CAR-T cells reduced differentiation and improved their efficacy in acute lymphoblastic leukemia (114). Reducing ex vivo expansion time enhances CAR-T cell durability. Castella et al. developed ARI-0001, a closed semi-automatic bioreactor system, to expedite CAR-T cell culture for hematological malignancies, with potential implications for solid tumor applications (115). Combination therapy with other drugs is also a viable approach. PD-L1 inhibitors are believed to reverse the rapid depletion of CAR-T cells (25, 116). Chemotherapeutic agents can enhance CAR-T cell trafficking, infiltration, and anti-tumor efficacy in solid tumors; however, the dosage and side effects of these agents must be carefully considered (71, 94, 117). Zheng et al. suggested that PI3K inhibitors could maintain the hypofunctional state of CAR-T cells without affecting T cell expansion, thereby improving their persistence and reducing tumor burden (118). Additionally, Lei et al. found that the histone deacetylase (HDAC) inhibitor SAHA enhances the anti-tumor activity of B7-H3-targeted CAR-T cells in solid tumors (119). Therefore, these drugs have the potential to function as immunomodulators and optimize CAR-T therapies.
Side effects associated with CAR-T cells in osteosarcoma include CRS, off-target toxicity, GVHD, and increased MDSCs (43, 56, 69). CRS is the most common adverse event following CAR-T cell therapy and is caused by the release of large amounts of inflammatory factors (IL-6, IL-1, GM-CSF, etc.), which can lead to fever and, in severe cases, heart failure and death (120). GVHD and MDSCs are also frequently observed in in vivo osteosarcoma trials. Although CAR-T cell therapy for osteosarcoma is still in pre-clinical research, the dangers associated with its side effects should be taken seriously. IL-6 is currently considered a key factor in the development of CRS, and the IL-6 inhibitor tocilizumab has been approved for the treatment of CRS (121). Other studies have shown that IL-1 inhibitors and GM-CSF inhibitors can reduce the incidence of CRS (122, 123). Ghahri-Saremi et al. proposed that CRISPR-Cas9 technology could be used to produce CAR-T cells that knock down inflammatory factors to reduce the incidence of side effects (124). Additionally, introducing suicide genes into CAR-T cells can be an effective approach. Specifically, the expression of suicide genes, such as herpes simplex thymidine kinase (HSV-TK) and induced caspase 9 (iCasp9), can lead to the selective destruction and ablation of CAR-T cells, thus reducing the occurrence of off-target toxicity and side effects (125). Glucocorticoids may also be effective in suppressing CRS due to their ability to inhibit inflammatory responses. However, it should be noted that glucocorticoids can also impact the efficacy of CAR-T cells (126). The current approach to addressing GVHD and increased MDSCs involves the infusion of allogeneic CAR-T cells and the application of ATRA. However, further exploration of additional solutions is warranted.
The TME refers to the unique environment created by tumor cells, consisting of tumor cells, supporting stromal cells, microvasculature, cytokines, and chemokines (127). The TME contains more suppressive immune factors (MDSCs, tumor-associated macrophages, and regulatory T cells) than normal tissues and can impede T cell activity (128). In addition, the intercellular matrix produced by CAFs in the periphery of the TME and the surrounding microvasculature together form a physical barrier that limits the penetration of CAR-T cells (129). The “chemical barrier” (suppressive immune factors) and “physical barrier” (interstitial cells and microvasculature) both diminish the capacity of CAR-T cells to penetrate and infiltrate, thereby reducing their efficacy. The unique bone microenvironment in osteosarcoma, characterized by a porous extracellular matrix and abundant blood supply, leads to substantial immune cell infiltration, notably tumor-associated macrophages (TAM) and T lymphocytes, fostering an immune-tolerant setting for tumor chemotaxis. This increases the challenge of overcoming the “chemical barrier” (130, 131). One strategy to address the “chemical barrier” is to decrease the presence of suppressive immune factors using immune factor antibodies, such as MDSC antibodies, TAM antibodies, and regulatory T cell antibodies. Another approach involves employing CRISPR-Cas9 gene editing to target immune factor receptors and remodel the TME (104, 132). Fourth-generation CAR-T cells are now considered to play a regulatory role in the TME owing to their ability to secrete cytokines that stimulate the immune response around tumor cells (133). Some researchers have suggested that a combination of immune checkpoint inhibitors (PD-L1 inhibitors) can increase the number of immune checkpoints in T cells, thus reducing immunosuppression (134). Chemotherapeutic agents can also reduce the levels of immune factors in the TME (117). As for the “physical barrier,” targets that act on the tumor mesenchyme or microvasculature (FAP, CSPG4, etc.) may also improve the penetration of CAR-T cells in the TME (135). Additionally, local injection of CAR-T cells increases their concentration and improves their infiltration capacity (136). Furthermore, Li et al. found that the use of porous microneedle patches during local injection could further improve infiltration without a loss of activity, thus providing a reference for practical clinical applications (137).
