 Wouter Claeys1,2,3,4Daan Verhaege3,4Griet Van Imschoot3,4
Wouter Claeys1,2,3,4Daan Verhaege3,4Griet Van Imschoot3,4 Elien Van Wonterghem3,4
Elien Van Wonterghem3,4 Lore Van Acker3,4Laura Amelinck3,4Federico F. De Ponti4,5
Lore Van Acker3,4Laura Amelinck3,4Federico F. De Ponti4,5 Charlotte Scott4,5Anja Geerts1,2,6Christophe Van Steenkiste7,8
Charlotte Scott4,5Anja Geerts1,2,6Christophe Van Steenkiste7,8 Lien Van Hoecke3,4†
Lien Van Hoecke3,4† Roosmarijn E. Vandenbroucke3,4*†
Roosmarijn E. Vandenbroucke3,4*†- 1Department of Internal Medicine and Paediatrics, Hepatology Research Unit, Ghent University, Ghent, Belgium
- 2Liver Research Center Ghent, Ghent University, Ghent University Hospital, Ghent, Belgium
- 3Barriers in Inflammation, VIB-UGent Center for Inflammation Research, VIB, Ghent, Belgium
- 4Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
- 5Laboratory of Myeloid Cell Biology in Tissue Damage and Inflammation, VIB–UGent Center for Inflammation Research, Ghent, Belgium
- 6Department of Gastroenterology and Hepatology, Ghent University Hospital, Ghent, Belgium
- 7Antwerp University, Department of Gastroenterology and Hepatology, Antwerp, Belgium
- 8Department of Gastroenterology and Hepatology, Maria Middelares Hospital, Ghent, Belgium
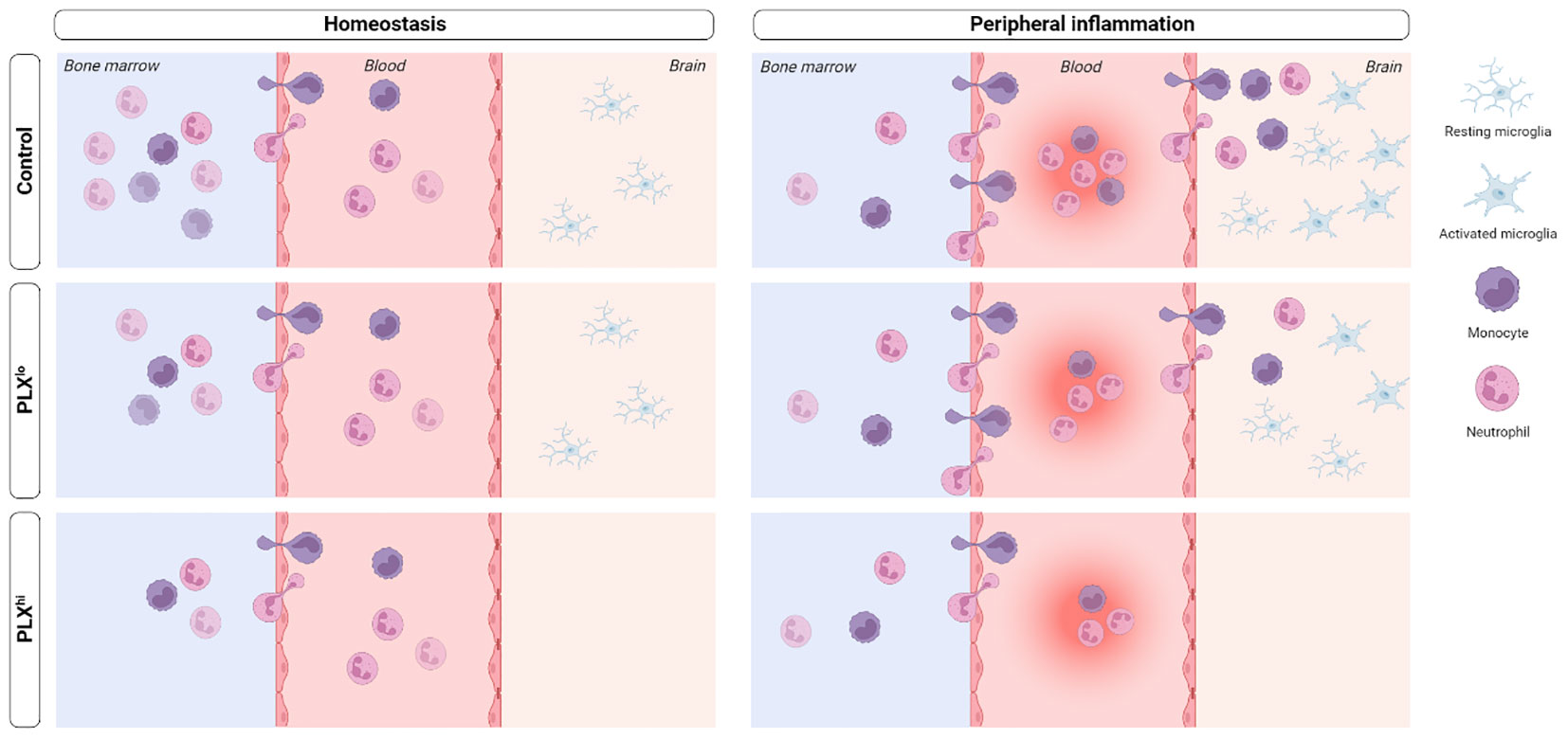
Microglia, the resident macrophages of the central nervous system (CNS), play a critical role in CNS homeostasis and neuroinflammation. Pexidartinib (PLX3397), a colony-stimulating factor 1 (CSF1) receptor inhibitor, is widely used to deplete microglia, offering flexible options for both long-term depletion and highly versatile depletion-repopulation cycles. However, the potential impact of PLX3397 on peripheral (immune) cells remains controversial. Until now, the microglia-specificity of this type of compounds has not been thoroughly evaluated, particularly in the context of peripherally derived neuroinflammation. Our study addresses this gap by examining the effects of PLX3397 on immune cells in the brain, liver, circulation and bone marrow, both in homeostasis and systemic inflammation models. Intriguingly, we demonstrate that PLX3397 treatment not only influences the levels of tissue-resident macrophages, but also affects circulating and bone marrow immune cells beyond the mononuclear phagocyte system (MPS). These alterations in peripheral immune cells disrupt the response to systemic inflammation, consequently impacting the phenotype irrespective of microglial depletion. Furthermore, we observed that a lower dose of PLX3397, which does not deplete microglia, demonstrates similar (non-)MPS effects, both in the periphery and the brain, but fails to fully replicate the peripheral alterations seen in the higher doses, questioning lower doses as a ‘peripheral control’ strategy. Overall, our data highlight the need for caution when interpreting studies employing this compound, as it may not be suitable for specific investigation of microglial function in the presence of systemic inflammation.
Graphical Abstract
Introduction
Tissue resident macrophages, monocytes, classical dendritic cells (cDCs) and their progenitors together form the mononuclear phagocyte system (MPS). Macrophages are specialized tissue phagocytes derived from embryonic progenitors, including yolk-sac macrophages and fetal liver monocytes (1–3). Macrophage proliferation and differentiation is controlled by signals from the colony-stimulating factor 1 receptor (CSF1R) (4). In the brain, parenchymal microglia are the main macrophage population. Microglia develop from yolk-sac precursors in a CSF1R dependent manner (5). In steady state conditions, they continuously scan their environment in search of pathogens or tissue damage (6–8). In disease conditions, microglia develop a so-called activated phenotype, accompanied by an amoeboid morphology and increased cyto/chemokine production (7). Often, this is associated with a significant influx of bone marrow-derived monocytes (8–11).
To investigate the role of microglia in homeostasis and disease, various microglial depletion strategies have been developed (reviewed in (12)). As microglia are thought to be particularly dependent on CSF1R signaling, many of these strategies target this receptor (13, 14). However, due to the general importance of CSF1R in macrophage biology, full body knockout strategies were plagued by a shortened lifespan, developmental deficits (15) and reduced tissue macrophage levels beyond the brain (15, 16). Inducible genetic depletion models on the other hand only allowed for short term microglial depletion (12). The development of blood-brain barrier (BBB) penetrant, small molecule CSF1R kinase inhibitors (CSF1Ri), initially PLX3397 (17) and later PLX5622 (18), has revolutionized the field of microglia research. Using these compounds, long-term microglial depletion can be achieved without evident side effects. Furthermore, due to the reversible nature of the inhibition, highly flexible microglial depletion and repopulation cycles can be achieved. A considerable body of literature emerged, showing that the effect of BBB penetrant CSF1Ri treatment is highly dependent on the specific disease context and the type of modulation employed, such as repopulation paradigms versus continued depletion (8, 19, 20).
In line with studies involving full-body knockout mice and CSF1R blocking antibodies (21), it was claimed that PLX3397/PLX5622 had no significant effects on hematopoiesis and circulating immune cells (18, 22–24). Additionally, it was hypothesized that, compared to other tissue macrophages, microglia may be particularly dependent on CSF1R for their survival, based on the extremely low brain concentrations of CSF1Ri needed to achieve complete microglial depletion (17, 18, 21). To control for potential off-target effects on peripheral macrophages, some authors suggested the inclusion of a control group treated with a lower dose CSF1Ri that has minimal central nervous system (CNS) presence and neglectable impact on microglia, although this strategy has scarcely been implemented (12, 17, 18). However, whether lower doses have similar peripheral effects despite vastly different plasma concentrations compared to the higher dose has not been studied. Additionally, the proposed control dose of PLX3397 [75 ppm in chow] can still be detected in brain tissue, raising the question whether CNS effects at this dose are really negligible (17, 18). Finally, subsequent studies challenged the notion that PLX3397 and PLX5622 have a negligible impact on peripheral tissue macrophages and especially circulating cells and bone marrow cells, some of which even do not express CSF1R, e.g. CD4+ T-cells (25–31).
Despite the prior belief that the brain is an ‘immune-privileged’ site, peripheral inflammation is now known to play a pivotal role in neuropathology, including via direct immune cell infiltration into the brain (8, 32, 33). In light of this periphery-brain communication, the potential non-microglial effects associated with BBB-penetrant CSF1Ri PLX3397 and PLX5622 have either been insufficiently explored or disregarded, possibly explaining the the sometimes very divergent results when employing different kinds of CSF1R modulation in similar disease contexts. To illustrate, in the 5xFAD model of Alzheimer’s disease, pharmacological microglial depletion and constitutive absence in Csf1rΔFIRE/ΔFIRE mice produce a quite divergent phenotype (18, 34). Recent studies using PLX5622 have demonstrated that this compound prolongs sickness behaviour after lipopolysaccharide (LPS) injection, ameliorates Western Nile virus encephalitis, and induces massive mortality in polymicrobial sepsis (31, 35, 36). The specific role of microglial depletion, compared to effects on peripheral MPS cells, was either limited or absent (35, 36). Furthermore, evidence suggests that the improved phenotype in Western Nile virus encephalitis may be directly attributed to effects of PLX5622 on bone marrow production of monocytes. While off-target effects of PLX3397 have been reported under steady state conditions (29), its impact in the context of peripherally-derived neuroinflammation remains unexplored.
Motivated by these observations, our study aimed to investigate the influence of PLX3397 treatment on the response to a peripheral inflammatory stimulus, with or without microglial presence. Our findings demonstrate that PLX3397 treatment significantly affects the response to acute and chronic inflammatory stimuli, associated with direct effects on the mature myeloid cell pool in the circulation and bone marrow, including cells not expressing CSF1R. Surprisingly, non-depleting doses of PLX3397 exhibit similar off-target toxicity, but still affect the brain, questioning its use as a ‘peripheral control’. These findings shed light on the complexities surrounding PLX3397 and its effects on the peripheral inflammatory response and circulating immune cells, highlighting the need for thorough consideration of its off-target effects when employing this compound.
Materials and methods
Animals
10 to 15-week-old male C57Bl/6j wild type (Janvier Labs, Le Genest-Saint-Isle, France) were used. Mice were housed with 14-to-10-hour light and dark cycles and free access to food and water in specific pathogen free (SPF) conditions. All experiments complied with the current laws of Belgium (Law of 14. August 1986 related to protection and welfare of animals) and EU directive 2010/63/EU, and were approved by the animal ethics committee of the Faculty of Sciences, Ghent University (EC 2022-070, EC2023-034, EC2023-018).
Microglial depletion
PLX3997 was provided by Medchemexpress (HY-16749). It was formulated in standard AIN-76A rodent chow at a concentration of 75 ppm (PLXlo) and 600 ppm (PLXhi) (Research Diets, New Brunswick, NJ) and provided ad libitum. PLX3397 diet was administered for 7 (steady state/LPS experiments) or 9 (BDL experiments) days consecutively. Control mice were fed withs standard AIN-76A diet ad libitum. Average daily food intake and average ingested PLX dose was assessed after 7 days of diet. Body weight was assessed daily.
