 Dan Xie
Dan Xie Shi Ouyang
Shi Ouyang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 October 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1279264
This article is part of the Research Topic Myeloid cells in health and liver disease View all 11 articles
Acute liver failure (ALF) is a severe liver disease caused by disruptions in the body’s immune microenvironment. In the early stages of ALF, Kupffer cells (KCs) become depleted and recruit monocytes derived from the bone marrow or abdomen to replace the depleted macrophages entering the liver. These monocytes differentiate into mature macrophages, which are activated in the immune microenvironment of the liver and polarized to perform various functions. Macrophage polarization can occur in two directions: pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages. Controlling the ratio and direction of M1 and M2 in ALF can help reduce liver injury. However, the liver damage caused by pyroptosis should not be underestimated, as it is a caspase-dependent form of cell death. Inhibiting pyroptosis has been shown to effectively reduce liver damage induced by ALF. Furthermore, macrophage polarization and pyroptosis share common binding sites, signaling pathways, and outcomes. In the review, we describe the role of macrophage polarization and pyroptosis in the pathogenesis of ALF. Additionally, we preliminarily explore the relationship between macrophage polarization and pyroptosis, as well as their effects on ALF.
The liver plays a crucial role in the immune system by serving as a vital organ responsible for removing toxins, producing immune proteins, and maintaining metabolic homeostasis (1). The first point to note is that the liver contains a high concentration of both innate and adaptive immune cells. These cells have the ability to trigger inflammation and liver damage in response to disease, but also have the capability of maintaining a state of tolerance during homeostasis. The liver is home to a variety of T cell subsets, including regulatory T cells and cytotoxic T lymphocytes, which play an essential role in maintaining liver tolerance (2–4). Not only that, innate immune cells, particularly liver-resident macrophages known as Kupffer cells (KCs), work together with acquired immune cells to eliminate common pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) in the body. Besides, KCs also have a crucial role in maintaining liver homeostasis by engaging in phagocytosis, eliminating dead and senescent cells, and promoting tissue repair (5–7). In addition, the liver has the highest concentration of macrophages, which are dispersed throughout a network of circulatory channels that can quickly detect pathogens in the hepatic portal system (8).
The liver has a unique anatomy because it receives blood from both the hepatic artery and portal vein. This dual blood supply nourishes to the diverse structures and cells within the liver (8). The hepatic arterial and portal circulation terminate in the liver sinusoidal endothelial cells (LSECs), which consist of a thin, porous network of special capillaries and complement KCs in the hepatic sinusoids to form a solid surveillance system (9, 10)(Figure 1). The blood flows in sinusoidal waves at a slow pace, which allows for prolonged exposure to antigens within the sinusoids. This facilitates the recognition and handling of antigens by both immune and non-immune cells. The portal vein supplies the liver with the majority of its blood supply (9). In addition to being rich in nutrients, the portal vein is also rich in pathogenic molecules such as lipopolysaccharide (LPS) (11). When intestinal epithelial damage or failure, it can lead to the entry of infections into the bloodstream. Then, these infections can travel from the portal vein to the liver, bypassing conventional immune organs such as the spleen and lymph nodes (8, 11, 12).
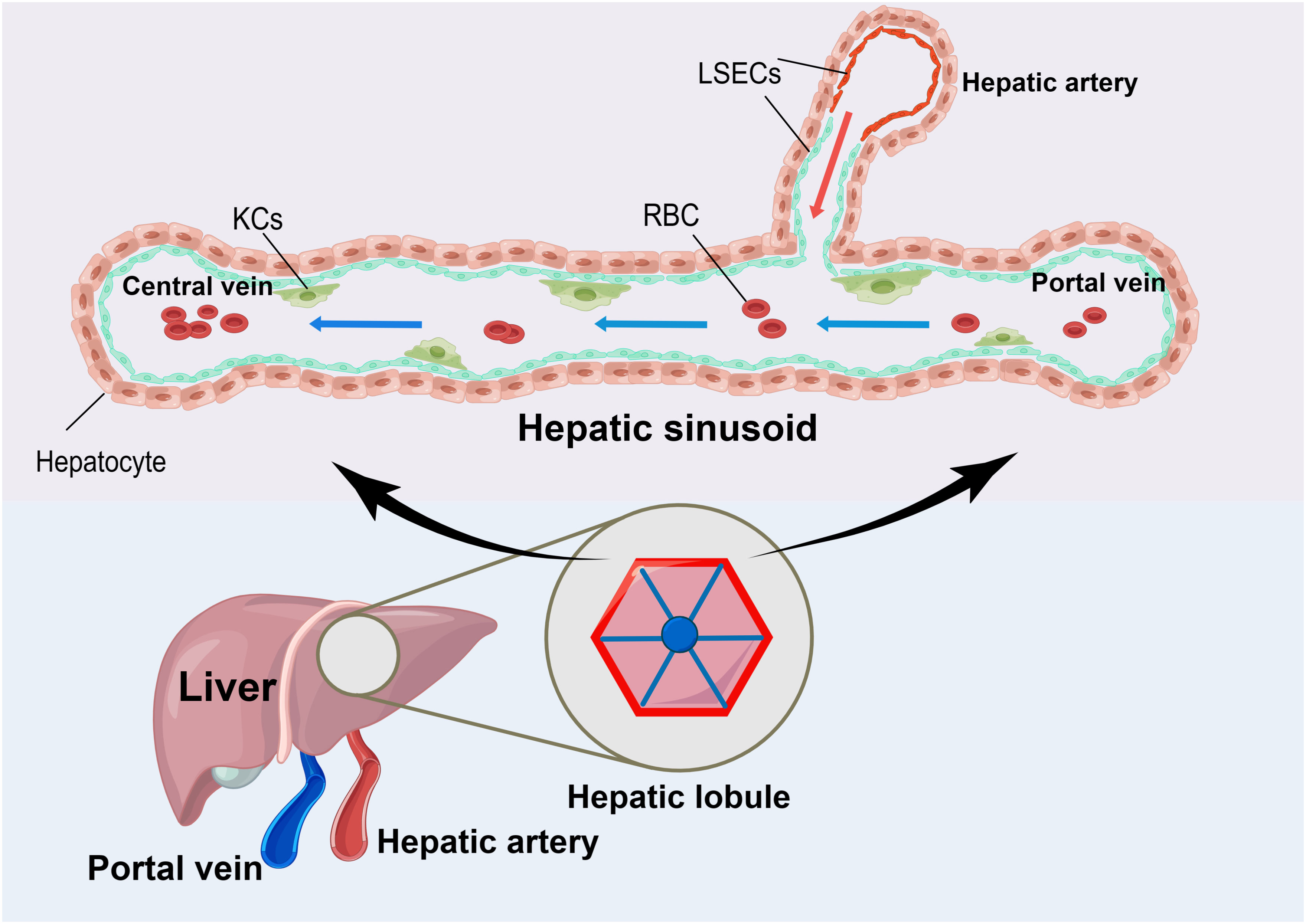
Figure 1 The Unique Anatomy of the Liver. The liver has a unique anatomy in that it receives blood from both the hepatic artery and the portal vein. Both sets of blood supplies end in liver sinusoidal endothelial cells (LSEC) with a large number of Kupffer cells (KCs) attached to their surface. When the blood flows through this area, it moves slowly in sinusoidal waves at a slow pace. This allows for the effective absorption of nutrients and nourishment of the liver tissues. Additionally, it enables the attached KCs to remove disease-causing substances, thereby maintaining the body’s homeostasis.
Meanwhile, the liver conducts circulating immune surveillance by mobilizing immune cells (e.g., KCs) within the liver to eliminate pathogens or toxins and maintain immune homeostasis in the liver and throughout the body. A study found that KCs crawling within the hepatic vascular system were able to effectively capture blood-borne disseminating Borrelia burgdorferi, thus creating an efficient surveillance and filtering system. Additionally, KCs can induce the formation of chemokine receptor (CXCR)3-dependent clusters of activated invariant natural killer T cells (iNKT cells) after ingesting Borrelia burgdorferi. This clustering prevents the spread of pathogens to organs, such as the joints (13). Therefore, KCs in the liver are considered the body’s first line of defense against any pathogens that are transmitted (8). The effect was similarly demonstrated in animal experiments by depleting KCs following intravenous administration of liposome-entrapped clodronate or using CRISPR/Cas9-mediated genome editing to prepare lacking liver immune receptor models. It has been discovered that in a mouse model, the depletion of KCs or immune receptors in the liver leads to 100% mortality from a sub-lethal dose of Listeria monocytogenes. However, removing the spleen did not have any impact on host immunity or survival (8, 14, 15). In short, this suggests that the liver plays a crucial role in detecting pathogens and defending the host. When the function or structure of the liver is compromised, especially for KCs, it becomes vulnerable to systemic diseases and can cause damage to multiple organs, resulting in disruptions to the immune microenvironment.
The main pathophysiological features of acute liver failure (ALF) are massive hepatocyte death and immune-inflammatory response (16). Among these, immune-mediated liver injury starts early in ALF and is primarily caused by innate immunity followed by an adaptive immune response that leads to further injury. The early activation of the innate immune system is specific to the activating substances, such as PAMPs and DAMPs. PAMPs play a more significant role in ALF induced by liver pathogens, while DAMPs are released from damaged cells and are crucial in ALF caused by hepatotoxic substances (17, 18). Monocytes and macrophages are essential components of the innate and adaptive immune response because they possess receptors on their cell surface that recognize PAMPs and DAMPs. Upon activation, they can modulate the immune response by producing reactive oxygen radicals and either anti-inflammatory or pro-inflammatory cytokines (17–19). Based on the anatomy of the liver, KCs are located in the hepatic blood sinusoids as an important defense device within the liver. They have a scavenging and filtering effect on the incoming and outgoing blood, effectively identifying toxic substances or pathogens in the blood (20, 21). In the diseased state, mononuclear macrophages recruited from outside the liver differentiate into various subsets of macrophage, leading to different functions. This process, known as polarization, determines changes in the local hepatic immune microenvironment and even the systemic immune state, especially in cases of ALF (11, 22).
Another major cause of damage in ALF is the destruction of hepatocytes by toxic substances and their death. In addition to necrosis and apoptosis, which are the accepted modes of cell death, several new modes of cell death have been identified and confirmed over the past decade. These include pyroptosis, necroptosis, and ferroptosis, which are available for study (16, 23–25). Pyroptosis is a newly discovered form of programmed cell death that specifically targets the innate immune defenses of intracellular bacteria. It plays a crucial role in defending against pathogens and danger signals (26, 27). However, excessive pyroptosis can lead to the development of ALF, as demonstrated in LPS/D-galactosamine(D-GalN)-induced ALF mice models (28). Therefore, inhibiting cellular pyroptosis has also been used as a research hotspot for ALF treatment in recent years.
At the same time, there has been interest in the relationship between cell death and the immune-inflammatory response. In ALF, dying hepatocytes release DAMPs that bind to evolutionarily conserved pattern recognition receptors of the innate immune system. These receptors are found in both liver-resident cells (e.g., KCs, LSECs) and cells that are recruited in response to injury (e.g., monocytes, macrophages, NK cells). This binding triggers the release of inflammatory mediators, including cytokines and chemokines. In turn, inflammatory mediators lead to further cell death, establishing a highly hepatotoxic feedforward cycle of inflammation and cell death (29). As important intrinsic immune cells, macrophages and their polarization play a crucial role in this process. However, there is a lack of clear description and discussion regarding the novel cell death modality known as pyroptosis. Therefore, this paper will discuss the roles and molecular mechanisms of macrophage polarization and hepatocyte pyroptosis in ALF. It will further explore how these processes alter the immune microenvironment of the liver, leading to immune dysfunction in the liver or even the entire circulatory system. Additionally, this paper will delve into the crosstalk between macrophage polarization and hepatocyte pyroptosis to provide a new theoretical basis for the pathogenesis and immunotherapy of ALF in the future.
