 Hongxia Zhang
Hongxia Zhang Jian Wang
Jian Wang Fengqi Li
Fengqi Li- 1Institute of Health and Medicine, Hefei Comprehensive National Science Center, Hefei, Anhui, China
- 2Department of Neurology, The First Affiliated Hospital of University of Science and Technology of China (USTC), Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui, China
Lung cancer is the leading cause of tumor-induced death worldwide and remains a primary global health concern. In homeostasis, due to its unique structure and physiological function, the lung microenvironment is in a state of immune tolerance and suppression, which is beneficial to tumor development and metastasis. The lung tumor microenvironment is a more complex system that further enhances the immunosuppressive features in the lungs. NK cells are abundantly located in the lungs and play crucial roles in lung tumor surveillance and antitumor immunity. However, the immunosuppressive microenvironment promotes significant challenges to NK cell features, leading to their hypofunction, exhaustion, and compromised antitumor activity. Thus, understanding the complex interactions among the lung microenvironment, lung tumor microenvironment, and NK cell exhaustion is critical for the development of effective cancer immunotherapeutic strategies. The present review will discuss NK cell hypofunction and exhaustion within the lung microenvironment and lung tumor microenvironment, focusing on lung tissue-specific factors, including key cytokines and unique environmental components, that modulate NK cell activation and function. Understanding the functional mechanisms of key factors would help to design strategies to reverse NK cell exhaustion and restore their antitumor function within the lung tumor microenvironment.
Introduction
Lung cancer is a disease with very high morbidity and mortality worldwide in both sexes (1). In addition, the lung is the primary site for disseminated tumor seeding (2). The continuous interaction on the open surface and rich capillary network of the lungs brings a higher chance of making tumors “seed” into the lungs, and the immunosuppressive microenvironment of the lungs provides “soil” for tumor growth, so the lung becomes a high incidence site of tumors in situ and metastases. For example, breast cancer with very high morbidity in the world has lung metastasis in 60-70% of relapsed patients (3). Natural killer (NK) cells are an important innate lymphocyte population that are abundant in the lungs and play critical roles in the antitumor immune response. NK cells mediate antitumor effects by producing effector cytokines or exerting direct cytotoxic activity (4). However, NK cells in the lungs display inactivated and hypofunctional features (5). When the tumor microenvironment is formed in the lungs, NK cells are getting exhausted (6, 7). This review summarized the roles of the main lung-specific components that can modulate NK cell hypofunction and exhaustion in both the lung microenvironment and lung tumor microenvironment. Furthermore, future studies on promising antitumor strategies will also be discussed.
The lung is a unique, complex, and delicate organ
As the main organ for oxygen and carbon dioxide exchange, the lung is essential for mammalian survival. The surface area of the lower respiratory tract is approximately one hundred square meters in a healthy adult. Furthermore, the alveoli are the terminal units of the respiratory tract tree, and they are highly vascularized and have thin-walled architecture. The surface area of capillaries encompassing the alveoli is approximately 140 square meters. Moreover, the basement membrane of the alveolocapillary, which is the interface of alveolar epithelium and capillary endothelium, is only approximately 0.3 μm in thickness. To meet the active metabolic demands of the body, the distal airway of a healthy human adult needs to filter approximately ten thousand liters of air every day, and the pulmonary capillaries need to handle approximately 5 liters of blood every minute. Meanwhile, the large thin tenuous lung barrier is persistently exposed to different kinds of allergens, particles, and pathogens inhaled from the air and carried from the blood (8). Thus, the unique physiological functions and structures determine that the lung is a complex and delicate organ.
The physiological functions and the complex environment of the lungs require that the immune response in the lungs must be fast and efficient but tightly controlled to protect the tenuous lung barrier from excessive immune responses and inflammation and maintain immune homeostasis (9–11). The lung possesses a unique immune regulatory network that consists of structural cells and resident immune cells. All these cells are involved in the mounting of the lung-specific microenvironment (12).
Lung immune cells in homeostasis and disease
The lung has a unique set of immune cells, including innate and adaptive immune cells that are located within the lung niche and mediate the lung-specific immune response in homeostasis and disease (12). In the lungs, the particles, allergens, and pathogens from the inhaled air are removed mainly by lung-resident macrophages and dendritic cells (DC). Alveolar macrophages (AMs) are found in the alveoli, where they are composed of 90-95% of immune cells in the steady state in mice (13). Another type of lung-resident macrophage is interstitial macrophages (IMs), which are divided into two subsets. Lyve1hi IMs are closely located with blood vessels and play a crucial role in maintaining blood vessel integrity and anti-fibrotic activity. Lyve1lo IMs are situated close to lung nerve bundles and display higher antigen-presentation capacities (14). Lung resident DCs are mainly located at the basolateral side of the epithelium and are important for initiating appropriate immune responses to inhaled antigens (13). In addition, different types of lymphocytes/lymphoid cells are found in the lungs at steady state, including T cells, B cells, NK cells, and innate lymphoid cells (ILCs). Increased evidence has shown that lung-resident memory T and B cells, approximately 80% of lymphocytes in total in mouse lungs, play an important role in protecting against respiratory reinfection (15). ILCs also mediate a protective immune response from pathogens and promote tissue repair and homeostasis after infections (16), although the total lung ILC population is approximately 20–30 thousand cells in naive mouse lungs and only 0.1% of all CD45+ cells in human lungs (17). Furthermore, as a critical part of lymphocytes, lung NK cells are crucial effector lymphocyte populations in antitumor and anti-infection immune responses (18, 19). In this review, we will focus on lung NK cells and discuss their phenotype and function in the lung microenvironment.
The lung microenvironment shapes the specificity of lung NK cells in homeostasis
As important innate lymphocytes, NK cells are widely distributed in lymphoid and non-lymphoid tissues and play a crucial role in immune surveillance (20, 21). The lung is an important barrier of the body and is full of interactions between immune cells and foreign pathogens. NK cells are abundant in the lungs of both humans and mice (22, 23), indicating the significance of lung NK cells. There are high percentages of NK cells among lymphocytes in the lungs, approximately 10% in mice and 10-20% in humans, compared to other lymphoid and non-lymphoid organs, such as lymph nodes, bone marrow, spleen, blood, and liver (24–26), and the cytokine interleukin (IL)-15 derived from epithelial cells and alveolar macrophages might contribute to the high percentages of NK cells in the lung microenvironment (27, 28).
In normal lungs, NK cells are located in the lung interstitium. NK cells infiltrate into the alveoli and are observed in the bronchoalveolar fluid (BAL) during respiratory infection and in lung inflammatory diseases (29). In 1980, the antitumor function of NK cells was proven by three independent groups at almost the same time (30–32). Talmadge et al. found that NK cells have an important function in controlling tumor growth and metastasis in an NK cell-deficient mouse model (30). Karre et al. reported that NK cell-deficient mice display low in vivo resistance to syngeneic leukemias (31). Roder et al. observed that patients with dysfunctional NK cells have a profound defect in their ability to spontaneously lyse various tumor cells in vitro (32). Since then, an increasing number of studies have confirmed that NK cells have antitumor effects in multiple different tumors (33). In lung cancer patients, a correlation between tumor-infiltrating NK cells and a better prognosis has been observed (34, 35). In the Kras-driven spontaneous lung cancer and lung tumor implantation mouse model, NK cells showed antitumor effects. In both studies, mice lacking NK cells showed a higher tumor burden (36, 37). In addition to their antitumor function, NK cells are involved in immune responses to lung infections. In humans, increased respiratory viral infections are associated with dysfunction of NK cells (38, 39). However, studies with mouse models show that lung NK cells have a protective role in controlling viral and bacterial burden after infection (40). On the other hand, NK cells also contribute to inflammation-mediated damage and exacerbate the pathology of infection (41). The different functions of NK cells are driven by the local lung tissue microenvironment.
