 Farooq Riaz
Farooq Riaz Zhihui Huang
Zhihui Huang Fan Pan
Fan Pan- Shenzhen Institute of Advanced Technology (SIAT), Chinese Academy of Sciences (CAS), Shenzhen, China
A healthy immune system is pivotal for the hosts to resist external pathogens and maintain homeostasis; however, the immunosuppressive tumor microenvironment (TME) damages the anti-tumor immunity and promotes tumor progression, invasion, and metastasis. Recently, many studies have found that Foxp3+ regulatory T (Treg) cells are the major immunosuppressive cells that facilitate the formation of TME by promoting the development of various tumor-associated cells and suppressing the activity of effector immune cells. Considering the role of Tregs in tumor progression, it is pivotal to identify new therapeutic drugs to target and deplete Tregs in tumors. Although several studies have developed strategies for targeted deletion of Treg to reduce the TME and support the accumulation of effector T cells in tumors, Treg-targeted therapy systematically affects the Treg population and may lead to the progression of autoimmune diseases. It has been understood that, nevertheless, in disease conditions, Foxp3 undergoes several definite post-translational modifications (PTMs), including acetylation, glycosylation, phosphorylation, ubiquitylation, and methylation. These PTMs not only elevate or mitigate the transcriptional activity of Foxp3 but also affect the stability and immunosuppressive function of Tregs. Various studies have shown that pharmacological targeting of enzymes involved in PTMs can significantly influence the PTMs of Foxp3; thus, it may influence the progression of cancers and/or autoimmune diseases. Overall, this review will help researchers to understand the advances in the immune-suppressive mechanisms of Tregs, the post-translational regulations of Foxp3, and the potential therapeutic targets and strategies to target the Tregs in TME to improve anti-tumor immunity.
Introduction
During the formation and growth of the tumor, immune cells not only suppress the tumor but also shape the immunogenicity of cancer cells in three distinct phases of cancer immunoediting, including the elimination phase (tumor cells show strong immunogenicity), equilibrium phase (tumor cells managed to survive to exhibit low immunogenicity), and escape phase (tumor cells escape immune recognition) (1). In the tumor microenvironment (TME), there is a continuous interaction between tumor cells and immune cells. Exploring the intrinsic mechanism demonstrating this interaction between the tumor and the immune cells is essential to find targeted therapeutic strategies. Meanwhile, several tumor-associated cell types, including tumor-associated macrophages (TAMs), tolerogenic dendritic cells (DCs), and myeloid-derived suppressive cells (MDSCs) have been seen in the TME, which play different functions compared to their normal cell types (2, 3). Besides, TME disrupts immune cell function by manipulating glucose metabolism, lactic acid metabolism, hypoxia, and tumor-associated cells. However, the involvement of various tumor-associated cell types and the interaction of immune and cancer cells in TME lowers the efficiency of a targeted drug to treat a specific tumor.
In the TME, several novel CD4+ T subsets have been identified through research in recent years. Among these subsets, one of the most important milestone is the discovery of regulatory T (Treg) cells, which are crucial for the immune system to deal with self-antigen and maintain homeostasis by suppressing the immune response to foreign pathogens or self-tissue response (4, 5). Tregs are characterized by constitutive expression of the transcription factor forkhead box protein P3 (Foxp3) (6). Foxp3 is a member of the forkhead box (Fox) family, subfamily P. This subfamily has four members in mammals, namely Foxp1-4 (7). Foxp3 is the most conserved across mammals. For instance, the similarity between the amino acid sequences of human and mouse Foxp3 is 91% (8). Foxp3, an oligomeric molecule and a critical transcription factor, comprises four key domains: an N-terminal domain (amino acids 1-97) for transcriptional control, a central zinc-finger domain (amino acids 197-222) involved in interactions and possibly oligomer formation, a leucine-zipper domain (amino acids 239-260) crucial for protein interactions, and a C-terminal forkhead domain (amino acids 337-423) which allows Foxp3 to bind specific DNA sequences, regulating gene expression. Mutations in these domains can lead to autoimmune disorders like IPEX syndrome, highlighting their collaborative role in the function of Tregs (9–12). The Foxp3 promoter is activated by a range of transcription factors, including NFAT and AP-1, in response to TCR signaling and co-stimulation pathways (4). Additionally, Forkhead box protein O (FOXO) proteins, specifically FOXO1 and FOXO3, have been observed binding to the Foxp3 promoter and other regulatory elements of the Foxp3 gene (13). CREB (cAMP response element binding protein)-activating transcription factor 1 (ATF1) complexes also contribute to Foxp3 promoter activation (14). Notably, Foxp3 promoter exhibits modest trans-activating potential, with its regulation heavily dependent on conserved enhancer regions. These interactions play a crucial role in the regulation of Foxp3 gene expression (4).
Foxp3 plays a central role in the maturation and immunosuppression function of Tregs by regulating the gene expression of the target gene through direct interaction and supporting the immunosuppressive microenvironment by expressing suppressive molecules of Tregs (6, 15). It is evident that loss or gain of Foxp3 also minimizes or maximizes the immunosuppression function of Tregs, which ultimately attenuates severe autoimmune diseases or tumor development (16). Besides the genetic manipulation of Foxp3 to regulate its functions, transcriptional and post-translational modifications (PTM) have also been recognized to regulate Foxp3 function (15, 17). The highly specific and versatile PTMs provide new insights into Foxp3 functions and therapeutic approaches. In this review, we will discuss PTMs of Foxp3 and emphasize the function and suppressive mechanisms of Tregs in tumors. Moreover, we will also highlight the potential of Treg-targeted immunotherapy.
Immunosuppressive mechanism of Treg in the tumor microenvironment
CD4+CD25+Foxp3+Tregs are the immunosuppressive cells that play pivotal roles in maintaining immune homeostasis. Tregs maintain their function through a variety of inhibitory pathways. However, Tregs not only play an active role in healthy humans but also plays a side effect in patients with tumors or inflammation by inhibiting the effector T, DC, and NK cells, thereby promoting the occurrence and development of tumors in patients, which results in poor prognosis (18). It has been illustrated that Granzyme B is not expressed in nTreg but is highly expressed in 5%-30% of tumor infiltrating Treg (19). Tumor-derived Tregs mediate immunosuppressive effects by expressing granzyme B and perforin to induce apoptosis of effector T and NK cells. At the same time, Tregs can mediate apoptosis by expressing FasL-Fas (Figure 1) (19–22). Interleukin 2 (IL-2) is necessary for the suppressive function and the survival of Tregs (23). Tregs produce a low amount of IL-2 cytokine but highly express IL-2R, of which CD25 is the α chain of IL-2R. IL-2 capture is dispensable for CD4+ T cells, but IL-2 deprivation can limit the activation of CD8+ T cells, which require IL-2 cytokine to activate and maintain their cytotoxic effect. Thus, large amounts of IL-2R in Tregs deprive effector T activation and enable the apoptosis-mediated inhibition effect of Tregs (24, 25).
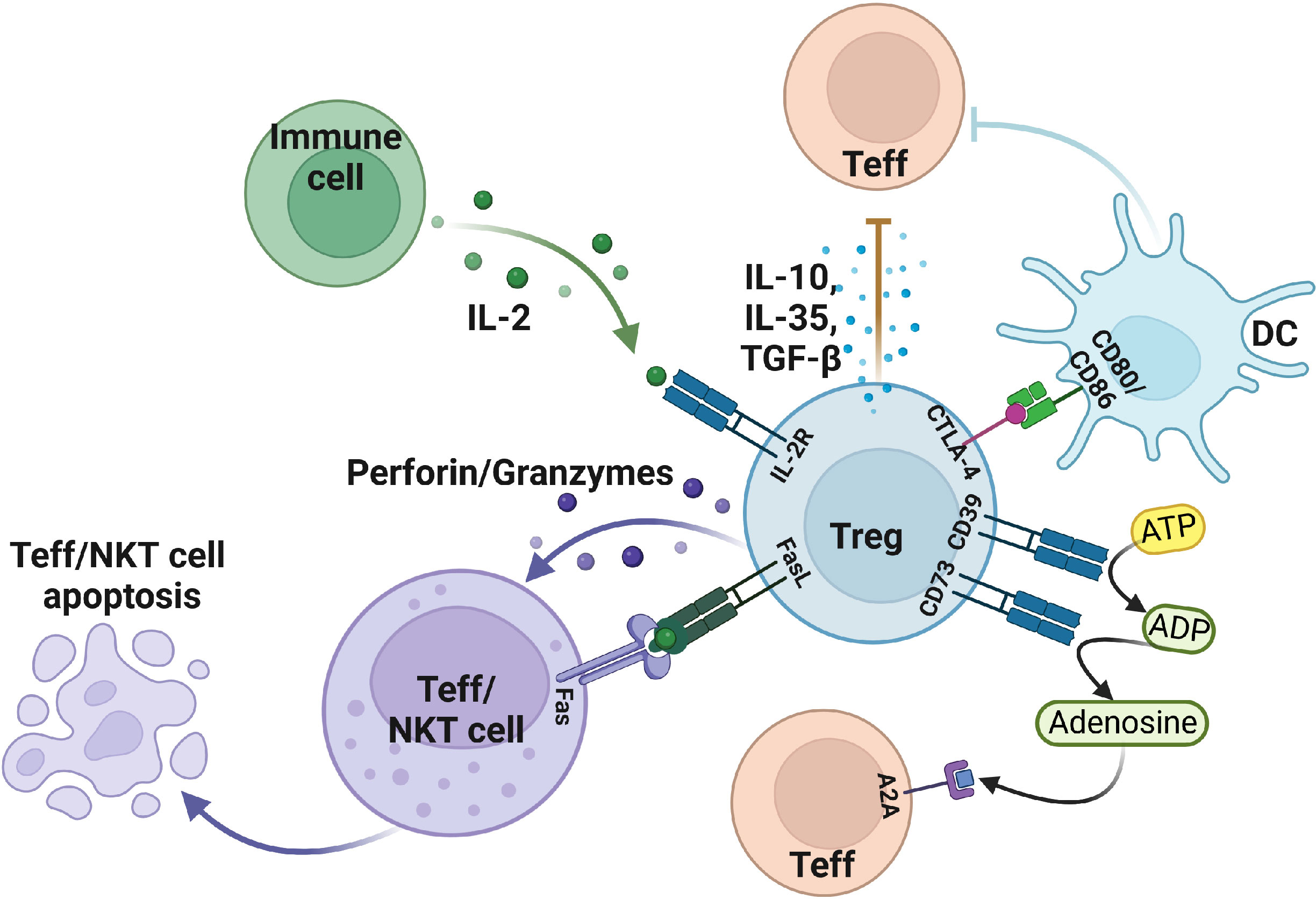
Figure 1 The suppressive function of Treg in the tumor microenvironment. Treg cells induce apoptosis of effector T and NKT cells by expressing FasL, granzyme, perforin, and IL-2R. In addition, Treg also secretes IL-10, TGF-β, IL-35, and other cytokines to make effector T cells less functional. Moreover, Treg cells mediate DC, and effector T cell status by expressing negative regulatory receptors. Figure was created with BioRender.com.
