Corrigendum: Advances in oncolytic herpes simplex virus and adenovirus therapy for recurrent glioma
 Mingming Hu
Mingming Hu XuLiang Liao
XuLiang Liao Yi Tao1,2†
Yi Tao1,2†- 1Institute of Thoracic Oncology, West China Hospital, Sichuan University, Chengdu, China
- 2Frontiers Science Center for Disease-Related Molecular Network, West China Hospital, Sichuan University, Chengdu, China
- 3Department of Thoracic Surgery, West China Hospital, Sichuan University, Chengdu, China
Recurrent glioma treatment is challenging due to molecular heterogeneity and treatment resistance commonly observed in these tumors. Researchers are actively pursuing new therapeutic strategies. Oncolytic viruses have emerged as a promising option. Oncolytic viruses selectively replicate within tumor cells, destroying them and stimulating the immune system for an enhanced anticancer response. Among Oncolytic viruses investigated for recurrent gliomas, oncolytic herpes simplex virus and oncolytic adenovirus show notable potential. Genetic modifications play a crucial role in optimizing their therapeutic efficacy. Different generations of replicative conditioned oncolytic human adenovirus and oncolytic HSV have been developed, incorporating specific modifications to enhance tumor selectivity, replication efficiency, and immune activation. This review article summarizes these genetic modifications, offering insights into the underlying mechanisms of Oncolytic viruses’ therapy. It also aims to identify strategies for further enhancing the therapeutic benefits of Oncolytic viruses. However, it is important to acknowledge that additional research and clinical trials are necessary to establish the safety, efficacy, and optimal utilization of Oncolytic viruses in treating recurrent glioblastoma.
1 Introduction
Gliomas, specifically glioblastomas (GBM), represent a majority of central nervous system malignancies and are the most common primary brain tumors. Gliomas originate from glial or progenitor cells and are classified into four grades by the World Health Organization (WHO). Glioblastoma (GBM), the most aggressive primary brain tumor in adults, accounts for 16% of all central nervous system tumors (1). It is classified as a grade 4 glioma according to the WHO grading system (2). The standard treatment for GBM includes surgical therapy, radiation therapy, and temozolomide (TMZ) therapy (3). Combining TMZ with radiation therapy can raise the 2-year survival rate to 26.5%, compared to a lower rate of 10.4% with radiation therapy alone (4). However, despite the use of surgical intervention, postoperative radiotherapy, and chemotherapy, GBM remains highly invasive, leading to metastasis, recurrence, and mortality (5). The median survival time for GBM patients is approximately 15 months (1), with minimal likelihood of resurgery for relapsed cases (6–9). Recurrent glioblastoma (rGBM) presents a significant challenge in neurooncology, characterized by increased tumor cell density, neovascularization, blood-brain barrier disruption, permeability, tortuous neovascularization, uneven thickness, and slow blood flow. The pathophysiological mechanism underlying pseudoprogression remains unclear; however, it is believed to involve vascular endothelial, blood-brain barrier, and oligodendrocyte damage, leading to local inflammation, increased blood-brain barrier permeability, and vasogenic edema, resulting in abnormal lesion enhancement on imaging. Surgical resection is often impractical for recurrent tumors, which demonstrate reduced therapy responsiveness and invasion into functional brain areas (4–10). Consequently, median overall survival (mOS) after relapse is approximately 6 months, with no established standard treatment for rGBM, leading to patient mortality within 12-15 months of initial diagnosis (10, 11). RGBM often exhibits resistance to temozolomide (TMZ, a DNA alkylating agent), the standard GBM chemotherapy agent (4). Despite advancements in genetic studies of GBM, no molecular targeting agent has been identified to prolong OS in patients with rGBM.
Tumor immunotherapy is a promising approach to activating specific immune responses against cancer cells within the body, offering the advantages of targeted, efficient treatment with reduced harm to healthy tissues. Unlike conventional methods like surgery, targeting, radiotherapy, and chemotherapy, immunotherapy does not directly eliminate cancer cells. Instead, it mobilizes immune cells capable of recognizing tumors, enhances the body’s immune system’s ability to combat cancer, and relies on these cells to indirectly control and eliminate cancer cells. This strategy has minimal side effects, ensuring safety and efficacy (12). Immunotherapy, including immune checkpoint inhibitors, has shown efficacy in clinical trials for various tumor types (13). However, its effectiveness in patients with recurrent glioma is limited due to factors such as the tumor heterogeneity of GBM, the presence of the blood-brain barrier, and the immunosuppressive nature of the tumor microenvironment (TME)[ (14). Glioma, a tumor with heterogeneous characteristics including proliferative potential, invasiveness, histological grade, and clinical behavior, presents a significant challenge for immunotherapy (15). Studies have highlighted the obstacles caused by glioma cells’ immune evasion mechanisms, such as antigen loss or downregulation, which hinder vaccine therapy and CAR-T cell therapy (16). Moreover, the glioma tumor microenvironment contains more immunosuppressive cells than immune effector cells, contributing to the establishment of an immunosuppressive state that promotes glioma growth, invasion, and metastasis (17). Conversely, oncolytic virus (OVs) therapy exhibits promise in early clinical trials for GBM. OVs can selectively infect and destroy tumor cells while modulating the immune system to enhance the anti-tumor response (18).
OVs represent a form of immunotherapy that selectively infect and destroy cancer cells, leading to the release of infectious virus particles that contribute to the destruction of residual tumors. These viruses can impede cancer cell replication or be genetically modified to specifically target and eliminate them. Moreover, OVs have the ability to activate the suppressed immune system, resulting in an adaptive anti-tumor immune response while suppressing tumor growth (19). Both preclinical and clinical trials have evaluated OVs derived from herpes simplex virus-1 (HSV-1), adenovirus (Ad), Newcastle disease virus (NDV), and reovirus (RV) for the treatment of rGBM, demonstrating promising therapeutic effects. However, further clinical trials are necessary to validate these findings.
This review aims to provide an overview of the oncolytic mechanisms of different OVs, including oHSV, CRAd, and others (Figure 1). Additionally, we discuss the survival benefits and safety profiles based on major preclinical and clinical trials of oncolytic viruses in glioma, specifically rGBM (Table 1). Currently, PVS-RIPO and DNX-2401 have received fast track designation from the US Food and Drug Administration (FDA), while G47Δ has been approved by the Japanese Ministry of Health, Labor and Welfare for the treatment of malignant glioma. Furthermore, numerous other OVs have demonstrated significant anti-tumor potential in both preclinical and clinical trials. Our goal is to provide a reference for researchers involved in the development of novel OVs, facilitate improved clinical trials for OVs, and offer valuable recommendations for the application of OVs in glioma treatment.
FIGURE 1
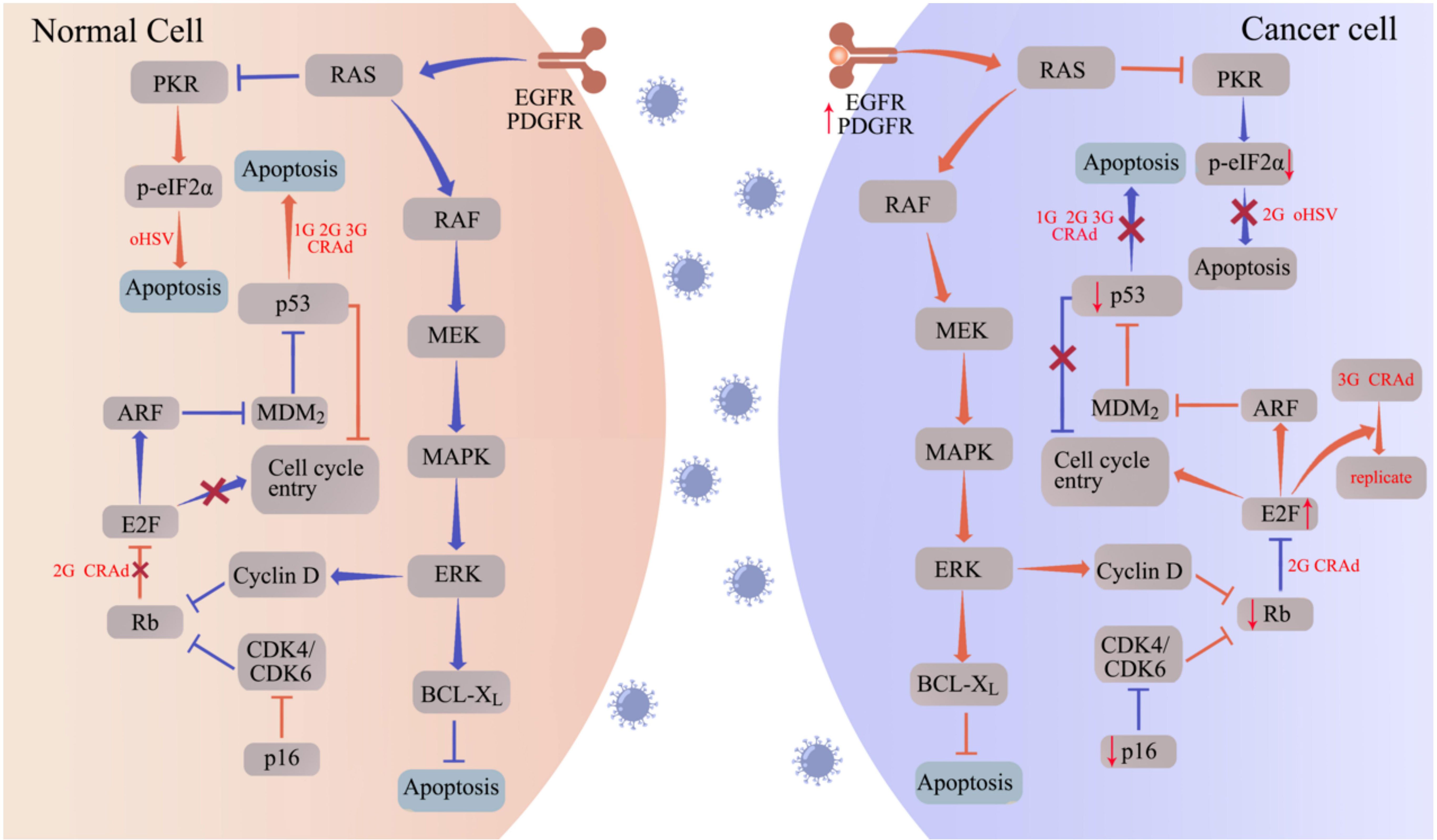
Figure 1 oHSV and CRAd replicate specifically in tumor cells by targeting tumor-associated pathways. Aberrantly expressed proteins in tumor cells promote the replication and oncolytic activity of oHSV and CRAd. In normal cells (left panel), the cell cycle is regulated by proteins such as protein kinase R (PKR), p16, retinoblastoma (Rb), and the tumor suppressor p53. Upon infection with oHSV and CRAd, these cell cycle regulators facilitate apoptosis, hindering viral replication. Conversely, cancer cells often exhibit disruptions in these cell cycle regulators, such as p53 and Rb mutations, to support uncontrolled proliferation. Consequently, when infected with oHSV and CRAd, cancer cells fail to initiate the apoptotic program, allowing for viral replication within the tumor cells. The abbreviations used in the figure are as follows: 1G/2G/3G oHSV refers to first generation, second generation, and third-generation oHSV respectively; 1G/2G/3G CRAd stands for first generation, second generation, and third-generation CRAD; CDK represents cyclin-dependent kinase; EGFR denotes epidermal growth factor receptor; ERK signifies extracellular signal-regulated kinase; MAPK refers to mitogen-activated protein kinase; MEK stands for MAPK/ERK kinase; PDGFR represents platelet-derived growth factor receptor.