Lung NK cells display unique features compared to NK cells in other tissues: i) Lung NK cells maintain a more mature phenotype. In human lungs, NK cells are found in the parenchyma, and most of them display a fully differentiated CD56dimCD16+ phenotype. However, NK cells in the liver and other lymphoid organs display a CD56brightCD16– phenotype (19, 42). Similarly, in mouse lungs, most NK cells exhibit a mature CD27-CD11b+ phenotype (24); ii) The majority of lung NK cells are not tissue-resident. In both humans and mice, only a few lung NK cells express CD49a, which is a tissue-resident NK marker, indicating that lung NK cells mainly circulate between blood and the lungs (5, 19); iii) Lung NK cells display an inhibitory phenotype. In mice, compared to NK cells isolated from the spleen and bone marrow, lung NK cells express lower levels of activation-related molecules, such as CD69, NKp46, and NKG2D, and higher levels of inhibitory receptors CD94 and NKG2A (24). In humans, lung NK cells also express killer Ig-like receptors (KIRs), indicating that lung NK cells are quiescent in a steady state (5); iv) Lung NK cells are hypofunctional. In the 1980s, human NK cells were found to be functionally important in the lungs (42). Lung NK cells showed impaired cytotoxic abilities compared to NK cells from peripheral blood. The main structural cells (lung epithelial cells) and resident immune cells (alveolar macrophages) in the lungs are involved in the process of NK cell hypofunction (43–46). The key environmental components in the lungs, such as prostaglandin E2 (PGE2), TGF-β, and pulmonary surfactant, are the main mediators and are responsible for impaired NK cell functions (46). Recently, further studies have shown that PGE2 produced by lung fibroblasts is the main mechanism to explain the inhibition of NK cell functions (2). However, the inhibitory phenotype of NK cells in homeostasis could be reversed after respiratory viral or bacterial infection, and NK cells have the potential to be activated rapidly and involved in immunopathology in an inflammatory lung microenvironment (24). Overall, the particular phenotype and special features of lung NK cells under physiological conditions are shaped by the unique lung microenvironment and are beneficial for avoiding unnecessary inflammation and maintaining immune homeostasis in the lungs.
The lung tumor microenvironment causes exhaustion of lung NK cells
NK cells play critical roles in tumor surveillance and antitumor immunity in the circulating system and multiple organs (47). NK cells can be activated directly to eliminate target cells without priming (20). NK cells perform their effector function in several different ways. After activation, NK cells can directly kill transformed cells or tumor cells by producing perforin and granzyme B (48, 49) or by inducing target cell death through tumor necrosis factor (TNF)-α, Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) (50–52). In addition, NK cells can eliminate transformed cells or tumor cells through antibody-dependent cell-mediated cytotoxicity (ADCC) in the presence of antibodies (53). Furthermore, NK cells can secrete different cytokines and chemokines that subsequently promote other immune cells to exert antitumor functions (54).
However, in the condition of lung cancer, NK cells display an exhaustion status with an altered phenotype and impaired effector function. In mice, NK cells could prevent lung tumor initiation in a Kras-driven lung cancer model. However, once the tumor enters the promotion and progression stage, NK cells exhibit an exhausted phenotype with low cytotoxicity and viability (36). Similarly, intratumor NK cells show an exhausted phenotype, lower IFN-γ production, and impaired cytotoxic function in patients with non-small cell lung carcinoma (NSCLC) (7, 55–57). Most recently, Tang et al. analyzed human NK cells from 716 patients across 24 tumor types by integrative single-cell sequencing and found that NK cells display tumor-type preferences and are associated with both intrinsic organ properties and factors from the tumor microenvironment. The proportions of CD56dimCD16hi NK cells are significantly decreased in lung cancer compared with noncancer lungs. Further transcriptomic feature analysis in this study has shown that a subset of tumor-associated NK cells is enriched in tumors and displays exhaustion features and impaired antitumor functions in multiple cancers, including lung cancer (58). Thus, understanding the mechanisms and key factors that promote NK cell exhaustion in the lung tumor microenvironment would provide new directions for antitumor strategies.
Unique components of the lung microenvironment and lung tumor microenvironment promote lung NK cell exhaustion
Based on the special structure and composition of structural cells and immune cells, the lungs have many unique intrinsic features. For example, the expression of PGE2 in the lungs is relatively higher than that in other tissues (2). Furthermore, the expression of TGF-β in the lungs is high during lung development (59). There are also other unique chemical conditions, such as high levels of oxygen (60, 61), low levels of glucose (62–64), and abundant surfactant protein and lipids (65) in the alveoli (Figure 1). All these special components are involved in shaping the inactivation and hypofunctional status of NK cells. When tumors grow in the lungs, there are more immunosuppressive cells infiltrating the lung tumor microenvironment, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) (66). These types of cells produce more PGE2 and TGF-β (67, 68) (Figure 2). Moreover, the physical and chemical environment is changing to hypoxia, low pH, and lower glucose (69–71) (Figures 1, 2). All these mediators further promote NK cells changing to exhaustion status with altered phenotype and impaired function.
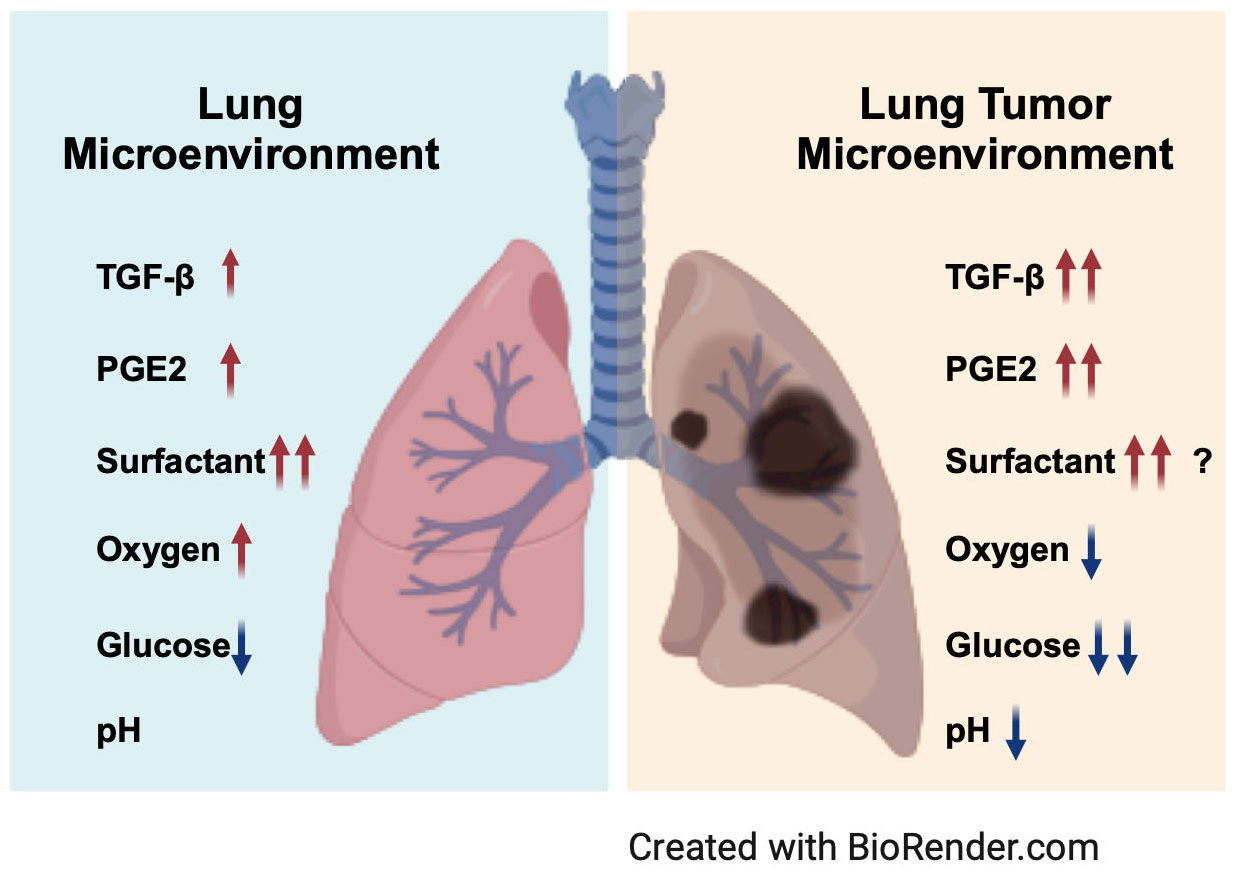
Figure 1 Key components from the lung microenvironment and lung tumor microenvironment. Abundant TGF-β, PGE2 and surfactant in the lung microenvironment are crucial for lung homeostasis. High oxygen and low glucose are special features of the lung microenvironment. When tumors grow in the lungs, more TGF-β and PGE2 are produced, and the physical and chemical environment changes to low oxygen, lower glucose, and low pH. Created with BioRender.com.
