 Ana Casado-García1,2†
Ana Casado-García1,2† Marta Isidro-Hernández1,2†
Marta Isidro-Hernández1,2† Silvia Alemán-Arteaga1,2
Silvia Alemán-Arteaga1,2 Belén Ruiz-Corzo1,2Susana Riesco3Pablo Prieto-Matos3Lucía Sánchez4
Belén Ruiz-Corzo1,2Susana Riesco3Pablo Prieto-Matos3Lucía Sánchez4 Isidro Sánchez-García1,2
Isidro Sánchez-García1,2 Carolina Vicente-Dueñas3*
Carolina Vicente-Dueñas3*- 1Experimental Therapeutics and Translational Oncology Program, Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Científicas (CSIC)/Universidad de Salamanca, Salamanca, Spain
- 2Institute for Biomedical Research of Salamanca (IBSAL), Salamanca, Spain
- 3Department of Pediatrics, Hospital Universitario de Salamanca, Institute for Biomedical Research of Salamanca (IBSAL), Salamanca, Spain
- 4School of Law, University of Salamanca, Salamanca, Spain
B-cell acute lymphoblastic leukemia (B-ALL) stands as the primary contributor to childhood cancer-related mortality on a global scale. The development of the most conventional forms of this disease has been proposed to be conducted by two different steps influenced by different types of risk factors. The first step is led by a genetic insult that is presumably acquired before birth that transforms a healthy cell into a preleukemic one, which is maintained untransformed until the second step takes place. This necessary next step to leukemia development will be triggered by different risk factors to which children are exposed after birth. Murine models that recap the stepwise progression of B-ALL have been instrumental in identifying environmental and genetic factors that contribute to disease risk. Recent evidence from these models has demonstrated that specific environmental risk factors, such as common infections or gut microbiome dysbiosis, induce immune stress, driving the transformation of preleukemic cells, and harboring genetic alterations, into fully transformed leukemic cells. Such models serve as valuable tools for investigating the mechanisms underlying preleukemic events and can aid in the development of preventive approaches for leukemia in child. Here, we discuss the existing knowledge, learned from mouse models, of the impact of genetic and environmental risk factors on childhood B-ALL evolution and how B-ALL prevention could be reached by interfering with preleukemic cells.
1 Introduction
B-cell Acute Lymphoblastic Leukemia (B-ALL), also known as B-cell precursor ALL, is one of the principal types of human leukemias. It is a clonal malignant disease that primarily affects children (1), characterized by the accumulation of blast cells that resemble the initial steps of normal B cell development.
B-ALL originates from a single cell and involves the abnormal enlargement of precursor B-cells, which exhibit phenotypic similarities to healthy precursor B-cells. There has been debate regarding the specific cell-of-origin for B-ALL, with some suggesting that it arises from a committed B cell (2). However, the exact origin of B-ALL is still a topic of ongoing discussion. Recent studies conducted in mouse models have provided insights into the development of B-cell leukemias showing that the restricted expression of an oncogene (ETV6-RUNX1 or BCR-ABL) to the hematopoietic stem/progenitor cell (HSPC) compartment of mice is capable of inducing the disease (3–5). These studies indicate that genetic modifications can act in a “hit-and-run” manner on the cell-of-origin, establishing a tumor cell character. This unique mechanism of cell transformation involves an “epigenetic priming” process initiated by the initial genetic lesion. Interestingly, this initial hit may not be necessary for the subsequent evolution and persistence of the tumor, as the epigenetic priming becomes the driving force behind tumor development (6, 7).
Despite the molecular heterogeneity of B-ALL, there are common biological characteristics shared by a significant percentage of childhood B-ALL cases. Firstly, B-ALL leukemia in child exhibits a distinct age scattering, with a highest incidence between 2 and 5 years of age, followed by a decline in rates (8, 9). Furthermore, B-ALLs generally respond well to chemotherapy, leading to significant improvements in survival rates (reaching nearly 90%) among affected children over the past five decades (1). However, it is important to note that existing treatments are often associated with significant toxicity and morbidity (10) and between 10% and 20% of patients, who have achieved complete remission after initial treatment for ALL, will relapse and will have worse outcomes (11). Prior to being able to present novel therapeutic strategies for children, or ideally, institute preventive measures to hinder the onset of leukemia, it is imperative to comprehend the biological processes underlying the disease, and to this ambition, modelling the disease in mice is crucial.
One of the most notable biological features of many childhoods B-ALL types is the presence of a latent silent preleukemic stage. During this phase, the initial event that triggers leukemia is present, yet the actual development of leukemia remains dormant and unapparent. This preleukemic situation can be found in up to 5% of healthy children, yet only a minority fraction (less than 1%) among the susceptible children will progress to develop B-ALL (12, 13). So, it is accepted that for the most common forms of B-ALL follow a “two-step” model of B-ALL development, from the preleukemic step to the leukemic one. It is likely that certain external or environmental factors play a role in triggering the progression from the preleukemic stage to full-blown disease (Figure 1). In recent decades, the incidence of B-ALL has shown an increase, which appears to be associated with modern lifestyle changes or exposures (14). Several epidemiological studies have shown that certain environmental factors, as is the case of exposure to infectious agents during infancy, are associated with childhood B-ALL (15–17). However, the practical prerequisites needed to obtain clear-cut solutions to the proposed epidemiological theories can only be met through experimentation in animal models. In this context, the creation of mouse models emulating the initiation of B-ALL has played a pivotal role in identifying the environmental and genetic elements that support disease risk.