As mentioned before, the liver is supplied by two blood supply systems: the hepatic artery and the portal vein. It has immune regulation and circulatory immune monitoring functions. The portal vein is rich in both nutrients and molecules that can cause diseases, such as LPS (11). The circulating blood flows through the portal system and directly into the liver, where it undergoes a detoxifying and removal process (30). The process is primarily carried out by immune cells in the liver, with hepatic macrophages playing a crucial role (21).
The source of macrophages in a healthy liver mainly consists of self-renewing tissue-resident macrophages, such as KCs. These cells are located in the hepatic sinusoids and account for 80% of systemic macrophages (22). They also make up 20-25% of non-parenchymal cells in the liver and are the largest population of innate immune cells in the liver. KCs maintain hepatic homeostasis by removing pathogens through the portal vein (31). This regulatory immune and clearance function plays a vital role in maintaining liver function and immune system balance. Firstly, the immunomodulatory function of macrophages is to regulate both innate and acquired immune responses by releasing pro- and anti-inflammatory mediators. This helps to maintain immune balance in the body. Secondly, macrophages are also able to function as antigen-presenting cells and regulate the adaptive immune response (32). Thirdly, macrophages also function to clear harmful substances from the blood and prevent infection. This includes the clearance of translocated gut microbiota (20). Particularly, KCs are capable of specifically phagocytosing particulate material larger than 200 nm (20, 33).
But, under normal circumstances, the intestinal flora also releases some LPS into the bloodstream. So, how does the liver maintain immune tolerance without causing local inflammation? This issue is closely intertwined with the LSECs to which KCs are attached. LSECs mainly form highly permeable capillaries without basement membranes in the hepatic sinusoids. They share similar functions with KCs as antigen-presenting cells and are involved in the process of phagocytosis. LSECs and KCs collaborate to phagocytose blood-borne pathogens and substances present in the hepatic arteries and portal veins, preventing their further systemic circulation and averting widespread inflammatory reactions. However, it is important to note that LSECs have a higher responsiveness to LPS compared to KCs. This is predominantly due to the presence of Toll-like receptor 4(TLR4) and cluster of differentiation 14 (CD14) on the surface of LSECs, which enables them to directly interact with LPS, a byproduct of bacterial degradation. This interaction triggers a decrease in the expression of CD54 molecules on the surface of LSECs, which in turn reduces the adherence of leukocytes to LSECs (34). Ultimately, this leads to a decrease in localized inflammatory responses and promotes immune tolerance. In addition, LSECs bind LPS and produce prostaglandins, including prostaglandin E2, which can inhibit downstream gene expression induced by TLR4 ligands through nuclear receptors (35). This mechanism promotes immunological tolerance to LPS in normal conditions by inhibiting leukocyte adhesion and local activation, thereby maintaining the integrity of the liver endothelial cell layer.
The regulation of LPS tolerance by LSECs is very delicate. Because LSECs can initially tolerate LPS at a certain concentration, but as the LPS concentration gradually increases, LSECs can overcome their initial tolerance and simultaneously activate KCs. This not only ensures immune tolerance within the physiological range, but also enables an accurate response to bacterial infection during this period (36). Mechanistically, when the concentration of LPS is too high, LSECs instead increase the expression level of CD54 on the surface. This leads to an increase in leukocyte adhesion and aggregation, facilitating the local clearance of toxic substances against pathogens. Meanwhile, it has been found that LSECs can directly respond to LPS stimulation in an inflammatory environment by altering the expression pattern of their chemokine genes, such as C-C motif chemokine ligand 2(CCL2), CCL3, CCL4, and CCL7 (37, 38). Specifically, CCL2 plays a role in recruiting inflammatory monocytes into the liver (37). Therefore, LSECs and KCs play a crucial role in maintaining local immune tolerance in the liver. They have a significant regulatory function in accurately detecting high levels of LPS and facilitating the recruitment of monocytes to enhance the initial signaling, thereby initiating the downstream inflammatory cascade response.
When a liver injury occurs, the macrophage population in the liver undergoes changes. This process is primarily caused by the release of pro-inflammatory mediators or chemokines from activated KCs into the bloodstream. This triggers the accumulation of peritoneal macrophages and monocyte-derived macrophages (MoMFs) in the liver and is implicated in the development of various liver diseases (21, 22, 39).
In the pathogenesis of ALF, hepatocytes are exposed to foreign toxic substances such as acetaminophen (APAP), pathogens, and LPS produced by bacteria, which can lead to a significant number of hepatocyte deaths in the liver (40, 41). The disease progression leads to the regeneration and repair of hepatocytes, but they may not be able to fully compensate for the damage caused by cell death. After that, the dead cells release DAMPs (39), which can bind to pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs), cytoplasmic Nod-like receptors (NLRs), Retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and C-type lectin receptors (CLRs) (42). PRRs are expressed on the surface of immune cells and upon binding cause the immune cells to transform their phenotype and become activated (43), which initiates an inflammatory response. This ultimately leads to changes in the immune microenvironment in the liver (44).
As the predominant intrinsic immune cells in the liver, activated KCs release inflammatory mediators and chemokines into the bloodstream, which recruit bone marrow-derived monocytes to develop into mature MoMFs. However, in the early stages of ALF, the liver-resident KCs are gradually depleted (22, 45–48). Recent research has found that the acute injury model induced by carbon tetrachloride (CCl4) in mice has three distinct phases: necroinflammation, early repair, and late repair. At the gene and protein level, the immune microenvironment of the liver was characterized by MoMFs-induced immune damage, with lower levels of KCs observed during the necroinflammation. This finding indicates a potential role for MoMFs in the phagocytosis of necrotic hepatocytes. However, the opposite cellular distribution was observed during the repair (48). Therefore, the majority of macrophages in ALF are replaced by MoMFs, which can perform either pro-inflammatory or anti-inflammatory functions.
The pro-inflammatory and anti-inflammatory effects described here are illustrated in two main ways. To begin with, the pro-inflammatory effect of MoMFs is due to their high expression of the C-C motif chemokine receptor 2(CCR2) and surface marker Ly-6C (CCR2+Ly-6Chigh MoMFs). Their main function is to clear toxic substances from the liver by releasing vasoactive and inflammatory mediators such as Term1 and S100 calcium binding protein A8 and A9 (S100A8/9) into the peripheral blood during the early phase of acute liver injury (11, 49). S100A8/9 is a novel molecule of DAMPs that can bind to TLR4 receptors, promoting inflammation propagation and activating other relevant immune cells (50, 51). During the disease’s repair phase, MoMFs and yolk sac-derived KCs undergo a transformation into anti-inflammatory MoMFs after being stimulated by macrophage-colony stimulating factor 1 (CSF1) (52, 53). The phenotypic transition from pro-inflammatory CCR2+Ly-6Chigh MoMFs to anti-inflammatory CCR2-Ly-6Clow MoMFs, which secrete anti-inflammatory factors, facilitate hepatic repair, suppresses inflammation, and maintain the stability of the hepatic immune microenvironment (11, 22, 49, 54). In addition, the release of anti-inflammatory factors from CCR2-Ly-6Clow MoMFs into the bloodstream contributes to the deactivation of functional monocytes and increases the risk of sepsis (22, 54). Briefly, it was observed that MoMFs had the ability to undergo differentiation towards either M1 macrophages, representing a classic proinflammatory phenotype, or M2 macrophages, representing an alternative anti-inflammatory phenotype in different phases of the disease (55, 56).
However, monocytes and other immune cells are recruited to the liver from the systemic circulation, resulting in a relative decrease in the number of immune cells and immunity in the systemic circulation. This can lead to an increased risk of systemic opportunistic infections (45, 57). In particular, the occurrence of bacterial translocation in the gut releases PAMPs, which can easily initiate systemic infections (43, 58) and enhances hepatocyte death by binding to TLRs (59). In conjunction with macrophage-derived mediators, they can also cause vascular endothelial dysfunction and microcirculatory disturbances. These disturbances can result in extrahepatic organ dysfunction (22), which is part of a larger process known as systemic inflammatory response syndrome (SIRS). If left untreated, SIRS can progress to sepsis, septicemia, or even multi-organ failure, ultimately leading to a poor prognosis for patients with ALF (60–62).
Therefore, there is a conflict regarding the role of macrophages in ALF. Some studies suggest that recruited monocytes develop into mature macrophages with an improved ability to clear hepatotoxic substances and alleviate liver damage. At the same time, some scholars believe that the pro-inflammatory capacity of macrophages in ALF will further exacerbate liver damage and induce SIRS. Moreover, simply eliminating or impairing the function of various immune cells will unavoidably cause a delay in the healing process of damaged tissue. This underscores the crucial role of the immune system in tissue repair. In general, liver macrophages cannot be restricted to a single role. Their phenotypes can change according to the altered immune microenvironment in the liver, and they perform different functions both in the liver and systemically to maintain the balance of the immune microenvironment. Such conflicting and mutually limiting roles also pose one of the main challenges in the development of ALF immunotherapies. This is because potential molecular targets may have varying local and systemic effects (22, 32, 54).
In addition to regulating the immune system and performing phagocytosis, macrophages are also highly diverse and adaptable. They can exhibit various functions depending on the stimuli or proteins present in the immune microenvironment and can differentiate into different subtypes through a process known as polarization (63–67). Macrophages can be classified into two phenotypes: pro-inflammatory (M1) and anti-inflammatory (M2). These phenotypes are determined by various factors, including microorganisms, tissue microenvironment, and cytokine signals (64, 68, 69). M1 macrophages are induced by various stimuli, including LPS, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α), which are Th1 cytokines. Additionally, inducible nitric oxide synthase (iNOS) and granulocyte-macrophage colony-stimulating factor (GM-CSF) can also induce M1 macrophages (31, 70). Primitive macrophages differentiate into M1 macrophages, which produce a large number of pro-inflammatory factors, such as interleukin (IL)-1β, reactive oxygen species (ROS), and TNF-α. These factors mediate antimicrobial defense, tissue destruction, and antitumor resistance (31, 70, 71). In contrast, M2 macrophages are induced by anti-inflammatory factors such as IL-4, IL-13 (which are Th2 cytokines), IL-10, and transforming growth factor-β (TGF-β). These macrophages produce anti-inflammatory factors. M2 macrophages are primarily involved in wound repair, angiogenesis, resistance to parasites, and promotion of tumor growth (67, 70–72). The underlying mechanisms are even more complex, involving multiple signaling pathways and associated regulatory factors (Figure 2).