In addition to the secretion of suppressive cytokines, Treg cells can exert suppressive functions through cell surface receptors. Treg cells generally express inhibitory cytotoxic T lymphocyte-associated protein 4 (CTLA-4) on the cell surface, whereas Tconv can also express it. CTLA-4 has two functions in Tregs: 1) protect against autoimmune diseases; 2) avoid excessive inflammatory responses. CTLA4+ Tregs can downregulate the CD80/CD86 in DC cells to inhibit their maturation which can eventually reduce the activation of effector T cells (26, 27). On the other hand, CTLA-4 interacts with CD80/CD86 and induces DC to express indoleamine 2,3 dioxygenase (IDO) catabolize tryptophan which can inhibit the response of effector T cells in vivo (28). Some studies found that DCs from interferon γ (IFN-γ)-receptor-deficient mice don’t affect the induction of IDO, indicating that IFN-γ is not necessary for Tregs (29).
The enzymatic activities of the CD39 and CD73 receptors also mediate Treg immunosuppression. Mechanistically, CD39/CD73 turns a pro-inflammatory extracellular microenvironment into an anti-inflammatory microenvironment through ADP/ATP to AMP and then AMP to adenosine, respectively. This adenosine binds to the adenosine receptor A2A and activates an immune regulatory pathway to mediate the differentiation of Th1 and Th2 (30, 31). Treg cells were discovered to transform DCs into a functional and phenotypically tolerogenic form by producing IL-10 and TGF-β (32). Tumor-derived Tregs induce the generation of tolerogenic DCs, which finally reduce the expression of co-stimulatory receptors and the secretion of pro-inflammatory cytokines, including IL-6, IL-12, and TGF-β. Therefore, tolerogenic DCs can’t effectively stimulate the other effector T cells, thus suggesting another Treg-mediated form of immunosuppression (33–36).
Similarly, Treg cells can inhibit pro-inflammatory macrophages and guide macrophages to differentiate into anti-inflammatory cytokine phenotypes through immunomodulatory effects. TGF-β and IL-10 are the key inhibitory cytokines in this process (37). In addition, tumors overexpressing IDO inhibit the recruitment and functional activation of myeloid-derived suppressor cells (MDSCs) after the systemic deletion of Foxp3+ Treg cells (38). Aryl-hydrocarbon receptor (AHR) plays a distinct role in immune cells, particularly in the development of Tregs in multiple diseases (39). Some research reports that tryptophan catabolism by IDO/TDO acts as AHR ligands and promotes immunosuppressive effects in different tumors. The immunosuppressive effects mediated by IDO-Kyn-AHR depend on the interaction of Treg and tumor-associated macrophages (TAMs). Selective inhibition of AHR combined with PD-1 therapy can effectively delay tumor growth (40). Collectively, the immunosuppressive mechanism of Tregs in TME is highly cross-linked and comprehensive, which further needs investigation.
Post-translational modifications of Foxp3
Post-translational modifications (PTMs) refer to cellular processes that manipulate the expression of a specific protein and significantly impact the overall characteristics of that particular protein, including the turnover, interaction, localization, etc. (41). Consequently, Foxp3 is also highly regulated by numerous processes involving PTMs, including acetylation, glycosylation, phosphorylation, ubiquitylation, and methylation (39). These PTMs of Foxp3 can restrict the function of Tregs in maintaining the immunosuppressive microenvironment (42). In this part, we will discuss recent studies highlighting the role of these PTMs on Foxp3 stability and function.
Acetylation modification of Foxp3
Acetylation is an important process that regulates the expression of Foxp3 (Figure 2A). In this process, the coordination of histone deacetylases (HDACs) and histone acetyltransferases (HTAs) introduces or removes an acetyl group to modify a protein (43). HTAs can stabilize protein structure that binds to the target gene to regulate its expression, while HDACs overturn this process (44). Acetylation has been well studied to influence the PTMs of Foxp3. It has been shown that Foxp3 is more stable under the influence of acetylation compared to deacetylation because the acetylation of Foxp3 hinders its degradation. Briefly, it was illustrated that proteasome-mediated ubiquitylation degradation is impaired during the acetylation of lysine residues, which allows the binding of Foxp3 to the chromatin, thus regulating the expression of the downstream gene (45–47).
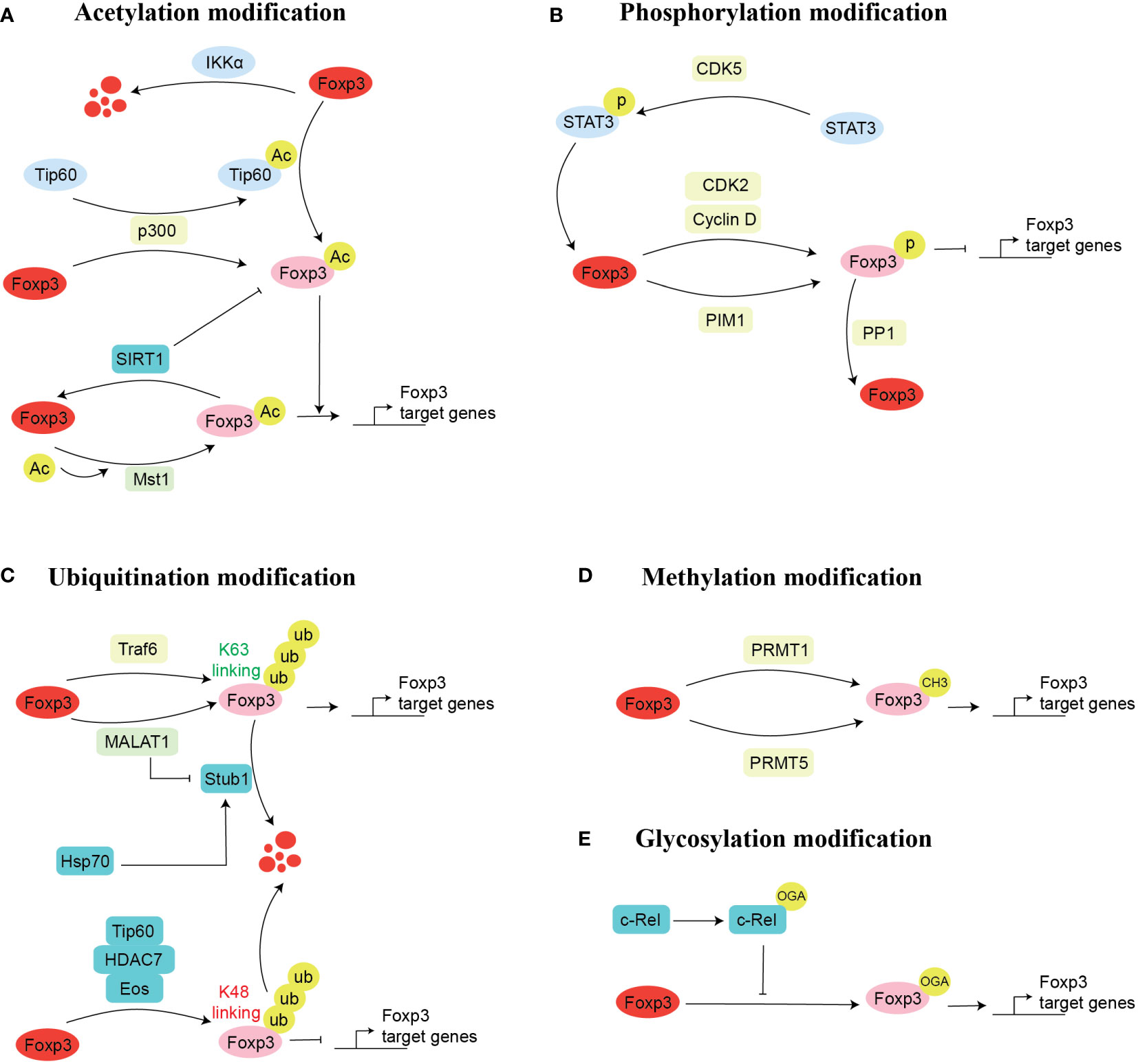
Figure 2 Mechanism of Foxp3 post-transcriptional modification. There are five possible ways for the post-transcriptional modifications of Foxp3. These are acetylation (A), phosphorylation (B), ubiquitination (C), methylation (D), and glycosylation (E). Here, we mentioned the enzymes and proteins and their cellular processes involved in post-transcriptional modifications of Foxp3. Acetylation modification: acetylation (Tip60, p300), deacetylation (SIRT1), promotes acetylation (p300) and inhibits acetylation (Mst1); Glycosylation modification: O-GlcNAcylation (c-Rel); Phosphorylation modification: phosphorylation (CDK2, PIM1, PIM2), dephosphorylation (PPP1); Ubiquitination modification: ubiquitination (TRAF6, Stub1), inhibits ubiquitination (MALAT1); Methylation modification: methylation (PRMT1, PRMT5).
