 Ana L. Portillo1,2,3
Ana L. Portillo1,2,3 Jonathan K. Monteiro1,2,3
Jonathan K. Monteiro1,2,3 Eduardo A. Rojas1,2Tyrah M. Ritchie1,2
Eduardo A. Rojas1,2Tyrah M. Ritchie1,2 Amy Gillgrass1,2,3
Amy Gillgrass1,2,3 Ali A. Ashkar1,2,3*
Ali A. Ashkar1,2,3*- 1Department of Medicine, McMaster University, Hamilton, ON, Canada
- 2McMaster Immunology Research Centre, McMaster University, Hamilton, ON, Canada
- 3Centre for Discovery in Cancer Research, McMaster University, Hamilton, ON, Canada
The ability to expand and activate natural Killer (NK) cells ex vivo has dramatically changed the landscape in the development of novel adoptive cell therapies for treating cancer over the last decade. NK cells have become a key player for cancer immunotherapy due to their innate ability to kill malignant cells while not harming healthy cells, allowing their potential use as an “off-the-shelf” product. Furthermore, recent advancements in NK cell genetic engineering methods have enabled the efficient generation of chimeric antigen receptor (CAR)-expressing NK cells that can exert both CAR-dependent and antigen-independent killing. Clinically, CAR-NK cells have shown promising efficacy and safety for treating CD19-expressing hematologic malignancies. While the number of pre-clinical studies using CAR-NK cells continues to expand, it is evident that solid tumors pose a unique challenge to NK cell-based adoptive cell therapies. Major barriers for efficacy include low NK cell trafficking and infiltration into solid tumor sites, low persistence, and immunosuppression by the harsh solid tumor microenvironment (TME). In this review we discuss the barriers posed by the solid tumor that prevent immune cell trafficking and NK cell effector functions. We then discuss promising strategies to enhance NK cell infiltration into solid tumor sites and activation within the TME. This includes NK cell-intrinsic and -extrinsic mechanisms such as NK cell engineering to resist TME-mediated inhibition and use of tumor-targeted agents such as oncolytic viruses expressing chemoattracting and activating payloads. We then discuss opportunities and challenges for using combination therapies to extend NK cell therapies for the treatment of solid tumors.
1 Introduction
Natural killer (NK) cells are a subset of innate lymphocytes that can directly kill target cells without prior antigen sensitization. NK cells were first recognized for this unique ability over five decades ago and have since been characterized as an integral part of the body’s innate immune defense against infection and cancer. NK cells are of particular interest as they can recognize and kill tumor cells while leaving healthy cells unharmed through recognition of HLA-class I molecules expressed on healthy cells (1). NK cell-based cancer immunotherapies leverage this unique ability and provide a safer alternative to other adoptive cell therapies like chimeric antigen receptor (CAR)-T cells which become activated against any cell expressing the target antigen. Indeed, various clinical trials have shown that ex vivo activated and expanded NK cells can achieve high clinical efficacy against hematologic malignancies with a lower risk for adverse effects such as graft vs host disease (2–4). Additionally, NK cells engineered to express a CAR have shown clinical efficacy against B-cell malignancies with limited therapy-induced toxicities such as cytokine release syndrome (5). CAR-NK cell therapies can also overcome tumor antigen escape problems seen with CAR-T cells due to complete loss or downregulation of antigen expression by target cells since CAR-NK cells can target tumors through CAR-independent mechanisms. These characteristics enable NK cells to be used as an effective allogenic “of-the-shelf” product reducing cost and time to treatment in comparison to CAR-T cell therapy. However, despite over a decade of pre-clinical and clinical development, the use of NK cell therapy against solid tumors remains limited. Solid tumors pose unique barriers to adoptive cell therapies including NK cells, by impairing immune cell entry into the tumor core and dampening anti-tumor effector functions within the immunosuppressive and metabolically hostile tumor microenvironment (TME). However, a more challenging hurdle is the tumor’s ability to block NK cell trafficking to the tumor core, leaving the highly efficient killers on the sidelines. In this review, we focus on summarizing the mechanisms blocking NK cell homing and infiltration to the solid tumor core and discuss various strategies that try to overcome these barriers and give access to NK cell adoptive cell therapy. This includes therapies that directly modulate the structural barriers by the tumor and that improve blood flow into the tumor. We also discuss strategies that modulate the chemokine-chemokine receptor axis to enhance the homing ability of NK cells to the tumor site. Lastly, we highlight how engineered oncolytic viruses (OVs) and nanotechnologies can offer unique advantages to other therapeutic modalities through their enhanced ability express or deliver immunomodulatory agents within the TME. OVs are of particular interest for synergizing with NK cells therapy as they offer a platform for expressing a wide range of chemoattracting and activation factors in the TME, while also contributing to tumor clearance through tumor debulking. The development of tumor selective therapies that are permissive to the tumor core and that can be modified to deliver NK cell recruitment and activation payloads will unleash the potential of NK cell adoptive therapies for solid tumors.
2 Natural Killer (NK) cell adoptive cell therapy
2.1 Ex vivo activation and genetic manipulation of NK cells for cancer immunotherapy
NK cells account for 10-15% of peripheral blood lymphocytes and play a key role in the first-line immune response against viral infection and malignancy (1). NK cells express a variety of different germ-line encoded activation receptors that recognize stress-induced ligands upregulated on the surface of target cells (6). Moreover, NK cells also express various inhibitory receptors such as the killer immunoglobulin-like receptors (KIRs) and NKG2A which prevent their activation towards normal cells overexpressing HLA-class I and HLA-E molecules (7). NK cell activation and their resultant effector function is orchestrated by a balance between activation and inhibitory signaling that occurs upon NK cell receptor engagement (7). NKG2D and Natural Cytotoxicity Receptors (NCRs; NKp30, NKp44, and NKp46) are major NK cell activation receptors for the detection and elimination of viral and malignant cells (7). NKG2D recognizes the ligands MIC-A, MIC-B, ULBP 1-6 proteins which are absent from the surface of healthy cells but are upregulated by infected or stressed cells (8). In addition to upregulation of stress ligands, virally infected cells and tumors can also downregulate HLA class I which render them sensitive to NK cell-mediated killing and untargeted by effector T cells. NK cells also mediate antibody-dependent cellular cytotoxicity (ADCC) by engagement of FcγRIII (CD16) (6). Upon activation, NK cells kill targets directly through the release of lytic granules including granzymes and perforin (6). NK cell activation also leads to the secretion of pro-inflammatory cytokines including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) which further shape innate and adaptive immune responses (6). NK cell can also induce tumor killing independent of perforin and granzyme by engagement of Fas Ligand (FasL) and Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL) on NK cells with cognate receptors, Fas or TRAIL, expressed on target cells (7). Human NK cells are generally defined as CD3-CD56+, and traditionally, the extent of CD56 expression has been used to classify NK cells into two distinct functional subsets. The two subsets can be defined as the regulatory and high cytokine-producing CD56brightCD16- subset and the cytotoxic CD56dimCD16+ subset. The CD56dim NK cell population is mature and accounts for 80-95% of NK cells in peripheral blood (9).
Developments in the ex vivo activation, expansion, and genetic engineering of NK cells over the last decade has enabled their use as a potent cancer immunotherapy. Different strategies have been developed for the clinical grade expansion of highly pure and activated NK cells including the use of cytokines (IL-2, IL-12, IL-15, and IL-21), or irradiated feeder cells such as K562 cells and plasma membrane particles expressing membrane bound (mb)-IL-21 or -IL-15 for co-stimulation (10–12). Small molecules for NK cell expansion and activation have also been investigated including the use of nicotinamide and a glycogen synthase kinase 3 inhibitor (13). NK cells from different sources including healthy donor or cancer patient peripheral blood, umbilical cord blood, NK cell lines and induced pluripotent stem cells (iPSC)-derived NK cells have been used to develop adoptive cell therapies (13). Developments in genetic engineering methods has allowed the efficient generation of scalable, clinical-grade NK cells products with superior anti-tumor functions. Various strategies are being pursued to increase NK cell cytotoxicity via gene manipulation including expression of exogenous cytokines, downregulation of inhibitory receptors, and expression of chimeric antigen receptors (CARs) or T cell receptors. Novel genetic engineering methods using clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein (Cas9) gene editing and transduction with viral vectors such as adeno-associated virus (AAV) have enabled simultaneous insertion of CAR transgenes with long-term expression and gene knockouts in primary NK cells (14, 15). This is beneficial as CAR expression is target to a specific site on the genome compared to viral-based engineering methods such as lentivirus or retroviruses in which the integration site is uncontrolled. Other methods that do not require CRISPR engineering have been recently described such as the use of an AAV carrying a Sleeping Beauty transposon and electroporation of an mRNA encoding a transposase for permanent transgene integration (16). This method potentially offers a higher safety profile than other viral-based methods such as retroviruses (17). CAR-NK cell therapies in particular hold immense promise in targeting heterogenous solid tumors due to their ability to exert both antigen-independent and CAR-directed killing while remaining safe against healthy cells (5, 18). The use of CARs to specifically redirect NK cells towards tumor targets has already demonstrated high clinical efficacy against CD19-positive hematological malignancies without the severe toxicities and graft-vs-host disease seen with CAR-T cell therapies (5). Due to a higher safety profile, NK cell-based immunotherapies have an unparalleled potential for an “off-the-shelf” product reducing the cost and time of treatment administration. Further, NK cells maybe overcome tumor escape problems due to loss of the CAR-specific antigen, as seen after CAR-T cell therapy, as NK cells maintain antigen-independent activity. While relapse due to CAR-NK cell self-recognition after trogocytosis of the tumor antigen is possible, co-expression of a traditional activating CAR and a CAR with the signal domain from an inhibitory KIR can limit NK cell self-targeting enhancing treatment efficacy (19).
Clinical trials using ex vivo expanded NK cells have achieved therapeutic efficacies against multiple hematological malignancies such as high-risk myeloid cancers (2–4). However, there are more limited studies using NK cell-based adoptive cell therapies against solid tumors. Although the frequency of NK cells within solid tumor sites is typically lower compared to other immune cells such as cytotoxic CD8+ T cells, they exert potent anti-tumor functions (20). Indeed, a recent meta-analysis and systematic review by Nersesian et al. demonstrates that infiltration of NK cells is a positive prognostic factor in solid tumors and is associated with improved overall survival (21). Importantly, a lower risk of death was highly related to the location of NK cells within the tumor, with a lower effect seen in tumors where NK cells primarily localized around the tumor margin or in the stromal compartment as opposed to the intra-tumoral epithelial region (21). These studies exemplify the importance of NK cell anti-tumor immunity not only in hematologic malignancies but also solid tumors. In various pre-clinical studies, K562 mb-IL21 expanded NK cells have shown to be highly efficient at preventing tumor establishment in human ovarian, lung, and breast cancer models (22–24). However, these activated NK cells are limited at eliminating already established primary tumors that are not easily accessible such as breast cancer. The real-time biodistribution of fluorescently labeled NK cells expanded with K562 feeder cells has been investigated after intravenous administration to non-tumor bearing and human hepatocellular carcinoma (HCC) bearing mice (25). Transferred NK cells are detected primarily within the lungs within the first 4 hours of injection which decreased after 24 hours, after which NK cells were found to accumulate within the liver and persisted for 14-21 days (25). Near-infrared real-time tracking has been used to examine the biodistribution of adoptively transferred ex vivo expanded NK cells in human MDA-MB-231 triple negative breast cancer (TNBC) xenograft models (26). Using this technique Thanh Uong et al. found that expanded NK cells mainly trafficked to the lungs and not the primary tumor site, reaching the lungs within 30 minutes of intravenous administration (26). While NK cells were detected in the tumor by 4h, immunohistochemistry showed that NK cells primarily localized around the tumor margin as opposed to the tumor core (26). Thus, combining adoptive transfer of NK cells with additional strategies is needed to enhance their therapeutic response.
2.2 Combination of NK cell therapy with other anti-cancer strategies
NK cell anti-tumor functions can be harnessed to improve efficacy of conventional anti-cancer treatments such as radiotherapy and chemotherapy, as well as checkpoint blockade and targeted antibody therapies. Checkpoint blockade involves blocking the cytotoxic T lymphocyte antigen-4 or programmed death-1/programmed death ligand 1 (PD-1/PD-L1) pathways to promote T cell anti-tumor responses (27, 28). NK cells also contribute to higher anti-tumor responses during PD-1/PD-L1 blockade (29). Pembrolizumab, a monoclonal antibody binding PD-1, is a first-line treatment for PD-L1 expressing non-small cell lung cancer (NSCLC). A phase II clinical trial has shown that combining NK cells and pembrolizumab significantly improved the overall survival of patients with NSCLC compared to checkpoint inhibitor monotherapy (30). Co-administration of monoclonal antibodies (mAbs) targeting tumor associated antigens (TAAs) and NK cells has also garnered clinical interest as a way to engage NK cell-mediated ADCC. The most common TAAs targeted in clinical trials for solid tumors are GD2, epidermal growth factor receptor (EGFR), and human epidermal growth factor receptor-2 (HER-2) (13). Completed clinical trials using trastuzumab (anti-HER2 mAb) or cetuximab (anti-EGFR mAb) in combination with NK cell therapy show these therapies are well tolerated (31, 32). In a phase I/II clinical trial, NK cell administration with cetuximab also significantly improved overall survival in patients with NSCLC (31).
NK cells can also be combined with traditional anti-cancer therapies. Patin et al. has shown that radiotherapy and inhibiting the DNA damage response pathway can synergize to enhance NK cell activity and tumor infiltration in a mouse model of head and neck squamous cell carcinoma (HNSCC) (33). Another recent study demonstrated that radiotherapy enhances the anti-tumor efficacy of a fused IL-2 and anti-PD-1 cytokine in HNSCC tumors (34). In this model, combination therapy enhanced NK cell activity leading to reduced tumor metastasis in the lungs. Another study showed that local irradiation of the MDA-MB-231 tumors can improve human expanded NK cell infiltration into the tumors. Combination therapy with radiation and NK cells led to significantly higher suppression of primary and metastatic tumor growth compared to the NK or irradiation only groups (35). Further, CD56-positive NK cells were only detected within primary tumor tissue in the group that received irradiation therapy prior to NK cell administration (35). While radiotherapy can enhance expression of NK cell activation ligands, it can also make tumor cells resistant to perforin-mediated apoptosis reducing their sensitivity to NK cell targeting 72h post-irradiation (36). Further, radiation can increase expression of PD-L1 and reduce NKG2D ligands on tumor cells inhibiting NK cell killing which is more pronounced with increased radiation dosing (37). Hence, determining optimal dosing and timing of radiotherapy regimens will be important when combining them with NK cell therapy. Pre-treatment with chemotherapies has also shown to sensitize tumor cells to NK cell-mediated killing. In a clinical study assessing adoptive transfer of NK cells with chemotherapy combination therapy, NK cell administration after chemotherapy significantly increased progression-free survival of patients with stage III advanced colon cancer (38). Overall, there are currently many ongoing clinical trials assessing non-engineered and engineered NK cells alone or in combination with other therapies for solid tumors and have been reviewed elsewhere (13, 39).
3 Barriers limiting NK cell trafficking, infiltration, and activation in the solid tumor
Despite the promising potential for combination of NK cells and already clinically approved anti-cancer therapies or other modalities, their efficient infiltration to the tumor core remains a challenge. There are various major factors that impair NK cell trafficking and infiltration into the solid tumor including the extensive extracellular matrix (ECM) barrier surrounding the tumor bed, the aberrant tumor vasculature leading to poor tumor perfusion, and a mismatch in the chemokine expression pattern within the TME to attract activated NK cells to the tumor. In this section we will summarize how these mechanisms work together to prevent efficient NK cell entry into the tumor bed and how the immunosuppressive TME impacts NK cell function inside the tumor. Enhancing NK cell infiltration into the tumor core and maintaining their anti-tumor function will be crucial to achieve durable responses with NK cell therapies for solid tumors.
3.1 The tumor stroma creates a rigid barrier around the tumor core
The solid tumor is a complex tissue made up tumor cells, a variety of immunosuppressive immune cells and the tumor stroma which consists of the abundant ECM, basement membrane, CAFs and endothelial cells. To enter the solid tumor site, immune cells first migrate towards the tumor through a cytokine/chemokine gradient. However, once they reach the tumor, they are shielded from the actual tumor core by the rigid and stiff ECM barrier (40). Thus, immune cells must migrate around the barrier and past ECM-adhesion sites to reach the tumor cells (40). Within the stroma, immune cells are unable to make direct contact with tumor cells and exert their anti-tumor functions. Tumor cells can themselves produce extracellular matrix components such as collagenases and ECM-modifying enzymes (41). Tumor cells also induce the differentiation of stromal cells into CAFs through secretion of the different growth factors including transforming growth factor β (TGF-β) and basic fibroblast growth factor (bFGF) (40). CAFs are abundant in solid tumors, making up to 80% of the tumor mass in some cancers (42). Within the tumor, CAFs are main contributors to ECM deposition, can produce TGF-β to support tumor growth and drive epithelial to mesenchymal transition leading to tumor invasiveness and metastasis (40). CAFs can be defined using a combination of morphological characteristics, genetic and protein biomarkers (43). For example, common biomarkers expressed by CAFs include alpha-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), and platelet-derived growth factor alpha (43). However, these markers are not specific as they are also expressed by mesenchymal cells. Further, the intra-tumoral heterogeneity of CAF populations with distinct phenotypes and functions have been described in murine and human tumors (44–46). This heterogeneity makes targeting CAFs as an anti-tumor treatment option more challenging.