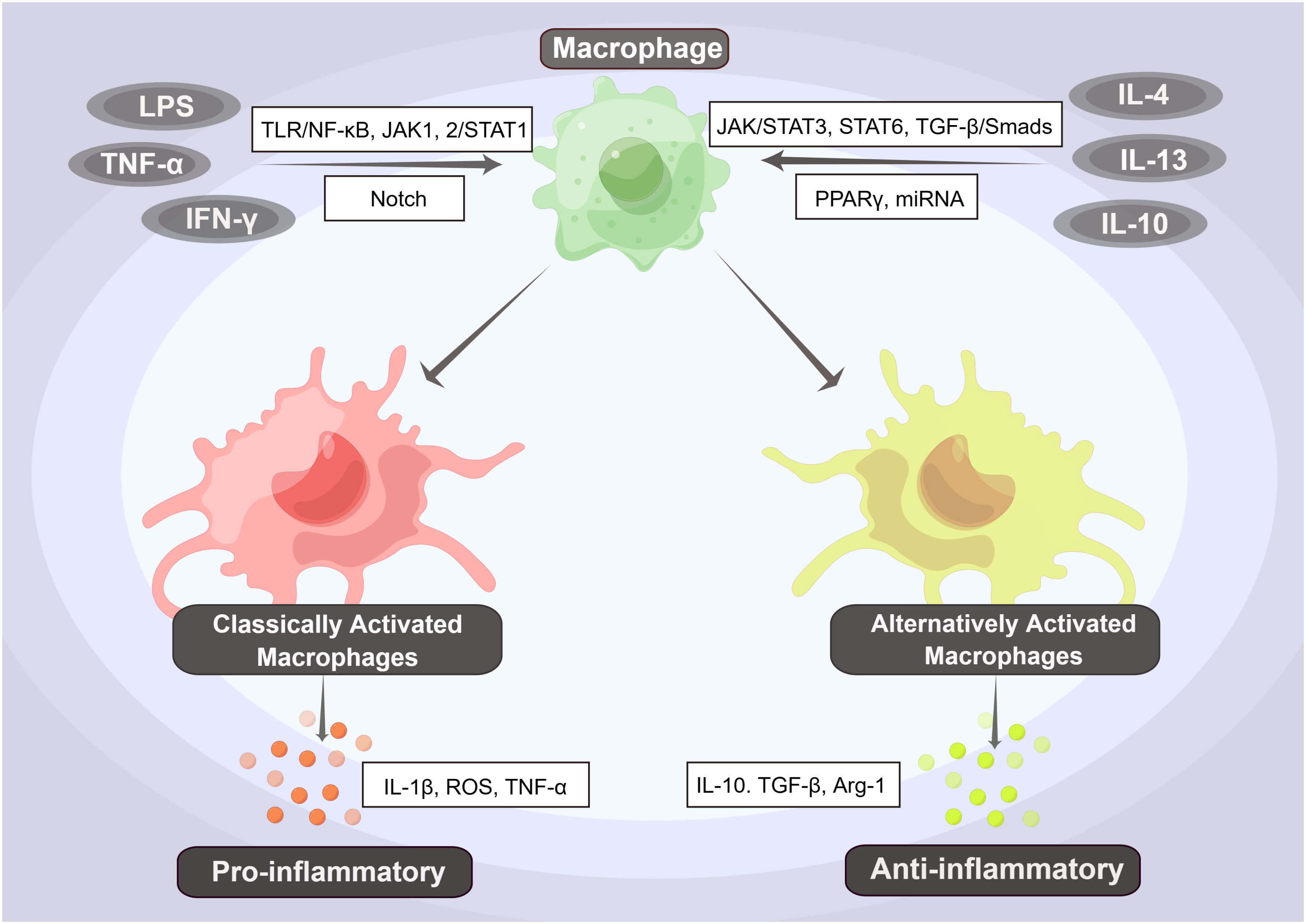
Figure 2 The Phenotypes and Pathways of Macrophage Polarization. Primary macrophages can differentiate into pro-inflammatory (M1) and anti-inflammatory (M2) macrophages by activating various factors and pathways. Among them, factors such as LPS, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) can induce the differentiation of primitive macrophages into M1-type macrophages, i.e., classically activated macrophages (CAMs), through activation of the TLR/NF-κB, JAK1,2/STAT1, and Notch signaling pathways. These M1-type macrophages produce a large number of pro-inflammatory factors, such as interleukin (IL)-1β, reactive oxygen species (ROS), and TNF-α, further inducing a proinflammatory response. In contrast, when anti-inflammatory factors such as IL-4, IL-13, and IL-10 activate the JAK/STAT3, STAT6, and TGF-β/Smads signaling pathways, they induce the differentiation of M2-type macrophages, also known as alternatively activated macrophages (AAMs). These macrophages produce anti-inflammatory factors such as IL-10, TGF-β, and Arg-1, which initiate an anti-inflammatory response.
M1 macrophages, also known as classically activated macrophages (CAMs), are characterized by the release of large amounts of inflammatory cytokines, Th1 chemokines, and ROS/RNS products. They also act as positive feedback to unpolarized macrophages (70, 73, 74). The regulation of M1 polarization is primarily controlled by the TLR/nuclear factor-kB (NF-kB) signaling pathway, the Janus kinase 1, 2 (JAK1, 2)/signal transducer and activator of transcription 1 (STAT1), and the Notch signaling pathway. LPS is the main factor for the activation of the TLR/NF-κB signaling pathway (75). It promotes the polarization of CAMs by binding to TLR4 receptors on the surface of initial macrophages and activating NF-κB via the MyD88-dependent pathway or interferon regulatory factor 3 (IRF3). This activation leads to the production of IL-6 and iNOS (76, 77), while the level of IL-10 decreases. Ultimately, this process mediates the formation of the pro-inflammatory phenotype of CAMs (71, 78, 79). Activation of NF-κB p65 is a marker of CAM activation (79). However, the binding of IFN-γ to its receptor (IFN-γR) activates JAK1 and JAK2, which are members of the tyrosine kinase family. This activation leads to the phosphorylation of STAT1, which then translocates into the nucleus to bind the conserved Gamma interferon activation site (GAS) DNA element. This binding activates the transcription of interferon-stimulated genes (ISGs), resulting in the formation of CAMs and the promotion of chemokine and antigen-presenting molecule production (80, 81) (Figure 3). Moreover, the JAK1,2/STAT1 signaling pathway and TLR/NF-κB signaling pathway have synergistic effects (67, 81, 82).
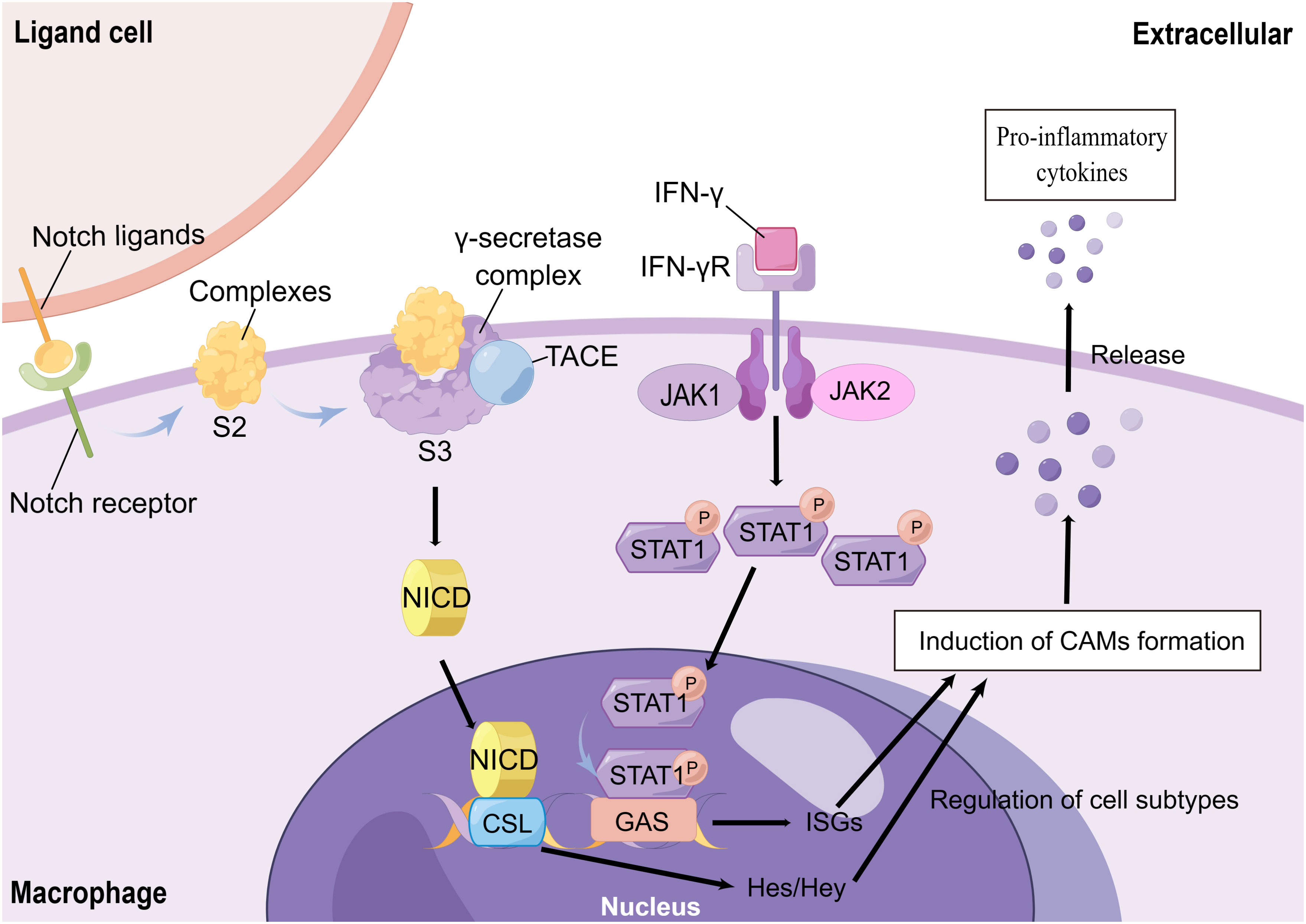
Figure 3 The Mechanisms of CAMs Formation. Notch receptors, which are expressed on the surface of macrophages, bind to neighboring cellular ligands. The ligand-receptor complex is formed and then exposed to hydrolysis site 2 (S2) in the extracellular proximal membrane region after endocytosis. After cleavage by tumor necrosis factor-α-converting enzyme (TACE) and hydrolysis of the γ-secretase complex (located at site S3 in the transmembrane region), a soluble Notch intracellular domain (NICD) is formed. This NICD then enters the cytoplasm and translocates to the nucleus, where it binds to the nuclear CSL transcription factor complex. This binding further activates Hes and Hey, induces the formation of CAMs, and mediates the release of inflammatory factors. On the other hand, the interferon-γ (IFN-γ) receptor (IFN-γR) on the surface of macrophages activates JAK1/JAK2 upon IFN-γ stimulation, which leads to the phosphorylation of intracellular STAT1. Phosphorylated STAT1 then enters the nucleus and binds to the Gamma interferon activation site (GAS) DNA element. This binding induces the transcription of interferon-stimulated genes (ISGs) and triggers the formation of CAMs.
Recently, the Notch signaling pathway has also received widespread attention in the polarization of CAMs. Macrophages stably express Notch ligands and Notch 1, 2, and 4 receptors on their surface, which can bind to ligands or receptors in adjacent cells. On the ligand cells, endocytosis of the ligand-receptor complexes leads to a change in the mechanical conformation of the endocytosed receptor. This change exposes the complexes to the hydrolysis site 2 (S2) in the extracellular near-membrane region. The complexes are cleaved by tumor necrosis factor-α-converting enzyme (TACE) and hydrolyzed by the γ-secretase complex (located at the S3 site in the transmembrane region). This process forms a soluble Notch intracellular domain (NICD) that enters the cytoplasm and translocates to the nucleus. NICD binds to the nuclear CSL transcription factor complex and activates the hairy enhancer of split (Hes) and Hes with YRPW motif (Hey) family members, which are classical Notch target genes. These genes induce CAMs and mediate the release of inflammatory factors (Figure 3). On the other hand, blocking Notch signaling could promote the polarization of M2 cells (66, 83–85). Not only that, but the Notch pathway regulates a variety of biological properties of macrophages that still need further exploration.