Motor function assessment
Motor function tests were performed 2 weeks before (baseline) and 6 days after diet start. Two tests (difficult beam traversal test and open field test (OFT)) were carried out to gain insights into motor function and exploratory behaviour. In short, in the OFT, mice were allowed to move freely in a clear open-field area (40x40x40 cm) for 5 min. Travelling distance, a measure of general activity, as well as time spent in the center and border zones of the open field, a measure of anxiety, were assessed. For the difficult beam traversal, mice were trained to traverse a beam of 1m with 4 sections of narrowing width (3.5, 2.5, 1.5 and 0.5 cm) placed on top of 2 inverted mouse cages. During the testing phase, a metal grid was placed on top of the beam. Time to traverse the beam was measured. Detailed protocols can be found in supplementary file 1. All tests were video recorded. To exclude the existence of olfactory clues, all objects were thoroughly cleaned with 20% ethanol after each trial. Analysis of OFT was performed using Ethovision XT 15 software (Noldus).
Models of peripheral inflammation
LPS from S. abortus equi (L-5886) was obtained from Sigma-Aldrich and diluted in sterile phosphate bufferred saline (PBS). For survival experiments, LPS was injected intraperitoneally (IP) at an LD50 dose (5.0 mg/kg). Body weight, temperature and mortality were recorded every 2 hours for the first 12 hours and 3 times per day afterwards. For all other experiments, LPS was injected IP at an LD0 dose (2.5 mg/kg). Body weight and temperature were followed up every 6 h. Mice were sacrificed 6 and 24 hours after injection. Control mice received an equivalent volume of sterile PBS and were sacrificed 24 hours after injection.
The bile duct ligation (BDL) and sham procedures were performed under sterile conditions as previously described (37). In short, under isoflurane inhalation anesthesia, a midline abdominal incision was made and the common bile duct was isolated from the flanking portal vein. Next, the common bile duct was occluded with a double ligature of a non-resorbable suture (Mersilk 5-0, Ethicon 682H) and cut in between ligatures to prevent recanalization. Mice received buprenorphine 0.1 mg/kg intraperitoneally for 72 hours to prevent postoperative pain and distress (38) and mice were sacrificed 14 days post-surgery. The study is reported in accordance with ARRIVE guidelines (39).
Tissue sample collection
Mice were sedated through IP injection with an overdose of ketamine (87.5 mg/kg) and xylazine (12.5 mg/kg). After disappearance of paw and tail reflexes, whole blood was isolated using heart puncture, collected in Microvette 500 K2 ethylenediaminetetraacetic acid (EDTA) tubes (Sarstedt, 20.1339.100) and stored at 4°C until analysis. Afterwards, mice were transcardially perfused using 10 ml 0.2% heparin (Sigma, H‐3125) in ice-cold D‐PBS (Gibco, 14190‐094) per mouse (4.50 ml/min), followed by brain, cerebrospinal fluid (CSF) and liver sample isolation.
Analysis of BBB and blood-CSF barrier integrity
Fifteen minutes before sacrifice, mice were injected intravenously (IV) with 250 mg/kg FITC-labelled dextran (4 kDa, Sigma). Mice were perfused with ice-cold D-PBS/heparin (0.2% heparin (5000 IU/ml, Wockhardt) to remove blood. Adequate perfusion was checked visually. CSF was obtained from the fourth ventricle via the cisterna magna puncture method as described previously (40). Briefly, borosilicate glass capillary tubes (Sutter Instruments, B100-75-15) were used to pull needles on the Sutter P-87 flaming micropipette puller (pressure 330 Pa, heat index 300). The incision site was sterilized with 70% ethanol. The cisterna magna was exposed by dissecting skin and muscle tissue on the posterior side of the skull. The head of the mouse was mounted at an angle of 135°, and CSF was collected from the fourth ventricle by puncturing the cisterna magna using the capillary needles. Samples were centrifuged for 5 min at 300 g at 4°C to assess blood contamination. To measure blood-CSF barrier leakage, CSF was diluted 1/100 in PBS and fluorescence was measured. Brain samples were lysed mechanically and subsequently incubated overnight at 37°C in formamide (47671; Sigma-Aldrich) while shaking and in the dark. The next day, 100 µl supernatant was collected after centrifugation at 400 g for 7 min and fluorescence level was assessed, (λex/λem = 485/520 nm) using the FLUOstar omega (Isogen LifeScience). Weight-corrected tissue fluorescence was used to determine BBB integrity.
Complete blood cell count analysis
EDTA-anticoagulated blood was kept on ice and complete blood cell count analysis was performed using the Vetscan HM5 Hematology Analyzer (Zoetis) according to the manufacturer’s instructions within 12 hours after isolation.
Plasma biomarker measurement
Plasma was isolated from whole blood by centrifugation and isolation of supernatant (10 min, 1300 g, 4°C followed by 15 min, 2400 g, 3°C). Plasma levels of alanine aminotransferase (ALT) and bilirubin were assessed using the Architect c16000 (Abbott). Plasma endotoxin levels were assessed using the mouse LPS ELISA kit (Cusabio, CSB-E13066m) according to the manufacturer’s instructions or the Limulus Amebocyte Lysate (LAL) assay (Associates of Cape Cod Inc.) according to the manufacturer’s instructions, depending on experiments.
Isolation of brain cells
For astrocyte and microglial isolation, whole brain samples were collected in ice-cold in 1 x HBSS -/- and cut to pieces approximately 1 mm³ in size using spring scissors. Brain slurry was dissociated into single cell suspensions using the Neural Tissue Dissociation Kit (P) (Miltenyi Biotec, 130‐092‐628) as described previously (41). In short, cells were enzymatically dissociated using activated enzyme (P) for 15 min at 37°C and enzyme (A) for 2x 10 min at 37°C under continuous nutation. Additionally, the samples were mechanically dissociated by trituration 10 times with a 5 ml serological pipet in between enzymatic dissociation steps. To stop the enzymatic reaction, samples were diluted with an excess of HBSS -/-. The samples were then passed through a 70 µM cell strainer (BD Falcon, 734‐0003) and mixed with 90% Percoll™ PLUS (equilibrated in HBSS‐/‐, pH7.4; Merck, GE17‐5445‐02) and DNase I amplification grade (10U/µl; Invitrogen, 18068‐015) to obtain a final concentration of 24% Percoll™ PLUS and 75U/ml DNase. Following, the samples were spun down at 300 g for 11 min at room temperature (RT) with a low acceleration and deceleration brake. The myelin layer and supernatans were aspirated and the pellet was resuspended in 50 µl of 0.5% BSA (Jackson ImmunoResearch, 001‐000‐162) in D‐PBS (Gibco 14190‐094). This process was repeated twice. Single cell suspensions were kept on ice until staining and cell sorting.
For immune cell phenotyping, the right brain hemisphere was collected in 1 ml of ice-cold RPMI 1640 (Gibco, 52400-025) and cut to pieces approximately 1 mm³ in size using spring scissors. Brain slurry was enzymatically dissociated as previously described (41). In short, brain halves were incubated with DNase I, collagenase I and collagenase IV at a final concentration of 30 U/ml, 10 U/ml and 400 U/ml respectively, for 30 min at 37°C, with mechanical dissociation using a p1000 micropipette every 10 min. Samples were passed twice through a 70 µM cell strainer (BD Falcon, 734‐0003), centrifuged and supernatant was removed. Cell pellets were resuspended in 10 ml of 30% Percoll™ PLUS (equilibrated in HBSS‐/‐, pH7.4; Merck, GE17‐5445‐02) and spun down at 600 g for 10 min at 4°C, with no acceleration and break. Myelin layers and supernatant were removed, and pellets were resuspended in FACS buffer (2 mM EDTA, 2% BSA in 1x HBSS -/-). Suspensions were kept on ice until staining and flow cytometry.
Isolation of bone marrow cells
For bone marrow isolation, one hind leg was collected in ice-cold RPMI. After thorough removal of skin and muscle tissue, knee and hip joints were cut from tibia and femur respectively, and bone marrow cells were isolated by centrifugation (1900 g, 1 min, RT). Samples were filtered over a 70 µm mesh (BD Falcon, 734‐0003) and red blood cells were lysed using ACK lysis (Westburg, 10-548E) for 1 min at RT. The reaction was stopped with an excess of 1x PBS, cells were centrifuged (400 g, 7 min, 4°C) and kept on ice before proceeding to antibody staining and flow cytometry.
Isolation of liver cells
Liver cells were isolated by liver perfusion and digestion as described previously (26). Briefly, after retrograde cannulation, livers were perfused for 1-2 min with an EGTA-containing solution, followed by a 5 min (6 ml/min) perfusion with 0.2 mg/ml collagenase A. Livers were then removed, minced and incubated for 20mins with 0.4 mg/ml collagenase A and 10 U/ml DNase at 37°C. All subsequent procedures were performed at 4°C. Samples were filtered over a 100 µm mesh filter and red blood cells were lysed. Samples were again filtered over a 40 µm mesh filter. After two centrifugation steps of 1 min at 50 g to isolate hepatocytes, remaining liver cells (leukocytes, LSECs and HSCs) were centrifuged at 400 g for 5 min before proceeding to antibody staining for flow cytometry.
Flow cytometry and cell sorting
For isolation of brain and BM cells, single cell suspensions were pre-incubated with Fc Block (1/100; BD Biosciences, 553142) for 10 min at 4°C and stained with appropriate antibodies at 4°C in the dark for 30 min. For isolation of liver cells 0.5-5x10 (6) cells were pre-incubated with 2.4G2 antibody (Bioceros) to block Fc receptors and stained with appropriate antibodies at 4°C in the dark for 30-45 min. Antibodies and dilutions are listed in Supplementary Table 1. Reactions were stopped by adding an excess of staining buffer, cells were spun down at 400 g for 7 min at 4°C. Pellets were resuspended in FACS buffer and transferred through a 35 µm mesh into a 5 ml Falco®Round‐Bottom Polystyrene Test Tubes with Cell Strainer Snap Cap (Fisher Scientific, 08‐771‐23). Cell viability was assessed using a Fixable Viability dye (eFluor506; Thermo Fisher, 65-0866-14).
Flow cytometry and cell sorting was performed on the BD FACSymphony S6. Cells were sorted into 2 ml eppendorfs containing 450 µl RLT Plus lysis buffer (QIAGEN) containing 1% BME. The samples were extensively vortexed and stored at ‐80°C until RNA isolation. Flow cytometry without cell sorting was performed on the BD FACSymphony A3 or A5. Flow cytometry plots were analyzed using FlowJo 10.8.1. Gating strategies for astrocyte isolation as well as brain, bone marrow and liver immune phenotyping can be found in Supplementary figure 1, 2 and 3 respectively.
RNA isolation and real-time qPCR
Selected brain regions (cortex, hippocampus) and liver tissue were isolated, snap frozen in liquid nitrogen and stored at -80°C until analysis. Liver samples for RNA analyses were submerged in an excess of RNA Later (Thermo Fischer Scientific) and kept overnight at 4°C, after which they were transferred to -80°C until further analysis.
Tissue RNA was isolated using the Aurum total RNA Mini Kit (Bio-Rad), according to the manufacturer’s instructions. RNA concentration and purity were determined spectrophotometrically using the Nanodrop ND-1000 (Nanodrop Technologies, Thermo Scientific). RNA from sorted cells was isolated using the RNeasy ®Plus Micro Kit (QIAGEN, 74034) according to the manufacturer’s instructions. The concentration and purity of the RNA was determined using the Agilent RNA 6000 Pico Kit (Agilent 5067-1513) and the Agilent 2100 Bio-Analyzer. A RIN value of 7 or higher was required to proceed. cDNA was synthesized with the SensiFAST™ cDNA Synthesis Kit (Bioline). Real time-qPCR was performed with the Light Cycler 480 system (Roche) using SensiFAST SYBR No-Rox (Bio-Line). Volumes were dispensed using the I.DOT (DISPENDIX). Expression levels were normalized to the expression of all stable (at least two) reference genes, determined using the geNorm Housekeeping Gene Selection Software (42). Sequences of forward and reverse primers can be found in Supplementary Table 2.
Histology
For microscopy analyses, liver and brain tissue fixed in 4% PFA overnight at 4°C. After dehydration steps, samples were embedded in paraffin at RT until further use.
Five µm liver tissue sections were cut (HM 340 E, Thermo Fischer Scientific) and stained with haematoxylin-eosin (Klinipath). Sections were imaged using Zeiss Axioscan Z.1 (x10 magnification, Zeiss, Germany). All images were stained simultaneously and evaluated in a blinded manner. Bile infarcts fractions were quantified using ImageJ (version 1.53c, National Institutes of Health). Images of at least 10 random low power fields were analyzed per liver section. Bile infarcts were quantified on H&E sections as a percentage necrotic are relative to the total section area.