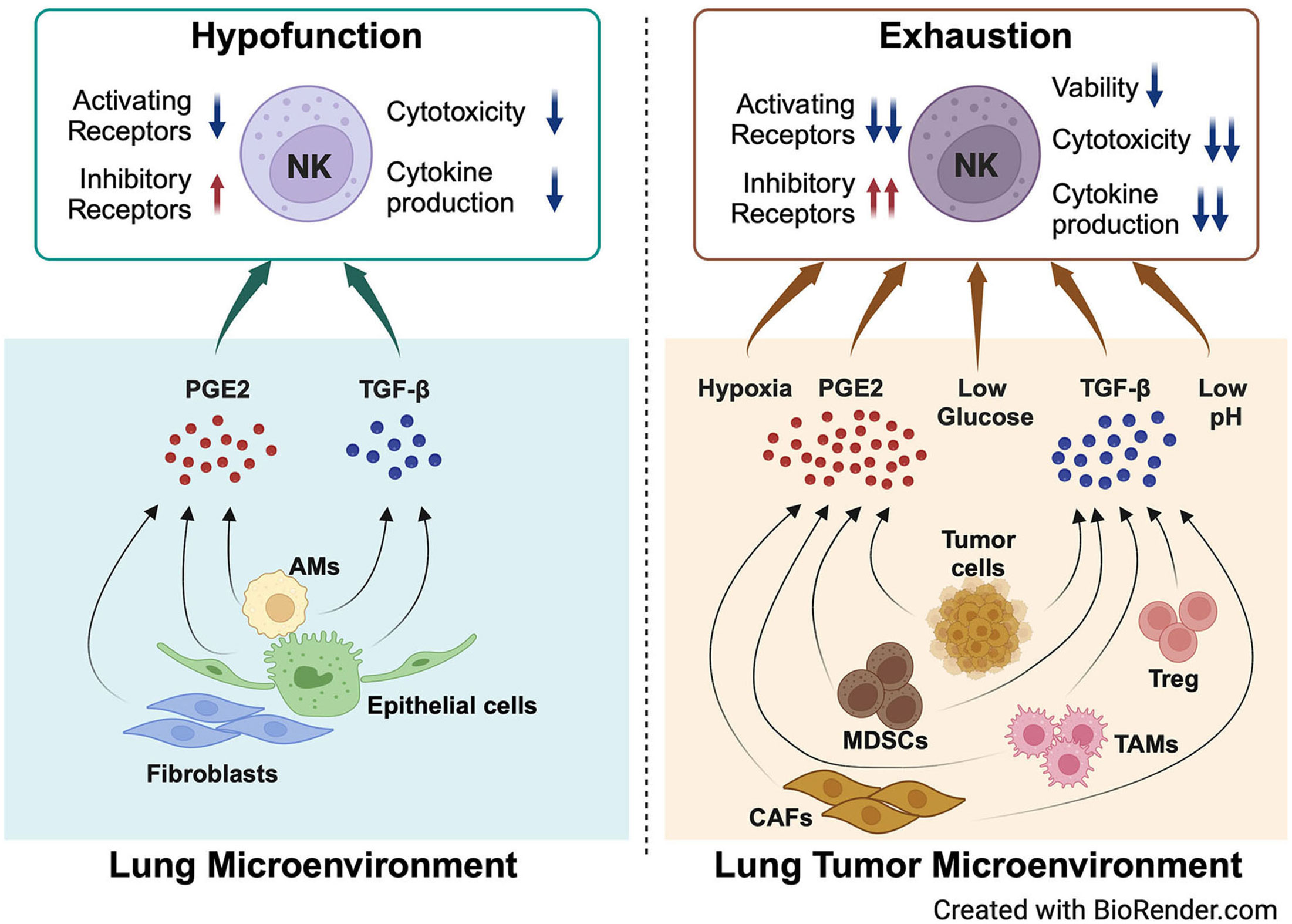
Figure 2 NK cell hypofunction and exhaustion are modulated by key components of the lung microenvironment and lung tumor microenvironment. PGE2 and TGF-β secreted by lung epithelial cells, AMs, and fibroblasts in the lung microenvironment modulate the hypofunction of lung NK cells in homeostasis. When tumors grow in the lungs, more PGE2 and TGF-β are secreted by multiple cell types, including tumor cells, MDSCs, TAMs, CAFs, and Tregs. The physical and chemical environment changes to hypoxia, lower glucose, and low pH. All these factors promote NK cell exhaustion in the lung tumor microenvironment. Created with BioRender.com.
TGF-β
TGF-β is a critical immunosuppressive cytokine of the lungs in a steady state and plays critical roles in driving AMs proliferation and differentiation and maintaining lung immune homeostasis (72). TGF-β is produced by multiple cell types in the normal lung microenvironment (12). In 1984, Robinson and colleagues found that NK cells isolated from lung tissue display impaired cytotoxic function (42). Later, several studies have proven that exogenous TGF-β could inhibit the IFN-γ production and activity of human NK cells in vitro, indicating the role of TGF-β in shaping special phenotypes and impaired function of lung NK cells (73–75).
As a biomarker of tumors and an extensive inducer of the tumor microenvironment, TGF-β is produced by different types of cells and plays multiple roles in tumor progression. Lung tumor cells themselves can secrete large amounts of TGF-β (76). TGF-β can induce tumor formation by promoting the process of epithelial-mesenchymal transformation (EMT) (77, 78). TGF-β also facilitates the transformation of fibroblasts into cancer-associated fibroblasts (CAFs), and CAFs can be the main source of TGF-β to further elevate the levels of TGF-β in the lung tumor microenvironment (79). TGF-β promotes CD4+ T cells to differentiate into Tregs and macrophages to polarize to M2 cells (80). Furthermore, with the formation of a tumor microenvironment, large numbers of TAMs, MDSCs, and Tregs appear in lung tumors (66). These cells could also secrete TGF-β, which further promotes NK cell exhaustion (Figure 2). TGF-β promotes NK cell exhaustion in several different ways. First, TGF-β alters the receptor-ligand interaction between NK cells and tumor cells. In response to TGF-β, NK cells downregulate the activating receptors NKG2D, NKp30, and NKp80 (81) and upregulate the inhibitory receptors NKG2A and PD1 (82). On the other hand, TGF-β also inhibits the expression of NKG2D ligands in human lung cancer cells (67). Second, TGF-β changes the metabolic features of NK cells. In the Kras-driven lung cancer mouse model, TGF-β inhibits glycolysis in NK cells by regulating the production of FBP1, which further mediates the exhaustion of NK cells (36). Finally, TGF-β induces microRNAs to silence NK cells. In human lung cancer, high levels of TGF-β deplete DAP12 by inducing microRNA-183 expression (83). Overall, in exposure to gradually increased TGF-β in the lung tumor microenvironment, lung NK cells are precisely regulated step by step and change themselves from hypofunction to exhaustion.