CAR-T cell therapy has shown effectiveness against osteosarcoma in pre-clinical studies, and promising results have been observed in clinical trials targeting certain antigens. However, challenges such as low antigen specificity, limited durability, significant side effects, and an unfavorable TME hinder the wider application of CAR-T therapy in osteosarcoma. To overcome these limitations, future advancements may include the development of novel CAR-T cells, combination therapies with drugs, and gene editing techniques, aiming to enhance the efficacy and safety of CAR-T cell therapies for osteosarcoma treatment.
SL: Software, Writing – original draft. HZ: Funding acquisition, Writing – review & editing. GS: Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. 1. Scientific research project of Liaoning Provincial Department of Education (LJKMZ20221189). 2. Shenyang Science and Technology Project (21-173-9-24). 3. China Postdoctoral Science Foundation (2021M693912). 4. 345 Talent Project of Shengjing Hospital (M1367). 5. 345 Talent Project of Shengjing Hospital (M1378).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Meltzer PS, Helman LJ. New horizons in the treatment of osteosarcoma. N Engl J Med (2021) 385(22):2066–76. doi: 10.1056/NEJMra2103423
2. Gill J, Gorlick R. Advancing therapy for osteosarcoma. Nat Rev Clin Oncol (2021) 18(10):609–24. doi: 10.1038/s41571-021-00519-8
3. Harris MA, Hawkins CJ. Recent and ongoing research into metastatic osteosarcoma treatments. Int J Mol Sci (2022) 23(7):3817. doi: 10.3390/ijms23073817
4. Young RM, Engel NW, Uslu U, Wellhausen N, June CH. Next-generation CAR T-cell therapies. Cancer Discovery (2022) 12(7):1625–33. doi: 10.1158/2159-8290.CD-21-1683
5. Yang YH, Liu JW, Lu C, Wei JF. CAR-T cell therapy for breast cancer: from basic research to clinical application. Int J Biol Sci (2022) 18(6):2609–26. doi: 10.7150/ijbs.70120
6. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359(6382):1361–5. doi: 10.1126/science.aar6711
7. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
8. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn BC. CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol (2022) 13:925985. doi: 10.3389/fimmu.2022.925985
9. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel) (2023) 15(3):663. doi: 10.3390/cancers15030663
10. Lin Z, Wu Z, Luo W. Chimeric antigen receptor T-cell therapy: the light of day for osteosarcoma. Cancers (Basel) (2021) 13(17):4469. doi: 10.3390/cancers13174469
11. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev (2014) 257(1):83–90. doi: 10.1111/imr.12125
12. Kim DW, Cho JY. Recent advances in allogeneic CAR-T cells. Biomolecules (2020) 10(2):263. doi: 10.3390/biom10020263
13. Asmamaw Dejenie T, Tiruneh GMM, Dessie Terefe G, Tadele Admasu F, Wale Tesega W, Chekol Abebe E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccin Immunother (2022) 18(6):2114254. doi: 10.1080/21645515.2022.2114254
14. Lu J, Jiang G. The journey of CAR-T therapy in hematological Malignancies. Mol Cancer (2022) 21(1):194. doi: 10.1186/s12943-022-01663-0
15. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer (2018) 17(1):58. doi: 10.1186/s12943-018-0782-4
16. Oh DY, Bang YJ. HER2-targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol (2020) 17(1):33–48. doi: 10.1038/s41571-019-0268-3