The long-term imbalance of Treg/Th17 causes multiple cancers and chronic inflammatory diseases, including cancer, metabolic diseases, and autoimmunity (48–51). Numerous acetylases, i.e., Tip60 (Histone acetyltransferase KAT5) (52), CBP/p300 (p300-CBP coactivator family) (53), and MST1 (Macrophage Stimulating 1) (54), have been found to modulate the Foxp3 acetylation in different ways, thus regulating the suppressive function of Tregs. Mechanistically, in rheumatoid arthritis (RA), naïve CD4+CD45RO- T produces fewer Foxp3 proteins, which results in the differentiation of naïve T cells into Th17 commitment, while leads to fewer Treg populations, which affects the Th17/Treg balance (52). Detailed molecular mechanism indicate that T cells in RA reduce their ability to produce enough histone acetyltransferase Tip60, leading to a decreased Foxp3 acetylation (52). The complex of Foxp3:Tip60 and Foxp3:HDAC7, and Foxp3:HDAC9 is necessary for the repression by Foxp3 (55). Foxp3 mutation also affects the function of Tregs. P.A384T IPEX mutation in patient-derived Treg cells has been associated with a decreased suppressive function of Tregs. Still, it maintains the ability of Foxp3 to suppress the production of inflammatory cytokines (56). This phenomenon is due to the restriction of Foxp3A384T binding with Tip60 (56). In addition, allosteric modifiers can enhance the Foxp3-Tip60 interaction and the activity of Foxp3 (56).
CBP, a p300 paralog, is crucial in regulating the differentiation of Tregs (57) and the expression of Foxp3 (53). CBP/p300 is closely related to acetyltransferase and transcriptional coactivators of Foxp3. Evidence indicates that Treg-specific deletion of CBP or p300 develops minimal autoimmune disease. In contrast, Treg-specific deletion of both CBP and p300 creates serious consequences and the death of mice in 3 to 4 weeks (53). However, using Garcinol, a natural p300 inhibitor, reduces the suppressive function of Treg cells and improves anti-tumor immunity (58). In addition, some studies have found that p300 activates Tip60 through acetylation, which acetylates Foxp3 and maintains its function and stability. At the same time, Foxp3 can also be directly acetylated by interaction with p300, which can avoid proteasome-mediated Foxp3 degradation (47).
The histone deacetylases (HDACs) can negatively regulate the Foxp3 expression and Treg function (59). Inhibition of HDACs elevates and stabilizes the expression of Foxp3 and sustains the suppressive function of Tregs (59–61). Recent data demonstrated that the acetylation of Foxp3 can be reversed by class III HDAC SIRT1 (Sirtuin1) (62). Briefly, using the major histocompatibility complex (MHC)-mismatched cardiac allografts model, researchers found that targeted deletion of SIRT1 in CD4+T cells or Treg cells extends the survival of mice. This demonstrated that SIRT1 could negatively regulate the Treg function (62). Meanwhile, in abdominal aortic aneurysms patients and Hashimoto’s thyroiditis patients, SIRT1 is highly expressed in infiltrated CD4+ T cells, and the frequency of Treg is significantly decreased in these patients (63, 64). Therefore, inhibition of SIRT1 inhibited the acetylation of Foxp3, which is essential for maintaining the suppressive function of Treg and the stable expression of Foxp3 (63, 64). Conversely, Mammalian Sterile 20-like Kinase 1(Mst1) is another serine/threonine kinase that plays an important role in immune cell development (65). In Tregs, the part of Mst1 is opposite to that of SIRT1. It is evident that Mst1 can interact with Foxp3 and enhances the activity of Foxp3 by stabilizing it in vitro and in vivo (54). Interestingly, Mst1 could attenuate the SIRT1-dependent deacetylation of Foxp3 by impeding the interaction of SIRT1 and Foxp3 (47, 54). In our recent study, we have optimized that IκB Kinase α (IKKα), which PPARα (Peroxisome proliferator–activated receptor α) transcriptionally controls, regulates the Th17 development by modifying the Foxp3 expression. We found that overexpression of IKKα enhances Th17 development and leads to the proteasomal degradation of Foxp3 (66). Likewise, nucleus accumbens-associated protein-1 (NAC1) enhances the deacetylation of various proteins by interacting with HDACs (67). Nevertheless, targeted depletion of NAC1 accelerated the acetylation of Foxp3, whereas overexpression of NAC1 can decrease the acetylation of Foxp3 and may lead to the progression of autoimmune disease (68). Overall, it is well-understood that various proteins influence the acetylation of Foxp3, which interferes with the immune suppressive function of Tregs by affecting the stability of Foxp3.
Glycosylation modification of Foxp3
Glycosylation-dependent PTMs significantly affect proteins’ activity, stability, conformation, folding, and distribution by attaching sugars (glycans and monosaccharides) to proteins through covalent binding (69). Changes in glycosylation are well associated with various cellular functions of T cells, including differentiation, development, activation, and apoptosis (70, 71). O-linked-N-acetylglucosaminylation (O-GlcNAcylation) is particular glycosylation that involves the attachment of a monosaccharide sugar, O-GlcNAc, to the serine or threonine residues (72, 73). During the process of O-GlcNAcylation, two enzymes, namely O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), catalyze the whole process by mediating the addition and removing the modification, respectively (74). Notably, O-GlcNAc is highly associated with Treg lineage stability, effector differentiation, and suppressive function (74). Mechanistically, several O-GlcNAcylation sites are present in Foxp3, which upon glycosylation mediates Foxp3 expression by regulating IL-2/STAT5 signaling pathway. Genetic ablation of OGT in Foxp3+ cells decreases Treg cell lineage stability, which can eventually elevate the severity of autoimmune diseases (74). Besides the direct role of O-GlcNAc on Foxp3, it can also influence the activity of Foxp3 binding partners. It was noted that c-Rel, a binding partner of Foxp3 (75), undergoes glycosylation, either by hyperglycemic- or chemically-induced O-GlcNAcylation, and reduces the binding of c-Rel with Foxp3 promoter; thus, and negatively regulates the immunosuppressive function of Tregs by reducing Foxp3 expression (76). Although a few studies have investigated the role of glycosylation in Tregs and Foxp3 regulation, the potential of O-GlcNAcylation targeted therapy in modulating the function of Tregs should be investigated to achieve anti-tumor and anti-autoimmune immunity (Figure 2E).
Phosphorylation modification of Foxp3
Foxp3 also undergoes phosphorylation in the amino-terminal domain of Foxp3, which is modified at several sites (77) (Figure 2B). Specific protein kinase phosphorylation can regulate Treg development and stabilize the suppressive function of Tregs. Among these, cyclin-dependent kinases (CDK2) (78), proto-oncogene serine/threonine-protein kinase-1 (PIM1) (79), and PIM2 (77) kinase phosphorylate the Foxp3, thus negatively regulating it’s expression. Alternatively, PP1 (protein phosphatase 1) kinase negatively regulates Foxp3 by dephosphorylation (80), while CDK5 kinase could regulate Foxp3 complex protein by phosphorylation STAT3 (signal transducer and activator of transcription 3) (81).