Confocal microscopy
Brain samples were cut into 5 μm paraffin sections (HM 340 E, Thermo Scientific). Sections were permeabilized in PBS containing 0.5% Triton X-100. Following blocking with blocking buffer (5% species matched serum, 0.5% Triton X-100 in PBS) at RT for 1 h, sections were incubated with primary antibodies in blocking buffer at 4°C overnight. Serum was matched to species of secondary antibodies. The same blocking buffer was used to dilute primary and secondary antibodies. After washing with PBS, sections were stained with fluorophore-conjugated secondary antibodies in blocking buffer at RT for 2 h. The full list of antibodies can be found in Supplementary Table 3. Counterstaining was done with DAPI 1/1000 in PBS. Confocal images were taken with a Zeiss LSM 780 (Zeiss, Germany), using a Plan-Apochromat 40 × 1.3 oil DIC UV-IR M27 objective or a Plan-Apochromat 25x 0.8 Imm Korr DIC M27 objective. Image analysis was performed using ImageJ (version 1.53c, National Institutes of Health).
Statistics
For comparison of two groups, unpaired t-test or Mann-Whitney test were used based on normality testing of data. For comparison of multiple groups, significance was determined using one-way ANOVA with Dunnett post-hoc testing (comparison with a single control group), Sidak post-hoc testing (comparison between all groups) or Kruskal-Wallis with Dunn post-hoc testing, based on normality distribution of residuals. For comparison of grouped data, 2-way ANOVA with Dunnett post-hoc testing was performed. ANOVA tables can be found in Supplementary Tables 4–9 PLXlo and PLXhi groups were only compared with AIN76A chow fed animals. For survival analysis, survival curves were compared using the log-rank test with Holm-Sidak multiple testing correction. The logrank test for trend was used to evaluate effect of different doses of PLX3397 on survival. All testing was two-sided. Differences were considered significant at p<0.05. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. All data is represented as mean + SEM. Graphpad 9.2 (LaJolla, California) was used for all statistical analyses.
Results
A high dose of PLX depletes microglia and peripheral macrophages without affecting tissue and circulating monocyte levels
We utilized PLX3397 mixed in AIN76A chow at two different doses: PLXhi (600 ppm), a dose reported to deplete microglia, and PLXlo (75 ppm), a dose that preserves microglial cell numbers (17). Chow containing the PLX dose was provided ad libitum for 7 days (Figure 1A). On average, daily PLX3397 intake was ~59 mg/kg body weight in the PLXhi group and ~8.5 mg/kg body weight in the PLXlo group. We observed weight loss in the PLXhi group, totalling 5% after 7 days (Figure S4A). This weight loss was likely due to reduced food intake (Figure S4B), and is in line with what is reported in conditional microglial depletion in rats (43). Additionally, in PLXhi mice, a slight reduction in activity in the open field was observed, which was not associated with anxiety-like behaviours nor apparent impairment in balance function (Figures S4C, D).
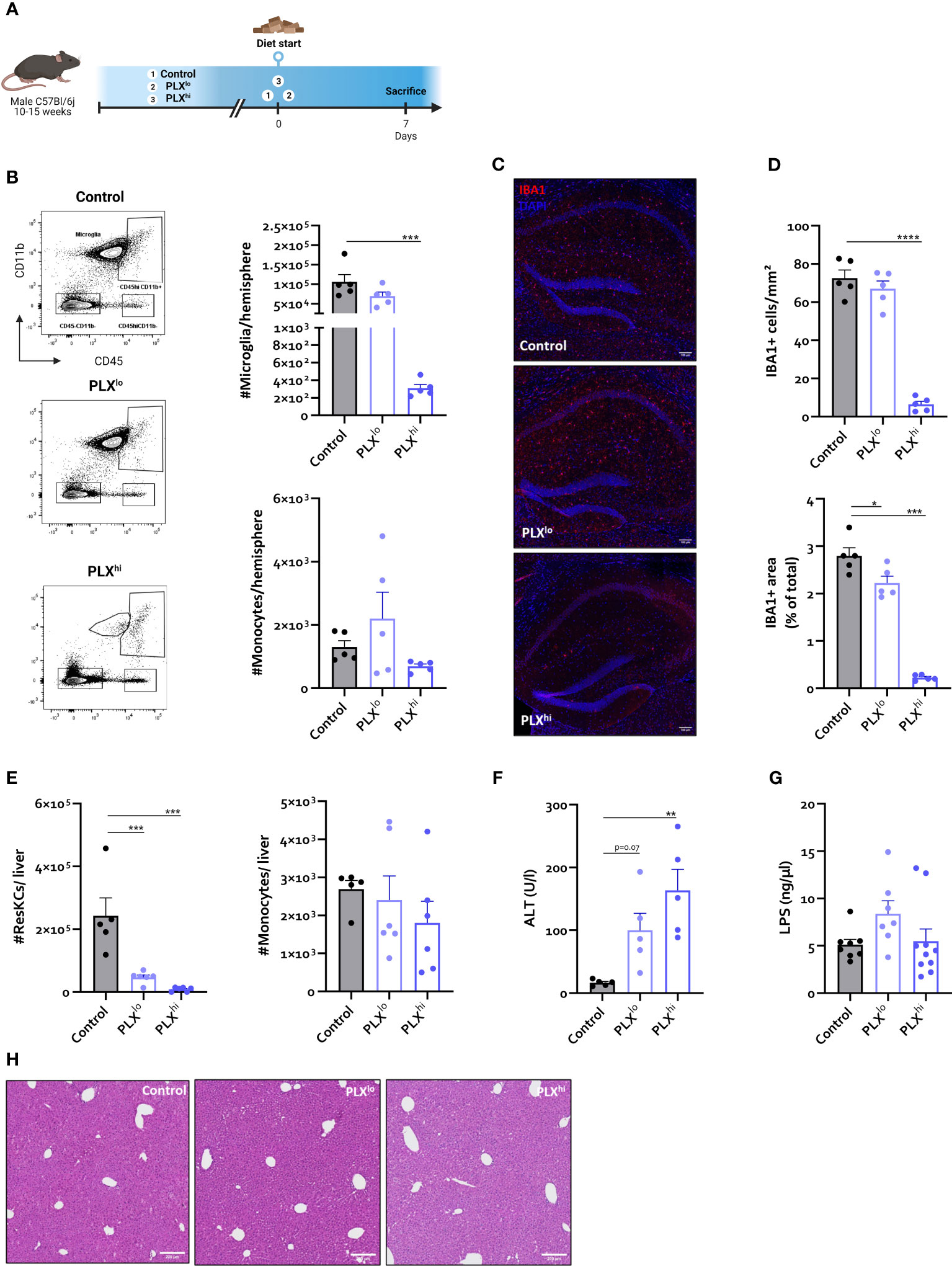
Figure 1 In steady state conditions. PLX3397 has dose dependent effects on tissue macrophages without affecting monocyte levels. (A) Experimental set-up. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to sacrifice and analysis. (B) Representative flow cytometry plots and absolute cell count of microglia and Ly6C+ monocytes per hemisphere. (C–D) Representative images of IBA1 staining in hippocampus (C) and quantification of IBA1+ cell number and stained area (D). Scale bar represents 100 µm. Data are derived from a single experiment with n=5/group. (E, F) Absolute cell count of resident Kupffer cells and Ly6C+ monocytes in mouse liver (E) and plasma ALT levels (F). Data are derived from a single experiment with n=5-6/group. (G) Plasma LPS levels. Data is derived from 2 independent repeats with n=7-10/group. (H) Representative H&E images of liver tissue. Scale bar represents 200 µm. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
As expected, flow cytometry confirmed ablation of >99% of all microglia within 7 days after PLXhi treatment, while PLXlo chow did not significantly affect microglial numbers. Brain monocyte numbers were not affected by either dose (Figure 1B). These results were confirmed on hippocampal ionized calcium-binding adapter molecule 1 (IBA1) immunofluorescence staining. Despite similar numbers of microglia, we observed that IBA1+ stained area was slightly reduced in PLXlo treated animals compared to controls (Figure 1D).
Given the dependence of non-CNS tissue macrophages on CSF1R for survival and the reported association of PLX3397 with liver injury in humans (44), we assessed levels of resident Kupffer cells (KCs), the liver resident macrophages, after PLX treatment. Both doses significantly reduced KC levels. Again, this was not associated with changes in tissue monocyte levels (Figure 1E). Although increased plasma ALT levels were detected, indicating potential liver damage, KC depletion did not lead to apparent liver injury on H&E staining. Furthermore, KC reductions did not affect hepatic bacterial clearance, based on similar plasma LPS levels in all treatment groups (Figure 1G, I). It has been reported that depleted microglia are not replaced by infiltrating monocytes (45), but the opposite is true for KCs (26, 46). We performed complete blood count analysis (CBC) to assess whether decreased circulating monocyte levels could account for this lack of infiltrating liver monocytes despite KC depletion. This did not reveal significant differences in levels of circulating monocytes, neutrophils or platelets. However, PLXhi but not PLXlo chow feeding induced a slight but significant decrease in blood lymphocyte and red blood cell (RBC) levels (Figure 2A, B).
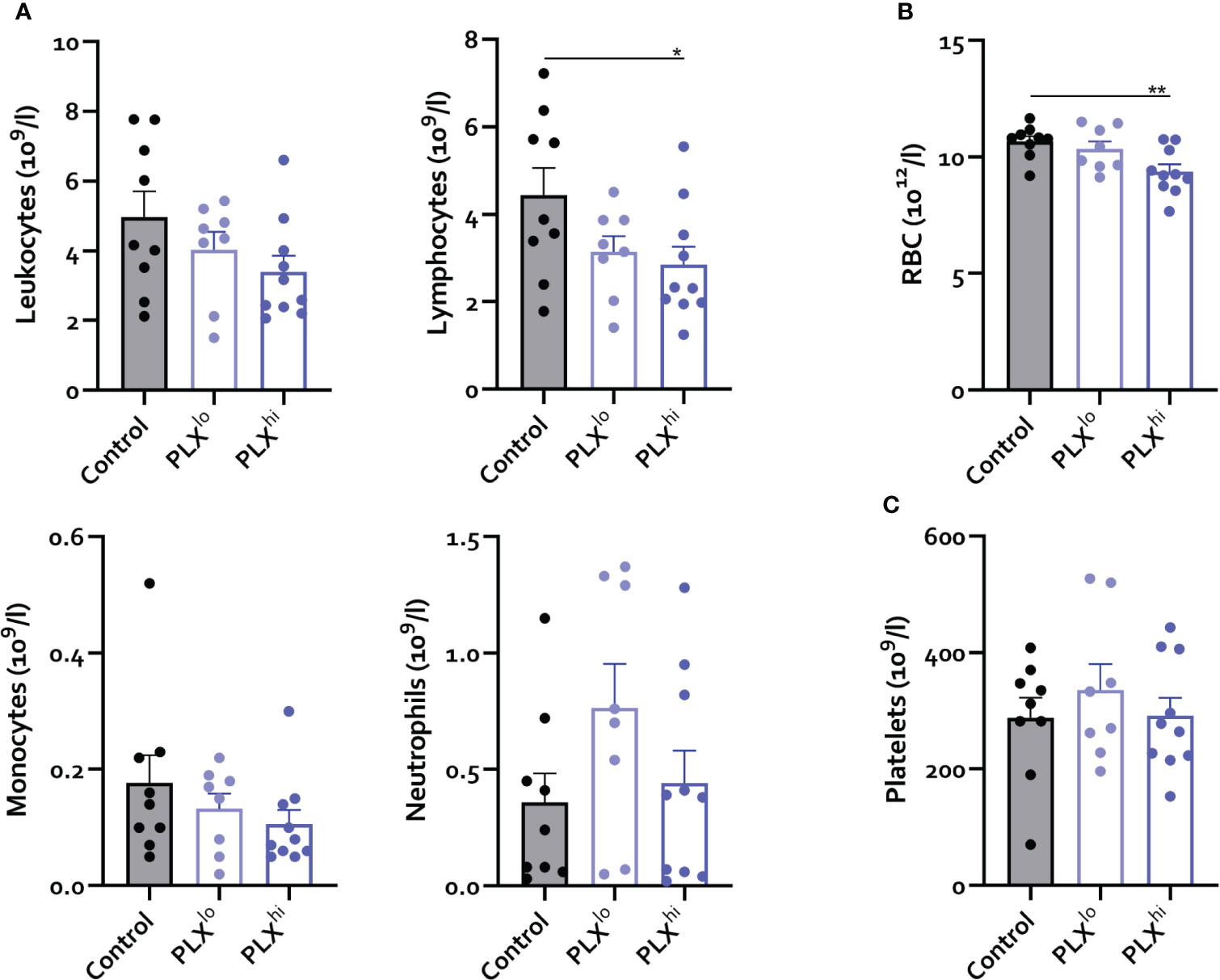
Figure 2 PLX3397 does not affect the levels of circulating monocytes. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to sacrifice and analysis. (A–C) Levels of circulating leukocytes, monocytes, neutrophils, lymphocytes (A), red blood cells (B) and platelets (C). Data is derived from 2 independent experiments with n=8-10/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
As we observed effects of PLX3397 treatment beyond microglia outside of the brain, we sought to investigate whether central nervous system (CNS) effects of PLX3397 were specific to microglia. Consistent with previous findings (17), a microglia-depleting dose of PLX3397 induced upregulation of the astrocyte reactivation marker glial fibrillary acidic protein (Gfap) in sorted astrocytes without affecting astrocyte number or GFAP immunoreactivity on staining (Figure S5A-E). Strikingly, this effect was also evident in PLXlo treated animals. On the other hand, oligodendrocyte and neuron staining did not reveal differences in cell number and stained area between groups (Figure S5F-I). We confirmed that PLX3397, at least when given in low doses, did not induce changes in blood-brain barrier (BBB) permeability. Nevertheless, we found that PLXlo treated mice exhibited a disturbed blood-cerebrospinal fluid (CSF) barrier, which is made up by epithelial cells in the choroid plexus (ChP), a region devoid of microglia (Figure S5J) (47).
