
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 January 2024
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1287512
This article is part of the Research TopicAdvances in Understanding Lung Injury and Repair during Immune ResponsesView all 3 articles
 Adriana Ribeiro Silva1
Adriana Ribeiro Silva1 Kauê Franscisco Correa de Souza e Souza2
Kauê Franscisco Correa de Souza e Souza2 Thamires Bandeira De Souza2,3
Thamires Bandeira De Souza2,3 Mauricio Younes-Ibrahim4
Mauricio Younes-Ibrahim4 Patrícia Burth3
Patrícia Burth3 Hugo Caire de Castro Faria Neto1
Hugo Caire de Castro Faria Neto1 Cassiano Felippe Gonçalves-de-Albuquerque1,2*
Cassiano Felippe Gonçalves-de-Albuquerque1,2*Acute respiratory distress syndrome (ARDS) is marked by damage to the capillary endothelium and alveolar epithelium following edema formation and cell infiltration. Currently, there are no effective treatments for severe ARDS. Pathologies such as sepsis, pneumonia, fat embolism, and severe trauma may cause ARDS with respiratory failure. The primary mechanism of edema clearance is the epithelial cells’ Na/K-ATPase (NKA) activity. NKA is an enzyme that maintains the electrochemical gradient and cell homeostasis by transporting Na+ and K+ ions across the cell membrane. Direct injury on alveolar cells or changes in ion transport caused by infections decreases the NKA activity, loosening tight junctions in epithelial cells and causing edema formation. In addition, NKA acts as a receptor triggering signal transduction in response to the binding of cardiac glycosides. The ouabain (a cardiac glycoside) and oleic acid induce lung injury by targeting NKA. Besides enzymatic inhibition, the NKA triggers intracellular signal transduction, fostering proinflammatory cytokines production and contributing to lung injury. Herein, we reviewed and discussed the crucial role of NKA in edema clearance, lung injury, and intracellular signaling pathway activation leading to lung inflammation, thus putting the NKA as a protagonist in lung injury pathology.
Acute respiratory distress syndrome (ARDS) is a lethal or disabling clinical syndrome triggered by sepsis, pneumonia, and severe trauma (1) with a high morbidity and mortality rate (2). The first report of patients with ARDS occurred in Denver, Colorado, in 1967 (3). According to the updated Berlin definition, ARDS is a syndrome with bilateral diffuse infiltrates on the chest followed by non-cardiogenic respiratory failure with mild, moderate, or severe oxygenation impairment (4). Also, ARDS pathology is characterized as damage to the capillary endothelium and alveolar epithelium with infiltrate accumulation in the alveolar space, forming interstitial and alveolar edema (5).
Fluid management is one of the most critical measures impacting ARDS, and dynamic monitoring of the lung fluid balance seems to influence clinical outcomes (6). Alveolar edema clearance depends on the vectorial transport of sodium and water across the alveolar epithelium. It happens, in part, through apically located sodium channels (ENaC) that drain the liquid into the lung interstitium via the basolaterally located Na/K-ATPase (NKA) and next to the capillary net through specific transcellular channels, as the aquaporins (7). Pathological changes in the alveolar-capillary barrier occur during pulmonary infection, altering NKA expression and ion channels in epithelial cells, causing edema and impairing alveolar fluid clearance (8).
Inhibitory NKA molecules are potential candidates to induce lung injury (9) because they block alveolar edema fluid clearance, which depends on the vectorial transport of sodium (7, 10). Molecules altering NKA activity may induce or increase lung edema and inflammation. Therefore, we link cell signaling induced by NKA to triggering lung inflammation.
The mammal’s respiratory system comprises the conductive and respiratory portions, including the respiratory bronchiole and alveoli (11). The nasal cavity is lined by pseudostratified columnar ciliated cells, the bronchioles lined by simple columnar, cuboidal epithelium, and the alveoli lined by a thin squamous epithelium (12) formed by squamous type I cells and cuboidal type II cells. Those cells are essential to homeostasis maintenance, antimicrobial control, host defense, pathogen recognition (13), and tissue repair (14).
The squamous type I cells are fragile and have large cytoplasmic extensions, ideal for gas exchange (15). Also, it plays an active role in water permeability and regulating alveolar fluid homeostasis through the NKA, ENaC, and water channels of the aquaporin 3 and 5 (AQP3 and AQP5) (16). Cuboidal type II cells play a crucial role in the lung immune response, participating in the tissue repair process after pulmonary injury, producing and secreting pulmonary surfactant, promoting transepithelial water movement, and expressing immunomodulatory proteins for host defense (15). In transepithelial water flux, these cells remove fluid from the alveolar space through sodium transport ions by NKA and ENaC channels (Figure 1) (17).
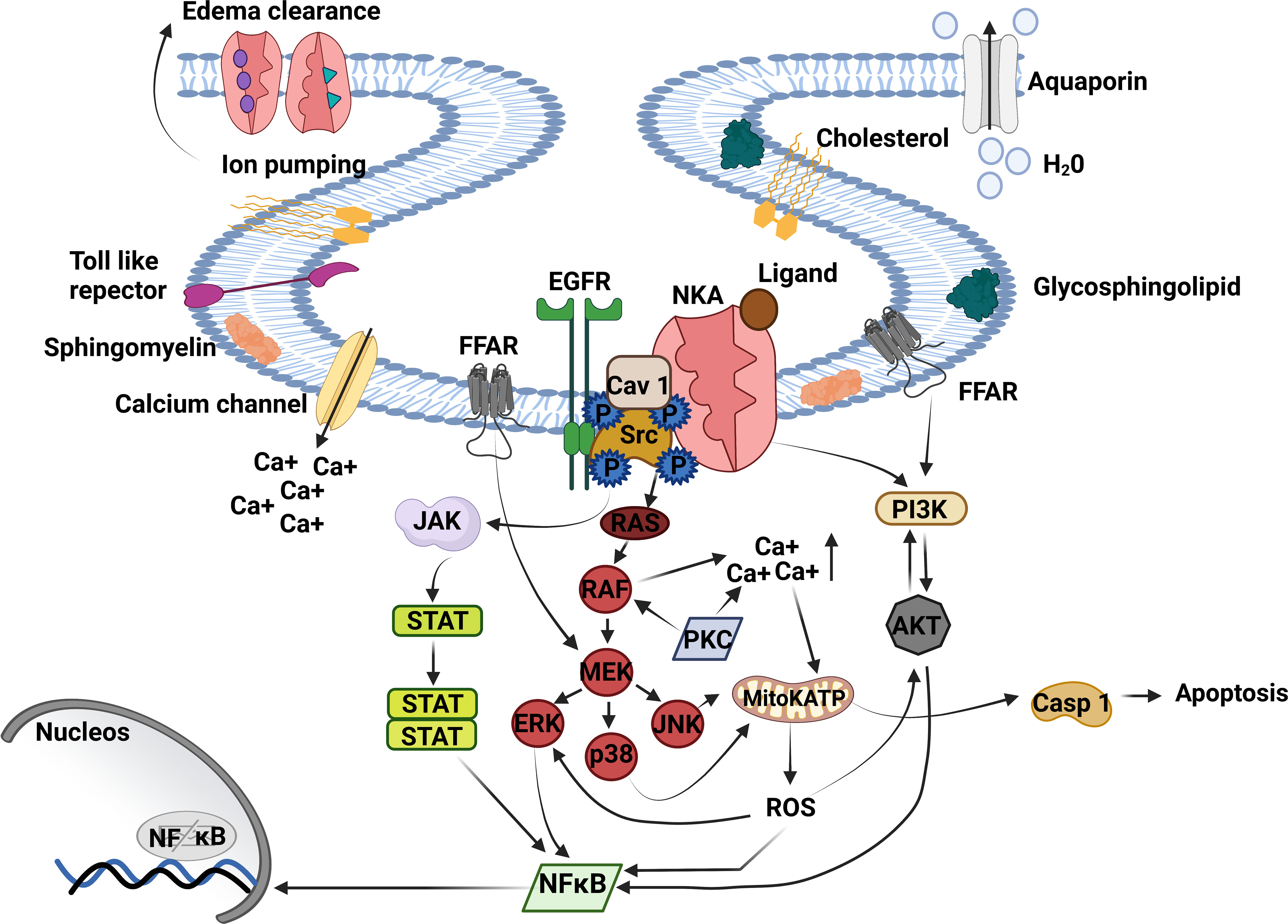
Figure 1 Na+/K+-ATPase signalosome. NKA acts as an ion pump and signal transducer, triggering lung inflammation. The binding of cardiac glycosides, GLP, oleic, and linoleic acid to NKA signalosome located in caveolae transduces signals to multiple pathways. The CG binding to NKA activates Src tyrosine kinase – EGFR complex is associated with inflammatory response and proinflammatory mediator production. Activated EGFR recruits’ protein adaptors that activate Ras - RAF (MAPK). RAF directly regulates MEK transactive JNK, p38, and ERK being the last activating NFκB. MAPK activation triggers the opening of mitochondrial ATP-sensitive potassium channels (mitoKATP) through increased intracellular Ca++, producing ROS production and casp 1 activation. ROS release stimulates ERK, AKT, and NFκB activation. Moreover, AKT can activate PI3K and NFκB or be phosphorylated by PI3K, essential to inflammation and edema formation. Another critical pathway that triggers lung injury is JAK/STAT3. JAK/STAT3 is regulated by Src upstream and ends with NFκB activation. NKA, Na+, K+ ATPase; Ca++, calcium; Src, non-receptor tyrosine kinases; EGFR, epithelial growth factor receptor; JAK, Janus kinase; STAT, signal transducer and activation of transcription; RAS, rat sarcoma virus; RAF, proto-oncogene serine; MAPK, mitogen-activated protein kinase; MEK, MAPK–ERK activating kinase; ERK, extracellular signal-regulated kinases; p38, mitogen-activated protein kinase; JNK, c-Jun n-terminal kinase; mitoKATP, mitochondrial ATP-sensitive potassium channel; ROS, reactive oxygen species; Casp-1, caspase-1; NFκB, factor nuclear kappa B; PI3K, phosphoinositide 3′ kinase; AKT, protein B kinase; FFAR, free fatty acid receptor. Created with BioRender.com.