Different mechanisms to regulate the phosphorylation of Foxp3 have been identified depending on the nature of protein kinases. For instance, CDK2 can bind to its partner cyclin E to phosphorylate Foxp3 at CDK motifs (S19, S88, T114, T175). This process could be inhibited by using the CDK inhibitor roscovitine (78). Similarly, PIM1 could negatively regulate Fopx3 expression by specific phosphorylation at Ser422. However, phosphorylation of Foxp3 at the Ser418 region could inhibit this phosphorylation at Ser422 (79). Interestingly, depletion of PIM1 increased Foxp3 binding activity and elevated the Foxp3-induced gene expression, such as CTLA-4, CD25, and GITR (Glucocorticoid-induced TNFR related protein), in human Tregs and decreased the expression of IL-2 gene, which ultimately improved the immunosuppression function of Tregs (79). In parallel to PIM1, PIM2, which also belongs to the PIM serine/threonine kinase family, was found to interact with Foxp3 in the human Tregs. Researchers found that the amino acid phosphorylation sites of PIM2 are located at Ser31 and Ser41. In addition, inhibition of PIM2 expression can increase the suppressive function of Treg cells and elevate the stabilization of Treg cell lineages (77).
Besides, CDK5 and PP1 regulate Foxp3 protein in different ways. As for PP1 can dephosphorylate Foxp3 at the Ser418 site in the C-terminal DNA-binding domain to decrease Treg-mediated suppressive function, particularly in the model of rheumatoid arthritis (82). Briefly, TNF-α produced in the inflamed synovium can induce the expression and enzymatic activity of PP1, which dephosphorylates Foxp3 and mediates a decrease in Treg suppressive function. Using a TNF-α specific antibody can restore the suppressive function of Treg in rheumatoid arthritis (82). In contrast, CDK5 phosphorylating Ser722 of STAT3, which promotes the transport of STAT3 to the nucleus and binds to the enhancer II region of Foxp3, increases the transcription and expression of Foxp3 and responds to the IL-6. Drug inhibition or targeted deletion of CDK5 can effectively attenuate Foxp3 expression and the suppressive ability of T cells (81). Thus, it can be suggested that phosphorylation of Foxp3 through various proteins can affect the function of Tregs.
Ubiquitylation modification of Foxp3
The ubiquitination process involves the addition of a 3.5kDa ubiquitin protein to the target protein, which is driven by the action of ubiquitinates, including E1(ubiquitin-activating enzyme), E2(ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) (83). In the process of ubiquitination, adding a ubiquitin-protein to a target protein is monoubiquitylation; however, adding a chain of ubiquitin proteins to a target protein is polyubiquitylation (84, 85). Ubiquitination has been found to regulate various cellular and biological processes, including the cell cycle, transcriptional regulation, apoptosis, inflammatory response, and cell differentiation by targeting different proteins (86).
Ample evidence suggests the modification of Foxp3 by ubiquitination, which eventually regulates the Foxp3 expression and plays a vital role in maintaining the suppressive function and stability of Treg cells (87) (Figure 2C). For instance, lysine 48(K48)-linked polyubiquitylation often acts on target proteins for their proteasomal degradation, while K63-linked polyubiquitylation often acts in signal transduction cascades in cells (88). Meanwhile, polyubiquitylation is also associated with other lysine residues, such as K6, K11, K27, K29, and K33 (89–91). Our previous finding suggests that K63-linked ubiquitination plays an essential role in regulating the immunosuppression of Treg cells and the expression of Foxp3. Briefly, we found that TNF receptor-associated factor 6 (TRAF6) is ubiquitin-ligase, which regulates the K63 linked ubiquitination is used by the Foxp3 for its nuclear localization and its improvement in the transcription factor gene regulation activity in Treg by K63-linked ubiquitination (90). Conversely, another group of scientists found that TRAF6 interacts with Foxp3 by ubiquitination modification at lysine residue 262, which is necessary for the suppressive function of Treg, and they observed dysfunctional Tregs in vivo in TRAF6 depleted mice. Importantly, Treg-specific TRAF6 depleted cells attenuated tumor size and boosted anti-tumor immunity (92). Overall, these studies urge the potential of targeting TRAF6 to regulate Treg function.
Numerous stress factors, inflammatory cytokines, and lipids decrease the expression of Foxp3. It was found that this decrease in Foxp3 is modified by ubiquitination through E3 ubiquitin ligase Stub1 (STIP1 homology and U-box containing protein 1) (93). Mechanistically, the interaction between Stub1 and Foxp3 relies on the stress indicator protein Hsp70 (heat shock protein 70). Hsp70 recruits Stub1 and acts as a subunit of the Foxp3 complex, and the silencing of Stub1 expression enhances Treg suppressive function in vitro and in vivo (93). Unlike the Stub1 protein, MALAT1 (metastasis associated lung adenocarcinoma transcript 1) can increase Foxp3 expression through another mechanism. MALAT1 generally covers the Stub1 interaction region of Foxp3 with modified proteins so that it cannot be modified by Stub1 ubiquitination, resulting in stable expression of Foxp3 (94). Apart from that, MALAT1 also plays a key role in the post-transcriptional modification of Foxp3, which affects the GINS1 (GINS Complex Subunit 1) transcription and ultimately leads to the progression of NSCLC (94).
Loss of Foxp3 expression is linked with the pathogenesis of multiple autoimmune diseases, including type 1 diabetes in NOD mice (95). This was further testified by Matthew et al. in a Foxp3gfp reporter mouse on NOD background mice. They illustrated that nTreg development and function were not significantly different in vitro in Foxp3gfp NOD and C57BL/6 mice, as autoimmune diabetes on an NOD background was accelerated (96). In contrast, Treg cell development was reduced under inflammation or induction by TGF-β. This was because Foxp3gfp could not interact with Tip60, HDAC7, and EOS, which reduced the stability of Foxp3. Correspondingly, the enhanced K48-linked polyubiquitination resulted in Foxp3 degradation, decreased Treg development, and immunosuppressive function (96–98). Collectively, it can be reported that the ubiquitylation of Foxp3 plays a significant role in the stability and function of Foxp3.
Methylation modification of Foxp3
In addition to the PTMs mentioned above, methylation of transcription factors or co-stimulatory factors is also important for the regulation of gene expression (99). Altered methylation of Foxp3 is linked with the onset of multiple diseases (100, 101) (Figure 2D). The main sites of methylation modification are arginine and lysine residues (102). Multiple pieces of evidence suggest that protein arginine methyltransferase (PRMT) family members PRMT1 and PRMT5 regulate the suppressive function of Treg cells by introducing methyl group to regulate Foxp3 transcription activity and exert a vital role in autoimmune and cancer diseases (103–106).
A deeper understanding revealed that the Foxp3 transcription factor is methylated at arginine residues at 48 and 51 via PRMT1 (105). Methylation at these two sites promotes Treg cell-mediated suppressive function. The use of MS023 (type I protein arginine methyltransferase (PRMT) inhibitor) confers Th1-like gene expression profiles to Foxp3-expressing T cells (105). Importantly, PRMT1 also plays a crucial role in the differentiation between Th17 and Treg. PRMT1 is associated with RORγt and regulates Th17 differentiation. It was found that overexpression of PRMT1 effectively promotes the Th17 expansion, but inhibition or deletion of PRMT1 reduced the Th17 differentiation, which ultimately leads to an increased Foxp3+ Tregs population (107). Therefore, it can be assumed that the methylation of transcription factors can effectively regulate gene expression through methyltransferases.
In addition, the mass spectrometric analysis showed that FOXP3 can be di-methylated at positions R27, R51, and R146 by PRMT5. Whereas arginine(R)51 is mutated to Lysine(K), leading to the loss of suppressive function in human CD4 T cells (106). These PRMT5 knockout mice can develop severe scurfy-like autoimmune diseases in mice by showing an impaired number of Treg cells in the spleen, while no change was observed in the Tregs from peripheral lymph nodes (106). However, the peripheral Treg with PMRT5 knockout exhibited decreased suppressive function (106). DS-437 (Pharmacological inhibitors for PMRT5) can enhance anti-erbB2/neu monoclonal antibody targeted therapy (106). In contrast, Zheng et al. described that PRMT5 inhibition in mice can resolve autoimmune diseases by elevating the number of Treg cells (108). These studies conclude that PRMT1 and PRMT5 are necessary for post-transcriptional methylation of Foxp3. Meanwhile, further studies are required to fully understand the role of PRMTs in PTMs and the function of Foxp3. Overall, these studies urge that targeting the methyltransferases may serve as potential therapeutic targets to achieve Treg-mediated immunotherapy.
Targeting Treg as a potential immunotherapy
Targeted deletion of Tregs in tumors is a fascinating and promising treatment; however, deleting Tregs can cause autoimmune diseases and affect the effectiveness of treatment. Thus, researchers are interested in developing therapies that selectively target the immunosuppressive Tregs in tumors without disrupting the normal inflammatory response, effector T cells, or causing autoimmune diseases. Since CD25 is a highly expressed surface marker of Treg cells, targeting CD25 to delete Treg cells is a suitable choice. In mice, the anti-CD25 antibody can effectively delete Treg in vivo and enhance anti-tumor immunity (109, 110). An earlier study in clinics demonstrated that using an anti-CD25 antibody along with daclizumab to delete Treg in breast cancer patients successively eliminates the CD4+CD25+Foxp3+ Treg cells in peripheral blood. Using daclizumab combined with a specific tumor antigen vaccine can effectively activate the production of toxic T lymphocytes (111). However, the effector T cells also express CD25 and require IL-2, and the use of daclizumab also affects their killing function.