The ECM consists of different matrix molecules including collagens, proteoglycans, hyaluronic acid and glycoproteins like laminins and elastin and increased levels of all these molecules have been correlated with poor prognosis (47–49). The most common feature of the ECM is high deposition of fibrillar collagen and upregulation of collagen processing enzymes, such as lysyl oxidase (LOX) (40). Secretion of LOX by tumor cells leads to cross linking of collagen and elastin and this highly crosslinked collagen matrix contributes to the extensive tumor rigidity and reduced oxygen supply to tumor (40, 50). Additionally, accumulation of proteoglycans and hyaluronic acid and their interactions with collagen within the tumor directly contribute to tumor stiffness by sequestering water (51, 52). As the tumor stiffens, there are mechanical forces that are generated from the surrounding healthy tissue pushing back on the growing tumor which induces solid stress (50). High solid stress can obstruct blood and lymphatic vessels leading to hypoxia and more interstitial fluid pressure which further acts to inhibit immune cell infiltration (53, 54). Altogether, the stiff ECM barrier prevents effective immune infiltration into the tumor core.
3.2 Hypoxia promotes the aberrant tumor vasculature and downregulation of adhesion molecules impairing immune cell infiltration
Other important factors that contribute to decreased intra-tumoral immune cell infiltration are tumor hypoxia and the aberrant tumor vasculature. Rapidly proliferating tumor cells have a high demand of oxygen, which is limited in the tumor. To support fast tumor growth, the tumor induces angiogenesis to increase nutrient and oxygen supply to the tumor. Hypoxia induces signalling of the major hypoxia inducible factor (HIF) pathway, which alters the balance in the production of pro- and anti-angiogenic factors promoting tumor angiogenesis (55). Specifically, activation of the hypoxia inducible transcription factor 1 (HIF-1) upregulates expression of vascular endothelial growth factor (VEGF) which can induce endothelial cell recruitment and blood vessel formation (55, 56). HIF-1 also upregulates MPPs, collagen modifying enzymes, and integrins which are all required for the formation of new blood vessels (56). Further, hypoxia and HIF-1α also induce production of angiopoietin-1, platelet-derived growth factor and TGF-β which drive vessel maturation through recruitment of supporting cells including pericytes to the newly formed vessels (57). Defective interactions between pericytes and endothelial cells can also contribute to tumor angiogenesis and formation of the disorganized neovasculature (58). For example, pericytes and endothelial cells are loosely associated in tumor capillaries in contrast to their tight associations in normal capillaries (59). Further, pericyte coverage on tumor vessels can vary from high to little or no coverage across different cancers (60). Low pericyte coverage can also result in increased vessel permeability, contributing to the vessel leakiness (60). The aberrant vasculature is also made up of anergic endothelial cells and is highly leaky leading to high interstitial fluid pressure causing vessels to collapse resulting in low perfusion into the tumor (61). Further, high levels of pro-angiogenic factors in the tumor can impair immune cell extravasation into the tumor site. Normally, immune cells including NK cells migrate towards an inflammation site through a chemokine gradient. Selectin-selectin interactions between NK cells and endothelial cells facilitates rolling along the endothelium (62). Chemokines present on the endothelium activate immune cells leading to integrin activation. The integrins, LFA-1 and VLA-4, expressed on the surface of NK cells bind to ICAM-1 and VCAM-1 respectively, facilitating firm interactions with the endothelium, followed by extravasation into the tissue (62). Increased levels of VEGF and bFGF can downregulate of expression of the adhesion molecules such as ICAM-1, VCMA-1 and E and P selectins, disrupting interactions with the endothelium (62). Overall, the resulting tumor vasculature is highly immature and disorganized, thus indirectly reducing infiltration of anti-tumor immune cells and promotes the immunosuppressive and hypoxic TME. Increases in pro-angiogenic factors due to hypoxic conditions induces lower immune cell and vessel wall interactions, also impairing immune cell infiltration into the tumor.
3.3 Chemokine receptor expression influencing NK cell tumoral infiltration
The two NK cell functional subsets express a variety of different chemokine receptors corresponding with their distribution within the body. While the CD56bright subset is abundant within the liver, secondary lymphoid tissues, uterus, adrenal glands and the kidney, CD56dim NK cells are prominent within the bone marrow, lung, spleen and breast tissue (48). The CD56bright NK cells express CCR7 (CCL19 and CCL21 receptor), CCR5 (CCL3, CCL4, and CCL5 receptor), and the adhesion molecule CD62L (63, 64). The perforin and granzyme expressing CD56dim subset express CX3CR1 (CX3CL1 receptor) (65). These CD56dim NK cells also uniformly express CXCR1 and low levels of CXCR2 (CXCL1, CXCL2, CXCL3, CXCL5, and CXCL7 receptors) (66). In addition to chemokine receptors, CD56dim NK cells also rely on sphingosine 1-phosphate (S1P) for trafficking (67). The CD56bright and CD56dim NK cells share expression of the CXCR4 (CXCL12 receptor) and CXCR3 (CXCL9, CXCL10 and CXCL11 receptor) chemokine receptors, however these are expressed to a lower extent in CD56dim NK cells (68).
Various chemokine and chemokine receptors directly modulate NK cell homing towards solid tumors. Human clinical data demonstrates that solid tumors can upregulate chemokine expression which favors the recruitment of the CD56brightCD16- poorly cytotoxic NK cell subset. In turn, it also downregulates chemokines that recruit the CD56dim cytotoxic subset (48, 69). Carrega et al. showed that in lung and breast cancer tissue, the percentage of CD56bright and perforinlow expressing NK cells were much higher when compared to the corresponding normal tissue (70). This correlated with an upregulation of the chemokines CCL5, CCL19, CXCL9, and CXCL10 expression from the tumor (70). Hypoxic conditions have also shown to modulate chemokine receptor expression in peripheral blood NK cells. While IL-2 stimulation transiently enhanced CXCR4 expression, hypoxia significantly up-regulated CXCR4 expression on CD56dim and CD56bright NK cells compared to normoxic conditions (71). Intra-tumoral TGF-β can also modulate chemokine receptor expression. Active TGF-β1 present in the TME can upregulate expression of CXCR4 and CXCR3 in both CD56bright and CD56 dim NK cells while also reducing CX3CR1 expression in CD56dim NK cells (72, 73). Work in murine lymphoma tumor models has shown that the expression of CXCR3 and CX3CR1 chemokine ligands in the TME are needed for intra-tumoral accumulation of NK cells (74, 75).
Cytokine activation can also modulate chemokine receptor expression in human NK cells (76). The CX3CR1 receptor becomes downregulated in ex vivo activated human NK cells after cytokine stimulation with IL-15 (65). There is a general trend for downregulation of CXCR3 during short term stimulation with cytokines such as IL-2, IL-12, and IL-15 (69). Interestingly, the opposite has been seen with long term stimulation with IL-2 and K562 feeder cell-based NK cell expansion. Wennerberg et al. found that CXCR3 expression was significantly upregulated in ex vivo expanded NK cells (77). In addition, other reports show that while freshly isolated NK cells exhibited >80% expression of CXCR1, expansion of NK cells using K562 feeder cells expressing mb-IL21, mbIL-15 and 4-1BBL completely downregulated CXCR1 expression to undetectable levels (66). Expression of CXCR2 is also lost after NK cell ex vivo expansion using irradiated Epstein-Barr Virus lymphoblastoid cell lines as feeder cells (78). While human TNBC and renal cell carcinoma (RCC) cells express high levels of CXCR2 ligands including CXCL1 and CXCL2, a lack of CXCR2 expression by activated NK cells can limit their migration towards these tumors (78, 79). Expanded NK cells also downregulate expression of CXCR4 which is an important chemokine receptor for trafficking into the bone marrow (80). Further, expansion of NK cells can promote expression of chemokine receptors that promote NK cell trafficking to the liver including CXCR6 and CCR5 (80). Enhanced accumulation in the liver would diminish the number of NK cells that can enter non-hepatic tumor sites.
3.4 Maintaining potent NK cell anti-tumor functions within the solid tumor microenvironment
Even though NK cells may be able to infiltrate tumors, factors such as poor nutrient availability and immunosuppressive signals in the solid tumor microenvironment (TME) can inhibit NK cell tumor clearance. Thus, overcoming NK cell immune dysfunction within the TME is also necessary to sustain effective anti-tumor responses when targeting solid tumors. Apart from tumor and structural cells, various immune cells such as tumor associated macrophages (TAMs), regulatory T-cells (T-regs), myeloid derived suppressor cells (MDSCs) and neutrophils contribute to release of immunosuppressive factors such TGF-β and prostaglandin E2 (PGE2) that inhibit NK cell anti-tumor functions at the tumor site. Further, rapid tumor cell proliferation results in depletion of available nutrients in the TME which can impair NK cell function. Here we will summarize the inhibitory effects of TGF-β and PGE2 on NK cell function and discuss how NK cell expansion and cytokine activation can overcome inhibition in the TME.
In the TME, TAMs, T-regs, and MDSCs express soluble and membrane bound TGF-β while neutrophils can express the matrix metalloprotease (MMP)-9 to activate latent TGF-β to active TGF-β1 (81). TGF-β inhibits NK cell mediated killing by impairing the expression and signaling of NK cell activation receptors, primarily NKG2D. TGF-β directly reduces endogenous NKG2D expression in NK cells by inducing the production of microRNA(miRNA)-1245 which targets NKG2D transcripts (82). Furthermore, TGF-β can decrease the expression of DAP10 at the mRNA and protein level, impairing the intracellular signaling cascade of NKG2D (83). Lastly, TGF-β can induce the expression of miR-183 which promotes the downregulation of DAP12 and impairs the signal transduction of the activating receptor NKp44 (84). Thus, TGF-β renders NK cells unable to recognize malignant cells effectively by impairing the signal transduction of their activation receptors. TGF-β can also inhibit NK cell function by reducing their metabolic capacity. In mouse NK cells, in vitro culturing with TGF-β can block the induction of the mTOR pathway by IL-15 and decreases granzyme B and perforin expression following IL-2 stimulation (85). TGF-β also significantly decreases IL-2 induced mitochondrial metabolism in human NK cells. It inhibits the rates of oxidative phosphorylation and maximal respiration, while having no effect on glycolytic basal rate but reducing the glycolytic capacity of the cells (86). This impaired metabolic activity correlates with decreased IFN-γ, granzyme B and TRAIL expression on NK cells. Interestingly, this effect was found to occur without affecting mTORC1 signaling as seen in murine NK cells (86). Therapies inhibiting TGF-β are currently in development and are being evaluated for clinical efficacy and safety. Treatments blocking the synthesis of TGF-β and TGF-β receptor 1 kinase inhibitors have shown promise in high-grade gliomas, while a small molecule TGF-β1 inhibitor is demonstrating promising efficacy in the treatment of HCC (87, 88). It would be important to determine if rescued NK cell activity is playing a role in the efficacy of targeting intra-tumoral TGF-β. However, one study has demonstrated that the anti-tumor function of tumor-infiltrating NK cells isolated from patients with glioblastoma multiforme could not be rescued with administration of a TGF-β receptor kinase inhibitor (89). These authors demonstrated that genetic deletion of TGF-β on NK cells significantly improved tumor control in vivo (89).
PGE2 is another factor that can impair NK cell function within the TME. The expression of PGE2 in human gastric and prostate cancer has been inversely correlated with overall patient survival (90, 91). Cancer cells and CAFs produce PGE2 through upregulation of cyclooxygenases COX1 and COX2 (92). The receptors of PGE2 (EP1-EP4) are present on multiple cell types and murine NK cells express all four PGE2 receptors (93). Signalling of PGE2 through the EP4 receptor is primarily responsible for the dysfunction of murine NK cells leading to the inhibition of IFN-γ and TNF-α. Blocking the EP4 receptor with a small molecule can rescue murine NK cell activity in vivo (94). In human NK cells, the EP2 and EP4 receptors are responsible for the suppression of human NK cell cytotoxicity mediated through NKG2D, CD16, and NCRs (95, 96). PGE2 can act through these receptors to inhibit the activation of the ERK and NF-κB pathways on NK cells, thus leading to a reduction in their cytotoxic activity and IFN-γ production (95). PGE2 can also act on human type 1 conventional dendritic cells (DCs) leading to anti-tumor dysfunction by downregulating their expression of T cell recruiting chemokine ligands such as CXCL9 and CXCL10 and production of IL-12 which could also impair NK cell function (97). Initial therapies targeting COX-2 in tumours to reduce the expression of PGE2 in the TME were effective in arresting tumour growth, however, the off-target inhibition of COX-1 caused serious adverse side effects affecting the gastro-intestinal and cardiovascular system (98). Current research efforts are focusing on developing a COX-2 specific inhibitor that would avoid any of the side effects related with COX-1 inhibition and could be beneficial in combination with NK cell-based immunotherapies. EP antagonists could also be combined with NK cell adoptive cell therapy to sustain NK cell function in the TME.
Tumor resistance to NK cell therapy can also be attributed to the immense metabolic requirements of rapidly proliferating tumor cells resulting in poor nutrient availability in the TME (99). Indeed, maintaining high metabolic fitness in the TME is necessary to sustain high CAR-NK cell function and prevent tumor escape (99). We have shown that expanding NK cells using K562 feeder cells expressing mb-IL-21 reprograms NK cells towards a metabolically flexible phenotype, which not only maintains, but also enhances their cytotoxicity within a hostile and nutrient deprived TME (100). Further arming CAR-NK cells with activating cytokines like IL-15 can enhance their in vivo persistence and improve their metabolic fitness, leading to enhanced tumor control (99). However, CAR-NK cells expressing IL-15 eventually succumbed to immune dysfunction losing their ability to kill target cells by day 35 post administration compared to the pre-infused product (99). This loss in long-term metabolic fitness may necessitate multiple CAR-NK cells doses to ensure durable responses (99). Further, deletion of the cytokine-inducible SH2-containing protein (CIS) in cord blood-derived CAR-NK cells expressing IL-15 significantly enhances their persistence and boosts their metabolic fitness (101). CIS acts as a negative regulator of cytokine signaling including IL-2 and IL-15 signaling and is an important checkpoint in NK cells (102). Knockout of the gene encoding for CIS (CISH) lowers the activation threshold of NK cells and enhances NK cell sensitivity to IL-15 improving their anti-tumor function (102). CISH knockout in iPSC-derived NK cells expanded with K562 mb-IL-21 feeder cells also enhances their metabolic fitness and persistence (103). Whether further engineering CAR-NK cells with a CISH knockout will sustain their long-term metabolic fitness within the TME in a clinical setting remains to be determined. Overall, providing IL-15 signaling may be necessary to maintain NK cell persistence, metabolic fitness, and enhanced anti-tumor functions in the TME. Table 1 summarizes the barriers posed by the solid tumor which prevent efficient NK cell infiltration and activation.
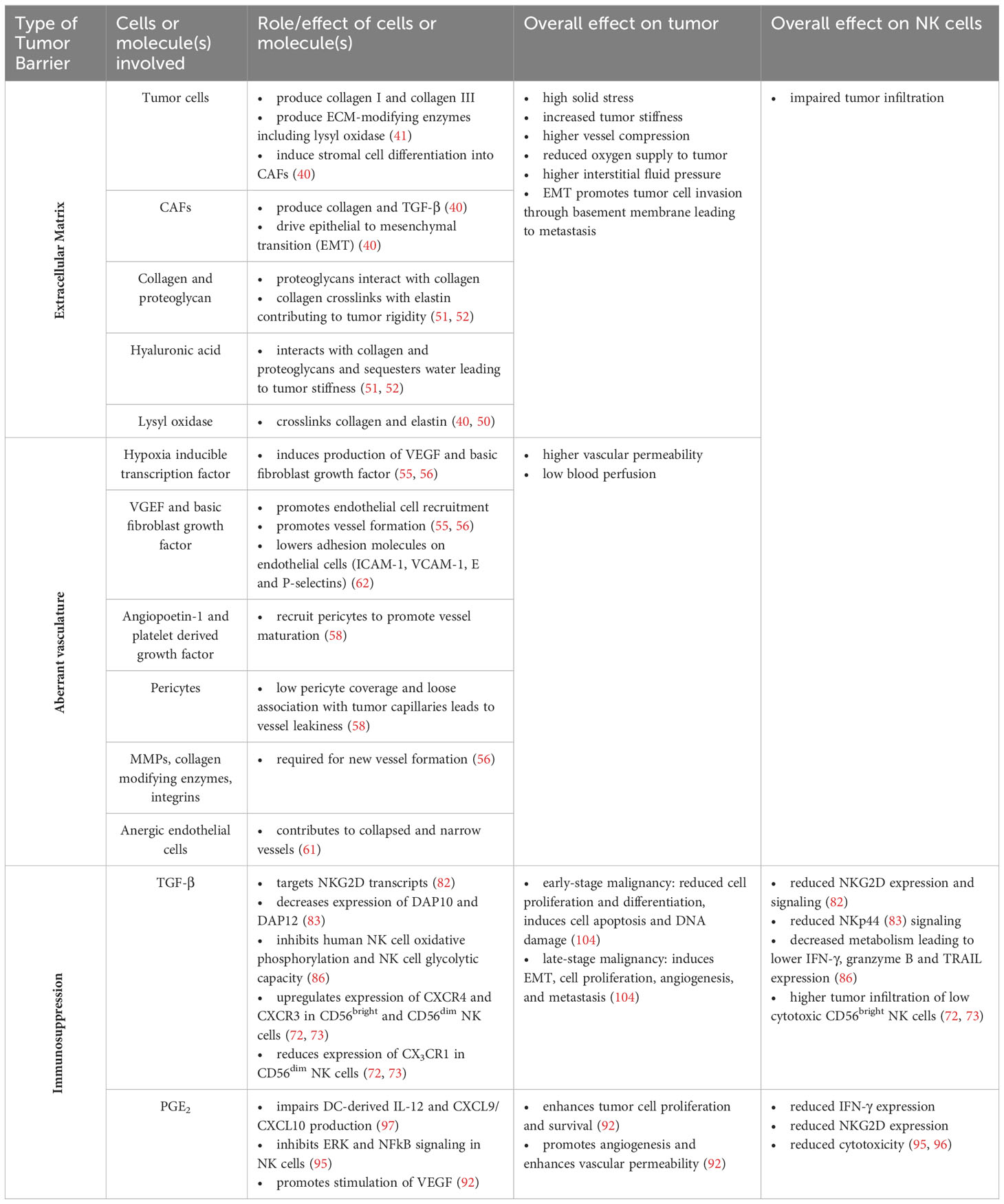
Table 1 Summary of barriers posed by the solid tumor that hinder NK cell infiltration and activation in the tumor site.