M2 macrophages are also known as alternatively activated macrophages (AAMs). AAMs are characterized by the production of large amounts of anti-inflammatory cytokines, Th2 chemokines, C-type lectins, clearance receptors, and polyamines (66, 78). It is mainly regulated by signaling pathways such as JAK/STAT3, STAT6, TGF-β/Smads, peroxisome proliferator-activated receptor γ (PPARγ), and certain miRNAs. Th2 cytokines IL-4 and IL-13 inhibit M1 polarization and promote AAM formation mainly through the corresponding receptors (IL-4Rα), which ultimately activate STAT6. Furthermore, STAT3 is equally important for the formation of AAMs, in addition to IL-4 and IL-13, other cytokines such as IL-10 are also activated (67, 78, 86, 87). Similarly, TGF-β induces phosphorylation of type I receptors by binding to type II receptors on the macrophage surface. This leads to the activation of Smad2 and Smad3 (88), which promotes the formation of AAMs and suppresses CAMs (67, 89). PPARγ is an essential transcription factor for cell differentiation (67, 90). It coordinates M1/M2 cell homeostasis with NF-κB and promotes the polarization of M2 cells (91). Activation of NF-κB p50 is critically important for the polarization of AAMs in vitro and in vivo (79). In recent years, the development of stem cell transplantation technology has raised significant interest in the immunomodulatory role of stem cells in various diseases, especially in the regulation of immune cells. Stem cells primarily exert their effects through the release of exosomes. It contains a large number of functional microRNAs (miRNAs) that regulate M1 and M2 macrophage polarization by targeting various transcription factors (74, 92, 93). Besides the classical signaling pathways mentioned above, the Wnt/β-catenin pathway and the PI3K/Akt/mTOR pathway have been demonstrated to have a significant impact on the regulation of macrophage polarization (94, 95). Nevertheless, M2 macrophage typing can be refined and further subdivided into M2a, M2b, M2c, and M2d subtypes, depending on their specific function and the genes they express (96).
The pathogenesis of ALF is complex and involves interactions between pathogenic agents and the host immune system. This interaction leads to the disorganization of the hepatic immune microenvironment and the simultaneous apoptosis of hepatocytes. Therefore, polarization is a dynamic process in ALF, as the polarized CAMs and AAMs still retain their plasticity and can interconvert again depending on the changing environment (22, 46). In the early stage of ALF, liver damage predominates due to immune injury. When the liver is invaded by foreign toxins or bacteria, or exposed to hepatotoxic substances, it can result in the death of hepatocytes. This, in turn, triggers the release of PAMPs by pathogens and DAMPs by dying hepatocytes. These PAMPs and DAMPs activate macrophages by binding to PR receptors (e.g., TLRs, NLRs) on the surface of macrophages. This activation prompts macrophages to shift towards the M1 pro-inflammatory phenotype and release inflammatory mediators into the bloodstream. Consequently, this process recruits and activates numerous inflammatory cells in the liver in order to eliminate pathogenic bacteria from the liver (97). However, simultaneously, it leads to a substantial infiltration of inflammatory cells and the formation of an excessive release of cytokines in the liver, initiating a “cascade activation” that results in a detrimental cycle (22). This phenomenon has also been observed during the early stages of injury in the model of ALF induced by APAP (31). Such an excessive inflammatory response not only exacerbates liver necrosis and injury but also increases the risk of extrahepatic and systemic infections. Hence, during the middle and late stages of ALF, the immune system of the body is functionally suppressed as a consequence of macrophages being excessively activated. This in turn leads to the suppression of both the functions of presenting antigens and pro-inflammatory functions. Consequently, a state of functional depletion arises. Both intrahepatic and extrahepatic immune components exhibit signs of immune paralysis (98). Furthermore, the remaining macrophages undergo a shift in function from the inflammatory M1 phenotype to the anti-inflammatory M2 phenotype, which facilitates tissue repair (99, 100). This is an antagonistic effect of the body aimed at protecting against an early, excessive inflammatory response (101). However, as a result of premature over-activation and depletion, the immune cells and their function become compromised, elevating the vulnerability to opportunistic infections. Consequently, the immune function of the body is further weakened. It is important to note that the direction of macrophage polarization during ALF is not absolute and relies on the influence of various cytokines and mediators in the immune microenvironment on primitive macrophages (99). Therefore, macrophage M1/M2 regulation can significantly modulate the systemic immune microenvironment and initiate a cascade of immune responses.
The imbalance of M1/M2 macrophage polarization is a key factor in the pathogenesis of ALF and plays a central role in the imbalance of the immune microenvironment in ALF (102). In the thioacetamide (TAA)-induced acute liver injury (TAA-ALI) mice model, the expression of senescence-associated secretory phenotype (SASP) was significantly increased, inducing M1 macrophage polarization. This exacerbates liver injury in ALI through repression of autophagy-related gene 5 (ATG5) (103). Similarly, the CC-5013, a TNF-α inhibitor, was able to significantly ameliorate liver damage in ALF by reducing the proportion of CAMs through the inhibition of the TNF-α/HMGB1 signaling pathway (104). Overall, CAMs have a positive effect on liver injury, making them a potential strategy for ALF treatment. Upregulating STAT6 by mesenchymal stem cells (MSCs) can increase the proportion of AAMs and significantly alleviate liver injury in a study, which demonstrates a positive therapeutic effect in ALF (105). Similarly, the overexpression of hepatocyte nuclear factor 4α (HNF4α) increased the transcription of IL-10 and promoted the polarization of AAMs through the IL-10/STAT3 pathway. This novel therapy for ALF resulted in the alleviation of ALF (106). In the D-GalN/LPS-induced ALF mice model, treatment with JWH-13, a cannabinoid receptor 2 (CB2) agonist, attenuated alanine aminotransferase (ALT) levels and reduced the production of pro-inflammatory cytokines, thereby protecting against ALF-associated death. Not only that, pretreatment of macrophages in vitro with JWH-133 significantly increased the secretion of the anti-inflammatory cytokine IL-10 in CAMs. It also enhanced the expression of AAMs markers, such as Arg and IL-10. These findings suggest that JWH-133 promotes the transformation of M1 to M2 macrophage phenotype, thereby improving ALF (107). At the same time, exosomes derived from human umbilical cord MSCs inhibited macrophage activation and the production of inflammatory cytokines in vitro and in vivo when exposed to LPS. This was achieved by releasing miR-455-3p, which resulted in reduced levels of serum inflammatory factors and improved IL-6-induced acute liver injury in ALF (108). In addition, stem cells can also regulate the direction of macrophage polarization by releasing cytokines (74). A study reported that treatment with MSCs in a D-GalN-induced ALF model induces the MSCs to secrete IL-4 in a paracrine manner. This secretion promotes the phenotypic conversion of inflammatory CAMs to anti-inflammatory AAMs, leading to improved ALF (109). Therefore, adjusting the proportion of M1/M2 macrophages has become a hot topic in ALF therapy. However, it also presents a new challenge for clinical application, as CAMs are essential cells for the clearance of toxic substances in the liver. The timing of their application still requires further experimental validation. This validation should consider the benefits and adverse consequences of reducing initial immune activation and its harmful downstream effects.
Pyroptosis, as a novel mode of cell death, has received much attention in recent years, and its specific mechanisms have been well explained (26). There are two activation pathways for pyroptosis: the canonical pathway, which depends on Caspase-1, and the non-canonical pathway, which depends on Caspase-4/5/11 activations (26, 27). When damage mediators enter the tissue, they induce the release of pro-inflammatory factors and the activation of immune cells, further stimulating the formation of the intracellular inflammasome, which can be found in various cells, such as macrophages, neutrophils, and hepatocytes (110). Inflammasomes, intracellular multiprotein complexes, consist of three parts: a cytosolic sensor, a bridging adaptor, and an effector (27, 111). The cytosolic sensor of the inflammasome is formed by nucleotide-binding oligomerization domain NLRs, with NLRP3 predominantly mediating pyroptosis. Apoptosis-associated speck-like proteins containing caspase recruitment domains (ASCs) act as bridging junctions for the inflammasome, with pro-caspase-1 serving as the effector. Therefore, NLPR3 and pro-caspase-1 form an inflammasome by binding to ASCs (27, 112, 113). The canonical pathway is initiated by the recognition of different endogenous and exogenous damage factors, such as DAMPs and PAMPs, by the inflammasome. This recognition triggers the activation pro-caspase-1, leading to its maturation into caspase-1, the effector molecule. It is released into the cytoplasm act on the NF-κB signaling pathway and promotes the cleavage of pro-IL-1β and pro-IL-18 into mature cytokines (27, 113). Activated Caspase-1 cleaves the gasdermin D (GSDMD) protein into N- and C-terminal fragments (114, 115). The GSDMD-N-terminal protein folds on the cell surface, forming a membrane pore. This pore allows for the release of cell contents, including IL-1β and IL-18 pro-inflammatory cytokines, outside the cell. As a result, the cell becomes swollen and highly permeable to the plasma membrane due to an imbalance of intra- and extracellular fluids. Eventually, the cell rapidly lyses, a process known as pyroptosis (27, 114–116).
In the non-canonical pathway, LPS, which is the primary stimulus in the non-classical pathway, enters the cell directly. Its Lipid A portion then binds to the CARD structural domain on the pro-Caspase-4/5/11, promoting the activation of mature Caspase-4/5/11 (117, 118). Caspase-4/5/11 not only cleaves GSDMD to form pore membranes like the canonical pathway that leads to pyroptosis, but it can also activate pannexin-1, the membrane channel for ATP, which induces the extracellular release of ATP (27). In the extracellular space, ATP binds to the P2X7 receptor through an autocrine or paracrine mechanism, causing the opening of the P2X7 pore and resulting in pyroptosis (119, 120). At the same time, Caspase-4/5/11 induces the formation of the NLRP3 inflammasome by promoting K-ion efflux and activating the classical scorch pathway (27, 119). Therefore, there is a distinction and link between the non-canonical and canonical pathways (Figure 4). The nature of pyroptosis is an effective immune defense against bacteria-infected cells in the internal environment. And IL-1β and IL-18, released from the cleaved cell by pyroptosis, are potent pro-inflammatory cytokines that can recruit innate immune cells to the site of infection and regulate acquired immune cells, aiding in the capture and clearance of pathogens (121, 122). This immune response towards pathogens facilitates the elimination of foreign microorganisms. However, if not well regulated, this excessive pro-inflammatory cascade response and host cell pyroptosis can be harmful to healthy tissue (27, 120, 123). Moreover, mature IL-18 can promote the production of IFN-γ and enhance the cytolytic activity of NK cells and T cells. (121, 122).