NKA maintains the electrochemical gradient and cell homeostasis (18) through ions across the cell membrane (19). Its structure is formed by three subunits, α subunit or catalytic (4 isoforms), β subunit or regulator (4 isoforms), and γ subunit (FXYD1-7 proteins) that is auxiliary to the holoenzyme complex with regulatory function (20). Like the NKA isoforms, each NKA subunit is expressed tissue- and cell-specific. For example, the α1 and β1 isoform NKA is the predominant isozyme expressed in alveolar epithelial cells type I and II, and the α2 subunit is present in alveolar epithelial type I cells (21). The β3 isoform is found in rat lungs, but its function remains unknown (22).
Alveolar cells NKA are located on the basolateral membrane and actively contribute to alveolar edema clearance (7). Additionally, ENaC and cystic fibrosis transmembrane conductance regulator (CFTR) are located apically (23, 24), contributing to edema clearance. Finally, the AQP5 transports water across the apical membrane in epithelial cells (16). However, the foremost mechanism of lung edema clearance is the transport of ion sodium and ion chloride across epithelial cells type I and II (7, 25), bringing water from the lungs into the capillaries (Figure 1).
NKA also has an essential role in regulation and the formation of tight junctions (26). Tight junctions are barriers between cells that control the passage of substances between alveolar and interstitial spaces through active transport (27, 28). Abnormal regulation of tight junction barrier allows the passage of pathogens and antigens through epithelial monolayers favoring disease development (28). Tight junctions also act as scaffolding platforms for cell signaling and docking stations for transport vesicles (29). Structurally, they comprise heteromeric occludin and claudin protein complexes that form a sealed interface between adjacent airway epithelial cells (30).
Together with the adapter proteins, the tight junctions can regulate tension, selective permeability, cell signaling, and gene expression (31, 32). However, insults such as lung injury (25), mechanical stress (33), fungal infection (34), viral and bacterial infection (35), and kinases activation (36) cause dysregulation tight endothelial junctions (37, 38) worsening lung disease.
The NKA β1 subunit overexpression upregulates tight epithelial junctions decreasing lung permeability independent of NKA pumping function (39). Also, NKA effects on tight junction formation depend on sodium concentration in the cell, preventing the formation of the stress-bundled fibers (40).
Sodium increase during NKA inhibition changes epithelial cells’ polarity (41). The disruption of tight junction complexes at the lung epithelial cells is deleterious and leads to lung edema formation in ARDS (42). In addition, establishing the tight junction drives epithelial polarization (43). Thus, the NKA influences the epithelial polarity, regulation, and tight junctions, strengthening the alveolar-epithelial barrier in a mechanism involving β1 subunit and myotonic dystrophy kinase-related cdc42-binding kinase α (39). Conversely, NKA inhibition leads to tight junctions’ integrity loss, increasing the alveolar permeability and worsening acute lung injury/ARDS.
Cardiac glycosides are known worldwide as classical inhibitors of the NKA (44). They were identified as a plant secondary metabolite, and later on as an endogenous mammalian substance produced by adrenal gland, hypothalamus, and hypophysis (45). The cardiac glycosides bind to the NKA α subunit, increasing the intracellular concentration of ions calcium and sodium (44), resulting in reactive oxygen species (ROS) production due to intracellular signaling activation (46). Also, they inhibit ion-pumping (7), decreasing edema fluid clearance (8). The cardiac glycoside concentration directly impacts NKA’s effect, triggering cell signaling at low concentrations and blocking the ionic pump at high concentrations (47). Our group recently reviewed the role of NKA in cell adhesion, motility, and migration in cancer cells (48). Cardiac glycosides induce apoptosis and autophagy in transformed cells, involving molecular pathways inducing deleterious effects on lung cancer cells (48). In addition, different cardiac glycoside concentrations can also have pro- or anti-inflammatory effects on lung (45).
In 1985, Tamura and co-workers showed that free unsaturated fatty acid such as linoleic acid (LA) and oleic acid (OA) in plasma inhibits NKA promoting increased extracellular fluid accumulation in hogs (49). Also, OA inhibits the NKA in vitro (50, 51). Furthermore, our group and others showed that lung NKA inhibition induces lung injury and an inflammatory microenvironment in the lungs (52–56). Ouabain, a cardiac glycoside, induced lung injury in mice (52). In addition, OA in vivo causes lung injury with immune cell accumulation, edema formation, inflammatory mediators production that amplify inflammatory response, such as tumor necrosis factor alfa (TNF-α), interleukin 1 beta (IL-1β), leukotriene B4-(LTB4), and intracellular pathways activation ((mitogen-activated protein kinase (MAPK), phosphoinositide 3’ kinase (PI3K), extracellular signal-regulated kinase 1/2 (ERK1/2), factor nuclear kappa B (NF-κB)) in lung tissues (56, 57). Thus, these data reinforce the importance of NKA activity for lung edema clearance and make it a potential molecular target for lung injury treatment (Figure 1).
Changes in the alveolar–capillary barrier happen during pulmonary infection and cause altered expression of epithelial NKA, decreasing alveolar fluid clearance. TGF-β, TNF-α, interferons, or IL-1β are produced after infection with Streptococcus pneumoniae, Klebsiella pneumoniae, Mycoplasma pneumoniae, influenza A virus, pathogenic coronaviruses, or respiratory syncytial virus and amplify alveoli-capillary disturbances and damage (8).
Cardiac glycosides activate the signalosome, a NKA protein complex restricted to the caveolae, triggering intracellular signaling pathways (19). The cell signaling from NKA occurs through the interaction of signalosome proteins such as NKA, caveolin-1, non-receptor tyrosine kinases (Src), rat sarcoma virus protein (RAS) binding of small guanosine triphosphate (GTP) (19), and epidermal growth factor receptor (EGFR) (58). EGFR is a transmembrane protein with cytoplasmic kinase activity that stimulates cell proliferation, differentiation, growth, and migration (59). Caveolin-1, an integral protein cell membrane, interacts with several tyrosine kinases, like EGFR, and connects Src kinase in the MAPK pathway activation. Src belongs to the Src family kinase (SFKs) (60). The caveolin-1 forms a spontaneous complex with NKA and interacts with Src kinase forming the active NKA-caveolin-1-Src Figure 1 (58).