4 Strategies to enhance NK cell homing and infiltration into the solid tumor
As summarized in Figure 1, the aberrant tumor vasculature is highly leaky while the solid stress generated by the growing tumor can further compress the tumor vasculature reducing blood flow into the tumor bed. This enhances hypoxic conditions and pro-angiogenic factors leading to less immune cell interactions with the vasculature. Further, the rigid ECM barrier prevents immune cell entry into the tumor core, inhibiting direct interactions with tumor cells. These factors can negatively impact delivery of anti-cancer drugs and infiltration of immune effector cells into solid tumors. To overcome this, various anti-stroma and vascular normalization strategies have been developed to enhance blood perfusion throughout the solid tumor core. In addition, genetic engineering of NK cells to express chemokine receptors or altering chemokine expression within the TME is being actively pursued.
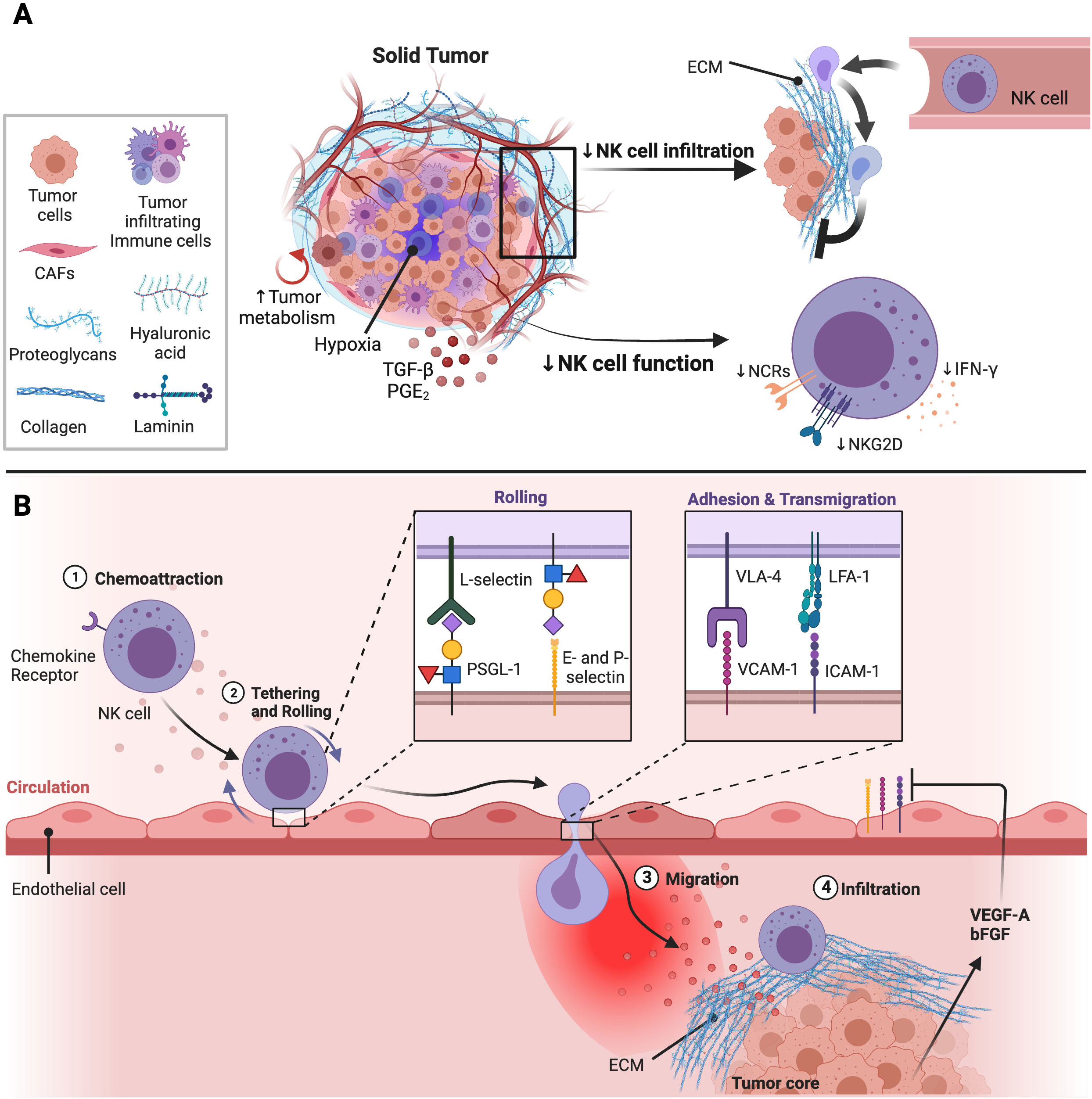
Figure 1 Impact of the TME on NK cell function and infiltration into solid tumor sites. (A) The solid tumor consists of tumor cells, immunosuppressive immune cells, and cancer associated fibroblasts (CAFs) which inhibit NK cell function and infiltration. CAFs are major producers of components that make the extracellular matrix (ECM). The ECM is made up different molecules including collagen, proteoglycans, hyaluronic acid, and laminin which contribute to tumor stiffness and solid stress. Solid stress generated can obstruct the tumor vasculature leading to hypoxic conditions in the TME. Further, the high energy demands of rapidly proliferating tumor cells leads to poor nutrient availability in the TME decreasing NK cell metabolic fitness and anti-tumor activity. Lastly, immunosuppressive factors enriched in the TME such as TGF-β and PGE2 also inhibit NK cell function through downregulation of activation receptor expression and signaling. Adapted from “Tumor Extracellular Matrix Reduces Therapeutic Efficiency in Solid Tumors”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates. (B) Production of pro-angiogenic factors including vascular endothelial growth factor (VEGF)-A and basic fibroblast growth factor (bFGF) induced by hypoxic conditions within the TME can downregulate expression of adhesion molecules on endothelial cells impairing NK cell extravasation into tumor sites. After NK cells migrate towards the tumor, NK cells are shielded from tumor cells by the dense and stiff ECM surrounding the tumor core. Adapted from “Leukocyte Extravasation - The Role of Glycans in Inflammation”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
4.1 Breaking down the ECM and normalizing the tumor vasculature to promote immune cell tumor infiltration
Directly targeting the tumor stroma can reduce ECM deposition and solid stress of the tumor. One such strategy involves the use of Angiotensin II receptor blockers (ARBs). ARBs are common anti-hypertensive drugs, but their anti-tumor effects have also been investigated as angiotensin II has been shown to contribute to tumor ECM formation (105). Fibroblasts such as CAFs also express the angiotensin II receptor type I (ARTI) (106). Further, patients with ovarian cancer exhibit elevated levels of the angiotensin converting enzymes which converts angiotensin I to the bioactive angiotensin II (107). In ovarian and breast cancer murine models, blocking ARTI signaling can downregulate collagen I expression, decompress vessels, increase vessel perfusion, and reduce solid stress (108, 109). This strategy can be used to enhance intra-tumoral delivery of other anti-cancer drugs such as chemotherapies. Specifically, Zhao et al. have demonstrated that while stroma normalization with Losartan, an FDA approved ARB, did not reduce tumor burden in human ovarian cancer models it did enhance the anti-tumor effects of the chemotherapy agent paclitaxel when used in combination (109). Here, combination therapy led to a significant reduction in the incidence and volume of ascites in the tumor bearing mice (109). Similarly, using a tumor selective ARB alone had no anti-tumor efficacy but when combined with checkpoint immune blockade there was a 66% response rate in a TNBC syngeneic mouse model (108). In addition, this combination therapy increased the number of intra-tumoral CD45+ immune cells and CD8+ cytotoxic T cells (108). Further, ARBs can also modulate chemokine expression in the TME. Nakamura et al. found that ARB treatment significantly lowered the expression of CXCL12 by ARTI-expressing CAFs in a murine model of colon cancer (110). CXCL12 is expressed in a variety of tumors including ovarian cancer, breast cancer, glioblastoma and pancreatic cancer (111). CXCL12 has been demonstrated to promote tumor cell proliferation and survival and can synergize with VEGF to induce neoangiogenesis in vivo (111–113). High levels of CXCL12 in the tumor can attract CXCR4-expressing immune cells, fibroblasts and endothelial cells to the tumor site further promote tumor growth (113). Recruitment of FAP-expressing CAFs via CXCR4/CXCL12 signaling can prevent accumulation of intra-tumoral cytotoxic T cells (114). Hence, targeting the CXCR4/CXCL12 axis to promote immune cell infiltration within tumors has been of interest. Chen et al. directly targeted the CXCR4/CXCL12 signaling axis in metastatic breast cancer in vivo models with the FDA approved drug plerixafor (115). They found that blocking the CXCR4 receptor expressed on α-SMA+ cells including CAF cells and pericytes led to decreased tumor fibrosis, less solid stress, higher degree of decompressed blood vessels and enhanced cytotoxic T cell infiltration (115). Similarly, remodeling of the stroma and vasculature also doubled the response to immune checkpoint blockade therapy (115). Targeting the ECM by administration of MMP inhibitors or enzymes that breakdown ECM components such as hyaluronan have also been tested with low success (116). A clinical trial with chemotherapy and pegylated recombinant human hyaluronidase combination treatment showed decreased overall survival in patients with metastatic pancreatic cancer due to treatment induced toxicity (117). Further, altering hyaluronan could also enhance the glucose metabolism of tumor cells by increasing their expression of the Glut1 glucose transporter, promoting their migration capacity (118). Directly targeting the tumor stroma through CAR-T cells is another strategy that has been investigated. Depletion of FAP-expressing CAF cells by CAR-T cells enhanced intra-tumoral CD8+ T cells and FN-γ production significantly impeding tumor growth (119). However, a similar strategy has demonstrated that directly targeting FAP-positive cells can lead to lethal bone marrow toxicity and muscle loss due to the expression of FAP in multipotent bone marrow stromal cells (120). Further, Xie et al. have shown that targeting the EIIB fibronectin splice variant which is mainly expressed in the tumor ECM and neovasculature can also delay tumor growth and improve survival in melanoma tumors (121). Directly targeting CAFs could also improve migration of immune cells to the tumor core by modulation of the CXCL10/CXCL9-CXCR3 axis. Primary CAFs isolated from pancreatic cancer samples have been shown to downregulate the expression of CXCR3 on human T cells leading to reduced T cell migratory capacity towards CXCL10 in vitro (122). Depletion of CAFs could enhance CXCR3 ligand availability in the TME. However, complete depletion of the tumor stromal components such as CAFs may not be feasible anti-tumor strategy due to possible toxicities and unwanted side effects. Instead, strategies that modulate the functions of specific CAF populations could lead to improved therapeutic responses. For example, TGF-β blockade has shown to reduce expression of genes involved in ECM and induced an IFN-response gene signature in murine and human CAFs, including upregulation of CXCR3 ligand expression (45). TGF-β blockade also led to a significant increase of tumor infiltrating cytotoxic T cells in a murine breast cancer model (45).
Vascular normalization strategies aim to remodel the highly disorganized tumor vasculature to resemble the more mature normal vessels which have higher pericyte coverage to improve blood perfusion (123). While the use of anti-angiogenic molecules such as VEGFR inhibitors have been widely investigated for their anti-tumor effects, monotherapy with angiogenic inhibitors has not yielded much clinical success (124, 125). Further, careful considerations for the dosing and timing of anti-angiogenic therapy are critical to avoid excessive vascular death which could prevent perfusion and further enhance hypoxia (126). Huang et al. demonstrated that a low dose of an antibody blocking the VGEF receptor (VGEFR), VGEFR2, combined with a cancer cell vaccine significantly inhibited the tumor growth in breast cancer models (127). Administration of the anti-VGEFR2 therapy led to higher perfusion of the tumor and improved distribution of perfused vessels. They also found significantly higher levels of infiltrating CD4+ and CD8+ T cells when combined with a vaccine (127). Another study utilizing dual PD-1 and VEGFR2 blockade significantly inhibited tumor growth and doubled survival in an orthotopic mouse model of hepatocellular carcinoma (128). The authors demonstrated that combination therapy increased the number of pericyte covered vessels and enhanced micro vessel density. They also noted an enhancement CD8+ T cell infiltration and downregulation of the M2 pro-tumor macrophages and T-regs (128). Interestingly, immunotherapy and checkpoint blockade therapies alone has also shown to promote tumor vessel normalization. Tian et al. showed that adoptive transfer of type 1 T helper cells (Th1) in a patient derived xenograft TNBC tumor model improved tumor perfusion and reduced vessel leakiness and hypoxia (129). Monotherapy with immune checkpoint blockade has been demonstrated to increase tumor vessel perfusion in multiple breast cancer mouse models through the enhanced expression of intra-tumor IFN-γ (130). IFN-γ acts on vessels to induce blood vessel death and can inhibit formation of new blood vessels through downregulation of VEGF (131, 132). Production of high levels of IFN-γ in the TME by expanded NK cells could act as a positive feedback loop to promote vascular normalization and further enhance immune infiltration. Although these studies have not directly examined whether stroma or vascular normalization can specifically enhance NK cell tumor infiltration, they provide evidence that therapies that enhance tumor perfusion could synergize with NK cell adoptive cell therapies to improve entry into the tumor bed. There is currently one phase II clinical trial in recruitment that will be assessing the efficacy and safety of a VGEFR2 tyrosine kinase inhibitor (Apatinib) and an anti-PD-1 (Pembrolizumab) antibody in combination with cord blood-derived NK cells for HCC (NCT05171309).
4.2 Directly modulating chemokine-chemokine receptor expression to enhance NK cell migration
Modulating chemokine expression within the tumor or altering the expression of chemokine receptors in NK cells can influence their migration into solid tumor sites. One strategy involves genetic engineering of NK cells to express chemokine receptors that correspond to the specific chemokines present within the tumor being targeted. For example, engineering the YTS NK cell line to co-express an anti-EGFRvIII CAR and CXCR4 led to enhanced NK cell infiltration and improved survival in glioblastoma xenograft models overexpressing CXCL12 (133). Similarly, human ex vivo expanded NK cells have also been engineered to co-express CXCR4 with a gain-of-function mutation and an anti-BMCA CAR using mRNA electroporation for treatment of multiple myeloma (134). While there was enhanced NK cell migration to the bone marrow and tumor control, the treatment did not completely abolish tumors leading to relapse which could be overcome through stable expression of the CAR (134). The human NK cell line, NK-92s, have also been engineered to overexpress CXCR4 and CCR7 to promote their migration in human colon cancer in vivo models (135). Other chemokine receptors have also been expressed on NK cells to target different tumors. For example, expression of CXCR2 in human NK cells using retroviral transduction significantly enhanced their migration towards CXCR2 recombinant ligands and RCC lines in vitro (78). Further, enhanced migration of CXCR2-expressing NK cells led to higher killing of target cells compared to the NK cells transduced with the control vector (78). Another study showed that overexpression of CXCR1 via electroporation significantly enhanced human expanded NK cell infiltration in a human HNSCC xenograft model (66). Specifically, using NIR fluorescently labeled NK cells they found that after 72h post intravenous injection the CXCR1-expressing NK cells localized to the subcutaneous tumor sites with a 10-fold increase in signal intensity compared to mock NK cells. Additionally, NK cells co-transfected with CXCR1 and an NKG2D-CAR significantly improved survival in a human ovarian cancer model (66). Another viable strategy to improve NK cell infiltration to tumor sites involves the downregulation of chemokine receptors that are involved in NK cells trafficking to the liver. For example, Levy et al. demonstrated that downregulating CCR5 expression on expanded NK cells using CRISPR/Cas9 significantly reduced homing to the liver after intravenous administration (80). Combining the deletion of CCR5 with overexpression of other chemokine receptors could enhance NK cell infiltration away from the liver and into the targeted tumor site. The concentration of chemokines within the TME can also be altered to improve NK cell infiltration. For example, administration of an irradiated tumor vaccine secreting CCL3 led to a 3-fold increase in the number of NK cells within murine CT26 colon tumors (136). Accumulation of NK cells within the tumors contributed to slowed tumor growth and augmented T cell infiltration through induction of CXCL9 and CXCL10 via NK cell-derived IFN-γ (136).