PGE2
PGE2 is a lipid compound generated from arachidonic acid via the catalysis of the enzyme cyclooxygenase (COX), including COX1 and COX2, and it plays multiple roles in regulating different stages of the immune response (68). In healthy lungs, PGE2 is mainly secreted by lung fibroblasts (2). The expression level of Ptgs2, encoding COX2, is much higher in lung fibroblasts than in lung epithelial cells, endothelial cells, lung CD45+ immune cells, and fibroblasts from other tissues, such as bone, heart, liver, spleen, and mammary gland, in mice (2). Similarly, high levels of PTGS2 expression were also detected in human lung tissues compared to other tissues through analyses of microarray data (2, 84). Gong et al. proved that the high level of PGE2 secreted by lung fibroblasts is responsible for the unique lung microenvironment, and the unique lung microenvironment endows the immunosuppressive phenotype of myeloid cells and the dysfunction of DCs in the lungs, which ultimately inhibits the cytotoxicity of NK cells (2) (Figure 2). The lung is the most common site for tumor metastasis (2). Deletion of Ptgs2 in lung fibroblasts or inhibition of the PGE2 receptor EP2 and EP4 could reverse the immunosuppression of resident myeloid cells and diminish lung metastasis in several breast cancer models, indicating that PGE2 is a critical factor in mediating premetastatic niche formation (2). As an important component of the premetastatic niche, neutrophils secrete the inflammatory cytokine IL-1β in the lungs, which facilitates lung fibroblasts to produce more PGE2 and promote the formation of the lung tumor microenvironment. Furthermore, TGF-β could increase the production of PGE2 by augmenting the expression of COX-2 through a noncanonical pathway (80).
In the tumor microenvironment, an increased amount of PGE2 is secreted by multiple cell types, including tumor cells, TAMs, CAFs, and MDSCs (76, 85–87). Increased evidence has shown that PGE2 can inhibit the antitumor activity of NK cells in lung cancer (88, 89). A high level of PGE2 could also induce immunosuppressive FOXP3+ Treg cells in lung cancer (90, 91). In PGE2-producing tumors, fewer NK cells are in the tumor, and they lose the ability to produce the cytokines CCL5 and CXCL1 (92). Moreover, PGE2 downregulates the expression of the activating receptors NKp46, NKp44, NKp30, and NKG2D by binding to the receptors EP2 and EP4 on NK cells (93), then promoting the amplification of cyclic AMP cascade protein kinase A signaling (94), and finally resulting in the exhaustion of NK cells (95, 96) (Figure 2). Thus, PGE2 plays an important role in inducing NK hypofunction in the lung microenvironment and NK exhaustion in the lung tumor microenvironment.
Oxygen
Since the critical physiological function of the lungs is to mediate the exchange of oxygen and carbon dioxide, the respiratory tract is a unique niche with relatively high levels of oxygen (61), which has a direct influence on lung immunity (60). Oxygen is transported by red blood cells from alveoli to different organs. Under physiological conditions, due to oxygen being delivered according to the metabolic requirements of each tissue, different tissues have their own special ‘physioxia’ status. The oxygen levels in the atmosphere, so-called normoxia, are 21.1% (160 mmHg). The oxygen levels are approximately 19% (150 mmHg) in the trachea, approximately 14.5% (110 mmHg) in the alveoli, and 13.2% in arterial blood, which is relatively higher than other tissues (61). Cells from different organs can sense oxygen levels and change their metabolic and transcriptional features. Moreover, inflammation and oxygen levels are tightly linked, and hypoxia can cause inflammation (97). In the lung microenvironment, hypoxia could induce fundamental biological actions in lung structure cells and alveolar macrophages. The decline in oxygen levels in the local environment leads to the expression and stabilization of the transcription factor hypoxia-inducible factor (HIF), which plays a key role in lung diseases (98). HIF1α and HIF2α can be expressed by pulmonary artery smooth muscle cells (PASMCs), endothelial cells, and epithelial cells (98). Furthermore, recent studies have shown that the Von Hippel− Lindau (VHL) protein, which is a negative regulator of HIF, directly controls the terminal differentiation, self-renewal, and function of alveolar macrophages in homeostasis (99, 100). All these changes induced by hypoxia in the lung microenvironment might modulate other immune cells, including NK cells.
When a tumor appears in the lungs, the oxygen levels are completely changed due to the rapid growth of tumor cells. Hypoxia can promote the progression of tumors in multiple ways (101). The hypoxia-induced pathway plays important roles in promoting EMT, maintaining tumor stem cells, promoting angiogenesis, and driving the activation and expansion of immune-suppressive stromal cells (101). After the formation of a typical tumor microenvironment, hypoxia could further induce NK cell exhaustion (102). HIF1α induced in NK cells suppresses the upregulation of activating receptors such as NKp46, NKp44, NKp30, and NKG2D in the presence of activating cytokines, and NK cells display impaired cytotoxic capacity to target cells (103). Hypoxia in NK cells induces lower cytokine production (104). Furthermore, NK cells from HIF1α KO mice increase cytokine secretion and display higher antitumor activity (105), further demonstrating the important role of HIF1α. Overall, the low levels of oxygen (hypoxia) in the lung tumor microenvironment could promote the phenotype and function of NK exhaustion, but the detailed mechanism is not clear, and more studies need to be done.
Glucose
In homeostasis, the glucose concentration in the fluid line with lung epithelial cells is more than 10 times lower than that in blood. The glucose concentration in blood is maintained at approximately 4.0-5.5 mmol/L, which is the optimal level for normal brain functions (106). The liquid from the lower airways of healthy human donors contains only approximately 0.4 mmol/L glucose (106), which is critical to limit bacterial growth in mice and human lungs (62–64). Glucose levels in the alveoli increase when airway homeostasis is disrupted by inflammation and viral infection, and high glucose levels in blood are observed in patients with diabetes or acute hyperglycemia (64, 106). Low glucose levels in the alveoli are maintained against a concentration gradient by the highly activated glucose transporters Glut1 and Glut 10 (62). Elevated airway glucose concentrations may increase the risk of respiratory bacterial infection, particularly methicillin-resistant Staphylococcus aureus (MRSA) (64). High airway glucose concentrations might also exacerbate lung disease by inducing local inflammation. As the critical carbon source, the effect of glucose on inflammation and infection is higher than that of other nutrients (63). Interestingly, the glycolytic capacity of alveolar macrophages is relatively low compared to that of macrophages from other tissues (107), which might be explained by the low glucose levels in the alveolar niche. Normally, aerobic glycolysis is vital for T, B, and NK cell activation by supporting the biosynthetic demands of these cells (108). Thus, the inactivated state of lymphocytes might be partially associated with glucose shortage in the lung microenvironment.
Glucose restriction is also a biomarker of the lung tumor microenvironment (71). Cong et al. found that NK cells isolated from the lung tumor microenvironment have lower levels of glycolytic capacity accompanied by attenuated cytotoxic function and cytokine production (36). They also found that the increased expression of fructose-1,6-bisphosphatase (FBP1) inhibits glycolysis in NK cells from a lung cancer mouse model. These data indicate that tumor-driven glucose restriction inhibits glycolysis in NK cells and impairs their antitumor activity (109). On the other hand, the increased lactate concentration is also a biomarker of the TME, which is due to tumor cells primarily using glucose for glycolytic metabolism. Elevated lactate levels or low pH have been proven to prevent the cytotoxic activity of NK cells (110). Stimulation of NK cells by PMA/ionomycin in the presence of lactic acid could block their IFN-γ production (111). Overall, the correlation between glucose metabolism and NK cell exhaustion in the lung tumor microenvironment is largely unknown, and more research needs to be done.