17. Dhritlahre RK, Saneja A. Recent advances in HER2-targeted delivery for cancer therapy. Drug Discovery Today (2021) 26(5):1319–29. doi: 10.1016/j.drudis.2020.12.014
18. Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer (2020) 126(19):4278–88. doi: 10.1002/cncr.33102
19. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet (2020) 396(10251):635–48. doi: 10.1016/S0140-6736(20)31288-5
20. Wang SL, Zhong GX, Wang XW, Yu FQ, Weng DF, Wang XX, et al. Prognostic significance of the expression of HER family members in primary osteosarcoma. Oncol Lett (2018) 16(2):2185–94. doi: 10.3892/ol.2018.8931
21. Zhang Q, Liu F, Wang B, Li Z, Zhou D, Yang Q, et al. HER-2 expression in biopsy and surgical specimen on prognosis of osteosarcoma: A systematic review and meta-analysis of 16 studies. Med (Baltimore) (2016) 95(23):e3661. doi: 10.1097/MD.0000000000003661
22. Ebb D, Meyers P, Grier H, Bernstein M, Gorlick R, Lipshultz SE, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children's oncology group. J Clin Oncol (2012) 30(20):2545–51. doi: 10.1200/JCO.2011.37.4546
23. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther (2009) 17(10):1779–87. doi: 10.1038/mt.2009.133
24. Rainusso N, Brawley VS, Ghazi A, Hicks MJ, Gottschalk S, Rosen JM, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther (2012) 19(3):212–7. doi: 10.1038/cgt.2011.83
25. Park JA, Cheung NV. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J Hematol Oncol (2020) 13(1):172. doi: 10.1186/s13045-020-01012-y
26. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi: 10.1200/JCO.2014.58.0225
27. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
28. Alfaro-Arnedo E, Lopez IP, Pineiro-Hermida S, Canalejo M, Gotera C, Sola JJ, et al. IGF1R acts as a cancer-promoting factor in the tumor microenvironment facilitating lung metastasis implantation and progression. Oncogene (2022) 41(28):3625–39. doi: 10.1038/s41388-022-02376-w
29. Ameline B, Kovac M, Nathrath M, Barenboim M, Witt O, Krieg AH, et al. Overactivation of the IGF signalling pathway in osteosarcoma: a potential therapeutic target? J Pathol Clin Res (2021) 7(2):165–72. doi: 10.1002/cjp2.191
30. Li S, Zhang H, Liu J, Shang G. Targeted therapy for osteosarcoma: a review. J Cancer Res Clin Oncol (2023) 149(9):6785–97. doi: 10.1007/s00432-023-04614-4
31. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatr Blood Cancer (2014) 61(3):452–6. doi: 10.1002/pbc.24605
32. Huang X, Park H, Greene J, Pao J, Mulvey E, Zhou SX, et al. IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PloS One (2015) 10(7):e0133152. doi: 10.1371/journal.pone.0133152
33. Forbes BE, Blyth AJ, Wit JM. Disorders of IGFs and IGF-1R signaling pathways. Mol Cell Endocrinol (2020) 518:111035. doi: 10.1016/j.mce.2020.111035
34. Kipps TJ. ROR1: an orphan becomes apparent. Blood (2022) 140(14):1583–91. doi: 10.1182/blood.2021014760
35. Zhao Y, Zhang D, Guo Y, Lu B, Zhao ZJ, Xu X, et al. Tyrosine kinase ROR1 as a target for anti-cancer therapies. Front Oncol (2021) 11:680834. doi: 10.3389/fonc.2021.680834
36. Dai B, Shen Y, Yan T, Zhang A. Wnt5a/ROR1 activates DAAM1 and promotes the migration in osteosarcoma cells. Oncol Rep (2020) 43(2):601–8. doi: 10.3892/or.2019.7424
37. Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, et al. Analysis of ROR1 protein expression in human cancer and normal tissues. Clin Cancer Res (2017) 23(12):3061–71. doi: 10.1158/1078-0432.CCR-16-2083