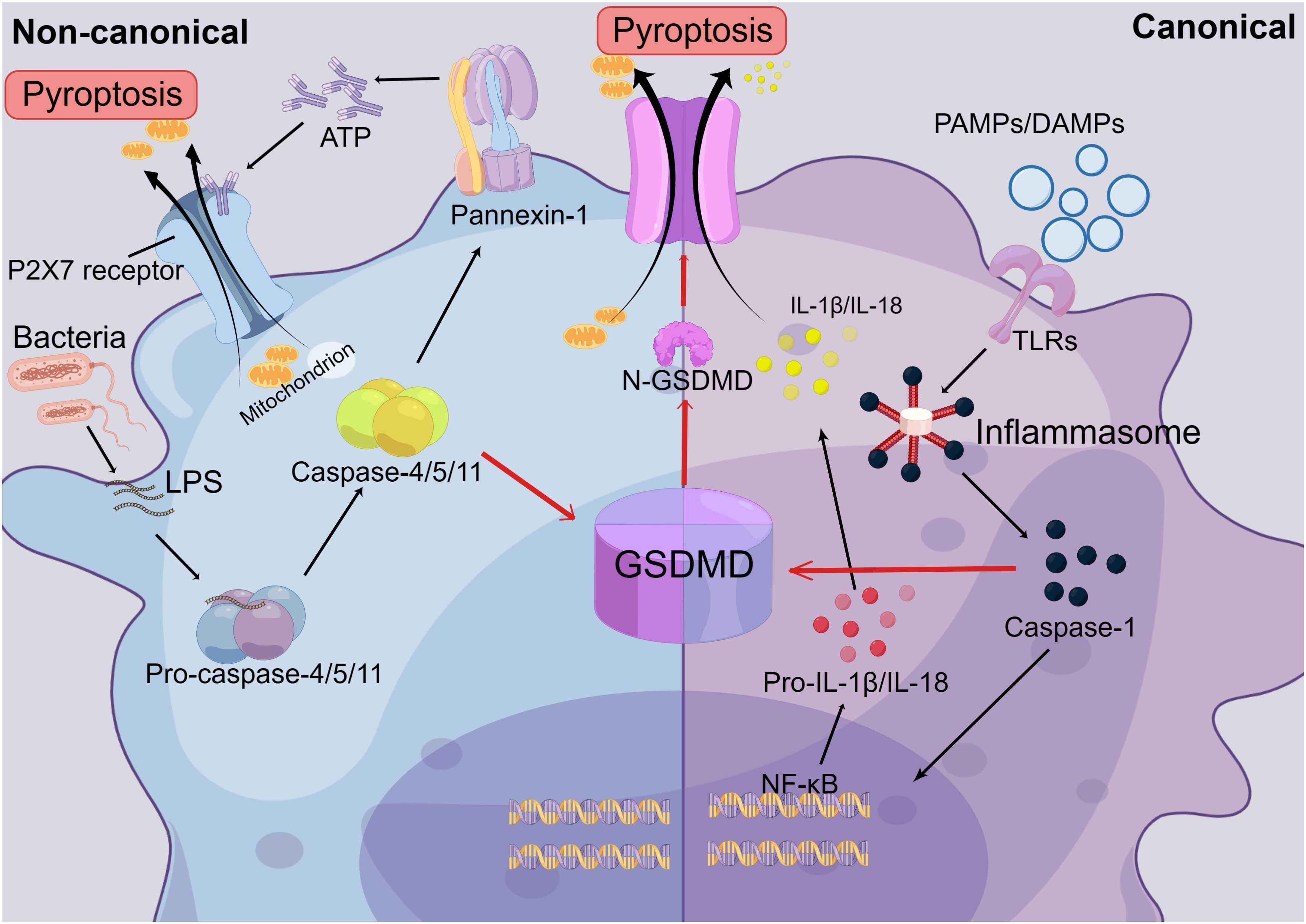
Figure 4 The Mechanisms of Pyroptosis. In the canonical pathway of pyroptosis, Toll-like receptors (TLRs) recognize extracellular pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), leading to the formation of inflammasomes. This, in turn, activates pro-caspase-1, causing it to mature into caspase-1. Caspase-1 is then released into the cytoplasm and acts on the NF-κB signaling pathway, promoting the maturation of IL-1β and IL-18. Secondly, activated caspase-1 cleaves the gasdermin D (GSDMD) protein into N-terminal and C-terminal fragments. The N-GSDMD fragments form pore membranes on the cell surface, allowing the release of IL-1β and IL-18 from the cell, thereby triggering inflammatory responses. Cell contents can also flow out through the pore membrane, resulting in an imbalance of intracellular and extracellular fluids and rapid cell lysis. In the non-canonical pathway, extracellular LPS can directly enter the cell and bind to the CARD domain on the intracellular Caspase-4/5/11 precursor, promoting the activation of mature Caspase-4/5/11. This activation leads to the formation of the N-GSDMD pore membrane, which triggers cellular pyroptosis. Additionally, it activates the ATP membrane channel, pannexin-1, which leads to the release of ATP from the cell. ATP binds to the P2X7 receptor through autocrine or paracrine mechanisms, resulting in the opening of the P2X7 pore. This leads to the release of cellular contents and ultimately triggers pyroptosis.
Inflammasome formation is also present in hepatocytes. Hepatocyte pyroptosis, induced by the activation of the specific NLRP3 inflammasome in hepatocytes, is considered to be a significant contributor to liver injury and liver fibrosis (27, 124). The production of NLRP3 inflammasome, cleavage of Caspase-1, and elevated levels of IL-1β factor have been observed in the concanavalin (ConA) and LPS/D-GalN induced liver failure models (27, 125, 126). In addition, the levels of proteins associated with pyroptosis, including caspase 1/4, GSDMD-N, IL-1β, IL-18, TNFα, and IFN-γ, were also detected in liver tissue from patients with ALF (127). Therefore, numerous studies have been conducted to alleviate liver damage in ALF by inhibiting key proteins or genes involved in pyroptosis. This is considered a potential therapeutic mechanism for treating ALF.
GSDMD, as a pyroptosis executioner (128), has been the primary focus of several studies. Using necrosulfonamide (NSA), an inhibitor of GSDMD, on the LPS/D-GalN-induced ALF mouse model resulted in a significant improvement in the pathophysiology and serology of liver damage. Additionally, it significantly decreased the levels of GSDMD, NLRP3, Caspase-1, and IL-1β (28). This study not only highlights the significance of pyroptosis in the progression of ALF but also demonstrates that inhibiting pyroptosis in vivo can effectively mitigate liver damage associated with ALF and yield therapeutic benefits. Similarly, limonin was able to inhibit LPS-induced pyroptosis by preventing cell membrane rupture and GSDMD activation. Additionally, limonin could prevent LPS-induced liver injury by primarily reducing the expression of NLRP3 and Caspase-1-related proteins, thereby inhibiting IL-1β (129). The 3,4-dihydroxyphenylethyl alcohol glycoside (DAG) isolated from Sargentodoxa cuneata has been shown to possess antioxidant, anti-apoptotic, and anti-inflammatory effects. Further studies have revealed that DAG reduces the levels of pyroptosis-related factors IL-1β, IL-18, and ROS. It also inhibits the expression of Caspase-1 and GSDMD in a dose-dependent manner, thereby inhibiting pyroptosis to treat APAP-induced ALF (130). The tyrosine-alanine (YA) peptide, which is a significant constituent of oyster (Crassostrea gigas) hydrolysate (OH), has demonstrated a hepatoprotective effect. It reduces the upregulation of GSDMD, the activation of caspase-1, and the cleavage of the C-terminus of GSDMD in mice injected with LPS/D-GalN (131).
Besides that, it has also been reported that PAMPs and DAMPs can directly or indirectly cause hepatocyte pyroptosis through cell-to-cell crosstalk (27). And it is not only hepatocyte pyroptosis, but macrophage pyroptosis also contributes to the development of liver disease (132, 133). In summary, pyroptosis is a promising therapeutic target for inflammatory diseases. This can be achieved by blocking related molecules such as NLRP3, Caspase-1, and GSDMD, which ultimately affects the progression of ALF. However, pyroptosis is an important defense mechanism against pathogenic invasion by its nature. Under physiological circumstances, moderate pyroptosis plays an important role in host defense against pathogenic microorganisms (134). Many pathogens have developed antimicrobial activity against macrophages, which are intrinsic immune cells. These pathogens can invade and replicate within macrophages, effectively isolating themselves from extracellular immune defenses and allowing them to escape the immune system. However, these pathogens cannot resist extracellular immunity (135). Bacteria, on the other hand, can activate the formation of various pyroptosis-associated inflammasomes, such as Listeriolysin released by Listeria monocytogenes, B. anthracis protease lethal factor (LF), Pneumolysin (PLY), and α-hemolysin released by Staphylococcus aureus. All of these are known to be activators of NLRP3 (136). LF was the first activator of pyroptosis to be identified and discovered. Mechanistically, LF is cleaved intracellularly and further degraded by the proteasome (137). The degradation product can then participate in the formation of CARD at the C-terminus of Caspase-1 (138, 139). This activation leads to the formation of inflammasomes and triggers the canonical pathway, inducing pyroptosis. As a result, infected cells rupture, releasing pathogens into the extracellular environment and initiating an immune response to eliminate the pathogens. In addition, the gene sequence of LPS released by Gram-negative bacteria, LipidA, is a highly conserved. LipidA binds to Caspase-11/4/5 and triggers the oligomerization and activation of caspases, leading to the induction of the non-canonical cellular pyroptosis (140, 141). Overall, this mechanism serves as a clearance mechanism for the organism to defend against invading pathogens and plays a crucial role in protecting the organism from such pathogens. Therefore, therapeutic interventions aimed at inhibiting pyroptosis may have inherent flaws. It can worsen the existing pathogenic invasion and increase the risk of opportunistic infections. Furthermore, when regulating the local signaling pathways or key proteins in the organism, it is necessary to consider the systemic response that it triggers. The pros and cons of this issue are unavoidable and will require extensive research before it can be translated into clinical treatment. It is indisputable that conducting comprehensive studies on pyroptosis is essential for understanding the pathogenesis of ALF and for the development of drugs targeting this process.
Macrophage polarization and pyroptosis are important for the development of ALF and share certain signaling pathways or regulatory mediators. Therefore, it is reasonable to speculate that there is an interaction between macrophage polarization and pyroptosis. However, there are fewer studies on the mutual regulation and crosstalk between macrophage polarization and pyroptosis. Cluster of Differentiation 38 (CD38) is a type II transmembrane protein that is widely expressed in immune cells. It controls the innate immune response and inflammatory pathways triggered by infection (122, 142). It was found that liver-injured mice with CD38 knockdown exhibited more severe pyroptosis and liver damage. By comparing protein expression in WT and CD38-deficient mice, researchers found elevated expression of M1 macrophage marker proteins such as TLR4, MyD88, and phosphorylated NF-κB p65 in CD38-deficient mice. Furthermore, the increased expression of pyroptosis-associated markers caused by CD38 knockdown could be reversed by TLR4 mutation. This suggests that the more severe liver damage and pyroptosis caused by CD38 deficiency are related to the TLR4 signaling pathway. However, further research is needed to elucidate the role of CD38 in M1 macrophages and pyroptosis through the TLR4 signaling pathway is not available (122). TLR receptor activation has been found to induce the production of the NLRP3 inflammasome and the development of pyroptosis (143). Therefore, we can speculate that CD38 inhibits pyroptosis by regulating the TLR4 signaling pathway. But it remains to be considered whether CD38 further regulates pyroptosis through the polarization of M1 macrophages.
High mobility group box protein 1 (HMGB1) is a nuclear DNA-binding protein that activates Caspase-1-dependent pyroptosis in hepatocytes, thereby exacerbating the inflammatory response and damage. This process can be ameliorated by HMGB1 inhibitors (144). In addition, HMGB1 also acts as a DAMP that easily translocates to the outside of cells in response to endogenous tissue damage or exogenous microbial invasion. It activates immune cells and releases pro-inflammatory factors, which cause an inflammatory response (145). When the DAMPs activate macrophages by binding to PRR on the surface of the macrophage, the activated macrophage will secrete the pro-inflammatory HMGB1 (22, 144). Once HMGB1 translocates to the outside of the cell membrane or is transported to target cells via extracellular vesicles, it binds to its receptor RAGE or TLR4 and initiates as a DAMP molecule. This leads to the activation of the NLRP3 inflammasome, inducing pyroptosis in recipient cells and provoking an inflammatory response (144, 146–148). Moreover, the HMGB1 outside the cell could activate the MyD88-dependent TLR4 signaling pathway and enhance NF-κB expression through TLR4 binding (122, 144). Therefore, macrophages following LPS induction will release HMGB1 to initiate hepatocyte pyroptosis. This process can also induce the formation of the NLRP3-inflammasome through activation of the TLR4/MyD88/NF-κB signaling pathway (144). As discussed in a previous paragraph, this pathway is also considered a classical pathway for M1 macrophage polarization. Although the role of macrophage polarization is not highlighted in this article, HMGB1 plays an important role in liver damage of ALF as a common mediator of both macrophage polarization and pyroptosis, and the therapeutic effect of HMGB1 inhibition on ALF has been demonstrated in several studies. Besides, in the LPS/D-GalN-induced ALF mouse model, it was discovered that lenalidomide (CC-5013) treatment significantly reduced the activation of the TNF-α/HMGB1 signaling pathway. This reduction resulted in a decrease in the number of M1 macrophages in both liver and kidney tissues, ultimately leading to a decrease in intra-tissue pyroptosis levels (104).