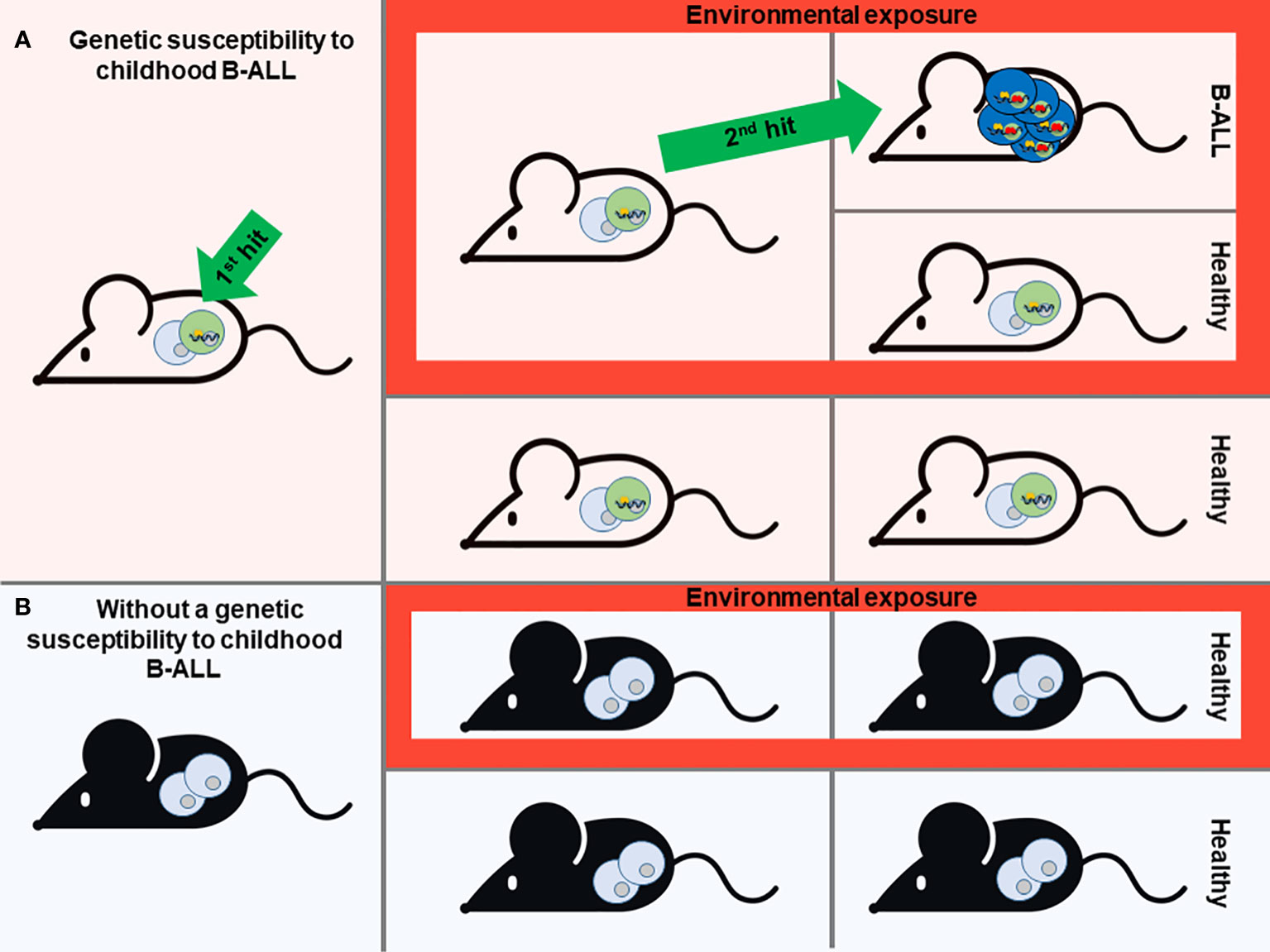
Figure 1 Modeling of Childhood B-ALL development. A prevailing theory for most B-ALL cases is the “Two-hit” model. This model suggests that the development of B-ALL involves two key genetic events. The first event, often acquired before birth (the first hit), initiates the formation of preleukemic cells. Subsequently, a second genetic mutation occurs after birth (the second hit), which drives the full development of B-ALL. Mouse models that mimic the stepwise progression of B-ALL are instrumental in identifying environmental and genetic factors that contribute to disease risk (A). Based on this scenario, different genetic susceptibilities to childhood leukemia (first hit) have been replicated in mice, giving rise to preleukemic cells that are susceptible to transformation (A). In the absence of this genetic susceptibility, mice will never develop B-ALL although they are exposed to environmental factors associated with this type of leukemia (B). However, not all individuals who carry the genetic susceptibility to childhood B-ALL will ultimately develop the disease, despite exposure to environmental triggers. This phenomenon remains enigmatic (A). Mouse modeling is essential to understand this issue and to identify environmental or other factors that have a direct association with the heightened susceptibility to this disease.
In vivo research using mouse models incorporates all levels of organization relevant to the occurrence of the disease, ranging from the intricate structure of organs and their various cell types to the overall physiological condition of the organism. Furthermore, the genetic similarity among mice in these models provides scientists with precise control over genome alterations. These animal models have played a significant role in advancing our comprehension of the natural progression of B-ALL, spanning from the preleukemic phase to the totally transformed phase. In this context, some experimental models cited in this review have demonstrated both similarities in physical characteristics (phenotype) and genetic traits (genotype) with the human disease they are replicating. However, some of them are of limited value for environmental risk factors identification as the disease incidence cannot be consistently increased or decreased by exposure to those external agents that this disease is associated with.
2 Risk factors and B-ALL initiation
As we have already mentioned, leukemia stands as the primary contributor to childhood cancer-related deaths globally, and within this category, B-ALL ranks as the most prevalent subtype (11, 18). While the precise origins of acute leukemias remain unclear, it is widely acknowledged that several factors increase susceptibility to these blood disorders. These influential elements encompass both genetic and environmental components (19, 20).
The aforementioned “two-step” model of B-ALL development serves as the essential foundation for modelling the illness in mice. Consequently, the model should encompass the initial triggering event, which can either be acquired during prenatal development through somatic mutations or as a constitutional germline genetic variant. The prenatal origin of this disease is particularly true for germline mutations and inferred for somatic mutations based on studies from monozygotic twins carrying identical genetic alterations (21). While the second event can also be incorporated into mouse models, it holds greater significance to utilize mouse models that solely harbor the first oncogenic event. This approach enables the evaluation of potential risk factors that facilitate the natural occurrence of additional events. (Figure 1).