PLX treatment increases mortality in severe peripheral inflammation
Based on the observed effects of PLX3397 on tissue resident macrophages in brain and liver and the absence of effects on circulating immune cells, we next investigated if PLX3397-treated mice respond differently to an LD50 dose of lipopolysaccharide (LPS) (Figure 3A). This endotoxemia model mimics the severe inflammatory response syndrome in sepsis without the presence of microbial infection (48). Interestingly, mortality rates were significantly increased in mice pretreated with PLXhi. The PLXlo pretreated mice showed an intermediary phenotype, albeit not significantly different from control chow treated animals. Nonetheless, when performing the log-rank test for trend, increasing mortality was found with increasing doses of PLX3397 (Figure 3B). Additionally, the LPS-induced temperature drop was both delayed and exacerbated in PLXhi treated mice compared to control chow fed mice, with higher body temperatures at 6 hours, but lower at 24 hours post LPS injection. Again, PLXlo treated mice exhibited an intermediary phenotype, with a trend towards lower body temperatures compared to control chow fed mice 24 hours after LPS. Only in PLX3397 pretreated mice, we observed significant differences in body temperature between the two timepoints (Figure 3C). The body weight drop 24 hours after LPS was slightly lower in PLX treated groups (Figure 3D).
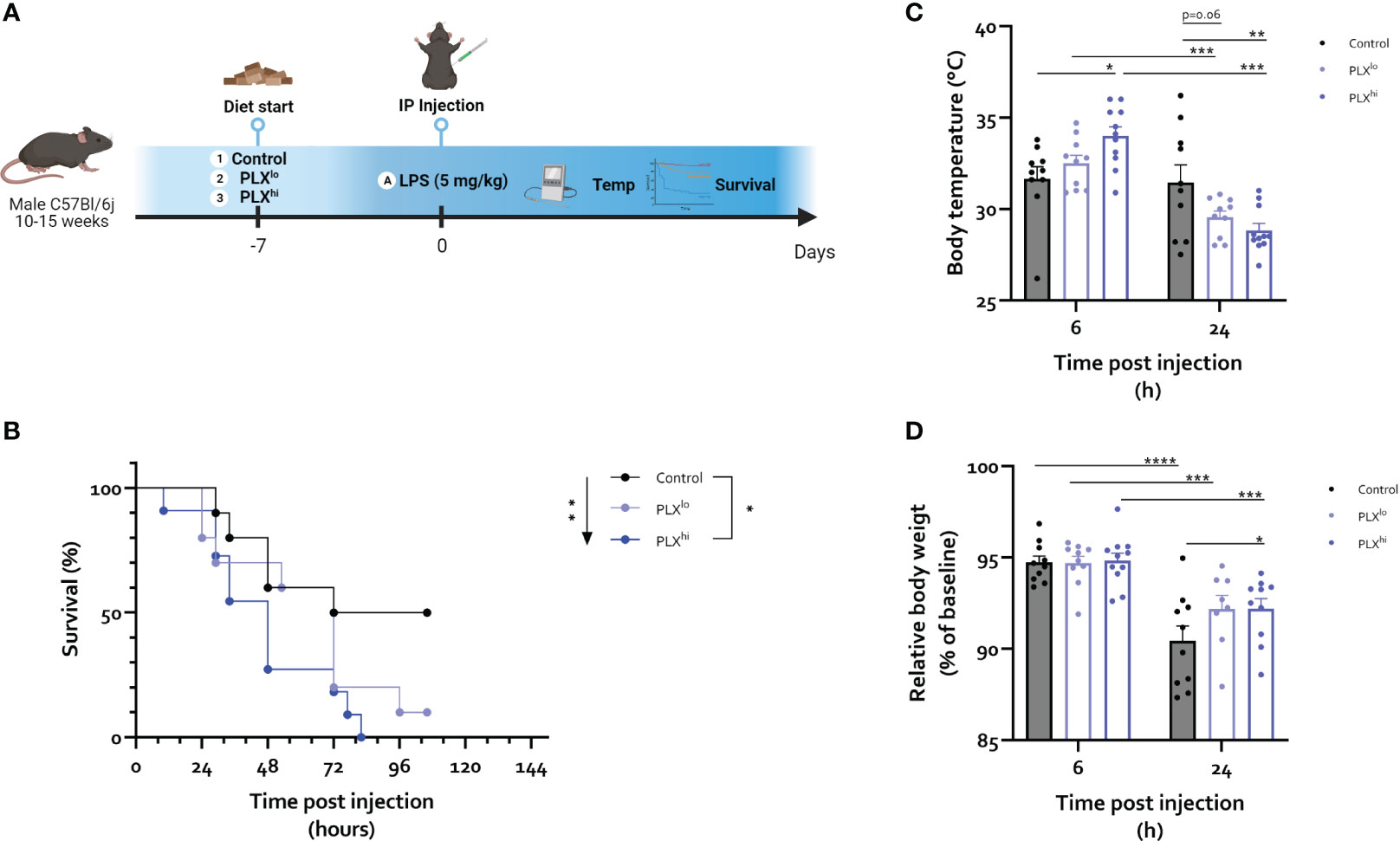
Figure 3 PLX3397 exacerbates the response to an LD50 dose of LPS. (A) Experimental set-up. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (5 mg/kg, IP). Survival, body weight and temperature were followed up. (B) Survival after LPS injections. Arrow indicates logrank test for trend, bar indicates log-rank test comparing individual groups. (C, D) Body temperature (C) and relative body weight (D) 6 and 24 hours after LPS injection. Data from a single repeat with n=10-11/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Next, we shifted to an LD0 of LPS (2.5 mg/kg IP) to investigate the mechanisms of this PLX3397-induced increased sensitivity for LPS in more detail (Figure 4A). This did not lead to mortality within 24 hours in any treatment group. Although no significant differences were observed between treatment groups, temperature differences following LPS followed a similar pattern to the LD50 dose experiments (Figure S6). To examine whether alterations in the cerebral inflammatory response could account for the aggravated phenotype, brain cyto/chemokine responses were evaluated. This revealed a significant decrease in microglia/macrophage markers Tmem119 and Csf1r in the PLXhi pretreated mice, both after LPS and PBS injection (Figure 4B; Table S4). Additionally, LPS-associated increases in Tnf and Il1b, but not Il6 and Ccl2, gene expression level were abrogated by PLXhi pretreatment (Figure 4C; Table S4). PLXlo treatment had no effect on transcription of microglia/macrophage marker genes nor cytokine or chemokine mRNA levels, suggesting an altered cerebral cytokine response does not explain the aggravated response to LPS at this dose.

Figure 4 Microglial depletion partially abrogates the cerebral inflammatory response. (A) Experimental set-up. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. (B, C) Hippocampal mRNA levels of the microglia/macrophage markers Csf1r, Tmem119 (B) and the inflammatory cytokines Tnf, Il1b, Il6 and chemokines Ccl2 (C). All data are derived from 4 independent repeats with n=8/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
We wanted to assess whether PLX3397 chow treatment after the primary inflammatory stimulus in a more chronic inflammation model would induce similar results. Considering the effect we observed on liver resident macrophages and the development of transaminase elevations in patients treated with pexidartinib (44), we opted for the BDL model, an archetypical model of inflammatory liver injury associated with secondary microglial activation and hepatic encephalopathy (38, 41, 49). PLX3397 chow treatment was commenced 5 days after BDL, until sacrifice at 14 days (Figure S7A). Mortality was only observed in PLX-treated mice (PLXlo: 20% at 14 days; PLXhi: 29% at 14 days) (Figure S7B) and BDL mice treated with PLXhi showed a more pronounced temperature drop (Figure S7C).
Exacerbation of liver injury after PLX3397 treatment is dependent on dose and model system
Next, we studied whether the heightened sensitivity of PLX3397-treated mice to systemic inflammation could be attributed to its effects on KCs and/or subsequent altered hepatic response, which (partly) dictates the systemic and neuroinflammatory response in both the BDL and LPS models. However, in the LPS endotoxemia model, we observed only limited effects of PLX3397 pretreatment on the hepatic response and these effects were unique to the PLXhi pretreated group. Specifically, we found a transient increase in ALT levels 6 hours after LPS, only in the PLXhi group (Figure 5A; Table S7). Bilirubin levels were below the detection limit suggesting liver function was preserved in all mice (data not shown). Hepatic mRNA levels of the macrophage marker Csf1r levels decreased after LPS, while myeloid marker Itgam transcription increased. These changes were ablated in PLXhi treated mice, while levels in PLXlo treated animals were comparable with control conditions. Of note, an intermediary reduction of Csf1r mRNA levels in PLXlo treated animals was observed in PBS injected animals (Figure 5D; Table S5). Regarding the cytokine response, we observed (non significant) reductions in levels of Tnf and Il1b in PLX3397 treated animals compared to controls, 6 hours after LPS injection. This was more pronounced in the PLXhi treated group. After 24 hours, levels of Tnf, Il1b, Il6 and Ccl2 were lower in PLXhi pretreated groups compared to control chow fed mice, although only reaching statistical significance for Il1b and a trend for Il6. Levels of Ccl2 and Il6 in PLXlo pretreated animals Were also decreased at this timepoint, albeit not significantly. Of note, Il1b and Il6 levels were already lower in PLXhi treated animals at baseline (Figure 5C; Table S5). The reduction in KC numbers did not significantly impact LPS clearance at either timepoint (Figure 5D; Table S7).
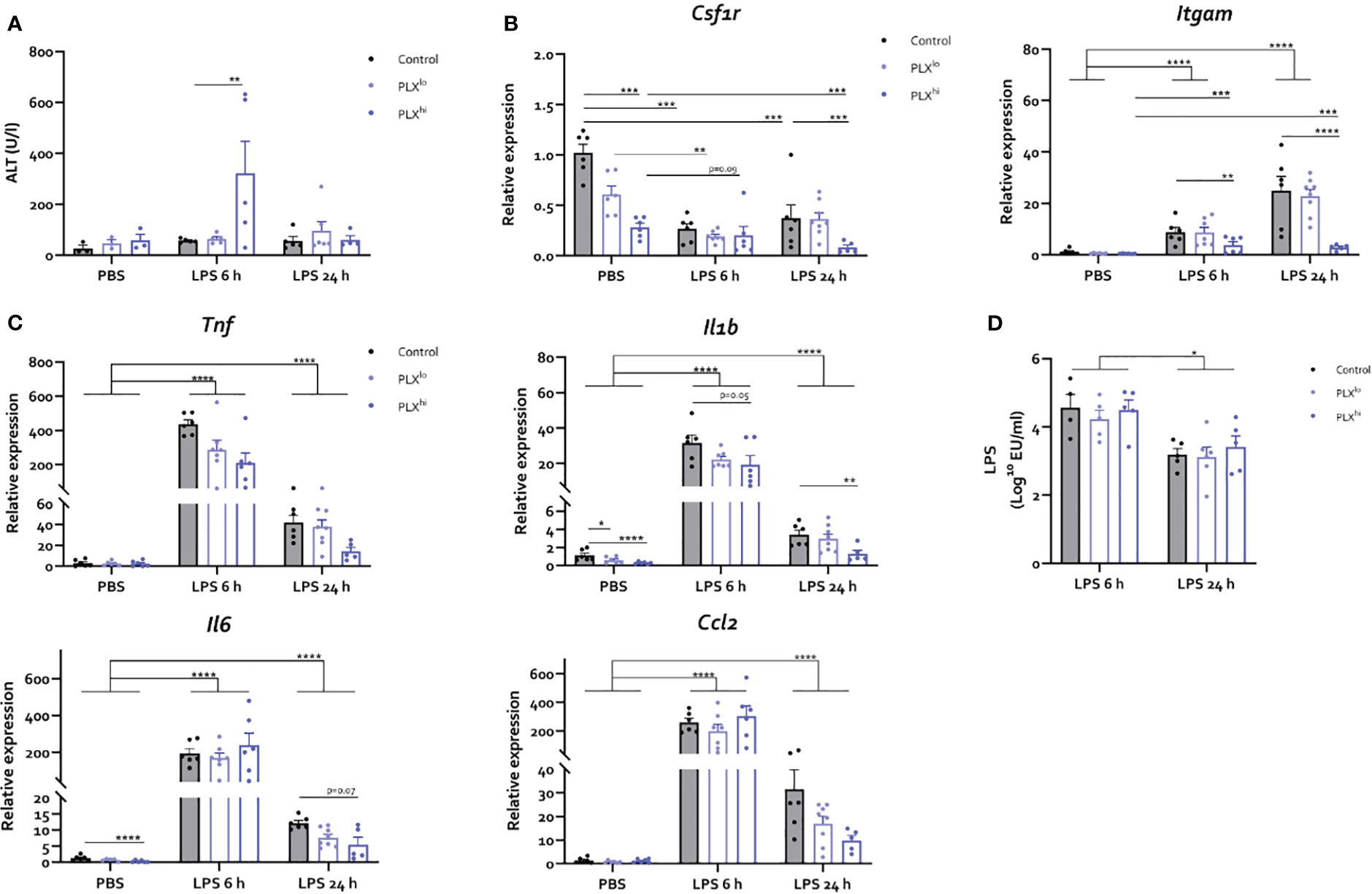
Figure 5 PLX3397 has limited and dose specific effects on the hepatic response to LPS. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. (A) Plasma ALT levels. (B, C) Liver mRNA levels of the macrophage marker Csf1r, the myeloid marker Itgam (B), the inflammatory cytokines Tnf, Il1b, and Il6 and the chemokine Ccl2 (C). (D) Plasma endotoxin levels after LPS injection. All data are derived from 4 independent repeats with n=3-8/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
In mice subjected to BDL, we observed an elevation of liver injury markers ALT and bilirubin, specifically in the PLXhi treated group (Figure S8A, B). Additionally, we noted heightened bile infarct area in the PLXhi treated group compared to untreated BDL mice (Figure S8C, D). BDL mice exhibit bacterial translocation. These bacteria are putatively cleared by KCs. Similar to our findings in the LPS model however, systemic endotoxemia as a result of bacterial translocation was similar in PLX3397 fed mice and chow fed BDL controls (Figure S8E). The hepatic mRNA expression levels of the macrophage marker Csf1r and the myeloid marker Itgam, both increased after BDL surgery, were significantly reduced when mice were treated with PLXhi. Of note, PLXlo treated mice exhibited an intermediary phenotype, not significantly different from untreated BDL controls (Figure S8F). Finally, while the increase in hepatic Tnf levels after BDL was abrogated in PLXhi treated mice, the levels of Il6, Il1b and Ccl2, although lower in PLXhi treated mice, were not significantly different from untreated BDL controls. PLXlo feeding did not affect the expression levels of these cyto/chemokines after BDL (Figure S8G).