TABLE 1
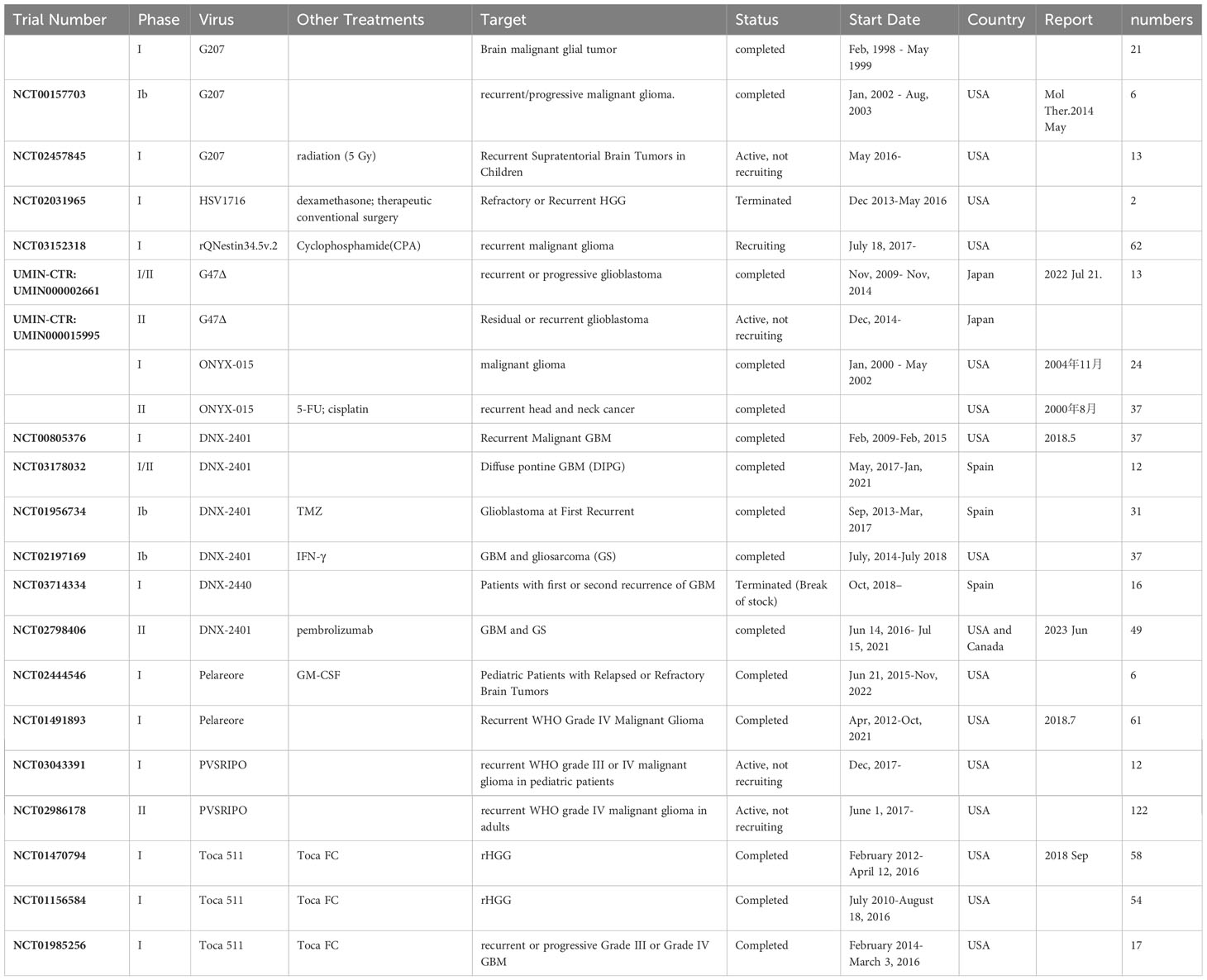
Table 1 OVs clinical trials.
2 Oncolytic herpes simplex virus (oHSV)
HSV-1, an enveloped double-stranded DNA virus belonging to the Herpesviridae family, primarily infects and replicates in nervous tissue. Its antitumor activity was first observed in the 1950s when cancer patients with concurrent viral infections experienced tumor reduction (20). As a result, researchers explored different newly discovered viruses as potential cancer treatments. However, these therapies often caused significant toxicity to healthy tissues alongside tumor reduction, leading to a decline in the pursuit of viral-based cancer treatments (21). In 1989, Robert Martuza reported the inactivation of HSV-1 virus using the thymidine kinase gene. Subsequent treatment of glioma-bearing mice with this modified virus demonstrated no encephalitis. This discovery marked a turning point, making OVs a viable therapeutic option for GBM (22). To enhance safety and specificity, successive generations of OVs have been developed through genetic modification of the original HSV-1 wild type virus (Figure 2).
FIGURE 2
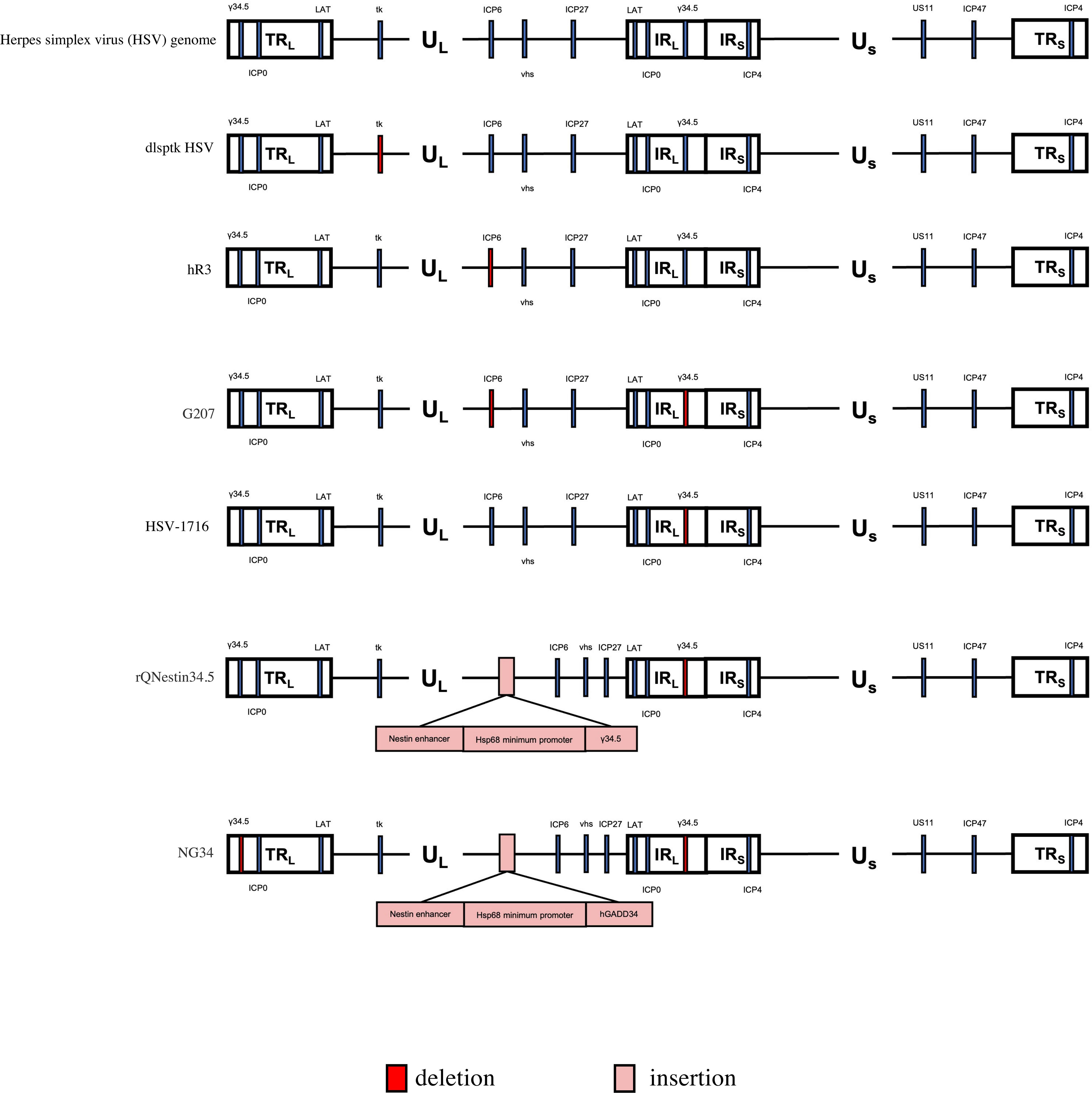
Figure 2 Genetic diagram of each generation of oncolytic HSV-1. The HSV-1 genome consists of long and short unique regions (UL and US) each bounded by terminal (T) and internal (I) repeat regions (RL, and Rs). TK, thymidine kinase protein (TK). US11, unique short 11 gene. hGM-CSF, human granulocyte-macrophage colony stimulating factor gene. hGADD34, human homologue GADD34 replaces ICP34.5 for PP1 binding and eIF2a dephosphorylation. Nestin enhancer ,a glioma-specific enhancer.
2.1 The first-generation oHSV lacking self-replication ability
2.1.1 dlsptk HSV
The first modified version of herpes simplex virus (HSV), called dlsptk HSV, was developed in 1989 by Coen (23) and Martuza published the use of HSV dlsptk in 1991 (22). This engineered virus lacks the thymidine kinase protein (TK), which is necessary for viral replication in non-dividing cells. Studies by T. Valyi-Nagy showed that dlsptk HSV significantly extended the survival of mice with subcutaneous and orthotopic tumor models (24). However, it’s important to note that this modified virus still poses a risk of infection in immunocompromised patients. To ensure patient safety, alternative modifications of HSV-1 have been explored for tumor therapy.
2.1.2 hrR3
The hrR3 mutant of HSV-1 was created by deleting ICP6 (UL39), the gene responsible for encoding the large subunit of viral ribonucleotide reductase. This enzyme plays a vital role in converting ribonucleotides to deoxyribonucleotides (dNMP) needed for viral genome synthesis. As a result, the absence of ICP6 in HSV-1 limits the availability of dNMP, thereby restricting the replication of hrR3 mutants to actively proliferating cells (25). Studies have shown that the mutant has considerable anti-cancer prospects. In previous studies, hrR3 showed a strong killing effect on human glioblastoma cell line cells, and in animal experiments, treatment with 5 × 106 hrR3 plaque forming units showed a significant inhibitory effect on tumor growth (26). Experiments in which mutant herpes simplex virus 1 (hrR3) was injected into gliomas implanted in the brain of rats showed the lack of efficacy of hrR3 in the eradication of cancer due to interference of the immune system (27). As oHSV is further modified, safer and more effective oHSVs are made.