In addition, Src kinases and NKA act together to transduce outside-in signaling by ouabain (19). Then, Src binds to the α-subunit of NKA and triggers the Src/EGFR-MAPK pathway signaling (61). Src activation affects different cellular processes such as adhesion, migration, cell differentiation (60), and ROS formation (61). EGFR activates Ras-proto-oncogene serine (Raf)-MAPK, PI3K (59), which increases ROS production (62), NFκB activation, and proinflammatory cytokine production (63). Activation of the PI3K pathway by ouabain can also result in the endocytosis of the NKA-Src complex (19), blocking NKA signaling in the myocyte. Also, ouabain triggers EGFR-Src-Ras-Raf-MAPK–ERK activating kinase (MEK)-extracellular signal-regulated kinases (ERK1/2) pathway and Src-independent activation of PI3K1A and Akt, leading to fibroblast proliferation (Figure 1) (64).
The cardiac glycosides activate Src and trigger the ERK 1/2 signaling through the Ras-Raf-MEK pathway-independent intracellular calcium fluctuations (65). ERK activation induces the production of proinflammatory mediators and can be activated by MEK-MAPK (66), EGFR (67), and ROS (68). Also, EGFR transactivation by Src activates ERK 1/2 through the activation of Ras-Raf-MEK cascade (69). In addition, the MAPK activation induces an increase in calcium concentration, favoring the opening of mitochondrial adenosine triphosphate (ATP)-sensitive K+ channels, which increases nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity and generates mitochondrial ROS (70).
The specific NKA cell signaling is related to disease etiology (71). Despite several studies denoting the link between NKA and intracellular signaling pathways activation (72), some details in the signaling cascade remain to be unveiled.
Summarizing what has been introduced before, the NKA activity on edema fluid clearance is crucial to lung injury (7, 9). The impairment of the alveoli-capillary barrier and damage to the alveolar epithelium occur during acute lung injury and ARDS, resulting in the accumulation of protein-rich edema fluid (73), impairing gas exchange (7). Epithelium cells type I and II, through NKA activity, remove salt and water from the alveoli (74). Furthermore, cell signaling from NKA of the epithelium cells type I and II activate proinflammatory pathways in the lung, which is harmful to the lung. Lung injury happens by inhibiting or triggering intracellular signaling. Interestingly the ion-pumping function increases due to NKA cell signaling activation (75). In addition, cardiac glycosides, OA, and LA in the lung inhibit NKA and induce injury through NKA signaling cascade activation (55, 76) (77) (Figure 1).
Besides cardiac glycosides, FXYD proteins modulate NKA activity. FXYD5 overexpression in mice damages the alveoli-epithelial barrier and causes lung inflammation. LPS stimulation of alveolar epithelial cells and mice lungs increased FXYD5 plasma membrane expression, NF-κB activation, and cytokine production. FXYD5-deficient cells have not responded to LPS. FXYD5 overexpression increased the monocyte migration to the lung, and in turn, FXYD5 silent mice showed less CCL2, monocytes, and protein extravasation after LPS stimulation. As it occurred in vitro, the FXYD5 effect depended on NF-κB, and they were not just involved in the LPS-inflammatory effect and cytokines, reinforcing the role of a NKA modulator in inflammation (78).
NKA activates Src and ERK. Activating Src and ERK leads to lung injury and inflammation (79). Src activates ERK signaling (80) and induces proinflammatory cytokine production (81). Also, neutrophil migration, edema, lipid body formation, and cytokine IL-6 production in lung injury depend on ERK activation in an OA-induced lung injury animal model (57). Furthermore, JNK, p38, p65 (77), and p50 phosphorylation (82) is linked to a decrease of lung alveolar permeability and neutrophil accumulation in the bronchoalveolar lavage fluid (BALF). Inhibiting those pathways reduces lung vascular leak and suppresses the NF-kB pathway (82).
ARDS patients that express high angiotensin-converting enzyme (ACE) in BALF have Ras phosphorylated (83, 84). Furthermore, it suggested that Ras protein regulates pulmonary vascular tone, which can contribute to the pathogenesis of ARDS (84). Raf and MEK phosphorylation and the release of IL-1β, IL-4, IL-6, and TNFα are increased in LPS-induced acute lung injury (85). In addition, RAF activates JNK and p38 and ERK, activating NF-κB, contributing to increased lung permeability and inflammation in ARDS (86).
The signaling of cardiac glycosides occurred through the NKA/α-1-Src kinase complex in immune cells to produce ROS and inflammatory cytokines. NKA α-1 knock-down or the specific inhibition of the NKA α-1-Src kinase complex by pNaKtide, or even the Src inhibitor PP2 impaired the inflammatory effect of cardiac glycoside telecinobufagin (TCB) in macrophages. TCB, ouabain, digoxin, and marinobufagenin activated NF-κB via NKA α-1-Src kinase complex (87).
PI3K, a NKA downstream signaling protein, is essential in lung inflammation and edema (88). Its inhibition may alleviate lung injury with lower leukocyte accumulation and edema formation in the lungs (56). PI3K, through AKT activation, regulates broad cellular processes, including apoptosis, proliferation, and differentiation (89).
Different pathways activated by NKA signaling are similar to LPS-triggered ones and crosstalk with pathways triggered by LPS during lung cell and immune cell stimulation. Ouabain activated NF-κB, leading to proinflammatory cytokine synthesis (MCP-1/CCL-2, TNF-α, IL-1β, and IL-6) by murine peritoneal macrophages and human monocyte-derived macrophages. Macrophages partially deficient in NKA lacked ouabain-induced NF-κB activation, and consequently the synthesis of proinflammatory mediators (90). Ouabain in fact may have either pro or anti-inflammatory role (76, 91, 92). Ouabain challenged the monocytes to produced IL-1β, TNF-α, IL-10 and VEGF, showing its immunomodulatory role (92). Ouabain also inhibits proinflammatory monocyte activation, downregulating membrane CD14 and CD16 in early time points (91). Others showed NKA binding of cardiac glycosides leads to IL-1β production and release. The mechanisms of the cardiac glycoside digoxin and others include activation of the NLRP3 inflammasome in macrophages at concentrations used in clinical treatment. The result is the induction of the programmed cell death pathway through caspase-1, called pyroptosis. Which causes inflammation and tissue damage showing the direct correlation of NKA signaling to the induction of IL-1β via inflammasome (93). In turn, inhibiting inflammasome activity reduced ischemic damage (94). IL-1β expression in the lungs is associated with neutrophil recruitment and induction of acute lung injury (95), acting as a biomarker of barrier integrity loss in the lungs via interaction with EGFR and claudins (96). Also, IL-1β stimulates the IL-6 and TNF-α release (97). The claudins are proteins that form intercellular junctions supporting the permeability and ion selectivity of paracellular pores of the large airways (98). Among them, cldn-18 is highly expressed in the alveolar epithelium, while cldn-4 is vital in regulating paracellular permeability during alveolar fluid clearance supporting the resolution edema (25). In addition, IL-18 controls the release of IL-1β (97), being both (IL-1β and IL-18) activated by caspase-1 (97), which is critical to the development of sepsis/ARDS (99).
The leptospiral endotoxin glycolipoprotein (GLP) binds to NKA and causes organ dysfunctions during leptospirosis. Metabolic alterations increase free fatty acid levels in the blood and lipotoxicity. During the infection, inflammasomes are formed, and NKA once again leads to pro-inflammatory and metabolic alterations and involves inflammasome activation. NKA is related to the severity and considered a therapeutic target (100).
Activation of NKA triggers JAK/STAT3 pathway signaling, an inductor of lung injury (101, 102). STAT activation induces IL-6 release (103), and mutation in STAT3 impairs IL-6 activity and recurrent infections in the lungs (104). Also, IL-6 and TNF-α release are regulated by JNK (105) and p38 (77, 106). The p38 causes deleterious effects and cell death (107). When p38 is activated, the lung neutrophils induce tissue damage through the secretion of ROS and IL-8 (108), promoting endothelial and epithelial barrier dysfunction (109), and worsening ARDS. ROS upregulates the expression of proinflammatory cytokines and adhesion molecules, amplifying the tissue damage and pulmonary edema (109). Once released, IL-8 decreased cell viabilityand inhibited the expression of surfactant proteins A and B (33). The absence of surfactant protein in the lung increases the surface tension, resulting in the alveolar and peripheral airway collapse (110). TNF-α also downregulates the surfactant protein (111), potentializes the release of chemokines and cytokines, increases lung vascular permeability, and modulates the recruitment and activation of neutrophils, contributing to lung injury. NKA signaling activation may induce multiple signaling cascades that may cause lung injury and cause the production of proinflammatory mediators by structural cells and recruited leukocytes that amplify the inflammatory response in the lung (Figure 1). Further research may unveil additional molecular mechanisms and other receptors involved in the lung injury processes.