Levels of CNS infiltrating and circulating myeloid cells are reduced in a dose dependent manner upon PLX3397 treatment
To investigate whether changes in the cellular immune response within the brain and periphery could account for the heightened response to peripheral inflammation, we examined levels of infiltrating immune cells in the CNS. Similar to our steady state experiments, only PLXhi pretreated mice exhibited microglial ablation (Figure 6A; Table S6). Furthermore, no significant differences were detected in brain monocytes or neutrophils in PBS injected animals when comparing chow regimens (Figure 6B; Table S6). We observed that PLXhi pretreated mice exhibited reduced levels of CNS-associated monocytes 6 and 24 hours after LPS injection, and reduced levels of CNS-associated neutrophils at 24 hours after LPS injections, compared to control chow fed animals. Interestingly, we observed that LPS induced microglial accumulation in control chow fed animals was diminished in PLXlo pretreated animals. Additionally, PLXlo too significantly reduced CNS accumulation of monocytes and neutrophils 24 hours after LPS, albeit to a lesser extent than in PLXhi pretreated mice (Figure 6A, B; Table S6). In the chronic BDL model, which, in line with earlier observations (41, 49), is not associated with microglial proliferation (Figure 6D), we observed similar patterns regarding CNS monocyte and neutrophil numbers (Figure 6E). Considering these striking findings, we performed crude CBC analysis, which revealed that 24 hours after LPS and upon BDL induced hepatic and systemic inflammation, both PLXlo and PLXhi treated mice exhibited reduced levels of circulating monocytes and neutrophils compared to control chow fed mice. All effects were more pronounced when using a higher dose, and in BDL experiments, the monocyte/neutrophil reduction in PLXlo treated animals was not significantly different from BDL animals fed control chow (Figure 6C, F; Table S7).
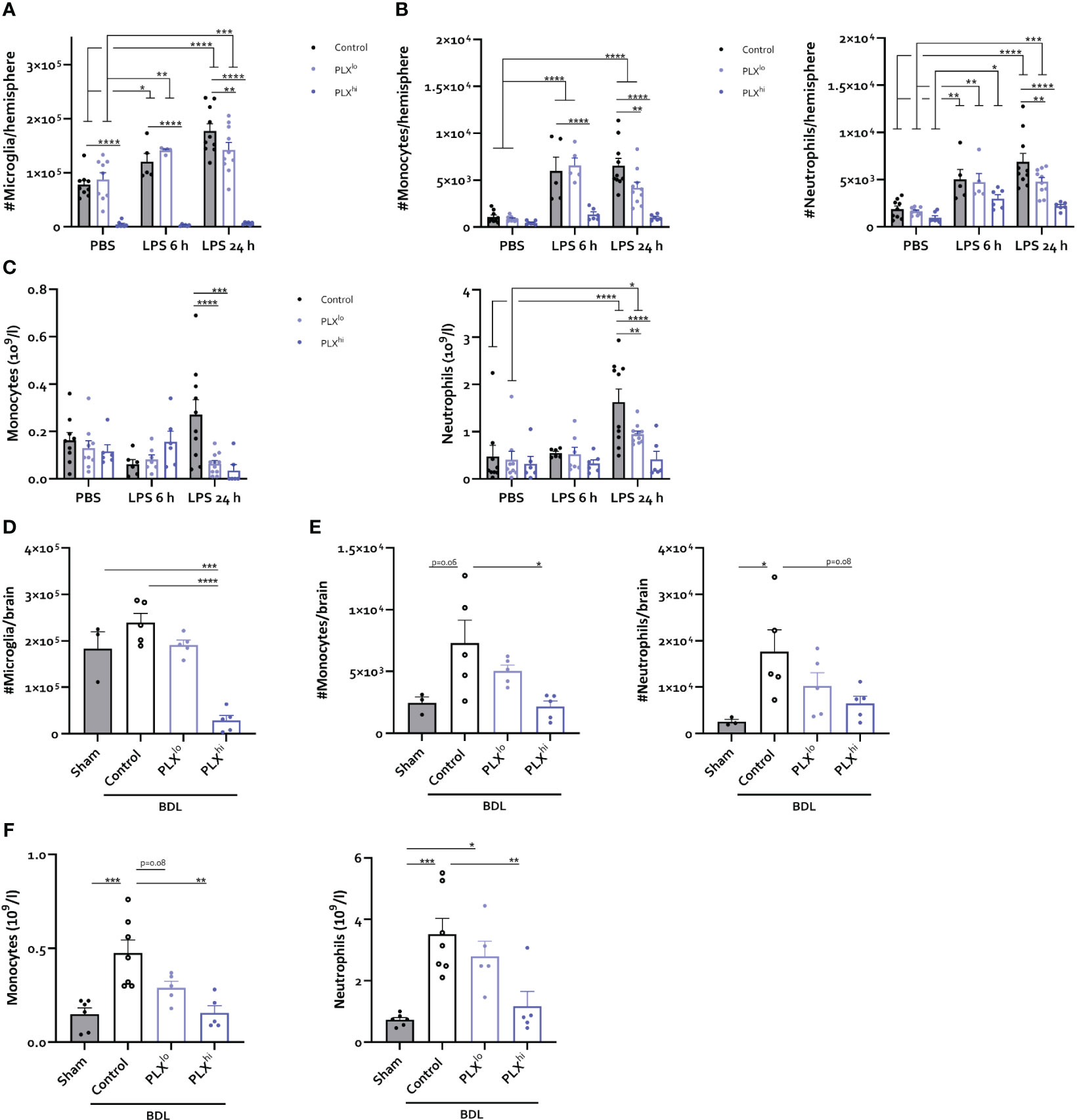
Figure 6 PLX3397 has dose dependent effects on CNS infiltrating and circulating myeloid cells. (A–C) Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. Absolute number of microglia (A), Ly6C+ Monocytes and Ly6G+ neutrophils (B) per hemisphere and levels of circulating monocytes and neutrophils (C). Data are derived from 4 independent experiments with n= 5-10/group. (D–F) Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 from 5 days after BDL surgery. Mice were sacrificed 14 days after induction surgery. Sham mice were used as controls. Absolute number of microglia (D), Ly6C+ monocytes and Ly6G+ neutrophils (E) in the brain and levels of circulating monocytes and neutrophils (F). Data are derived from 2 independent experiments with n= 3-7/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
In line with our previous data (41), a small but non-significant increase in brain cDC counts was observed after BDL. This was not observed in response to LPS. Notably, a decreased cDC count, more pronounced with increasing dose, was detected in PBS-injected mice as well as in the PLX3397-treated BDL mice (Figure S9B, G; Table S6). Analysis of brain lymphoid cells did not reveal any brain influx in either disease model, nor consistent effects of PLX3397 (pre)treatment (Figure S9A, F; Table S6). Circulating lymphocytes were significantly reduced in PLX3397-preteated animals at either dose, 24 hours after LPS injection (Figure S9C; Table S7). In addition, BDL-induced lymphopenia was similar in all animals regardless of CSF1Ri treatment or dose (Figure S9H). As for platelets, we observed development of thrombopenia in LPS-injected mice, which was less severe in case of PLX3397 pretreatment at either dose after 24 hours (Figure S9D). In the BDL model, we rather observed thrombocytosis, which was reduced by PLX3397 treatment in a dose-dependent manner (Figure S9I). In line with data in the steady state experiments, RBC count was significantly lower in PLXhi treated animals compared to untreated controls in either model and at all assessed timepoints (Figure S9D; Table S7). A trend towards decreasing RBC numbers was also found in PLXlo treated animals after BDL (Figure S9J). Finally, LPS induced a transient increase in RBC count after 6 h, but only in control chow and PLXlo fed mice (Figure S9E; Table S7).
PLX induces myeloid and NK cell depletion in the bone marrow
Given the striking effects of PLX3397 treatment at both doses on myeloid cell levels in the brain and circulation, we sought to investigate whether these effects were mediated through PLX3397-induced bone marrow alterations. As illustrated in Figure 7A-D, LPS triggered recruitment of myeloid cells from the bone marrow, leading to extensive decreases in the number of monocytes and neutrophils. At either timepoint after LPS injection, we did not find significant differences between the PLX3397 treatment groups. However, PBS-injected PLXhi pretreated mice exhibited a substantial reduction in the levels of both monocytes and neutrophils compared to control chow fed mice. Significant decreases in these cell types were also observed in the PLXlo treated groups, although to a lesser extent, mirroring the 7-fold difference in ingested doses between both PLX3397 treated groups (Figure 7A, B; Table S8). To evaluate whether reduced monocyte and neutrophil numbers could additionally be attributable to reduced efflux from (and/or production of myeloid cells in) the bone marrow in response to an inflammatory stimulus, we assessed the relative amounts of monocytes and neutrophils in LPS versus PBS controls. A trend towards increased relative monocyte levels compared to control chow treated mice was detected in PLXlo pretreated animals, 24 hours after LPS injection (Figure 7C; Table S9). Relative levels of neutrophils after LPS injection were comparable between PLX treatment groups (Figure 7D; Table S9). Evaluating lymphoid cells and cDCs, we observed reductions of absolute cell numbers in the bone marrow upon LPS injection, similarly indicating recruitment to the blood and other tissues (Figure S10A, B). Strikingly, in the PBS condition, we found reductions of not only cDC counts, but also NK cell counts in both PLXlo and PLXhi treated animals. Similar to our findings in monocytes and neutrophils, relative reduction of cell counts was not affected by PLXhi pretreatment. In PLXlo fed mice, trends for increased residual cell proportions after LPS injection were detected in NK cells, CD8+ T-cells and B-cells, only at 24 hours after LPS injection. This was not the case in cDCs or CD4+ T-cells (Figure S10C, D; Table S9).