38. Lee BK, Wan Y, Chin ZL, Deng L, Deng M, Leung TM, et al. Developing ROR1 targeting CAR-T cells against solid tumors in preclinical studies. Cancers (Basel) (2022) 14(15):3618. doi: 10.3390/cancers14153618
39. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell (2021) 39(2):193–208 e110. doi: 10.1016/j.ccell.2020.11.005
40. Xiao T, Xiao Y, Wang W, Tang YY, Xiao Z, Su M. Targeting ephA2 in cancer. J Hematol Oncol (2020) 13(1):114. doi: 10.1186/s13045-020-00944-9
41. Wilson K, Shiuan E, Brantley-Sieders DM. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene (2021) 40(14):2483–95. doi: 10.1038/s41388-021-01714-8
42. Giordano G, Merlini A, Ferrero G, Mesiano G, Fiorino E, Brusco S, et al. EphA2 expression in bone sarcomas: bioinformatic analyses and preclinical characterization in patient-derived models of osteosarcoma, ewing's sarcoma and chondrosarcoma. Cells (2021) 10(11):2893. doi: 10.3390/cells10112893
43. Hsu K, Middlemiss S, Saletta F, Gottschalk S, McCowage GB, Kramer B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther (2021) 28(3-4):321–34. doi: 10.1038/s41417-020-00221-4
44. Posthumadeboer J, Piersma SR, Pham TV, van Egmond PW, Knol JC, Cleton-Jansen AM, et al. Surface proteomic analysis of osteosarcoma identifies EPHA2 as receptor for targeted drug delivery. Br J Cancer (2013) 109(8):2142–54. doi: 10.1038/bjc.2013.578
45. Espie D, Donnadieu E. New insights into CAR T cell-mediated killing of tumor cells. Front Immunol (2022) 13:1016208. doi: 10.3389/fimmu.2022.1016208
46. Nicolosi PA, Dallatomasina A, Perris R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics (2015) 5(5):530–44. doi: 10.7150/thno.10824
47. Riccardo F, Tarone L, Iussich S, Giacobino D, Arigoni M, Sammartano F, et al. Identification of CSPG4 as a promising target for translational combinatorial approaches in osteosarcoma. Ther Adv Med Oncol (2019) 11:432496765. doi: 10.1177/1758835919855491
48. Rolih V, Barutello G, Iussich S, De Maria R, Quaglino E, Buracco P, et al. CSPG4: a prototype oncoantigen for translational immunotherapy studies. J Transl Med (2017) 15(1):151. doi: 10.1186/s12967-017-1250-4
49. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer (2014) 2:25. doi: 10.1186/2051-1426-2-25
50. Harrer DC, Dorrie J, Schaft N. CSPG4 as target for CAR-T-cell therapy of various tumor entities-merits and challenges. Int J Mol Sci (2019) 20(23):5942. doi: 10.3390/ijms20235942
51. Young O, Ngo N, Lin L, Stanbery L, Creeden JF, Hamouda D, et al. Folate receptor as a biomarker and therapeutic target in solid tumors. Curr Probl Cancer (2023) 47(1):100917. doi: 10.1016/j.currproblcancer.2022.100917
52. Yang R, Kolb EA, Qin J, Chou A, Sowers R, Hoang B, et al. The folate receptor alpha is frequently overexpressed in osteosarcoma samples and plays a role in the uptake of the physiologic substrate 5-methyltetrahydrofolate. Clin Cancer Res (2007) 13(9):2557–67. doi: 10.1158/1078-0432.CCR-06-1343
53. Zhu J, Simayi N, Wan R, Huang W. CAR T targets and microenvironmental barriers of osteosarcoma. Cytotherapy (2022) 24(6):567–76. doi: 10.1016/j.jcyt.2021.12.010