2.1 Genetic risk factors modelled in mice
Numerous pieces of evidence point to the involvement of genetic factors in the development of acute leukemias. Although the process of leukemogenesis remains partially incomplete, in recent years there have been substantial advancements in understanding the mechanisms responsible for the malignant conversion of hematopoietic precursor cells. However, despite these strides, much of this knowledge has yet to translate into direct benefits for patients. Utilizing mouse models that faithfully replicate the genetic predisposition to childhood leukemia has increased our knowledge of the malignant transformation mechanism behind this disease and can significantly contribute to unravelling the intricate connections between leukemia and environmental factors.
The genomic landscape of pediatric leukemia patients has undergone comprehensive characterization (22–25). In this context, in pursuit of comprehending the disease’s etiology, numerous mouse models have been created, each harboring distinct genetic alterations (germline or prenatally acquired somatic mutations) analogous to those found in humans (7).
An increasing array of genetic changes impacting B-cell transcription factors has been uncovered. These changes either directly trigger or make individuals more susceptible to the development of B-ALL. (22–24, 26–29). One illustrative instance is the PAX5 gene, a pivotal transcription factor governing B-cell identity; approximately 30% of pediatric B-ALL cases exhibit somatic mutations or deletions impacting this gene (22, 24). Accordingly, several models have been developed altering the expression of Pax5 in mice that subsequently develop B-ALL (30–36). Somatic alterations involving Pax5, emulated in mice (e.g. PAX5-JAK2, PAX5-ELN or PAX5ETV6/+), achieve almost complete penetrance of nearly 100% (33, 34, 36). Interestingly, germline Pax5 mutations modeled in mice (e.g. Pax5+/- mice) display an incomplete penetrance (32), mirroring the scenario observed in humans (26, 27, 29). Similarly, IL7R activating mutations (which is essential for lymphoid development) are also frequent in B-ALL. Mice harbouring these mutations have been made and acquire B-ALL with also a variable penetrance depending on the cell-of-origin in which the mutation is expressed (37–39). When the IL7R mutational activation occurred from the CLP stage and in homozygosis the incidence in nearly 100% (37).
Chromosomal translocation stands as one of the most prevalent genetic aberrations observed in B-ALL, with the ETV6-RUNX1 fusion gene (t(12;21)(p13;q22)) being the most frequently encountered (40). While attempts to model the ETV6-RUNX1 translocation in mice have been made, the majority of these models have failed to induce leukemia due to the expression of the fusion gene in committed B-cells (41–46). However, restricting the expression of ETV6-RUNX1 to more immature, malleable hematopoietic cells, particularly murine stem cells, can lead to the development of childhood B-ALL under environmental pressures, such as common infection exposures (3). B-ALL instigated by either the E2A-PBX1 (t(1;19)(q23;p13)) or the BCR-ABL (t(9;22)(q34;q11)) fusion genes has also been successfully modeled in mice (3, 47–51), albeit with varying penetrance. The discrepancy in penetrance observed among studies, despite expressing the same genetic alteration, relies on the fact that each study used different promoters and the cell of origin in which this promoter is active can differ a lot. The best mouse model will be the one that expresses the mutation in the right cell of origin and mimics the human pathology despite not having a complete penetrance.
The creation of these mouse models underscores the paramount importance of not only precisely identifying the genetic mutations existing in humans but also determining the specific cell-of-origin responsible for the emergence of B-ALL. This process aids in tailoring targeted interventions, ultimately culminating in the generation of an authentic and valuable mouse model for childhood B-ALL.
2.2 Environmental risk factors modelled in mice
In the most common forms of B-ALL, a genetic alteration, either acquired in utero or as a constitutional germline genetic variant, will give rise to the creation of preleukemic clones. These preleukemic cells could be present in healthy carriers for their whole life, but in fewer cases, further postnatal alterations will trigger the second hit and transform the preleukemic cells into leukemic ones (19, 52). There is a huge number of epidemiological studies on environmental/perinatal risk factors (e.g. exposure to common infections, low doses of ionizing radiation, pesticide exposure, living in proximity to nuclear facilities, maternal intake of fertility treatment, high birth weight (≥4000 g), caesarean delivery etc.) that are linked to ALL (53–58), but few of them are established as risk factors due to the lack of biological pieces of evidence.
There are only a few instances in which solid biological or mechanistic evidence supports the epidemiological connection between a risk factor and paediatric B-ALL. The first recognized environmental risk factor for childhood B-ALL is the exposure to higher levels of ionizing radiation (IR). The primary support for this correlation originates from studies involving atomic bomb survivors in Hiroshima and Nagasaki (59, 60), as well as research into the consequences of IR utilization for both therapy and diagnostics (61).
The second, and so far, the last environmental risk factor case with robust biological or mechanistic evidence, is the exposure to infectious agents and the role of immune function as a risk factor for B-ALL development. In the context of this multi-step disease, the second step occur perinatally or during infancy and can be prompted by infectious agents challenging the already dysregulated immune system due to the presence of a genetic predisposition, which constitutes the first step. In this scenario, the biological evidence that endorses the epidemiological association came from the use of mouse models that faithfully mimic B-ALL (3, 32). Two mouse models has been used to prove the interaction between infection and B-ALL development, the Pax5+/- and the Sca1-ETV6-RUNX1 mice (3, 32) that replicate the initial phase responsible for the predisposition to B-ALL observed in certain human patients. Consistent with the "two-step" model, Pax5+/- and the Sca1-ETV6-RUNX1 mice housed exclusively in a specific pathogen-free (SPF) environment never developed B-ALL despite harbouring the genetic predisposition; however, when these predisposed mice were moved to a conventional facility where they are exposure to infectious agents, 22% of Pax5+/- and 10% of Sca1-ETV6-RUNX1 mice developed B-ALL (3, 32). These discoveries offer the initial scientific evidence that exposure to infections could serve as a catalyst for the development of B-ALL in individuals with genetic predispositions. Moreover, the specific genetic changes linked to the progression to leukemia in these mouse models closely resemble those observed in human patients (3, 26, 27, 29, 34), further supporting the idea that these models faithfully recapitulate the human pathology and how valuable they are to test environmental risk factors. In relation to this environmental role in leukemia development, mouse model studies must be very exhaustive when detailing the housing conditions to which the animals are exposed since this can greatly affect the final result and reproducibility.