Targeting CCR8 has also been presented as an effective therapy to target the Tregs. It has been illustrated that usage of anti-CCR8 alone or in combination with other immune checkpoint inhibitors promotes the anti-tumor immune response by depleting the intratumoral Tregs (112–115). Meanwhile, Sugiyama et al. reported that targeting CCR4 in Tregs by using anti-CCR4 can selectively deplete the Tregs and augment the response of tumor-specific CD8+ T cells, thereby improving the anti-tumor immune response (116). Another attractive therapeutic target site is CTLA-4. Technically, the competitive binding of CTLA-4 on CTLs with CD80/CD86 on APCs bypasses CD28 on APCs and suppresses hyperactivated cytotoxic T cells (117). But using immune checkpoint mAbs, ipilimumab, and tremelimumab (118), avoids a decline in immune responses, increases the number of activated T cells, and promotes T cell–tumor cell interactions (119). Some studies have found that ipilimumab can induce Fc-mediated antibody-dependent cytotoxicity (ADCC)-mediated Treg cell reduction in Pancreatic cancer due to the infiltration of FcγR-dense myeloid cells in the tumor, which increases the sensitivity to CTLA-4 mAbs (120, 121). In contrast, tremelimumab does not bind to the FcγR of human leukocytes to release the IL-2 cytokine, avoiding cytokine release syndrome. However, the antibody can enhance IL-2 cytokine production by T cells in healthy individuals or patients with tumors, including ovarian, renal, prostate, and rectal cancers (122). Programmed cell death protein 1 (PD-1), an inhibitory receptor for tumor-specific T cells, can bind to the PD-L1 ligand produced by tumor cells to inhibit T cell effector function. CTLA-4 and PD-1 inhibitory immune checkpoints can suppress the activation of effector T cells. Interestingly, combined treatment with CTLA-4 mAbs (Ipilimumab) and PD-1 (nivolumab) mAbs can kill tumor cells to the greatest extent and delay the occurrence and development of tumors, including metastatic melanoma, metastatic renal cell carcinoma, colorectal cancer with MSI-H and MMR aberrations (123–125). Interestingly, it has been validated that use of anti–PD-1 decreases the tumor infiltrating Tregs (126). Meanwhile, the use of anti-CTLA-4 doesn’t influence the population of Foxp3+ Tregs in the tumors (127). These new findings suggest that CTLA-4 and PD-1 may serve as novel target for the future in different tumor therapy.
In addition to the receptors mentioned above, numerous other molecules are also expressed on the surface of Treg cells, which may serve as target sites for Treg deletion. GITR belongs to glucocorticoid-induced TNF-receptor family-related protein, which is highly expressed on the surface of naïve Treg cells and intermediately expressed on the surface of naïve CD4/CD8, naïve myeloid cells. Some researchers have shown that the anti-GITR monoclonal antibody DTA-1 can effectively destabilize Treg, leading to the loss of Foxp3 expression, which is essential for maintaining Treg suppressive function (128–131). However, another study reported that GITR-dependent therapies are context-dependent as GITR modulates the balance between effector CD4+ T cells and Tregs by elevating their proliferation of both populations in parallel (132, 133). OX40 is another co-stimulatory molecule of the TNF receptor family, expressed in activated CD4/CD8 T cells and constitutively expressed on most Tregs (134, 135). It appears to act on Treg in a different mechanism compared to GITR. Conjugation of OX40 to agonist OX40-specific reagents can effectively enhance anti-tumor immunity and regulate the generation of effector and memory T cells (136, 137). Using agonist OX40 can effectively delete intertumoral Treg while combining chemotherapy can directly modulate the inhibitory effect of intertumoral Treg (138, 139). Some preclinical experiments have shown that OX40 ligation can weaken the suppressive function of Treg. Thus, GITR and OX40 are novel immunotherapy targets currently being tested in clinical studies (140).
Similarly, WO2017011559A1 (anti-CCL20 Abs) can bind to CCL20 and inhibit the interaction between CCR6 and CCL20, thereby mediating the inhibition of Treg/Th17 recruitment to the tumors, which can inhibit cancer stem cell activity and the tumorigenesis (141). However, inhibiting CCR6 will also affect the recruitment of other anti-tumor cells or lead to the onset of autoimmune diseases in vivo because Tregs are also recruited to other tissues by CCR6 to play an immunosuppressive role (142). Alternatively, research studies have shown the detrimental effects of Tregs-targeted immunotherapy on the body and other organs. Therefore, more efforts should be put into achieving Treg-targeted therapy with minimal adverse effects and prolonging the survival of the patients.
Targeting PTMs in Tregs as a potential therapeutic strategy
Meanwhile, we also summarized the mechanisms defining the suppressive function of Tregs, particularly through PTMs, such as phosphorylation, glycosylation, ubiquitination, acetylation, and methylation. We demonstrated that these PTMs might contribute to fine-tuning Treg function, stability, differentiation, and lineage development. Considering the diverse links between Tregs and TME regulation, various potential drugs targeting PTMs have demonstrated their efficacy in in-vivo and clinical studies, which influence the function and differentiation of Tregs. For instance, pharmacological downregulation of PRMT5 by using DS-437 can decrease the overall population of Tregs by significantly reducing Foxp3 methylation (106). Conversely, a new study investigating the impact of D1ManPrup3, a synthetic glycodendropeptides, on the Treg-dependent immune tolerance and desensitization suggests that the use of D1ManPrup3 may lead to methylation changes in Foxp3 and alternatively influence the function of Tregs (143). Meanwhile, a recently recognized drug Qi-Dong-Huo-Xue-Yin (QD), which is a traditional Chinese medicine widely used in treating COVID-19 patients, showed a positive role in the development of Tregs by enlightening the Foxp3 acetylation in CD4+ T cells (144).
Although various studies have identified the drugs involved in diminishing the population of Tregs in tumors, how to advance Treg-specific targeting remains challenging. In recent years, strategies to target Tregs systematically or locally by using ligand-directed toxins or monoclonal antibodies have been adopted (145). Nevertheless, not all healthcare problems respond effectively to such treatment methods, particularly autoimmune disorders and other inflammation-related disorders. In order to target Tregs specifically, similar to other cell-type-specific medications, a computational tool and platform must be developed. The most efficient initial step in accomplishing this is to avoid tackling the isoforms of Foxp3. In clinics, four HDAC inhibitors are used to achieve anti-tumor responses in the USA (59). For instance, the usage of Trichostain A (TSA), a class I/II HDAC inhibitor, enhanced the proportion of Treg cells and their immunosuppressive activity (146), which was mainly associated with the inhibition of class IIb HDACs (59). Since HDAC-6 and -10 are members of class IIb HDAC and have been shown to have more significant roles in Treg activities in vivo, an inhibitor specifically designed to target HDAC6 may eventually provide the best immunotherapeutic response (59, 147). In parallel, targeting E3 ligases is a crucial additional strategy for achieving Treg-specific targeted therapy (148). The use of nanoparticles to specifically target Tregs is also fascinating. Recent investigations have employed uniquely designed particles to deliver antigens or medications to Treg cells (149–152), implying that targeting PTMs through small-molecule inhibitors may also be employed in developing new therapeutic strategies.
A recently developed approach, Proteolysis Targeting Chimaeras (PROTACs), represents an innovative technique in the realm of pharmacology, offering a solution to the challenge of targeting proteins that were previously considered hard to target. While conventional medications typically bind to target proteins, PROTACs function as molecular orchestrators, initiating the specific degradation of particular proteins within cells (153). Comprising a ligand for the protein of interest (POI), a ligand for an E3 ubiquitin ligase, and a connecting linker, PROTACs instigate a cascade of events (153, 154). They facilitate rapid ubiquitination and subsequent proteasomal degradation of the target protein by effectively bridging the POI and the E3 ligase, bringing them into close proximity (154, 155). In a recent study, anti-apoptotic B-cell lymphoma extra-large (BCL-XL) was identified as a potential therapeutic target within tumor-infiltrating Tregs. The authors used a specialized PROTAC molecule, which induced the BCL-XL degradation, leading to Treg apoptosis within the tumor microenvironment, suggesting a novel strategy to boost anti-tumor immune responses (156). Similarly, a previous study uncovered a process involving the degradation of the Foxp3 protein in response to LPS stimuli facilitated by the E3 ligase STUB1 and the heat shock protein HSP70. This degradation effectively impairs the suppressive function of Tregs (93). Although studies using the PROTACs in targeting Tregs are very limited, we believe that this innovative approach holds great promise in drug development, offering a precise means of modulating cellular processes by selectively eliminating Foxp3, with potential in anti-tumor immunity.
Conclusion and perspectives
Over the past decades, researchers have been working to understand the suppressive function of Tregs in the TME and the factors regulating these suppressive functions of Tregs and the Foxp3 expression. So far, the complex interaction between Treg and various intertumoral cells hasn’t been completely studied. It is still unknown which Treg inhibitory mechanism plays the main role or whether multiple inhibitory mechanisms work together. Here, we have also summarized the role of various factors in mitigating the Treg accumulation in tumors and the immunosuppressive mechanisms of Treg. Finally, we recapitulated the advances in Treg-targeted immunotherapy including CD25, CTLA-4, PD-1, OX-4O, GITR, chemokines receptors, and ligands.