54. Lu Y, Xu LC, Parker N, Westrick E, Reddy JA, Vetzel M, et al. Preclinical pharmacokinetics, tissue distribution, and antitumor activity of a folate-hapten conjugate-targeted immunotherapy in hapten-immunized mice. Mol Cancer Ther (2006) 5(12):3258–67. doi: 10.1158/1535-7163.MCT-06-0439
55. Amato RJ, Shetty A, Lu Y, Ellis R, Low PS. A phase I study of folate immune therapy (EC90 vaccine administered with GPI-0100 adjuvant followed by EC17) in patients with renal cell carcinoma. J Immunother (2013) 36(4):268–75. doi: 10.1097/CJI.0b013e3182917f59
56. Lu YJ, Chu H, Wheeler LW, Nelson M, Westrick E, Matthaei JF, et al. Preclinical evaluation of bispecific adaptor molecule controlled folate receptor CAR-T cell therapy with special focus on pediatric Malignancies. Front Oncol (2019) 9:151. doi: 10.3389/fonc.2019.00151
57. Kontos F, Michelakos T, Kurokawa T, Sadagopan A, Schwab JH, Ferrone CR, et al. B7-H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res (2021) 27(5):1227–35. doi: 10.1158/1078-0432.CCR-20-2584
58. Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci (2020) 16(11):1767–73. doi: 10.7150/ijbs.41105
59. Nunes-Xavier CE, Karlsen KF, Tekle C, Pedersen C, Oyjord T, Hongisto V, et al. Decreased expression of B7-H3 reduces the glycolytic capacity and sensitizes breast cancer cells to AKT/mTOR inhibitors. Oncotarget (2016) 7(6):6891–901. doi: 10.18632/oncotarget.6902
60. Liu H, Tekle C, Chen YW, Kristian A, Zhao Y, Zhou M, et al. B7-H3 silencing increases paclitaxel sensitivity by abrogating Jak2/Stat3 phosphorylation. Mol Cancer Ther (2011) 10(6):960–71. doi: 10.1158/1535-7163.MCT-11-0072
61. Xie C, Liu D, Chen Q, Yang C, Wang B, Wu H. Soluble B7-H3 promotes the invasion and metastasis of pancreatic carcinoma cells through the TLR4/NF-kappaB pathway. Sci Rep (2016) 6:27528. doi: 10.1038/srep27528
62. Xie L, Chen C, Liang X, Xu J, Sun X, Sun K, et al. Expression and clinical significance of various checkpoint molecules in advanced osteosarcoma: possibilities for novel immunotherapy. Orthop Surg (2023) 15(3):829–38. doi: 10.1111/os.13620
63. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res (2019) 25(8):2560–74. doi: 10.1158/1078-0432.CCR-18-0432
64. Talbot LJ, Chabot A, Funk A, Nguyen P, Wagner J, Ross A, et al. A novel orthotopic implantation technique for osteosarcoma produces spontaneous metastases and illustrates dose-dependent efficacy of B7-H3-CAR T cells. Front Immunol (2021) 12:691741. doi: 10.3389/fimmu.2021.691741
65. Zhang Q, Zhang Z, Liu G, Li D, Gu Z, Zhang L, et al. B7-H3 targeted CAR-T cells show highly efficient anti-tumor function against osteosarcoma both in vitro and in vivo. BMC Cancer (2022) 22(1):1124. doi: 10.1186/s12885-022-10229-8
66. Prapa M, Chiavelli C, Golinelli G, Grisendi G, Bestagno M, Di Tinco R, et al. GD2 CAR T cells against human glioblastoma. NPJ Precis Oncol (2021) 5(1):93. doi: 10.1038/s41698-021-00233-9
67. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
68. Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer (2014) 120(4):548–54. doi: 10.1002/cncr.28461
69. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res (2016) 4(10):869–80. doi: 10.1158/2326-6066.CIR-15-0230
70. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4(1):e954829. doi: 10.4161/21624011.2014.954829
71. Chulanetra M, Morchang A, Sayour E, Eldjerou L, Milner R, Lagmay J, et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am J Cancer Res (2020) 10(2):674–87.
72. Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res (2017) 23(14):3499–509. doi: 10.1158/1078-0432.CCR-16-2138
73. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol (2018) 51:55–61. doi: 10.1016/j.coi.2018.02.004
74. Fuertes MB, Domaica CI, Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol (2021) 12:713158. doi: 10.3389/fimmu.2021.713158
75. Frazao A, Rethacker L, Messaoudene M, Avril MF, Toubert A, Dulphy N, et al. NKG2D/NKG2-ligand pathway offers new opportunities in cancer treatment. Front Immunol (2019) 10:661. doi: 10.3389/fimmu.2019.00661
76. Fernandez L, Valentin J, Zalacain M, Leung W, Patino-Garcia A, Perez-Martinez A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Lett (2015) 368(1):54–63. doi: 10.1016/j.canlet.2015.07.042
77. Fernandez L, Metais JY, Escudero A, Vela M, Valentin J, Vallcorba I, et al. Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin Cancer Res (2017) 23(19):5824–35. doi: 10.1158/1078-0432.CCR-17-0075
78. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.CIR-18-0572
79. VanSeggelen H, Hammill JA, Dvorkin-Gheva A, Tantalo DG, Kwiecien JM, Denisova GF, et al. T cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther (2015) 23(10):1600–10. doi: 10.1038/mt.2015.119
80. Sentman ML, Murad JM, Cook WJ, Wu MR, Reder J, Baumeister SH, et al. Mechanisms of acute toxicity in NKG2D chimeric antigen receptor T cell-treated mice. J Immunol (2016) 197(12):4674–85. doi: 10.4049/jimmunol.1600769
81. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res (2019) 7(1):100–12. doi: 10.1158/2326-6066.CIR-18-0307
82. Yang Y, Sanders AJ, Dou QP, Jiang DG, Li AX, Jiang WG. The clinical and theranostic values of activated leukocyte cell adhesion molecule (ALCAM)/CD166 in human solid cancers. Cancers (Basel) (2021) 13(20):5187. doi: 10.3390/cancers13205187
83. Ferragut F, Vachetta VS, Troncoso MF, Rabinovich GA, Elola MT. ALCAM/CD166: A pleiotropic mediator of cell adhesion, stemness and cancer progression. Cytokine Growth Factor Rev (2021) 61:27–37. doi: 10.1016/j.cytogfr.2021.07.001
84. Darvishi B, Boroumandieh S, Majidzadeh AK, Salehi M, Jafari F, Farahmand L. The role of activated leukocyte cell adhesion molecule (ALCAM) in cancer progression, invasion, metastasis and recurrence: A novel cancer stem cell marker and tumor-specific prognostic marker. Exp Mol Pathol (2020) 115:104443. doi: 10.1016/j.yexmp.2020.104443
85. Federman N, Chan J, Nagy JO, Landaw EM, McCabe K, Wu AM, et al. Enhanced growth inhibition of osteosarcoma by cytotoxic polymerized liposomal nanoparticles targeting the alcam cell surface receptor. Sarcoma (2012) 2012:126906. doi: 10.1155/2012/126906
86. Cardenes B, Clares I, Bezos T, Toribio V, Lopez-Martin S, Rocha A, et al. ALCAM/CD166 is involved in the binding and uptake of cancer-derived extracellular vesicles. Int J Mol Sci (2022) 23(10):5753. doi: 10.3390/ijms23105753
87. Wang Y, Yu W, Zhu J, Wang J, Xia K, Liang C, et al. Anti-CD166/4-1BB chimeric antigen receptor T cell therapy for the treatment of osteosarcoma. J Exp Clin Cancer Res (2019) 38(1):168. doi: 10.1186/s13046-019-1147-6
88. Michel L, Grasmuck C, Charabati M, Lecuyer MA, Zandee S, Dhaeze T, et al. Activated leukocyte cell adhesion molecule regulates B lymphocyte migration across central nervous system barriers. Sci Transl Med (2019) 11(518):eaaw0475. doi: 10.1126/scitranslmed.aaw0475
89. Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature (2017) 552(7683):110–5. doi: 10.1038/nature24676
90. Nishina T, Deguchi Y, Ohshima D, Takeda W, Ohtsuka M, Shichino S, et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat Commun (2021) 12(1):2281. doi: 10.1038/s41467-021-22450-3