However, not all B-ALL genetic predisposition situations adhere to this mechanism, as demonstrated by the situation of B-ALLs containing the BCR-ABLp190 oncogene. In mouse models, the development of BCR-ABLp190+ B-ALL occurs independently of infection (4), this aligns with the observation that in humans, this particular subtype of B-ALL is rarely seen in children (62, 63). Moreover, chromosomal translocations linked to distinct infant leukemias, such as MLL, appear capable of inducing full-fledged leukemic progression without the requirement of subsequent events. Instead, alternative molecular mechanisms seem to be employed to achieve these outcomes and seems to be independent of environmental factors (64–66).
3 Mechanisms of action of B-ALL risk factors
3.1 Epigenetic priming as cell transformation process
A recently accepted justification has shed light on the relationship between the distinct immunophenotype observed in childhood ALL (B- or T-ALL) and the precise underlying genetic disorder responsible for its development. B-ALL and T-ALL exhibit unique genetic alterations that are exclusive to each of these biological entities, resulting in a strong connection between genotype and phenotype. Traditionally, it was believed that the pivotal genetic event occurred in a committed B- or T-cell, explaining the correlation with the immunophenotype. However, current findings propose an alternative perspective, suggesting that the initial genetic disruption has the ability to induce genetic priming in the cell-of-origin, thereby dictating the definitive phenotype of the transformed cell. In this novel notion of the transformation mechanism, the first abnormal genetic alteration imposes a specific cell-differentiation program in the cancer cell-of-origin, determining whether the resulting tumor cells will exhibit a B-cell or T-cell phenotype. Importantly, the primary oncogenic modification, while essential for tumor beginning, becomes dispensable once the differentiation program is activated (3–5, 67). Consequently, in later stages of transformation, the oncogene loses its necessity and does not play a critical role in tumor progression. As a consequence, targeting this oncogene for therapies may not be effective since it lacks significance during the later phases of tumor development. This epigenetic reprogramming phenomenon has been demonstrated in various animal cancer models that closely resemble human disease quite well (6).
An open question is whether epigenetic priming may also respond to specific environmental factors in the context of particular genetic susceptibility. In this context, a “gene–environment interaction” will increase the sensitivity to a given environmental exposure as a result of which a second hit will appear in a preleukemic cell that will drive leukemia formation. Mouse models will be instrumental in deciphering this issue.
3.2 Immune stress
We have previously described that the presence of infectious stimuli promotes leukemia in genetically engineered murine models that faithfully recapitulate childhood B-ALL caused by specific genetic predisposition (Pax5+/- and Sca1-ETV6-RUNX1 mice) (3, 32). The infection exposure promotes the immune stress capable of triggering preleukemia-to-leukemia conversion within an immune-dysregulated system. The immune stress is defined as the stress experienced during immune activation (e.g. as a result of exposure to infections) that boosts innate and adaptive immune responses. This includes changes in hematopoietic cell balance as well as local and systemic production of cytokines. Consequently, Pax5+/- mice exhibit a significant higher proportion of (pro and pre)-B and immature B cells in the bone marrow (BM), coupled with a reduced amount of total B cells in the peripheral blood (PB) (32). Interestingly, Pax5+/- BM precursors display a remarkable sensitivity to IL-7 cytokine withdrawal, underscoring a potential cell vulnerability. These findings imply that having only one functional copy of the Pax5 gene (Pax5 heterozygosity) promotes the emergence of an abnormal B-cell precursor compartment in the BM. This impairs their ability to develop into mature peripheral blood B cells. Moreover, the immune stress incited by infection stimuli further fosters the acquisition of secondary mutations within this vulnerable B cell population.
Similarly, in the Sca1-ETV6-RUNX1 mouse model, exposure to common pathogens induces a temporal expansion of an aberrant B-cell precursor compartment within the BM (3). These amplified preleukemic cells display heightened expression of Rag1 and Rag2 (RAGs: recombination-activating genes, critical for generating mature B cells and T cells) consequently promoting the acquisition of secondary mutations (3).
However, this immune stress can be promoted not only by infectious stimuli but also by other mechanisms. An example of this is the gut microbiome dysbiosis. Recent data from both Pax5+/- and ETV6-RUNX1+ mice show that the genetic predisposition is firmly associated with changes in the gut microbiome as the genetic predisposition in each case can be predicted just based on the murine gut microbiome composition, irrespective of whether the animals develop B-ALL or not (68). The gut microbiome might serve as an integration hub for environmental signals that interact with the immune system, consequently influencing the risk of developing B-ALL. Experiments conducted on predisposed mice have provided evidence that perturbation of the gut microbiome caused by antibiotic treatment promotes B-cell ALL development, even in the absence of infectious stimuli (68). This indicates a protective role for the gut microbiome and suggests that its dysbiosis could constitute another form of immune stress capable of triggering B-ALL within a genetically predisposed context.
As a result, we have learned from mouse models that different immunological stressors are linked to the conversion of preleukemic B cells to B-ALL. Recent experiments involving a murine model have dived deeper into this mechanism of immune stress, indicating that dysregulation of innate immune signaling within preleukemic precursor B cells plays a pivotal role in driving this immune stress, ultimately culminating in leukemia development (69). Specifically, Myd88, a central player in immune cell activation via Toll-like receptors (TLRs), undergoes downregulation in immune-stressed pre-malignant cells, and through an inflammation-dependent mechanism, contributes to the onset of leukemia (69).
To sum up, we have learned from murine models that some environmental factors stimulate immune stress that favors the full transformation of a preleukemic cell (a healthy cell with a genetic predisposition) by perturbing innate immune signaling regulation.
4 Targeting B-ALL promotion: toward B-ALL prevention
It is difficult to avoid exposure to common environmental factors such as exposure to infections, but eliminating the preleukemic cells susceptible to transformation becomes plausible to prevent the development of B-ALL.
A fundamental aspect to bear in mind toward B-ALL prevention is that the evolution from preleukemic cells to leukemic ones is led by the acquisition of secondary mutations as a result of environmental factors. The exposures do not facilitate the expansion of preleukemic clones already containing one or more second hits as has been demonstrated in previous mouse studies (32, 70). The second mutation appears just before B-ALL transformation, indeed if the first and the second hit are induced at the same time in the cell (e.g. Pax5 and Jak3 mutations), B-ALL development is immediate and as a consequence cannot be preventable (71). As the external factors initiate the emergence of these secondary mutations, this process could be prevented by eliminating the preleukemic cells before they fully transform (14, 72). In this regard, it is essential to explore methods for precisely targeting these preleukemic cells (Figure 2).