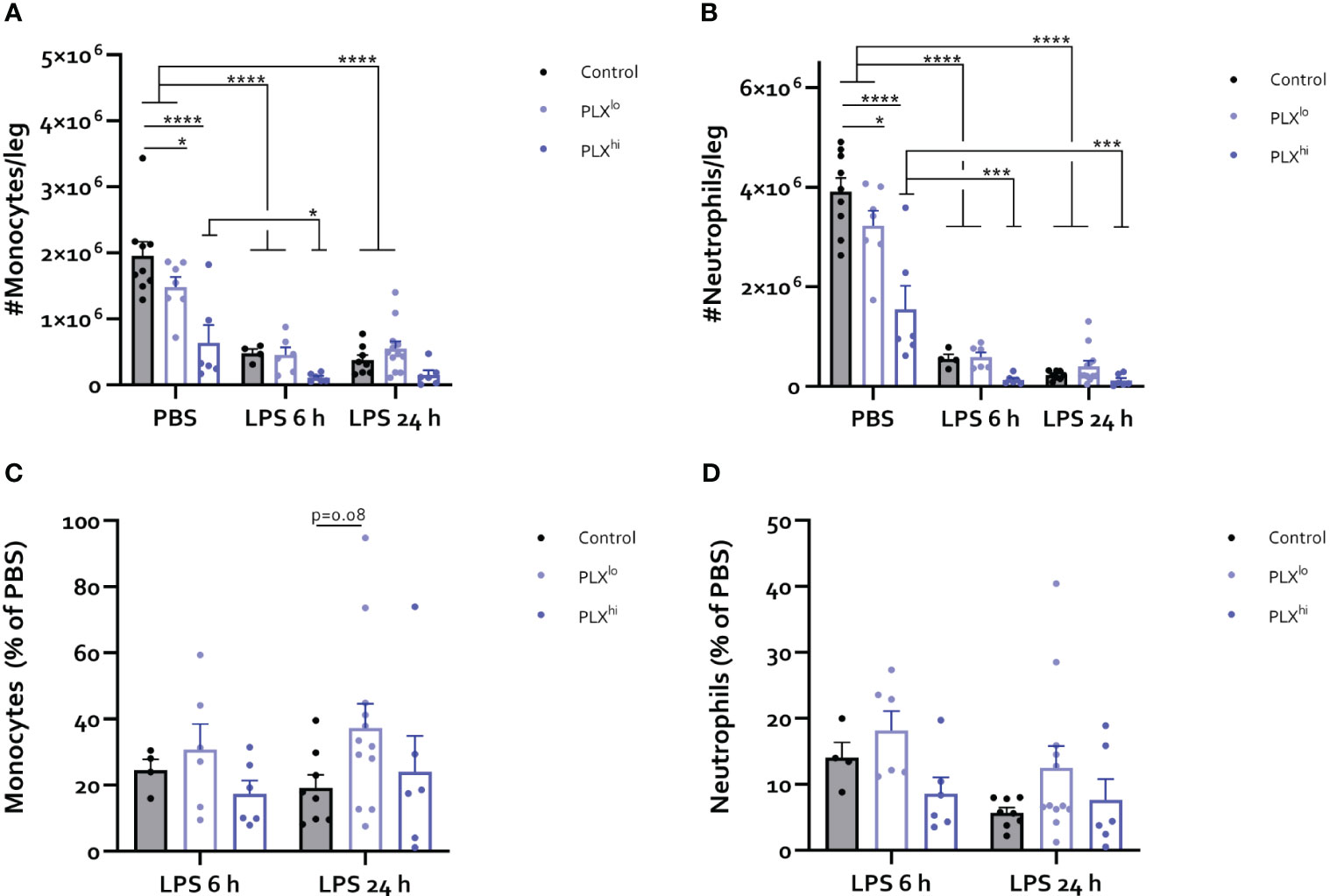
Figure 7 PLX3397 depletes bone marrow monocyte and neutrophil pool. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. (A-D) Absolute number of Ly6C+ monocytes (A) and Ly6G+ neutrophils (B) per leg. Relative amount of Ly6C+ monocytes (C) and Ly6G+ neutrophils (D) after LPS injection, compared to respective PBS controls. Data are derived from 4 independent experiments with n= 4-12/group. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Discussion
The mononuclear phagocyte system (MPS) comprises a group of related cell types including monocytes, tissue macrophages, classical dendritic cell (cDCs) and their progenitors. These cells share the expression of colony-stimulating factor 1 receptor (CSF1R), a tyrosine kinase that plays a crucial role in maturation, proliferation and survival, regulated by its ligands IL-34 and CSF1 (50). Consequently, inhibiting CSF1R signalling can have profound effects on MPS cell abundance and function (15). Blood-brain barrier (BBB) penetrant small molecule CSF1R inhibitors (CSF1Ri) are of particular interest for the study of microglia, the brain resident mononuclear phagocyte. Due to the reversible nature of the inhibition, they make versatile modulation possible (12). However, it is important to recognize the impact of these compounds on non-microglial cells. Unfortunately, the examination of such effects is often inadequate, despite their potential relevance in disease settings when using BBB penetrant CSF1Ri. A comprehensive understanding of these compounds’ broader implications is vital for accurately interpreting experimental outcomes.
Here, we investigated the effects of PLX3397, a commonly used compound for studying microglia in health and disease, on the MPS in the brain, liver, circulation and bone marrow. We confirmed previous finding that a high dose of PLX3397 (600 ppm in chow; PLXhi) depleted microglia within 7 days of administration, while a lower dose (75 ppm; PLXlo) had no effect on microglial cell numbers (17, 18). In addition, we did not find an influx of monocytes into the brain upon pharmacological microglial depletion (17, 45). Interestingly, the impact of PLX3397 extended beyond the central nervous system (CNS) for both doses, leading to a dose-dependent reduction in Kupffer cells (KCs) in the liver, consistent with reversible transaminase increases seen in patients (44, 51) and findings in mice treated with PLX5622, a compound derived from PLX3397, but with higher specificity for the CSF1R over related kinases Fms like tyrosine kinase 3 (Flt3) and c-Kit (25, 30, 52). Confirming data from general macrophage depletion paradigms, ALT elevations did not coincide with clear histopathological liver injury, suggesting a role for KCs in transaminase clearance (53). Although KC depletion is usually associated with a rapid influx of bone marrow-derived monocytes and the development of monocyte-derived KCs (26, 46), we did not observe changes in hepatic or circulating monocyte numbers, suggesting defective recruitment from the bone marrow. Indeed, we found that treatment with PLX3397 induces dose-dependent reductions in bone marrow monocyte numbers, even at a low dose, indicating that there is no dose can selectively deplete microglia without significant bone marrow toxicity. Similarly, PLX5622 reduced the number of bone marrow-derived inflammatory monocytes in Western Nile virus encephalitis without affecting microglial cell counts at a dose of 200 ppm (31). These findings highlight the complexities of using PLX3397 and PLX5622 in microglial studies and underscore the need for careful consideration of their off-target effects on peripheral immune cells.
While under steady state conditions, the impact of PLX3397 on the peripheral MPS may seem less relevant, our data indicate that this is not the case in pathological conditions. In the LPS endotoxemia model, we observed increased mortality in mice upon PLX3397 pretreatment, which is consistent with findings of prolonged sickness behaviour after LPS injection (35) and increased mortality in polymicrobial sepsis (36) in mice pretreated with PLX5622. Additionally, the temperature response was exacerbated. The body weight drop was slightly less pronounced in the PLXhi pretreated group, possibly due to a lower absolute starting body weight. Effects on KCs could only partly account for this increased mortality, as LPS-induced ALT elevations were limited and only slightly exacerbated in high dose pretreated mice. Moreover, LPS clearance was similar in all treatment groups and effects on the hepatic inflammatory response were relatively limited. The inflammatory response in the brain was also decreased, but only in the high dose treatment group, again only partly accounting for the altered phenotype. Interestingly however, at both doses, we found evidence of impaired recruitment of monocytes and neutrophils to the circulation and the brain, linked to reduced bone marrow pool for these cell types. The fact that this was detectable in the low dose treatment group indicates this is a compound-specific effect which can additionally explain the intermediary increase of mortality after LPS injection in the low dose treatment group. Additionally, this indicates that in this set-up, it is impossible to distinguish the role of microglial presence on the myeloid cell infiltration to the brain.
Interestingly, we observed PLX3397-associated reductions in several unexpected cell types. In case of monocytes and cDCs, reduced numbers can be explained by their CSF1R expression (29, 30, 50, 54) and are in line with previous reports using PLX3397 and PLX5622 (28–31). In contrast, neutrophils are believed to not express CSF1R protein, and therefore should not be affected by its inhibition (50, 55). Although one study has reported a similar effect (52), this contradicts most reports using PLX5622 (31, 56), CSF1R blocking monoclonal antibodies (mAbs), and Csf1rΔFIRE/ΔFIRE mice (16, 57). The discrepancy between PLX3397 and PLX5622 suggests that non-CSF1R-related toxicity, which is more likely in PLX3397, might be responsible for these effects, similar to what has been described for oligodendrocyte precursors (58). The reduction in NK cells, while unexpected, aligns with previous reports (28, 29) and might be linked with decreased myeloid cell-derived IL-15, which is necessary for NK cell survival (59).
The finding of increased mortality after LPS when pretreating with PLX3397 was surprising. Monocyte/macrophage and NK cell reductions or depletion experiments have rather been associated with an attenuated response to the LPS model of endotoxemia, which lacks microbial infection (60, 61). Furthermore, the LPS-induced inflammatory response, which putatively drives organ dysfunction and mortality, was not exacerbated, but reduced in our experiments. As neutrophil depletion has been shown to increase mortality in LPS-induced endotoxemia (62), we speculate that this effect on neutrophils partially explains the exacerbated phenotype we observe despite the attenuated cytokine response in PLXhi mice. We additionally can’t exclude that effects of PLX3397 on microglia directly affects the systemic inflammatory response, dampening the systemic inflammation feedback loop. Future research could elucidate the operant mechanism of this exacerbated phenotype.
We further confirmed our findings in the bile duct ligation (BDL) model, which is characterized by liver inflammation with secondary microglial activation and influx of immune cells in the brain (41, 49). Here, we observed significant worsening of liver injury in the PLXhi treated group after BDL, consistent with non-microglial effects of CSF1R inhibition and showing that the PLX off-target effects depend on the disease model studied. This exacerbated liver injury is consistent with findings using other non-specific monocyte/macrophage depletion methods (63, 64), but not KC-specific depletion (65), again implying broad-spectrum effects of PLX3397. Differences in the effect of PLX3397 on LPS/BDL-induced platelet and lymphocyte changes in the blood further underscore that the disease model partly dictates the off-target effects of PLX3397. However, we again observed reduced accumulation of myeloid cells in the BDL brain and circulation at both doses, suggesting that this mechanism is of universal importance when using PLX3397 at any dose in the context of peripheral inflammation.
Our findings offer crucial insights into how bone marrow alterations may play a role in the observed dynamics of myeloid cell responses when using PLX3397-dependent microglia depletion in combination with peripheral inflammation. Of note, using crude CBC analyses, differences were only noticeable when employing an inflammatory trigger, possibly explaining why some studies employing limited evaluation in non-inflammatory conditions could not detect changes in immune cell populations (23, 24). Furthermore, we are the first to perform extensive phenotyping of the bone marrow mature immune cell pool using PLX3397. Our data confirms findings from studies utilizing PLX5622, showing significant impairment of bone marrow monocyte production (30, 31). Our data collectively suggest that PLX3397 can directly or indirectly affect the cell survival of myeloid and NK cells, thus compromising their ability to cope with a proinflammatory stimulus. It’s worth nothing that PLX3397 is known to inhibit kinases similar to CSF1R, namely c-kit and Flt3 (66). These kinases are necessary for normal hematopoiesis and inhibition might consequently contribute to the decreased myeloid cell pool in the bone marrow. Interestingly, a recent report indicates that after 14 days of PLXhi treatment resulted in the expansion, rather than supression, of bone marrow common myeloid and granulocyte-monocyte progenitors, which give rise to monocytes, neutrophils (45). However, it’s worth noting that neutrophil depletion can trigger ‘emergency granulopoiesis’, leading to progenitor expansion (67). This suggests that there is an insufficient ‘emergency hematopoiesis’ response to cope with the reduced mature cell pool, which could explain our seemingly contradictory findings compared to those of Hohsfield et al. ( 45).
Furthermore, our data shed light on the use of PLXlo as a ‘peripheral control’. While PLXlo treatment in some regards produced similar effects on liver, blood and bone marrow as PLXhi, these were consistently less pronounced. For instance, only PLXhi treated mice showed exacerbated liver injury after BDL. Additionally, PLXlo treatment seemed effective in inhibiting LPS-induced microglial accumulation, similar to other small molecule CSF1R kinase inhibitors (68). Moreover, astrocytic glial fibrillary acidic protein (Gfap) upregulation and decreased ionized calcium-binding adapter molecule 1 (IBA1) stained area in PLXlo treated animals might be related to the development of a phagocytic astrocyte phenotype aimed at clearing microglial debris (69, 70), and suggests some degree of microglial cell death even at this dose. It is worth noting that PLXlo treatment did not affect BBB permeability, but we did observe increased permeability of the blood-CSF barrier, which may be attributed to effects on choroid plexus (ChP) stromal macrophages or epiplexus cells. Hence, PLXlo’s impact on the brain cannot be overlooked, while the peripheral effects seen at higher doses are also not entirely replicated. In summary, PLXlo can at best be considered as a partial peripheral control.
An important limitation of our study concerns the dosing of PLX3397. Typically, it is administered in chow at a dose of 290 ppm, about half of the PLXhi dose we employed. At 290 ppm, microglial depletion is achieved within 21 days after the chow administration (17) although differences are reported depending on the chow formulation and animal gender (28). In contrast, the PLXhi dose employed in our study generally leads to rapid and more extensive microglia depletion within 7 days (45, 71). Some of the unique effects observed at the PLXhi dose, such as the reduction in red blood cells (RBCs), might be linked to c-kit inhibition rather than CSF1R blockade. This suggests that the PLXhi dose likely induces relevant effects on hematopoiesis through c-Kit inhibition. These hematopoietic effects have not been observed with the standard 290 ppm dose but have been reported at a dose of 400 ppm (28).