M2 macrophages play a crucial role in protecting the liver in ALF. They exhibit hepatoprotective effects by releasing the anti-inflammatory cytokine IL-10 and pro-fibrosis (149). Additionally, M2 macrophages exert hepatoprotective effects by expressing the Galectin-3 (GAL3) gene, which inhibits the expression of pyroptosis signaling proteins in ALF mice (150). Surely, the one-way regulatory mechanism is incomplete. Some studies have suggested that hepatocyte pyroptosis mediated by GSDMD can recruit macrophages to release inflammatory mediators through the upregulation of the monocyte chemotactic protein 1/CC chemokine receptor-2 (MCRP1/CCR2) signaling pathway, leading to the spread of the inflammatory response. Furthermore, immunohistochemistry of the liver showed a significant decrease in the expression of the macrophage-specific protein F4/80 in the D-GalN/LPS ALF mouse model with GSDMD knockout, compared to the wild type(WT) D-GalN/LPS ALF model (127). This phenomenon suggests that inhibiting GSDMD-induced pyroptosis can significantly decrease macrophage infiltration in ALF liver tissue. In a wore, all of this evidence suggests a positive feedback loop between macrophage polarization and hepatocyte pyroptosis, which can induce an inflammatory cascade response in ALF. Given these observations, further research is needed to understand the cellular crosstalk between macrophage polarization and hepatocyte pyroptosis and its contribution to the progression of ALF. Additionally, the complex signaling pathways between these two processes should be explored and confirmed through additional experiments (Figure 5).
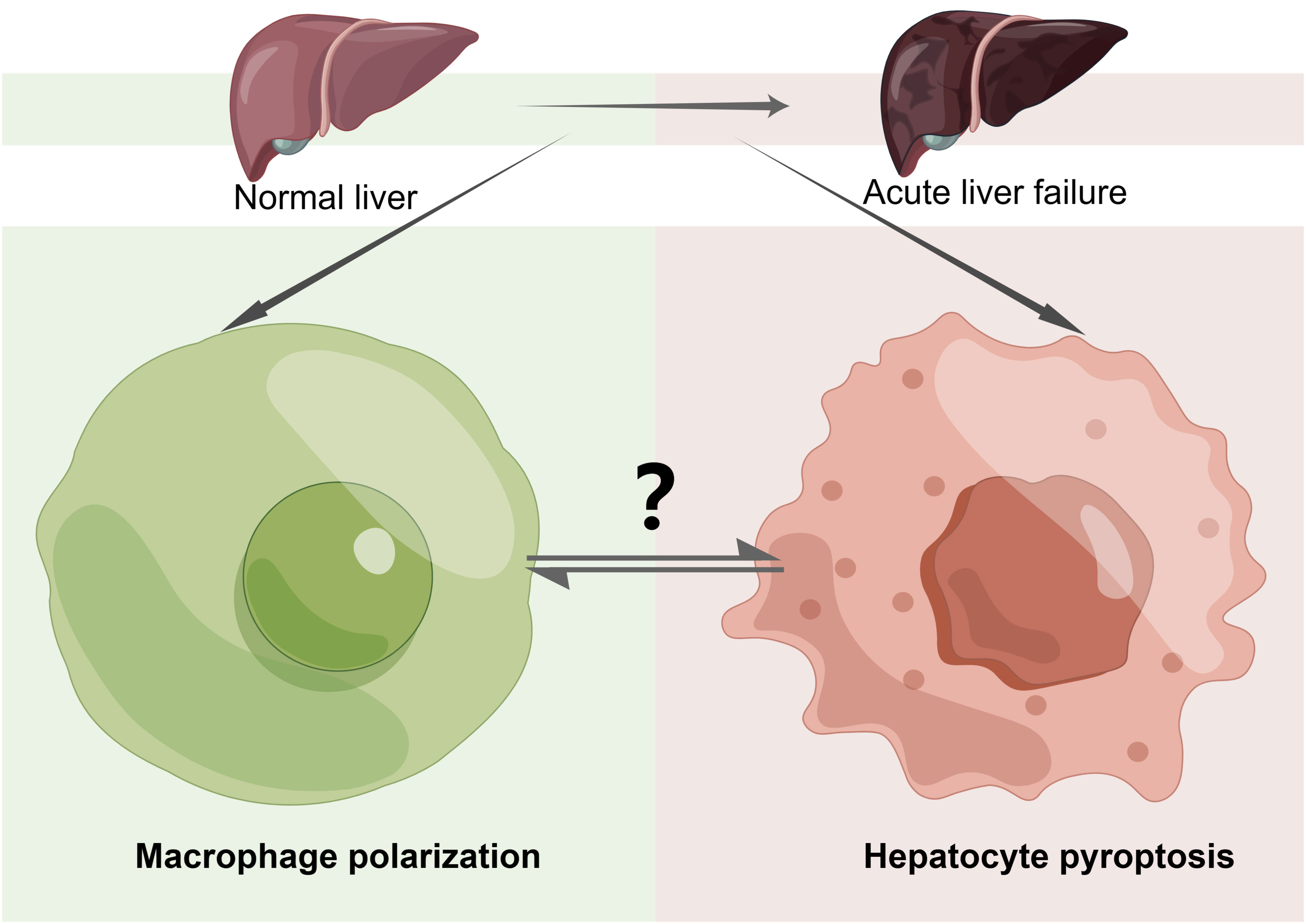
Figure 5 The Crosstalk of Pyroptosis and Macrophage Polarization in ALF. Macrophage polarization and hepatocyte pyroptosis play important roles in acute liver failure (ALF). The mutual crosstalk and regulation between these two processes can impact the progression of the disease. However, the specific mechanisms involved require further study and exploration. “?”: there are many unknown relationships between macrophage polarization and hepatocyte pyroptosis in ALF that need to be further explored and investigated in the future, including the impact of their mutual regulation and constraints on disease progression in ALF as well as the specific pathways of action.
The study of the hepatocyte pyroptosis pathway in ALF is still in its preliminary stage. All factors that induce liver injury led to pyroptosis, and the late stage of hepatocyte pyroptosis, accompanied by the release of inflammatory factors such as IL-1β and IL-18, is central to the exacerbation of ALF (132, 151). The inflammatory factors released from hepatocyte pyroptosis enhance the activation of the NF-κB signaling pathway in hepatocytes and macrophages, thereby further promoting the inflammatory response. At the same time, the release of pro-inflammatory factors can recruit mononuclear macrophages in the body, leading to a more severe inflammatory response and ultimately causing massive hepatocyte death (22, 152). The suggestion of improving liver damage and inflammatory response by inhibiting pyroptosis in ALF has also been the focus of several studies. However, inhibiting pyroptosis is equivalent to compromising the system’s ability to clear microorganisms, which can potentially lead to opportunistic infections and even the development of adverse outcomes like bacteremia and sepsis. Therefore, further exploration is needed in the therapeutic strategy of treating ALF by inhibiting pyroptosis. This is important to consider due to the local intrahepatic damage and the systemic state. The aim is to find more precise methods of targeting intrahepatic pyroptosis.
The role of hepatic macrophages in ALF has been controversial. It is mentioned in our paper that macrophage activation is influenced by various factors in the immune microenvironment and can polarize in either a pro-inflammatory or an anti-inflammatory direction. However, polarized macrophages can still switch their polarization depending on the cytokines and mediators present in the immune microenvironment. Depending on the stage of ALF disease, macrophages can either perpetuate inflammation or promote its remission. These contradictions and inconsistencies are attributed to the diversity of macrophage subtypes. For this reason, regulating the direction of macrophage polarization has become a therapeutic approach in the study of ALF. However, therapies targeted at macrophages for the treatment of ALF carry certain risks and challenges. Firstly. However, promoting the anti-inflammatory effects of macrophages, they can improve liver damage and disease progression in ALF, but at the same time, they can also facilitate the spread of pathogens in the system and increase susceptibility to infection. Secondly, by promoting the pro-inflammatory effects of macrophages, it can effectively inhibit opportunistic infections and remove foreign pathogens. However, it can also easily induce a severe inflammatory response, resulting in the release of inflammatory mediators that are dispersed throughout the system via the bloodstream. This can lead to multi-organ damage and even failure. The contradictions of the approaches described for macrophage-targeted therapy highlight the challenges of immunotherapeutic strategies in ALF. The local hepatic immune microenvironment and the systemic immune microenvironment mutually regulate and influence each other. Anti-inflammatory therapy in the local hepatic immune microenvironment is effective in alleviating liver damage and repairing tissue. However, it has the opposite effect in controlling systemic pathogen dissemination. The timing of targeting application to induce different macrophage polarization directions needs to be precisely controlled, especially in order to clarify the function and composition of macrophages at different stages of ALF in humans.
Combining the signaling pathways and mechanisms of macrophage polarization and pyroptosis mentioned above, it is clear that several signaling pathways are shared between the two. For example, the TLR/NF-κB pathway not only plays an important role in M1 polarization but also induces cell pyroptosis (27, 67). In addition, several studies have also implicitly suggested that there is a reciprocal regulation between macrophage polarization and pyroptosis. For example, the knockdown of CD38 promotes the expression of molecules related to M1 polarization and also triggers more severe cell pyroptosis. CD38 serves as a common regulatory mediator of both processes (122). The interaction between macrophage polarization and pyroptosis still requires further investigation and exploration. In particular, the roles of both factors are crucial to the development of the disease. However, it is unclear whether they are mutually independent, whether they regulate each other, or if they even trigger a more severe inflammatory response through positive feedback. This assessment cannot be made without further experiments and studies to confirm.
In summary, this paper describes the mechanisms and regulatory pathways of macrophage polarization and pyroptosis in ALF. It also explores the interplay between macrophage polarization and pyroptosis. Fundamentally, macrophage polarization and pyroptosis in ALF are significant factors that have been extensively studied in recent years. However, there are still numerous unanswered questions that require further exploration. It is hoped that future research will address these questions, leading to new insights into the pathogenesis and therapeutic strategies for ALF.
DX: Conceptualization, Writing – original draft. SO: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81803884), Plan on enhancing scientific research in GMU (02-410-2302092XM), 2022 Guangzhou Medical University discipline construction projects (02-410-2206013), the 2022 Student Innovation Capacity Enhancement Program Project of Guangzhou Medical University (02-408-2304-19064XM), 2023 City school (college) enterprise joint funding projects (2023A03J0421), the Undergraduate Capacity Enhancement Innovation Project of Guangzhou Medical University (2022JXA003), the 2023 Artificial Liver Special Fund (iGandanF-1082023-RGG023), the Key Medical Discipline of Guangzhou [2021-2023] and the Key Laboratory of Guangdong Higher Education Institutes (2021KSYS009).