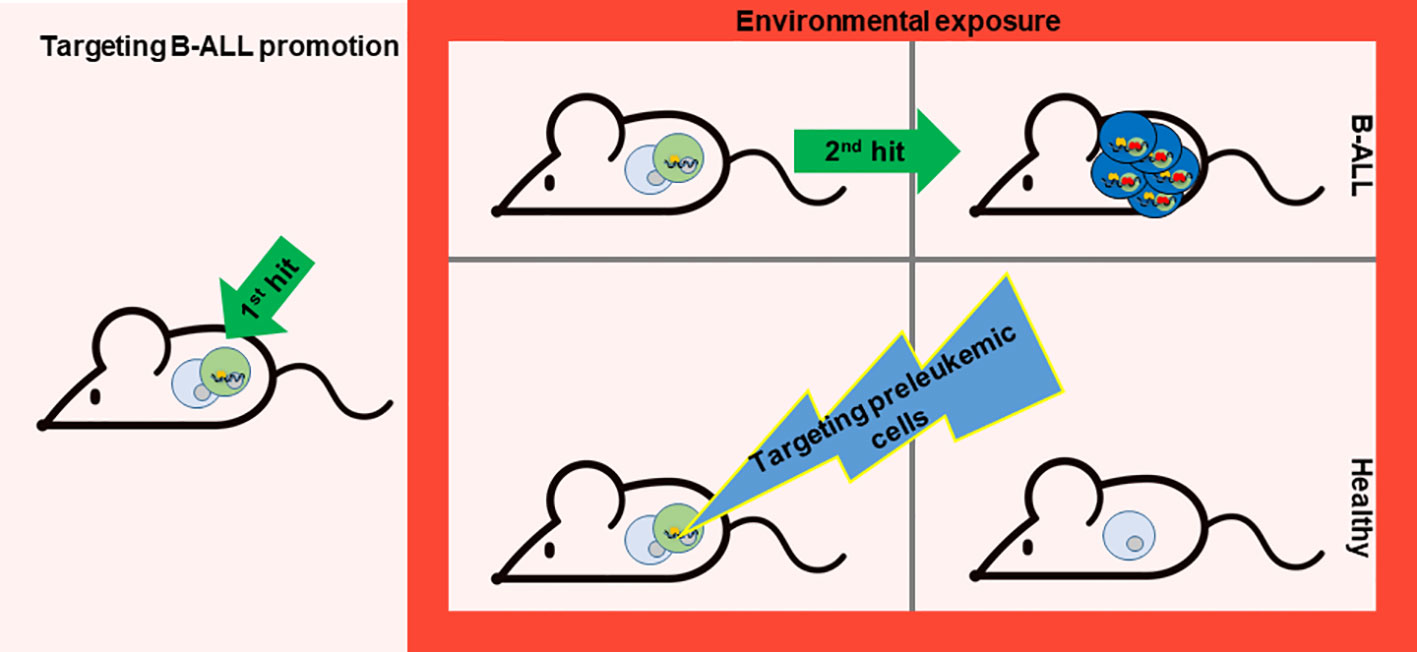
Figure 2 Targeting B-ALL progression. While evading exposure to environmental factors is challenging, the possibility of preventing B-ALL development lies in the potential elimination of preleukemic cells prone to transformation. Nonetheless, the key lies in discovering a method to specifically target these preleukemic cells. A recent study has paved the way for such a therapeutic approach (71). This proof-of-concept experiment was carried out in a mouse model that mirrors the characteristics of a leukemia-predisposition syndrome (Pax5+/- mice), which develops B-ALL following exposure to common infections. Temporary administration of the Jak1/2 inhibitor ruxolitinib to Pax5+/− mice significantly diminishes the risk of leukemia occurrence, owing to the reduction of preleukemic cells.
A recent study has opened the path for this kind of therapy. This proof-of-principle experiment has been conducted in a mouse model that recapitulates the phenotype of a leukemia-predisposition syndrome (Pax5+/- mice) that develop B-ALL upon exposure to common infections. Short-term administration of the Jak1/2 inhibitor ruxolitinib to Pax5+/− mice substantially decreases the likelihood of leukemia development by reducing the population of preleukemic cells (71). This approach is effective because Pax5+/− mice are especially dependent on the cytokine interleukin-7 (IL-7) for their survival, and impeding IL-7-induced signaling using the JAK1/2 inhibitor ruxolitinib led to augmented cell death in vitro (32). Based on these findings, Pax5+/− mice treated with ruxolitinib-containing chow for 28 days early in life eliminate preleukemic cells in vivo and thus avoid B-ALL development compared to mice fed with control chow (71) (Figure 2). All the mice in the experiment were exposed to common mouse pathogens thus the oncogenic environment persist, as this factor is difficult to avoid for prevention. Interestingly, in vivo, the administration of ruxolitinib exhibited a higher affinity for eliminating Pax5+/− B-cell progenitors compared to their wild-type (WT) counterparts (71). Thus, this is the first prevention approach for leukemia development as it is able to selectively eliminate preleukemic cells before transformation.
This strategy of eliminating preleukemic cells by the JAK-STAT inhibition using ruxolitinib indicates the potential utility of a similar strategy for children who are susceptible to developing B-ALL due to PAX5 genetic susceptibility or other inherited mutations that render the preleukemic cells reliant on this signaling pathway. Given that some patients with germline mutations remain healthy into late age (26, 27, 29) the identification of biomarkers that can identify those healthy carriers that will develop leukemia will be essential to select those high-risk carriers who will benefit from interventional preventive therapies. In this regard, mouse models will be instrumental in identifying those biological markers that distinguish between those carriers that will develop the disease from the ones that will remain healthy despite carrying the genetic predisposition.