This review also summarizes various PTMs that are vital in regulating Foxp3 protein stability and immunosuppressive functions. These PTMs, which can occur at different sites, help in initiating, terminating, or fine-tuning the Treg-dependent immune responses. Importantly, protein complexes create a dynamic network of PTMs that govern the function of Foxp3 by altering the physical and chemical properties, affecting its conformation, function, and interactions with Foxp3 transcription factor complexes. Additionally, Foxp3 PTMs may play a role in context-specific gene regulation by altering the stability of Foxp3 transcription factor complexes. However, multiple key challenges persist in targeting PTMs in achieving anti-tumor immunity. The hurdles and uncertainties include: (1) how metabolic-linked PTMs modulate Treg function; (2) hard to forecast the effect of specific modifications on the development and function of Tregs; (3) the effects of various endogenous factors, such as microbiota in TME, on the Treg function is not known; (4) the factors which influence the role of Foxp3 transcription factor complexes between transcriptional repressor and activator are not fully known; (5) the mechanism by which transcription factor complexes lead to the PTMs of Foxp3 are not entirely understood; (6) an efficient and effective delivery and evaluation of drugs targeting PTMs in Tregs is necessary to determine the potential and cost-effectiveness of Treg-targeted anticancer treatment.
The significance of this discovery lies in the critical role of Tregs in maintaining immune balance, where both their reduction and elevation can, respectively, contribute to autoimmune diseases and cancers. Consequently, developing drugs that target the interaction of Foxp3 with its binding partners, either restrict or enhance their interaction, holds promise for the development of Treg-specific treatments with potential applications in immune-related disorders and cancer therapy. Overall, understanding the complex signaling complexes that are involved in crosstalk between Foxp3 and several PTMs, and clarifying the association of these complex networks will highlight the Treg biology in immune health and diseases. Also, this PTM machinery will help us identify the novel targets for developing Treg-targeted drugs to achieve anti-tumor and anti-autoimmune immune responses.
Author contributions
FR: Conceptualization, Writing – original draft, Writing – review & editing. ZH: Writing – original draft. FP: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China (2022YFC2403000) and (2021YFC2400500), the National Natural Science Foundation of China (Grant 32170925), Shenzhen Science and Technology Program (KQTD20210811090115019), the Shenzhen Science and Technology Program (JCYJ2022081800807016), the startup fund of SIAT and CAS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
2. Wang H, Franco F, Ho PC. Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer (2017) 3(8):583–92. doi: 10.1016/j.trecan.2017.06.005
3. Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer (2021) 9(7):e002591. doi: 10.1136/jitc-2021-002591
4. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
5. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer (2010) 127(4):759–67. doi: 10.1002/ijc.25429
6. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30(5):636–45. doi: 10.1016/j.immuni.2009.04.010
7. Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer (2013) 13(7):482–95. doi: 10.1038/nrc3539
8. Jackson BC, Carpenter C, Nebert DW, Vasiliou V. Update of human and mouse forkhead box (FOX) gene families. Hum Genomics (2010) 4(5):345–52. doi: 10.1186/1479-7364-4-5-345
9. Xie X, Stubbington MJ, Nissen JK, Andersen KG, Hebenstreit D, Teichmann SA, et al. The regulatory T cell lineage factor foxp3 regulates gene expression through several distinct mechanisms mostly independent of direct DNA binding. PloS Genet (2015) 11(6):e1005251. doi: 10.1371/journal.pgen.1005251
10. Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, et al. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol (2006) 177(5):3133–42. doi: 10.4049/jimmunol.177.5.3133
11. Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol (2007) 19(7):825–35. doi: 10.1093/intimm/dxm043
12. Deng G, Xiao Y, Zhou Z, Nagai Y, Zhang H, Li B, et al. Molecular and biological role of the FOXP3 N-terminal domain in immune regulation by T regulatory/suppressor cells. Exp Mol Pathol (2012) 93(3):334–8. doi: 10.1016/j.yexmp.2012.09.013
13. Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11(7):618–27. doi: 10.1038/ni.1884
14. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204(7):1543–51. doi: 10.1084/jem.20070109
15. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17(11):703–17. doi: 10.1038/nri.2017.75
16. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
17. Luo CT, Li MO. Transcriptional control of regulatory T cell development and function. Trends Immunol (2013) 34(11):531–9. doi: 10.1016/j.it.2013.08.003
18. Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, et al. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol (2010) 136(10):1585–95. doi: 10.1007/s00432-010-0816-9
19. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
20. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol (2005) 174(4):1783–6. doi: 10.4049/jimmunol.174.4.1783
21. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21(4):589–601. doi: 10.1016/j.immuni.2004.09.002
22. Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol (2009) 182(3):1469–80. doi: 10.4049/jimmunol.182.3.1469
23. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O'Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol (2010) 185(11):6426–30. doi: 10.4049/jimmunol.0903940
24. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
25. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
26. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
27. Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood (2012) 119(22):5155–63. doi: 10.1182/blood-2011-11-388918
28. Miwa N, Hayakawa S, Miyazaki S, Myojo S, Sasaki Y, Sakai M, et al. IDO expression on decidual and peripheral blood dendritic cells and monocytes/macrophages after treatment with CTLA-4 or interferon-gamma increase in normal pregnancy but decrease in spontaneous abortion. Mol Hum Reprod (2005) 11(12):865–70. doi: 10.1093/molehr/gah246
29. Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol (2004) 16(10):1391–401. doi: 10.1093/intimm/dxh140
30. Yang J, Yang Y, Fan H, Zou H. Tolerogenic splenic IDO (+) dendritic cells from the mice treated with induced-Treg cells suppress collagen-induced arthritis. J Immunol Res (2014) 2014:831054. doi: 10.1155/2014/831054
31. Farias AS, Spagnol GS, Bordeaux-Rego P, Oliveira CO, Fontana AG, de Paula RF, et al. Vitamin D3 induces IDO+ tolerogenic DCs and enhances Treg, reducing the severity of EAE. CNS Neurosci Ther (2013) 19(4):269–77. doi: 10.1111/cns.12071
32. Sharma A, Rudra D. Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00883
33. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med (2013) 19(6):355–67. doi: 10.1016/j.molmed.2013.03.005
34. Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC. CD39 and control of cellular immune responses. Purinergic Signal (2007) 3(1-2):171–80. doi: 10.1007/s11302-006-9050-y
35. Mahnke K, Johnson TS, Ring S, Enk AH. Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J Dermatol Sci (2007) 46(3):159–67. doi: 10.1016/j.jdermsci.2007.03.002
36. Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol (2004) 172(8):4676–80. doi: 10.4049/jimmunol.172.8.4676
37. Lin J, Li M, Wang Z, He S, Ma X, Li D. The role of CD4+CD25+ regulatory T cells in macrophage-derived foam-cell formation. J Lipid Res (2010) 51(5):1208–17. doi: 10.1194/jlr.D000497
38. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep (2015) 13(2):412–24. doi: 10.1016/j.celrep.2015.08.077
39. Riaz F, Pan F, Wei P. Aryl hydrocarbon receptor: The master regulator of immune responses in allergic diseases. Front Immunol (2022) 13:1057555. doi: 10.3389/fimmu.2022.1057555
40. Campesato LF, Budhu S, Tchaicha J, Weng CH, Gigoux M, Cohen IJ, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun (2020) 11(1):4011. doi: 10.1038/s41467-020-17750-z
41. Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol (2010) 17(6):666–72. doi: 10.1038/nsmb.1842
42. Golzari-Sorkheh M, Zúñiga-Pflücker JC. Development and function of FOXP3+ regulators of immune responses. Clin Exp Immunol (2023) 213(1):13–22. doi: 10.1093/cei/uxad048
43. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol (2019) 20(3):156–74. doi: 10.1038/s41580-018-0081-3
44. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene (2007) 26(37):5310–8. doi: 10.1038/sj.onc.1210599
45. van Loosdregt J, Coffer PJ. Post-translational modification networks regulating FOXP3 function. Trends Immunol (2014) 35(8):368–78. doi: 10.1016/j.it.2014.06.005
46. Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, et al. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci U.S.A. (2008) 105(37):14023–7. doi: 10.1073/pnas.0806726105
47. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi: 10.1182/blood-2009-02-207118
48. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol (2018) 15(5):458–69. doi: 10.1038/s41423-018-0004-4
49. Lee GR. The balance of th17 versus Treg cells in autoimmunity. Int J Mol Sci (2018) 19(3):730. doi: 10.3390/ijms19030730
50. Zhang S, Gang X, Yang S, Cui M, Sun L, Li Z, et al. The alterations in and the role of the th17/Treg balance in metabolic diseases. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.678355