Fatty acids, such as the NKA ligands LA and OA, also bind to free fatty acid receptors (FFARs). Four FFARs have physiological importance in biological processes (FFAR1, FFAR2, FFAR3, and FFAR4, formerly known as GPR40, 43, 41, and 120) (112, 113). The long-chain free fatty acid (FFA) preferentially binds to FFAR1 and FFAR4, while short-chain fatty acid binds to FFAR2 and FFAR3 (114). Also, FFARs are similarly expressed in metabolic tissues and immune cells, regulating energetic metabolism and inflammatory responses (115). The FFARs exhibit overlapping functions through signaling pathways involving the activation of Ca+2, cAMP, or ERK1/2 responses, with G protein-dependent or independent pathways acting more as modulators rather than initiators of biological processes (116). For instance, OA generates an aggressive phenotype in prostate cancer cells via the PI3K/Akt signaling pathway depending on FFA1/GPR40 (117).
Non-Esterified Fatty Acids (NEFA) are FFA with elevated levels in obese individuals, and long-chain FFAs act as endogenous ligands of the FFAR1. OA, LA, and GW9508 (FFAR1/FFAR4 dual agonist) induced human airway smooth muscle cell proliferation, dependent on p70S6K phosphorylation through MEK/ERK and PI3K/Akt signaling pathways (118). Saturated fatty acids (SFA) are thought to reduce vascular reactivity by decreasing insulin signaling via vasodilator pathways (PI3K/Akt/endothelial nitric oxide synthase (eNOS)) and enhancing pro-inflammatory pathways. In comparison, OA promotes signaling via the PI3K/Akt/eNOS pathway (119). Fatty acids induced IKKβ activity, with palmitic acid showing greater activation than OA and LA, reducing nitric oxide production (120). FFAR1 and FFAR4 are expressed in the lungs (113). FFAR4 is highly expressed in murine lungs and appears to be restricted to the airway epithelium, mainly on mucous-secreting goblet cells and ciliate columnar epithelial cells (121, 122). Saturated and unsaturated long-chain fatty acids may bind FFAR1 and FFAR4 and activate intracellular signaling, such as PI3K, ERK, and PKC (116, 123). Thus, FFAR1 and FFAR 4 activation may activate intracellular pathways with crosstalk with NKA signalosome.
Interestingly, FFAR4 activation promotes bronchial epithelial repair after epithelial injury, and this event possibly occurs by fatty acid-specific induction through FFAR4 (124). High levels of FFAR4 expression in the lungs will be linked with airway function and dysfunction (123), so the specific type of FFA (saturated, unsaturated, or polyunsaturated) that binds and activates FFAR4 may have a positive or a negative impact on lung injury.
Several preventive therapies were tested unsuccessfully in patients with lung injury at the initial stages aiming to prevent acute lung injury and the progression of ARDS (125). Previous studies showed the effectiveness of the treatments based on exogenous surfactant, inhaled nitric oxide, intravenous prostaglandin E1, glucocorticoids, ketoconazole, lisofylline, n-acetylcysteine, and activated protein C. However, effectiveness could not be confirmed when these potential treatments were advanced to phase III clinical studies (126). Therefore, new therapeutic approaches have been suggested for acute lung injury/ARDS treatment, and NKA has been indicated as a possible therapeutic target (127). Some studies had already signaled the edema clearance improvement through stimulation of NKA activity by aldosterone (128), growth factors (129), catecholamines (130, 131), β-adrenergic agonist (132), β-adrenergic receptor overexpression (133), dopamine (134), vasopressin (135) and rosuvastatin (127). This effect is expected because NKA of alveolar epithelial cell types I and II support edema clearance (136).
On the other hand, gene therapy studies with the overexpression of the NKA β1 subunit upregulated the expression of tight junction proteins, leading to increased alveolar epithelial barrier function (39). Machado-Aranda and coworkers (2005) tested gene therapy using electroporation of the NKA β1 subunit to increase alveolar fluid reabsorption (137). Similarly, a study with transfection with adenoviruses carrying genes encoding the α1 and the β1 subunit of NKA showed a decrease of ARDS in C57/BL6 mice (138). Additionally, the gene transfer of the NKA α2 subunit by adenovirus also increased NKA activity in rat alveolar epithelial cells and adenocarcinoma human alveolar basal epithelial cells (A549 cells) and improved the basal lung fluid clearance rate (139).
The importance of other ionic channels for edema clearance, such as ENaC (140) and AQPs (141) has also been demonstrated in ARDS. Although important, ENaC and AQPs are not essential for the alveolar fluid removal (142, 143), conversely, the NKA which is identified as the primary edema clearance agent (143). Therefore, new therapeutic approaches, including gene therapy targeting NKA, can be an effective alternative to improve edema clearance, favoring lesion recovery in acute lung injury and ARDS
Acute lung injury/ARDS is a respiratory failure syndrome marked by the disruption of alveolar endothelial and epithelial barriers and accumulation of edema fluid within the alveolus and interstitium (73). Edema clearance is crucial for resolving these lung injuries and can be accomplished by NKA activity in type I and II epithelial cells. Nevertheless, NKA may trigger lung injury through signaling pathways independent of ion pumping inhibition. Interestingly, the Src-EGFR-Ras-Raf-MAPK signaling cascade is the main NKA pathway and the responsible for triggering lung injury, fostering neutrophil influx into the lungs (79), edema and lipid body formation, NF-κB transactivation (63), IL-1β, IL-4, IL-6, TNFα and ROS production (57, 144). Therefore, NKA is a protagonist in edema clearance but may also induce lung injury by triggering cell signaling independent of its pumping activity.
Additionally, the overexpression of the NKA β1 subunit avoids lung injury. This occurs because of the improvement in edema clearance by NKA pumping activity, decreasing alveolar fluid accumulation. Besides, the integrity of the alveolar epithelial barrier between alveolar and interstitial spaces depends on the NKA activity. Further studies are necessary to fully elucidate the specific pathways triggered by NKA on lung injury and develop alternative therapeutic strategies targeting NKA for acute lung injury/ARDS treatment.
AS: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. KSS: Writing – original draft. TS: Writing – original draft. MY-I: Funding acquisition, Writing – original draft. PB: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. HCFN: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. CG-d-A: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Universidade Federal Fluminense (PROPPI/UFF), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Grant [001], Programa de Biotecnologia da Universidade Federal Fluminense (UFF), Programa de Pós Graduação em Biologia Molecular Celular (UNIRIO), Universidade Federal do Estado do Rio de Janeiro (UNIRIO), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) Grants (E-26/010.000983/2019, E-26/203.290/2017, and E-26/2010.592/2019, E-26/201.448/2021), FINEP Grant (01.23.0139.00), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Instituto Oswaldo Cruz, FIOCRUZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chaikajornwat J, Rattanajiajaroen P, Srisawat N, Kawkitinarong K. Leptospirosis manifested with severe pulmonary haemorrhagic syndrome successfully treated with venovenous extracorporeal membrane oxygenation. BMJ Case Rep (2020) 13(1):e230075. doi: 10.1136/bcr-2019-230075
2. Sedhai YR, Yuan M, Ketcham SW, Co I, Claar DD, McSparron JI, et al. Validating measures of disease severity in acute respiratory distress syndrome. Ann Am Thorac Soc (2021) 18(7):1211–8. doi: 10.1513/AnnalsATS.202007-772OC
3. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet (1967) 2(7511):319–23. doi: 10.1016/s0140-6736(67)90168-7
4. Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA (2012) 307(23):2526–33. doi: 10.1001/jama.2012.5669
5. Huang D, Ma H, Xiao Z, Blaivas M, Chen Y, Wen J, et al. Diagnostic value of cardiopulmonary ultrasound in elderly patients with acute respiratory distress syndrome. BMC Pulm Med (2018) 18(1):136. doi: 10.1186/s12890-018-0666-9
6. Vignon P, Evrard B, Asfar P, Busana M, Calfee CS, Coppola S, et al. Fluid administration and monitoring in ARDS: which management? Intensive Care Med (2020) 46(12):2252–64. doi: 10.1007/s00134-020-06310-0
7. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med (2017) 377(6):562–72. doi: 10.1056/NEJMra1608077
8. Peteranderl C, Sznajder JI, Herold S, Lecuona E. Inflammatory responses regulating alveolar ion transport during pulmonary infections. Front Immunol (2017) 8:446. doi: 10.3389/fimmu.2017.00446
9. Schuster DP. ARDS: clinical lessons from the oleic acid model of acute lung injury. Am J Respir Crit Care Med (1994) 149(1):245–60. doi: 10.1164/ajrccm.149.1.8111590
10. Zhang JL, Zhuo XJ, Lin J, Luo LC, Ying WY, Xie X, et al. Maresin1 stimulates alveolar fluid clearance through the alveolar epithelial sodium channel Na,K-ATPase via the ALX/PI3K/Nedd4-2 pathway. Lab Invest (2017) 97(5):543–54. doi: 10.1038/labinvest.2016.150