2.2 The second generation oHSV designed to target tumor cells with PKR-eIF2α mutations
The presence of the γ34.5 protein is crucial for assessing the neuropathogenicity of HSV. In normal cells, HSV infection triggers the phosphorylation of eIF2α by the cell’s protein kinase R (PKR), which prevents viral protein synthesis. However, the γ34.5 complex of HSV counteracts this process by dephosphorylating eIF2α, allowing wild-type HSV to replicate effectively in these cells. On the other hand, if γ34.5 is deleted from HSV, the modified virus loses its ability to replicate in normal cells. Interestingly, in cancer cells with a defective antiviral PKR-eIF2α pathway, deleting γ34.5 enables selective replication of HSV in these cells (28, 29). ICP6 is a large subunit of ribonucleotide reductase (RR) that is critical for viral replication and growth in nondividing cells. By deleting γ34.5 and inactivating ICP6, a safer second-generation oHSV was generated. Proliferation of the second-generation oHSV was restricted to tumor cells with PKR-eIF2α mutations.
2.2.1 G207
G207 was generated by inserting the Escherichia coli lacZ gene into the coding sequence of viral ICP6 (UL39) and deleting two copies of the γ34.5 (RL1) locus in the viral genome (30). The ICP6 gene encodes the vital RR subunit essential for viral replication in non-dividing cells. Removing these genes restricts HSV-1 G207 proliferation to tumor cells (30). In preclinical animal models, the efficacy of G207 has been extensively demonstrated. Administration of 109 plaque forming units (pfu) of G207 in BALB/c mice and Aotus nancymai owl monkeys’ brains showed no adverse effects (31). Remarkably, whereas 103 pfu of wild-type HSV-1 proves lethal for Aotus nancymai owl monkeys, G207 has exhibited safety. Magnetic resonance imaging and histopathological evaluation of these primates revealed no central nervous system abnormalities, cell structure alterations, or presence of HSV immune response cells. These findings preliminarily verify the safety of G207 (32).
Markert et al. conducted a phase I clinical trial with 21 adults having advanced brain gliomas, demonstrating the safety of G207 without significant adverse effects. Tumor growth was suppressed in eight patients after one month of vaccination, and two patients achieved disease-free survival exceeding five years (33). These findings provide a strong basis for further clinical trials to explore the therapeutic potential of G207 in adults with recurrent brain gliomas. In another phase I trial implemented by James M. Markert et al., G207 combined with radiation therapy was investigated in nine patients with progressive, relapsed glioblastoma (34). The trial confirmed the safety of single-dose oncolytic HSV therapy augmented with radiation for treating malignant glioma patients. Despite observing reductions in tumor size and improved survival time, the absence of a control group necessitates additional clinical trials (NCT00157703) to establish the clinical therapeutic effect of G207.
G207 has demonstrated efficacy in treating adult patients with rGBM in multiple clinical trials. However, current research is mainly focused on assessing its effectiveness in pediatric patients with rGBM. Gregory K. Friedman conducted a phase I clinical trial (NCT02457845) to evaluate the safety, efficacy, and immune response of G207 in children with recurrent or progressive cerebellar brain tumors. The study aimed to enroll 15 participants and provide preliminary insights into using G207 to treat pediatric brain gliomas. The results showed that Intratumoral G207, alone or combined with radiation, had an acceptable adverse-event profile and exhibited evidence of responses in patients with recurrent or progressive high-grade glioma in pediatric cases. Additionally, G207 treatment converted immunologically “cold” tumors to “hot”. However, the loss of γ34.5 in G207 improved safety but impaired viral replication in glioblastoma stem cells (GSC) (35).
2.2.2 HSV-1716
HSV-1716 is an example of an HSV variant that has undergone a 759-bp deletion in both copies of the ICP 34.5 gene, resulting in reduced neurotoxicity compared to the wild-type virus (36, 37). Additionally, the deletion of γ34.5 significantly limits the replication potential of HSV-1716 specifically in tumor cells.
HSV-1716 is worth noting that the antitumor effect of HSV-1716 is not solely attributed to its oncolytic activity but also to its direct anti-angiogenic properties. Through in vitro and in vivo experiments, Fabian Benencia et al. have confirmed the direct anti-angiogenic effects of the oHSV: HSV-1716 (38). One study demonstrates that HSV-1716 can specifically inhibit pediatric high-grade glioma (pHGG) and diffuse intrinsic pontine glioma (DIPG) migration and invasion, highlighting a novel mechanism of action for an OV against a principal hallmark of cancer. HSV1716 was also evaluated in this study, as it has previously been applied in early-phase trials for high-grade gliomas (39). Nine patients with high-grade glioma who had relapsed after curative treatment were treated with HSV-1716 injection to evaluate the safety of 1716 in patients with recurrent malignant glioma. The safety of HSV-1716 in the treatment of gliomas was demonstrated without toxic effects at intratumoral doses up to 105 p.f.u (40).. Another study involved 12 patients with rGBM who received tumor injections of 105 HSV1716 p.f.u. Viral replication and an immune response to HSV1716 were detected after vaccination, proving that HSV1716 replicates in rGBM and does not trigger a toxic reaction in the patient (41). S Harrowet al. enrolled 12 patients with advanced gliomas for intratumoral injection of HSV1716 to observe the treatment of HSV1716. The results showed a promising improvement in the survival of GBM patients after HSV1716 inoculation compared to the expected median survival of GBM patients (42). An additional clinical trial of HSV-1716 for glioma included two patients with recurrent pediatric glioma who underwent surgical resection. However, the results of this trial (NCT02031965) have not yet been reported.
2.2.3 rQNestin34.5 and NG34
Deletion of two copies of γ34.5 in HSV restricts its replication in tumor cells, albeit with a significant reduction in overall replicative capacity. To address this issue, Hiromasa Kambara et al. developed rQNestin34.5, a novel selective mutant of HSV-1 (43). This mutant reintroduces a single copy of γ1 34.5 into the viral genome under the control of the nestin gene enhancer (a glioma-specific enhancer) and the hsp68 promoter.In vitro and in vivo experiments demonstrated robust replication and oncolytic activity of rQNestin34.5 specifically in glioma cells (44). Safety evaluations conducted in immunized and immunodeficient mice further supported its safety profile (43). Notably, the Dana-Farber Cancer Institute is currently sponsoring a Phase I clinical trial (NCT03152318) investigating the therapeutic potential of rQnestine 34.5V. 2, a transgenic HSV-1 virus, in recurrent glioblastoma alongside cyclophosphamide-based immunoregulation.
In the absence of active nestin enhancer, transcriptional leakage and minimal functionality from the hsp68 promoter in normal CNS neuronal cells may generate a small amount of ICP34.5, potentially leading to neurotoxicity induced by rQNestin34.5 (44, 45). NG34, an enhanced version of rQNestin34.5, employs GADD34 as a substitute for ICP34.5 to bind PP1 and dephosphorylate eIF2α (46). This replacement eliminates the beclin-1 binding motif responsible for neurotoxicity and autophagy inhibition in ICP34.5 (47). In human in situ GBM mice, NG34 demonstrates comparable efficacy to QNestin34.5 while exhibiting reduced neurotoxicity.
Tumor cells commonly express PD-1 to suppress immune cell destruction (48, 49). Intra-tumor administration of an oHSV carrying a scFv (single chain fragment variable) antibody against PD-1 can enhance the anti-tumor immune response without compromising the virus’s oncolytic efficacy. NG34scFvPD-1, obtained by modifying NG34 with CMV-controlled scFvPD-1 cDNA, has shown expression of a single-chain antibody against mouse PD-1 in animal experiments. NG34scFvPD-1 exhibits comparable oncolytic properties to NG34 in vitro and improved survival rates in immunoactive mice. Furthermore, immunocompetent mice develop anti-tumor immune memory, protecting against tumor metastasis (50).
2.2.4 G47Δ
The G47Δ vector, a modified version of HSV-1 based on the G207 vector, was constructed by further deleting the α47 gene. Because the expression of the α47 gene inhibits the antigen presenting (TAP) associated transporter, this deletion leads to increased MHC class I expression in infected cells, resulting in enhanced activation against tumor T cells. Additionally, G47Δ incorporates the late US11 gene under the control of the early α47 promoter, effectively suppressing the growth properties of the γ34.5 deficient mutant (51). It is noteworthy that G47Δ has obtained approval in Japan for glioblastoma treatment.
The University of Tokyo conducted a clinical study (UMIN-CTR: UMIN000002661) on G47Δ in patients with recurrent brain gliomas for the first time, specifically glioblastoma (52). This open-label, single-armed phase I-II trial aimed to assess the safety of intracranial administration of G47Δ. Subsequently, IMSUT Hospital initiated a Phase II clinical trial (UMIN-CTR: UMIN000015995) to evaluate the effectiveness and safety of G47Δ in patients with residual or recurrent glioblastoma who had previously received radiation therapy and TMZ chemotherapy. The trial involved intratumoral and repeated administration of G47Δ in 19 adult patients with rGBM. After excluding those with IDH1 mutations, among the remaining 13 patients, the one-year survival rate after G47Δ initiation was 84.2%, with a median OS of 20.2 months and a median PFS of 4.7 months. In comparison, treatment with chemotherapeutic agents resulted in a median OS of 5.0 months and a median PFS of 1.8 months. G47Δ demonstrated superior survival benefits and a favorable safety profile for treating rGBM (53). These pivotal trial findings led to the conditional and time-limited approval of G47Δ for GBM by the Ministry of Health, Labour and Welfare (MHLW) on June 11, 2021, positioning G47Δ as a potential breakthrough in glioblastoma treatment, offering improved survival outcomes and the possibility of a cure for a subset of patients. It may become the first new drug since TMZ and the first new treatment since TTF.
G47Δ is currently undergoing preclinical and clinical studies for stomach cancer, prostate cancer, and other types of cancer, where it has demonstrated superior anti-tumor efficacy compared to G207 and excellent safety characteristics. At lower multiple infection rates, G47Δ exhibits enhanced killing effects in human prostate cancer cell lines LNCaP and DU145, resulting in a 22-fold increase in viral production. Treatment with G47Δ in a mouse model of prostate cancer reduces tumor growth in established s.c. TRAMP and HONDA tumors, as well as inhibiting the recurrence of HONDA tumors previously regressed by androgen ablation therapy (54).
Furthermore, G47Δ shows promising therapeutic potential for human gastric cancer. In vitro experiments confirm favorable cytopathic effects and replication of G47Δ in nine tested human gastric cancer cell lines. In vivo intratumoral inoculation of G47Δ (at 2×105 or 1×106 pfu) significantly suppresses subcutaneous tumor growth in MKN45, MKN74, and 44As3 models (55).
In summary, these findings indicate that G47Δ may possess potent inhibitory effects on various tumor types beyond brain gliomas.
3 Adenovirus -based oncolytic viruses
Oncolytic adenovirus (also known as conditionally replicating adenovirus or CRAd) is a natural selection or genetically engineered adenovirus. Utilizing the distinguishing characteristics of tumor cells and normal cells in terms of structure and metabolic pathways, oncolytic adenovirus selectively proliferates and replicates within tumor cells, resulting in their lysis. Specifically, wild-type adenovirus has been enhanced to replicate within tumor cells and effectively lyse the target cells while minimizing its toxicity toward normal cells. At present, the third-generation of adenovirus modification for glioma treatment has been achieved. Genetically engineered or screened conditionally replicating viruses utilize aberrant molecular/genetic pathways within tumors, ensuring non-toxicity in normal cells. They are designed to replicate efficiently solely within cancer cells, leading to the lysis and destruction of the infected cancer cells (56).