5 Future perspective
As mentioned earlier, there are various mouse models designed to replicate B-ALL through the manipulation of genetic mutations observed in humans. However, only a limited number of these models have proven effective in identifying environmental risk factors associated with the disease. This is primarily because most models do not inherently emulate the “multi-stage” progression characteristic of the majority of B-ALL subtypes, nor do they accurately represent the specific cellular origins where these genetic mutations occur.
Furthermore, B-ALL is marked by genetic diversity in the preleukemic cells’ predisposition, and the subsequent events that trigger disease progression also exhibit considerable variation. As a result, mouse models aiming to simulate this disease should take into account these crucial elements. B-ALL mouse models fulfilling these tasks will serve as valuable tools to assess preventive therapies to avoid leukemia development. Moreover, these models will offer valuable insights into the reasons behind the presence of healthy individuals who carry preleukemic cells but never progress to develop the disease. They will also aid in elucidating whether specific environmental factors, within the context of specific genetic susceptibilities, can initiate an epigenetic priming process that ultimately propels leukemia formation.
Author contributions
AC-G: Writing – review & editing, Writing – original draft. MI-H: Writing – review & editing, Writing – original draft. SA-A: Writing – review & editing, Writing – original draft. BR-C: Writing – review & editing, Writing – original draft. SR: Writing – review & editing, Writing – original draft. PP-M: Writing – review & editing, Writing – original draft. LS: Writing – review & editing, Writing – original draft. IS-G: Writing – review & editing, Funding acquisition, Writing – original draft. CV-D: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Research in CV-D group has been funded by Instituto de Salud Carlos III (ISCIII) through the project PI22/00379 and co-funded by the European Union (European Regional Development Fund (ERDF)/European Social Fund (ESF)). Research in IS-G group is partially supported by FEDER and by SAF2015-64420-R MINECO/FEDER, UE; by RTI2018-093314-B-I00 MCIU/AEI/FEDER, UE; by PID2021-122185OB-I00 MCIU/AEI/FEDER, UE, and by Junta de Castilla y León (UIC-017, CSI001U16, CSI234P18, and CSI144P20), by the Fundacion Unoentrecienmil (CUNINA project), and by the Fundación Científica de la Asociación Española contra el Cáncer (PRYCO211305SANC). AC-G (CSI067-18), MI-H (CSI021-19) and BR-C are supported by FSE-Conserjería de Educación de la Junta de Castilla y León 2019, 2020, and 2022 (ESF, European Social Fund) fellowship, respectively. SA-A is supported by an Ayuda para Contratos predoctorales para la formación de doctores (PRE2019-088887). LS is supported by a scholarship from the University of Salamanca co-financed by Banco Santander and ESF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet (2013) 381(9881):1943–55. doi: 10.1016/S0140-6736(12)62187-4
2. le Viseur C, Hotfilder M, Bomken S, Wilson K, Rottgers S, Schrauder A, et al. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell (2008) 14(1):47–58. doi: 10.1016/j.ccr.2008.05.015
3. Rodriguez-Hernandez G, Hauer J, Martin-Lorenzo A, Schafer D, Bartenhagen C, Garcia-Ramirez I, et al. Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia via impaired H3K4 demethylases. Cancer Res (2017) 77(16):4365–77. doi: 10.1158/0008-5472.CAN-17-0701
4. Martin-Lorenzo A, Auer F, Chan LN, Garcia-Ramirez I, Gonzalez-Herrero I, Rodriguez-Hernandez G, et al. Loss of pax5 exploits sca1-BCR-ABL(p190) susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res (2018) 78(10):2669–79. doi: 10.1158/0008-5472.CAN-17-3262
5. Vicente-Duenas C, Gonzalez-Herrero I, Sehgal L, Garcia-Ramirez I, Rodriguez-Hernandez G, Pintado B, et al. Dnmt1 links BCR-ABLp210 to epigenetic tumor stem cell priming in myeloid leukemia. Leukemia (2019) 33(1):249–78. doi: 10.1038/s41375-018-0192-z
6. Raboso-Gallego J, Casado-Garcia A, Isidro-Hernandez M, Vicente-Duenas C. Epigenetic priming in childhood acute lymphoblastic leukemia. Front Cell Dev Biol (2019) 7:137. doi: 10.3389/fcell.2019.00137
7. Isidro-Hernandez M, Aleman-Arteaga S, Casado-Garcia A, Ruiz-Corzo B, Riesco S, Prieto-Matos P, et al. Childhood B-cell preleukemia mouse modeling. Int J Mol Sci (2022) 23(14):7562. doi: 10.3390/ijms23147562
8. Greaves MF, Pegram SM, Chan LC. Collaborative group study of the epidemiology of acute lymphoblastic leukaemia subtypes: background and first report. Leuk Res (1985) 9(6):715–33. doi: 10.1016/0145-2126(85)90281-4
9. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
10. Yen HJ, Eissa HM, Bhatt NS, Huang S, Ehrhardt MJ, Bhakta N, et al. Patient-reported outcomes in survivors of childhood hematologic Malignancies with hematopoietic stem cell transplant. Blood (2020) 135(21):1847–58. doi: 10.1182/blood.2019003858
11. Pui CH, Yang JJ, Bhakta N, Rodriguez-Galindo C. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc Health (2018) 2(6):440–54. doi: 10.1016/S2352-4642(18)30066-X
12. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U.S.A. (2002) 99(12):8242–7. doi: 10.1073/pnas.112218799
13. Schafer D, Olsen M, Lahnemann D, Stanulla M, Slany R, Schmiegelow K, et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood (2018) 131(7):821–6. doi: 10.1182/blood-2017-09-808402
14. Cobaleda C, Vicente-Duenas C, Sanchez-Garcia I. Infectious triggers and novel therapeutic opportunities in childhood B cell leukaemia. Nat Rev Immunol (2021) 21(9):570–81. doi: 10.1038/s41577-021-00505-2
16. Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia (1988) 2(2):120–5.