Surfactant
The alveoli are coated with a layer of surfactant, a highly surface-active phospholipid-rich material, which is essential for respiratory function in mammals (112). Pulmonary surfactant is a complex mixture of approximately 90% lipids and 8-10% protein, which is essential for reducing surface tension at the air-liquid interface of the terminal airways and preventing the alveolus collapse upon expiration (113, 114). Surfactant is produced in type II alveolar epithelial cells, where it is assembled into lamellar bodies, densely packed membranous acidic organelles (112, 115). Type II alveolar epithelial cells and alveolar macrophages are the main cells responsible for surfactant uptake and degradation (114). When inflammation or lung injury occurs, some of the surfactant components might leak into the blood. The appearance of surfactant proteins in plasma could be used as an early marker of lung injury (114).
Surfactant protein
There are four surfactant proteins (SP), SP-A, SP-B, SP-C, and SP-D in the alveoli. SP-B and SP-C are small and very hydrophobic proteins that are essential for forming a monolayer at the air-liquid interface and have lower surface tension. SP-B deficiency or gene mutation is associated with respiratory distress syndrome in neonates (116). Furthermore, gene mutation of SP-C induces pulmonary alveolar proteinosis-like disease in some cases (117). SP-A and SP-D are larger hydrophilic-related proteins of the collectin family, which have important roles in innate immunity and local immune modulation (118). SP-A and SP-D can directly bind respiratory pathogens, allergens, and particles through their collectin binding domain and mediate phagocytosis by lung-resident cells, which are important for pathogen clearance in the early stage of infection (119–121). Studies have shown that SP-A could increase AM alternative activation (122, 123), indicating that SP-A might be involved in mounting the anti-inflammatory microenvironment in the lungs. However, SP-A has also been reported to increase activated NK cell numbers and prevent lung cancer progression by promoting the polarization of M1 tumor-associated macrophages (124). SP-D was found to prevent lung cancer progression by downregulating epidermal growth factor signaling (125). Although the role of surfactant protein in lung cancer has been studied, its importance in NK cells remains largely unclear. More research is needed to investigate the direct role of surfactant protein in influencing NK cell functions.
Surfactant lipids
Approximately 75–80% of phospholipids are phosphatidylcholines. The most abundant component of surfactant lipids is saturated dipalmitoylphosphatidylcholine (DPPC), approximately 40% of the total lipids. DPPC contains two saturated palmitic chains (16:0/16:0 PC). In addition, other anionic phospholipids, such as phosphatidylglycerol (PG, approximately 7%), phosphatidylethanolamine (PE, approximately 3.7%), phosphatidylinositol and phosphatidylserine (5.4%), and sphingomyelin (6.8%), appear as components of surfactant (126, 127). Decreased levels of surfactant lipids have been described in smokers and COPD patients (128, 129). Surfactant phospholipids reduce alternative activation and proliferation of alveolar macrophages by decreasing the activation of PI3K-Akt-mTORC1 signaling (130, 131). Studies have shown that lipids purified from the BAL fluid of healthy donors inhibit NK cell cytotoxicity in vitro (132). However, different lipid components have distinct roles in NK cell activation. PG was found to suppress, while PE could augment, the cytotoxic function of NK cells (132). Recently, Gong et al. reported that lipid-laden lung mesenchymal cells facilitate breast cancer lung metastasis via metabolic reprogramming of tumor cells and NK cells. They found that lipid-laden mesenchymal cells could transport their lipids to tumor cells and NK cells via exosome-like vesicles, which leads to tumor cell proliferation and NK cell exhaustion (133). Despite the high abundance of surfactant lipids in the alveoli, their roles in inflammation and cancer are unknown, and more studies are necessary to understand the importance of each phospholipid in shaping the function of NK cells.
Perspective
The effector function of NK cells is regulated by the fine balance between activating and inhibitory signals. Blockage of checkpoint receptors has displayed the potential to reverse NK cell exhaustion in tumors and boost their antitumor function. As a promising immunotherapy strategy for tumors, NK cell-based checkpoint blockade immunotherapy has displayed potential roles in improving current T-cell-based tumor immunotherapy. Recently, the therapeutic potential of many inhibitory receptors on NK cells, such as NGK2A, KIRs, TIM-3, TIGIT, CD96, and PD-1, has been proven by animal experiments or some clinical trials (102, 134). However, NK cell-based checkpoint blockade did not fully exhibit its antitumor function because the immunosuppressive tumor microenvironment plays a critical role in regulating the antitumor functions of NK cells. Thus, combining NK cell-based checkpoint blockade and targeting the vital specific components of the lung microenvironment would be a better solution for lung cancer therapy.
As the key immunosuppressive components of the lung tumor microenvironment, TGF-β and PGE2 have been widely studied. Targeting TGF-β or PGE2 in several studies has been shown to restore NK cell function in tumor therapy. However, the direct role of other components of the lung tumor microenvironment (glucose metabolism, hypoxia, and surfactant protein and lipids) is largely unknown. More basic research is needed to fully understand the relationship between these components and NK cell exhaustion. Notably, insights in the field of the lung tumor microenvironment and NK cell exhaustion will allow and facilitate the development of rationally designed combination immunotherapeutic strategies for lung cancers.
Author contributions
HZ: Writing – original draft. JW: Writing – review & editing. FL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Institute of Health and Medicine, Hefei Comprehensive National Science Center (2022KYQD010, DJK-LX-2022005), the R&D Program of Guangzhou Laboratory (SRPG22-006), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0490000), the National Key Research and Development Program of China (2022YFC2304102), the Natural Science Foundation of China (82271827), and the Fundamental Research Funds for the Central Universities (WK3520000016).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature (2018) 553(7689):446–54. doi: 10.1038/nature25183
2. Gong Z, Li Q, Shi J, Wei J, Li P, Chang CH, et al. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity (2022) 55(8):1483–500 e9. doi: 10.1016/j.immuni.2022.07.001
3. Medeiros B, Allan AL. Molecular mechanisms of breast cancer metastasis to the lung: clinical and experimental perspectives. Int J Mol Sci (2019) 20(9):2272. doi: 10.3390/ijms20092272
4. Crinier A, Narni-Mancinelli E, Ugolini S, Vivier E. Snapshot: natural killer cells. Cell (2020) 180(6):1280– e1. doi: 10.1016/j.cell.2020.02.029
5. Cong J, Wei H. Natural killer cells in the lungs. Front Immunol (2019) 10:1416. doi: 10.3389/fimmu.2019.01416
6. Guillerey C, Smyth MJ. Nk cells and cancer immunoediting. Curr Top Microbiol Immunol (2016) 395:115–45. doi: 10.1007/82_2015_446
7. Cremer I, Fridman WH, Sautes-Fridman C. Tumor microenvironment in nsclc suppresses nk cells function. Oncoimmunology (2012) 1(2):244–6. doi: 10.4161/onci.1.2.18309