Other strategies which do not rely on genetic engineering of NK cells with chemokine receptors to improve infiltration of NK cells in the tumor have also been investigated. One novel strategy involves the use of an NK cell recruiting protein-conjugated antibody (NRP-body) that can be selectively activated within the TME. Lee et al. demonstrated that an NRP-body specific for mesothelin and conjugated with a cleavable soluble form of CXCL16 is released in the local tumor environment through cleavage by furins that are highly expressed on the surface pancreatic cancer cells (137). This NRP-body significantly enhanced the infiltration of human expanded NK cells to primary and metastatic tumor sites in a preclinical pancreatic ductal cell carcinoma mouse model (137). In glioblastoma cancer models, inhibition of the b-galactoside-binding lectin, galectin-1 has shown to enhance expression of CXCL10, CXCL12 and CCL5 resulting in recruitment of Gr-1+/CD11b+/CCR2+ myeloid cells which promoted the recruitment of tumor-infiltrating NK cells (138).
Although the impact of NK cell intra-tumoral infiltration with stroma or vascular normalization therapies has not been thoroughly investigated, enhanced perfusion to the tumor by reducing vessel compression has the potential to improve adoptive NK cell immunotherapy. Further, strategies that directly modulate the chemokine/chemokine receptor axis have shown to specifically enhance NK cell trafficking and infiltration to tumor sites in various tumor models.
5 Strategies for tumor-targeted delivery of immunomodulatory agents to pull and boost NK cells in the TME
While the strategies discussed above and summarized in Figure 2 are promising, there are limitations that can preclude their efficacy including: 1) potential unwanted side effects associated with systemic administration of anti-stroma and anti-angiogenic strategies 2) modulating NK cells based on the chemokines endogenously expressed by specific tumors would limit the therapeutic potential of these strategies across various tumor types. Alternatively, tumor-targeted delivery of agents that promote stroma and vascular normalization as well as recruit and activate NK cells to the tumor site would address some of these limitations. Oncolytic viruses (OVs) are an attractive platform for combination therapy with NK cells given their ability to selectively replicate in a wide range of tumor types and their capacity to express a wide range of immunomodulatory agents (139). In contrast to strategies discussed in the previous section, engineered OVs have the added advantage of limiting the effects of the payload to the tumor site. In this section we summarize how OVs can be used to enhance NK cell infiltration and activation in the TME. We also discuss the challenges of applying OVs for cancer therapy and discuss other methods available to deliver immunomodulatory therapies to the TME.
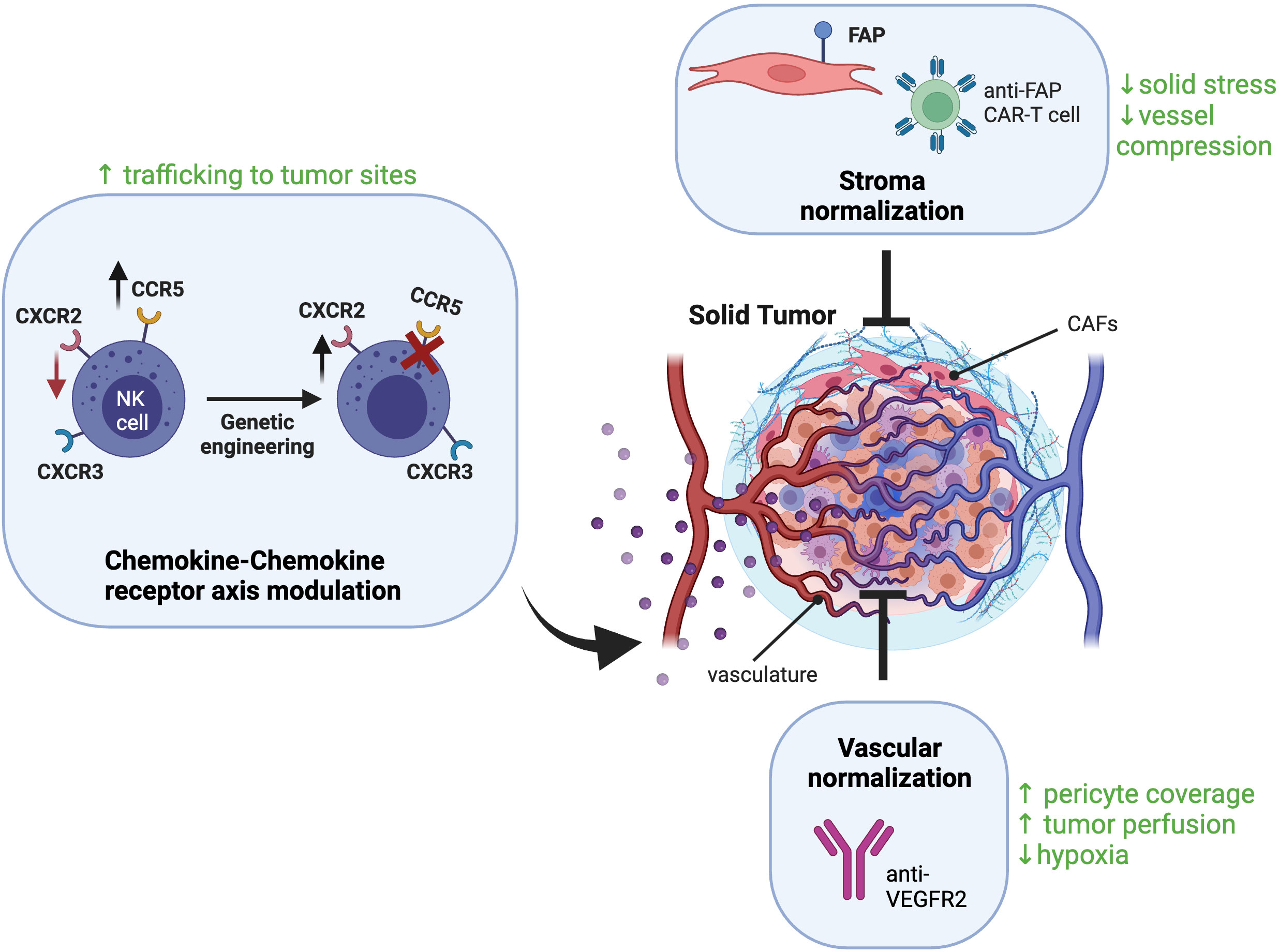
Figure 2 Strategies to enhance NK cell infiltration into solid tumors. Different CAF‐targeting strategies are being explored for stroma normalization including elimination of CAFs using CAR-T cells against CAF surface markers like fibroblast activation protein (FAP). These strategies can relive solid stress of the tumor, reduce ECM formation, and reduce vessel compression to improve tumor perfusion. Vascular normalization strategies aim to improve tumor perfusion by reducing vessel formation and increasing pericyte recruitment, one example is blockade of the VEGF receptor VEGFR2. To enhance NK cell trafficking to the tumor site, NK cells can be engineered to overexpress chemokine receptors corresponding to chemokines expressed by the tumor. Created with BioRender.com.
5.1 Oncolytic viruses trigger direct tumor oncolysis and induce NK cell recruitment and activation
Oncolytic virotherapy for cancer treatment harnesses the ability of OVs to effectively infect and replicate in cancer cells. This is attributed to the fact that unlike healthy cells, cancer cells have dysregulated IFN signaling, which typically activates host antiviral responses to shut down virus replication (140). OVs subsequently exert direct anti-tumor activity through direct oncolysis and through the stimulation of innate and adaptive anti-tumor immunity (141). A variety of different OVs have been investigated for oncolytic virotherapy, including herpes simplex virus (HSV), adenoviruses, vesicular stomatitis virus (VSV), measles virus (MV), vaccinia virus (VV), and Newcastle Disease virus (NDV) (140). Currently, the only fully US Food and Drug Administration approved oncolytic virotherapy is talimogene laherparepvec (T-VEC), while there have been three other OVs approved around the world (142, 143). T-VEC is an engineered OV derived from HSV-1 expressing human granulocyte macrophage colony-stimulating factor (GM-CSF) used to treat unresectable melanoma (142). However, other OVs are currently being explored in clinical trials for a wide variety of solid tumors.
While OVs can trigger direct lysis of infected tumor cells, their primary anti-tumor function is maintained by their ability to elicit high levels of inflammatory immune responses. OVs can induce tumor cell death that can stimulate an immune response, termed immunogenic cell death (ICD). After OV replication and oncolysis, dying cancer cells undergo cell death pathways which cause the release of virus-derived pathogen-associated molecular patterns (PAMPs) and cell host-derived damage-associated molecular patterns (DAMPs) (141). DAMPs typically induced by ICD include the release of cellular contents such as ATP high-mobility group box 1 (HMGB1) and heat shock proteins (HSP) such as HSP90 (144). OVs are thought to activate type II ICD, which is characterized by induction of immunogenic apoptosis through targeting of the ER (144). Each OV has a unique ability to induce ICD based on its ability to infect and lyse tumor cells (145). OV infection leads to the local release of type 1 IFNs and chemokines, which attract DCs to the tumor site and enhance their expression of MHC molecules and the uptake of tumor cell debris which promotes recruitment and expansion of anti-tumor lymphocytes (146). In addition, mature DCs produce IL-12, IL-18, and type I IFNs to activate NK cells (147). The pro-inflammatory environment and immune cell-attracting chemokines generated at the tumor site promotes the trafficking of activated NK cells and tumor-specific CD8+ T cells (146). Additionally, NK cell-derived chemokines including CCL5 and XCL1 can also mediated recruitment of DCs into the tumor leading to improved tumor control in murine solid tumor models (148). The ability of OVs to promote robust immune mediated anti-tumor responses is critical to achieve sustained anti-tumor efficacy with studies suggesting that the immunogenicity of an OV is more important than early replication in tumors, and that their efficacy is primarily due to lymphocyte infiltration and recruitment to the tumor. This allows them to be leveraged as candidates for combination with adoptive cell therapies, as they can promote homing of adoptively transferred immune cells and enhance their activation.
Several studies demonstrate that OVs have the capacity to activate and enhance NK cell infiltration in various cancer models. For example, administration of one to five doses of oncolytic reovirus to patients with colorectal cancer led to type 1 IFN production and NK cell activation, marked by elevated expression of the activation marker CD69 on these cells (149). In addition, infection of TNBC cells by VSV virus can enhance expression of immune cell attracting chemokines, leading to enhanced migration of NK cells in vitro (150). OV infection of tumors themselves also induces stress ligand expression on tumor cells as a mechanism of conventional antiviral defense to elicit successful NK cell responses, enhancing NK cell activation. Work has shown that the immunogenic effect of OVs can be enhanced by using secondary drug therapeutics, such as histone deacetylase inhibitors, to enhance the extent of the inflammatory response. Indeed, combining the HDACi valproic acid with T-VEC enhances GM-CSF production induced by the virus, and as well enhances tumor killing by increasing expression of NK cell activating ligands and endogenous tumor antigens on tumor cells (151). Additionally, various studies using Orf virus (OrfV) as an oncolytic agent have demonstrated that its anti-tumor efficacy relies heavily on NK cells (152, 153). OrfV is a dsDNA virus of the Parapoxvirus genus that naturally infects sheep and goats but causes mild infection in humans (154). Work in metastatic lung tumor models have shown that OrfV can significantly reduce lung metastasis formation in a mechanism dependent on NK cells (153). Further, OrfV administration significantly enhanced the proportion of IFN-γ and granzyme positive NK cells in the spleen, blood, and lungs of OrfV treated mice (153). In a study analyzing postoperative NK cell dysfunction, preoperative administration of OrfV significantly reduced formation of lung nodules in a 4T1 mammary carcinoma tumor model and enhanced the cytotoxic function of these dysfunctional NK cells (155). OrfV has also been used in a murine end-stage ovarian cancer model showing efficacy as a monotherapy through stimulation of NK cell responses leading to enhanced recruitment of CD8+ CXCR3-expressing T cells through upregulation of CXCR3 ligands (156). The significant reliance of OrfV on NK cells makes it an attractive OV to engineer to enhance NK cell infiltration into tumor sites. Moreover, since OrfV primarily infects sheep there is likely to be an existing immune response against the virus that could shut down its ability to infect tumor cells which is the case with other OVs such as adenoviruses (157). However, OrfV has a limited tumor tropism with low replication capacity in various human cancer cells which would hinder its clinical use against a wide range of cancer types (152, 153).
5.2 Engineering oncolytic viruses to enhance NK cell tumor infiltration and function in TME
OVs can also be engineered to express exogenous immunostimulatory molecules to enhance NK cell activation and function in the immunosuppressive TME. In a murine breast cancer model, combination therapy with VV and a 4-1BB agonist significantly increased levels of intra-tumoral NK cells by nine days post administration and depletion of NK cells reduced therapeutic efficacy of the treatment (158). Administration of VV expressing a murine IL-15 and IL15 receptor α (IL-15/IL-Rα) complex induced gene expression of NKp46 and NKG2D in murine ovarian cancer tumors suggesting enhanced NK cell infiltration in the tumor (159). However, depletion of NK cells had no impact on tumor progression or survival in this study. Co-expression of IL-12 and IL-15/IL-Rα using oHSV-1 has also been demonstrated to promote efficient tumor clearance in syngeneic colon and sarcoma models (160). This strategy resulted in a slight, albeit not significant, enhancement in the percentage of NK cells in treated CT26 tumors (160). There are currently still very limited pre-clinical studies employing the combination of OVs and NK cell adoptive cell therapy for solid tumors. A previous study utilizing VV expressing the CCL5 chemokine was used to promote NK cell migration leading to over 50% complete response rates in human colon cancer models (161). However, this effect was only seen when mice were also administered engineered CCR5-expressing NK cells (161). Other studies have used oHSV-1 and CAR-NK cell combination therapies for the treatment of glioblastoma (162, 163). Administration of oHSV-1 expressing the IL-15/IL-Rα complex (OV-IL15C) and primary anti-EGFR CAR-NK cells led to significantly longer survival than the monotherapies alone (163). Further, OV-IL15C increased persistence of the EGFR CAR-NK cells in the brain compared to EGFR CAR-NK cells alone, yet they did not exhibit an exhausted phenotype within the TME (163). This suggests that OVs can be used for both immune cell recruitment to solid tumors, while also enhancing their survival and cytotoxic function once within the tumor. While neurotoxicity that results from inflammation during OV infection is a concern, new work in humans has shown that direct administration of an oncolytic adenovirus into the brain can prolong survival with limited toxicity (164). OVs can also be engineered to express other immunostimulatory molecules to stimulate intra-tumoral NK cells as a combination therapy. A recent study demonstrated that OVs can redirect and activate NK cells towards the tumor through expression of a bispecific killer engager (BiKE) which possess a tumor antigen binding domain and NK cell binding domain. Floerchinger et al. generated a BiKE-encoding MV and found that tumors infected with the MV-BiKE construct significantly enhanced the degranulation capacity of human NK cells present in the patient-derived pancreatic cancer tumor tissue (165). Further, MV-BiKE treatment was also able to enhance the degranulation, IFN-γ production and killing capacity of allogenic human NK cells in a co-culture model with primary pancreatic cancer cells. However, the enhanced killing varied and was dependent on the degree of permissiveness of the patient tumor sample to MV infection (165). Other immunomodulatory factors that could be expressed by OVs to activate NK cells include the bifunctional TGF-βRII and IL-15/IL-15Ralpha protein domain which has been shown to promote NK cell infiltration into murine melanoma tumors (166). A recent study has also shown that VV can be engineered to express a TGF-βRII inhibitor to outcompete local TGF-β in the TME (167). This engineered OV rescued anti-tumor efficacy and significantly increased survival in a murine melanoma tumor model resistant to wild type VV treatment (167). This anti-tumor effect was correlated with a less suppressive T-reg phenotype but could also synergize with other immune modalities sensitive to TGF-β inhibition such as NK cells. Several OVs that are similarly engineered with cytokines and other recombinant proteins are currently in early clinical trials as therapies for numerous types of solid tumors (Table 2). Common candidates for engineering include IL-12, IL-15, IL-21, type 1 IFN, and other immune stimulatory agents such as 4-1BBL. Many of these approaches combine cytokine expressing OVs with checkpoint inhibitors, that increase lymphocyte infiltration and activity within the TME, and a few also combine these OVs with T cell therapies. Overall, co-expression of these cytokines or other activating molecules that can specifically boost NK cell anti-tumor activity and chemoattracting factors will be advantageous when considering OVs and NK cell combination therapies.