11. Khan YS, Lynch DT. Histology, Lung. In: StatPearls. Treasure Island (FL:StatPearls Publishing (2021).
12. Kia'i N, Bajaj T. Histology, Respiratory Epithelium. In: StatPearls. Treasure Island (FL:StatPearls Publishing (2021).
13. Leiva-Juarez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol (2018) 11(1):21–34. doi: 10.1038/mi.2017.71
14. Joshi N, Walter JM, Misharin AV. Alveolar macrophages. Cell Immunol (2018) 330:86–90. doi: 10.1016/j.cellimm.2018.01.005
15. Kasper M, Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci Rep (2017) 37(6):BSR20171301. doi: 10.1042/BSR20171301
16. Pires-Neto RC, Del Carlo Bernardi F, Alves de Araujo P, Mauad T, Dolhnikoff M. The expression of water and ion channels in diffuse alveolar damage is not dependent on DAD etiology. PloS One (2016) 11(11):e0166184. doi: 10.1371/journal.pone.0166184
17. Olajuyin AM, Zhang X, Ji HL. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discovery (2019) 5:63. doi: 10.1038/s41420-019-0147-9
18. Sen AK, Post RL. Stoichiometry and localization of adenosine triphosphate-dependent sodium and potassium transport in the erythrocyte. J Biol Chem (1964) 239:345–52. doi: 10.1016/S0021-9258(18)51788-9
19. Cui X, Xie Z. Protein interaction and Na/K-ATPase-mediated signal transduction. Molecules (2017) 22(6):990. doi: 10.3390/molecules22060990
20. Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol (2017) 8:371. doi: 10.3389/fphys.2017.00371
21. Schneeberger EE, McCarthy KM. Cytochemical localization of Na+-K+-ATPase in rat type II pneumocytes. J Appl Physiol (1986) 60(5):1584–9. doi: 10.1152/jappl.1986.60.5.1584
22. Arystarkhova E, Sweadner KJ. Tissue-specific expression of the Na,K-ATPase beta3 subunit. The presence of beta3 in lung and liver addresses the problem of the missing subunit. J Biol Chem (1997) 272(36):22405–8. doi: 10.1074/jbc.272.36.22405
23. Martin SL, Saint-Criq V, Hwang TC, Csanady L. Ion channels as targets to treat cystic fibrosis lung disease. J Cyst Fibros (2018) 17(2S):S22–7. doi: 10.1016/j.jcf.2017.10.006
24. Reihill JA, Douglas LEJ, Martin SL. Modulation of ion transport to restore airway hydration in cystic fibrosis. Genes (Basel) (2021) 12(3):453. doi: 10.3390/genes12030453
25. Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch (2017) 469(1):135–47. doi: 10.1007/s00424-016-1917-3
26. Lin X, Barravecchia M, Kothari P, Young JL, Dean DA. beta1-Na(+),K(+)-ATPase gene therapy upregulates tight junctions to rescue lipopolysaccharide-induced acute lung injury. Gene Ther (2016) 23(6):489–99. doi: 10.1038/gt.2016.19
27. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science (2018) 359(6380):1118–23. doi: 10.1126/science.aam6603
28. Citi S. The mechanobiology of tight junctions. Biophys Rev (2019) 11(5):783–93. doi: 10.1007/s12551-019-00582-7
29. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med (2018) 50(8):1–9. doi: 10.1038/s12276-018-0126-x
30. Zhou J, Zhou XD, Xu R, Du XZ, Li Q, Li B, et al. The degradation of airway epithelial tight junctions in asthma under high airway pressure is probably mediated by piezo-1. Front Physiol (2021) 12:637790. doi: 10.3389/fphys.2021.637790
31. Balda MS, Matter K. Tight junctions as regulators of tissue remodelling. Curr Opin Cell Biol (2016) 42:94–101. doi: 10.1016/j.ceb.2016.05.006
32. Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res (2017) 120(1):179–206. doi: 10.1161/CIRCRESAHA.116.306534
33. Yang J, Pan X, Wang L, Yu G. Alveolar cells under mechanical stressed niche: critical contributors to pulmonary fibrosis. Mol Med (2020) 26(1):95. doi: 10.1186/s10020-020-00223-w
34. Heyen L, Muller U, Siegemund S, Schulze B, Protschka M, Alber G, et al. Lung epithelium is the major source of IL-33 and is regulated by IL-33-dependent and IL-33-independent mechanisms in pulmonary cryptococcosis. Pathog Dis (2016) 74(7):ftw086. doi: 10.1093/femspd/ftw086
35. Nickol ME, Ciric J, Falcinelli SD, Chertow DS, Kindrachuk J. Characterization of host and bacterial contributions to lung barrier dysfunction following co-infection with 2009 pandemic influenza and methicillin resistant staphylococcus aureus. Viruses (2019) 11(2):116. doi: 10.3390/v11020116
36. Ouyang J, Zhang ZH, Zhou YX, Niu WC, Zhou F, Shen CB, et al. Up-regulation of tight-junction proteins by p38 mitogen-activated protein kinase/p53 inhibition leads to a reduction of injury to the intestinal mucosal barrier in severe acute pancreatitis. Pancreas (2016) 45(8):1136–44. doi: 10.1097/MPA.0000000000000656
37. Sapoznikov A, Gal Y, Falach R, Sagi I, Ehrlich S, Lerer E, et al. Early disruption of the alveolar-capillary barrier in a ricin-induced ARDS mouse model: neutrophil-dependent and -independent impairment of junction proteins. Am J Physiol Lung Cell Mol Physiol (2019) 316(1):L255–68. doi: 10.1152/ajplung.00300.2018
38. Gon Y, Hashimoto S. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int (2018) 67(1):12–7. doi: 10.1016/j.alit.2017.08.011
39. Bai H, Zhou R, Barravecchia M, Norman R, Friedman A, Yu D, et al. The Na+, K+-ATPase beta1 subunit regulates epithelial tight junctions via MRCKalpha. JCI Insight (2021) 6(4):e134882. doi: 10.1172/jci.insight.134881
40. Rajasekaran SA, Palmer LG, Quan K, Harper JF, Ball WJ Jr., Bander NH, et al. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell (2001) 12(2):279–95. doi: 10.1091/mbc.12.2.279
41. Yeaman C, Grindstaff KK, Nelson WJ. New perspectives on mechanisms involved in generating epithelial cell polarity. Physiol Rev (1999) 79(1):73–98. doi: 10.1152/physrev.1999.79.1.73
42. Herrero R, Sanchez G, Lorente JA. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann Transl Med (2018) 6(2):32. doi: 10.21037/atm.2017.12.18
43. Otani T, Nguyen TP, Tokuda S, Sugihara K, Sugawara T, Furuse K, et al. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J Cell Biol (2019) 218(10):3372–96. doi: 10.1083/jcb.201812157
44. Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends Cardiovasc Med (2016) 26(7):585–95. doi: 10.1016/j.tcm.2016.03.011
45. Cavalcante-Silva LHA, Lima EA, Carvalho DCM, de Sales-Neto JM, Alves AKA, Galvao J, et al. Much more than a cardiotonic steroid: modulation of inflammation by ouabain. Front Physiol (2017) 8:895. doi: 10.3389/fphys.2017.00895
46. Feng S, Chen W, Cao D, Bian J, Gong FY, Cheng W, et al. Involvement of Na(+), K (+)-ATPase and its inhibitors in HuR-mediated cytokine mRNA stabilization in lung epithelial cells. Cell Mol Life Sci (2011) 68(1):109–24. doi: 10.1007/s00018-010-0444-1
47. Xie Z, Cai T. Na+-K+–ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv (2003) 3(3):157–68. doi: 10.1124/mi.3.3.157