9. Hintzen G, Ohl L, del Rio ML, Rodriguez-Barbosa JI, Pabst O, Kocks JR, et al. Induction of tolerance to innocuous inhaled antigen relies on a ccr7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J Immunol (2006) 177(10):7346–54. doi: 10.4049/jimmunol.177.10.7346
10. de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med (2004) 200(1):89–98. doi: 10.1084/jem.20040035
11. Lo B, Hansen S, Evans K, Heath JK, Wright JR. Alveolar epithelial type ii cells induce T cell tolerance to specific antigen. J Immunol (2008) 180(2):881–8. doi: 10.4049/jimmunol.180.2.881
12. Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol (2008) 8(2):142–52. doi: 10.1038/nri2236
13. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol (2015) 16(1):36–44. doi: 10.1038/ni.3052
14. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science (2019) 363(6432):eaau0964. doi: 10.1126/science.aau0964
15. Humphries DC, O'Connor RA, Larocque D, Chabaud-Riou M, Dhaliwal K, Pavot V. Pulmonary-resident memory lymphocytes: pivotal orchestrators of local immunity against respiratory infections. Front Immunol (2021) 12:738955. doi: 10.3389/fimmu.2021.738955
16. Xu J, Xiao N, Zhou D, Xie L. Disease tolerance: A protective mechanism of lung infections. Front Cell Infect Microbiol (2023) 13:1037850. doi: 10.3389/fcimb.2023.1037850
17. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi: 10.1031/ni.2131
18. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
19. Marquardt N, Kekalainen E, Chen P, Kvedaraite E, Wilson JN, Ivarsson MA, et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional cd69(-)Cd56(Dim) cells. J Allergy Clin Immunol (2017) 139(4):1321–30 e4. doi: 10.1016/j.jaci.2016.07.043
20. Kiessling R, Klein E, Pross H, Wigzell H. "Natural" Killer cells in the mouse. Ii. Cytotoxic cells with specificity for mouse moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5(2):117–21. doi: 10.1002/eji.1830050209
21. Tomasello E, Yessaad N, Gregoire E, Hudspeth K, Luci C, Mavilio D, et al. Mapping of nkp46(+) cells in healthy human lymphoid and non-lymphoid tissues. Front Immunol (2012) 3:344. doi: 10.3389/fimmu.2012.00344
22. Castriconi R, Carrega P, Dondero A, Bellora F, Casu B, Regis S, et al. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front Immunol (2018) 9:2324. doi: 10.3389/fimmu.2018.02324
23. Lepretre F, Gras D, Chanez P, Duez C. Natural killer cells in the lung: potential role in asthma and virus-induced exacerbation? Eur Respir Rev (2023) 32(169):230036. doi: 10.1183/16000617.0036-2023
24. Wang J, Li F, Zheng M, Sun R, Wei H, Tian Z. Lung natural killer cells in mice: phenotype and response to respiratory infection. Immunology (2012) 137(1):37–47. doi: 10.1111/j.1365-2567.2012.03607.x
25. Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, et al. The trafficking of natural killer cells. Immunol Rev (2007) 220(1):169–82. doi: 10.1111/j.1600-065X.2007.00563.x
26. Hervier B, Russick J, Cremer I, Vieillard V. Nk cells in the human lungs. Front Immunol (2019) 10:1263. doi: 10.3389/fimmu.2019.01263
27. Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol (2011) 11(10):658–71. doi: 10.1038/nri3065
28. Ge N, Nishioka Y, Nakamura Y, Okano Y, Yoneda K, Ogawa H, et al. Synthesis and secretion of interleukin-15 by freshly isolated human bronchial epithelial cells. Int Arch Allergy Immunol (2004) 135(3):235–42. doi: 10.1159/000081309
29. Franklin M, Connolly E, Hussell T. Recruited and tissue-resident natural killer cells in the lung during infection and cancer. Front Immunol (2022) 13:887503. doi: 10.3389/fimmu.2022.887503
30. Talmadge JE, Meyers KM, Prieur DJ, Starkey JR. Role of nk cells in tumour growth and metastasis in beige mice. Nature (1980) 284(5757):622–4. doi: 10.1038/284622a0
31. Karre K, Klein GO, Kiessling R, Klein G, Roder JC. Low natural in vivo resistance to syngeneic leukaemias in natural killer-deficient mice. Nature (1980) 284(5757):624–6. doi: 10.1038/284624a0
32. Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, et al. A new immunodeficiency disorder in humans involving nk cells. Nature (1980) 284(5756):553–5. doi: 10.1038/284553a0
34. Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, et al. Prognostic significance of tumor infiltrating natural killer cells subset cd57 in patients with squamous cell lung cancer. Lung Cancer (2002) 35(1):23–8. doi: 10.1016/s0169-5002(01)00292-6
35. Jin S, Deng Y, Hao JW, Li Y, Liu B, Yu Y, et al. Nk cell phenotypic modulation in lung cancer environment. PloS One (2014) 9(10):e109976. doi: 10.1371/journal.pone.0109976
36. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of natural killer cells by fbp1-induced inhibition of glycolysis during lung cancer progression. Cell Metab (2018) 28(2):243–55.e5. doi: 10.1016/j.cmet.2018.06.021
37. Takeda K, Nakayama M, Sakaki M, Hayakawa Y, Imawari M, Ogasawara K, et al. Ifn-gamma production by lung nk cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J Leukoc Biol (2011) 90(4):777–85. doi: 10.1189/jlb.0411208
38. Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology (2009) 128(2):151–63. doi: 10.1111/j.1365-2567.2009.03167.x
39. Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect (2002) 4(15):1545–58. doi: 10.1016/s1286-4579(02)00038-2
40. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene ncr1. Nat Immunol (2006) 7(5):517–23. doi: 10.1038/ni1322
41. Li F, Zhu H, Sun R, Wei H, Tian Z. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J Virol (2012) 86(4):2251–8. doi: 10.1128/jvi.06209-11
42. Robinson BW, Pinkston P, Crystal RG. Natural killer cells are present in the normal human lung but are functionally impotent. J Clin Invest (1984) 74(3):942–50. doi: 10.1172/JCI111513
43. Bordignon C, Villa F, Allavena P, Introna M, Biondi A, Avallone R, et al. Inhibition of natural killer activity by human bronchoalveolar macrophages. J Immunol (1982) 129(2):587–91. doi: 10.4049/jimmunol.129.2.587
44. Weissman DN, deShazo RD, Banks DE. Modulation of natural killer cell function by human alveolar macrophages. J Allergy Clin Immunol (1986) 78(4 Pt 1):571–7. doi: 10.1016/0091-6749(86)90073-4
45. Michel T, Poli A, Domingues O, Mauffray M, Theresine M, Brons NH, et al. Mouse lung and spleen natural killer cells have phenotypic and functional differences, in part influenced by macrophages. PloS One (2012) 7(12):e51230. doi: 10.1371/journal.pone.0051230
46. Lauzon W, Lemaire I. Alveolar macrophage inhibition of lung-associated nk activity: involvement of prostaglandins and transforming growth factor-beta 1. Exp Lung Res (1994) 20(4):331–49. doi: 10.3109/01902149409064391
47. Malmberg KJ, Carlsten M, Bjorklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol (2017) 31:20–9. doi: 10.1016/j.smim.2017.08.002
48. Clement MV, Haddad P, Soulie A, Legros-Maida S, Guillet J, Cesar E, et al. Involvement of granzyme B and perforin gene expression in the lytic potential of human natural killer cells. Res Immunol (1990) 141(6):477–89. doi: 10.1016/0923-2494(90)90017-s
49. Trapani JA. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust N Z J Med (1995) 25(6):793–9. doi: 10.1111/j.1445-5994.1995.tb02883.x
50. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ (2003) 10(1):45–65. doi: 10.1038/sj.cdd.4401189
51. Arase H, Arase N, Saito T. Fas-mediated cytotoxicity by freshly isolated natural killer cells. J Exp Med (1995) 181(3):1235–8. doi: 10.1084/jem.181.3.1235
52. Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, et al. Expression and function of tnf-related apoptosis-inducing ligand on murine activated nk cells. J Immunol (1999) 163(4):1906–13. doi: 10.4049/jimmunol.163.4.1906