91. Lewis VO. IL-11Ralpha: a novel target for the treatment of osteosarcoma. Adv Exp Med Biol (2014) 804:285–9. doi: 10.1007/978-3-319-04843-7_15
92. Lewis VO, Devarajan E, Cardo-Vila M, Thomas DG, Kleinerman ES, Marchio S, et al. BMTP-11 is active in preclinical models of human osteosarcoma and a candidate targeted drug for clinical translation. Proc Natl Acad Sci U.S.A. (2017) 114(30):8065–70. doi: 10.1073/pnas.1704173114
93. Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res (2012) 72(1):271–81. doi: 10.1158/0008-5472.CAN-11-2778
94. Jiang J, Wang R, Yang L, Sha Y, Zhao S, Guo J, et al. IL-11Ralpha-targeted nanostrategy empowers chemotherapy of relapsed and patient-derived osteosarcoma. J Control Release (2022) 350:460–70. doi: 10.1016/j.jconrel.2022.08.048
95. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101(1):147–76. doi: 10.1152/physrev.00048.2019
96. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer (2020) 146(4):895–905. doi: 10.1002/ijc.32193
97. Bughda R, Dimou P, D'Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther (2021) 10:313–23. doi: 10.2147/ITT.S291767
98. Shahvali S, Rahiman N, Jaafari MR, Arabi L. Targeting fibroblast activation protein (FAP): advances in CAR-T cell, antibody, and vaccine in cancer immunotherapy. Drug Delivery Transl Res (2023) 13(7):2041–56. doi: 10.1007/s13346-023-01308-9
99. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
100. Zhang L, Yang L, Xia ZW, Yang SC, Li WH, Liu B, et al. The role of fibroblast activation protein in progression and development of osteosarcoma cells. Clin Exp Med (2020) 20(1):121–30. doi: 10.1007/s10238-019-00591-6
101. Miao L, Zhang Z, Ren Z, Tang F, Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front Immunol (2021) 12:687822. doi: 10.3389/fimmu.2021.687822
102. Koksal H, Muller E, Inderberg EM, Bruland O, Walchli S. Treating osteosarcoma with CAR T cells. Scand J Immunol (2019) 89(3):e12741. doi: 10.1111/sji.12741
103. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2018) 20(4):506–18. doi: 10.1093/neuonc/nox182
104. Daei Sorkhabi A, Mohamed Khosroshahi L, Sarkesh A, Mardi A, Aghebati-Maleki A, Aghebati-Maleki L, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol (2023) 14:1113882. doi: 10.3389/fimmu.2023.1113882
105. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell (2019) 35(3):489–503 e488. doi: 10.1016/j.ccell.2019.02.003
106. Lee YG, Marks I, Srinivasarao M, Kanduluru AK, Mahalingam SM, Liu X, et al. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res (2019) 79(2):387–96. doi: 10.1158/0008-5472.CAN-18-1834
107. Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T cell in adoptive cancer immunotherapy: challenges and ways forward. Front Immunol (2020) 11:702. doi: 10.3389/fimmu.2020.00702
108. Zhou Z, Tao C, Li J, Tang JC, Chan AS, Zhou Y. Chimeric antigen receptor T cells applied to solid tumors. Front Immunol (2022) 13:984864. doi: 10.3389/fimmu.2022.984864
109. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi: 10.1172/JCI83416
110. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res (2022) 10(1):86. doi: 10.1186/s40364-022-00434-9
111. DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19(+) Malignancies take autologous CAR T cells 'off the shelf'? Nat Rev Clin Oncol (2021) 18(4):195–6. doi: 10.1038/s41571-021-00485-1
112. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer (2022) 21(1):78. doi: 10.1186/s12943-022-01559-z
113. van der Stegen SJC, Lindenbergh PL, Petrovic RM, Xie H, Diop MP, Alexeeva V, et al. Generation of T-cell-receptor-negative CD8alphabeta-positive CAR T cells from T-cell-derived induced pluripotent stem cells. Nat BioMed Eng (2022) 6(11):1284–97. doi: 10.1038/s41551-022-00915-0
114. Ghassemi S, Nunez-Cruz S, O'Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res (2018) 6(9):1100–9. doi: 10.1158/2326-6066.CIR-17-0405
115. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-of-care CAR T-cell production (ARI-0001) using a closed semi-automatic bioreactor: experience from an academic phase I clinical trial. Front Immunol (2020) 11:482. doi: 10.3389/fimmu.2020.00482
116. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. doi: 10.1172/JCI83092
117. Wang AX, Ong XJ, D'Souza C, Neeson PJ, Zhu JJ. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front Immunol (2023) 14:1140541. doi: 10.3389/fimmu.2023.1140541
118. Zheng W, O'Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia (2018) 32(5):1157–67. doi: 10.1038/s41375-017-0008-6
119. Lei X, Ou Z, Yang Z, Zhong J, Zhu Y, Tian J, et al. A pan-histone deacetylase inhibitor enhances the antitumor activity of B7-H3-specific CAR T cells in solid tumors. Clin Cancer Res (2021) 27(13):3757–71. doi: 10.1158/1078-0432.CCR-20-2487
120. Chen H, Wang F, Zhang P, Zhang Y, Chen Y, Fan X, et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front Med (2019) 13(5):610–7. doi: 10.1007/s11684-019-0714-8
121. Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol (2019) 15(8):813–22. doi: 10.1080/1744666X.2019.1629904
122. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
123. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood (2019) 133(7):697–709. doi: 10.1182/blood-2018-10-881722
124. Ghahri-Saremi N, Akbari B, Soltantoyeh T, Hadjati J, Ghassemi S, Mirzaei HR. Genetic modification of cytokine signaling to enhance efficacy of CAR T cell therapy in solid tumors. Front Immunol (2021) 12:738456. doi: 10.3389/fimmu.2021.738456
125. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer (2019) 18(1):125. doi: 10.1186/s12943-019-1057-4
126. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med (2014) 6(224):224ra225. doi: 10.1126/scitranslmed.3008226
127. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi: 10.1038/nm.3394
128. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis (2018) 9(3):282. doi: 10.1038/s41419-018-0278-6
129. Santi A, Kugeratski FG, Zanivan S. Cancer associated fibroblasts: the architects of stroma remodeling. Proteomics (2018) 18(5-6):e1700167. doi: 10.1002/pmic.201700167
130. Lu Y, Zhang J, Chen Y, Kang Y, Liao Z, He Y, et al. Novel immunotherapies for osteosarcoma. Front Oncol (2022) 12:830546. doi: 10.3389/fonc.2022.830546
131. Tian H, Cao J, Li B, Nice EC, Mao H, Zhang Y, et al. Managing the immune microenvironment of osteosarcoma: the outlook for osteosarcoma treatment. Bone Res (2023) 11(1):11. doi: 10.1038/s41413-023-00246-z
132. Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res (2018) 24(12):2920–34. doi: 10.1158/1078-0432.CCR-17-1365
133. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics (2018) 8:41–51. doi: 10.1016/j.omto.2017.12.003
134. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7(23):34341–55. doi: 10.18632/oncotarget.9114
135. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
136. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med (2020) 26(5):712–9. doi: 10.1038/s41591-020-0821-8
Keywords: CAR-T, osteosarcoma, immunotherapy, targeted therapy, tumor microenvironment
Citation: Li S, Zhang H and Shang G (2023) Current status and future challenges of CAR-T cell therapy for osteosarcoma. Front. Immunol. 14:1290762. doi: 10.3389/fimmu.2023.1290762
Received: 08 September 2023; Accepted: 07 December 2023;
Published: 22 December 2023.
Edited by:
Ru Zhou, University of North Carolina at Charlotte, United StatesReviewed by:
Nancy Gordon, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Li, Zhang and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guanning Shang, Y211c2hhbmdnbkAxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.