48. Gonçalves-de-Albuquerque CF, Silva AR, Silva CI, Castro-Faria-Neto HC, Burth P. Na/K Pump and Beyond: Na/K-ATPase as a Modulator of Apoptosis and Autophagy. Molecules (2017) 22(4):578. doi: 10.3390/molecules22040578
49. Tamura M, Kuwano H, Kinoshita T, Inagami T. Identification of linoleic and oleic acids as endogenous Na+,K+-ATPase inhibitors from acute volume-expanded hog plasma. J Biol Chem (1985) 260(17):9672–7. doi: 10.1016/S0021-9258(17)39291-8
50. Younes-Ibrahim M, Burth P, Castro-Faria M, Cheval L, Buffin-Meyer B, Marsy S, et al. Effect of Leptospira interrogans endotoxin on renal tubular Na,K-ATPase and H,K-ATPase activities. Ann N Y Acad Sci (1997) 834:684–6. doi: 10.1111/j.1749-6632.1997.tb52350.x
51. Burth P, Younes-Ibrahim M, Goncalez FH, Costa ER, Faria MV. Purification and characterization of a Na+, K+ ATPase inhibitor found in an endotoxin of Leptospira interrogans. Infect Immun (1997) 65(4):1557–60. doi: 10.1128/iai.65.4.1557-1560.1997
52. Goncalves-de-Albuquerque CF, Burth P, Silva AR, de Moraes IM, de Jesus Oliveira FM, Santelli RE, et al. Oleic acid inhibits lung Na/K-ATPase in mice and induces injury with lipid body formation in leukocytes and eicosanoid production. J Inflammation (Lond) (2013) 10(1):34. doi: 10.1186/1476-9255-10-34
53. Vadasz I, Morty RE, Kohstall MG, Olschewski A, Grimminger F, Seeger W, et al. Oleic acid inhibits alveolar fluid reabsorption: a role in acute respiratory distress syndrome? Am J Respir Crit Care Med (2005) 171(5):469–79. doi: 10.1164/rccm.200407-954OC
54. Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol (2013) 305(10):L665–81. doi: 10.1152/ajplung.00232.2013
55. Goncalves-de-Albuquerque CF, Silva AR, Burth P, Castro-Faria MV, Castro-Faria-Neto HC. Acute respiratory distress syndrome: role of oleic acid-triggered lung injury and inflammation. Mediators Inflammation (2015) 2015:260465. doi: 10.1155/2015/260465
56. Feng H, Choi YH, Cui C, X. H, Jiang J, Yan G, et al. Effects of polydatin on oleic acid-induced acute respiratory distress syndrome in rats. Int J Clin Exp Med (2018) 11(11):8.
57. Goncalves-de-Albuquerque CF, Silva AR, Burth P, Moraes IMde, Oliveira FM, Younes-Ibrahim M, et al. Oleic acid induces lung injury in mice through activation of the ERK pathway. Mediators Inflammation (2012), 2012:956509. doi: 10.1155/2012/956509
58. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun (2016) 7:11190. doi: 10.1038/ncomms11190
59. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) (2017) 9(5):52. doi: 10.3390/cancers9050052
60. Espada J, Martin-Perez J. An update on src family of nonreceptor tyrosine kinases biology. Int Rev Cell Mol Biol (2017) 331:83–122. doi: 10.1016/bs.ircmb.2016.09.009
61. Bejcek J, Jurasek M, Spiwok V, Rimpelova S. Quo vadis cardiac glycoside research? Toxins (Basel) (2021) 13(5):344. doi: 10.3390/toxins13050344
62. Xie Z, Askari A. Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem (2002) 269(10):2434–9. doi: 10.1046/j.1432-1033.2002.02910.x
63. Liu L, Wu J, Kennedy DJ. Regulation of cardiac remodeling by cardiac na(+)/K(+)-ATPase isoforms. Front Physiol (2016) 7:382. doi: 10.3389/fphys.2016.00382
64. Askari A. The other functions of the sodium pump. Cell Calcium (2019) 84:102105. doi: 10.1016/j.ceca.2019.102105
65. Wong RW, Lingwood CA, Ostrowski MA, Cabral T, Cochrane A. Cardiac glycoside/aglycones inhibit HIV-1 gene expression by a mechanism requiring MEK1/2-ERK1/2 signaling. Sci Rep (2018) 8(1):850. doi: 10.1038/s41598-018-19298-x
66. Lu N, Malemud CJ. Extracellular signal-regulated kinase: A regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression. Int J Mol Sci (2019) 20(15):3792. doi: 10.3390/ijms20153792
67. Shan X, Zhang Y, Chen H, Dong L, Wu B, Xu T, et al. Inhibition of epidermal growth factor receptor attenuates LPS-induced inflammation and acute lung injury in rats. Oncotarget (2017) 8(16):26648–61. doi: 10.18632/oncotarget.15790
68. Nakashima K, Sato T, Shigemori S, Shimosato T, Shinkai M, Kaneko T. Regulatory role of heme oxygenase-1 in silica-induced lung injury. Respir Res (2018) 19(1):144. doi: 10.1186/s12931-018-0852-6
69. Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem (2000) 275(36):27832–7. doi: 10.1074/jbc.M002951200
70. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
71. Khalaf FK, Dube P, Mohamed A, Tian J, Malhotra D, Haller ST, et al. Cardiotonic steroids and the sodium trade balance: new insights into trade-off mechanisms mediated by the na(+)/K(+)-ATPase. Int J Mol Sci (2018) 19(9):2576. doi: 10.3390/ijms19092576
72. Kobayashi SD, Malachowa N, DeLeo FR. Influence of microbes on neutrophil life and death. Front Cell Infect Microbiol (2017) 7:159. doi: 10.3389/fcimb.2017.00159
73. Brodie D, Slutsky AS, Combes A. Extracorporeal life support for adults with respiratory failure and related indications: A review. JAMA (2019) 322(6):557–68. doi: 10.1001/jama.2019.9302
74. Huppert LA, Matthay MA. Alveolar fluid clearance in pathologically relevant conditions: in vitro and in vivo models of acute respiratory distress syndrome. Front Immunol (2017) 8:371. doi: 10.3389/fimmu.2017.00371
75. Orlov SN, Klimanova EA, Tverskoi AM, Vladychenskaya EA, Smolyaninova LV, Lopina OD. Na(+)i,K(+)i-dependent and -independent signaling triggered by cardiotonic steroids: facts and artifacts. Molecules (2017) 22(4):635. doi: 10.3390/molecules22040635
76. Souza E. S. K. F. C., Moraes BPT, Paixao I, Burth P, Silva AR, Goncalves-de-Albuquerque CF. Na(+)/K(+)-ATPase as a target of cardiac glycosides for the treatment of SARS-coV-2 infection. Front Pharmacol (2021) 12:624704. doi: 10.3389/fphar.2021.624704
77. Wang H, Wang T, Yuan Z, Cao Y, Zhou Y, He J, et al. Role of receptor for advanced glycation end products in regulating lung fluid balance in lipopolysaccharide-induced acute lung injury and infection-related acute respiratory distress syndrome. Shock (2018) 50(4):472–82. doi: 10.1097/SHK.0000000000001032
78. Brazee PL, Soni PN, Tokhtaeva E, Magnani N, Yemelyanov A, Perlman HR, et al. FXYD5 is an essential mediator of the inflammatory response during lung injury. Front Immunol (2017) 8:623. doi: 10.3389/fimmu.2017.00623
79. Toumpanakis D, Vassilakopoulou V, Sigala I, Zacharatos P, Vraila I, Karavana V, et al. The role of Src & ERK1/2 kinases in inspiratory resistive breathing induced acute lung injury and inflammation. Respir Res (2017) 18(1):209. doi: 10.1186/s12931-017-0694-7
80. Jang EJ, Jeong HO, Park D, Kim DH, Choi YJ, Chung KW, et al. Src tyrosine kinase activation by 4-hydroxynonenal upregulates p38, ERK/AP-1 signaling and COX-2 expression in YPEN-1 cells. PloS One (2015) 10(10):e0129244. doi: 10.1371/journal.pone.0129244