All figures created by Figdraw.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cheng ML, Nakib D, Perciani CT, MacParland SA. The immune niche of the liver. Clin Sci (Lond) (2021) 135(20):2445–66. doi: 10.1042/CS20190654
2. Nakano T, Lai CY, Goto S, Hsu LW, Kawamoto S, Ono K, et al. Immunological and regenerative aspects of hepatic mast cells in liver allograft rejection and tolerance. PloS One (2012) 7(5):e37202. doi: 10.1371/journal.pone.0037202
3. Herkel J. Regulatory T cells in hepatic immune tolerance and autoimmune liver diseases. Dig Dis (2015) 33 Suppl 2:70–4. doi: 10.1159/000440750
4. Drescher HK, Bartsch LM, Weiskirchen S, Weiskirchen R. Intrahepatic T(H)17/T(Reg) cells in homeostasis and disease-it's all about the balance. Front Pharmacol (2020) 11:588436. doi: 10.3389/fphar.2020.588436
5. Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13(3):267–76. doi: 10.1038/cmi.2016.3
6. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
7. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity (2019) 51(4):655–670.e658. doi: 10.1016/j.immuni.2019.09.002
8. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14(10):996–1006. doi: 10.1038/ni.2691
9. McDonald B, Kubes P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology (2016) 151(6):1087–95. doi: 10.1053/j.gastro.2016.09.048
10. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol (2017) 27(21):R1147–51. doi: 10.1016/j.cub.2017.09.019
11. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
12. Son G, Kremer M, Hines IN. Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract (2010) 2010:1–13. doi: 10.1155/2010/453563
13. Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol (2010) 11(4):295–302. doi: 10.1038/ni.1855
14. Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, et al. The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int (1999) 49(6):519–32. doi: 10.1046/j.1440-1827.1999.00910.x
15. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. ALK1 signaling is required for the homeostasis of Kupffer cells and prevention of bacterial infection. J Clin Invest (2022) 132(3):e150489. doi: 10.1172/JCI150489
16. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol (2018) 15(12):738–52. doi: 10.1038/s41575-018-0065-y
17. Chung RT, Stravitz RT, Fontana RJ, Schiodt FV, Mehal WZ, Reddy KR, et al. Pathogenesis of liver injury in acute liver failure. Gastroenterology (2012) 143(3):e1–7. doi: 10.1053/j.gastro.2012.07.011
18. Dong V, Nanchal R, Karvellas CJ. Pathophysiology of acute liver failure. Nutr Clin Pract (2020) 35(1):24–9. doi: 10.1002/ncp.10459
19. Kumagai K, Mawatari S, Ido A. [Pathophysiology of acute liver failure from onset to liver regeneration]. Nihon Shokakibyo Gakkai Zasshi (2020) 117(9):750–5. doi: 10.11405/nisshoshi.117.750
20. Abdullah Z, Knolle PA. Liver macrophages in healthy and diseased liver. Pflugers Arch (2017) 469(3-4):553–60. doi: 10.1007/s00424-017-1954-6
21. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
22. Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front Immunol (2018) 9:2948. doi: 10.3389/fimmu.2018.02948
23. Linkermann A, Green DR. Necroptosis. N Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
24. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol (2016) 26(13):R568–72. doi: 10.1016/j.cub.2016.02.019
25. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
26. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
27. Wu J, Lin S, Wan B, Velani B, Zhu Y. Pyroptosis in liver disease: new insights into disease mechanisms. Aging Dis (2019) 10(5):1094–108. doi: 10.14336/AD.2019.0116
28. Wu YL, Ou WJ, Zhong M, Lin S, Zhu YY. Gasdermin D inhibitor necrosulfonamide alleviates lipopolysaccharide/D-galactosamine-induced acute liver failure in mice. J Clin Transl Hepatol (2022) 10(6):1148–54. doi: 10.14218/JCTH.2021.00560
29. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol (2013) 59(3):583–94. doi: 10.1016/j.jhep.2013.03.033
30. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol (2017) 14(1):55–66. doi: 10.1038/nrgastro.2016.168
31. Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol (2019) 10:3112. doi: 10.3389/fimmu.2019.03112
32. Shan Z, Ju C. Hepatic macrophages in liver injury. Front Immunol (2020) 11:322. doi: 10.3389/fimmu.2020.00322
33. Kempka G, Kolb-Bachofen V. Binding, uptake, and transcytosis of ligands for mannose-specific receptors in rat liver: an electron microscopic study. Exp Cell Res (1988) 176(1):38–48. doi: 10.1016/0014-4827(88)90118-8
34. Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, et al. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol (2005) 77(5):626–33. doi: 10.1189/jlb.0604332
35. Rieder H, Ramadori G, Allmann KH, Meyer zum Buschenfelde KH. Prostanoid release of cultured liver sinusoidal endothelial cells in response to endotoxin and tumor necrosis factor. Comparison with umbilical vein endothelial cells. J Hepatol (1990) 11(3):359–66. doi: 10.1016/0168-8278(90)90222-d
36. Kastenbauer S, Ziegler-Heitbrock HW. NF-kappaB1 (p50) is upregulated in lipopolysaccharide tolerance and can block tumor necrosis factor gene expression. Infect Immun (1999) 67(4):1553–9. doi: 10.1128/IAI.67.4.1553-1559.1999
37. Gschwandtner M, Derler R, Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
38. Papaioannou S, See JX, Jeong M, de la Torre C, Ast V, Reiners-Koch PS, et al. Liver sinusoidal endothelial cells orchestrate NK cell recruitment and activation in acute inflammatory liver injury. Cell Rep (2023) 42(8):112836. doi: 10.1016/j.celrep.2023.112836
39. Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res (2017) 3(Suppl 1):170–88. doi: 10.18053/jctres.03.2017S1.003
40. Ramachandran A, Jaeschke H. Acetaminophen hepatotoxicity. Semin Liver Dis (2019) 39(2):221–34. doi: 10.1055/s-0039-1679919
41. Stravitz RT, Lee WM. Acute liver failure. Lancet (2019) 394(10201):869–81. doi: 10.1016/S0140-6736(19)31894-X
42. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
43. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
44. Mihm S. Danger-associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci (2018) 19(10):3104. doi: 10.3390/ijms19103104
45. Gale RP, Sparkes RS, Golde DW. Bone marrow origin of hepatic macrophages (Kupffer cells) in humans. Science (1978) 201(4359):937–8. doi: 10.1126/science.356266
46. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol (2016) 65(4):758–68. doi: 10.1016/j.jhep.2016.05.037
47. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut (2018) 67(2):333–47. doi: 10.1136/gutjnl-2016-313615
48. Flores Molina M, Abdelnabi MN, Mazouz S, Villafranca-Baughman D, Trinh VQ, Muhammad S, et al. Distinct spatial distribution and roles of Kupffer cells and monocyte-derived macrophages in mouse acute liver injury. Front Immunol (2022) 13:994480. doi: 10.3389/fimmu.2022.994480
49. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology (2016) 64(5):1667–82. doi: 10.1002/hep.28682
50. Wu R, Zhang Y, Xiang Y, Tang Y, Cui F, Cao J, et al. Association between serum S100A9 levels and liver necroinflammation in chronic hepatitis B. J Transl Med (2018) 16(1):83. doi: 10.1186/s12967-018-1462-2
51. Bai L, Kong M, Duan Z, Liu S, Zheng S, Chen Y. M2-like macrophages exert hepatoprotection in acute-on-chronic liver failure through inhibiting necroptosis-S100A9-necroinflammation axis. Cell Death Dis (2021) 12(1):93. doi: 10.1038/s41419-020-03378-w
52. Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology (2015) 149(7):1896–1909.e1814. doi: 10.1053/j.gastro.2015.08.053
53. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol (2017) 66(6):1300–12. doi: 10.1016/j.jhep.2017.02.026
54. Yang T, Wang H, Wang X, Li J, Jiang L. The dual role of innate immune response in acetaminophen-induced liver injury. Biol (Basel) (2022) 11(7):1057. doi: 10.3390/biology11071057
55. Zhou D, Yang K, Chen L, Wang Y, Zhang W, Xu Z, et al. Macrophage polarization and function: new prospects for fibrotic disease. Immunol Cell Biol (2017) 95(10):864–9. doi: 10.1038/icb.2017.64
56. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol (2020) 11:1169. doi: 10.3389/fimmu.2020.01169
57. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell (2016) 165(3):668–78. doi: 10.1016/j.cell.2016.03.009
58. Tujios S, Stravitz RT, Lee WM. Management of acute liver failure: update 2022. Semin Liver Dis (2022) 42(3):362–78. doi: 10.1055/s-0042-1755274
59. Filliol A, Piquet-Pellorce C, Raguenes-Nicol C, Dion S, Farooq M, Lucas-Clerc C, et al. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J Hepatol (2017) 66(6):1205–13. doi: 10.1016/j.jhep.2017.01.005
60. Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology (2000) 32(4 Pt 1):734–9. doi: 10.1053/jhep.2000.17687
61. Choudhury A, Kumar M, Sharma BC, Maiwall R, Pamecha V, Moreau R, et al. Systemic inflammatory response syndrome in acute-on-chronic liver failure: Relevance of 'golden window': A prospective study. J Gastroenterol Hepatol (2017) 32(12):1989–97. doi: 10.1111/jgh.13799
62. Yang R, Zou X, Tenhunen J, Tonnessen TI. HMGB1 and extracellular histones significantly contribute to systemic inflammation and multiple organ failure in acute liver failure. Mediators Inflamm (2017) 2017:5928078. doi: 10.1155/2017/5928078
63. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology (2018) 154(2):186–95. doi: 10.1111/imm.12910
64. Thapa B, Lee K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep (2019) 52(6):360–72. doi: 10.5483/BMBRep.2019.52.6.140
65. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
66. Chen W, Liu Y, Chen J, Ma Y, Song Y, Cen Y, et al. The Notch signaling pathway regulates macrophage polarization in liver diseases. Int Immunopharmacol (2021) 99:107938. doi: 10.1016/j.intimp.2021.107938
67. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
68. Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci (2021) 23(1):144. doi: 10.3390/ijms23010144
69. Lampiasi N. Macrophage polarization: learning to manage it. Int J Mol Sci (2022) 23(13):7208. doi: 10.3390/ijms23137208
70. Yao Y, Xu XH, Jin L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol (2019) 10:792. doi: 10.3389/fimmu.2019.00792
71. Bosco MC. Macrophage polarization: Reaching across the aisle? J Allergy Clin Immunol (2019) 143(4):1348–50. doi: 10.1016/j.jaci.2018.12.995
72. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
73. Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al. Macrophage polarization in chronic inflammatory diseases: killers or builders? J Immunol Res (2018) 2018:8917804. doi: 10.1155/2018/8917804
74. Arabpour M, Saghazadeh A, Rezaei N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cell-derived exosomes. Int Immunopharmacol (2021) 97:107823. doi: 10.1016/j.intimp.2021.107823
75. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci (2021) 78(4):1233–61. doi: 10.1007/s00018-020-03656-y
76. Chen XX, Tang L, Fu YM, Wang Y, Han ZH, Meng JG. Paralemmin-3 contributes to lipopolysaccharide-induced inflammatory response and is involved in lipopolysaccharide-Toll-like receptor-4 signaling in alveolar macrophages. Int J Mol Med (2017) 40(6):1921–31. doi: 10.3892/ijmm.2017.3161
77. Gong J, Li J, Dong H, Chen G, Qin X, Hu M, et al. Inhibitory effects of berberine on proinflammatory M1 macrophage polarization through interfering with the interaction between TLR4 and MyD88. BMC Complement Altern Med (2019) 19(1):314. doi: 10.1186/s12906-019-2710-6
78. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
79. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
80. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol (2017) 18(4):374–84. doi: 10.1038/ni.3691
81. Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
82. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
83. Suresh S, Irvine AE. The NOTCH signaling pathway in normal and Malignant blood cell production. J Cell Commun Signal (2015) 9(1):5–13. doi: 10.1007/s12079-015-0271-0