17. Kinlen L. Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet (1988) 2(8624):1323–7. doi: 10.1016/S0140-6736(88)90867-7
18. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. New Engl J Med (2004) 350(15):1535–48. doi: 10.1056/NEJMra023001
19. Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer (2018) 18(8):471–84. doi: 10.1038/s41568-018-0015-6
20. Schmidt JA, Hornhardt S, Erdmann F, Sanchez-Garcia I, Fischer U, Schuz J, et al. Risk factors for childhood leukemia: radiation and beyond. Front Public Health (2021) 9:805757. doi: 10.3389/fpubh.2021.805757
21. Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci U.S.A. (1998) 95(8):4584–8. doi: 10.1073/pnas.95.8.4584
22. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature (2007) 446(7137):758–64. doi: 10.1038/nature05690
23. Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematol Am Soc Hematol Educ Program (2014) 2014(1):174–80. doi: 10.1182/asheducation-2014.1.174
24. Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet (2019) 51(2):296–307. doi: 10.1038/s41588-018-0315-5
25. Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol (2019) 16(4):227–40. doi: 10.1038/s41571-018-0136-6
26. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet (2013) 45(10):1226–31. doi: 10.1038/ng.2754
27. Auer F, Ruschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia (2014) 28(5):1136–8. doi: 10.1038/leu.2013.363
28. Yazdanparast S, Khatami SR, Galehdari H, Jaseb K. One missense mutation in exon 2 of the PAX5 gene in Iran. Genet Mol Res (2015) 14(4):17768–75. doi: 10.4238/2015.December.22.1
29. Duployez N, Jamrog LA, Fregona V, Hamelle C, Fenwarth L, Lejeune S, et al. Germline PAX5 mutation predisposes to familial B-cell precursor acute lymphoblastic leukemia. Blood (2021) 137(10):1424–8. doi: 10.1182/blood.2020005756
30. Heltemes-Harris LM, Willette MJ, Ramsey LB, Qiu YH, Neeley ES, Zhang N, et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med (2011) 208(6):1135–49. doi: 10.1084/jem.20101947
31. Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood (2015) 125(23):3609–17. doi: 10.1182/blood-2015-02-626127
32. Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of pax5-inherited susceptibility. Cancer Discovery (2015) 5(12):1328–43. doi: 10.1158/2159-8290.CD-15-0892
33. Smeenk L, Fischer M, Jurado S, Jaritz M, Azaryan A, Werner B, et al. Molecular role of the PAX5-ETV6 oncoprotein in promoting B-cell acute lymphoblastic leukemia. EMBO J (2017) 36(6):718–35. doi: 10.15252/embj.201695495
34. Jamrog L, Chemin G, Fregona V, Coster L, Pasquet M, Oudinet C, et al. PAX5-ELN oncoprotein promotes multistep B-cell acute lymphoblastic leukemia in mice. Proc Natl Acad Sci U.S.A. (2018) 115(41):10357–62. doi: 10.1073/pnas.1721678115
35. Boast B, Helian K, Andrews TD, Li X, Cho V, Mosquera AC, et al. Dysregulation of PAX5 causes uncommitted B cell development and tumorigenesis in mice. bioRxiv (2021). doi: 10.1101/2021.01.29.428877
36. Jurado S, Fedl AS, Jaritz M, Kostanova-Poliakova D, Malin SG, Mullighan CG, et al. The PAX5-JAK2 translocation acts as dual-hit mutation that promotes aggressive B-cell leukemia via nuclear STAT5 activation. EMBO J (2022) 41(7):e108397. doi: 10.15252/embj.2021108397
37. Almeida ARM, Neto JL, Cachucho A, Euzebio M, Meng X, Kim R, et al. Interleukin-7 receptor alpha mutational activation can initiate precursor B-cell acute lymphoblastic leukemia. Nat Commun (2021) 12(1):7268. doi: 10.1038/s41467-021-27197-5
38. Geron I, Savino AM, Fishman H, Tal N, Brown J, Turati VA, et al. An instructive role for Interleukin-7 receptor alpha in the development of human B-cell precursor leukemia. Nat Commun (2022) 13(1):659. doi: 10.1038/s41467-022-28218-7
39. Thomas KR, Allenspach EJ, Camp ND, Wray-Dutra MN, Khim S, Zielinska-Kwiatkowska A, et al. Activated interleukin-7 receptor signaling drives B-cell acute lymphoblastic leukemia in mice. Leukemia (2022) 36(1):42–57. doi: 10.1038/s41375-021-01326-x
40. Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia (1995) 9(12):1985–9.
41. Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet (2001) 130(2):93–104. doi: 10.1016/S0165-4608(01)00518-0
42. Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood (2004) 103(10):3890–6. doi: 10.1182/blood-2003-10-3695
43. Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc Natl Acad Sci U.S.A. (2004) 101(22):8443–8. doi: 10.1073/pnas.0402063101
44. Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, Roscher S, et al. Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene (2005) 24(51):7579–91. doi: 10.1038/sj.onc.1208931
45. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-beta pathway in early B lineage progenitor cells. J Clin Invest (2009) 119(4):826–36. doi: 10.1172/JCI36428
46. Kantner HP, Warsch W, Delogu A, Bauer E, Esterbauer H, Casanova E, et al. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia (2013) 15(11):1292–300. doi: 10.1593/neo.131310
47. Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature (1990) 344(6263):251–3. doi: 10.1038/344251a0
48. Castellanos A, Pintado B, Weruaga E, Arevalo R, Lopez A, Orfao A, et al. A BCR-ABL(p190) fusion gene made by homologous recombination causes B-cell acute lymphoblastic leukemias in chimeric mice with independence of the endogenous bcr product. Blood (1997) 90(6):2168–74. doi: 10.1182/blood.V90.6.2168
49. Bijl J, Sauvageau M, Thompson A, Sauvageau G. High incidence of proviral integrations in the Hoxa locus in a new model of E2a-PBX1-induced B-cell leukemia. Genes Dev (2005) 19(2):224–33. doi: 10.1101/gad.1268505
50. Virely C, Moulin S, Cobaleda C, Lasgi C, Alberdi A, Soulier J, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia (2010) 24(6):1200–4. doi: 10.1038/leu.2010.63