81. Krotova K, Khodayari N, Oshins R, Aslanidi G, Brantly ML. Neutrophil elastase promotes macrophage cell adhesion and cytokine production through the integrin-Src kinases pathway. Sci Rep (2020) 10(1):15874. doi: 10.1038/s41598-020-72667-3
82. Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang M, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-kappaB signaling pathways. Sci Rep (2016) 6:27911. doi: 10.1038/srep27911
83. Idell S, Kueppers F, Lippmann M, Rosen H, Niederman M, Fein A. Angiotensin converting enzyme in bronchoalveolar lavage in ARDS. Chest (1987) 91(1):52–6. doi: 10.1378/chest.91.1.52
84. Imai Y, Kuba K, Penninger JM. The renin-angiotensin system in acute respiratory distress syndrome. Drug Discovery Today Dis Mech (2006) 3(2):225–9. doi: 10.1016/j.ddmec.2006.06.012
85. Hsieh YH, Deng JS, Chang YS, Huang GJ. Ginsenoside rh2 ameliorates lipopolysaccharide-induced acute lung injury by regulating the TLR4/PI3K/akt/mTOR, raf-1/MEK/ERK, and keap1/nrf2/HO-1 signaling pathways in mice. Nutrients (2018) 10(9):1208. doi: 10.3390/nu10091208
86. Fang W, Cai SX, Wang CL, Sun XX, Li K, Yan XW, et al. Modulation of mitogenactivated protein kinase attenuates sepsisinduced acute lung injury in acute respiratory distress syndrome rats. Mol Med Rep (2017) 16(6):9652–8. doi: 10.3892/mmr.2017.7811
87. Khalaf FK, Dube P, Kleinhenz AL, Malhotra D, Gohara A, Drummond CA, et al. Proinflammatory Effects of Cardiotonic Steroids Mediated by NKA alpha-1 (Na+/K+-ATPase alpha-1)/Src Complex in Renal Epithelial Cells and Immune Cells. Hypertension (2019) 74(1):73–82. doi: 10.1161/HYPERTENSIONAHA.118.12605
88. Jiang Y, Xia M, Xu J, Huang Q, Dai Z, Zhang X. Dexmedetomidine alleviates pulmonary edema through the epithelial sodium channel (ENaC) via the PI3K/Akt/Nedd4-2 pathway in LPS-induced acute lung injury. Immunol Res (2021) 69(2):162–75. doi: 10.1007/s12026-021-09176-6
89. Zhao YR, Wang D, Liu Y, Shan L, Zhou JL. The PI3K/Akt, p38MAPK, and JAK2/STAT3 signaling pathways mediate the protection of SO2 against acute lung injury induced by limb ischemia/reperfusion in rats. J Physiol Sci (2016) 66(3):229–39. doi: 10.1007/s12576-015-0418-z
90. Chen Y, Huang W, Yang M, Xin G, Cui W, Xie Z, et al. Cardiotonic steroids stimulate macrophage inflammatory responses through a pathway involving CD36, TLR4, and Na/K-ATPase. Arterioscler Thromb Vasc Biol (2017) 37(8):1462–9. doi: 10.1161/ATVBAHA.117.309444
91. Valente RC, Araujo EG, Rumjanek VM. Ouabain inhibits monocyte activation in vitro: prevention of the proinflammatory mCD14(+)/CD16(+) subset appearance and cell-size progression. J Exp Pharmacol (2012) 4:125–40. doi: 10.2147/JEP.S35507
92. Teixeira MP, Rumjanek VM. Ouabain affects the expression of activation markers, cytokine production, and endocytosis of human monocytes. Mediators Inflammation (2014) 2014:760368. doi: 10.1155/2014/760368
93. LaRock DL, Sands JS, Ettouati E, Richard M, Bushway PJ, Adler ED, et al. Inflammasome inhibition blocks cardiac glycoside cell toxicity. J Biol Chem (2019) 294(34):12846–54. doi: 10.1074/jbc.RA119.008330
94. Zhu M, Sun H, Cao L, Wu Z, Leng B, Bian J. Role of Na(+)/K(+)-ATPase in ischemic stroke: in-depth perspectives from physiology to pharmacology. J Mol Med (Berl) (2022) 100(3):395–410. doi: 10.1007/s00109-021-02143-6
95. Patton LM, Saggart BS, Ahmed NK, Leff JA, Repine JE. Interleukin-1 beta-induced neutrophil recruitment and acute lung injury in hamsters. Inflammation (1995) 19(1):23–9. doi: 10.1007/BF01534377
96. Ma X, Yu X, Zhou Q. The IL1beta-HER2-CLDN18/CLDN4 axis mediates lung barrier damage in ARDS. Aging (2020) 12(4):3249–65. doi: 10.18632/aging.102804
97. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery (2020) 6(1):36. doi: 10.1038/s41421-020-0167-x
98. Monaco A, Ovryn B, Axis J, Amsler K. The epithelial cell leak pathway. Int J Mol Sci (2021) 22(14):7677. doi: 10.3390/ijms22147677
99. Peukert K, Fox M, Schulz S, Feuerborn C, Frede S, Putensen C, et al. Esicm lives 2020. Intensive Care Med Exp (2020) 8(Suppl 2):73. doi: 10.1186/s40635-020-00354-8
100. Goncalves-de-Albuquerque CF, Cunha C, Castro LVG, Martins CA, Barnese MRC, Burth P, et al. Cellular pathophysiology of leptospirosis: role of Na/K-ATPase. Microorganisms (2023) 11(7):1695. doi: 10.3390/microorganisms11071695
101. Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol (2015) 34:75–82. doi: 10.1016/j.coi.2015.02.008
102. Jordan SC, Choi J, Kim I, Wu G, Toyoda M, Shin B, et al. Interleukin-6, A cytokine critical to mediation of inflammation, autoimmunity and allograft rejection: therapeutic implications of IL-6 receptor blockade. Transplantation (2017) 101(1):32–44. doi: 10.1097/TP.0000000000001452
103. Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, et al. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol (2004) 286(6):L1282–92. doi: 10.1152/ajplung.00349.2003
104. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol (2015) 16(5):448–57. doi: 10.1038/ni.3153
105. Kim MS, Han JY, Kim SH, Kim HY, Jeon D, Lee K. Polyhexamethylene guanidine phosphate induces IL-6 and TNF-alpha expression through JNK-dependent pathway in human lung epithelial cells. J Toxicol Sci (2018) 43(8):485–92. doi: 10.2131/jts.43.485
106. Oh K, Lee OY, Park Y, Seo MW, Lee DS. IL-1beta induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer (2016) 16(1):724. doi: 10.1186/s12885-016-2746-7
107. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol (2021) 22(5):346–66. doi: 10.1038/s41580-020-00322-w
108. Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol (1997) 15:675–705. doi: 10.1146/annurev.immunol.15.1.675
109. Kellner M, NooNepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol (2017) 967:105–37. doi: 10.1007/978-3-319-63245-2_8
110. Echaide M, Autilio C, Arroyo R, Perez-Gil J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochim Biophys Acta Biomembr (2017) 1859(9 Pt B):1725–39. doi: 10.1016/j.bbamem.2017.03.015
111. Natarajan K, Gangam K, Meganathan V, Gottipati KR, Mitchell C, Boggaram V. Organic dust inhibits surfactant protein expression by reducing thyroid transcription factor-1 levels in human lung epithelial cells. Innate Immun (2019) 25(2):118–31. doi: 10.1177/1753425919827360
112. Miyamoto J, Hasegawa S, Kasubuchi M, Ichimura A, Nakajima A, Kimura I. Nutritional signaling via free fatty acid receptors. Int J Mol Sci (2016) 17(4):450. doi: 10.3390/ijms17040450
113. Hopkins MM, Meier KE. Free fatty acid receptors and cancer: from nutrition to pharmacology. Handb Exp Pharmacol (2017) 236:233–51. doi: 10.1007/164_2016_48
114. Secor JD, Fligor SC, Tsikis ST, Yu LJ, Puder M. Free fatty acid receptors as mediators and therapeutic targets in liver disease. Front Physiol (2021) 12:656441. doi: 10.3389/fphys.2021.656441
115. Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol (2014) 5:236. doi: 10.3389/fphar.2014.00236
116. Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free fatty acid receptors in health and disease. Physiol Rev (2020) 100(1):171–210. doi: 10.1152/physrev.00041.2018
117. Liotti A, Cosimato V, Mirra P, Cali G, Conza D, Secondo A, et al. Oleic acid promotes prostate cancer Malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J Cell Physiol (2018) 233(9):7367–78. doi: 10.1002/jcp.26572
118. Matoba A, Matsuyama N, Shibata S, Masaki E, Emala CW, Mizuta K. The free fatty acid receptor 1 promotes airway smooth muscle cell proliferation through MEK/ERK and PI3K/Akt signaling pathways. Am J Physiol Lung Cell Mol Physiol (2018) 314(3):L333–48. doi: 10.1152/ajplung.00129.2017
119. Jackson KG, Newens KJ, Fry MJ, Thompson AK, Williams CM. Differential effects of single fatty acids and fatty acid mixtures on the phosphoinositide 3-kinase/Akt/eNOS pathway in endothelial cells. Eur J Nutr (2022) 61:2463–73. doi: 10.1007/s00394-022-02821-2
120. Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol (2005) 25(5):989–94. doi: 10.1161/01.ATV.0000160549.60980.a8
121. Miyauchi S, Hirasawa A, Iga T, Liu N, Itsubo C, Sadakane K, et al. Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedebergs Arch Pharmacol (2009) 379(4):427–34. doi: 10.1007/s00210-008-0390-8
122. Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med (2004) 350(6):560–9. doi: 10.1056/NEJMoa021218
123. Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: pharmacology and therapeutic opportunities. Trends Pharmacol Sci (2017) 38(9):809–21. doi: 10.1016/j.tips.2017.06.006
124. Lee KP, Park SJ, Kang S, Koh JM, Sato K, Chung HY, et al. omega-3 Polyunsaturated fatty acids accelerate airway repair by activating FFA4 in club cells. Am J Physiol Lung Cell Mol Physiol (2017) 312(6):L835–44. doi: 10.1152/ajplung.00350.2016
125. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
126. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Delivery (2010) 23(4):243–52. doi: 10.1089/jamp.2009.0775
127. Xu HR, Yang Q, Xiang SY, Zhang PH, Ye Y, Chen Y, et al. Rosuvastatin enhances alveolar fluid clearance in lipopolysaccharide-induced acute lung injury by activating the expression of sodium channel and Na,K-ATPase via the PI3K/AKT/Nedd4-2 pathway. J Inflammation Res (2021) 14:1537–49. doi: 10.2147/JIR.S299267
128. Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol Respir Environ Exerc Physiol (1984) 57(6):1809–16. doi: 10.1152/jappl.1984.57.6.1809
129. Hamill OP, McBride DW Jr. The pharmacology of mechanogated membrane ion channels Pharmacol Rev (1996) 48(2):231–52
130. Saldias FJ, Lecuona E, Comellas AP, Ridge KM, Sznajder JI. Dopamine restores lung ability to clear edema in rats exposed to hyperoxia. Am J Respir Crit Care Med (1999) 159(2):626–33. doi: 10.1164/ajrccm.159.2.9805016
131. Azzam ZS, Saldias FJ, Comellas A, Ridge KM, Rutschman DH, Sznajder JI. Catecholamines increase lung edema clearance in rats with increased left atrial pressure. J Appl Physiol (1985) (2001) 90(3):1088–94. doi: 10.1152/jappl.2001.90.3.1088
132. Saldias FJ, Lecuona E, Comellas AP, Ridge KM, Rutschman DH, Sznajder JI. beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med (2000) 162(1):282–7. doi: 10.1164/ajrccm.162.1.9809058
133. McGraw DW, Fukuda N, James PF, Forbes SL, Woo AL, Lingrel JB, et al. Targeted transgenic expression of beta(2)-adrenergic receptors to type II cells increases alveolar fluid clearance. Am J Physiol Lung Cell Mol Physiol (2001) 281(4):L895–903. doi: 10.1152/ajplung.2001.281.4.L895
134. Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol (2006) 290(4):L710–22. doi: 10.1152/ajplung.00486.2004
135. Guetta J, Klorin G, Tal R, Berger G, Ismael-Badarneh R, Bishara B, et al. Vasopressin-2 receptor antagonist attenuates the ability of the lungs to clear edema in an experimental model. Am J Respir Cell Mol Biol (2012) 47(5):583–8. doi: 10.1165/rcmb.2012-0117OC
136. Hollenhorst MI, Richter K, Fronius M. Ion transport by pulmonary epithelia. J BioMed Biotechnol (2011) 2011:174306. doi: 10.1155/2011/174306
137. MaChado-Aranda D, Adir Y, Young JL, Briva A, Budinger GR, Yeldandi AV, et al. Gene transfer of the Na+,K+-ATPase beta1 subunit using electroporation increases lung liquid clearance. Am J Respir Crit Care Med (2005) 171(3):204–11. doi: 10.1164/rccm.200403-313OC
138. Stern M, Ulrich K, Robinson C, Copeland J, Griesenbach U, Masse C, et al. Pretreatment with cationic lipid-mediated transfer of the Na+K+-ATPase pump in a mouse model in vivo augments resolution of high permeability pulmonary oedema. Gene Ther (2000) 7(11):960–6. doi: 10.1038/sj.gt.3301193
139. Ridge KM, Olivera WG, Saldias F, Azzam Z, Horowitz S, Rutschman DH, et al. Alveolar type 1 cells express the alpha2 Na,K-ATPase, which contributes to lung liquid clearance. Circ Res (2003) 92(4):453–60. doi: 10.1161/01.RES.0000059414.10360.F2
140. Gotts JE, Matthay MA. Mesenchymal stem cells and the stem cell niche: a new chapter. Am J Physiol Lung Cell Mol Physiol (2012) 302(11):L1147–9. doi: 10.1152/ajplung.00122.2012
141. Ma T, Fukuda N, Song Y, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-5 knockout mice. J Clin Invest (2000) 105(1):93–100. doi: 10.1172/JCI8258
142. Dobbs LG, Gonzalez R, Matthay MA, Carter EP, Allen L, Verkman AS. Highly water-permeable type I alveolar epithelial cells confer high water permeability between the airspace and vasculature in rat lung. Proc Natl Acad Sci U.S.A. (1998) 95(6):2991–6. doi: 10.1073/pnas.95.6.2991
143. Mutlu GM, Sznajder JI. Mechanisms of pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol (2005) 289(5):L685–95. doi: 10.1152/ajplung.00247.2005
Keywords: Na/K-ATPase, lung inflammation, ARDS, oleic acid, cardiac glycosides
Citation: Silva AR, Souza e Souza KFC, Souza TB, Younes-Ibrahim M, Burth P, Castro Faria Neto HC and Gonçalves-de-Albuquerque CF (2024) The Na/K-ATPase role as a signal transducer in lung inflammation. Front. Immunol. 14:1287512. doi: 10.3389/fimmu.2023.1287512
Received: 01 September 2023; Accepted: 26 December 2023;
Published: 17 January 2024.
Edited by:
Huaiyong Chen, Tianjin Haihe Hospital, ChinaReviewed by:
Luiz Henrique Agra Cavalcante-Silva, Federal University of Paraíba, BrazilCopyright © 2024 Silva, Souza e Souza, Souza, Younes-Ibrahim, Burth, Castro Faria Neto and Gonçalves-de-Albuquerque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cassiano Felippe Gonçalves-de-Albuquerque, Y2Fzc2lhbm8uYWxidXF1ZXJxdWVAdW5pcmlvLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.