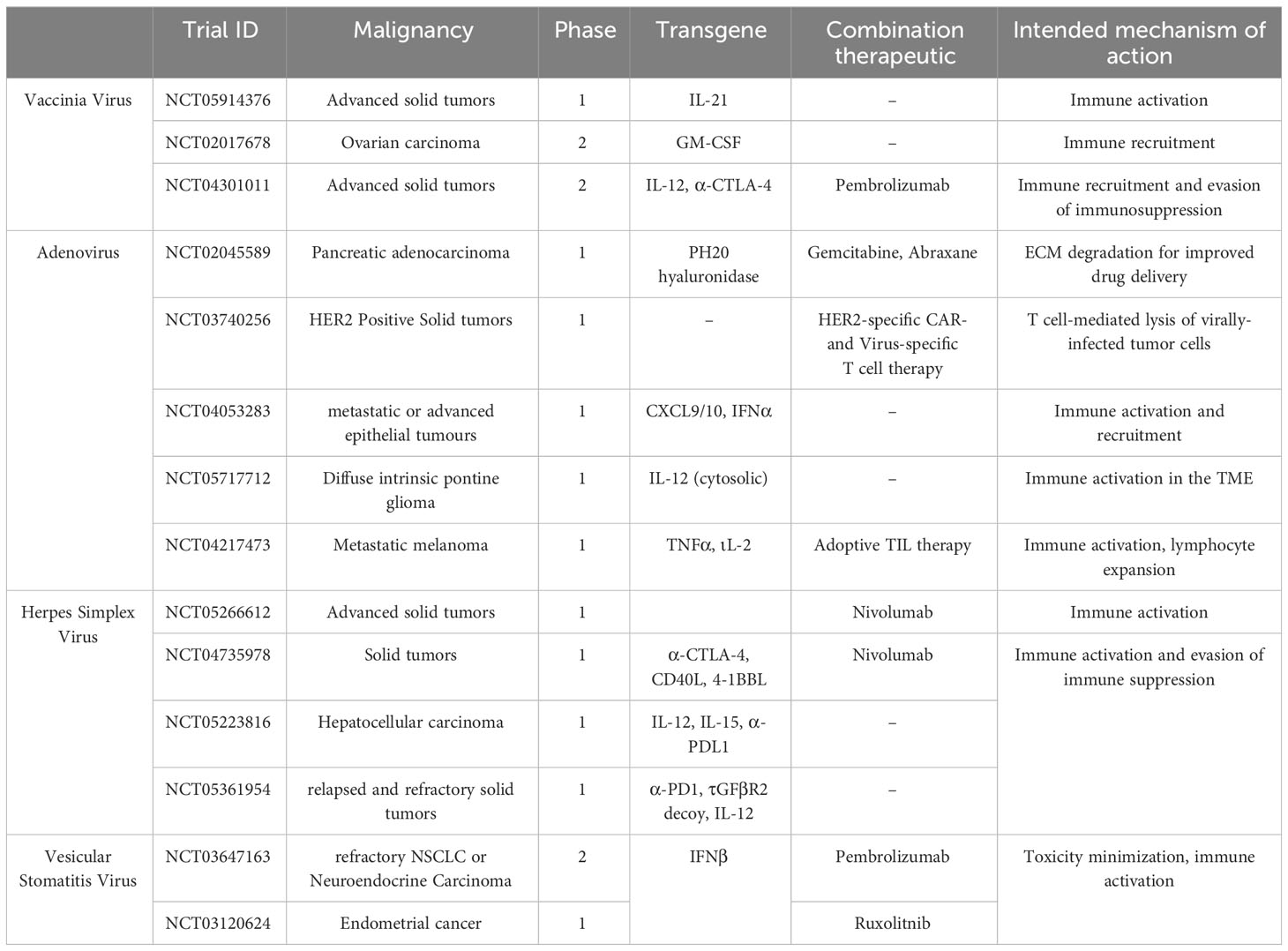
Table 2 Clinical trials using engineered oncolytic viruses to enhance immune recruitment and activation for solid tumor therapy.
5.3 Implications of OV interactions in the solid tumor and therapeutic timing for OV and NK cell combination therapy efficacy
Although arming OVs with immunomodulatory agents that could enhance NK cell intra-tumoral infiltration and activation is promising, the anti-tumor efficacy of oncolytic virotherapy is also impacted by the TME. For example, several studies have shown that OVs can have anti-angiogenic effects by directly infecting tumor vascular endothelial cells leading to vascular shutdown (168). While this phenomenon was previously regarded as beneficial for virotherapy due to sequestering of the OV in the tumor, complete collapse of the vasculature can have a negative effect on immune cell infiltration and drug delivery into the solid tumor (168). OVs such as VSV, VV, and oHSV-1 have been shown to directly infect and replicate in endothelial cells, leading to decreased blood perfusion to the tumor core and increased hypoxic conditions (169–172). This has been mainly attributed to high levels of VEGF in the TME which can sensitize OV replication in vascular endothelial cells (168). To overcome the vascular shutdown induced by OVs, vascular normalization strategies prior to OV infection can be administered. For example, Matuszewska et al. demonstrated that administration of a protein containing the three homologous thrombospondin type 1 repeat domains (3TSR) prior to NDV infection rescued vascular shutdown caused by NDV in ovarian cancer tumors (173). 3TSR binds to the CD36 receptor on endothelial cells preventing endothelial cell proliferation and reduces VEGF production, leading to vascular normalization and enhancing tumor perfusion (173, 174). Indeed, combination therapy with 3TSR and NDV significantly enhanced infiltration of activated NK cells and T cells to the tumor core and led to tumor regression in an advanced-stage ovarian cancer model (173). This work provides further evidence that preventing complete vascular shutdown may be crucial to ensure high immune mediated anti-tumor efficacy and that OVs which do not infect vascular endothelial cells would be preferred candidates for combination with adoptive cell therapies.
OVs can be delivered via systemic or intra-tumoral routes, with systemic administration being preferred when treating hard to reach solid tumors or after cancer metastasis. However, the dense tumor stroma can also be a barrier to tumor cell infection by OVs which could limit OV efficacy during systemic administration. To overcome this, OVs have been engineered to directly target the tumor stroma. Jing et al. demonstrated that using a modified version of MV that can infect and replicate in both stromal and tumor cells resulted in 69% reduction in tumor volume and improved survival in a colon xenograft tumor model (175). Adenoviruses have also been engineered to express proteins like relaxin and decorin which downregulate collagen expression in tumor tissue, enhancing viral spread in the tumor and improving anti-tumor effects (176–178). Co-administration of bacterial collagenase with HSV-1 improved virus distribution and tumor infection in a melanoma model by disrupting fibrillar collagen (179). Other methods to enhance oncolytic viral spread within the tumor core is by targeting collagen through expression of matrix metalloproteinases such as MMP-9 (180). In addition, a phase I clinical trial using an oncolytic adenovirus expressing human hyaluronidase to target hyaluronic acid, facilitating ECM breakdown, showed this strategy reduced tumor stiffness in patients with pancreatic cancer (NCT02045589) (181). Further, OVs can also package proteins that modulate effector cell cytotoxicity against stromal cells. Bi-specific T cell engagers (biTEs) have been designed to specifically direct cytotoxic T cells towards a tumor target. Both, adenoviruses and VV have been used to express biTEs binding to CD3 and mouse or human FAP proteins (182, 183). Locally targeting T cells to FAP+ stromal cells led to high infiltration of T cells to the tumor sites in mouse melanoma and human lung and breast cancer models (182, 183). Further, combination of the biTE secreting adenovirus and intravenous injection of pre-activated T cells led to significant enhancement in survival compared to the monotherapies (182). Using OVs to express and release biTEs in the local tumor environment can be a successful way to overcome potential off-target safety concerns as seen in anti-FAP CAR-T cell strategies. Given the recent development of bi- and tri-functional NK cell engagers (NKCEs) which bind to NK cell activation receptors, there is potential to engineer OVs with NKCEs targeting stromal components similarly to the biTEs discussed (184). Overall, these strategies may not only enhance OV distribution in the tumor enabling systemic administration when tumors are not physically accessible but could also increase intra-tumoral infiltration of adoptively transferred NK cells.
The timing of NK cell administration is an important consideration during combination therapy as NK cells could impede oncolytic virotherapy through elimination of virally infected cells, limiting viral replication and spread. Mathematical modelling was performed to determine the highest efficacy between oHSV-1 and the proteasome inhibitor, Bortezomib, in combination with NK cell therapy (185). The modelling determined that the highest anti-tumor efficacy would be achieved when endogenous NK cells are depleted or when higher numbers of exogenous NK cells are added (185). In addition, they found that higher anti-tumor efficacy against intracranial glioma tumors was achieved when there was a delay between OV infection and exogenous NK cell administration (185). Performing simulations such as these could be useful to inform the rational development of engineered OVs and NK cell combination therapies to maximize the ability of OVs to promote infiltration of NK cells into the tumor.
5.4 Other tools for the delivery of immune recruiting and activation agents to the TME
While OVs are one method to induce chemokine production in the tumor, new avenues have emerged which can penetrate the tumor core more efficiently. One strategy involves the use of nanomedicines which are therapeutic carriers that are nanometers in size, typically lower than 100 nm (186, 187). An example of these are lipid nanoparticles (LNPs) or extracellular vesicles such as exosomes which can be loaded to carry similar cargo as OVs to induce the production of chemokines and cytokines from tumors to mediate immune cell recruitment (186, 188). These nanomedicines were originally formulated as a mechanism of delivering chemotherapeutic agents directly to tumors, to prevent off-tumor toxicity (189, 190). However, the widespread use of mRNA based COVID-19 vaccines delivered via nanoparticles has led to an increased interest in investigating how LNPs can be exploited to promote anti-tumor immunity. LNPs are typically manufactured using a profile of lipids, cholesterol, and stabilizing agents such as polyethylene glycol (PEG). After complexing with genetic material, such as mRNA, miRNA, or siRNA, they can be systemically administered to promote therapeutic gene manipulation. These nanomedicines are endocytosed by cells, after which they release their genetic cargo into cells following endosomal maturation allowing efficient delivery of therapeutic cargo (187, 191).
As they often lack tissue specificity, there has been a focus on investigating methods for targeted delivery of these nanoparticles to different tissue sites, including tumors. While direct, intratumoral delivery of such nanomedicines is possible, research has been focused on advancing ease of tumor permeability of systemically delivered nanoparticles to enhance their efficacy (187, 192). Such approaches usually involve conjugating nanoparticles with antibodies against ligands expressed on tumors, to increase tumor targeting and decrease competition for uptake by other organs. Other strategies leverage high expression of certain proteins within the tumor (193–195). For example, nanoparticles can be designed to become active in response to MMP-2, a metalloproteinase highly expressed within the tumor bed (196, 197). Other than passive transport into the tumor bed through blood vessels, nanoparticles can also be actively transported through tumor endothelial cells (198). Further, entry into the tumor by nanoparticles can also be inhibited by the basement membrane of the solid tumor. However, unlike immune cells, nanoparticles that are larger in size (>30 nm) have enhanced entry into the tumor through their uptake by TAMs, which are abundant in the TME and can migrate within the tumor core (199–203). Recent research shows that nanoparticles can exit the tumor through the tumor-draining lymph nodes and re-enter the circulation allowing for re-entry into the tumor (204). This discovery will allow for further modification of nanoparticles to enhance tumor accumulation.
Various candidate therapeutics have been explored for the delivery of different cargoes including cytokines, chemokines, mAbs, and other forms of engineered antibodies such as nanobodies, and other immune-stimulatory molecules such as stimulator of interferon genes (STING) agonists (205–209). STING agonists have emerged as a strategy to activate the cyclic GMP-AMP synthase (cGAS) STING pathway to enhance CD8+ T cell and NK cell infiltration and activation in the TME. The pathway is triggered by sensing of cytosolic double stranded DNA (dsDNA) by cGAS which catalyzes the production of the secondary messenger, cyclic 2’3’-GMP-AMP (cGAMP) (210). cGAMP is the endogenous cyclic dinucleotide (CDN) ligand for STING (210). Pharmacologic activation of the STING pathway has mainly been focused on intra-tumoral administration of STING ligands such as natural CDNs like cGAMP, synthetic CDNs, and non-CDN molecules.
STING agonists induce potent local and systemic anti-tumor immunity through STING activation in tumor cells, CAFs and tumor infiltrating DCs and macrophages (211, 212). STING activation in these cells leads to the production of type 1 IFNs and CXCL9 and CXCL10 chemokines, enhancing the migration of CXCR3-expressing CD8+ T cells and NK cells (209, 213–216). Additionally, type 1 IFNs can promote cross-presentation of antigens to CD8+ T cells and induce expression of IL-15 and the IL-15 receptor α (IL-15Rα) by DCs leading to NK cell activation (217). Given the upregulation of CXCR3 on ex vivo expanded NK cells and their ability to migrate towards tumors expressing exogenous CXCL10, STING agonists could enhance their tumor homing (77). STING therapy also can normalize the aberrant tumor vasculature through activation of STING on endothelial cells, leading to reduced tumor vessel density and improving pericyte coverage thereby increasing intratumoral CD8+ T cell infiltration (218). STING agonist therapy is currently being evaluated preclinically as a combination therapy with adoptive cell therapies such as CAR-T cell and NK cells. Xu et al. demonstrated that subcutaneous delivery of STING agonists enhanced the infiltration and persistence of CAR-T cells in an orthotopic mouse breast tumor model through increased expression of CXCL9 and CXCL10 by myeloid cells in the TME (219). However, STING agonists can be toxic to human T cells which will limit their combination (220). Interestingly, the same effect is not observed with human naïve or expanded NK cells which may be partly due to rapid degradation of STING after STING agonist stimulation (221). Da et al. showed that cGAMP can directly activate the human NK-92 cell line and increase their anti-tumor function against pancreatic tumors and enhanced migration of CCR5-expressing NK-92 cells in vitro (222). Knelson et al. showed that STING agonist therapy can also enhance migration and cytotoxicity of primary NK cells in a more physiological relevant model using patient-derived organotypic tumor spheroids (221). The enhanced tumor targeting and migration which was dependent on release of CXCR3 ligands by STING agonist activated tumor cells. STING agonists can also be combined with cytokine therapy to improve overall NK cell responses. Administration of a high affinity IL-2 cytokine after intra-tumoral CDN injection induced higher expression of cytotoxic effector molecules by tumor infiltrating NK cells in HLA class 1 deficient tumors and enhanced infiltration of NK cells to the distal tumor (223).
Using nanocarriers as a delivery method for STING-agonists is under active investigation to enhance their half-life by preventing their degradation by proteases and decreasing non-specific uptake which can cause apoptosis of STING agonist sensitive cells like T cells and B cells (224–226). In addition, delivery using nanocarriers improves target cell uptake since CDNs are hydrophilic and negatively charged they do not passively enter the cellular membrane and are instead transported using important channels (227, 228). For example, systemic delivery of CDNs using PEGylated lipid nanodiscs (LNDs) has shown improved CDN delivery in the tumor and uptake by DCs in tumor draining lymph nodes enhancing STING-agonist mediated anti-tumor immunity (229). Although STING agonist delivery using OVs has been recently reported, enhanced tumor entry to the tumor core by nanocarriers is an advantage (230). Overall, STING agonists have the potential to improve the efficacy of NK cell-based therapies by promoting their infiltration into the tumor and further activating the anti-tumor responses without affecting their viability.
A few studies have previously tried to leverage the use of engineered nanotherapeutics to promote tumor immune cell infiltration. One study had described the delivery of a CCL2-binding single-domain antibody to the TME of TNBC tumors in mice (231). CCL2 is of interest as tumor-associated adipocytes, a major component of the tumor stroma in TNBC, can secrete CCL2 to promote infiltration of other pro-tumoral cells, such as MDSCs, into the TME (232). The CCL2 antibody was encoded in a plasmid encapsulated in a lipid-protamine-DNA nanoparticle and was delivered intravenously. This led to a higher anti-tumor efficacy than an anti-CCL2 monoclonal antibody alone and was able to significantly inhibit TNBC tumor progression. CCL2 blockade also led to an increase in CD3+ lymphocyte recruitment by promoting an increase in production of CXCL9 and CXCL10, as well as other inflammatory cytokines like IL-12 and IFN-γ (232). Importantly, this strategy was relatively tumor-specific and did not lead to tissue damage to other organs.
An alternative approach is the expression of surface ligands on nanomedicines to improve the efficacy of immunotherapies. For example, some studies leverage the expression of surface ligands on nanoparticles to trigger expansion and activation of cells in vivo. One study has shown that complexing an IL-15 super-agonist complex selectively expands T cells in tumors and enabled higher administration of IL-15 to boost CAR-T cell efficacy in mice (233). Membrane-bound cytokines, such as IL-15 and IL-21, in combination with adoptive immunotherapy could improve the activation and persistence of NK cell immunotherapies. Further, coating nanoparticles with other molecules such as membrane-bound chemokines and cytokines or ligands that block immune-checkpoint ligands has been investigated. One study conjugated anti-PD-L1 antibodies to promote T cell expansion within the tumor. As ICB therapy is often limited by the penetration of these antibodies into the tumor, this strategy acts to effectively trigger anti-PD-L1 engagement at a deeper level inside the tumor bed (234–236). Another study showed nanoparticle solid tumor penetration when using an anti-CCR7-loaded nanoparticle to sequester CCR7. CCR7 is known to be involved in mediating TNBC metastasis to the lymph nodes (237, 238). This strategy prevented metastasis in a murine syngeneic melanoma model (237, 238). While an anti-CCR7 antibody elicited immunosuppression by counteracting immune cell migration, CCR7-loaded nanoparticles are not limited in the same way (237, 238). CCR7-loaded nanoparticles would limit their effects directly to the tumor, decreasing bystander effects of the therapy compared an anti-CCR7 antibody which works systemically. Overall, when considering strategies to induce immune cell infiltration into the tumor bed, nanoparticles hold promise due to their ability to enter the tumor bed and ability to be targeted to tumors.