51. Duque-Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, et al. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J Clin Invest (2015) 125(9):3667–80. doi: 10.1172/JCI81158
52. Fregona V, Bayet M, Gerby B. Oncogene-induced reprogramming in acute lymphoblastic leukemia: towards targeted therapy of leukemia-initiating cells. Cancers (Basel) (2021) 13(21):5511. doi: 10.3390/cancers13215511
53. Milne E, Greenop KR, Metayer C, Schuz J, Petridou E, Pombo-de-Oliveira MS, et al. Fetal growth and childhood acute lymphoblastic leukemia: findings from the childhood leukemia international consortium. Int J Cancer (2013) 133(12):2968–79. doi: 10.1002/ijc.28314
54. O'Neill KA, Murphy MF, Bunch KJ, Puumala SE, Carozza SE, Chow EJ, et al. Infant birthweight and risk of childhood cancer: international population-based case control studies of 40 000 cases. Int J Epidemiol (2015) 44(1):153–68. doi: 10.1093/ije/dyu265
55. Paltiel O, Tikellis G, Linet M, Golding J, Lemeshow S, Phillips G, et al. Birthweight and childhood cancer: preliminary findings from the international childhood cancer cohort consortium (I4C). Paediatr Perinat Epidemiol (2015) 29(4):335–45. doi: 10.1111/ppe.12193
56. Schuz J, Erdmann F. Environmental exposure and risk of childhood leukemia: an overview. Arch Med Res (2016) 47(8):607–14. doi: 10.1016/j.arcmed.2016.11.017
57. Jimenez-Hernandez E, Fajardo-Gutierrez A, Nunez-Enriquez JC, Martin-Trejo JA, Espinoza-Hernandez LE, Flores-Lujano J, et al. A greater birthweight increases the risk of acute leukemias in Mexican children-experience from the Mexican Interinstitutional Group for the Identification of the Causes of Childhood Leukemia (MIGICCL). Cancer Med (2018) 7(4):1528–36. doi: 10.1002/cam4.1414
58. Onyije FM, Olsson A, Baaken D, Erdmann F, Stanulla M, Wollschlager D, et al. Environmental risk factors for childhood acute lymphoblastic leukemia: an umbrella review. Cancers (Basel) (2022) 14(2):382. doi: 10.3390/cancers14020382
59. Preston DL, Kusumi S, Tomonaga M, Izumi S, Ron E, Kuramoto A, et al. Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma 1950-1987. Radiat Res (1994) 137(2 Suppl):S68–97.
60. Walsh L, Kaiser JC. Multi-model inference of adult and childhood leukaemia excess relative risks based on the Japanese A-bomb survivors mortality data, (1950-2000). Radiat Environ Biophys (2011) 50(1):21–35. doi: 10.1007/s00411-010-0337-6
61. Wakeford R. The risk of childhood leukaemia following exposure to ionising radiation–a review. J Radiol Prot (2013) 33(1):1–25. doi: 10.1088/0952-4746/33/1/1
62. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature (2008) 453(7191):110–4. doi: 10.1038/nature06866
63. Pui CH, Roberts KG, Yang JJ, Mullighan CG. Philadelphia chromosome-like acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk (2017) 17(8):464–70. doi: 10.1016/j.clml.2017.03.299
64. Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol (2011) 152(2):141–54. doi: 10.1111/j.1365-2141.2010.08459.x
65. Sanjuan-Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, et al. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood (2015) 126(25):2676–85. doi: 10.1182/blood-2015-09-667378
66. Hyrenius-Wittsten A, Pilheden M, Sturesson H, Hansson J, Walsh MP, Song G, et al. De novo activating mutations drive clonal evolution and enhance clonal fitness in KMT2A-rearranged leukemia. Nat Commun (2018) 9(1):1770. doi: 10.1038/s41467-018-04180-1
67. Garcia-Ramirez I, Bhatia S, Rodriguez-Hernandez G, Gonzalez-Herrero I, Walter C, Gonzalez de Tena-Davila S, et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J (2018) 37(14):(e98783. doi: 10.15252/embj.201798783
68. Vicente-Duenas C, Janssen S, Oldenburg M, Auer F, Gonzalez-Herrero I, Casado-Garcia A, et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood (2020) 136(18):2003–17. doi: 10.1182/blood.2019004381
69. Isidro-Hernandez M, Casado-Garcia A, Oak N, Aleman-Arteaga S, Ruiz-Corzo B, Martinez-Cano J, et al. Immune stress suppresses innate immune signaling in preleukemic precursor B-cells to provoke leukemia in predisposed mice. Nat Commun (2023) 14(1):5159. doi: 10.1038/s41467-023-40961-z
70. Rodriguez-Hernandez G, Opitz FV, Delgado P, Walter C, Alvarez-Prado AF, Gonzalez-Herrero I, et al. Infectious stimuli promote Malignant B-cell acute lymphoblastic leukemia in the absence of AID. Nat Commun (2019) 10(1):5563. doi: 10.1038/s41467-019-13570-y
71. Casado-Garcia A, Isidro-Hernandez M, Oak N, Mayado A, Mann-Ran C, Raboso-Gallego J, et al. Transient inhibition of the JAK/STAT pathway prevents B-ALL development in genetically predisposed mice. Cancer Res (2022) 82(6):1098–109. doi: 10.1158/0008-5472.CAN-21-3386
Keywords: childhood leukemia, B-ALL, mouse models, risk factors, genetic predisposition, environmental factors
Citation: Casado-García A, Isidro-Hernández M, Alemán-Arteaga S, Ruiz-Corzo B, Riesco S, Prieto-Matos P, Sánchez L, Sánchez-García I and Vicente-Dueñas C (2023) Lessons from mouse models in the impact of risk factors on the genesis of childhood B-cell leukemia. Front. Immunol. 14:1285743. doi: 10.3389/fimmu.2023.1285743
Received: 30 August 2023; Accepted: 02 October 2023;
Published: 12 October 2023.
Edited by:
Juan M. Zapata, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Cyril Broccardo, INSERM U1037 Centre de Recherche en Cancérologie de Toulouse, FranceCopyright © 2023 Casado-García, Isidro-Hernández, Alemán-Arteaga, Ruiz-Corzo, Riesco, Prieto-Matos, Sánchez, Sánchez-García and Vicente-Dueñas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Vicente-Dueñas, Y3ZkQHVzYWwuZXM=
†These authors have contributed equally to this work and share first authorship