In summary, our data demonstrate that PLX3397 has significant effects on the MPS in the brain, peripheral tissues, and bone marrow. The observed neutrophil depletion and RBC reduction also suggest non-CSF1R mediated effects when using this compound. Giving these off-target effects, results obtained using systemically administered PLX3397 (and PLX5622) for studying microglia in CNS pathology should be interpreted with caution, specifically of CNS disease is driven by peripheral inflammation. However, in other neuroinflammatory diseases, it is crucial to carefully characterize these effects to distinguish microglial-specific effects from generalized CSF1R inhibition effects. Considering the recent identification of the skull bone marrow as a separate important contributor to CNS-infiltrating myeloid cells compared to the blood, effects of PLX3397/PLX5622 on the skull bone marrow should also be evaluated (72, 73). Lower doses of PLX3397, which fail to deplete microglia, can at best serve as partial controls, and their potential (in)direct effects on the CNS should be taken into consideration. Researchers may explore alternative strategies, such as non-BBB penetrant compounds or mAbs, to control for peripheral effects, while considering differences in specificity and activity against CSF1R and related kinases. Another potential approach involves repeated intracerebroventricular injection of PLX3397, as this has been shown to deplete microglia without affecting peripheral tissues or bone marrow (29, 74). Further efforts should be focused on developing long-term and modulatable depletion and repopulation paradigms that are truly microglia-specific to advance our understanding of microglial function in health and disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by Animal Ethics Committee, Faculty of Sciences, Ghent University, Ghent, Belgium. The study was conducted in accordance with the local legislation and institutional requirements. All experiments complied with the current laws of Belgium (Law of 14. August 1986 related to protection and welfare of animals) and EU directive 2010/63/EU, and were approved by the animal ethics committee of the Faculty of Sciences, Ghent University (EC 2022-070, EC2023-034, EC2023-018).
Author contributions
WC: Writing – original draft, Conceptualization, Formal Analysis, Investigation, Methodology, Visualization. DV: Investigation, Writing – review & editing, Methodology. GV: Investigation, Writing – review & editing. EW: Investigation, Writing – review & editing. LV: Investigation, Writing – review & editing. LA: Investigation, Writing – review & editing. FD: Investigation, Writing – review & editing. CS: Methodology, Writing – review & editing. AG: Funding acquisition, Writing – review & editing. CV: Funding acquisition, Methodology, Supervision, Writing – original draft. LV: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft. RV: Conceptualization, Funding Acquisition, Methodology, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. WC, LH, DV and FP are supported by a grants from the Research Foundation – Flanders (11A6420N, 1268823N, 11L2122N, 11D0522N). CS is a Francqui Foundation Professor and supported by an ERC starting grant (MyeFattyLiver, 851908). AG is a senior clinical researcher of the Research Foundation – Flanders (1805718N). RV is supported by a FWO junior research project grant (G055121N). GI is supported by a grant from the Stichting voor Alzheimer onderzoek (SAO) (20200032). This project is supported by VIB.
Acknowledgments
We thank the VIB Flow Cytometry Core, Gert Van Isterdael, Julie Van Duyse and Elien Ruyssinck for training, expert technical assistance and access to the instrument park. We thank the VIB BioImaging core for training and access to the instrument park. We thank Aimée Bugler-Lamb for help with the bone marrow isolation experiments. We thank Joyce Foroozandeh and Pieter Dujardin for technical assistance during sampling. All figures were created using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1283711/full#supplementary-material
Supplementary File 1 | Detailed protocols for behavioural evaluation
Supplementary Figure 1 | Gating strategy for brain cells. (A) Strategy for immune cell phenotyping in brain of sham/BDL mice and PBS/LPS mice. * denotes strategy only employed in BDL/sham mouse brains. (B) Strategy for isolation of astrocytes. Cells are pregated on live singlets.
Supplementary Figure 2 | Gating strategy for bone marrow cells.
Supplementary Figure 3 | Gating strategy for isolation of liver immune cells.
Supplementary Figure 4 | High dose PLX3397 induces anorexia and reduced activity in the open field. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to sacrifice and analysis. (A) Body weight evolution after diet start. Data derived from 2 independent repeats with n=10-11/group. (B) Average daily food intake 7 days after diet start. Data derived from 6 independent repeats with n=11-16/group. (C) Distance travelled in the open field test 6 days after diet start compared to baseline levels and time spent in the center of the open field. (D) Average beam traversal time 6 days after diet start compared to baseline. Behaviour data are derived from 2 independent repeats with n=10-11/group.
Supplementary Figure 5 | PLX3397 affects astrocytes and the blood-CSF barrier. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to sacrifice and analysis. (A-C) Representative flow cytometry plots (A), absolute cell counts of ACSA2+ astrocytes (B) and Gfap mRNA levels in sorted astrocytes (C). (D-I) Representative images and quantification of GFAP (D, E), Olig2 (F, G) and NeuN (H, I) staining in hippocampus. Data are derived from a single experiment with n=5/group. (F) Relative barrier permeability of the BBB and the blood-CSF barrier as assessed by fluorescence intensity in cortex, hippocampus, cerebellum and CSF after IV injection of 4kDa FITC-Dextran. Data are derived from a single experiment with n=6-7/group.
Supplementary Figure 6 | PLX3397 does not affect body weight or temperature response to an LD0 dose of LPS. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. (A-B) Body temperature (A) and relative body weight (B). Data are derived from 4 independent repeats with n=8-13/group.
Supplementary Figure 7 | PLX3397 exacerbates the response to BDL. (A) Experimental set-up. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 from 5 days after BDL surgery. Mice were sacrificed 14 days after induction surgery. Sham mice were used as controls. (B) Survival graphs. (C) Body temperature at sacrifice. Data are derived from 2 independent repeats with n=5-7/group.
Supplementary Figure 8 | High dose PLX3397 exacerbates liver injury in BDL mice. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 from 5 days after BDL surgery. Mice were sacrificed 14 days after induction surgery. Sham mice were used as controls. (A-B) Plasma levels of ALT (A) and bilirubin (B). (C-D) Bile infarctions with representative images of H&E stained sections (C) and quantification of bile infarct area (D). Scale bar represents 100 µm. (E) Plasma LPS levels. (F-G) Hepatic expression level of the macrophage related gene Csf1r, myeloid related gene Itgam (F), inflammatory cytokines (Tnf, Il1b, Il6) and chemokines (Ccl2) (G). All data are derived from 3 independent experiments with n=2-11/group depending on the outcome measure.
Supplementary Figure 9 | Effects of PLX3397 on lymphoid cells, cDCs, red blood cells and platelets are model dependent. (A-E) Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. Absolute counts of CD4+ T-cells, CD8+ T-cells, NK cells (A) and cDCs per hemisphere (B). Circulating levels of lymphocytes (C), platelets (D) and RBCs (E). Data are derived from 4 independent experiments with n= 5-10/group. (F-J) Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 from 5 days after BDL surgery. Mice were sacrificed 14 days after induction surgery. Sham mice were used as controls. Absolute counts of T-cells, B-cells (F) and cDCs per brain (G). Circulating levels of lymphocytes (H), platelets (I) and RBCs (J). Data are derived from 2 independent experiments with n= 3-7/group.
Supplementary Figure 10 | PLX3397 depletes cDCs and NK cells in the bone marrow. Mice were treated with AIN76A chow containing no (control), low (75 ppm; PLXlo) or high (600 ppm; PLXhi) concentrations of PLX3397 for 7 days prior to injection of LPS (2.5 mg/kg, IP). Mice were sacrificed 6 and 24 hours after injection. (A, B) Absolute number of CD4+ T-cells, CD8+ T-cells, B-cells, NK cells (A) and cDCs per leg (B). (C, D) Relative amount of CD4+ T-cells, CD8+ T-cells, B-cells, NK cells (C) and cDCs (D) after LPS injection, compared to respective PBS controls. Data are derived from 4 independent experiments with n= 4-12/group.
Supplementary Table 1 | Antibodies used for flow cytometry
Supplementary Table 2 | Primer sequences used for qPCR
Supplementary Table 3 | Antibodies used for immunofluorescence
Supplementary Table 4 | ANOVA table of brain gene expression levels in PBS/LPS injected mice pretreated with different doses PLX3397 chow
Supplementary Table 5 | ANOVA table of liver gene expression levels in PBS/LPS injected mice pretreated with different doses PLX3397 chow
Supplementary Table 6 | ANOVA table of absolute counts of brain immune cells in PBS/LPS injected mice pretreated with different doses PLX3397 chow
Supplementary Table 7 | ANOVA table of absolute counts of circulating cells, plasma ALT and LPS in PBS/LPS injected mice pretreated with different doses PLX3397 chow
Supplementary Table 8 | ANOVA table of absolute counts of bone marrow immune cells in PBS/LPS injected mice pretreated with different doses PLX3397 chow
Supplementary Table 9 | ANOVA table of relative amounts bone marrow immune cells LPS injected mice pretreated with different doses PLX3397 chow, compared to PBS controls
Abbreviations
ALT, Alanine aminotransferase; BBB, Blood-brain barrier; BDL, Bile duct ligation; CBC, Complete blood count; cDC, Classical dendritic cell; ChP, Choroid plexus; CNS, Central nervous system; CSF, Cerebrospinal Fluid; CSF1, Colony-stimulating factor 1; CSF1R, Colony-stimulating factor 1 receptor; CSF1Ri, Colony-stimulating factor 1 receptor kinase inhibitor; Flt3, Fms like tyrosine kinase 3; GFAP, Glial fibrillary acidic protein; IBA1, Ionized calcium-binding adapter molecule 1; IP, Intraperitoneal; IV, Intravenous; KC, Kupffer cell; LPS, Lipopolysaccharide; MPS, Mononuclear phagocyte system; mAb, Monoclonal antibody; NK Cell, Natural killer cell; OFT, Open field test; PBS, Phosphate buffered saline; PLXhi, PLX3397 (600 ppm); PLXlo, PLX3397 (75 ppm); RBC, Red blood cell; RT, Room temperature; SEM, Standard error of the mean.