Adenovirus is a nonenveloped virus with icosahedral capsid containing a 36kD double-stranded linear genome. The genome of the virus can be categorized into two distinct regions: the early gene region (E1-E4) and the late gene region. The former primarily governs viral replication and transcriptional regulation, while the latter plays a pivotal role in the synthesis of structural proteins (57). Within the early gene region, viral regulatory proteins are encoded, which play a crucial role in controlling the expression of late genes. Notably, the initial expression of E2 products, including those stemming from the E1 gene, is essential for adenovirus genome replication, virus packaging, and protein translation processes (58). Due to this significance, current genetic modification strategies for adenovirus primarily concentrate on targeting the E1 region (Figure 3).
FIGURE 3
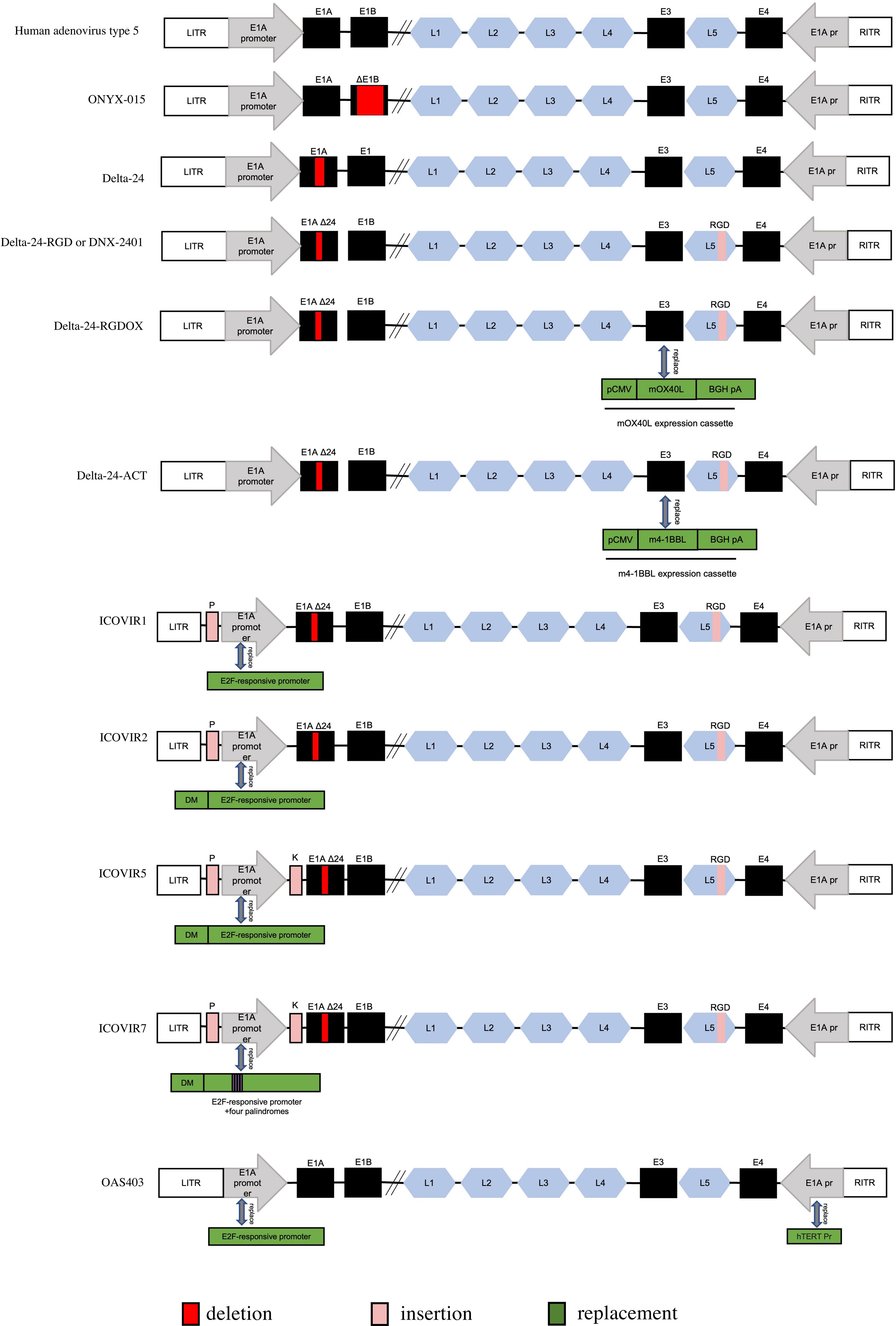
Figure 3 Genetic diagram of each generation of Oncolytic adenovirus. ITR, inverted terminal repeat. E1AΔ24: a deletion of 24 base pairs within the E1A region. DM, insulator DM-1. E2Fp, E2F-responsive promoter. K, a Kozak sequence. P, E2F-responsive palindromes (8 E2F-binding sites). RGD, an RGD integrin-binding motif in the HI loop of the fiber. pCMV, the cytomegalovirus promoter. mOX40L, mouse OX40L cDNA. BGH PA, bovine growth hormone poly-adenylation signal. The mOX40L expression cassette replaces the E3 region in Delta-24-RGDOX. hTERT Pr, human telomerase reverse transcriptase promoter.
3.1 The first-generation CRAds specifically targeting p53 mutated cancer cells
The wild-type p53 protein acts as a tumor suppressor, preventing abnormal cell proliferation and eliminating cells with abnormalities. Mutated p53, on the other hand, loses its regulatory function in controlling the normal cell cycle (59, 60). GBM is an aggressive brain cancer with a poor prognosis, and the frequency of p53 mutations varies between primary and secondary GBM. Approximately 30% of primary cases and 65% of secondary cases exhibit p53 mutations (61). In wild-type adenovirus infection, p53-mediated apoptosis is activated during the S phase. However, adenovirus E1B protein inactivates p53, inhibiting p53-mediated apoptosis and promoting viral replication (62). To address this, the first conditional adenovirus was developed by introducing E1B deficiency, exemplified by ONYX-015.
3.1.1 ONYX-015
ONYX-015 is a genetically modified CRAd derived from a human type 2/5 chimeric adenovirus with an E1B deletion. This modification allows ONYX-015 to selectively replicate in cancer cells lacking functional p53 while sparing normal cells with intact p53 (63–65). Initial reports indicated that ONYX-015 selectively killed p53-deficient tumor cells (62) (66),. The NABTT CNS Consortium conducted a Phase I trial to evaluate the efficacy and safety of ONYX-015 in recurrent brain gliomas. Twenty-four adult patients with recurrent gliomas received multiple injections of ONYX-015 at the margins of resected recurrent gliomas. The median time to progression after treatment was 46 days, and the median survival time was 6.2 months. No serious adverse events occurred in any of the 24 patients, validating the therapeutic safety of ONYX-015. The NABTT CNS Consortium conducted a Phase I trial to evaluate the efficacy and safety of ONYX-015 in recurrent brain gliomas. Twenty-four adult patients with recurrent gliomas received multiple injections of ONYX-015 at the margins of resected recurrent gliomas. The median time to progression after treatment was 46 days, and the median survival time was 6.2 months. No serious adverse events occurred in any of the 24 patients, validating the therapeutic safety of ONYX-015 (67). However, ONYX-015 did not exhibit significant antitumor effects on recurrent gliomas, suggesting that p53 may not play a key role in the tumor selectivity of ONYX-015. A Phase II clinical trial combining intratumor ONYX-015 therapy with standard intravenous cisplatin and 5-fluorouracil (5-FU) chemotherapy in patients with recurrent squamous cell carcinoma of the head and neck yielded promising results, with tumors not progressing at 6 months in the combination group (68).
Recent studies have shown that ONYX-015 replication is not strictly restricted to p53-mutated tumor cells and can replicate and eliminate cells even when the p53 pathway is intact (64, 65) (69–72),. The tumor selectivity of ONYX-015 relies not only on the p53 protein but also on the RNA export function of E1B-55K provided by tumor cells. Additionally, ONYX-015 exhibits limited replication and toxicity in tumor cells without p53 mutations (68). Despite these findings, ONYX-015 currently does not have any registered clinical trials in the United States following the withdrawal of a Phase I trial by the Dana-Farber Cancer Institute in 2013.
3.2 The second-generation CRAd specifically targeting cancer cells with mutations in the Rb pathway
ONYX-015’s limited progress in clinical trials is attributed to its untargeted viral replication and inability to infect CAR (Coxsackie adenovirus receptor)-deficient tumor cells. However, the presence of multiple adenovirus genes targeting cell cycle regulators provides an opportunity to develop oncolytic adenoviruses that can target alternative pathways. Since abnormalities in the p16/Rb/E2F pathway are present in most gliomas, viral therapies targeting the Rb pathway were developed as the second-generation of CRAd (73).
3.2.1 DNX-2401
A typical example of a second-generation CRAd targeting the Rb pathway is the Delta-24 adenovirus derived from human adenovirus type 5. Delta-24 carries a 24-base pair deletion in the Rb-binding domain of the E1A gene, resulting in a mutant E1A (mE1A) protein. Normally, the E1A protein binds to the Rb protein, releasing E2F and allowing cell progression into the S phase. In Delta-24, however, mE1A fails to effectively bind the Rb protein, leading to limited E2F release. As a result, Delta-24 cannot replicate in normal cells since mE1A is unable to bind the Rb protein and release E2F (74). Notably, Stolarek, R et al. demonstrated that Delta-24 exhibits replicative capability and cytotoxicity against medulloblastoma cells (75).
Delta-24 has demonstrated the ability to sensitize glioma cells to the camptothecin analogue irinotecan (CPT-11), both in vitro and in vivo. This effect is achieved through the upregulation of topoisomerase I expression and the induction of cancer cell accumulation in the S phase. The sequential administration of Delta-24 and CPT-11 has shown a significant extension in the survival time of animals with glioma. Therefore, the combination of adenovirus therapy and chemotherapy enhances its anticancer effect (76).
To enter host cells, Delta-24 first binds to the coxsackievirus and adenovirus receptor (CAR) on the cell surface. However, certain cancers, including glioblastoma, exhibit low levels of CAR, which greatly limits the infectivity of Delta-24. The internalization of adenovirus into host cells is facilitated by secondary interactions between the RGD motif on the Penton-based protein ring and integrins (αvβ5 and αvβ3) (77). In order to address this limitation, the gene encoding the arginine-glycine aspartic acid (RGD) peptide was introduced into the viral fiber knob receptor of Delta-24, resulting in the second-generation Delta-24-RGD or DNX-2401. Integrins are commonly overexpressed on cancer cells (78). Consequently, the infection rate of Delta-24-RGD in glioblastoma was significantly increased (79, 80).
Lang et al. conducted a Phase I dose escalation trial of DNX-2401 in 37 patients with rGBM. Group A (n=25) received eight dose levels of DNX-2401 via a single intratumoral injection to assess safety and reactivity, while Group B underwent intratumoral injection using a permanently implanted catheter, followed by en bloc resection after 14 days to obtain post-treatment specimens. The results revealed that 20% of patients in Group A experienced survival beyond three years, with at least three patients exhibiting over 95% reduction in enhanced tumor survival, resulting in more than three years of PFS. Evaluation of post-treatment specimens demonstrated virus replication and spread within the tumor in Group B, indicating direct virus-induced oncolysis. These clinical trial findings suggest that DNX-2401 can achieve prolonged survival through its direct oncolytic effect and induction of immune-mediated anti-glioma response (NCT00805376) (81). These observations agreed with preclinical studies showing that Delta-24-RGD infection induces autophagy and immunogenic cell death in glioblastoma (82, 83).