51. Riaz F, Wei P, Pan F. Fine-tuning of regulatory T cells is indispensable for the metabolic steatosis-related hepatocellular carcinoma: A review. Front Cell Dev Biol (2022) 10. doi: 10.3389/fcell.2022.949603
52. Su Q, Jing J, Li W, Ma J, Zhang X, Wang Z, et al. Impaired Tip60-mediated Foxp3 acetylation attenuates regulatory T cell development in rheumatoid arthritis. J Autoimmun (2019) 100:27–39. doi: 10.1016/j.jaut.2019.02.007
53. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol (2014) 34(21):3993–4007. doi: 10.1128/MCB.00919-14
54. Li J, Du X, Shi H, Deng K, Chi H, Tao W. Mammalian sterile 20-like kinase 1 (Mst1) enhances the stability of forkhead box P3 (Foxp3) and the function of regulatory T cells by modulating foxp3 acetylation. J Biol Chem (2015) 290(52):30762–70. doi: 10.1074/jbc.M115.668442
55. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci USA. (2007) 104(11):4571–6. doi: 10.1073/pnas.0700298104
56. Bin Dhuban K, d'Hennezel E, Nagai Y, Xiao Y, Shao S, Istomine R, et al. Suppression by human FOXP3(+) regulatory T cells requires FOXP3-TIP60 interactions. Sci Immunol (2017) 2(12):eaai9297. doi: 10.1126/sciimmunol.aai9297
57. Castillo J, Wu E, Lowe C, Srinivasan S, McCord R, Wagle MC, et al. CBP/p300 drives the differentiation of regulatory T cells through transcriptional and non-transcriptional mechanisms. Cancer Res (2019) 79(15):3916–27. doi: 10.1158/0008-5472.CAN-18-3622
58. Du T, Nagai Y, Xiao Y, Greene MI, Zhang H. Lysosome-dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp Mol Pathol (2013) 95(1):38–45. doi: 10.1016/j.yexmp.2013.04.003
59. Wang L, Beier UH, Akimova T, Dahiya S, Han R, Samanta A, et al. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am J Transplant (2018) 18(7):1596–603. doi: 10.1111/ajt.14749
60. Choi SW, Gatza E, Hou G, Sun Y, Whitfield J, Song Y, et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood (2015) 125(5):815–9. doi: 10.1182/blood-2014-10-605238
61. Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL, et al. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol (2010) 136(3):348–63. doi: 10.1016/j.clim.2010.04.018
62. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol (2011) 31(5):1022–9. doi: 10.1128/MCB.01206-10
63. Yang X, Lun Y, Jiang H, Liu X, Duan Z, Xin S, et al. SIRT1-regulated abnormal acetylation of FOXP3 induces regulatory T-cell function defect in Hashimoto's thyroiditis. Thyroid (2018) 28(2):246–56. doi: 10.1089/thy.2017.0286
64. Jiang H, Xin S, Yan Y, Lun Y, Yang X, Zhang J. Abnormal acetylation of FOXP3 regulated by SIRT-1 induces Treg functional deficiency in patients with abdominal aortic aneurysms. Atherosclerosis (2018) 271:182–92. doi: 10.1016/j.atherosclerosis.2018.02.001
65. Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol (2015) 210(6):871–82. doi: 10.1083/jcb.201507005
66. Wei P, Kou W, Fu J, Chen Z, Pan F. Pparα knockout in mice increases the Th17 development by facilitating the IKKα/RORγt and IKKα/Foxp3 complexes. Commun Biol (2023) 6(1):721. doi: 10.1038/s42003-023-05104-6
67. Zhang Y, Ren YJ, Guo LC, Ji C, Hu J, Zhang HH, et al. Nucleus accumbens-associated protein-1 promotes glycolysis and survival of hypoxic tumor cells via the HDAC4-HIF-1α axis. Oncogene (2017) 36(29):4171–81. doi: 10.1038/onc.2017.51
68. Yang JM, Ren Y, Kumar A, Xiong X, Das JK, Peng HY, et al. NAC1 modulates autoimmunity by suppressing regulatory T cell-mediated tolerance. Sci Adv (2022) 8(26):eabo0183. doi: 10.1126/sciadv.abo0183
69. Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol (2019) 15(6):346–66. doi: 10.1038/s41581-019-0129-4
70. Daniels MA, Hogquist KA, Jameson SC. Sweet 'n' sour: the impact of differential glycosylation on T cell responses. Nat Immunol (2002) 3(10):903–10. doi: 10.1038/ni1002-903
71. Earl LA, Baum LG. CD45 glycosylation controls T-cell life and death. Immunol Cell Biol (2008) 86(7):608–15. doi: 10.1038/icb.2008.46
72. Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem (2019) 294(7):2211–31. doi: 10.1074/jbc.AW119.003226
73. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem (1984) 259(5):3308–17. doi: 10.1016/S0021-9258(17)43295-9
74. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature (2007) 446(7139):1017–22. doi: 10.1038/nature05815
75. Loizou L, Andersen KG, Betz AG. Foxp3 interacts with c-Rel to mediate NF-κB repression. PloS One (2011) 6(4):e18670. doi: 10.1371/journal.pone.0018670
76. de Jesus TJ, Tomalka JA, Centore JT, Staback Rodriguez FD, Agarwal RA, Liu AR, et al. Negative regulation of FOXP3 expression by c-Rel O-GlcNAcylation. Glycobiology (2021) 31(7):812–26. doi: 10.1093/glycob/cwab001
77. Deng G, Nagai Y, Xiao Y, Li Z, Dai S, Ohtani T, et al. Pim-2 kinase influences regulatory T cell function and stability by mediating foxp3 protein N-terminal phosphorylation. J Biol Chem (2015) 290(33):20211–20. doi: 10.1074/jbc.M115.638221
78. Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem (2013) 288(34):24494–502. doi: 10.1074/jbc.M113.467704
79. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem (2014) 289(39):26872–81. doi: 10.1074/jbc.M114.586651
80. Li Z, Li D, Tsun A, Li B. FOXP3+ regulatory T cells and their functional regulation. Cell Mol Immunol (2015) 12(5):558–65. doi: 10.1038/cmi.2015.10
81. Lam E, Choi SH, Pareek TK, Kim BG, Letterio JJ. Cyclin-dependent kinase 5 represses Foxp3 gene expression and Treg development through specific phosphorylation of Stat3 at Serine 727. Mol Immunol (2015) 67(2 Pt B):317–24. doi: 10.1016/j.molimm.2015.06.015
82. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19(3):322–8. doi: 10.1038/nm.3085
83. Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol (2011) 12(1):35–48. doi: 10.1038/nri3111
84. Ronai Z. Monoubiquitination in proteasomal degradation. Proc Natl Acad Sci (2016) 113(32):8894–6. doi: 10.1073/pnas.1610186113
85. Sadowski M, Sarcevic B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div (2010) 5:19. doi: 10.1186/1747-1028-5-19
86. Damgaard RB. The ubiquitin system: from cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ (2021) 28(2):423–6. doi: 10.1038/s41418-020-00703-w
87. Barbi J, Pardoll DM, Pan F. Ubiquitin-dependent regulation of Foxp3 and Treg function. Immunol Rev (2015) 266(1):27–45. doi: 10.1111/imr.12312
88. Wang G, Gao Y, Li L, Jin G, Cai Z, Chao JI, et al. K63-linked ubiquitination in kinase activation and cancer. Front Oncol (2012) 2. doi: 10.3389/fonc.2012.00005
89. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res (2016) 26(4):457–83. doi: 10.1038/cr.2016.40
90. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J (2019) 38(9). doi: 10.15252/embj.201899766
91. Michel MA, Swatek KN, Hospenthal MK, Komander D. Ubiquitin linkage-specific affimers reveal insights into K6-linked ubiquitin signaling. Mol Cell (2017) 68(1):233–246.e5. doi: 10.1016/j.molcel.2017.08.020
92. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med (2003) 197(4):403–11. doi: 10.1084/jem.20021633
93. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity (2013) 39(2):272–85. doi: 10.1016/j.immuni.2013.08.006
94. Li M, Shi M, Hu C, Chen B, Li S. MALAT1 modulated FOXP3 ubiquitination then affected GINS1 transcription and drived NSCLC proliferation. Oncogene (2021) 40(22):3870–84. doi: 10.1038/s41388-021-01816-3
95. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445(7129):766–70. doi: 10.1038/nature05479
96. Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity (2012) 36(5):717–30. doi: 10.1016/j.immuni.2012.03.020
97. Dong Y, Yang C, Pan F. Post-translational regulations of foxp3 in Treg cells and their therapeutic applications. Front Immunol (2021) 12:626172. doi: 10.3389/fimmu.2021.626172
98. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem (2001) 70:503–33. doi: 10.1146/annurev.biochem.70.1.503
99. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology (2013) 38(1):23–38. doi: 10.1038/npp.2012.112
100. Ma L, Fang X, Zhang A. The hypermethylation of FOXP3 gene as an epigenetic marker for the identification of arsenic poisoning risk. Hum Exp Toxicol (2022) 41:9603271221142819. doi: 10.1177/09603271221142819
101. Vecellio M, Wu H, Lu Q, Selmi C. The multifaceted functional role of DNA methylation in immune-mediated rheumatic diseases. Clin Rheumatol (2021) 40(2):459–76. doi: 10.1007/s10067-020-05255-5
102. Le Menn G, Jablonska A, Chen Z. The effects of post-translational modifications on Th17/Treg cell differentiation. Biochim Biophys Acta Mol Cell Res (2022) 1869(6):119223. doi: 10.1016/j.bbamcr.2022.119223
103. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer (2013) 13(1):37–50. doi: 10.1038/nrc3409
104. Srour N, Khan S, Richard S. The influence of arginine methylation in immunity and inflammation. J Inflammation Res (2022) 15:2939–58. doi: 10.2147/JIR.S364190
105. Kagoya Y, Saijo H, Matsunaga Y, Guo T, Saso K, Anczurowski M, et al. Arginine methylation of FOXP3 is crucial for the suppressive function of regulatory T cells. J Autoimmun (2019) 97:10–21. doi: 10.1016/j.jaut.2018.09.011
106. Nagai Y, Ji MQ, Zhu F, Xiao Y, Tanaka Y, Kambayashi T, et al. PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185(erbB2/neu) tumor immunotherapy. Front Immunol (2019) 10:174. doi: 10.3389/fimmu.2019.00174
107. Sen S, He Z, Ghosh S, Dery KJ, Yang L, Zhang J, et al. PRMT1 plays a critical role in th17 differentiation by regulating reciprocal recruitment of STAT3 and STAT5. J Immunol (2018) 201(2):440–50. doi: 10.4049/jimmunol.1701654
108. Zheng Y, Huang L, Ge W, Yang M, Ma Y, Xie G, et al. Protein arginine methyltransferase 5 inhibition upregulates foxp3(+) regulatory T cells frequency and function during the ulcerative colitis. Front Immunol (2017) 8:596. doi: 10.3389/fimmu.2017.00596
109. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999) 59(13):3128–33.