6 Concluding remarks
NK cell-based cancer immunotherapies have emerged as a promising cancer treatment option due to NK cell’s natural ability to recognize and kill cancer cells. Ex vivo activated and expanded NK cells have shown high clinical efficacies against hematologic malignancies. Further, advancements in NK cell genetic engineering methods have enabled the efficient generation of highly potent and safe CAR-NK cells which have shown promising safety and efficacy for treating CD19-expressing cancers. However, extending NK cell immunotherapies against solid tumors has been met with various challenges including impaired NK cell infiltration into solid tumor sites and NK cell dysfunction in the TME. While NK cell expansion using K562-mb-IL-21 feeder cells and IL-15 stimulation can enhance NK cell metabolic fitness allowing them to thrive in the TME, efficient homing of adoptively transferred NK cells to the tumor remains to be addressed. The tumor promotes the recruitment of structural and immune cells, which are reprogrammed into a suppressive phenotype within the tumor bed and support tumor growth. Physical barriers posed by the tumor also prevent recruitment of immune effector cells and prevent their permeation into the tumor core to elicit their activity. Various strategies have been developed to target the dense tumor stroma and aberrant tumor vasculature for improved drug delivery and immune cell infiltration into solid tumors. However, few studies directly explore the impact that these strategies have on NK cells, and instead focus generally on lymphocyte recruitment. In addition, the therapeutic efficacy of such strategies relies on stimulation of immune cells other than NK cells, particularly tumor specific T cells, and there are still very limited studies combining these strategies with NK cell adoptive cell therapies. As shown in Figure 3, reprogramming the TME with immunomodulatory agents to promote adoptive cell therapy recruitment is a promising strategy to enhance NK cell infiltration and anti-tumor activity. The ability of OVs to selectively express high concentrations of immunomodulatory agents within the tumor make them an attractive platform for developing novel engineered OVs to enhance intra-tumoral infiltration of adoptively transferred NK cells. An advantage of OVs is that expression of their therapeutic payload is restricted to their replication within the TME, which can limit toxicities seen with systemic administration of immunomodulatory agents. This strategy has been shown to promote enhanced recruitment and prolonged tumor clearance when combined with engineered NK cell therapies, through OV expression of chemokines and activating cytokines such as IL-15. Further, OVs without the capacity to induce complete vascular shutdown that are engineered to express stroma normalization agents and NK cell-specific chemokines could drastically enhance access of adoptively transferred NK cell to the tumor bed. However, careful considerations must be taken for rational design of such combination therapies given the complex interplay between OVs and NK cells. For example, optimizing the timing of OV and NK cell administration will be important due to NK cell’s ability to destroy infected tumor cells and prevent viral spread. Further, many translational barriers remain for production of clinically applicable OVs with regards to viral delivery, spread, and antiviral immunity. Recent advancements have shown that it is possible to engineer OVs such as VV to regulate their replication and control the timing and dose of therapeutic payload using inducible expression systems to enhance safety of OVs (239). Additionally, immunomodulatory proteins can also be introduced into the tumor core using nanomedicines such as NPs and may be safer than OV administration. While antiviral immunity would limit the repeated use of OVs, NPs can be used for the delivery of multiple proteins at different points during a therapeutic regimen. However, non-specific uptake of NPs by many different organs including the reticuloendothelial system in the liver can limit targeted tumor delivery. While this can be overcome through engineering NPs with tumor specific antibodies or proteins, efficient tumor delivery can also be hampered by the ECM. The ability to engineer both NK cells and tumor-targeting modalities such as OVs and NPs enables unlimited modifications to fully harness the anti-tumor functions of NK cell therapies against solid tumors with limited treatment options.
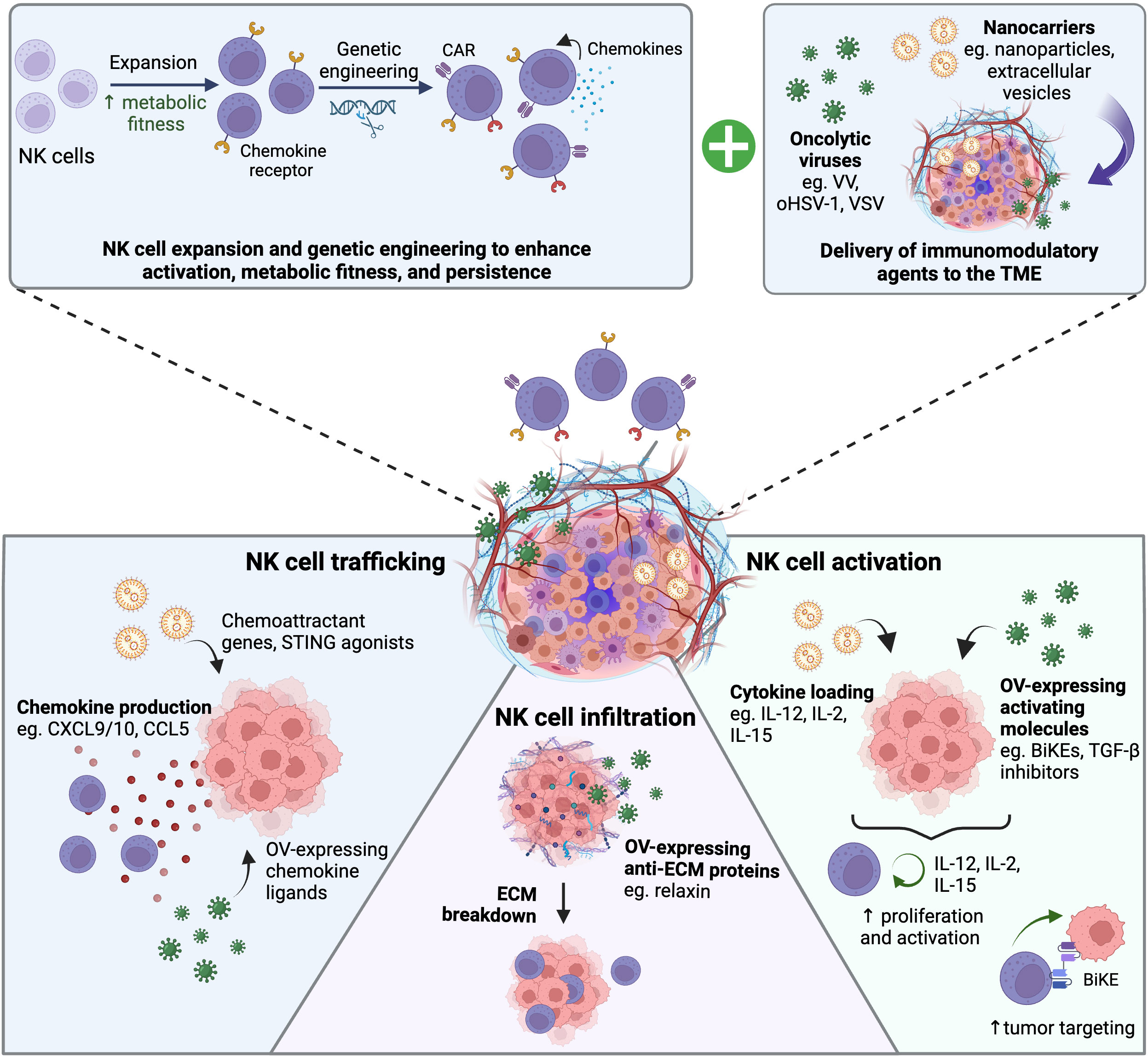
Figure 3 Delivery of immunomodulatory agents to the TME using oncolytic viruses or nanocarriers to pull and boost NK cell-based adoptive cell therapies. Schematic showing how engineered oncolytic viruses (OVs) or nanocarriers can synergize with NK cell-based adoptive cell therapies to enhance NK cell trafficking, infiltration, and activation in the solid tumor. Highlighted are different immunomodulatory agents that can be delivered in the tumor to induce chemokine production in the tumor, break down the extracellular matrix (ECM), or promote NK cell anti-tumor responses. Created with BioRender.com.
Author contributions
AP: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. JM: Writing – original draft, Writing – review & editing. ER: Writing – original draft. TR: Writing – review & editing. AG: Supervision, Writing – review & editing. AA: Funding acquisition, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Canadian Institutes of Health Research (CIHR) AAA is also a recipient of a CIHR Tier 1 Canada Research Chair. ALP is supported by a CIHR Doctoral Canada Graduate Scholarship.
Acknowledgments
All figures in this review were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol (2013) 132:536–44. doi: 10.1016/j.jaci.2013.07.006
2. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood (2017) 130:1857–68. doi: 10.1182/blood-2017-05-785659
3. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med (2016) 8:357ra123–357ra123. doi: 10.1126/scitranslmed.aaf2341
4. Tanaka J, Tanaka N, Wang YH, Mistuhashi K, Ryuzaki M, Iizuka Y, et al. Phase I study of cellular therapy using ex vivo expanded NK cell from autologous peripheral blood mononuclear cells combined with rituximab-containing chemotherapy for relapsed CD20-positive Malignant lymphoma patients. Haematologica (2020) 105:e190. doi: 10.3324/haematol.2019.226696
5. Liu E, Marin D, Banerjee P, MacApinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
6. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
7. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
8. Schmiedel D, Mandelboim O. NKG2D ligands–critical targets for cancer immune escape and therapy. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02040
9. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res (2015) 62:341–56. doi: 10.1007/s12026-015-8664-y
10. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol (2015) 6:578. doi: 10.3389/fimmu.2015.00578
11. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS One (2012) 7:e30264. doi: 10.1371/journal.pone.0030264
12. Oyer JL, Pandey V, Igarashi RY, Somanchi SS, Zakari A, Solh M, et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: Clinical implications for cancer treatment. Cytotherapy (2016) 18:653–63. doi: 10.1016/J.JCYT.2016.02.006
13. Lamers-Kok N, Panella D, Georgoudaki AM, Liu H, Özkazanc D, Kučerová L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol (2022) 15:1–55. doi: 10.1186/S13045-022-01382-5
14. Jo D-H, Kaczmarek S, Shin O, Wang L, Cowan J, McComb S, et al. Simultaneous engineering of natural killer cells for CAR transgenesis and CRISPR-Cas9 knockout using retroviral particles. Mol Ther Methods Clin Dev (2023) 29:173–84. doi: 10.1016/j.omtm.2023.03.006
15. Naeimi Kararoudi M, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep Methods (2022) 2:100236. doi: 10.1016/J.CRMETH.2022.100236
16. Ye L, Lam SZ, Yang L, Suzuki K, Zou Y, Lin Q, et al. AAV-mediated delivery of a Sleeping Beauty transposon and an mRNA-encoded transposase for the engineering of therapeutic immune cells. Nat Biomed Eng (2023), 1–17. doi: 10.1038/s41551-023-01058-6
17. Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest (2008) 118:3132–42. doi: 10.1172/JCI35700
18. Portillo AL, Hogg R, Poznanski SM, Rojas EA, Cashell NJ, Hammill JA, et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience (2021) 24:102619. doi: 10.1016/J.ISCI.2021.102619
19. Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med (2022) 28:2133–44. doi: 10.1038/s41591-022-02003-x
20. Garner WL, Minton JP, James AG, Hoffmann CC. Human breast cancer and impaired NK cell function. J Surg Oncol (1983) 24:64–6. doi: 10.1002/JSO.2930240115
21. Nersesian S, Schwartz SL, Grantham SR, MacLean LK, Lee SN, Pugh-Toole M, et al. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Transl Oncol (2021) 14:100930. doi: 10.1016/J.TRANON.2020.100930
22. Poznanski SM, Nham T, Chew MV, Lee AJ, Hammill JA, Fan IY, et al. Expanded CD56superbrightCD16+ NK cells from ovarian cancer patients are cytotoxic against autologous tumor in a patient-derived xenograft murine model. Cancer Immunol Res (2018) 6:1174–85. doi: 10.1158/2326-6066.CIR-18-0144
23. Shenouda MM, Gillgrass A, Nham T, Hogg R, Lee AJ, Chew MV, et al. Ex vivo expanded natural killer cells from breast cancer patients and healthy donors are highly cytotoxic against breast cancer cell lines and patient-derived tumours. Breast Cancer Res (2017) 19:76. doi: 10.1186/s13058-017-0867-9
24. Poznanski SM, Ritchie TM, Fan IY, El-Sayes A, Portillo AL, Ben-Avi R, et al. Expanded human NK cells from lung cancer patients sensitize patients’ PDL1–negative tumors to PD1-blockade therapy. J Immunother Cancer (2021) 9:1933. doi: 10.1136/jitc-2020-001933
25. Uong TNT, Yoon MS, Lee KH, Hyun H, Nam TK, Min JJ, et al. Live cell imaging of highly activated natural killer cells against human hepatocellular carcinoma in vivo. Cytother (2021) 23:799–809. doi: 10.1016/J.JCYT.2020.11.004
26. Uong TNT, Lee KH, Ahn SJ, Kim KW, Min JJ, Hyun H, et al. Real-time tracking of ex vivo-expanded natural killer cells toward human triple-negative breast cancers. Front Immunol (2018) 9:825. doi: 10.3389/fimmu.2018.00825
27. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
28. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350. doi: 10.1126/SCIENCE.AAR4060
29. Shaver KA, Croom-Perez TJ, Copik AJ. Natural killer cells: the linchpin for successful cancer immunotherapy. Front Immunol (2021) 12:679117/BIBTEX. doi: 10.3389/FIMMU.2021.679117
30. Lin M, Luo H, Liang S, Chen J, Liu A, Niu L, et al. Pembrolizumab plus allogeneic NK cells in advanced non–small cell lung cancer patients. J Clin Invest (2020) 130:2560–9. doi: 10.1172/JCI132712
31. Liang S, Lin M, Niu L, Xu K, Wang X, Liang Y, et al. Cetuximab combined with natural killer cells therapy: an alternative to chemoradiotherapy for patients with advanced non-small cell lung cancer (NSCLC). Am J Cancer Res (2018) 8:879.
32. Lee SC, Shimasaki N, Lim JSJ, Wong A, Yadav K, Yong WP, et al. Phase I trial of expanded, activated autologous NK-cell infusions with trastuzumab in patients with HER2-positive cancers. Clin Cancer Res (2020) 26:4494–502. doi: 10.1158/1078-0432.CCR-20-0768
33. Patin EC, Dillon MT, Nenclares P, Grove L, Soliman H, Leslie I, et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage–response inhibition and immune checkpoint blockade. J Immunother Cancer (2022) 10:e004306. doi: 10.1136/JITC-2021-004306
34. Gadwa J, Amann M, Bickett TE, Knitz MW, Darragh LB, Piper M, et al. Selective targeting of IL2Rβγ combined with radiotherapy triggers CD8- and NK-mediated immunity, abrogating metastasis in HNSCC. Cell Rep Med (2023) 4:101150. doi: 10.1016/J.XCRM.2023.101150
35. Kim KW, Jeong JUK, Lee KH, Uong TNT, Rhee JH, Ahn SJ, et al. Combined NK cell therapy and radiation therapy exhibit long-term therapeutic and antimetastatic effects in a human triple negative breast cancer model. Int J Radiat Oncol Biol Phys (2020) 108:115–25. doi: 10.1016/J.IJROBP.2019.09.041
36. Tuomela K, Mukherjee D, Ambrose AR, Harikrishnan A, Mole H, Hurlstone A, et al. Radiotherapy transiently reduces the sensitivity of cancer cells to lymphocyte cytotoxicity. Proc Natl Acad Sci USA (2022) 119:e2111900119. doi: 10.1073/PNAS.2111900119
37. Shen MJ, Xu LJ, Yang L, Tsai Y, Keng PC, Chen Y, et al. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6-MEK/Erk signaling pathway. Oncotarget (2017) 8:80506–20. doi: 10.18632/ONCOTARGET.19193
38. Li L, Li W, Wang C, Yan X, Wang Y, Niu C, et al. Adoptive transfer of natural killer cells in combination with chemotherapy improves outcomes of patients with locally advanced colon carcinoma. Cytotherapy (2018) 20:134–48. doi: 10.1016/J.JCYT.2017.09.009
39. Piccinelli S, Romee R, Shapiro RM. The natural killer cell immunotherapy platform: An overview of the landscape of clinical trials in liquid and solid tumors. Semin Hematol (2023) 60:42–51. doi: 10.1053/J.SEMINHEMATOL.2023.02.002
40. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
41. Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife (2014) 2014:e01308. doi: 10.7554/ELIFE.01308
42. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of Malignancy. Genes Dev (2016) 30:1002. doi: 10.1101/GAD.279737.116
43. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
44. Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res (2019) 79:5367–81. doi: 10.1158/0008-5472.CAN-19-0454
45. Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun (2020) 11:1–17. doi: 10.1038/s41467-020-19920-5
46. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res (2017) 23:1710–21. doi: 10.1158/1078-0432.CCR-15-2851
47. Bergamaschi A, Tagliabue E, Sørlie T, Naume B, Triulzi T, Orlandi R, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J Pathol (2008) 214:357–67. doi: 10.1002/PATH.2278
48. Castriconi R, Carrega P, Dondero A, Bellora F, Casu B, Regis S, et al. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front Immunol (2018) 9:2324. doi: 10.3389/fimmu.2018.02324
49. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer (2009) 9:874–85. doi: 10.1038/NRC2761
50. Walker C, Mojares E, Hernández A del R. Role of extracellular matrix in development and cancer progression. Int J Mol Sci (2018) 19:3028. doi: 10.3390/IJMS19103028
51. Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors 1. Cancer Res (2000) 60:2497–503.