References
1. Hume DA. The mononuclear phagocyte system. Curr Opin Immunol (2006) 18:49–53. doi: 10.1016/j.coi.2005.11.008
2. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
3. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
4. Hume DA, Caruso M, Ferrari-Cestari M, Summers KM, Pridans C, Irvine KM. Phenotypic impacts of CSF1R deficiencies in humans and model organisms. J Leukocyte Biol (2020) 107:205–19. doi: 10.1002/JLB.MR0519-143R
5. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi: 10.1126/science.1194637
6. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
7. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18:225–42. doi: 10.1038/nri.2017.125
8. Spiteri AG, Wishart CL, Pamphlett R, Locatelli G, King NJC. Microglia and monocytes in inflammatory CNS disease: integrating phenotype and function. Acta Neuropathol (2022) 143:179–224. doi: 10.1007/s00401-021-02384-2
9. Ju H, Park KW, Kim I, Cave JW, Cho S. Phagocytosis converts infiltrated monocytes to microglia-like phenotype in experimental brain ischemia. J Neuroinflamm (2022) 19:190. doi: 10.1186/s12974-022-02552-5
10. Honarpisheh P, Lee J, Banerjee A, Blasco-Conesa M. P, Honarpisheh P, d’Aigle J, et al. Potential caveats of putative microglia-specific markers for assessment of age-related cerebrovascular neuroinflammation. J Neuroinflamm (2020) 17:366. doi: 10.1186/s12974-020-02019-5
11. Grassivaro F, Menon R, Acquaviva M, Ottoboni L, Ruffini F, Bergamaschi A, et al. Convergence between microglia and peripheral macrophages phenotype during development and neuroinflammation. J Neurosci (2020) 40:784–95. doi: 10.1523/JNEUROSCI.1523-19.2019
12. Green KN, Crapser JD, Hohsfield LA. To kill a microglia: A case for CSF1R inhibitors. Trends Immunol (2020) 41:771–84. doi: 10.1016/j.it.2020.07.001
13. Kondo Y, Duncan ID. Selective reduction in microglia density and function in the white matter of colony-stimulating factor-1-deficient mice. J Neurosci Res (2009) 87:2686–95. doi: 10.1002/jnr.22096
14. Wang Y, Szretter K. J, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol (2012) 13:753–60. doi: 10.1038/ni.2360
15. Dai X-M, Ryan G. R, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi: 10.1182/blood.V99.1.111
16. Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun (2019) 10:3215. doi: 10.1038/s41467-019-11053-8
17. Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
18. Spangenberg E, Severson P. L, Hohsfield L. A, Crapser J, Zhang J, Burton E. A, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
19. Barca C, Foray C, Hermann S, Herrlinger U, Remory I, Laoui D, et al. The colony stimulating factor-1 receptor (CSF-1R)-mediated regulation of microglia/macrophages as a target for neurological disorders (Glioma, stroke). Front Immunol (2021) 12:787307. doi: 10.3389/fimmu.2021.787307
20. Han J, Chitu V, Stanley ER, Wszolek ZK, Karrenbauer VD, Harris RA. Inhibition of colony stimulating factor-1 receptor (CSF-1R) as a potential therapeutic strategy for neurodegenerative diseases: opportunities and challenges. Cell Mol Life Sci (2022) 79:219. doi: 10.1007/s00018-022-04225-1
21. Green KN, Hume DA. On the utility of CSF1R inhibitors. Proc Natl Acad Sci USA (2021) 118:e2019695118. doi: 10.1073/pnas.2019695118
22. Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep (2014) 9:2124–38. doi: 10.1016/j.celrep.2014.11.018
23. Groh J, Klein D, Berve K, West BL, Martini R. Targeting microglia attenuates neuroinflammation-related neural damage in mice carrying human PLP1 mutations. Glia (2019) 67:277–90. doi: 10.1002/glia.23539
24. Ma D, Klein D, Berve K, West BL, Martini R. A novel hydrogel-based treatment for complete transection spinal cord injury repair is driven by microglia/macrophages repopulation. Biomaterials (2020) 237:119830. doi: 10.1016/j.biomaterials.2020.119830
25. Santamaria-Barria JA, Zeng S, Greer JB, Beckman MJ, Seifert AM, Cohen NA, et al. Csf1r or Mer inhibition delays liver regeneration via suppression of Kupffer cells. PloS One (2019) 14:e0216275. doi: 10.1371/journal.pone.0216275
26. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity (2019) 51:638–654.e9. doi: 10.1016/j.immuni.2019.08.017
27. Merry TL, Brooks AES, Masson SW, Adams SE, Jaiswal J. K, Jamieson SMF, et al. The CSF1 receptor inhibitor pexidartinib (PLX3397) reduces tissue macrophage levels without affecting glucose homeostasis in mice. Int J Obes (2020) 44:245–53. doi: 10.1038/s41366-019-0355-7
28. Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med (2019) 216:2546–61. doi: 10.1084/jem.20190980
29. Han J, Fan Y, Zhou K, Zhu K, Blomgren K, Lund H, et al. Underestimated peripheral effects following pharmacological and conditional genetic microglial depletion. IJMS (2020) 21:8603. doi: 10.3390/ijms21228603
30. Lei F, Cui N, Zhou C, Chodosh J, Vavvas D. G, Paschalis E. I. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc Natl Acad Sci USA (2020) 117:23336–8. doi: 10.1073/pnas.1922788117
31. Spiteri AG, Ni D, Ling ZL, Macia L, Campbell IL, Hofer MJ, et al. PLX5622 reduces disease severity in lethal CNS infection by off-target inhibition of peripheral inflammatory monocyte production. Front Immunol (2022) 13:851556. doi: 10.3389/fimmu.2022.851556
32. Coulibaly AP. Neutrophil modulation of behavior and cognition in health and disease: The unexplored role of an innate immune cell. Immunol Rev (2022) 311:177–86. doi: 10.1111/imr.13123
33. Xie J, Van Hoecke L, Vandenbroucke RE. The impact of systemic inflammation on alzheimer’s disease pathology. Front Immunol (2022) 12:796867. doi: 10.3389/fimmu.2021.796867
34. Kiani Shabestari S, Morabito S, Danhash EP, McQuade A, Sanchez JR, Miyoshi E, et al. Absence of microglia promotes diverse pathologies and early lethality in Alzheimer’s disease mice. Cell Rep (2022) 39:110961. doi: 10.1016/j.celrep.2022.110961
35. Vichaya EG, Malik S, Sominsky L, Ford B. G, Spencer SJ, Dantzer R. Microglia depletion fails to abrogate inflammation-induced sickness in mice and rats. J Neuroinflamm (2020) 17:172. doi: 10.1186/s12974-020-01832-2
36. Chung H-Y, Wickel J, Hahn N, Mein N, Schwarzbrunn M, Koch P, et al. Microglia mediate neurocognitive deficits by eliminating C1q-tagged synapses in sepsis-associated encephalopathy. Sci Adv (2023) 9:eabq7806. doi: 10.1126/sciadv.abq7806
37. Van Campenhout S, Van Vlierberghe H, Devisscher L. Common bile duct ligation as model for secondary biliary cirrhosis. In: Vinken M, editor. Experimental cholestasis research, vol. 1981 . Springer New York (2019). p. 237–47.
38. Tag C, Weiskirchen S, Hittatiya K, Tacke F, Tolba R, Weiskirchen R. Induction of experimental obstructive cholestasis in mice. Lab Anim (2015) 49:70–80. doi: 10.1177/0023677214567748
39. Percie du Sert N, Ahluwalia A, Alam S, Avey M. T, Baker M, Browne WJ, et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PloS Biol (2020) 18:e3000411. doi: 10.1371/journal.pbio.3000411
40. Steeland S, Gorlé N, Vandendriessche C, Balusu S, Brkic M, Van Cauwenberghe C, et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol Med (2018) 10:e8300. doi: 10.15252/emmm.201708300
41. Claeys W, Van Hoecke L, Lernout H, De Nolf C, Van Imschoot G, Van Wonterghem E, et al. Experimental hepatic encephalopathy causes early but sustained glial transcriptional changes. J Neuroinflamm (2023) 20:130. doi: 10.1186/s12974-023-02814-w
42. Vandesompele J, Preter KD, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol RESEARCH0034 (2002) 3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034
43. De Luca SN, Sominsky L, Soch A, Wang H, Ziko I, Rank MM, et al. Conditional microglial depletion in rats leads to reversible anorexia and weight loss by disrupting gustatory circuitry. Brain Behavior Immun (2019) 77:77–91. doi: 10.1016/j.bbi.2018.12.008
44. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay J-Y, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet (2019) 394:478–87. doi: 10.1016/S0140-6736(19)30764-0
45. Hohsfield LA, Najafi AR, Ghorbanian Y, Soni N, Crapser J, Figueroa Velez D. X, et al. Subventricular zone/white matter microglia reconstitute the empty adult microglial niche in a dynamic wave. eLife (2021) 10:e66738. doi: 10.7554/eLife.66738.sa2
46. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
47. Vandenbroucke E. R. A hidden epithelial barrier in the brain with a central role in regulating brain homeostasis. Implications for aging. Ann ATS (2016) 13:S407–10. doi: 10.1513/AnnalsATS.201609-676AW
48. Lewis AJ, Seymour CW, Rosengart MR. Current murine models of sepsis. Surg Infections (2016) 17:385–93. doi: 10.1089/sur.2016.021
49. Claeys W, Van Hoecke L, Geerts A, Van Vlierberghe H, Lefere S, Van Imschoot G, et al. A mouse model of hepatic encephalopathy: bile duct ligation induces brain ammonia overload, glial cell activation and neuroinflammation. Sci Rep (2022) 12:17558. doi: 10.1038/s41598-022-22423-6
50. Grabert K, Sehgal A, Irvine KM, Wollscheid-Lengeling E, Ozdemir DD, Stables J, et al. A transgenic line that reports CSF1R protein expression provides a definitive marker for the mouse mononuclear phagocyte system. J Immunol (2020) 205:3154–66. doi: 10.4049/jimmunol.2000835
51. Benner B, Good L, Quiroga D, Schultz TE, Kassem M, Carson WE, et al. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. DDDT (2020) 14:1693–704. doi: 10.2147/DDDT.S253232
52. Warden AS, Triplett TA, Lyu A, Grantham EK, Azzam MM, DaCosta A, et al. Microglia depletion and alcohol: Transcriptome and behavioral profiles. Addict Biol (2021) 26(2). doi: 10.1111/adb.12889
53. Radi ZA, Koza-Taylor PH, Bell RR, Obert LA, Runnels HA, Beebe JS, et al. Increased serum enzyme levels associated with kupffer cell reduction with no signs of hepatic or skeletal muscle injury. Am J Pathol (2011) 179:240–7. doi: 10.1016/j.ajpath.2011.03.029
54. Louis C, Cook AD, Lacey D, Fleetwood AJ, Vlahos R, Anderson GP, et al. Specific contributions of CSF-1 and GM-CSF to the dynamics of the mononuclear phagocyte system. J Immunol (2015) 195:134–44. doi: 10.4049/jimmunol.1500369
55. Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ, et al. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J Leukocyte Biol (2007) 82:111–23. doi: 10.1189/jlb.1206713
56. Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja-Capdevila N, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol (2019) 137:321–41. doi: 10.1007/s00401-018-1954-4
57. MacDonald KPA, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood (2010) 116:3955–63. doi: 10.1182/blood-2010-02-266296
58. Liu Y, Dickson EL, Owens GP, Macklin WB, Bennett JL. Concentration-dependent effects of CSF1R inhibitors on oligodendrocyte progenitor cells ex vivo and. vivo. Exp Neurol (2019) 318:32–41. doi: 10.1016/j.expneurol.2019.04.011
59. Beffinger M, De Lara PT, Tugues S, Vermeer M, Montagnolo Y, Ohs I, et al. CSF1R-dependent myeloid cells are required for NK−mediated control of metastasis. JCI Insight (2018) 3:e97792. doi: 10.1172/jci.insight.97792
60. Guo Y, Luan L, Wang J, Patil N, Bohannon J, Rabacal W, et al. Interleukin-15 enables septic shock by maintaining natural killer cell integrity and function. J Immunol (2017) 198:208.15–5. doi: 10.4049/jimmunol.198.Supp.208.15
61. Xiang B, Zhang G, Guo L, Li X-A, Morris AJ, Daugherty A, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation. via cyclooxygenase 1 signalling pathway. Nat Commun (2013) 4:2657. doi: 10.1038/ncomms3657
62. Reber LL, Gillis CM, Starkl P, Jönsson F, Sibilano R, Marichal T, et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med (2017) 214:1249–58. doi: 10.1084/jem.20161238
63. Gehring S, Dickson EM, San Martin ME, Van Rooijen N, Papa EF, Harty MW, et al. Kupffer cells abrogate cholestatic liver injury in mice. Gastroenterology (2006) 130:810–22. doi: 10.1053/j.gastro.2005.11.015
64. Osawa Y, Seki E, Adachi M, Suetsugu A, Ito H, Moriwaki H, et al. Role of acid sphingomyelinase of Kupffer cells in cholestatic liver injury in mice. Hepatology (2010) 51:237–45. doi: 10.1002/hep.23262
65. De Muynck K, Vanderborght B, De Ponti FF, Gijbels E, Van Welden S, Guilliams M, et al. Kupffer cells contested as early drivers in the pathogenesis of primary sclerosing cholangitis. Am J Pathol (2023) 193:366–79. doi: 10.1016/j.ajpath.2022.12.008
66. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
67. Cain DW, Snowden PB, Sempowski GD, Kelsoe G. Inflammation triggers emergency granulopoiesis through a density-dependent feedback mechanism. PloS One (2011) 6:e19957. doi: 10.1371/journal.pone.0019957
68. Olmos-Alonso A, Schetters STT, Sri S, Askew K, Mancuso R, Vargas-Caballero M, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain (2016) 139:891–907. doi: 10.1093/brain/awv379
69. Konishi H, Okamoto T, Hara Y, Komine O, Tamada H, Maeda M, et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J (2020) 39(22):e104464. doi: 10.15252/embj.2020104464
70. Zhou T, Li Y, Li X, Zeng F, Rao Y, He Y, et al. Microglial debris is cleared by astrocytes via C4b-facilitated phagocytosis and degraded via RUBICON-dependent noncanonical autophagy in mice. Nat Commun (2022) 13:6233. doi: 10.1038/s41467-022-33932-3
71. Najafi AR, Crapser J, Jiang S, Ng W, Mortazavi A, West B. L, et al. A limited capacity for microglial repopulation in the adult brain. Glia (2018) 66:2385–96. doi: 10.1002/glia.23477
72. Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science (2021) 373:eabf7844. doi: 10.1126/science.abf7844
73. Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci (2018) 21:1209–17. doi: 10.1038/s41593-018-0213-2
Keywords: microglia, bile duct ligation (BDL), endotoxemia, lipopolysaccharide (LPS), PLX3397, peripheral inflammation, neuroinflammation
Citation: Claeys W, Verhaege D, Van Imschoot G, Van Wonterghem E, Van Acker L, Amelinck L, De Ponti FF, Scott C, Geerts A, Van Steenkiste C, Van Hoecke L and Vandenbroucke RE (2023) Limitations of PLX3397 as a microglial investigational tool: peripheral and off-target effects dictate the response to inflammation. Front. Immunol. 14:1283711. doi: 10.3389/fimmu.2023.1283711
Received: 26 August 2023; Accepted: 23 October 2023;
Published: 22 November 2023.
Edited by:
Wassim Elyaman, Columbia University, United StatesReviewed by:
Min-Soo Kwon, CHA University, Republic of KoreaJohannes Steffen, Otto-von-Guericke-Universität, Germany
Copyright © 2023 Claeys, Verhaege, Van Imschoot, Van Wonterghem, Van Acker, Amelinck, De Ponti, Scott, Geerts, Van Steenkiste, Van Hoecke and Vandenbroucke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roosmarijn E. Vandenbroucke, Um9vc21hcmlqbi5WYW5kZW5icm91Y2tlQGlyYy5WSUItVUdlbnQuYmU=
†These authors have contributed equally to this work