110. Couper KN, Lanthier PA, Perona-Wright G, Kummer LW, Chen W, Smiley ST, et al. Anti-CD25 antibody-mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol (2009) 182(7):3985–94. doi: 10.4049/jimmunol.0803053
111. Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci (2009) 1174:99–106. doi: 10.1111/j.1749-6632.2009.04939.x
112. Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, et al. Fc-optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res (2021) 81(11):2983–94. doi: 10.1158/0008-5472.CAN-20-3585
113. Wang T, Zhou Q, Zeng H, Zhang H, Liu Z, Shao J, et al. CCR8 blockade primes anti-tumor immunity through intratumoral regulatory T cells destabilization in muscle-invasive bladder cancer. Cancer Immunol Immunother (2020) 69(9):1855–67. doi: 10.1007/s00262-020-02583-y
114. Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci U.S.A. (2022) 119(7):e2114282119. doi: 10.1073/pnas.2114282119
115. Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer (2021) 9(2):e001749. doi: 10.1136/jitc-2020-001749
116. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U.S.A. (2013) 110(44):17945–50. doi: 10.1073/pnas.1316796110
117. Phan GQ, Weber JS, Sondak VK. CTLA-4 blockade with monoclonal antibodies in patients with metastatic cancer: surgical issues. Ann Surg Oncol (2008) 15(11):3014–21. doi: 10.1245/s10434-008-0104-y
118. Cranmer LD, Hersh E. The role of the CTLA4 blockade in the treatment of Malignant melanoma. Cancer Invest (2007) 25(7):613–31. doi: 10.1080/07357900701522315
119. Boasberg P, Hamid O, O'Day S. Ipilimumab: unleashing the power of the immune system through CTLA-4 blockade. Semin Oncol (2010) 37(5):440–9. doi: 10.1053/j.seminoncol.2010.09.004
120. Kumar P, Saini S, Prabhakar BS. Cancer immunotherapy with check point inhibitor can cause autoimmune adverse events due to loss of Treg homeostasis. Semin Cancer Biol (2020) 64:29–35. doi: 10.1016/j.semcancer.2019.01.006
121. Sandin LC, Eriksson F, Ellmark P, Loskog AS, Tötterman TH, Mangsbo SM. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology (2014) 3(1):e27614. doi: 10.4161/onci.27614
122. Comin-Anduix B, Escuin-Ordinas H, Ibarrondo FJ. Tremelimumab: research and clinical development. Onco Targets Ther (2016) 9:1767–76. doi: 10.2147/OTT.S65802
123. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res (2019) 38(1):255. doi: 10.1186/s13046-019-1259-z
124. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol (2016) 13(8):473–86. doi: 10.1038/nrclinonc.2016.58
125. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol (2014) 4:385. doi: 10.3389/fonc.2014.00385
126. Yoshida K, Okamoto M, Sasaki J, Kuroda C, Ishida H, Ueda K, et al. Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer (2020) 20(1):25. doi: 10.1186/s12885-019-6499-y
127. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
128. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PloS One (2010) 5(5):e10436. doi: 10.1371/journal.pone.0010436
129. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
130. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27:1–7. doi: 10.1016/j.coi.2013.12.005
131. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
132. van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol (2009) 182(12):7490–500. doi: 10.4049/jimmunol.0802751
133. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol (2013) 43(9):2421–9. doi: 10.1002/eji.201343451
134. Ndhlovu LC, Takeda I, Sugamura K, Ishii N. Expanding role of T-cell costimulators in regulatory T-cell function: recent advances in accessory molecules expressed on both regulatory and nonregulatory T cells. Crit Rev Immunol (2004) 24(4):251–66. doi: 10.1615/CritRevImmunol.v24.i4.30
135. Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol (2004) 172(6):3580–9. doi: 10.4049/jimmunol.172.6.3580
136. Redmond WL, Weinberg AD. Targeting OX40 and OX40L for the treatment of autoimmunity and cancer. Crit Rev Immunol (2007) 27(5):415–36. doi: 10.1615/CritRevImmunol.v27.i5.20
137. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol (2004) 4(6):420–31. doi: 10.1038/nri1371
138. Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med (2009) 206(5):1103–16. doi: 10.1084/jem.20082205
139. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med (2008) 205(4):825–39. doi: 10.1084/jem.20071341
140. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med (2019) 25(5):759–66. doi: 10.1038/s41591-019-0420-8
141. Ranasinghe R, Eri R. Modulation of the CCR6-CCL20 axis: A potential therapeutic target in inflammation and cancer. Medicina (Kaunas) (2018) 54(5):88. doi: 10.3390/medicina54050088
142. Meitei HT, Jadhav N, Lal G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun Rev (2021) 20(7):102846. doi: 10.1016/j.autrev.2021.102846
143. Núñez R, Rodríguez MJ, Lebrón-Martín C, Martín-Astorga MDC, Ramos-Soriano J, Rojo J, et al. A synthetic glycodendropeptide induces methylation changes on regulatory T cells linked to tolerant responses in anaphylactic-mice. Front Immunol (2023) 14:1165852. doi: 10.3389/fimmu.2023.1165852
144. Zhao T, Wang L, Zhang Y, Ye W, Liu J, Wu H, et al. Qi-Dong-Huo-Xue-Yin balances the immune microenvironment to protect against LPS induced acute lung injury. Front Pharmacol (2023) 14:1200058. doi: 10.3389/fphar.2023.1200058
145. Tanaka A, Sakaguchi S. Targeting Treg cells in cancer immunotherapy. Eur J Immunol (2019) 49(8):1140–6. doi: 10.1002/eji.201847659
146. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13(11):1299–307. doi: 10.1038/nm1652
147. Oehme I, Linke JP, Böck BC, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U.S.A. (2013) 110(28):E2592–601. doi: 10.1073/pnas.1300113110
148. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer (2006) 6(5):369–81. doi: 10.1038/nrc1881
149. Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, et al. Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. J Autoimmun (2018) 89:112–24. doi: 10.1016/j.jaut.2017.12.010
150. LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, et al. Tolerogenic nanoparticles induce antigen-specific regulatory T cells and provide therapeutic efficacy and transferrable tolerance against experimental autoimmune encephalomyelitis. Front Immunol (2018) 9:281. doi: 10.3389/fimmu.2018.00281
151. Liu Q, Wang X, Liu X, Kumar S, Gochman G, Ji Y, et al. Use of polymeric nanoparticle platform targeting the liver to induce Treg-mediated antigen-specific immune tolerance in a pulmonary allergen sensitization model. ACS Nano (2019) 13(4):4778–94. doi: 10.1021/acsnano.9b01444
152. Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol (2015) 62(6):1349–56. doi: 10.1016/j.jhep.2015.01.006
153. Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discovery (2022) 21(3):181–200. doi: 10.1038/s41573-021-00371-6
154. Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics (2003) 2(12):1350–8. doi: 10.1074/mcp.T300009-MCP200
155. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U.S.A. (2001) 98(15):8554–9. doi: 10.1073/pnas.141230798
Keywords: regulatory T cells, Foxp3, immunotherapy, tumor microenvironment, anti-tumor immunity, autoimmune diseases, post-translational modifications
Citation: Riaz F, Huang Z and Pan F (2023) Targeting post-translational modifications of Foxp3: a new paradigm for regulatory T cell-specific therapy. Front. Immunol. 14:1280741. doi: 10.3389/fimmu.2023.1280741
Received: 21 August 2023; Accepted: 09 October 2023;
Published: 23 October 2023.
Edited by:
Peng Liu, Sun Yat-sen University, ChinaReviewed by:
John Andersson, Karolinska Institutet (KI), SwedenXiaoxiao Wan, Washington University School of Medicine, United States
Kai Yang, Indiana University School of Medicine - Lafayette, United States
Kenji Ichiyama, Osaka University, Japan
Shuxun Liu, Second Military Medical University, China
Copyright © 2023 Riaz, Huang and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Pan, ZmFuLnBhbkBzaWF0LmFjLmNu