84. Wu G, Wilson G, George J, Qiao L. Modulation of Notch signaling as a therapeutic approach for liver cancer. Curr Gene Ther (2015) 15(2):171–81. doi: 10.2174/1566523214666141224100319
85. Aval SF, Lotfi H, Sheervalilou R, Zarghami N. Tuning of major signaling networks (TGF-beta, Wnt, Notch and Hedgehog) by miRNAs in human stem cells commitment to different lineages: Possible clinical application. BioMed Pharmacother (2017) 91:849–60. doi: 10.1016/j.biopha.2017.05.020
86. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
87. Porta C, Riboldi E, Ippolito A, Sica A. Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol (2015) 27(4):237–48. doi: 10.1016/j.smim.2015.10.003
88. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
89. Wang L, Li Y, Wang X, Wang P, Essandoh K, Cui S, et al. GDF3 protects mice against sepsis-induced cardiac dysfunction and mortality by suppression of macrophage pro-inflammatory phenotype. Cells (2020) 9(1):120. doi: 10.3390/cells9010120
90. Marion-Letellier R, Savoye G, Ghosh S. Fatty acids, eicosanoids and PPAR gamma. Eur J Pharmacol (2016) 785:44–9. doi: 10.1016/j.ejphar.2015.11.004
91. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
92. Essandoh K, Li Y, Huo J, Fan GC. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock (2016) 46(2):122–31. doi: 10.1097/SHK.0000000000000604
93. Lo Sicco C, Reverberi D, Balbi C, Ulivi V, Principi E, Pascucci L, et al. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects: endorsement of macrophage polarization. Stem Cells Transl Med (2017) 6(3):1018–28. doi: 10.1002/sctm.16-0363
94. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198(3):1006–14. doi: 10.4049/jimmunol.1601515
95. Feng Y, Ren J, Gui Y, Wei W, Shu B, Lu Q, et al. Wnt/beta-catenin-promoted macrophage alternative activation contributes to kidney fibrosis. J Am Soc Nephrol (2018) 29(1):182–93. doi: 10.1681/ASN.2017040391
96. Huang X, Li Y, Fu M, Xin HB. Polarizing macrophages in vitro. Methods Mol Biol (2018) 1784:119–26. doi: 10.1007/978-1-4939-7837-3_12
97. Jaeschke H, Ramachandran A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem Toxicol (2020) 138:111240. doi: 10.1016/j.fct.2020.111240
98. Wasmuth HE, Kunz D, Yagmur E, Timmer-Stranghoner A, Vidacek D, Siewert E, et al. Patients with acute on chronic liver failure display "sepsis-like" immune paralysis. J Hepatol (2005) 42(2):195–201. doi: 10.1016/j.jhep.2004.10.019
99. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
100. Chan MWY, Viswanathan S. Recent progress on developing exogenous monocyte/macrophage-based therapies for inflammatory and degenerative diseases. Cytotherapy (2019) 21(4):393–415. doi: 10.1016/j.jcyt.2019.02.002
101. Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology (2002) 35(5):1093–103. doi: 10.1053/jhep.2002.33162
102. Possamai LA, Thursz MR, Wendon JA, Antoniades CG. Modulation of monocyte/macrophage function: a therapeutic strategy in the treatment of acute liver failure. J Hepatol (2014) 61(2):439–45. doi: 10.1016/j.jhep.2014.03.031
103. Liu R, Cui J, Sun Y, Xu W, Wang Z, Wu M, et al. Autophagy deficiency promotes M1 macrophage polarization to exacerbate acute liver injury via ATG5 repression during aging. Cell Death Discov (2021) 7(1):397. doi: 10.1038/s41420-021-00797-2
104. Wang Y, Zhang H, Chen Q, Jiao F, Shi C, Pei M, et al. TNF-alpha/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif (2020) 53(6):e12829. doi: 10.1111/cpr.12829
105. Li Y, Sheng Q, Zhang C, Han C, Bai H, Lai P, et al. STAT6 up-regulation amplifies M2 macrophage anti-inflammatory capacity through mesenchymal stem cells. Int Immunopharmacol (2021) 91:107266. doi: 10.1016/j.intimp.2020.107266
106. Yu Y, Zhang Q, Wu N, Xia L, Cao J, Xia Q, et al. HNF4alpha overexpression enhances the therapeutic potential of umbilical cord mesenchymal stem/stromal cells in mice with acute liver failure. FEBS Lett (2022) 596(24):3176–90. doi: 10.1002/1873-3468.14453
107. Tomar S, Zumbrun EE, Nagarkatti M, Nagarkatti PS. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J Pharmacol Exp Ther (2015) 353(2):369–79. doi: 10.1124/jpet.114.220368
108. Shao M, Xu Q, Wu Z, Chen Y, Shu Y, Cao X, et al. Exosomes derived from human umbilical cord mesenchymal stem cells ameliorate IL-6-induced acute liver injury through miR-455-3p. Stem Cell Res Ther (2020) 11(1):37. doi: 10.1186/s13287-020-1550-0
109. Li YW, Zhang C, Sheng QJ, Bai H, Ding Y, Dou XG. Mesenchymal stem cells rescue acute hepatic failure by polarizing M2 macrophages. World J Gastroenterol (2017) 23(45):7978–88. doi: 10.3748/wjg.v23.i45.7978
110. Gan C, Cai Q, Tang C, Gao J. Inflammasomes and pyroptosis of liver cells in liver fibrosis. Front Immunol (2022) 13:896473. doi: 10.3389/fimmu.2022.896473
111. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12(7):387–400. doi: 10.1038/nrgastro.2015.94
112. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
113. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
114. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol (2017) 27(9):673–84. doi: 10.1016/j.tcb.2017.05.005
115. Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv Immunol (2017) 135:81–117. doi: 10.1016/bs.ai.2017.02.002
116. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
117. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
118. Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
119. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43(5):923–32. doi: 10.1016/j.immuni.2015.10.009
120. You R, He X, Zeng Z, Zhan Y, Xiao Y, Xiao R. Pyroptosis and its role in autoimmune disease: A potential therapeutic target. Front Immunol (2022) 13:841732. doi: 10.3389/fimmu.2022.841732
121. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
122. Zhang H, Du Y, Guo Y, Wang Z, Li H, Lv Z, et al. TLR4-NLRP3-GSDMD-mediated pyroptosis plays an important role in aggravated liver injury of CD38(-/-) sepsis mice. J Immunol Res (2021) 2021:6687555. doi: 10.1155/2021/6687555
123. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med (2017) 45(2):253–62. doi: 10.1097/CCM.0000000000002074
124. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol (2021) 74(1):156–67. doi: 10.1016/j.jhep.2020.07.041
125. Luan J, Zhang X, Wang S, Li Y, Fan J, Chen W, et al. NOD-like receptor protein 3 inflammasome-dependent IL-1beta accelerated conA-induced hepatitis. Front Immunol (2018) 9:758. doi: 10.3389/fimmu.2018.00758
126. Wang Y, Chen Q, Jiao F, Shi C, Pei M, Wang L, et al. Histone deacetylase 2 regulates ULK1 mediated pyroptosis during acute liver failure by the K68 acetylation site. Cell Death Dis (2021) 12(1):55. doi: 10.1038/s41419-020-03317-9
127. Li H, Zhao XK, Cheng YJ, Zhang Q, Wu J, Lu S, et al. Gasdermin D-mediated hepatocyte pyroptosis expands inflammatory responses that aggravate acute liver failure by upregulating monocyte chemotactic protein 1/CC chemokine receptor-2 to recruit macrophages. World J Gastroenterol (2019) 25(44):6527–40. doi: 10.3748/wjg.v25.i44.6527
128. Liu M, He J, Zheng S, Zhang K, Ouyang Y, Zhang Y, et al. Human umbilical cord mesenchymal stem cells ameliorate acute liver failure by inhibiting apoptosis, inflammation and pyroptosis. Ann Transl Med (2021) 9(21):1615. doi: 10.21037/atm-21-2885
129. Yang R, Yu H, Chen J, Zhu J, Song C, Zhou L, et al. Limonin attenuates LPS-induced hepatotoxicity by inhibiting pyroptosis via NLRP3/gasdermin D signaling pathway. J Agric Food Chem (2021) 69(3):982–91. doi: 10.1021/acs.jafc.0c06775
130. Liu T, Yang L, Gao H, Zhuo Y, Tu Z, Wang Y, et al. 3,4-dihydroxyphenylethyl alcohol glycoside reduces acetaminophen-induced acute liver failure in mice by inhibiting hepatocyte ferroptosis and pyroptosis. PeerJ (2022) 10:e13082. doi: 10.7717/peerj.13082
131. Siregar AS, Nyiramana MM, Kim EJ, Cho SB, Woo MS, Lee DK, et al. Oyster-derived tyr-ala (YA) peptide prevents lipopolysaccharide/D-galactosamine-induced acute liver failure by suppressing inflammatory, apoptotic, ferroptotic, and pyroptotic signals. Mar Drugs (2021) 19(11):614. doi: 10.3390/md19110614
132. Shu B, Zhou YX, Li H, Zhang RZ, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discov (2021) 7(1):368. doi: 10.1038/s41420-021-00756-x
133. Yang W, Tao K, Zhang P, Chen X, Sun X, Li R. Maresin 1 protects against lipopolysaccharide/d-galactosamine-induced acute liver injury by inhibiting macrophage pyroptosis and inflammatory response. Biochem Pharmacol (2022) 195:114863. doi: 10.1016/j.bcp.2021.114863
134. Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao C, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics (2022) 12(9):4310–29. doi: 10.7150/thno.71086
135. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol (2013) 16(3):319–26. doi: 10.1016/j.mib.2013.04.004
136. Greaney AJ, Leppla SH, Moayeri M. Bacterial exotoxins and the inflammasome. Front Immunol (2015) 6:570. doi: 10.3389/fimmu.2015.00570
137. Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem (1986) 261(16):7123–6. doi: 10.1016/S0021-9258(17)38364-3
138. Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1B inflammasome. Science (2019) 364(6435):82–5. doi: 10.1126/science.aau1208
139. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
140. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
141. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
142. Glaria E, Valledor AF. Roles of CD38 in the immune response to infection. Cells (2020) 9(1):228. doi: 10.3390/cells9010228
143. Kinra M, Nampoothiri M, Arora D, Mudgal J. Reviewing the importance of TLR-NLRP3-pyroptosis pathway and mechanism of experimental NLRP3 inflammasome inhibitors. Scand J Immunol (2022) 95(2):e13124. doi: 10.1111/sji.13124
144. Hou W, Wei X, Liang J, Fang P, Ma C, Zhang Q, et al. HMGB1-induced hepatocyte pyroptosis expanding inflammatory responses contributes to the pathogenesis of acute-on-chronic liver failure (ACLF). J Inflamm Res (2021) 14:7295–313. doi: 10.2147/JIR.S336626
145. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis (2021) 26(3-4):152–62. doi: 10.1007/s10495-021-01663-3
146. Dong W, Zhu Q, Yang B, Qin Q, Wang Y, Xia X, et al. Polychlorinated biphenyl quinone induces caspase 1-mediated pyroptosis through induction of pro-inflammatory HMGB1-TLR4-NLRP3-GSDMD signal axis. Chem Res Toxicol (2019) 32(6):1051–7. doi: 10.1021/acs.chemrestox.8b00376
147. Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis (2019) 10(10):778. doi: 10.1038/s41419-019-2021-3
148. Sun Z, Nyanzu M, Yang S, Zhu X, Wang K, Ru J, et al. VX765 attenuates pyroptosis and HMGB1/TLR4/NF-kappaB pathways to improve functional outcomes in TBI mice. Oxid Med Cell Longev (2020) 2020:7879629. doi: 10.1155/2020/7879629
149. Wang J, Ren H, Yuan X, Ma H, Shi X, Ding Y. Interleukin-10 secreted by mesenchymal stem cells attenuates acute liver failure through inhibiting pyroptosis. Hepatol Res (2018) 48(3):E194–202. doi: 10.1111/hepr.12969
150. Bai L, Lu W, Tang S, Tang H, Xu M, Liang C, et al. Galectin-3 critically mediates the hepatoprotection conferred by M2-like macrophages in ACLF by inhibiting pyroptosis but not necroptosis signalling. Cell Death Dis (2022) 13(9):775. doi: 10.1038/s41419-022-05181-1
151. de Carvalho Ribeiro M, Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol (2022) 17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529
Keywords: acute liver failure (ALF), macrophage, polarization, pyroptosis, immune
Citation: Xie D and Ouyang S (2023) The role and mechanisms of macrophage polarization and hepatocyte pyroptosis in acute liver failure. Front. Immunol. 14:1279264. doi: 10.3389/fimmu.2023.1279264
Received: 17 August 2023; Accepted: 16 October 2023;
Published: 26 October 2023.
Edited by:
Balachandran Ravindran, Institute of Life Sciences (ILS), IndiaReviewed by:
Santosh Panda, Washington University in St. Louis, United StatesCopyright © 2023 Xie and Ouyang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi Ouyang, b3V5YW5nc2hpQGd6aG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.