53. Zhang M, Wen B, Anton OM, Yao Z, Dubois S, Ju W, et al. Il-15 enhanced antibody-dependent cellular cytotoxicity mediated by nk cells and macrophages. Proc Natl Acad Sci U.S.A. (2018) 115(46):E10915–E24. doi: 10.1073/pnas.1811615115
54. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi: 10.1126/science.1198687
55. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral nk cell phenotype and function in lung carcinoma. Cancer Res (2011) 71(16):5412–22. doi: 10.1158/0008-5472.CAN-10-4179
56. Chen X, Chen Y, Xin Z, Lin M, Hao Z, Chen D, et al. Tissue-resident cd69(+) cxcr6(+) natural killer cells with exhausted phenotype accumulate in human non-small cell lung cancer. Eur J Immunol (2022) 52(12):1993–2005. doi: 10.1002/eji.202149608
57. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in cd56 bright cd16(-) cells and display an impaired capability to kill tumor cells. Cancer (2008) 112(4):863–75. doi: 10.1002/cncr.23239
58. Tang F, Li J, Qi L, Liu D, Bo Y, Qin S, et al. A pan-cancer single-cell panorama of human natural killer cells. Cell (2023) 186(19):4235–4251.e20. doi: 10.1016/j.cell.2023.07.034
59. Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest (2004) 125(2):754–65. doi: 10.1378/chest.125.2.754
60. Zenewicz LA. Oxygen levels and immunological studies. Front Immunol (2017) 8:324. doi: 10.3389/fimmu.2017.00324
61. Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med (2011) 15(6):1239–53. doi: 10.1111/j.1582-4934.2011.01258.x
62. Pezzulo AA, Gutierrez J, Duschner KS, McConnell KS, Taft PJ, Ernst SE, et al. Glucose depletion in the airway surface liquid is essential for sterility of the airways. PloS One (2011) 6(1):e16166. doi: 10.1371/journal.pone.0016166
63. Gill SK, Hui K, Farne H, Garnett JP, Baines DL, Moore LS, et al. Increased airway glucose increases airway bacterial load in hyperglycaemia. Sci Rep (2016) 6:27636. doi: 10.1038/srep27636
64. Philips BJ, Redman J, Brennan A, Wood D, Holliman R, Baines D, et al. Glucose in bronchial aspirates increases the risk of respiratory mrsa in intubated patients. Thorax (2005) 60(9):761–4. doi: 10.1136/thx.2004.035766
65. Morissette MC, Shen P, Thayaparan D, Stampfli MR. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur Respir J (2015) 46(5):1451–60. doi: 10.1183/09031936.00216914
66. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer (2019) 19(1):9–31. doi: 10.1038/s41568-018-0081-9
67. Lee YS, Choi H, Cho HR, Son WC, Park YS, Kang CD, et al. Downregulation of nkg2dls by tgf-beta in human lung cancer cells. BMC Immunol (2021) 22(1):44. doi: 10.1186/s12865-021-00434-8
68. Nakanishi M, Rosenberg DW. Multifaceted roles of pge2 in inflammation and cancer. Semin Immunopathol (2013) 35(2):123–37. doi: 10.1007/s00281-012-0342-8
69. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis (2018) 7(1):10. doi: 10.1038/s41389-017-0011-9
70. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol (2020) 82:103–26. doi: 10.1146/annurev-physiol-021119-034627
71. Navratilova J, Hankeova T, Benes P, Smarda J. Low-glucose conditions of tumor microenvironment enhance cytotoxicity of tetrathiomolybdate to neuroblastoma cells. Nutr Cancer (2013) 65(5):702–10. doi: 10.1080/01635581.2013.789118
72. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine tgf-beta promotes the development and homeostasis of alveolar macrophages. Immunity (2017) 47(5):903–12.e4. doi: 10.1016/j.immuni.2017.10.007
73. Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol (1986) 136(10):3916–20. doi: 10.4049/jimmunol.136.10.3916
74. Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of nk cell functions by tgf-beta 1. J Immunol (1995) 155(3):1066–73. doi: 10.4049/jimmunol.155.3.1066
75. Malygin AM, Meri S, Timonen T. Regulation of natural killer cell activity by transforming growth factor-beta and prostaglandin E2. Scand J Immunol (1993) 37(1):71–6. doi: 10.1111/j.1365-3083.1993.tb01667.x
76. Zalfa C, Paust S. Natural killer cell interactions with myeloid derived suppressor cells in the tumor microenvironment and implications for cancer immunotherapy. Front Immunol (2021) 12:633205. doi: 10.3389/fimmu.2021.633205
77. Saito A, Horie M, Nagase T. Tgf-beta signaling in lung health and disease. Int J Mol Sci (2018) 19(8):2460. doi: 10.3390/ijms19082460
78. Ahmadi A, Najafi M, Farhood B, Mortezaee K. Transforming growth factor-beta signaling: tumorigenesis and targeting for cancer therapy. J Cell Physiol (2019) 234(8):12173–87. doi: 10.1002/jcp.27955
79. Yoon H, Tang CM, Banerjee S, Delgado AL, Yebra M, Davis J, et al. Tgf-beta1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis (2021) 10(2):13. doi: 10.1038/s41389-021-00302-5
80. Farhood B, Khodamoradi E, Hoseini-Ghahfarokhi M, Motevaseli E, Mirtavoos-Mahyari H, Eleojo Musa A, et al. Tgf-beta in radiotherapy: mechanisms of tumor resistance and normal tissues injury. Pharmacol Res (2020) 155:104745. doi: 10.1016/j.phrs.2020.104745
81. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of nkp30 and nkg2d receptors: consequences for the nk-mediated killing of dendritic cells. Proc Natl Acad Sci U.S.A. (2003) 100(7):4120–5. doi: 10.1073/pnas.0730640100
82. Lee JC, Lee KM, Kim DW, Heo DS. Elevated tgf-beta1 secretion and down-modulation of nkg2d underlies impaired nk cytotoxicity in cancer patients. J Immunol (2004) 172(12):7335–40. doi: 10.4049/jimmunol.172.12.7335
83. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. Tgf-beta-inducible microrna-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U.S.A. (2014) 111(11):4203–8. doi: 10.1073/pnas.1319269111
84. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia (2004) 6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2
85. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
86. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate nk cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U.S.A. (2009) 106(49):20847–52. doi: 10.1073/pnas.0906481106
87. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
88. Park A, Lee Y, Kim MS, Kang YJ, Park YJ, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (Nk) cell cytotoxicity and nk cell differentiation. Front Immunol (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
89. Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via ep4-betaarrestin1-C-src signalsome. Mol Cancer Res (2010) 8(4):569–77. doi: 10.1158/1541-7786.MCR-09-0511
90. Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of foxp3 expression and cd4+ Cd25+ T regulatory cell activities in lung cancer. Cancer Res (2005) 65(12):5211–20. doi: 10.1158/0008-5472.CAN-05-0141
91. Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating foxp3+ Regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol (2010) 5(5):585–90. doi: 10.1097/JTO.0b013e3181d60fd7
92. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162(6):1257–70. doi: 10.1016/j.cell.2015.08.015
93. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (Pge (2)) suppresses natural killer cell function primarily through the pge(2) receptor ep4. Cancer Immunol Immunother (2011) 60(11):1577–86. doi: 10.1007/s00262-011-1064-9
94. Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, et al. Cooperation of adenosine and prostaglandin E2 (Pge2) in amplification of camp-pka signaling and immunosuppression. Cancer Immunol Immunother (2008) 57(11):1611–23. doi: 10.1007/s00262-008-0494-5
95. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood (2008) 111(3):1327–33. doi: 10.1182/blood-2007-02-074997
96. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res (2012) 72(6):1407–15. doi: 10.1158/0008-5472.CAN-11-2544
97. Corcoran SE, O'Neill LA. Hif1alpha and metabolic reprogramming in inflammation. J Clin Invest (2016) 126(10):3699–707. doi: 10.1172/JCI84431
98. Urrutia AA, Aragones J. Hif oxygen sensing pathways in lung biology. Biomedicines (2018) 6(2):68. doi: 10.3390/biomedicines6020068
99. Zhang W, Li Q, Li D, Li J, Aki D, Liu YC. The E3 ligase vhl controls alveolar macrophage function via metabolic-epigenetic regulation. J Exp Med (2018) 215(12):3180–93. doi: 10.1084/jem.20181211
100. Izquierdo HM, Brandi P, Gomez MJ, Conde-Garrosa R, Priego E, Enamorado M, et al. Von hippel-lindau protein is required for optimal alveolar macrophage terminal differentiation, self-renewal, and function. Cell Rep (2018) 24(7):1738–46. doi: 10.1016/j.celrep.2018.07.034
101. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol (2017) 8:1887. doi: 10.3389/fimmu.2017.01887
103. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in nk-cell-mediated target cell killing without affecting adcc. Eur J Immunol (2013) 43(10):2756–64. doi: 10.1002/eji.201343448
104. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human nk cells, modulates their immunoregulatory profile, and influences nk cell subset migration. Front Immunol (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
105. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-cell rna sequencing of tumor-infiltrating nk cells reveals that inhibition of transcription factor hif-1alpha unleashes nk cell activity. Immunity (2020) 52(6):1075–87.e8. doi: 10.1016/j.immuni.2020.05.001
106. Baker EH, Wood DM, Brennan AL, Clark N, Baines DL, Philips BJ. Hyperglycaemia and pulmonary infection. Proc Nutr Soc (2006) 65(3):227–35. doi: 10.1079/pns2006499
107. Svedberg FR, Brown SL, Krauss MZ, Campbell L, Sharpe C, Clausen M, et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat Immunol (2019) 20(5):571–80. doi: 10.1038/s41590-019-0352-y
108. Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol (2015) 68(2 Pt C):513–9. doi: 10.1016/j.molimm.2015.07.034
109. Terren I, Orrantia A, Vitalle J, Zenarruzabeitia O, Borrego F. Nk cell metabolism and tumor microenvironment. Front Immunol (2019) 10:2278. doi: 10.3389/fimmu.2019.02278
110. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and nk cells. J Immunol (2013) 191(3):1486–95. doi: 10.4049/jimmunol.1202702
111. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. Ldha-associated lactic acid production blunts tumor immunosurveillance by T and nk cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
113. Veldhuizen R, Nag K, Orgeig S, Possmayer F. The role of lipids in pulmonary surfactant. Biochim Biophys Acta (1998) 1408(2-3):90–108. doi: 10.1016/s0925-4439(98)00061-1
114. Olmeda B, Martinez-Calle M, Perez-Gil J. Pulmonary surfactant metabolism in the alveolar airspace: biogenesis, extracellular conversions, recycling. Ann Anat (2017) 209:78–92. doi: 10.1016/j.aanat.2016.09.008
116. Yin X, Meng F, Wang Y, Xie L, Kong X, Feng Z. Surfactant protein B deficiency and gene mutations for neonatal respiratory distress syndrome in China han ethnic population. Int J Clin Exp Pathol (2013) 6(2):267–72.
117. Tredano M, Griese M, Brasch F, Schumacher S, de Blic J, Marque S, et al. Mutation of sftpc in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet A (2004) 126A(1):18–26. doi: 10.1002/ajmg.a.20670
118. Nogee LM. Genetic causes of surfactant protein abnormalities. Curr Opin Pediatr (2019) 31(3):330–9. doi: 10.1097/MOP.0000000000000751
119. Sorensen GL. Surfactant protein D in respiratory and non-respiratory diseases. Front Med (Lausanne) (2018) 5:18. doi: 10.3389/fmed.2018.00018
120. LeVine AM, Elliott J, Whitsett JA, Srikiatkhachorn A, Crouch E, DeSilva N, et al. Surfactant protein-D enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol (2004) 31(2):193–9. doi: 10.1165/rcmb.2003-0107OC
121. Nathan N, Taytard J, Duquesnoy P, Thouvenin G, Corvol H, Amselem S, et al. Surfactant protein A: A key player in lung homeostasis. Int J Biochem Cell Biol (2016) 81(Pt A):151–5. doi: 10.1016/j.biocel.2016.11.003
122. Minutti CM, Garcia-Fojeda B, Saenz A, de Las Casas-Engel M, Guillamat-Prats R, de Lorenzo A, et al. Surfactant protein a prevents ifn-gamma/ifn-gamma receptor interaction and attenuates classical activation of human alveolar macrophages. J Immunol (2016) 197(2):590–8. doi: 10.4049/jimmunol.1501032
123. Minutti CM, Jackson-Jones LH, Garcia-Fojeda B, Knipper JA, Sutherland TE, Logan N, et al. Local amplifiers of il-4ralpha-mediated macrophage activation promote repair in lung and liver. Science (2017) 356(6342):1076–80. doi: 10.1126/science.aaj2067
124. Mitsuhashi A, Goto H, Kuramoto T, Tabata S, Yukishige S, Abe S, et al. Surfactant protein a suppresses lung cancer progression by regulating the polarization of tumor-associated macrophages. Am J Pathol (2013) 182(5):1843–53. doi: 10.1016/j.ajpath.2013.01.030
125. Hasegawa Y, Takahashi M, Ariki S, Asakawa D, Tajiri M, Wada Y, et al. Surfactant protein D suppresses lung cancer progression by downregulation of epidermal growth factor signaling. Oncogene (2015) 34(7):838–45. doi: 10.1038/onc.2014.20
126. Pryhuber GS. Regulation and function of pulmonary surfactant protein B. Mol Genet Metab (1998) 64(4):217–28. doi: 10.1006/mgme.1998.2722
127. Castillo-Sanchez JC, Cruz A, Perez-Gil J. Structural hallmarks of lung surfactant: lipid-protein interactions, membrane structure and future challenges. Arch Biochem Biophys (2021) 703:108850. doi: 10.1016/j.abb.2021.108850
128. Agudelo CW, Kumley BK, Area-Gomez E, Xu Y, Dabo AJ, Geraghty P, et al. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (Copd). PloS One (2020) 15(2):e0228279. doi: 10.1371/journal.pone.0228279
129. Higenbottam T. Lung lipids and disease. Respiration (1989) 55 Suppl 1:14–27. doi: 10.1159/000195747
130. Casals C, García-Fojeda B, Tenreiro P, Minutti CM. Surfactant lipids inhibit pi3k-dependent signaling pathways induced by il-4 in alveolar macrophages. J Immunol (2023) 210(1_Supplement):72.32–2. doi: 10.4049/jimmunol.210.Supp.72.32
131. García-Fojeda B, Minutti C, Landoni M, Tenreiro P, Casals C. Lung surfactant lipids reduce the alternative activation and proliferation of alveolar macrophages induced by il-4. ERJ Open Res (2022) 8(suppl 8):251. doi: 10.1183/23120541.Lsc-2022.251
132. Wilsher ML, Hughes DA, Haslam PL. Immunomodulatory effects of pulmonary surfactant on natural killer cell and antibody-dependent cytotoxicity. Clin Exp Immunol (1988) 74(3):465–70.
133. Gong Z, Li Q, Shi J, Li P, Hua L, Shultz LD, et al. Immunosuppressive reprogramming of neutrophils by lung mesenchymal cells promotes breast cancer metastasis. Sci Immunol (2023) 8(80):eadd5204. doi: 10.1126/sciimmunol.add5204
Keywords: natural killer cells, hypofunction, exhaustion, lung microenvironment, lung tumor microenvironment
Citation: Zhang H, Wang J and Li F (2023) Modulation of natural killer cell exhaustion in the lungs: the key components from lung microenvironment and lung tumor microenvironment. Front. Immunol. 14:1286986. doi: 10.3389/fimmu.2023.1286986
Received: 01 September 2023; Accepted: 23 October 2023;
Published: 06 November 2023.
Edited by:
Yugang Guo, Zhejiang University, ChinaReviewed by:
Jiacheng Bi, Chinese Academy of Sciences (CAS), ChinaTaisuke Kondo, National Institutes of Health (NIH), United States
Copyright © 2023 Zhang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengqi Li, ZmVuZ3FpLmxpQGlobS5hYy5jbg==; Jian Wang, dXN0Y3dqQHVzdGMuZWR1LmNu