In a pre-clinical study by Martinez-Velez N, the anti-glioma effects of Delta-24-RGD were assessed in pediatric pHGG and DIPGs models. The experimental data indicated significant antitumor effects of Delta-24-RGD in both cell lines and mouse models of pHGG and DIPG. Additionally, Delta-24-RGD administration triggered an anti-tumor immune response alongside its oncolytic effects. These promising preclinical findings lay the foundation for a Phase I/II clinical trial investigating DNX-2401 (NCT03178032) (84).
3.2.2 DNX-2401 and chemotherapy
In 2017, a Phase I trial (NCT01956734) evaluated the combination of DNX-2401 and TMZ for treating rGBM. Likewise, Clinica Universidad de Navarra conducted a Phase 1b, randomized, multicenter, open-label study (TARGET-I, NCT02197169) from 2014 to 2018, investigating conditional replicative adenovirus (DNX-2401) and interferon gamma (IFN-γ) for recurrent glioblastoma. However, no published results are available from these trials, necessitating additional clinical trials to demonstrate DNX-2401’s efficacy in treating rGBM.
In a completed Phase II trial (NCT02798406), the efficacy and safety of DNX-2401 in combination with Pembrolizumab for treating rGBM were investigated. The study included 49 glioma patients who received intratumoral treatment with various doses of DNX-2401 viral particles (vp) (5*108 vp, 5*109 vp, and 5*108 vp DNX-2401) alongside Pembrolizumab. The median OS was 12.5 months (10.7 to 13.5 months). Additionally, 56.2% (95%CI 41.1-70.5%) of patients achieved clinical benefit, defined as disease stabilization or improvement. Importantly, no toxic effects were observed with DNX-2401, even at a maximum dose of 5*1010 vp. These findings demonstrate the safety and significant survival benefits of combining DNX-2401 and Pembrolizumab for selected patients with recurrent brain gliomas (85). Clinical trials have shown a survival benefit when using DNX-2401 in combination with chemotherapy. However, it is worth noting that these trials lacked a negative control group that used chemotherapy alone. Additional clinical trials are required to confirm whether the combination of DNX-2401 and chemotherapy can indeed extend patient survival.
3.2.3 DNX-2401 and radiotherapy
Radiotherapy (RT) is commonly used in managing gliomas, but its effectiveness is limited to temporary clinical improvements. Thus, researchers evaluated the feasibility, safety, and efficacy of combining Delta-24-RGD with RT for pHGGs and DIPGs. This combination significantly improved survival rates and increased immune cell infiltration at the tumor site in treated mice (86). These promising findings suggest the potential of Delta-24-RGD and RT combination therapy for clinical use in pHGGs and DIPGs.
3.2.4 Delta-24-ACT
Delta-24-ACT, a modified oncolytic adenovirus, incorporates the 4-1BB ligand (4-1BBL) to enhance its therapeutic capabilities. Infected glioma cells express elevated levels of 4-1BB ligand, which binds to TNFRSF9 (CD137; 4-1BB), a co-stimulatory receptor. This interaction activates T cells and immune cells, augmenting the oncolytic effect of the adenovirus. The antitumor effects and induction of T cell activation by Delta-24-ACT have been validated in glioma cell lines. In CT-2A tumor-bearing glioma mouse models, both Delta-24-RGD and Delta-24-ACT improved median survival, with Delta-24-ACT exhibiting slightly superior efficacy over Delta-24-RGD treatment (87). Preclinical models have demonstrated the potent antitumor effects of 4-1BB agonists. However, in clinical trials, their use in cancer treatment has been hindered by notable hepatotoxicity (88, 89). To overcome this challenge, one potential approach is specifically targeting Delta-24-ACT to tumor cells. This allows bypassing the systemic administration of 4-1BB agonists and improving safety. By delivering Delta-24-ACT directly into the tumor, it can disrupt microenvironment tolerance observed in DIPG, triggering proinflammatory changes that activate T cells and generate immune memory (90). The safety and preclinical efficacy of Delta-24-ACT have been well-established. Yet, further clinical trials are necessary to evaluate its oncolytic effect and induction of anti-tumor immune response, as it represents a potential novel oncolytic virus.
3.2.5 Delta-24-RGDOX (DNX-2440)
Delta-24-RGDOX (DNX-2440), an improved version of Delta-24-RGD, stimulates immunostimulating OX40 ligand (OX40L) expression on infected tumor cells, activating T cells recognizing tumor cell antigens. In immunologically competent mouse glioma models, Delta-24-RGDOX induces more effective in situ autologous cancer vaccination than Delta-24-RGD, resulting in a lasting tumor-specific therapeutic effect (91). Currently, an ongoing Phase I clinical trial (NCT03714334) at Clinica Universidad de Navarra investigates stereotactic injection therapy using OVs DNX-2440 for patients experiencing their first or second recurrence of GBM.
3.3 The third-generation CRAds utilizing the human E2F-1 promoter
Transcription factors of the E2F family play an important role in entry into the S phase of the cell cycle (92, 93). E2F function is inhibited upon binding to the retinoblastoma tumor suppressor protein (pRb). Binding of E2F factors to nonphosphorylated pRb prevents E2F-mediated transactivation, but this complex also actively represses transcription when bound to the promoter. It is thought that all RB pathways in tumors have changes that inhibit the binding of pRb to E2F, which leads to an increase in free E2F (94, 95). the third-generation CRAds were obtained by replacing The E1A promoter of the second-generation CRAds with the human E2F-1 promoter. E2F-1 promoter can selectively replicate. The third-generation CRAds in tumor cells and reduce hepatotoxicity.
3.3.1 ICOVIR-1、ICOVIR-2、ICOVIR-5
To enhance adenovirus selectivity for glioma cancer cells, Majem et al. replaced the native E1A promoter in Delta-24-RGD with the human E2F-1 promoter, resulting in ICOVIR-1 (96). They further introduced the DM-1 element upstream of the E2F-1 promoter to create ICOVIR-2, which acted as an enhancer blocking insulator to reduce activity against normal cells. Cell experiments demonstrated that both ICOVIR-1 and ICOVIR-2 effectively prevented E1A expression in normal human cells, leading to reduced viral replication (97).
Based on ICOVIR-2, ICOVIR-5 was optimized by inserting the CCACC sequence (Kozak sequence) before the first codon of the E1a gene. This alteration aimed to enhance transcription of the heteroe2F-1 promoter (98). In 2013, Garcia et al. conducted a Phase I clinical trial with injectable ICOVIR-5 in 14 melanoma patients. While ICOVIR-5 could reach melanoma metastases after single intravenous administration, tumor regression was not observed in the evaluated patients. These findings support ICOVIR-5’s potential for treating disseminated cancer. Currently, no clinical trials are investigating ICOVIR-5 for glioma treatment (99). Nevertheless, as a safer and more potent conditional adenovirus, ICOVIR-5 holds significant promise for clinical applications in glioma treatment.
3.3.2 OAS403
The oncolytic adenovirus OAS403 utilizes a human adenovirus type 5 vector with the incorporation of the E2F-1 promoter. This promoter regulates the early region E1A in OAS403, while a human telomerase reverse transcriptase promoter controls the E4 region, which encodes toxic viral proteins responsible for cell damage (100). The inclusion of the human telomerase reverse transcriptase promoter in the E4 region allows selective expression of these toxic proteins specifically in cancer cells (101, 102). In a mouse tumor model, a single intravenous injection of OAS403 at a dosage of 3×1012 vp/kg showed significant antitumor efficacy. Particularly, in a preestablished LNCaP prostate tumor model, systemic administration of OAS403 resulted in complete tumor regression in over 80% of cases at tolerable doses. Moreover, an increase in site-specific viral replication within the tumor was observed, with no discernible growth in the liver. Additionally, combining OAS403 treatment with Adriamycin significantly enhanced its efficacy (103). OAS403 shows promise for treating various human cancers, including recurrent glioma. An alternative variant, ICOVIR-7, incorporates an additional E2F response site palindrome within the insulated E2F-1 promoter to exert greater control over E1A-δ24 and enhance E2F-dependent E1A gene expression (104).
4 Other oncolytic viruses
In addition to oHSV and CRAd, various other OVs such as reovirus, poliovirus, and retrovirus have been modified and explored for their potential in treating brain glioma. Notably, Pelareore (reovirus), PVSRIPO (poliovirus), and Toca 511 (retrovirus) have made significant clinical advancements.
4.1 Pelareore
Reovirus is a non-encapsulated icosahedral double-stranded RNA virus that selectively targets cells with activated Ras signaling pathways. It has therapeutic potential for various solid and hematological tumors, including pancreatic, colorectal, thyroid, and lung cancers, as well as acute myeloid leukemia (105–107). REOLYSIN® (Pelareore), derived from reovirus strain type 3 Dearing virus, is an FDA-designated fast track treatment for metastatic breast cancer and metastatic pancreatic ductal adenocarcinoma. Safety of reovirus has been demonstrated in two Phase I trials involving adult brain tumor patients, without reaching the MTD (108, 109). Studies on mice have shown the efficacy of the GM-CSF (sargramostim)/intravenous REOLYSIN® regimen for brain glioma (110). A Phase I trial on six pediatric patients with recurrent or refractory advanced brain tumors has explored the combination of GM-CSF and Pelareore, but the MTD remains undetermined (NCT02444546). Regrettably, despite the induction of an immune response in patients using the combination of GM-CSF and pelareoreep, no complete or partial tumor response was observed. Instead, all patients experienced disease progression within 60 days (111). The limited clinical efficacy of pelareore in glioma may be attributed to factors like the blood-brain barrier and immune clearance. To optimize the antitumor effect of pelareore, future clinical trials could explore the use of multiple para-tumor injections in combination with GM-CSF. This approach shows potential for enhancing the therapeutic outcomes of pelareore and requires further investigation.
4.2 Recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO)
PVSRIPO, also known as lerapolturev, is a modified version of poliovirus type 1 (Mahoney), where the internal ribosome entry site is replaced with the human rhinovirus type 2 I.E. element. While maintaining its affinity for the CD155 receptor, PVSRIPO exhibits reduced virulence compared to the wild-type poliovirus (112, 113). The FDA recognized its potential and granted PVSRIPO breakthrough therapy designation in 2016. The efficacy of PVSRIPO has been confirmed in a Phase I clinical trial involving 61 adult patients with recurrent WHO grade IV malignant glioma. In this trial, intratuminal infusion of PVSRIPO established a safe dose (3.3×109 TCID50) for direct delivery to intracranial tumors. The results showed significantly higher OS rates at 24 and 36 months in the PVSRIPO immunotherapy group compared to historical controls (21% vs 14% at 24 months; 21% vs 4% at 36 months) (NCT01491893) (114). The median survival in children with recurrent glioma is typically less than 6 months. However, a Phase I clinical trial administered polio-rhinovirus chimera lerapolturev to patients, resulting in one patient (1/8) surviving beyond 22 months. This finding suggests the potential of PVSRIPO (poliovirus) to prolong the lifespan of rGBM. Nonetheless, further validation through extensive studies is necessary to affirm these trial results conclusively (115). Building on this progress, Duke University is currently conducting a Phase II clinical study involving 122 adult patients with glioblastoma. The study aims to investigate the safety, efficacy (antitumor response, and survival) of PVSRIPO (NCT02986178). Moreover, preclinical studies have shown that combining PVSRIPO with immune checkpoint inhibitors can effectively target tumors. Consequently, a Phase II trial combining PVSRIPO and the immune checkpoint inhibitor Pembrolizumab is underway, involving 30 patients with recurrent glioblastoma (NCT04479241).