52. Gkretsi V, Stylianopoulos T. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Front Oncol (2018) 8:145. doi: 10.3389/FONC.2018.00145
53. Sarntinoranont M, Rooney F, Ferrari M. Interstitial stress and fluid pressure within a growing tumor. Ann Biomed Eng (2003) 31:327–35. doi: 10.1114/1.1554923
54. Barkovskaya A, Buffone A Jr., Žídek M, Weaver VM. Proteoglycans as mediators of cancer tissue mechanics. Front Cell Dev Biol (2020) 8:569377. doi: 10.3389/FCELL.2020.569377
55. Zimna A, Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. BioMed Res Int (2015) 2015:549412. doi: 10.1155/2015/549412
56. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun (2020) 11:1–19. doi: 10.1038/s41467-020-18794-x
57. Muz B, de la PP, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83. doi: 10.2147/HP.S93413
58. Sun R, Kong X, Qiu X, Huang C, Wong P-P. The emerging roles of pericytes in modulating tumor microenvironment. Front Cell Dev Biol (2021) 0:676342. doi: 10.3389/FCELL.2021.676342
59. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol (2002) 160:985. doi: 10.1016/S0002-9440(10)64920-6
60. Ribeiro AL, Okamoto OK. Combined effects of pericytes in the tumor microenvironment. Stem Cells Int (2015) 2015:868475. doi: 10.1155/2015/868475
61. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy article. Cell Death Dis (2018) 9:115. doi: 10.1038/s41419-017-0061-0
62. Harjunpää H, Asens ML, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
63. Luetke-Eversloh M, Killig M, Romagnani C. Signatures of human NK cell development and terminal differentiation. Front Immunol (2013) 0:499. doi: 10.3389/FIMMU.2013.00499
64. Lima M, Leander M, Santos M, Santos AH, Lau C, Queirós ML, et al. Chemokine receptor expression on normal blood CD56+ NK-cells elucidates cell partners that comigrate during the innate and adaptive immune responses and identifies a transitional NK-cell population. J Immunol Res (2015) 2015:839684. doi: 10.1155/2015/839684
65. Sechler JM, Barlic J, Grivel JC, Murphy PM. IL-15 alters expression and function of the chemokine receptor CX3CR1 in human NK cells. Cell Immunol (2004) 230:99–108. doi: 10.1016/j.cellimm.2004.10.001
66. Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics (2020) 16:75–85. doi: 10.1016/J.OMTO.2019.12.006
67. Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol (2007) 8:1337–44. doi: 10.1038/ni1523
68. Bernardini G, Antonangeli F, Bonanni V, Santoni A. Dysregulation of chemokine/chemokine receptor axes and NK cell tissue localization during diseases. Front Immunol (2016) 7:402. doi: 10.3389/fimmu.2016.00402
69. Yao X, Matosevic S. Chemokine networks modulating natural killer cell trafficking to solid tumors. Cytokine Growth Factor Rev (2021) 59:36–45. doi: 10.1016/j.cytogfr.2020.12.003
70. Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, et al. CD56 bright Perforin low Noncytotoxic Human NK Cells Are Abundant in Both Healthy and Neoplastic Solid Tissues and Recirculate to Secondary Lymphoid Organs via Afferent Lymph. J Immunol (2014) 192:3805–15. doi: 10.4049/jimmunol.1301889
71. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
72. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
73. Regis S, Caliendo F, Dondero A, Casu B, Romano F, Loiacono F. TGF-beta1 downregulates the expression of CX3CR1 by inducing miR-27a-5p in primary human NK cells. Front Immunol (2017) 8:868. doi: 10.3389/fimmu.2017.00868
74. Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-γ and CXCR3 ligands. Cancer Res (2008) 68:8437–45. doi: 10.1158/0008-5472.CAN-08-1440
75. Lavergne E, Combadière B, Bonduelle O, Iga M, Gao J-L, Maho M, et al. Fractalkine mediates natural killer-dependent antitumor responses in vivo. Cancer Res (2003) 63:7468–74.
76. Araujo JM, Gomez AC, Aguilar A, Salgado R, Balko JM, Bravo L, et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci Rep (2018) 8:1–9. doi: 10.1038/s41598-018-23099-7
77. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother (2015) 64:225–35. doi: 10.1007/s00262-014-1629-5
78. Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer (2017) 5:73. doi: 10.1186/s40425-017-0275-9
79. SenGupta S, Hein LE, Xu Y, Zhang J, Konwerski JR, Li Y, et al. Triple-negative breast cancer cells recruit neutrophils by secreting TGF-β and CXCR2 ligands. Front Immunol (2021) 12:659996. doi: 10.3389/fimmu.2021.659996
80. Levy ER, Clara JA, Reger RN, Allan DSJ, Childs RW. RNA-seq analysis reveals CCR5 as a key target for CRISPR gene editing to regulate in vivo NK cell trafficking. Cancers (2021) 13:872. doi: 10.3390/CANCERS13040872
81. Germann M, Zangger N, Sauvain M-O, Sempoux C, Bowler AD, Wirapati P, et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFβ. EMBO Mol Med (2020) 12:e10681. doi: 10.15252/EMMM.201910681
82. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica (2012) 97:1295–303. doi: 10.3324/haematol.2011.058529
83. Lee J-C, Lee K-M, Ahn Y-O, Suh B, Heo DS. A possible mechanism of impaired NK cytotoxicity in cancer patients: Down-regulation of DAP10 by TGF-$β$1. Tumori J (2011) 97:350–7. doi: 10.1177/030089161109700316
84. Donatelli SS, Zhou J-M, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-$β$–inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
85. Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9:ra19–9. doi: 10.1126/scisignal.aad1884
86. Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-$β$ signaling pathway represses human NK cell metabolism. J Immunol (2018) 200:3934–41. doi: 10.4049/jimmunol.1701461
87. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol (2010) 13:132–42. doi: 10.1093/neuonc/noq142
88. Faivre S, Santoro A, Kelley RK, Gane E, Costentin CE, Gueorguieva I, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int (2019) 39:1468–77. doi: 10.1111/liv.14113
89. Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest (2021) 131:e142116. doi: 10.1172/JCI142116
90. Zhao J, Wen S, Wang X, Zhang Z. Helicobacter pylori modulates cyclooxygenase-2 and 15-hydroxy prostaglandin dehydrogenase in gastric cancer. Oncol Lett (2017) 14:5519–25. doi: 10.3892/ol.2017.6843
91. Ko CJ, Lan SW, Lu YC, Cheng TS, Lai PF, Tsai CH, et al. Inhibition of cyclooxygenase-2-mediated matriptase activation contributes to the suppression of prostate cancer cell motility and metastasis. Oncogene (2017) 36:4597–609. doi: 10.1038/onc.2017.82
92. Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and cancer: insight into tumor progression and immunity. Biol (Basel) (2020) 9:1–26. doi: 10.3390/BIOLOGY9120434
93. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE (2)) suppresses natural killer cell function primarily through the PGE(2) receptor EP4. Cancer Immunol Immunother (2011) 60:1577–86. doi: 10.1007/s00262-011-1064-9
94. Ma X, Holt D, Kundu N, Reader J, Goloubeva O, Take Y, et al. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology (2013) 2:e22647. doi: 10.4161/ONCI.22647
95. Park A, Lee Y, Kim MS, Kang YJ, Park Y-J, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
96. Martinet L, Jean C, Dietrich G, Fournié JJ, Poupot R. PGE2 inhibits natural killer and γδ T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol (2010) 80:838–45. doi: 10.1016/j.bcp.2010.05.002
97. Felix Bayerl A, Meiser P, Donakonda S, Buchholz VR, Schraml BU, Bö ttcher Correspondence JP, et al. Tumor-derived prostaglandin E2 programs cDC1 dysfunction to impair intratumoral orchestration of anti-cancer T cell responses. Immunity (2023) 56:1341–58. doi: 10.1016/j.immuni.2023.05.011
98. Rayar A-M, Lagarde N, Ferroud C, Zagury J-F, Montes M, Sylla-Iyarreta Veitia M. Update on COX-2 selective inhibitors: Chemical classification, side effects and their use in cancers and neuronal diseases. Curr Top Med Chem (2017) 17:2935–56. doi: 10.2174/1568026617666170821124947
99. Li L, Mohanty V, Dou J, Huang Y, Banerjee PP, Miao Q, et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci Adv (2023) 9:eadd6997. doi: 10.1126/sciadv.add6997
100. Poznanski SM, Singh K, Ritchie TM, Aguiar JA, Fan IY, Portillo AL, et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab (2021) 33:1205–1220.e5. doi: 10.1016/j.cmet.2021.03.023
101. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood (2021) 137:624–36. doi: 10.1182/BLOOD.2020007748
102. Delconte RB, Guittard G, Goh W, Hediyeh-Zadeh S, Hennessy RJ, Rautela J, et al. NK cell priming from endogenous homeostatic signals is modulated by CIS. Front Immunol (2020) 11:75. doi: 10.3389/FIMMU.2020.00075
103. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell (2020) 27:224. doi: 10.1016/J.STEM.2020.05.008
104. Baba AB, Rah B, Bhat GR, Mushtaq I, Parveen S, Hassan R, et al. Transforming growth factor-beta (TGF-β) signaling in cancer-A betrayal within. Front Pharmacol (2022) 13:791272. doi: 10.3389/fphar.2022.791272
105. Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med (2017) 9:eaan5616. doi: 10.1126/scitranslmed.aan5616
106. George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: Old dog, new tricks. Nat Rev Cancer (2010) 10:745–59. doi: 10.1038/nrc2945
107. Beyazit F, Ayhan S, Celik HT, Gungor T. Assessment of serum angiotensin-converting enzyme in patients with epithelial ovarian cancer. Arch Gynecol Obstet (2015) 292:415–20. doi: 10.1007/s00404-015-3661-x
108. Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, et al. Reprogramming the microenvironment with tumorselective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci USA (2019) 166:10674–80. doi: 10.1073/pnas.1819889116
109. Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci USA (2019) 116:2210–9. doi: 10.1073/pnas.1818357116
110. Nakamura K, Yaguchi T, Ohmura G, Kobayashi A, Kawamura N, Iwata T, et al. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci (2018) 109:54–64. doi: 10.1111/cas.13423
111. Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol (2007) 292:987–95. doi: 10.1152/AJPCELL.00406.2006
112. Kryczek I, Lange A, Mottram P, Alvarez X, Cheng P, Hogan M, et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res (2005) 65:465–72.
113. Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene (2015) 35:816–26. doi: 10.1038/onc.2015.139
114. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
115. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci USA (2019) 116:4558–66. doi: 10.1073/pnas.1815515116
116. Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther (2018) 17:1147. doi: 10.1158/1535-7163.MCT-17-0646
117. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol (2019) 37:1062–9. doi: 10.1200/JCO.18.01295
118. Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell (2018) 175:117. doi: 10.1016/J.CELL.2018.08.017
119. Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
120. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CCR, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210:1065–8. doi: 10.1084/jem.20130110
121. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA (2019) 116:7624–31. doi: 10.1073/pnas.1817147116
122. Gorchs L, Oosthoek M, Yucel-Lindberg T, Moro CF, Kaipe H. Chemokine receptor expression on T cells is modulated by CAFs and chemokines affect the spatial distribution of T cells in pancreatic tumors. Cancers (Basel) (2022) 14:3826. doi: 10.3390/CANCERS14153826/S1
123. Goel S, Wong AHK, Jain RK. Vascular normalization as a therapeutic strategy for Malignant and nonmalignant disease. Cold Spring Harb Perspect Med (2012) 2:a006486. doi: 10.1101/cshperspect.a006486
124. Hayes DF. Bevacizumab treatment for solid tumors: boon or bust? JAMA (2011) 305:506–8. doi: 10.1001/JAMA.2011.57
125. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol (2006) 3:24–40. doi: 10.1038/NCPONC0403
126. Jain RK. Cancer cell antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
127. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
128. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (2020) 71:1247–61. doi: 10.1002/hep.30889
129. Tian L, Goldstein A, Wang H, Lo HC, Kim IS, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature (2017) 544:250–4. doi: 10.1038/nature21724
130. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest (2018) 128:2104–15. doi: 10.1172/JCI96582
131. Lu Y, Yang W, Qin C, Zhang L, Deng J, Liu S, et al. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J Immunol (2009) 183:6413–21. doi: 10.4049/JIMMUNOL.0901073
132. Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer (2011) 128:371–8. doi: 10.1002/IJC.25350
133. Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific Chimeric antigen receptor to Overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J Immunother (2015) 38:197–210. doi: 10.1097/CJI.0000000000000082
134. Ng YY, Du Z, Zhang X, Chng WJ, Wang S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther (2021) 29:475–83. doi: 10.1038/s41417-021-00365-x
135. Yang L, Huang C, Wang C, Zhang S, Li Z, Zhu Y. Overexpressed CXCR4 and CCR7 on the surface of NK92 cell have improved migration and anti-tumor activity in human colon tumor model. Anticancer Drugs (2020) 31:333–44. doi: 10.1097/CAD.0000000000000868
136. Allen F, Bobanga ID, Rauhe P, Barkauskas D, Teich N, Tong C, et al. CCL3 augments tumor rejection and enhances CD8+ T cell infiltration through NK and CD103+ dendritic cell recruitment via IFNγ. Oncoimmunology (2017) 7:e1393598. doi: 10.1080/2162402X.2017.1393598
137. Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res (2019) 7:219–29. doi: 10.1158/2326-6066.CIR-18-0317
138. Baker GJ, Chockley P, Zamler D, Castro MG, Lowenstein PR. Natural killer cells require monocytic Gr-1+/CD11b+ myeloid cells to eradicate orthotopically engrafted glioma cells. Oncoimmunology (2016) 5:e1163461. doi: 10.1080/2162402X.2016.1163461
139. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer (2018) 18:419–32. doi: 10.1038/s41568-018-0009-4
140. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: A new class of immunotherapy drugs. Nat Rev Drug Discov (2015) 14:642–62. doi: 10.1038/nrd4663
141. Lemos de Matos A, Franco LS, McFadden G. Oncolytic viruses and the immune system: the dynamic duo. Mol Ther Methods Clin Dev (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001
142. Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol (2010) 6:941–9. doi: 10.2217/fon.10.66
143. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol (2023) 20:160–77. doi: 10.1038/s41571-022-00719-w
144. Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol Ther (2014) 22:251–6. doi: 10.1038/mt.2013.220
145. Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis (2020) 11:1–15. doi: 10.1038/s41419-020-2236-3
146. Wan PKT, Ryan AJ, Seymour LW. Beyond cancer cells: Targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol Ther (2021) 29:1668–82. doi: 10.1016/j.ymthe.2021.04.015
147. Bhat R, Rommelaere J. Emerging role of natural killer cells in oncolytic virotherapy. Immunotargets Ther (2015) 4:65–77. doi: 10.2147/ITT.S55549
148. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172:1022–1037.e14. doi: 10.1016/J.CELL.2018.01.004
149. El-Sherbiny YM, Holmes TD, Wetherill LF, Black EVI, Wilson EB, Phillips SL, et al. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo. Clin Exp Immunol (2015) 180:98–107. doi: 10.1111/CEI.12562
150. Niavarani S-R, Lawson C, Boudaud M, Simard C, Tai L-H. Oncolytic vesicular stomatitis virus-based cellular vaccine improves triple-negative breast cancer outcome by enhancing natural killer and CD8 + T-cell functionality. J Immunother Cancer (2020) 8:465. doi: 10.1136/jitc-2019-000465
151. Jennings VA, Scott GB, Rose AMS, Scott KJ, Migneco G, Keller B, et al. Potentiating oncolytic virus-induced immune-mediated tumor cell killing using histone deacetylase inhibition. Mol Ther (2019) 27:1139–52. doi: 10.1016/j.ymthe.2019.04.008
152. Fiebig H-H, Siegling A, Volk H-D, Friebe A, Knolle P, Limmer A, et al. Inactivated orf virus (Parapoxvirus ovis) induces antitumoral activity in transplantable tumor models. Anticancer Res (2011) 31:4185–90.