4.3 Toca 511
Toca 511 is a modified non-soluble mouse leukemia virus vector that incorporates the cytosine deaminase (CD) enzyme gene, allowing for selective infection of tumor cells (116–119). Selective infection of tumor cells by Toca 511 results in the expression of the CD enzyme, which facilitates the conversion of the prodrug 5-fluorocytosine (Toca FC) into 5-FU within these cells, enabling targeted chemotherapy (120, 121). Toca 511 causes direct cytotoxicity and proinflammatory state of cancer cells via 5-FU. In a Phase I clinical study (NCT01470794), 43 patients with recurrent glioblastoma were treated with Toca 511, leading to a median survival of 13.6 months, compared to 7.1 months for untreated patients (122). Two additional Phase I studies (NCT01156584 and NCT01985256) demonstrated that intratumoral and intravenous administrations of Toca 511 as standalone treatments for rHGG is safe and tolerable.
However, a Phase III clinical trial (NCT02414165) involving 403 patients with recurrent glioblastoma and anaplastic astrocytoma treated with Toca 511/FC did not demonstrate a significant advantage over the standard of care (SOC) group. The Toca 511/FC group showed a median survival of 11.1 months, while the SOC group exhibited a median survival of 12.22 months (122). Further clinical trials are needed to provide robust evidence regarding the efficacy of Toca 511/FC in the treatment of recurrent glioma.
4.4 Zika virus
Zika virus (ZIKV) belongs to the Flaviviridae family and is an RNA virus. Its viral genome encodes a single polyprotein through a sole open reading frame, subsequently cleaved by cellular and viral proteases into ten proteins (123). Since 2015, ZIKV infection in pregnant women has emerged as a global public health emergency due to its association with microcephaly and other congenital diseases (124). Recent studies have identified ZIKV’s specific targeting of GSCs and its oncolytic activity (125). Moreover, ZIKV has been found to participate in viral endocytosis mediated by SOX2 and integrin avb5, which play roles in immune response suppression, GBM progression, and GSC maintenance (126–128). Notably, integrin avb5, typically expressed at low levels in normal tissues, exhibits heightened expression in tumors (128). Consequently, ZIKV has garnered attention as a potential oncolytic virus for treating GBM.
A safer live-attenuated Zika virus vaccine, ZIKV-LAV, was developed to enhance sensitivity to the host’s innate immune response. This vaccine is characterized by a 10-nucleotide deletion in the 3’ untranslated region (3’UTR) of the viral genome (129). Administration of ZIKV-LAV via brain injection in mice showed no detectable behavioral abnormalities, neurovirulence, or organ damage, confirming its high safety profile. Furthermore, ZIKV-LAV-treated mice exhibited significantly longer median survival times compared to the mock group (130, 131).
Contrarily, glioma slice cultures exhibit resistance to Zika virus infection unlike NPC, which is attributed to interferon-beta secretion by myeloid cells in the glioblastoma tumor microenvironment (132, 133). The combination of CDK4/6 inhibitors with ZIKV-LAV enhances the selective replication of the vaccine, resulting in significant inhibition of tumor growth and prolonged survival in glioma mice. Although ZIKV-LAV is currently in the preclinical stage, the combination of ZIKV-LAV with immunosuppressants shows promise as a novel immunotherapy for glioma.
5 Discussion
5.1 Current status
In the past 20 years, oncolytic viruses have achieved exciting results in the treatment of glioma. OVs have displayed promising results in the treatment of glioma over the past two decades, thanks to the development and application of genetic engineering technologies. These advancements have rendered OVs more specific, effective, and safe. Numerous oncolytic virus studies are currently undergoing phase I, II, and III clinical trials. While the first-generation of OVs, including dlsptk HSV and ONYX-015, demonstrated promise in preclinical trials, they were ultimately eliminated due to safety concerns and non-significant oncolytic effects during clinical trials. Subsequent modifications gave rise to second/third-generation oHSV and CRad, which exhibit great potential in treating glioma. Notably, DNX 2401 combined with PD-1 significantly prolonged the survival time of patients with glioma, and G47Δ has received approval for glioma treatment in Japan. Furthermore, ICOVIR-7 has shown promise in preclinical trials by displaying lower toxicity and increased antitumor efficacy compared to ICOVIR-5 in a subcutaneous xenograft mouse model (134). A comprehensive summary of ongoing and completed human trials investigating OVs in glioma can be found in Table 1. Despite the favorable progress achieved in most current OVs clinical trials, several limitations persist. These trials often impose strict inclusion criteria driven primarily by safety concerns, resulting in a limited patient population and random trial outcomes. Moreover, the evaluation of OVs efficacy and potential long-term consequences in GBM remains inadequate. The reliance on historical survival as a control further emphasizes the necessity for prospective randomized trials to effectively assess the effectiveness of OVs.
5.2 Future directions
To ensure the consistency between preclinical and clinical test results, conducting animal experiments is crucial for evaluating the safety and effectiveness of OVs-related products prior to clinical trials. However, it should be noted that the antitumor effect of OVs is partially mediated by inducing an immune response against the virus. Therefore, using immunodeficient mice in the PDX model to evaluate the oncolytic effects of OVs on brain gliomas disregards the impact of the immune response. Utilizing an induced glioma model can provide a more comprehensive assessment of OV’s therapeutic efficacy.
Despite promising results in preclinical and clinical trials, OVs have not yet demonstrated improved patient outcomes compared to standard care modalities like surgery, radiotherapy, and chemotherapy. This lack of improvement can be attributed to factors such as tumor heterogeneity, immune evasion, therapy resistance, limitations of the blood-brain barrier (BBB), and TME. To enhance therapeutic efficacy, it is recommended to classify OVs based on specific brain glioma types and target the p53 and Rb pathways according to their respective mutation types.
Numerous studies have investigated different routes of administration for OVs in glioma treatment, including intravenous injection, arterial injection, inhalation, intratumoral injection, and convection-enhanced delivery (CED). Intratumoral injection and CED are preferred due to their ability to provide local drug delivery to tumor lesions without opening the blood-brain barrier, although surgery is required (135). Alternatively, direct systemic administration can utilize adjunctive measures to actively open the blood-brain barrier, avoiding surgery (136). Developing new delivery routes that penetrate the blood-brain barrier and target tumors precisely is critical. Nanoparticles or cell-based carriers present non-invasive alternatives for patients who cannot undergo neurosurgical procedures. Improved imaging models are clinically needed to assess patient responses to OVs treatment and guide subsequent therapy. Additionally, determining the maximum safe dose of each OV is essential to maximize killing effect while ensuring patient safety.
The immune response, responsible for clearing OVs and hindering their replication, is a major reason for the effectiveness of oncolytic virotherapy. Combining OVs with immunosuppressants, such as anti-PD1 antibodies, has shown promise in clinical trials (137). Achieving a balance between immune response and clearance through the use of immunosuppressants and multi-dose OVs delivery requires further investigation. Combining oncolytic virotherapy with T cell therapy can help proliferate T cells in the local tumor microenvironment for optimal efficacy. Further understanding of immune mechanisms can aid in the development of improved OVs and expand their potential.
5.3 Conclusions and perspective
Recurrent gliomas, grade 4 tumors with a poor prognosis, do not see significant improvement in survival rates despite standard treatments like surgery, radiation therapy, and TMZ chemotherapy (138). New treatment methods are therefore necessary to improve patient outcomes.
One such promising approach is oncolytic virus therapy. This viral genome-based treatment selectively replicates in tumor cells while targeting them specifically, making it a hopeful treatment option for patients with recurrent tumors. Advancements in oncolytic virus genome modification have led to improvements in safety and anticancer efficacy. First-generation OVs include dlsptk HSV and ONYX-015, while second-and third-generation OVs consist mainly of oHSV and CRAd, respectively. Although recurrent glioma has a high mortality rate and poor prognosis, oncolytic virus therapy shows promise in improving patient outcomes due to its favorable safety profile. By directly targeting glioma cells and inducing cell death through selective replication and immune stimulation via acting as antigens, oncolytic viruses hold potential as a treatment option. However, safety and efficacy concerns remain, emphasizing the importance of developing safer and more effective oncolytic virus vectors. Additionally, optimizing virus delivery routes, enhancing specificity to tumor cells, limiting antiviral responses, enhancing anti-tumor immunity, reprogramming and reshaping the tumor immune microenvironment, and identifying drugs with similar anticancer effects are viable ways to improve oncolytic virus therapy.
Author contributions
MH: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. XL: Methodology, Writing – review & editing. YT: Data curation, Formal Analysis, Writing – original draft. YC: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.This work was supported by the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (Y.C., ZYJC21015).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev (2014) 23(10):1985–96. doi: 10.1158/1055-9965.EPI-14-0275
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
3. Linz U. Commentary on Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466). Cancer (2010) 116(8):1844–6. doi: 10.1002/cncr.24950
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
5. Wilson TA, Karajannis MA, Harter DH. Glioblastoma multiforme: State of the art and future therapeutics. Surg Neurol Int (2014) 5:64. doi: 10.4103/2152-7806.132138
6. Kumar AA, Abraham Koshy A. Regression of recurrent high-grade glioma with temozolomide, dexamethasone, and levetiracetam: case report and review of the literature. World Neurosurg (2017) 108:990.e911–990.e916. doi: 10.1016/j.wneu.2017.08.136
7. McNeill KA. Epidemiology of brain tumors. Neurol Clin (2016) 34(4):981–98. doi: 10.1016/j.ncl.2016.06.014
8. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol (2017) 18(6):e315–29. doi: 10.1016/S1470-2045(17)30194-8
9. Laub CK, Stefanik J, Doherty L. Approved treatments for patients with recurrent high-grade gliomas. Semin Oncol Nurs (2018) 34(5):486–93. doi: 10.1016/j.soncn.2018.10.005
10. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma–are we there yet? Neuro Oncol (2013) 15(1):4–27. doi: 10.1093/neuonc/nos273
11. Campos B, Olsen LR, Urup T, Poulsen HS. A comprehensive profile of recurrent glioblastoma. Oncogene (2016) 35(45):5819–25. doi: 10.1038/onc.2016.85
12. Xu S, Tang L, Li X, Fan F, Liu Z. Immunotherapy for glioma: Current management and future application. Cancer Lett (2020) 476:1–12. doi: 10.1016/j.canlet.2020.02.002
13. Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer (2018) 6(1):8. doi: 10.1186/s40425-018-0316-z
14. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol (2019) 20(9):1100–9. doi: 10.1038/s41590-019-0433-y
15. Vartanian A, Singh SK, Agnihotri S, Jalali S, Burrell K, Aldape KD, et al. GBM's multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro Oncol (2014) 16(9):1167–75. doi: 10.1093/neuonc/nou035
16. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
17. Mirzaei R, Sarkar S, Yong VWT. Cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol (2017) 38(2):104–15. doi: 10.1016/j.it.2016.11.005
18. Achard C, Surendran A, Wedge ME, Ungerechts G, Bell J, Ilkow CS. Lighting a fire in the tumor microenvironment using oncolytic immunotherapy. EBioMedicine (2018) 31:17–24. doi: 10.1016/j.ebiom.2018.04.020
19. Li J, Meng Q, Zhou X, Zhao H, Wang K, Niu H, et al. Gospel of Malignant Glioma: Oncolytic virus therapy. Gene (2022) 818:146217. doi: 10.1016/j.gene.2022.146217
20. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther (2007) 15(4):651–9. doi: 10.1038/sj.mt.6300108
21. Huebner RJ, Rowe WP, Schatten WE, Smith RR, Thomas LB. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer (1956) 9(6):1211–8. doi: 10.1002/1097-0142(195611/12)9:6<1211::aid-cncr2820090624>3.0.co;2-7
22. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (1991) 252(5007):854–6. doi: 10.1126/science.1851332
23. Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, et al. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U.S.A. (1989) 86(12):4736–40. doi: 10.1073/pnas.86.12.4736
24. Valyi-Nagy T, Gesser RM, Raengsakulrach B, Deshmane SL, Randazzo BP, Dillner AJ, et al. A thymidine kinase-negative HSV-1 strain establishes a persistent infection in SCID mice that features uncontrolled peripheral replication but only marginal nervous system involvement. Virology (1994) 199(2):484–90. doi: 10.1006/viro.1994.1150
25. Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol (1988) 62(1):196–205. doi: 10.1128/JVI.62.1.196-205.1988
26. Mineta T, Rabkin SD, Martuza RL. Treatment of Malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res (1994) 54(15):3963–6.
27. Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res (2006) 66(4):2314–9. doi: 10.1158/0008-5472.CAN-05-2661
28. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U.S.A. (1997) 94(3):843–8. doi: 10.1073/pnas.94.3.843
29. Cassady KA, Gross M, Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol (1998) 72(9):7005–11. doi: 10.1128/JVI.72.9.7005-7011.1998
30. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of Malignant gliomas. Nat Med (1995) 1(9):938–43. doi: 10.1038/nm0995-938
31. Sundaresan P, Hunter WD, Martuza RL, Rabkin SD. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J Virol (2000) 74(8):3832–41. doi: 10.1128/jvi.74.8.3832-3841.2000
32. Hunter WD, Martuza RL, Feigenbaum F, Todo T, Mineta T, Yazaki T, et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J Virol (1999) 73(8):6319–26. doi: 10.1128/JVI.73.8.6319-6326.1999
33. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of Malignant glioma: results of a phase I trial. Gene Ther (2000) 7(10):867–74. doi: 10.1038/sj.gt.3301205
34. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther (2009) 17(1):199–207. doi: 10.1038/mt.2008.228
35. Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med (2021) 384(17):1613–22. doi: 10.1056/NEJMoa2024947
36. MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the 'a' sequence. J Gen Virol (1991) 72(Pt 3):631–9. doi: 10.1099/0022-1317-72-3-631
37. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science (1990) 250(4985):1262–6. doi: 10.1126/science.2173860
38. Benencia F, Courreges MC, Conejo-Garcia JR, Mohamed-Hadley A, Zhang L, Buckanovich RJ, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther (2005) 12(5):789–802. doi: 10.1016/j.ymthe.2005.03.026
39. Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J (2012) 18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9
40. Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent Malignant glioma. Gene Ther (2000) 7(10):859–66. doi: 10.1038/sj.gt.3301184
41. Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human Malignant glioma: a proof of principle study. Gene Ther (2002) 9(6):398–406. doi: 10.1038/sj.gt.3301664
42. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther (2004) 11(22):1648–58. doi: 10.1038/sj.gt.3302289
43. Chiocca EA, Nakashima H, Kasai K, Fernandez SA, Oglesbee M. Preclinical toxicology of rQNestin34.5v.2: an oncolytic herpes virus with transcriptional regulation of the ICP34.5 neurovirulence gene. Mol Ther Methods Clin Dev (2020) 17:871–93. doi: 10.1016/j.omtm.2020.03.028
44. Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res (2005) 65(7):2832–9. doi: 10.1158/0008-5472.CAN-04-3227
45. Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol (2014) 16(3):334–51. doi: 10.1093/neuonc/not310
46. Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ, Martuza RL, Wakimoto H, et al. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol (2012) 86(8):4420–31. doi: 10.1128/JVI.00017-12
47. He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol (1997) 71(8):6049–54. doi: 10.1128/JVI.71.8.6049-6054.1997
48. Reardon DA, Freeman G, Wu C, Chiocca EA, Wucherpfennig KW, Wen PY, et al. Immunotherapy advances for glioblastoma. Neuro Oncol (2014) 16(11):1441–58. doi: 10.1093/neuonc/nou212
49. Cully M. Combinations with checkpoint inhibitors at wavefront of cancer immunotherapy. Nat Rev Drug Discovery (2015) 14(6):374–5. doi: 10.1038/nrd4648
50. Passaro C, Alayo Q, De Laura I, McNulty J, Grauwet K, Ito H, et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res (2019) 25(1):290–9. doi: 10.1158/1078-0432.CCR-18-2311
51. Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U.S.A. (2001) 98(11):6396–401. doi: 10.1073/pnas.101136398
52. Todo T, Ino Y, Ohtsu H, Shibahara J, Tanaka M. A phase I/II study of triple-mutated oncolytic herpes virus G47Δ in patients with progressive glioblastoma. Nat Commun (2022) 13(1):4119. doi: 10.1038/s41467-022-31262-y
53. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med (2022) 28(8):1630–9. doi: 10.1038/s41591-022-01897-x
54. Fukuhara H, Martuza RL, Rabkin SD, Ito Y, Todo T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin Cancer Res (2005) 11(21):7886–90. doi: 10.1158/1078-0432.CCR-05-1090
55. Sugawara K, Iwai M, Yajima S, Tanaka M, Yanagihara K, Seto Y, et al. Efficacy of a third-generation oncolytic herpes virus G47Delta in advanced stage models of human gastric cancer. Mol Ther Oncolytics (2020) 17:205–15. doi: 10.1016/j.omto.2020.03.022
56. Sze DY, Reid TR, Rose SC. Oncolytic virotherapy. J Vasc Interv Radiol (2013) 24(8):1115–22. doi: 10.1016/j.jvir.2013.05.040
57. Matthews DA, Russell WC. Adenovirus core protein V is delivered by the invading virus to the nucleus of the infected cell and later in infection is associated with nucleoli. J Gen Virol (1998) 79(Pt 7):1671–5. doi: 10.1099/0022-1317-79-7-1671
58. Vayda ME, Rogers AE, Flint SJ. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res (1983) 11(2):441–60. doi: 10.1093/nar/11.2.441
59. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell (1997) 88(3):323–31. doi: 10.1016/s0092-8674(00)81871-1
60. Hernandez Borrero LJ, El-Deiry WS. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer (2021) 1876(1):188556. doi: 10.1016/j.bbcan.2021.188556
61. Zhang Y, Dube C, Gibert M Jr., Cruickshanks N, Wang B, Coughlan M, et al. The p53 pathway in glioblastoma. Cancers (Basel) (2018) 10(9):297. doi: 10.3390/cancers10090297
62. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science (1996) 274(5286):373–6. doi: 10.1126/science.274.5286.373
63. Edwards SJ, Dix BR, Myers CJ, Dobson-Le D, Huschtscha L, Hibma M, et al. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(ARF) tumor suppressor genes. J Virol (2002) 76(24):12483–90. doi: 10.1128/jvi.76.24.12483-12490.2002
64. Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol (1998) 72(12):9470–8. doi: 10.1128/JVI.72.12.9470-9478.1998
65. Geoerger B, Grill J, Opolon P, Morizet J, Aubert G, Terrier-Lacombe MJ, et al. Oncolytic activity of the E1B-55 kDa-deleted adenovirus ONYX-015 is independent of cellular p53 status in human Malignant glioma xenografts. Cancer Res (2002) 62(3):764–72.
66. Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med (1997) 3(6):639–45. doi: 10.1038/nm0697-639
67. Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent Malignant gliomas, in the adjuvant setting. Mol Ther (2004) 10(5):958–66. doi: 10.1016/j.ymthe.2004.07.021
68. Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, et al. a controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med (2000) 6(8):879–85. doi: 10.1038/78638
69. Dix BR, O'Carroll SJ, Myers CJ, Edwards SJ, Braithwaite AW. Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res (2000) 60(10):2666–72.
70. Goodrum FD, Ornelles DA. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol (1998) 72(12):9479–90. doi: 10.1128/JVI.72.12.9479-9490.1998
71. Hall AR, Dix BR, O'Carroll SJ, Braithwaite AW. p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat Med (1998) 4(9):1068–72. doi: 10.1038/2057
72. Turnell AS, Grand RJ, Gallimore PH. The replicative capacities of large E1B-null group A and group C adenoviruses are independent of host cell p53 status. J Virol (1999) 73(3):2074–83. doi: 10.1128/JVI.73.3.2074-2083.1999
73. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect. vivo. Oncogene (2000) 19(1):2–12. doi: 10.1038/sj.onc.1203251
74. Ene CI, Fueyo J, Lang FF. Delta-24 adenoviral therapy for glioblastoma: evolution from the bench to bedside and future considerations. Neurosurg Focus (2021) 50(2):E6. doi: 10.3171/2020.11.FOCUS20853
75. Stolarek R, Gomez-Manzano C, Jiang H, Suttle G, Lemoine MG, Fueyo J. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther (2004) 11(11):713–20. doi: 10.1038/sj.cgt.7700731
76. Gomez-Manzano C, Alonso MM, Yung WK, McCormick F, Curiel DT, Lang FF, et al. Delta-24 increases the expression and activity of topoisomerase I and enhances the antiglioma effect of irinotecan. Clin Cancer Res (2006) 12(2):556–62. doi: 10.1158/1078-0432.CCR-05-1892
77. Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst (2003) 95(9):652–60. doi: 10.1093/jnci/95.9.652
78. Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell (1993) 73(2):309–19. doi: 10.1016/0092-8674(93)90231-e
79. Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst (2007) 99(18):1410–4. doi: 10.1093/jnci/djm102
80. Stepanenko AA, Sosnovtseva AO, Valikhov MP, Chernysheva AA, Cherepanov SA, Yusubalieva GM, et al. Superior infectivity of the fiber chimeric oncolytic adenoviruses Ad5/35 and Ad5/3 over Ad5-delta-24-RGD in primary glioma cultures. Mol Ther Oncolytics (2022) 24:230–48. doi: 10.1016/j.omto.2021.12.013
81. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent Malignant glioma. J Clin Oncol (2018) 36(14):1419–27. doi: 10.1200/JCO.2017.75.8219
82. Jiang H, Fueyo J. Healing after death: antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology (2014) 3(7):e947872. doi: 10.4161/21624011.2014.947872
83. Jiang H, Gomez-Manzano C, Rivera-Molina Y, Lang FF, Conrad CA, Fueyo J. Oncolytic adenovirus research evolution: from cell-cycle checkpoints to immune checkpoints. Curr Opin Virol (2015) 13:33–9. doi: 10.1016/j.coviro.2015.03.009
84. Martinez-Velez N, Garcia-Moure M, Marigil M, Gonzalez-Huarriz M, Puigdelloses M, Gallego Perez-Larraya J, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun (2019) 10(1):2235. doi: 10.1038/s41467-019-10043-0
85. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med (2023) 29(6):1370–8. doi: 10.1038/s41591-023-02347-y
86. Kiyokawa J, Wakimoto H. Preclinical and clinical development of oncolytic adenovirus for the treatment of Malignant glioma. Oncolytic Virother (2019) 8:27–37. doi: 10.2147/OV.S196403
87. Puigdelloses M, Garcia-Moure M, Labiano S, Laspidea V, Gonzalez-Huarriz M, Zalacain M, et al. CD137 and PD-L1 targeting with immunovirotherapy induces a potent and durable antitumor immune response in glioblastoma models. J Immunother Cancer (2021) 9(7):e002644. doi: 10.1136/jitc-2021-002644
88. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood (2018) 131(1):49–57. doi: 10.1182/blood-2017-06-741041
89. Yonezawa A, Dutt S, Chester C, Kim J, Kohrt HE. Boosting cancer immunotherapy with anti-CD137 antibody therapy. Clin Cancer Res (2015) 21(14):3113–20. doi: 10.1158/1078-0432.CCR-15-0263
90. Laspidea V, Puigdelloses M, Labiano S, Marrodan L, Garcia-Moure M, Zalacain M, et al. Exploiting 4-1BB immune checkpoint to enhance the efficacy of oncolytic virotherapy for diffuse intrinsic pontine gliomas. JCI Insight (2022) 7(7):e154812. doi: 10.1172/jci.insight.154812
91. Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res (2017) 77(14):3894–907. doi: 10.1158/0008-5472.CAN-17-0468
92. Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev (1998) 8(1):28–35. doi: 10.1016/s0959-437x(98)80058-0
93. Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci (1998) 3:d447–448. doi: 10.2741/a291
94. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev (1998) 12(15):2245–62. doi: 10.1101/gad.12.15.2245
95. Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet (2001) 10(7):699–703. doi: 10.1093/hmg/10.7.699
96. Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res (2001) 7(1):120–6.