153. Rintoul JL, Lemay CG, Tai LH, Stanford MM, Falls TJ, De Souza CT, et al. ORFV: A novel oncolytic and immune stimulating parapoxvirus therapeutic. Mol Ther (2012) 20:1148–57. doi: 10.1038/mt.2011.301
154. Haig DM, McInnes CJ. Immunity and counter-immunity during infection with the parapoxvirus orf virus. Virus Res (2002) 88:3–16. doi: 10.1016/S0168-1702(02)00117-X
155. Tai L-H, de SCT, Bélanger S, Ly L, Alkayyal AA, Zhang J, et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res (2013) 73:97–107. doi: 10.1158/0008-5472.CAN-12-1993
156. Van Vloten JP, Matuszewska K, Minow MAA, Minott JA, Santry LA, Pereira M, et al. Oncolytic Orf virus licenses NK cells via cDC1 to activate innate and adaptive antitumor mechanisms and extends survival in a murine model of late-stage ovarian cancer. J Immunother Cancer (2022) 10:e004335. doi: 10.1136/JITC-2021-004335
157. Wang R, Wang Y, Liu F, Luo S. Orf virus: A promising new therapeutic agent. Rev Med Virol (2019) 29:e2013. doi: 10.1002/rmv.2013
158. John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res (2012) 72:1651–60. doi: 10.1158/0008-5472.CAN-11-2788
159. Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther (2018) 26:2476–86. doi: 10.1016/J.YMTHE.2018.07.013
160. Chouljenko DV, Ding J, Lee I-F, Murad YM, Bu X, Liu G, et al. Induction of durable antitumor response by a novel oncolytic herpesvirus expressing multiple immunomodulatory transgenes. Biomedicines (2020) 8:1–21. doi: 10.3390/BIOMEDICINES8110484
161. Li F, Sheng Y, Hou W, Sampath P, Byrd D, Thorne S, et al. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer (2020) 8:e000131. doi: 10.1136/jitc-2019-000131
162. Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget (2016) 7:27764–77. doi: 10.18632/oncotarget.8526
163. Ma R, Lu T, Li Z, Teng K-Y, Mansour AG, Yu M, et al. An oncolytic virus expressing IL15/IL15Rα Combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res (2021) 81:3635–48. doi: 10.1158/0008-5472.CAN-21-0035
164. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med (2023) 29:1370–8. doi: 10.1038/s41591-023-02347-y
165. Floerchinger A, Klein JE, Finkbeiner MSC, Schäfer TE, Fuchs G, Doerner J, et al. A vector-encoded bispecific killer engager to harness virus-activated NK cells as anti-tumor effectors. Cell Death Dis (2023) 14:1–15. doi: 10.1038/s41419-023-05624-3
166. Liu B, Zhu X, Kong L, Wang M, Spanoudis C, Chaturvedi P, et al. Bifunctional TGF-β trap/IL-15 protein complex elicits potent NK cell and CD8+ T cell immunity against solid tumors. Mol Ther (2021) 29:2949–62. doi: 10.1016/j.ymthe.2021.06.001
167. DePeaux K, Rivadeneira DB, Lontos K, Dean VG, Gunn WG, Watson MJ, et al. An oncolytic virus-delivered TGFβ inhibitor overcomes the immunosuppressive tumor microenvironment. J Exp Med (2023) 220:e20230053. doi: 10.1084/JEM.20230053/276162
168. Santry LA, van Vloten JP, Knapp JP, Matuszewska K, McAusland TM, Minott JA, et al. Tumour vasculature: Friend or foe of oncolytic viruses? Cytokine Growth Factor Rev (2020) 56:69–82. doi: 10.1016/j.cytogfr.2020.07.007
169. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res (2013) 73:1265–75. doi: 10.1158/0008-5472.CAN-12-2687
170. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther (2011) 19:886–94. doi: 10.1038/mt.2011.26
171. Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, et al. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci USA (1990) 87:7095–9. doi: 10.1073/pnas.87.18.7095
172. Cinatl J, Michaelis M, Driever PH, Cinatl J, Hrabeta J, Suhan T, et al. Multimutated herpes simplex virus G207 is a potent inhibitor of angiogenesis. Neoplasia (2004) 6:725–35. doi: 10.1593/neo.04265
173. Matuszewska K, Santry LA, van Vloten JP, AuYeung AWK, Major PP, Lawler J, et al. Combining vascular normalization with an oncolytic virus enhances immunotherapy in a preclinical model of advanced-stage ovarian cancer. Clin Cancer Res (2019) 25:1624–38. doi: 10.1158/1078-0432.CCR-18-0220
174. Russell S, Duquette M, Liu J, Drapkin R, Lawler J, Petrik J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J (2015) 29:576–88. doi: 10.1096/fj.14-261636
175. Jing Y, Chavez V, Khatwani N, Ban Y, Espejo AP, Chen X, et al. In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses. Cancer Gene Ther (2020) 27:910–22. doi: 10.1038/s41417-020-0171-1
176. Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther (2010) 17:190–201. doi: 10.1038/gt.2009.142
177. Jung BK, Ko HY, Kang H, Hong J, Ahn HM, Na Y, et al. Relaxin-expressing oncolytic adenovirus induces remodeling of physical and immunological aspects of cold tumor to potentiate PD-1 blockade. J Immunother Cancer (2020) 8:e000763. doi: 10.1136/JITC-2020-000763
178. Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst (2006) 98:1482–93. doi: 10.1093/jnci/djj397
179. McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res (2006) 66:2509–13. doi: 10.1158/0008-5472.CAN-05-2242
180. Schäfer S, Weibel S, Donat U, Zhang Q, Aguilar RJ, Chen NG, et al. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer (2012) 12:1–9. doi: 10.1186/1471-2407-12-366/FIGURES/5
181. Bazan-Peregrino M, Garcia-Carbonero R, Laquente B, Álvarez R, Mato-Berciano A, Gimenez-Alejandre M, et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J Immunother Cancer (2021) 9:e003254. doi: 10.1136/JITC-2021-003254
182. De Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer (2019) 7:1–15. doi: 10.1186/s40425-019-0505-4
183. Yu F, Hong B, Song X-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl Med (2017) 3:122. doi: 10.4103/ctm.ctm_13_17
184. Gauthier L, Morel A, Anceriz N, Morel Y, Narni-Mancinelli E, Vivier E. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell (2019) 177:1701–1713.e16. doi: 10.1016/j.cell.2019.04.041
185. Kim Y, Yoo JY, Lee TJ, Liu J, Yu J, Caligiuri MA, et al. Complex role of NK cells in regulation of oncolytic virus–bortezomib therapy. Proc Natl Acad Sci USA (2018) 115:4927–32. doi: 10.1073/pnas.1715295115
186. Kim BYS, Rutka JT, Chan WCW. Nanomedicine. N Engl J Med (2010) 363:2434–43. doi: 10.1056/NEJMRA0912273
187. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol (2020) 20:321–34. doi: 10.1038/s41577-019-0269-6
188. Alavi M, Hamidi M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab Pers Ther (2019) 34:10.1515/dmpt-2018-0032. doi: 10.1515/DMPT-2018-0032
189. Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat Commun (2017) 8:1–12. doi: 10.1038/s41467-017-01830-8
190. Zhang Y, Li N, Suh H, Irvine DJ. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat Commun (2018) 9:1–15. doi: 10.1038/s41467-017-02251-3
191. Petros RA, Desimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov (2010) 9:615–27. doi: 10.1038/nrd2591
192. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater (2016) 1:1–12. doi: 10.1038/natrevmats.2016.14
193. Yang L, Mao H, Andrew Wang Y, Cao Z, Peng X, Wang X, et al. Single chain epidermal growth factor receptor antibody conjugated nanoparticles for in vivo tumor targeting and imaging. Small (2009) 5:235–43. doi: 10.1002/SMLL.200800714
194. Chen F, Hong H, Zhang Y, Valdovinos HF, Shi S, Kwon GS, et al. In vivo tumor targeting and image-guided drug delivery with antibody-conjugated, radiolabeled mesoporous silica nanoparticles. ACS Nano (2013) 7:9027–39. doi: 10.1021/nn403617j
195. Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther (2010) 18:1650–6. doi: 10.1038/mt.2010.136
196. Liu Y, Wang J, Zhang J, Marbach S, Xu W, Zhu L. Targeting tumor-associated macrophages by MMP2-sensitive apoptotic body-mimicking nanoparticles. ACS Appl Mater Interfaces (2020) 12:52402–14. doi: 10.1021/acsami.0c15983
197. Zhu L, Wang T, Perche F, Taigind A, Torchilin VP. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc Natl Acad Sci USA (2013) 110:17047–52. doi: 10.1073/pnas.1304987110
198. Kingston BR, Lin ZP, Ouyang B, Macmillan P, Ngai J, Syed AM, et al. Specific endothelial cells govern nanoparticle entry into solid tumors. ACS Nano (2021) 15:14080–94. doi: 10.1021/ACSNANO.1C04510/ASSET/IMAGES/LARGE/NN1C04510_0005.JPEG
199. Chan WCW. Principles of nanoparticle delivery to solid tumors. BME Front (2023) 4:0016. doi: 10.34133/BMEF.0016
200. Lin ZP, Nguyen LNM, Ouyang B, Macmillan P, Ngai J, Kingston BR, et al. Macrophages actively transport nanoparticles in tumors after extravasation. ACS Nano (2022) 16:6080–92. doi: 10.1021/ACSNANO.1C11578/SUPPL_FILE/NN1C11578_SI_002.PDF
201. Miller MA, Chandra R, Cuccarese MF, Pfirschke C, Engblom C, Stapleton S, et al. Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci Transl Med (2017) 9:eaal0225. doi: 10.1126/SCITRANSLMED.AAL0225
202. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun (2015) 6:1–13. doi: 10.1038/ncomms9692
203. Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y, et al. Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano (2018) 12:8423–35. doi: 10.1021/ACSNANO.8B03900
204. Nguyen LNM, Lin ZP, Sindhwani S, MacMillan P, Mladjenovic SM, Stordy B, et al. The exit of nanoparticles from solid tumours. Nat Mater (2023) 22:1–12. doi: 10.1038/s41563-023-01630-0
205. Wang B, Tang M, Yuan Z, Li Z, Hu B, Bai X, et al. Targeted delivery of a STING agonist to brain tumors using bioengineered protein nanoparticles for enhanced immunotherapy. Bioact Mater (2022) 16:232–48. doi: 10.1016/J.BIOACTMAT.2022.02.026
206. Yang K, Han W, Jiang X, Piffko A, Bugno J, Han C, et al. Zinc cyclic di-AMP nanoparticles target and suppress tumours via endothelial STING activation and tumour-associated macrophage reinvigoration. Nat Nanotechnol (2022) 17:1322–31. doi: 10.1038/s41565-022-01225-x
207. Guimarães PPG, Gaglione S, Sewastianik T, Carrasco RD, Langer R, Mitchell MJ. Nanoparticles for immune cytokine TRAIL-based cancer therapy. ACS Nano (2018) 12:912–31. doi: 10.1021/acsnano.7b05876
208. Liu JQ, Zhang C, Zhang X, Yan J, Zeng C, Talebian F, et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. J Controlled Release (2022) 345:306–13. doi: 10.1016/J.JCONREL.2022.03.021
209. Nakamura T, Sato T, Endo R, Sasaki S, Takahashi N, Sato Y, et al. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer (2021) 9:e002852. doi: 10.1136/JITC-2021-002852
210. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Sci (1979) (2013) 339:826–30. doi: 10.1126/science.1229963
211. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41:830–42. doi: 10.1016/J.IMMUNI.2014.10.017
212. Zheng J, Mo J, Zhu T, Zhuo W, Yi Y, Hu S, et al. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol Cancer (2020) 19:1–19. doi: 10.1186/S12943-020-01250-1
213. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep (2015) 11:1018–30. doi: 10.1016/J.CELREP.2015.04.031
214. Watkins-Schulz R, Tiet P, Gallovic MD, Junkins RD, Batty C, Bachelder EM, et al. A microparticle platform for STING-targeted immunotherapy enhances natural killer cell- and CD8+ T cell-mediated anti-tumor immunity. Biomaterials (2019) 205:94–105. doi: 10.1016/J.BIOMATERIALS.2019.03.011
215. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31:711. doi: 10.1016/J.CCELL.2017.04.003
216. Jiang X, Wang J, Zheng X, Liu Z, Zhang X, Li Y, et al. Original research: Intratumoral administration of STING-activating nanovaccine enhances T cell immunotherapy. J Immunother Cancer (2022) 10:3960. doi: 10.1136/JITC-2021-003960
217. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi: 10.1016/J.IMMUNI.2007.03.006
218. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest (2019) 129:4350–64. doi: 10.1172/JCI125413
219. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med (2021) 218:e20200844. doi: 10.1084/JEM.20200844/211644
220. Kuhl N, Linder A, Philipp N, Nixdorf D, Fischer H, Veth S, et al. STING agonism turns human T cells into interferon-producing cells but impedes their functionality. EMBO Rep (2023) 24:e55536. doi: 10.15252/EMBR.202255536
221. Knelson EH, Ivanova EV, Tarannum M, Campisi M, Lizotte PH, Booker MA, et al. Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol Res (2022) 10:947–61. doi: 10.1158/2326-6066.CIR-22-0017
222. Da Y, Liu Y, Hu Y, Liu W, Ma J, Lu N, et al. STING agonist cGAMP enhances anti-tumor activity of CAR-NK cells against pancreatic cancer. Oncoimmunology (2022) 11:2054105. doi: 10.1080/2162402X.2022.2054105
223. Wolf NK, Blaj C, Picton LK, Snyder G, Zhang L, Nicolai CJ, et al. Synergy of a STING agonist and an IL-2 superkine in cancer immunotherapy against MHC I–deficient and MHC I + tumors. Proc Natl Acad Sci (2022) 119:e2200568119. doi: 10.1073/PNAS.2200568119
224. Tang CHA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-mediated activation of STING induces apoptosis in Malignant B cells. Cancer Res (2016) 76:2137–52. doi: 10.1158/0008-5472.CAN-15-1885
225. Jang SC, Economides KD, Moniz RJ, Sia CL, Lewis N, McCoy C, et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun Biol (2021) 4:1–17. doi: 10.1038/s42003-021-02004-5
226. McAndrews KM, Che SPY, LeBleu VS, Kalluri R. Effective delivery of STING agonist using exosomes suppresses tumor growth and enhances antitumor immunity. J Biol Chem (2021) 296:100523. doi: 10.1016/J.JBC.2021.100523
227. Cordova AF, Ritchie C, Böhnert V, Li L. Human SLC46A2 Is the Dominant cGAMP Importer in Extracellular cGAMP-Sensing Macrophages and Monocytes. ACS Cent Sci (2021) 7:1073–88. doi: 10.1021/acscentsci.1c00440
228. Ritchie C, Cordova AF, Hess GT, Bassik MC, Li L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol Cell (2019) 75:372. doi: 10.1016/J.MOLCEL.2019.05.006
229. Dane EL, Belessiotis-Richards A, Backlund C, Wang J, Hidaka K, Milling LE, et al. STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity. Nat Mater (2022) 21:710–20. doi: 10.1038/s41563-022-01251-z
230. Whelan JT, Singaravelu R, Wang F, Pelin A, Tamming LA, Pugliese G, et al. CRISPR-mediated rapid arming of poxvirus vectors enables facile generation of the novel immunotherapeutic STINGPOX. Front Immunol (2023) 13:1050250. doi: 10.3389/fimmu.2022.1050250
231. Liu Y, Tiruthani K, Wang M, Zhou X, Qiu N, Xiong Y, et al. Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer. Nanoscale Horiz (2021) 6:319–29. doi: 10.1039/D0NH00588F
232. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature (2014) 515:130–3. doi: 10.1038/nature13862
233. Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie YQ, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol (2018) 36:707–16. doi: 10.1038/nbt.4181
234. Bu J, Nair A, Iida M, Jeong WJ, Poellmann MJ, Mudd K, et al. An avidity-based PD-L1 antagonist using nanoparticle-antibody conjugates for enhanced immunotherapy. Nano Lett (2020) 20:4901–9. doi: 10.1021/acs.nanolett.0c00953
235. Moon Y, Shim MK, Choi J, Yang S, Kim J, Yun WS, et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics (2022) 12:1999. doi: 10.7150/THNO.69119
236. Reda M, Ngamcherdtrakul W, Nelson MA, Siriwon N, Wang R, Zaidan HY, et al. Development of a nanoparticle-based immunotherapy targeting PD-L1 and PLK1 for lung cancer treatment. Nat Commun (2022) 13:1–11. doi: 10.1038/s41467-022-31926-9
237. An S, Tiruthani K, Wang Y, Xu L, Hu M, Li J, et al. Locally trapping the C-C chemokine receptor type 7 by gene delivery nanoparticle inhibits lymphatic metastasis prior to tumor resection. Small (2019) 15:1805182. doi: 10.1002/SMLL.201805182
238. Mburu YK, Wang J, Wood MA, Walker WH, Ferris RL. CCR7 mediates inflammation-associated tumor progression. Immunol Res (2006) 36:61–72. doi: 10.1385/IR:36:1:61/METRICS
Keywords: natural killer cells, cancer immunotherapy, solid tumors, tumor microenvironment, oncolytic virus, nanomedicine
Citation: Portillo AL, Monteiro JK, Rojas EA, Ritchie TM, Gillgrass A and Ashkar AA (2023) Charting a killer course to the solid tumor: strategies to recruit and activate NK cells in the tumor microenvironment. Front. Immunol. 14:1286750. doi: 10.3389/fimmu.2023.1286750
Received: 31 August 2023; Accepted: 23 October 2023;
Published: 08 November 2023.
Edited by:
Scott McComb, National Research Council Canada (NRC), CanadaReviewed by:
Arsalan Jalili, Royan institute for Stem Cell Biology and Technology (RI-SCBT), IranYunbin Ye, Fujian Cancer Hospital, China
Copyright © 2023 Portillo, Monteiro, Rojas, Ritchie, Gillgrass and Ashkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali A. Ashkar, YXNoa2FyYUBtY21hc3Rlci5jYQ==