97. Majem M, Cascallo M, Bayo-Puxan N, Mesia R, Germa JR, Alemany R. Control of E1A under an E2F-1 promoter insulated with the myotonic dystrophy locus insulator reduces the toxicity of oncolytic adenovirus Ad-Delta24RGD. Cancer Gene Ther (2006) 13(7):696–705. doi: 10.1038/sj.cgt.7700940
98. Alonso MM, Cascallo M, Gomez-Manzano C, Jiang H, Bekele BN, Perez-Gimenez A, et al. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect. vivo. Cancer Res (2007) 67(17):8255–63. doi: 10.1158/0008-5472.CAN-06-4675
99. Garcia M, Moreno R, Gil-Martin M, Cascallo M, de Olza MO, Cuadra C, et al. A phase 1 trial of oncolytic adenovirus ICOVIR-5 administered intravenously to cutaneous and uveal melanoma patients. Hum Gene Ther (2019) 30(3):352–64. doi: 10.1089/hum.2018.107
100. Tauber B, Dobner T. Adenovirus early E4 genes in viral oncogenesis. Oncogene (2001) 20(54):7847–54. doi: 10.1038/sj.onc.1204914
101. Gorziglia MI, Lapcevich C, Roy S, Kang Q, Kadan M, Wu V, et al. Generation of an adenovirus vector lacking E1, e2a, E3, and all of E4 except open reading frame 3. J Virol (1999) 73(7):6048–55. doi: 10.1128/JVI.73.7.6048-6055.1999
102. Halbert DN, Cutt JR, Shenk T. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J Virol (1985) 56(1):250–7. doi: 10.1128/JVI.56.1.250-257.1985
103. Ryan PC, Jakubczak JL, Stewart DA, Hawkins LK, Cheng C, Clarke LM, et al. Antitumor efficacy and tumor-selective replication with a single intravenous injection of OAS403, an oncolytic adenovirus dependent on two prevalent alterations in human cancer. Cancer Gene Ther (2004) 11(8):555–69. doi: 10.1038/sj.cgt.7700735
104. Cascallo M, Alonso MM, Rojas JJ, Perez-Gimenez A, Fueyo J, Alemany R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol Ther (2007) 15(9):1607–15. doi: 10.1038/sj.mt.6300239
105. White CL, Twigger KR, Vidal L, De Bono JS, Coffey M, Heinemann L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther (2008) 15(12):911–20. doi: 10.1038/gt.2008.21
106. Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science (1998) 282(5392):1332–4. doi: 10.1126/science.282.5392.1332
107. Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res (2009) 15(13):4374–81. doi: 10.1158/1078-0432.CCR-09-0334
108. Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent Malignant gliomas. Mol Ther (2008) 16(3):627–32. doi: 10.1038/sj.mt.6300403
109. Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent Malignant gliomas in adults. Mol Ther (2014) 22(5):1056–62. doi: 10.1038/mt.2014.21
110. Ilett E, Kottke T, Donnelly O, Thompson J, Willmon C, Diaz R, et al. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol Ther (2014) 22(10):1851–63. doi: 10.1038/mt.2014.118
111. Schuelke MR, Gundelach JH, Coffey M, West E, Scott K, Johnson DR, et al. Phase I trial of sargramostim/pelareorep therapy in pediatric patients with recurrent or refractory high-grade brain tumors. Neurooncol Adv (2022) 4(1):vdac085. doi: 10.1093/noajnl/vdac085
112. Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U.S.A. (1996) 93(6):2370–5. doi: 10.1073/pnas.93.6.2370
113. Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro Oncol (2004) 6(3):208–17. doi: 10.1215/S1152851703000577
114. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med (2018) 379(2):150–61. doi: 10.1056/NEJMoa1716435
115. Thompson EM, Landi D, Brown MC, Friedman HS, McLendon R, Herndon JE 2nd, et al. Recombinant polio-rhinovirus immunotherapy for recurrent paediatric high-grade glioma: a phase 1b trial. Lancet Child Adolesc Health (2023) 7(7):471–8. doi: 10.1016/S2352-4642(23)00031-7
116. Perez OD, Logg CR, Hiraoka K, Diago O, Burnett R, Inagaki A, et al. Design and selection of Toca 511 for clinical use: modified retroviral replicating vector with improved stability and gene expression. Mol Ther (2012) 20(9):1689–98. doi: 10.1038/mt.2012.83
117. Huang TT, Parab S, Burnett R, Diago O, Ostertag D, Hofman FM, et al. Intravenous administration of retroviral replicating vector, Toca 511, demonstrates therapeutic efficacy in orthotopic immune-competent mouse glioma model. Hum Gene Ther (2015) 26(2):82–93. doi: 10.1089/hum.2014.100
118. Ostertag D, Amundson KK, Lopez Espinoza F, Martin B, Buckley T, Galvao da Silva AP, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol (2012) 14(2):145–59. doi: 10.1093/neuonc/nor199
119. Tai CK, Wang WJ, Chen TC, Kasahara N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther (2005) 12(5):842–51. doi: 10.1016/j.ymthe.2005.03.017
120. Twitty CG, Diago OR, Hogan DJ, Burrascano C, Ibanez CE, Jolly DJ, et al. Retroviral replicating vectors deliver cytosine deaminase leading to targeted 5-fluorouracil-mediated cytotoxicity in multiple human cancer types. Hum Gene Ther Methods (2016) 27(1):17–31. doi: 10.1089/hgtb.2015.106
121. Logg CR, Robbins JM, Jolly DJ, Gruber HE, Kasahara N. Retroviral replicating vectors in cancer. Methods Enzymol (2012) 507:199–228. doi: 10.1016/B978-0-12-386509-0.00011-9
122. Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med (2016) 8(341):341ra75. doi: 10.1126/scitranslmed.aad9784
123. Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, et al. The 3.8 A resolution cryo-EM structure of Zika virus. Science (2016) 352(6284):467–70. doi: 10.1126/science.aaf5316
124. Heymann DL, Hodgson A, Sall AA, Freedman DO, Staples JE, Althabe F, et al. Zika virus and microcephaly: why is this situation a PHEIC? Lancet (2016) 387(10020):719–21. doi: 10.1016/S0140-6736(16)00320-2
125. Zhu Z, Gorman MJ, McKenzie LD, Chai JN, Hubert CG, Prager BC, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med (2017) 214(10):2843–57. doi: 10.1084/jem.20171093
126. Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell (2014) 157(3):580–94. doi: 10.1016/j.cell.2014.02.030
127. Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells (2009) 27(1):40–8. doi: 10.1634/stemcells.2008-0493
128. Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin alpha(v)beta(5) Axis. Cell Stem Cell (2020) 26(2):187–204.e110. doi: 10.1016/j.stem.2019.11.016
129. Shan C, Muruato AE, Nunes BTD, Luo H, Xie X, Medeiros DBA, et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat Med (2017) 23(6):763–7. doi: 10.1038/nm.4322
130. Chen Q, Wu J, Ye Q, Ma F, Zhu Q, Wu Y, et al. Treatment of human glioblastoma with a live attenuated zika virus vaccine candidate. mBio (2018) 9(5):e01683-18. doi: 10.1128/mBio.01683-18
131. Nair S, Mazzoccoli L, Jash A, Govero J, Bais SS, Hu T, et al. Zika virus oncolytic activity requires CD8+ T cells and is boosted by immune checkpoint blockade. JCI Insight (2021) 6(1). doi: 10.1172/jci.insight.144619
132. Bulstrode H, Girdler GC, Gracia T, Aivazidis A, Moutsopoulos I, Young AMH, et al. Myeloid cell interferon secretion restricts Zika flavivirus infection of developing and Malignant human neural progenitor cells. Neuron (2022) 110(23):3936–3951.e3910. doi: 10.1016/j.neuron.2022.09.002
133. Alvarado AG, Kornblum HI. Interfer(on)ing with zika virus. Neuron (2022) 110(23):3853–4. doi: 10.1016/j.neuron.2022.10.027
134. Rojas JJ, Cascallo M, Guedan S, Gros A, Martinez-Quintanilla J, Hemminki A, et al. A modified E2F-1 promoter improves the efficacy to toxicity ratio of oncolytic adenoviruses. Gene Ther (2009) 16(12):1441–51. doi: 10.1038/gt.2009.103
135. D'Amico RS, Aghi MK, Vogelbaum MA, Bruce JN. Convection-enhanced drug delivery for glioblastoma: a review. J Neurooncol (2021) 151(3):415–27. doi: 10.1007/s11060-020-03408-9
136. Li J, Zhao J, Tan T, Liu M, Zeng Z, Zeng Y, et al. Nanoparticle drug delivery system for glioma and its efficacy improvement strategies: A comprehensive review. Int J Nanomedicine (2020) 15:2563–82. doi: 10.2147/IJN.S243223
137. Kardani K, Sanchez Gil J, Rabkin SD. Oncolytic herpes simplex viruses for the treatment of glioma and targeting glioblastoma stem-like cells. Front Cell Infect Microbiol (2023) 13:1206111. doi: 10.3389/fcimb.2023.1206111
Keywords: oncolytic viruses, cancer therapy, recurrent gliomas, oncolytic herpes simplex virus, adenovirus therapy
Citation: Hu M, Liao X, Tao Y and Chen Y (2023) Advances in oncolytic herpes simplex virus and adenovirus therapy for recurrent glioma. Front. Immunol. 14:1285113. doi: 10.3389/fimmu.2023.1285113
Received: 29 August 2023; Accepted: 18 October 2023;
Published: 02 November 2023.
Edited by:
Björn L. Frendéus, BioInvent, SwedenReviewed by:
Marc Garcia-Moure, University of Texas MD Anderson Cancer Center, United StatesRui Guo, Jilin University, China
Gao Zhang, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2023 Hu, Liao, Tao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaohui Chen, bWNiLmNoZW55YW9odWlAZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship