 Christo Tsilifis
Christo Tsilifis Mary A. Slatter
Mary A. Slatter Andrew R. Gennery
Andrew R. Gennery
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 31 October 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1279201
This article is part of the Research TopicEmerging Talents in Primary Immunodeficiencies: 2022View all 7 articles
Primary immune regulatory disorders (PIRDs) are inborn errors of immunity caused by a loss in the regulatory mechanism of the inflammatory or immune response, leading to impaired immunological tolerance or an exuberant inflammatory response to various stimuli due to loss or gain of function mutations. Whilst PIRDs may feature susceptibility to recurrent, severe, or opportunistic infection in their phenotype, this group of syndromes has broadened the spectrum of disease caused by defects in immunity-related genes to include autoimmunity, autoinflammation, lymphoproliferation, malignancy, and allergy; increasing focus on PIRDs has thus redefined the classical ‘primary immunodeficiency’ as one aspect of an overarching group of inborn errors of immunity. The growing number of genetic defects associated with PIRDs has expanded our understanding of immune tolerance mechanisms and prompted identification of molecular targets for therapy. However, PIRDs remain difficult to recognize due to incomplete penetrance of their diverse phenotype, which may cross organ systems and present to multiple clinical specialists prior to review by an immunologist. Control of immune dysregulation with immunosuppressive therapies must be balanced against the enhanced infective risk posed by the underlying defect and accumulated end-organ damage, posing a challenge to clinicians. Whilst allogeneic hematopoietic stem cell transplantation may correct the underlying immune defect, identification of appropriate patients and timing of transplant is difficult. The relatively recent description of many PIRDs and rarity of individual genetic entities that comprise this group means data on natural history, clinical progression, and treatment are limited, and so international collaboration will be needed to better delineate phenotypes and the impact of existing and potential therapies. This review explores pathophysiology, clinical features, current therapeutic strategies for PIRDs including cellular platforms, and future directions for research.
Primary immune regulatory disorders (PIRDs) are a group of inborn errors of immunity (IEI) defined by excessive inflammation, autoimmunity frequently targeting multiple tissues, lymphoproliferation, and malignancy, resulting from loss or gain of function in immunity-related genes associated with the regulatory mechanism of the inflammatory or immune response. Additionally, patients may be susceptible to severe, recurrent or opportunistic infection from impaired cellular or humoral immunity, from accumulation of end-organ damage, or from immunosuppressive therapy used to control immune dysregulation. In contrast, immune dysregulation covers a combination of autoinflammation, autoimmunity, and lymphoproliferation alongside susceptibility to severe infections, and can manifest by a number of different genetic conditions in which loss of the regulatory mechanism is not the primary defect. IEIs in which immune dysregulation may feature include, but are not limited to, chronic granulomatous disease and Wiskott-Aldrich syndrome. The distinction is important, because the dysfunction of a single molecule in PIRDs makes them potentially amenable to targeted therapy using small molecules or specific molecular inhibitors.
The diversity of clinical manifestations in PIRDs, which frequently present to non-immunologists, may lead to delayed recognition of the immunological and genetic diagnoses, and thus this patient group can pose a significant therapeutic challenge. Prompt recognition and molecular diagnosis is important to prevent multiple organ morbidities and side effects of prolonged immunosuppression, which underlie initial management for PIRDs; this is increasingly relevant as targeted therapies become available for specific molecular entities.
The number of genetically defined PIRDs has grown significantly as our understanding of mechanisms of immune tolerance, and access to genomic medicine has expanded. The concept of PIRDs as a disease entity was seeded from identification of the association between immunodeficiency and autoimmunity over 5 decades ago (1). This idea initially challenged understanding of IEI syndromes, given the apparent contradiction of excessive immune activity, demonstrated by autoimmunity and lymphoproliferation, yet recurrent infections and malignancy suggesting a degree of immunoparesis. It was recognized that even “classical primary immunodeficiencies” can manifest autoimmune features (2). Proposed theories to explain this included incomplete clearance of pathogens in immunodeficient patients causing a suboptimal, chronic immune response and thereby damage to ‘bystander’ tissues (2). In 1982, a case series describing a family where 17 male infants died of diarrhea associated with early-onset eczema and autoimmunity directed against multiple endocrine glands led to the description of the prototypical PIRD – immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome (3) – and subsequent link with regulatory T-lymphocytes (Tregs) (4). Other disorders of this lymphocyte family constitute a subclassification of PIRDs. Many other PIRDs have since been described. Within the past decade, the number of PIRD genes listed in the International Union of Immunological Societies (IUIS) phenotypic classification of IEIs has grown from 21 (5) to 54 (6), shaping our understanding of immune function.
In this review, we discuss common manifestations of PIRDs, sitting at the crossroads of infection and autoimmunity, and the challenges of describing this heterogeneous group; disease subgroups including disorders of T regulatory lymphocytes, familial hemophagocytic lymphohistiocytosis (fHLH) syndromes, IEIs associated with very early onset inflammatory bowel disease, and diseases of autoimmunity and lymphoproliferation. We explore current and future directions for therapy such as targeted molecular treatment, allogeneic hematopoietic stem cell transplantation, and other cellular therapies.
Immune dysregulation may occur from disorders of a number of mechanisms spanning the breadth of immune function. However, PIRDs arise specifically from disordered regulation of immunity and inflammation.
Instrumental to the prevention of autoimmunity in a healthy individual are the processes of central and peripheral immune tolerance. Central tolerance occurs within the thymus: during T-lymphocyte development, progenitor T-lymphocytes migrate from the bone marrow to the thymic cortex to undergo proliferation, maturation, rearrangement of their T-cell receptors (TCR), and, in the thymic medulla, differentiation into mature T-lymphocytes that may enter the peripheral circulation (Figure 1A). These mature T-lymphocytes must be capable of recognizing and reacting to pathogens, virus-infected cells, and malignant cells through binding of antigen to the TCR, whilst crucially being tolerant to self-antigens; this is central tolerance. Central tolerance develops through two sequential stages, whereby immature double-negative (DN) T-lymphocytes that express the CD3-TCR complex but lack either CD4 or CD8 interact with thymic cortical epithelial cells to positively select TCRs that bind to the major histocompatibility receptor I and II expressed by ‘self’ cells, or else undergo apoptosis; this is termed positive selection, leading to a population of CD4+CD8+ (double positive, DP) lymphocytes. DP lymphocytes undergo negative selection in the thymic medulla, where lymphocytes reactive to self-antigens expressed by medullary thymic epithelial cells (mTEC) are deleted (7). Expression of this restricted set of tissue antigens by mTECs relies on the transcription factor autoimmune regulator, encoded by the AIRE gene (8). Impairment of central tolerance by inherited defects of thymic development leads to syndromes including the autoimmune lymphoproliferative syndromes (ALPS), caused by failure of the extrinsic activation-induced cell death pathways (mutations in FAS, FASLG, and FADD), and autoimmune polyglandular syndrome (APS) 1, caused by mutations in AIRE.
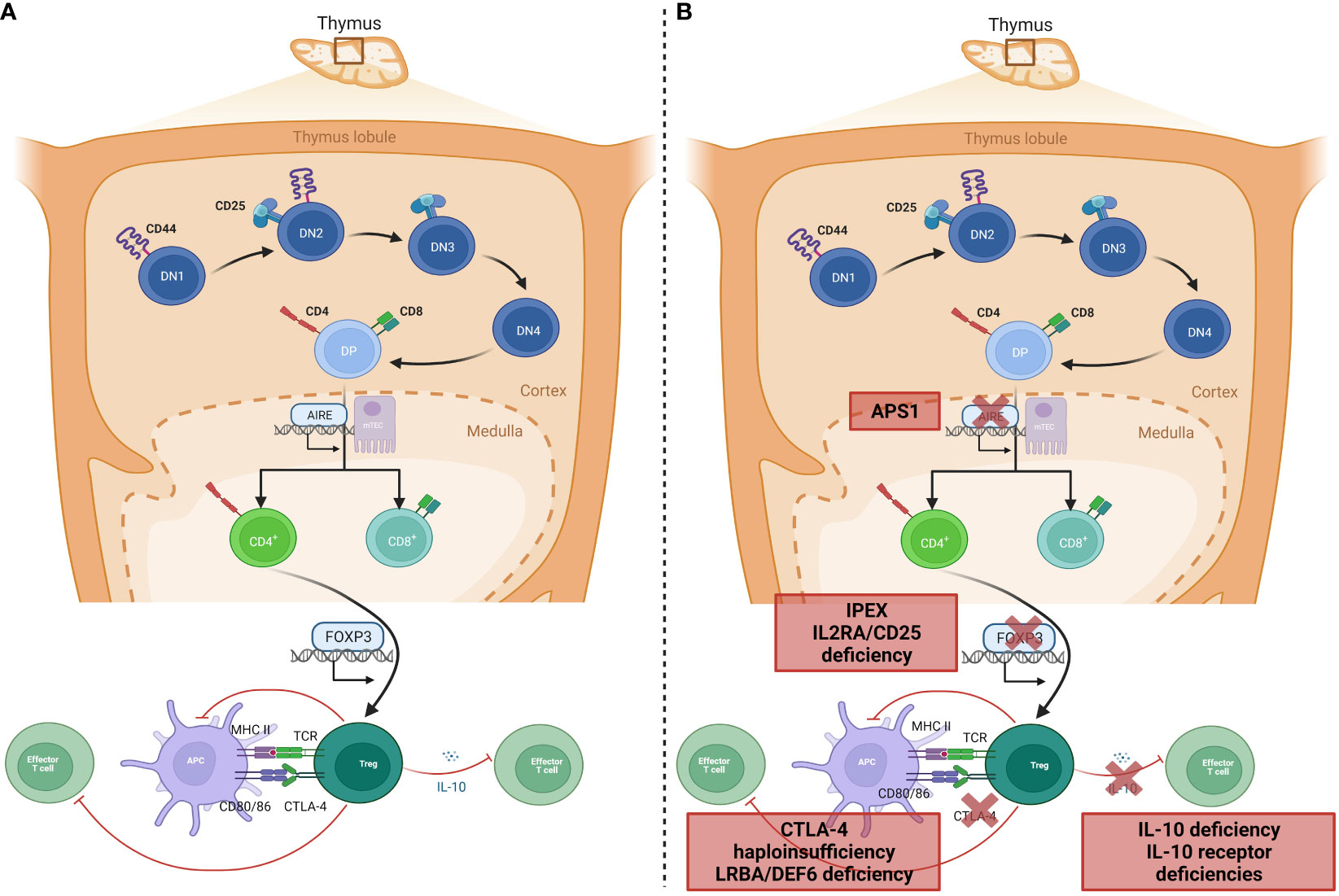
Figure 1 Genetic aetiology of specific PIRDs. (A) Normal T-lymphocyte development within the thymus results in removal of T-lymphocytes that bind self-antigen expressed by AIRE, on mTEC cells in the thymic medulla. A subset of CD4+ lymphocytes develop into Tregs with suppressor functions on effector T-lymphocytes through inhibition of co-stimulation (via CTLA-4 binding), or IL-10 production. (B) Monogenic defects of proteins involved in tolerance mechanisms of Treg development or function result in specific PIRDs. Adapted from “T-cell development in thymus 2” and “Tregs suppress dendritic cells and effector T cells”, by BioRender.com (2023). Retrieved from https://biorender.com/biorender-templates.
Once single-positive mature T-lymphocytes expressing either CD4 or CD8 are in the peripheral circulation, the effector functions are checked by Tregs, which maintain peripheral tolerance by upregulation of cell-suppressing surface markers such as cytotoxic lymphocyte antigen-4 (CTLA-4) and production of inhibitory cytokines including IL-10 (9, 10). The repertoire of Tregs extends beyond peripheral tolerance, and includes suppression of allergic inflammation, induction of tolerance to dietary antigens and to the fetus during pregnancy, and protection of the microbiome (11). It is therefore unsurprising that Tregopathies frequently feature an allergic preponderance. Tregs develop in the thymus as natural Tregs (nTregs) or following antigen exposure peripherally as inducible Tregs (iTregs). Treg development and function is dependent on the forkhead box protein-3 (FOXP3) transcription factor, deficiency of which abrogates Treg development and causes IPEX syndrome with absent or dysfunctional Tregs (9) (Figure 1B). A phenotypically similar syndrome is caused by deficiency of CD25 (the α-chain of the IL-2 receptor) (12, 13), which is universally expressed on Tregs. Functional Treg defects are seen in defects of surface markers and intracellular proteins such as CTLA-4 (14) and the closely-related lipopolysaccharide-responsive beige-like anchor (LRBA, Figure 1B) (15). Additionally, dysfunction of suppressor cytokine pathways involving IL-2 (15, 16), IL-10 (17) either by deficiency of receptors or altered downstream signaling molecules such as JAK1 (18), STAT1 (19), STAT3 (20), and STAT5b (21), renders ineffective the common mechanisms by which Tregs usually restrain inflammation.
PIRDs may also manifest in hyperactivation or hyperinflammation that functioning peripheral tolerance mechanisms cannot restrain: this typically manifests as HLH, a syndrome of fever, hepatosplenomegaly, and consumption of hematopoietic-derived cells in the peripheral blood, bone marrow and central nervous system (hemophagocytes) due to excessive pro-inflammatory cytokine release from activated macrophages. This process is usually prevented by the inhibitory effect of NK and CD8+ lymphocytes, which insert pores into macrophage cell membranes to deliver cytotoxic granules and cause cell death, through the intrinsic cell death pathway. Monogenic defects in pore formation, cytotoxic granule production, and their delivery cause familial HLH (22). However, a range of additional immune disorders may predispose to HLH due to NK cell deficiency or impairment, suboptimal handling of viral infections such as EBV, or dysregulated inflammasome control. HLH seen in the context of these other IEI is termed primary HLH (23).
PIRDs pose a diagnostic and therapeutic challenge for clinicians due to the broad spectrum of potential manifestations involving multiple organs, which may therefore present to various medical specialities. Autoimmunity may be antibody- or cell-mediated, and can target hematopoietic cells causing cytopenias, endocrine organs causing type 1 diabetes mellitus (T1DM) or thyroiditis, skin and connective tissue causing dermatitis, vitiligo, alopecia or arthritis, visceral organs causing hepatitis or glomerulonephritis, and the epithelial barrier causing uveitis or inflammatory bowel disease. Individually, these manifestations may be common: immune thrombocytopenia purpura has an estimated incidence of 1.9-6.4 per 100,000 children each year (24), whilst T1DM has an incidence of 22.9/100,000 per year (25). This makes early diagnosis of potential PIRDs difficult until multi-organ autoimmunity develops, or unless additional features such as a positive family history or severe or recurrent infection are present. Allergic disorders such as food or drug allergy, rhinitis, and asthma may also manifest. Impaired control of lymphoid cells may interact with abnormal handling of viruses such as Epstein-Barr virus (EBV) and predispose to lymphoproliferation and development of lymphoma.
Given the multi-organ nature of many PIRDs, understanding and quantifying the burden of immune dysregulation is important, and may be done through disease activity scoring systems. At an individual patient level, these scoring systems allow clinicians to track response to therapies, whilst at a population level, they aid understanding natural history and progression of different monogenic diseases. The heterogeneity between different PIRDs and failure to identify a monogenic defect in every patient with immune dysregulation features makes such scoring systems challenging to design. The Immune Deficiency and Dysregulation Activity (IDDA) score is a composite of the presence and severity of different clinical parameters, graded 0–4 depending on the impact of each manifestation. It is altered by other factors such as chronic infection and duration of hospitalization. This was initially conceived to provide a comparison of phenotype in patients before and after HSCT for LRBA deficiency (26), but has since been updated to encompass all PIRDs (IDDA2.1 (27)). It can be used to longitudinally track disease activity in individual patients and assess response to treatment, and used visually represent different disease phenotypes through a ‘kaleidoscope’ function (27).
Historically, challenges in obtaining a molecular diagnosis for PIRDs has led to patients receiving prolonged immunosuppression, particularly with corticosteroids. Chronic corticosteroid use carries significant morbidity due to weight gain, risk of osteoporotic fractures, and immunosuppression; the latter may be particularly important in these diseases where there is infection susceptibility in addition to autoimmunity.
Allogeneic HSCT may be considered diametrically opposed to targeted therapy; rather than selecting a specific pathway to modulate, HSCT offers the opportunity to replace recipient stem cells carrying a disease-causing allele with those from a healthy donor, thereby affecting all bone marrow-derived cells. The benefits of HSCT must be balanced against the limitations of its use: firstly, HSCT may not correct the underlying defect in all PIRDs, particularly in cases where there is pleiotropic expression of the mutated protein (such as in STAT3-GOF), or where the defect is of thymic origin (such as APS1). Secondly, HSCT carries a significant and variable risk of mortality. This is typically associated with early transplant-related complications arising from preparative conditioning, from the period of aplasia prior to engraftment of donor cells and subsequent immune reconstitution, and from alloreactivity from donor lymphocytes causing graft-versus-host disease (GvHD). Thirdly, HSCT may not be available in every healthcare setting, particularly when considering the differences between HSCT experience in IEI patients as opposed to more common indications such as hematological malignancy, and even if available, is expensive and reliant on availability of a well-matched donor. These limitations in availability have led to the subset of patients undergoing HSCT in historic series representing a multi-morbid, therapy-resistant cohort who were transplanted without a genetic diagnosis, late in their disease course with accumulated end-organ damage and treatment side effects. An additional challenge in HSCT for PIRDs as opposed to ‘classical’ immunodeficiency is the need to attain sufficient myeloablation to remove recipient lymphocytes which may facilitate graft rejection, whilst minimizing associated toxicities. A degree of alloreactivity is also required so that hyperactivated recipient cells may be destroyed by emerging donor immunity. HSCT for PIRDs thus represents the intersection of philosophies for treatment of hematological malignancy, where conditioning regimens favor myeloablation and where alloreactivity may aid a graft-versus-leukemia effect, and of treatment of classical immunodeficiency, where pre-HSCT infection and organ-damage is common and conditioning intensity aims to minimize associated toxicity. Together, these challenges result in poorer HSCT outcomes for PIRDs than for other monogenic IEIs such as chronic granulomatous disease or Wiskott-Aldrich syndrome, with particularly high rates of graft rejection or GvHD (28, 29). The role of autologous ex-vivo gene therapy for these diseases is evolving; whilst successful engraftment of corrected HSCs still mandates conditioning to create a marrow ‘niche’, advantages include ability to harvest from the patient and not rely on a matched donor, and no risk of GvHD (30). Gene therapy platforms have been established and trials are in clinical phases for severe combined immunodeficiency (SCID; IL2RG, RAG1, DCLRE1C) and non-SCID IEI including IPEX, Wiskott-Aldrich syndrome, and X-linked chronic granulomatous disease (30, 31). Gene therapy trials for familial HLH genes (UNC13D, PRF1) are currently in pre-clinical phases (32, 33). Whilst attractive, application of this science to the clinical setting is in early stages, with little long-term outcome data and concerns regarding cost and availability (34).
The ability to maintain remission of autoimmunity with minimal side-effects is therefore an attractive alternative to both chronic corticosteroid use, and allogeneic HSCT. Today, with several agents being either disease mechanism-specific (such as abatacept) or pathway-specific (such as JAK inhibitors), control of underlying immune dysregulation may be better balanced against side-effect profile, and thus the treatment paradigm may be changed. Diseases affecting CTLA-4 expression (CTLA-4 haploinsufficiency and LRBA/DEF6 deficiencies) may be treated with replacement of CTLA-4 protein in the form of abatacept or belatacept, with good clinical response, particularly for enteropathy or lymphoid cell-infiltrative disease of the lungs or nervous system (35, 36). A multicenter trial exploring its efficacy and safety is currently underway (37). For several diseases where there is a quantitative or qualitative Treg defect and subsequent hyperactivation of effector T lymphocytes, mTOR inhibition using sirolimus has proven to be an effective steroid-sparing agent (35, 38–40). It may also be useful in ALPS (41). A range of agents antagonizing common pro-inflammatory mediators such as IL-1 (anakinra) and IFN-γ (emapalumab) may be employed to aid remission in primary HLH, whilst specific aetiologies where IL-18 hyperactivation is implicated may benefit from IL-18 binding protein analogues (42–45). B-lymphocyte directed therapy, such as rituximab, may benefit diseases where EBV infection causes HLH (such as SAP and XIAP deficiencies) or where autoantibody production is problematic (such as APS1, or CTLA-4 haploinsufficiency) (35, 46). Anakinra, which is a recombinant IL-1 receptor antagonist, may remit the multi-system inflammation seen in disease states where IL-1 antagonism is impaired such as deficiency of endogenous IL-1RA and the closely-related IL-36RA (DIRA and DITRA syndromes, respectively) (47, 48). For PIRDs caused by hyperactivation of a specific protein, use of small molecules to inhibit activity and return it to baseline is attractive and has been trialed for activated phosphoinositide 3-kinase-δ syndrome (49). Given the broad clinical and immune phenotype of PIRDs, and that individual diseases may cause symptoms through different pathways, it is unsurprising that there is no ‘silver bullet’ that can remit all aspects of a disease. Furthermore, the novelty of several of these agents and small patient population means long-term data on efficacy and safety are limited, and the financial and clinical impact of very long-term treatment is yet to be determined. Collection of ongoing longitudinal data is required to fully understand whether these treatments will serve as long-term maintenance therapy, a tool for inducing remission, or a bridge to definitive cure with HSCT or other cellular therapies.
We will discuss specific PIRD entities, defined either by genotype or by phenotype, and their specific clinical and immunological characteristics and current understanding of different treatments. Table 1 summarizes disease entities and strategies for treatment, including targeted therapy and results of HSCT or gene therapy.
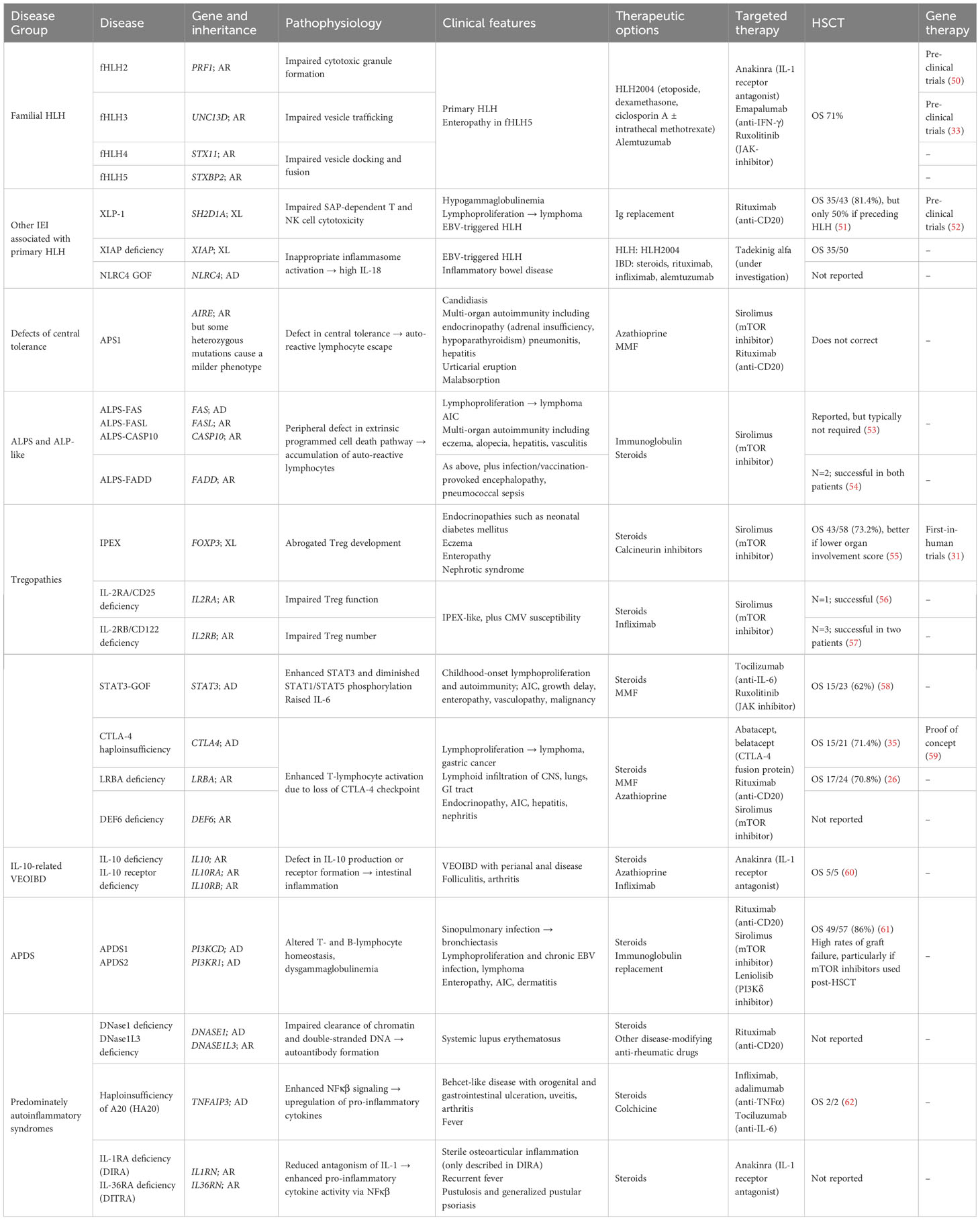
Table 1 Summary of PIRDs discussed in this review, with genetic and clinical features, and options for treatment including mechanism-specific therapies, HSCT, and gene therapy.
Biallelic LOF mutations in AIRE, which are autosomally inherited, cause a defect in central tolerance with failure to delete auto-reactive lymphocytes in the thymus, manifesting in a syndrome characterized by chronic mucocutaneous candidiasis and multi-organ autoimmunity, particularly against parathyroid and adrenal glands (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, APECED, also called APS1) (63). The syndrome was first reported in 1929 (64), with its genetic aetiology identified by two groups in 1997 (65, 66), and has a prevalence that varies by population, with certain founder mutations found in relative high numbers of the Persian Jewish, Sardinian, and Finnish populations (67). There is poor genotype-phenotype correlation, with significant intra-familial variability despite sharing identical AIRE mutations (68). However, some features appear enriched within an American cohort of patients, particularly a non-pruritic urticarial eruption which, in this cohort, was as prevalent as CMC. The diagnostic criteria therefore expanded to also include urticarial eruption, enamel hypoplasia, and intestinal malabsorption (69). Recently, heterozygous AIRE mutations exerting negative dominance have been identified in a syndrome with milder autoimmunity and incomplete penetrance (70).
Whilst the classic triad of hypoparathyroidism, Addison’s disease and CMC define the syndrome, the phenotype has expanded in recent years to include other endocrine autoimmunity impacting pancreatic islet cells, thyroid, and gonad; visceral autoimmunity including pneumonitis, hepatitis, gastritis and intestinal malabsorption; non-infectious epithelial features such as urticarial eruption, alopecia, vitiligo, nail dystrophy and enamel hypoplasia; and a Sjogren’s-like sicca syndrome (67). Whilst the infective manifestations have classically been restricted to epithelial-site Candida infection only, the recent emergence of the SaRS-CoV-2 pandemic and subsequent discovery of anti-type-I interferon antibodies underlying mortality risk from pneumonitis in both APECED patients (71) and the general population (72). Some patients develop epithelial-site malignancy associated with chronic Candida infection.
These infective complications relate to autoimmunity: failure of AIRE to project an “immunological self-shadow” (73) within the thymus causes escape of auto-reactive T lymphocytes into the circulation, which then infiltrate organs and cause autoimmunity either by a direct cellular effect, or by induction of autoantibody formation, possibly in tandem with auto-reactive B lymphocytes (46, 67). Multiple autoantibodies have been associated with APECED features, including against NACHT leucine-rich-repeat protein 5 and the calcium-sensing receptor (hypoparathyroidism), 17-α and 21-hydroxylases (adrenal insufficiency), intrinsic factor (pernicious anemia), and cytokines such as the IL-17 family (CMC), IL-22, and type-I interferons -α, -β, -λ, and -ω (SaRS-CoV-2 pneumonitis) (74); interestingly, anti-interferon-α antibodies have been posited to provide protection from development of T1DM in some APECED patients (75). The AIRE defect may also impair Th17 lymphocyte development, possibly explaining the Candida diathesis in APECED patients without detectable IL-17 autoantibodies (76).
Treatment of APECED commonly involves specialists from multiple disciplines including endocrinology, immunology, gastroenterology/hepatology and dermatology. CMC may be managed with azole antifungals, or amphotericin B in cases of azole resistance; patients should be screened for asplenia, which may co-occur in ~10% of patients. Autoimmunity may be managed with azathioprine, mycophenolate mofetil, or mTOR inhibitors such as sirolimus; B-lymphocyte depletion with rituximab has been described in cases of recalcitrant autoantibody-mediated organ disease and in pneumonitis (77). Allogeneic HSCT does not correct this disease, which is caused by a thymic defect.
Autoimmune lymphoproliferative syndrome (ALPS) and ALP-like syndromes are defects of lymphocyte homeostasis caused by impairment of the FAS-mediated apoptosis pathway. Classical ALPS was first described in 1967 and given the eponym Canale-Smith syndrome after the paper’s authors (78), as a syndrome of non-malignant lymphoproliferation with autoimmunity. Subsequently, a predisposition to lymphomatous transformation was also identified, and the syndrome was associated with deleterious variants in FAS (79) followed by FASL (80), CASP10 (81) and somatic FAS (82) mutations. These genes encode the proteins of the FAS-mediated apoptosis pathway: FAS, a cell-surface receptor, binds to its ligand (FAS-L) leading to conformational change and recruitment of intracellular proteins (caspase-8 and caspase-10, and FAD-associated death domain) to herald programmed cell death. This process is upregulated in T-lymphocytes which react to self-antigen, restraining proliferation of autoreactive lymphocytes: thus, failure of this mechanism causes an accumulation of autoreactive double-negative (TCRαβ+CD4-CD8-) T-lymphocytes (DNTs) with a significant proliferative potential (83). Along with clinical features and DNTs, biomarkers such as soluble FAS-L, vitamin B12, and IL-10 comprise the diagnostic criteria for ALPS (83).
Mutations in FAS are most commonly dominant and in the germline, with somatic mutations representing 15-20% of cases. However, some FAS mutations behave in a recessive manner, with heterozygous probands being minimally symptomatic. Similarly, the much rarer ALPS-FAS-L usually requires a biallelic mutation to cause disease (83, 84). The incomplete clinical penetrance of ALPS may be partially explained by a ‘double hit’ being necessary to impair FAS-related apoptosis (85). Patients lacking a mutation in the FAS-associated genes are categorized as ALPS-U; advances in availability of high-throughput genomic testing have identified mutations in >20 distinct IEI genes in ALPS-U patients, including other PIRDs such as Tregopathies (STAT3-GOF, CTLA-4 haploinsufficiency, LRBA deficiency) (86), highlighting how rapidly this field is expanding.
Clinical manifestations of ALPS typically begin in early childhood. Lymphoproliferation of DNTs causes multi-focal lymphadenopathy, particularly of cervical nodes, and splenomegaly. These demonstrate DNT infiltration and may remit in adulthood. Autoimmunity in ALPS is commonly directed against erythrocytes, platelets and neutrophils, with >80% of patients manifesting with hematological cytopenias (84). Organ-specific autoimmunity caused by autoantibody formation, possibly by disturbed B-lymphocyte homeostasis, may include uveitis, eczema, alopecia, hepatitis, Guillain-Barre syndrome and vasculitis (84); patients frequently have clonal IgG expansion. Lymphomatous transformation may occur, particularly into Hodgkin-type (87). Historically, splenectomy has been trialed to reduce the burden of lymphoproliferation; however, the risks of encapsulated bacterial infection compounded by attenuated polysaccharide vaccine responses do not support this treatment strategy (88). Autoimmune cytopenias (AICs) may respond in the first instance to immunomodulation with high dose intravenous immunoglobulin (1 – 2g/kg), though is commonly refractory and requires second-line therapy. Lymphoproliferation and autoimmunity may both respond to immunosuppression, typically steroids though mTOR inhibitors such as sirolimus have shown favorable results with minimal infection risk, possibly due to this pathway being hyperactivated in ALPS (40, 41). Following induction of remission, maintenance therapy may be continued to prevent relapse. In rare cases, due to uncontrolled life-threatening lymphoproliferation, HSCT may be considered but experience is limited to few reports (53, 88, 89). In a systemic review collating 12 patients with FAS or FASL mutations and ALPS who underwent HSCT, survival was seen in 11/12 (91.7%) (89). Transplant course was complicated by graft rejection necessitating second HSCT in two patients, and graft-versus-host disease (89).
Syndromes caused by mutations in caspase-8 (CASP8) or FAD-associated death domain (FADD), previously categorized as ALPS, behave differently. Caspase-8 deficiency may cause very early-onset inflammatory bowel disease (VEOIBD) presenting before age 6 years or other infiltrative autoimmune disease, contrasting with ALPS, or infection susceptibility (90, 91). FADD deficiency overlaps ALPS with the addition of infection- or vaccination-provoked encephalopathy and seizures, ocular findings, and overwhelming pneumococcal sepsis (54, 92–94). HSCT has been successful in 2 FADD-deficient patients (54).
Mutations in the FOXP3 gene, on the X chromosome, cause IPEX syndrome with abrogated Treg development. This syndrome typically presents in the first few weeks of life with intractable diarrhea, enteropathy and resultant failure to thrive, eczema, and T1DM (3), though this triad may only occur in ~60% of patients (55, 95); other features include food allergies, nephrotic syndrome, other endocrinopathies, AIC and serious infections such as sepsis and meningitis. Immunologically, patients have low or absent Tregs with raised IgE and eosinophilia, and characteristic autoantibodies against renal and gut epithelial proteins (96, 97). The impact of mutation type and site on FOXP3 protein expression vary; mutations that confer small changes to the protein with intact expression appear to result in a milder phenotype (95). Certain phenotypes and outcomes cluster to specific FOXP3 protein domains, such as poorer outcome in repressor domain mutations (98), but there is significant extra-familial variability amongst patients with the same variant, suggesting influence of other genetic or environmental factors on phenotype. More recently, emphasis has been placed on atypical presentations of IPEX: these include intrauterine or late onset, mild course, or IPEX-like syndromes in patients with some features of the syndrome but no FOXP3 mutation. Intrauterine IPEX may present with hydrops, ultrasonographic features such as hyperechoic bowel, and poor perinatal outcome including stillbirth or neonatal mortality (99), and appears to cluster in families. Relatively late-onset IPEX may present towards the end of the first decade of life with an IBD-like phenotype (100). Reports of mild IPEX with either single-organ involvement, or rapid response to immunosuppression, have been associated with non-coding region mutations and near-intact Treg compartments (100). In IPEX-like patients with intact FOXP3 function, mutations in other IEI genes such as LRBA, STAT3 (GOF), CTLA4, IL2RA and STAT5B have been implicated (95).
Treatment of IPEX centers on nutritional support and immunomodulation such as steroids and adjunctive therapy, usually calcineurin inhibitors or sirolimus; the latter may offer better disease control by restoring Treg function (38). Retrospective data highlight that whilst immunosuppression may ameliorate the disease, it unsurprisingly does not reverse end-organ damage and may only temporize accrual of disease manifestations, particularly as follow-up extends into the second or third decade after diagnosis (55). Allogeneic HSCT offers a better disease-free survival than immunosuppression alone, though the risk of mortality (~25%) with HSCT is stratified by pre-HSCT morbidity, suggesting that prompt treatment or aggressive optimization of patient condition is critical for this treatment modality. Interestingly, mixed rather than full donor chimerism does not negatively affect outcome and leads to functional donor Treg production, suggesting that reduced intensity conditioning strategies may provide a balance between transplant-related morbidity and immunological outcome (55). A phase 1, first-in-human trial of restoration of the Treg compartment by induction of CD4+ cell development into Tregs following lentiviral transfer of wild-type FOXP3 is currently in recruitment (NCT05241444) (31).
First identified in humans in 2014, autosomal dominant mutations in CTLA4, causing haploinsufficiency of its encoded protein, cause a complex PIRD with incomplete penetrance (14, 101). CTLA-4, expressed on Tregs, acts as an immune checkpoint by outcompeting CD28 for binding to CD80/CD86 on antigen presenting cells, prompting transendocytosis of the complex and thereby terminating the co-stimulatory second signal required for T-lymphocyte activation (102). Following the first description in seven patients from four kindreds, understanding of CTLA-4 haploinsufficiency has expanded by publication of international, multicenter cohorts (36). Whilst mutations may not be completely penetrant, leading to divergent or absent symptoms within families sharing the same mutation, affected patients commonly have multiorgan involvement with lymphoid infiltration into lungs, the central nervous system, and gastrointestinal tract along with autoimmune endocrinopathies including diabetes, arthritis, cytopenias, and infections (36). Lymphoproliferation may be symptomatic in its own right, or transform into lymphoid malignancy, often driven by EBV, with a lifetime risk of approximately 1 in 6 patients (103, 104). Penetrance of CTLA-4 haploinsufficiency appears to be independent of the specific mutation (35). Patients may develop primary hypogammaglobulinemia as CTLA-4 is involved in differentiation of follicular helper T-lymphocytes, and this may be compounded by immunosuppression (36, 105). CXCR5+PD1+ follicular helper T-lymphocytes may act as a biomarker for CTLA-4 disease; other immunophenotype changes appear to be variable across the CTLA-4 haploinsufficient population (106).
These clinical features are shared with three other genetic diseases. Monoallelic deletions of the 2q33.2 – 2q33.3 locus, where CTLA4 sits, present as phenotypic CTLA-4 haploinsufficiency (107). Recessive mutations affecting the lipopolysaccharide responsive beige-like anchor protein (LRBA) were identified in 2012 prior to CTLA-4 haploinsufficiency, in kindreds with childhood-onset hypogammaglobulinemia, impaired B-lymphocyte compartments, and autoimmunity (15). The overlap between these two diseases is explained by the role of LRBA in protecting CTLA-4 from lysosomal degradation, and indeed LRBA deficiency causes reduced cell-surface CTLA-4 expression (108). Also impacting CTLA-4 surface trafficking, homozygous mutations in DEF6 impair regulation of the GTPase RAB11 and thereby reduce the intracellular trafficking of CTLA-4-containing vesicles towards the cell-surface (109, 110). Initially conceived for LRBA deficiency, a multiorgan scoring system (IDDA) may be used for assessing longitudinal response to therapy (26). Following its use in a cohort of 76 patients with LRBA deficiency, the IDDA score has been revised to incorporate other features of immune dysregulation such as hemophagocytes (IDDA2.1), and its inclusion in the ESID registry will help expand its use in describing the phenotype of PIRDs beyond LRBA deficiency (27).
Treatment for CTLA-4 haploinsufficiency and related disorders centers on immunomodulation. There is variability in organ response to different therapies. Corticosteroids are effective in remitting granulomatous lymphocytic interstitial lung disease and gastrointestinal inflammation, they appear less effective for neurological involvement. Steroid-sparing agents, such as mTOR inhibitors, TNFα inhibitors, and B-lymphocyte directed therapy with rituximab are commonly employed to provide multimodal immunosuppression and reduce the adverse effects associated with chronic corticosteroid use. The aetiology of immune cytopenias likely relates to marrow infiltration by autoreactive T-lymphocytes, rather than peripheral sequestration or destruction, explaining the poor clinical response following splenectomy (no response in 10/14 patients) (35). Immunoglobulin replacement effectively reduces infection frequency. Replacement of insufficient CTLA-4 in these diseases, by intravenous or subcutaneous infusion of the CTLA-4 fusion proteins abatacept or belatacept, may be considered (26, 35, 111). Response rates appear promising, particularly for neurological and gastrointestinal disease, although existing tissue destruction such as that causing insulin-dependent diabetes mellitus cannot be reversed. Long-term efficacy and safety data are lacking. Surveillance for oncogenic viruses such as EBV may be important, as chronic viraemia is common, malignancy is frequently EBV-related, and there have been reports of herpesvirus reactivations with CTLA-4 fusion protein therapy (103, 112, 113). A phase IIa prospective multicenter trial (ABACHAI) aims to answer questions surrounding safety and efficacy in adult patients with CTLA-4 haploinsufficiency by use of a novel morbidity score (CHAI), which could also be employed in future study of other therapies in this disease (37). Whilst HSCT has a role in these diseases, further data to understand the true extent of its efficacy and which patients should be transplanted, and when, are required. For LRBA deficiency, twenty-four patients underwent HSCT with a median follow-up of 20 months and OS of 70.8%, with all deaths being transplant related. Interval to HSCT and disease burden, particularly pulmonary involvement, affected odds of survival (26). Surviving patients had significantly lower IDDA scores than those on conventional therapies, and 70.6% were in immunosuppression-free remission. In CTLA-4 haploinsufficiency, two HSCT series were updated in a large multicenter study recently, and along with one other series a total of 21 transplanted patients have been reported in the literature, with OS 15/21 (71.4%) (35, 36, 114, 115). Whilst disease activity appears improved in HSCT survivors, donor Tregs do not appear to have the same survival advantage seen in HSCT for IPEX and consequently, immune dysregulation may continue in the setting of mixed chimerism (114, 116). Questions regarding optimum HSCT strategy, and the role of CTLA-4 fusion therapy as a bridge to transplant, may be answered by an ongoing study of the Inborn Errors Working Party of EBMT. Autologous gene therapy using a homology-directed repair approach in human CTLA4+/- CD4+ lymphocytes has successfully restored CTLA-4 activity in an in vitro study with good efficiency, but requires translation to an HSC platform (59).
Whilst dominant negative mutations in signal transduction and activator of transcription (STAT) 3 cause a complex multisystem immunodeficiency with recurrent staphylococcal and fungal infection, eczema, and connective tissue disease (STAT3 hyper IgE syndrome) (117, 118), germline activating mutations lead to exaggerated transcriptional activity of STAT3 and impaired signaling through the other STAT molecules (20). STAT3 transduces multiple cytokine signals, including IL-6, IL-10, and IL-23 (119, 120). The resultant immunological phenotype of STAT3-GOF is of a quantitative and qualitative Treg defect. Clinically, patients display an incompletely-penetrant phenotype of childhood-onset multiorgan autoimmunity and lymphoproliferation, along with stunted growth (20, 21, 121). Whilst the most common manifestations are lymphoproliferation with raised DN T-lymphocytes, two-thirds have autoimmune cytopenias and end-organs affected by autoimmunity include the gastrointestinal tract, endocrine organs, liver causing hepatitis, and lung disease. The immune deficiency was initially reported as modest, though a large series of 191 patients found infections in three-quarters of patients with both humoral and cellular arms impacted (58). Similarly to STAT3-HIES, data do not support a genotype-phenotype correlation and indeed, mutations affecting the same codon may cause both STAT3-GOF and STAT3-HIES diseases in different patients (58, 122).
Treatment is challenging, with incomplete response to individual agents and most patients require multiple (>5) lines of therapy to sustain a clinical response (58). The ability to target the JAK-STAT cascade using JAK inhibitors such as ruxolitinib provides an attractive treatment option in STAT3-GOF, with efficacious response although long-term data are lacking (58, 123, 124). Other targeted therapies include IL-6 blockade (tocilizumab) (123). The use of small molecules to inhibit STAT3 activity is the subject of several pre-clinical trials, focusing primarily on somatic STAT3-mutated malignancies (125). Whether these may translate to therapy for germline activating mutations remains to be seen. Data on outcomes of HSCT are limited. Initial reports were dismal, with death in 4/5 patients, though subsequent series have shown improved survival up to 62% in a summary of 23 transplanted patients (58, 126). HSCT in childhood may not normalize growth velocity. A more detailed exploration of indication, transplant strategy, and morbidity and mortality beyond individual case reports is yet to be published, particularly as HSCT appears to be reserved for treatment-resistant cases with significant pre-HSCT morbidity. The pleiotropic nature of STAT3 suggests that, as in STAT3-HIES, HSCT for STAT3-GOF may not reverse the signaling defect in extra-hematopoietic tissues.
Whilst deficiency of the γ-chain of the IL-2 receptor causes T-B+NK- SCID (127, 128), deficiency of the α or β chains, encoded by IL2RA and IL2RB respectively, cause immune dysregulation (129). The α chain (IL-2RA, CD25) exclusively binds IL-2, in contrast to the γ chain, which forms receptors that bind interleukins -4, -7, -9, -15, and -21. CD25 is expressed at high levels on Tregs, and binding of IL-2 upregulates FOXP3 expression and therefore enhances Treg activity in response to immune stimulus (130). Meanwhile, the β chain (IL-2RB, CD122) forms a receptor with the γ chain which binds IL-2 and IL-15 (131). IL2-RA and IL2-RB deficiencies are rare, with few published cases. Their shared phenotype includes autoimmunity, with erythroderma, enteropathy, and alopecia, similar to IPEX. However, in contrast, these patients also exhibit a significant susceptibility to herpesvirus infections such as CMV (12, 13, 57). Phosphorylation of STAT5 following IL-2 stimulation is reduced or absent, depending on whether the mutation is null or hypomorphic, in both syndromes (57). In IL-2RA deficiency, Treg numbers are low-normal, but the cells are non-functional; in IL-2RB deficiency, Tregs are low-absent (129). Autoreactive CD8+ cells may infiltrate organs causing autoimmunity in IL-2RA deficiency (12). Interestingly, some patients with hypomorphic IL-2RG mutations display a phenotype akin to IL-2RA and IL-2RB mutations, with immune dysregulation and lymphoproliferation (129).
HSCT has been reported in one patient with IL-2RA deficiency, who survived (132). In IL-2RB deficiency, two cases have been published and one additional, unpublished case was performed at our center, with survival and cure in two patients and death from CMV in the third (57). mTOR inhibition has been reported to improve autoimmune symptoms in one case (133). Pooling of data for these rare monogenic diseases is needed to better understand their manifestations and natural history.
Hemophagocytic lymphohistiocytosis (HLH) describes a syndrome of immune hyperactivation caused by sustained activation of cytotoxic T-lymphocytes and resultant release of inflammatory cytokines, leading to persistent fever, hepatosplenomegaly, cytopenias, and coagulopathy (23). Whilst identification of hemophagocytes in bone marrow or cerebrospinal fluid specimens may aid diagnosis, absence does not exclude HLH (134). HLH may be classified by as primary (driven by monogenic IEI) or secondary (driven by environmental or acquired triggers, such as malignancy, infection, or inflammatory disease) (23). In the setting of rheumatological disease, typically systemic-onset juvenile idiopathic arthritis, HLH may be termed ‘macrophage activation syndrome.’ HLH is diagnosed using criteria defined by the Histocyte Society, including fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytes, low/absent NK-cell activity, hyperserotonemia and high soluble-IL-2-receptor levels; at least five criteria must be met in order to diagnose HLH (135). The treatment strategy set out by the HLH-2004 trial includes combination chemotherapy with etoposide, dexamethasone, ciclosporin A ± intrathecal methotrexate and corticosteroids (135, 136). In recent years, other therapeutic strategies have emerged, primarily as second-line or salvage therapy, including lymphodepletion with alemtuzumab (137, 138), blockade of inflammatory cytokines such as IL-1 (anakinra), IL-6 (tocilizumab), and IFN-γ (emapalumab) (43), and JAK-inhibition (ruxolitinib) (42, 139). Identification of whether HLH is primary or secondary is important, as patients with primary HLH should progress to allogeneic HSCT; this may rely on molecular or genetic studies as some infectious triggers, such as EBV, may cause either primary or secondary HLH.
Primary HLH may be subdivided into a collection of diseases impacting lymphocyte cytotoxicity, including the five ‘familial’ HLH (FHLH) syndromes where HLH is the primary disease manifestation, and those where impaired control of infection or dysregulated inflammasome activation leading to immune hyperactivation are seen in addition to other clinical manifestations. FHLH results from defective trafficking of vesicles containing cytotoxic molecules such as perforin and granzymes to a target cell, preventing its destruction and prolonging immune activation (140). Whilst the genetic aetiology of FHLH1 is unknown, the other syndromes relate to defects in vesicle content (FHLH2, PRF1), priming (FHLH3, UNC132), or docking and fusion to the target cell membrane (FHLH4, STX11 and FHLH5, STXBP2) (22, 140). The largest cohort published to date, from the HLH-2004 study, demonstrated 5-year overall survival of 71% in children with FHLH (141).
Aside from these familial syndromes, other IEI affecting lymphocyte granule-mediated cytotoxicity include pigmentary disorders such as Chediak-Higashi syndrome caused by mutations in LYST, Griscelli syndrome type 2, caused by RAB27A mutations, and less frequently in Hermansky-Pudlak syndrome type 2, caused by AP3B1 mutations (142–144). Unfortunately, HSCT does not arrest the neurodevelopmental sequelae seen in Chediak-Higashi syndrome (145). Susceptibility to EBV-driven HLH may be seen in mutations of SH2D1A, causing X-linked lymphoproliferative syndrome (XLP) 1 and manifesting in hypogammaglobulinemia, and development of EBV-driven lymphoma, as well as other IEIs rendering patients unable to handle this virus effectively, such as deficiencies of CD27, CTPS1, ITK, and CD70 (146, 147). A multi-center study reporting the outcome of HSCT for XLP1 recommended that transplantation is undertaken in all patients who develop HLH, as the outcome without HSCT is poor (overall survival 18.8%) although HLH manifestations associated with higher mortality during HSCT (51). Autologous T-lymphocyte gene therapy is under investigation, with evidence of correction of the T-lymphocyte defect in vitro (52). NK cell deficiency, such as that seen in MCM4 defects, also predisposes to primary HLH due to inability to clear virus-infected cells (148, 149). Other genetic aetiologies of primary HLH include genes regulating inflammasome activity, such as XIAP and NLRC4. XIAP-deficient males may develop HLH following EBV infection in addition to splenomegaly and inflammatory bowel disease (150, 151). Whilst termed XLP2, unlike XLP1 there does not appear to be an increased risk of EBV-driven lymphoma, and HLH episodes may occur more frequently (152). The incidence of HLH in XIAP deficiency is approximately 60%, whilst half have splenomegaly and a quarter develop Crohn’s disease-like colitis. Allogeneic HSCT remains the only curative treatment for XIAP deficiency; whilst the odds of survival have improved drastically between patients transplanted before and after 2015 (41% versus 89%), outcomes are poorer following myeloablative conditioning and in patients with active HLH at HSCT. It is not fully understood whether patients with milder disease should be offered HSCT as the outcomes for patients managed conservatively vary (153). For XIAP patients with significant bowel inflammation, HSCT appears to resolve inflammatory bowel disease (154). Future targeted therapy to aid remission of autoinflammation in XIAP and NLCR4-related disease, which is driven by high IL-18 levels, may come in the form of tadekinig alfa, which mimics endogenous neutralizing IL-18 binding protein; whilst its use in XIAP deficiency has been reported in a single patient, further evidence on efficacy may come from a randomized trial (NCT03113760), which has completed recruitment and is ending soon (44, 155).
Whilst the genetic landscape of primary HLH has grown with increasing understanding of interactions leading to cell-mediated cytotoxicity, a high index of clinical suspicion is needed to identify possible HLH and institute appropriate initial management. Use of specialist investigations such as NK degranulation and perforin expression may shorten the time taken to differentiate primary from secondary HLH and allow earlier definitive therapy (156).
Children diagnosed with inflammatory bowel disease before the age of 6 years form a subset of patients that is enriched for underlying IEI (157). This group of patients may also be prone to failure of conventional immunomodulation, and for patients with underlying IEI the balance between immunosuppression to achieve symptom control versus infection risk may pose a challenge. Identified monogenic defects associated with VEOIBD include chronic granulomatous disease, IPEX syndrome, XIAP deficiency, Wiskott-Aldrich syndrome, and defects of IL-10 signaling. In 2009, mutations in IL10RA and IL10RB encoding the two subunits of the IL-10 receptor, were identified in four patients with VEOIBD; subsequent to this, loss-of-function mutations in IL-10 were also identified in association with intestinal inflammation (60, 158). This is consistent with previous findings showing IL-10 to be a key suppressor of T lymphocyte activation and effector function, particularly in the gastrointestinal tract (159). IL-10 defect-associated VEOIBD appears to be fully penetrant and presents with perianal disease alongside additional features such as folliculitis and arthritis. Whilst reported cases are few, allogeneic HSCT appears to be curative (60, 160, 161). Identification of a genetic mutation associated with VEOIBD is important, as not all monogenic causes relate to the hematopoietic system and thus allogeneic HSCT may not resolve VEOIBD caused by epithelial defects or tricho-hepatico-enteric syndromes (162, 163).
Loss of function mutations in RIPK1, encoding a serine/threonine-protein kinase involved in cell death regulation, also cause VEOIBD in tandem with combined immunodeficiency and arthritis, which may be rescued by allogeneic HSCT (164).
Activated phosphoinositide 3-kinase-δ syndrome (APDS) results from either gain-of-function mutations in PIK3CD encoding the catalytic subunit p110δ (APDS1), or by loss-of-function variants in PIK3R1 encoding the regulatory subunits p85α, p55α, or p50α (APDS2) (165). APDS was first identified in 2013 in seven kindreds with recurrent respiratory infection progressing to bronchiectasis, and lymphopenia (166). The result of these dominant mutations is disruption of the tight balance in phosphoinositide 3-kinase (PI3K) activity, with hyperactivation of this pathway impacting T and B lymphocyte homeostasis (165). Patients with APDS manifest a heterogeneous immunophenotype, including reduced naïve and increased senescent CD8+ T-lymphocyte populations; reduced class-switched memory B-lymphocyte cells with increased transitional B-lymphocytes; and dysgammaglobulinemia, typically with elevated serum IgM with self-reactive antibody production but reduced IgA and IgG (167). Though not classified as a PIRD in the latest IUIS classification, this IEI shares many features with other PIRDs, as highlighted by the recent publication of an ESID registry study analyzing 170 patients (168). In addition to recurrent sinopulmonary infections, around 25% of patients have chronic EBV infection with high rates of lymphoproliferation (86%) and lymphoma (14%). AIC may occur, but less frequently than in CTLA-4 haploinsufficiency or STAT3-GOF, as with enteropathy and skin manifestations. Patients frequently develop bronchiectasis in childhood, and earlier onset of symptoms predicts worsening disease severity. Rare, but described manifestations include arthritis, pancreatic islet β-cell destruction causing diabetes, and neuroinflammation, which are all more common in other dominantly inherited PIRDs such as CTLA-4 haploinsufficiency. Phenotypically, APDS1 and APDS2 differ by the former being relatively enriched for AIC and skin disease, whilst the latter typically includes syndromic features such as short stature (167–169). Strategies to reduce infection frequency are antimicrobial prophylaxis and immunoglobulin replacement. Immune dysregulation may be treated with steroids, B-lymphocyte depleting agents, or mTOR inhibitors. mTOR inhibition is attractive given the high response rate of lymphoproliferation, enteropathy, and AIC, possibly explained by the hyperactivation of mTOR signaling downstream of Akt, which is enhanced by increased PI3K activity (168, 170). A retrospective study of 57 patients who underwent HSCT for APDS demonstrates the challenges in treating this disease with transplantation. Whilst 2-year overall survival probability was good (86%), there was a high rate of graft failure necessitating further cellular infusions, which rose from 10% at 1 year post-HSCT to 17% at 2 and 3 years after transplant (61). Strikingly, the incidence of graft failure was even higher in patients who received post-HSCT mTOR inhibitors, at 15-42% (1-3 years post-HSCT) compared to 9% in patients who did not receive mTOR inhibition, possibly due to giving remnant recipient lymphocytes a survival advantage over the reconstituting recipient cells (61). Targeted inhibition of PI3K with leniolisib is now possible, following first approval for treatment of APDS in adults and children aged 12 years and above in in the USA in 2023; the randomized controlled trial leading to its approval demonstrated high efficacy in reduction of lymphoproliferation and normalization of the APDS immunophenotype (49, 171). Further data, including exploration of leniolisib as a ‘bridge’ to HSCT, are warranted.
Systemic lupus erythematosus (SLE) has a strong heritable component indicated by the association of its phenotype with variants in multiple candidate genes in genome-wide association studies (172). In addition to population-level variation, several genes have been identified to correlate strongly with development of SLE in specific pedigrees despite low allele frequency in the general population, suggestive of a monogenic aetiology of SLE in this small subgroup. Consistent with the finding that DNA may act as an autoantigen in SLE, abnormal clearance of circulating DNA may cause tissue damage by provoking formation of autoantibodies. The primary enzyme mediating clearance of chromatin is DNase1. Low serum DNase1 activity is associated with development of autoimmune hepatitis, and higher SLE disease activity (173, 174). Monoallelic null mutations of DNASE1 were found in two unrelated patients with SLE (175). Homozygous null mutations in DNASE1L3, preventing the expression of this protein which is closely related to DNase1, may also cause a fully-penetrant form of monogenic lupus (176).The role of deficiency of these enzymes in provoking autoimmunity suggests augmenting the activity of DNase may help neutralize errant chromatin and interfere with the pathogenesis of autoimmunity in murine models (177).
In 2016, heterozygous loss-of-function mutations in TNFAIP3 leading to haploinsufficiency of its encoded protein, A20, were identified as a monogenic cause of Behçet disease (178). Haploinsufficiency of A20 (HA20) results in increased NFκβ signaling and subsequent upregulation of pro-inflammatory cytokines such as IL-1, IL-6, and TNFα; consequently, use of anakinra and anti-TNFα monoclonal antibodies such as infliximab are efficacious for controlling disease manifestations (178, 179). Symptoms of HA20 include Behçet disease-like features such as orogenital ulceration, arthritis, and uveitis. However, unlike polygenic Behçet disease, onset is typically in childhood and patients have recurrent febrile episodes (178, 179). HSCT has been reported in two patients with HA20, who both remitted completely (62).
IL-1 is a powerful inducer of fever and inflammation; upon binding of this cytokine to its receptor, transcription of pro-inflammatory cytokines is upregulated via MyD88, IRAK-4, and NFκβ (180). This process is regulated by IL-1RA, which competes with IL-1α and IL-1β for binding with the IL-1 receptor; recombinant IL-1RA (anakinra) is licensed for several autoimmune diseases including rheumatoid arthritis. Whilst impaired IL-1 results in immunodeficiency characterized by susceptibility to Staphylococcus aureus, Streptococcus pneumoniae, and Pseudomonas aeruginosa infections (181), deficiency of IL-1Ra leads to a syndrome of sterile multifocal osteomyelitis, periostitis, and pustulosis caused by unrestrained IL-1-mediated inflammation (DIRA) (47). These patients respond rapidly to replacement therapy with anakinra (47). Similarly, deficiency of IL-36RA, which shares 44% of its homology with IL-1RA, causes increased IL-1-related signaling and a clinical syndrome of infection-provoked generalized pustular psoriasis, fever and asthenia, which may progress to death from overwhelming infection (48, 182). This syndrome, labelled Deficiency of Interleukin-Thirty-six-receptor Antagonist, DITRA), does not appear to cause sterile osteoarticular inflammation seen in DIRA, but is responsive to anakinra (48).
The result of improved diagnostics and ability to differentiate patients with a PIRD phenotype into distinct genetic aetiologies is that understanding of how our immune system is regulated and how disruptions of this cause disease has grown, particularly over the past decade. Understanding these mechanisms has enabled development of targeted therapies that aim to ‘switch off’ a deleterious process whilst minimizing off-target effects. Many of these agents show significant promise in improving disease control and quality of life in patients with specific PIRDs, although long-term efficacy, and side effects are unknown. However, the inevitable conclusion of our ability to differentiate this patient group is that sample sizes are diluted, and our ability to fully understand the epidemiology, clinical and immunological phenotype, and treatment options for a given disease becomes more challenging and reliant on large, multicenter international efforts. Whilst time- and resource-consuming, these collaborations are necessary to further our understanding of these diseases, particularly for studies with a longitudinal component, such as assessing treatment efficacy or natural history. Setup and maintenance of prospective disease registries, including adaptations of existing projects such as addition of the IDDA2.1 score to the ESID registry, or formation of the German multi-organ AutoImmunity Network, www.g-a-i-n.de), will provide databases to be interrogated for future studies (27, 183).
Increasing attention has been placed on collection of quality-of-life data in patients with IEI, with recent publication of studies exploring the impact of chronic granulomatous disease, Wiskott-Aldrich syndrome, common variable immunodeficiency, and patients with SCID who have undergone HSCT (184–187). Whilst we intuitively understand that recurrent infection, hospitalization, and end-organ damage might negatively impact quality-of-life, it is important to explore associated factors, particularly when considering the impact of starting new treatments or offering HSCT. Few existing studies can relate to the diverse manifestations of PIRDs, and abstraction from studies that focus on a specific manifestation of immune dysregulation, such as colitis, do not account for the cumulative burden of having multiple organ system disease. Quality of life data pertaining to specific PIRDs are scant; one study of Finnish patients with APS1 identified poor general health and worsening fatigue and well-being with a longer interval from diagnosis to data collection (188). Use of quality-of-life instruments should form part of collaborative efforts to characterize clinical aspects of these diseases.
Intrinsic to discussions regarding HSCT in IEI is which patients should be offered HSCT, and at what stage of their disease course this treatment should be considered. For several IEI, the decision to offer HSCT early prior to accumulation of organ damage is justified by large multicenter studies, for example in SCID and chronic granulomatous disease (29, 189). However, for diseases with a variable natural history, or incomplete penetrance, the decision to offer HSCT exchanges a potential risk of subsequent poor health over a period of years-decades for a concentrated period of high risk around HSCT and during the initial period of immune reconstitution. Whilst the decision to undergo HSCT must come jointly from clinician and patients and their families, the potential risks and benefits of this treatment must be informed by good quality clinical data, requiring use of registries such as those coordinated by the European Blood and Marrow Transplant, European Society for Immunodeficiencies, and Stem Cell Transplantation for Immunodeficiencies in Europe groups.
Conceptually, PIRDs provide a fascinating lens through which we can observe how alterations in immune regulatory genes lead to diverse clinical phenotypes. However, for patients, these diseases are far from fascinating. Their multi-organ manifestations may lead to delay in reaching a unifying diagnosis, and the chronicity of ill health, need for long-term treatment, and absence of natural history data for many, cast uncertainty on patient lives. Collective efforts to better understand these diseases and the therapies we may offer patients must continue.
CT: Conceptualization, Resources, Writing – original draft, Writing – review & editing. MS: Conceptualization, Writing – review & editing. AG: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. CT is supported by the Job Research Foundation, who were not involved in the generation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Good RA, Yunis E. Association of autoimmunity, immunodeficiency and aging in man, rabbits, and mice. Fed Proc (1974) 33(9):2040–50.
2. Arkwright PD, Abinun M, Cant AJ. Autoimmunity in human primary immunodeficiency diseases. Blood (2002) 99(8):2694–702. doi: 10.1182/blood.V99.8.2694
3. Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr (1982) 100(5):731–7. doi: 10.1016/S0022-3476(82)80573-8
4. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
5. Bousfiha AA, Jeddane L, Ailal F, Al Herz W, Conley ME, Cunningham-Rundles C, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol (2013) 33(6):1078–87. doi: 10.1007/s10875-013-9901-6
6. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol (2022) 42(7):1508–20. doi: 10.1007/s10875-022-01352-z
7. Janeway CJ, Travers P, Walport M. Immunobiology: the immune system in health and disease. New York: Garland Science (2001). Available at: https://www.ncbi.nlm.nih.gov/books/NBK10757/.
8. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity (2005) 23(2):227–39. doi: 10.1016/j.immuni.2005.07.005
9. Sakaguchi S. The origin of FOXP3-expressing CD4+ regulatory T cells: Thymus or periphery. J Clin Invest. (2003) 112(9):1310–2. doi: 10.1172/JCI200320274
10. Nishimura E, Sakihama T, Setoguchi R, Tanaka K, Sakaguchi S. Induction of antigen-specific immunologic tolerance by in vivo and in vitro antigen-specific expansion of naturally arising Foxp3+CD25+CD4+ regulatory T cells. Int Immunol (2004) 16(8):1189–201. doi: 10.1093/intimm/dxh122
11. Corthay A. How do regulatory t cells work? Scand J Immunol (2009) 70(4):326–36. doi: 10.1111/j.1365-3083.2009.02308.x
12. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Mannurita SC, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (2013) 146(3):248–61. doi: 10.1016/j.clim.2013.01.004
13. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi: 10.1016/j.jaci.2006.10.007
14. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Sci (80- ) (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
15. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet (2012) 90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015
16. Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature (2006) 441(7095):890–3. doi: 10.1038/nature04790
17. Engelhardt KR, Grimbacher B. IL-10 in humans: Lessons from the Gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol (2014) 380:1–18. doi: 10.1007/978-3-662-43492-5_1
18. Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol (2017) 139(6):2016–2020.e5. doi: 10.1016/j.jaci.2016.12.957
19. Toubiana J, Okada S, Hiller J, Oleastro M, Gomez ML, Becerra JCA, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood (2016) 127(25):3154–64. doi: 10.1182/blood-2015-11-679902
20. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood (2015) 125(4):591–9. doi: 10.1182/blood-2014-09-602763
21. Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol (2018) 473:166–77. doi: 10.1016/j.mce.2018.01.016
22. Meeths M, Bryceson YT. Genetics and pathophysiology of haemophagocytic lymphohistiocytosis. Acta Paediatr (2021) 110(11):2903–11. doi: 10.1111/apa.16013
23. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood (2020) 135(16):1332–43. doi: 10.1182/blood.2019000936
24. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol (2010) 85(3):174–80. doi: 10.1002/ajh.21616
25. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet (2018) 391(10138):2449–62. doi: 10.1016/S0140-6736(18)31320-5
26. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol (2020) 145(5):1452–63. doi: 10.1016/j.jaci.2019.12.896
27. Seidel MG, Tesch VK, Yang L, Hauck F, Horn AL, Smolle MA, et al. The immune deficiency and dysregulation activity (IDDA2.1 ‘Kaleidoscope’) score and other clinical measures in inborn errors of immunity. J Clin Immunol (2022) 42(3):484–98. doi: 10.1007/s10875-021-01177-2
28. Albert MH, Slatter MA, Gennery AR, Güngör T, Bakunina K, Markovitch B, et al. Hematopoietic stem cell transplantation for Wiskott-Aldrich syndrome: an EBMT Inborn Errors Working Party analysis. Blood (2022) 139(13):2066–79. doi: 10.1182/blood.2021014687
29. Chiesa R, Wang J, Blok HJ, Hazelaar S, Neven B, Moshous D, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood (2020) 136(10):1201–11. doi: 10.1182/blood.2020005590
30. Fischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat Rev Immunol (2023) 23(6):397–408. doi: 10.1038/s41577-022-00800-6
31. Borna S, Lee E, Sato Y, Bacchetta R. Towards gene therapy for IPEX syndrome. Eur J Immunol (2022) 52(5):705–16. doi: 10.1002/eji.202149210
32. Ghosh S, Drexler I, Bhatia S, Gennery AR, Borkhardt A. Interleukin-2-inducible T-cell kinase deficiency-new patients, new insight? Front Immunol (2018) 9(MAY):1–7. doi: 10.3389/fimmu.2018.00979
33. Soheili T, Rivière J, Ricciardelli I, Durand A, Verhoeyen E, Derrien A-C, et al. Gene-corrected human Munc13-4–deficient CD8+ T cells can efficiently restrict EBV-driven lymphoproliferation in immunodeficient mice. Blood (2016) 128(24):2859–62. doi: 10.1182/blood-2016-07-729871
35. Egg D, Rump IC, Mitsuiki N, Rojas-Restrepo J, Maccari ME, Schwab C, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol (2022) 149(2):736–46. doi: 10.1016/j.jaci.2021.04.039
36. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 CTLA-4-insufficient individuals. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
37. Krausz M, Uhlmann A, Rump IC, Ihorst G, Goldacker S, Sogkas G, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: A non controlled phase 2 clinical trial. Contemp Clin trials Commun (2022) 30:101008. doi: 10.1016/j.conctc.2022.101008
38. Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, et al. Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol (2020) 145(4):1262–1271.e13. doi: 10.1016/j.jaci.2019.11.043
39. Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol (2008) 28(5):581–7. doi: 10.1007/s10875-008-9196-1
40. Klemann C, Esquivel M, Magerus-Chatinet A, Lorenz MR, Fuchs I, Neveux N, et al. Evolution of disease activity and biomarkers on and off rapamycin in 28 patients with autoimmune lymphoproliferative syndrome. Haematologica. Italy; (2017) 102:e52–6. doi: 10.3324/haematol.2016.153411
41. Völkl S, Rensing-Ehl A, Allgäuer A, Schreiner E, Lorenz MR, Rohr J, et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood (2016) 128(2):227–38. doi: 10.1182/blood-2015-11-685024
42. Summerlin J, Wells DA, Anderson MK, Halford Z. A review of current and emerging therapeutic options for hemophagocytic lymphohistiocytosis. Ann Pharmacother (2022) 57(7):867–79. doi: 10.1177/10600280221134719
43. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med (2020) 382(19):1811–22. doi: 10.1056/NEJMoa1911326
44. Geerlinks AV, Dvorak AM, Jordan MB, Schiffrin EJ, Behrens EM, Marsh R, et al. A case of XIAP deficiency successfully managed with tadekinig alfa (rhIL-18BP). J Clin Immunol (2022) 42(4):901–3. doi: 10.1007/s10875-022-01236-2
45. Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol (2017) 139(5):1698–701. doi: 10.1016/j.jaci.2016.10.022
46. Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc Natl Acad Sci (2008) 105(35):13009–14. doi: 10.1073/pnas.0806874105
47. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med (2009) 360(23):2426–37. doi: 10.1056/NEJMoa0807865
48. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei X-Y, Fraitag S, et al. Interleukin-36–receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med (2011) 365(7):620–8. doi: 10.1056/NEJMoa1013068
49. Rao VK, Webster S, Šedivá A, Plebani A, Schuetz C, Shcherbina A, et al. A randomized, placebo-controlled phase 3 trial of the PI3Kδ inhibitor leniolisib for activated PI3Kδ syndrome. Blood (2023) 141(9):971–83. doi: 10.1182/blood.2022018546
50. Ghosh S, Carmo M, Calero-Garcia M, Ricciardelli I, Bustamante Ogando JC, Blundell MP, et al. T-cell gene therapy for perforin deficiency corrects cytotoxicity defects and prevents hemophagocytic lymphohistiocytosis manifestations. J Allergy Clin Immunol (2018) 142(3):904–13. doi: 10.1016/j.jaci.2017.11.050
51. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood (2011) 117(1):53–62. doi: 10.1182/blood-2010-06-284935
52. Panchal N, Houghton B, Diez B, Ghosh S, Ricciardelli I, Thrasher AJ, et al. Transfer of gene-corrected T cells corrects humoral and cytotoxic defects in patients with X-linked lymphoproliferative disease. J Allergy Clin Immunol (2018) 142(1):235–45. doi: 10.1016/j.jaci.2018.02.053
53. Sleight BJ, Prasad VS, DeLaat C, Steele P, Ballard E, Arceci RJ, et al. Correction of autoimmune lymphoproliferative syndrome by bone marrow transplantation. Bone Marrow Transplant. (1998) 22(4):375–80. doi: 10.1038/sj.bmt.1701306
54. Savic S, Parry D, Carter C, Johnson C, Logan C, Gutierrez BM, et al. A new case of Fas-associated death domain protein deficiency and update on treatment outcomes. J Allergy Clin Immunol (2015) 136(2):502–505.e4. doi: 10.1016/j.jaci.2015.02.002
55. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol (2018) 141(3):1036–1049.e5. doi: 10.1016/j.jaci.2017.10.041
56. Bezrodnik L, Caldirola MS, Seminario AG, Moreira I, Gaillard MI. Follicular bronchiolitis as phenotype associated with CD25 deficiency. Clin Exp Immunol (2014) 175(2):227–34. doi: 10.1111/cei.12214
57. Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, et al. Human interleukin-2 receptor b mutations associated with defects in immunity and peripheral tolerance. J Exp Med (2019) 216(6):1311–27. doi: 10.1084/jem.20182304
58. Leiding JW, Vogel TP, Santarlas VGJ, Mhaskar R, Smith MR, Carisey A, et al. Monogenic early-onset lymphoproliferation and autoimmunity: Natural history of STAT3 gain-of-function syndrome. J Allergy Clin Immunol (2023) 151(4):1081–95. doi: 10.1016/j.jaci.2022.09.002
59. Fox TA, Houghton BC, Petersone L, Waters E, Edner NM, McKenna A, et al. Therapeutic gene editing of T cells to correct CTLA-4 insufficiency. Sci Transl Med (2022) 14(668):eabn5811. doi: 10.1126/scitranslmed.abn5811
60. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143(2):347–55. doi: 10.1053/j.gastro.2012.04.045
61. Dimitrova D, Nademi Z, Maccari ME, Ehl S, Uzel G, Tomoda T, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol (2022) 149(1):410–21. doi: 10.1016/j.jaci.2021.04.036
62. Shiraki M, Williams E, Yokoyama N, Shinoda K, Nademi Z, Matsumoto K, et al. Hematopoietic cell transplantation ameliorates autoinflammation in A20 haploinsufficiency. J Clin Immunol (2021) 41(8):1954–6. doi: 10.1007/s10875-021-01124-1
63. Björses P, Aaltonen J, Horelli-Kuitunen N, Yaspo ML, Peltonen L. Gene defect behind APECED: A new clue to autoimmunity. Hum Mol Genet (1998) 7(10):1547–53. doi: 10.1093/hmg/7.10.1547
64. Thorpe ES, Handley HE. Chronic tetany and chronic mycelial stomatitis in a child aged four and one-half years. Am J Dis Child. (1929) 38(2):328–38. doi: 10.1001/archpedi.1929.01930080104011
65. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
66. Aaltonen J, Björses P, Perheentupa J, Horelli–Kuitunen N, Palotie A, Peltonen L, et al. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet (1997) 17(4):399–403. doi: 10.1038/ng1297-399
67. Constantine GM, Lionakis MS. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev (2019) 287(1):103–20. doi: 10.1111/imr.12714
68. Bruserud Ø, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Curr Opin Immunol (2016) 43:8–15. doi: 10.1016/j.coi.2016.07.003
69. Ferre EMN, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight (2016) 1(13). doi: 10.1172/jci.insight.88782
70. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity (2015) 42(6):1185–96. doi: 10.1016/j.immuni.2015.04.021
71. Bastard P, Orlova E, Sozaeva L, Lévy R, James A, Schmitt MM, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med (2021) 218(7). doi: 10.1084/jem.20210554
72. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 4585(September). doi: 10.1126/science.abd4585
73. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298(5597):1395–401. doi: 10.1126/science.1075958
74. Ferré EMN, Schmitt MM, Lionakis MS. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Front Pediatr (2021) 9:723532. doi: 10.3389/fped.2021.723532
75. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Kärner J, et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell (2016) 166(3):582–95. doi: 10.1016/j.cell.2016.06.024
76. Dobeš J, Ben-Nun O, Binyamin A, Stoler-Barak L, Oftedal BE, Goldfarb Y, et al. Extrathymic expression of Aire controls the induction of effective TH17 cell-mediated immune response to Candida albicans. Nat Immunol (2022) 23(7):1098–108. doi: 10.1038/s41590-022-01247-6
77. Ferré EMN, Break TJ, Burbelo PD, Allgäuer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med (2019) 11(495):1–33. doi: 10.1126/scitranslmed.aav5597
78. Canale VC, Smith CH. Chronic lymphadenopathy simulating Malignant lymphoma. J Pediatr (1967) 70(6):891–9. doi: 10.1016/S0022-3476(67)80262-2
79. Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science (1995) 268(5215):1347–9. doi: 10.1126/science.7539157
80. Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. (1996) 98(5):1107–13. doi: 10.1172/JCI118892
81. Wang J, Zheng L, Lobito A, Chan FK-M, Dale J, Sneller M, et al. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell (1999) 98(1):47–58. doi: 10.1016/S0092-8674(00)80605-4
82. Holzelova E, Vonarbourg C, Stolzenberg M-C, Arkwright PD, Selz F, Prieur A-M, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med (2004) 351(14):1409–18. doi: 10.1056/NEJMoa040036
83. Consonni F, Gambineri E, Favre C. ALPS. FAS, and beyond: from inborn errors of immunity to acquired immunodeficiencies. Ann Hematol (2022) 101(3):469–84. doi: 10.1007/s00277-022-04761-7
84. Rieux-Laucat F, Magérus-Chatinet A, Neven B. The autoimmune lymphoproliferative syndrome with defective FAS or FAS-ligand functions. J Clin Immunol (2018) 38(5):558–68. doi: 10.1007/s10875-018-0523-x
85. Magerus-Chatinet A, Neven B, Stolzenberg M-C, Daussy C, Arkwright PD, Lanzarotti N, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. (2011) 121(1):106–12. doi: 10.1172/JCI43752
86. López-Nevado M, González-Granado LI, Ruiz-García R, Pleguezuelo D, Cabrera-Marante O, Salmón N, et al. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol (2021) 12(August). doi: 10.3389/fimmu.2021.671755
87. Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood (2001) 98(1):194–200. doi: 10.1182/blood.V98.1.194
88. Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood (2011) 118(18):4798–807. doi: 10.1182/blood-2011-04-347641
89. Hafezi N, Zaki-Dizaji M, Nirouei M, Asadi G, Sharifinejad N, Jamee M, et al. Clinical, immunological, and genetic features in 780 patients with autoimmune lymphoproliferative syndrome (ALPS) and ALPS-like diseases: A systematic review. Pediatr Allergy Immunol (2021) 32(7):1519–32. doi: 10.1111/pai.13535
90. Lehle AS, Farin HF, Marquardt B, Michels BE, Magg T, Li Y, et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology (2019) 156(1):275–8. doi: 10.1053/j.gastro.2018.09.041
91. Niemela J, Kuehn HS, Kelly C, Zhang M, Davies J, Melendez J, et al. Caspase-8 deficiency presenting as late-onset multi-organ lymphocytic infiltration with granulomas in two adult siblings. J Clin Immunol (2015) 35(4):348–55. doi: 10.1007/s10875-015-0150-8
92. Kohn LA, Long JD, Trope EC, Kuo CY. Novel compound heterozygote variations in FADD identified to cause FAS-associated protein with death domain deficiency. J Clin Immunol (2020) 40(4):658–61. doi: 10.1007/s10875-020-00779-6
93. Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet (2010) 87(6):873–81. doi: 10.1016/j.ajhg.2010.10.028
94. Meer E, Solanes F, Kohn L, Kuo CY, Wong DA, Pineles S, et al. Ocular findings associated with FADD deficiency resemble familial exudative vitreoretinopathy. Am J Ophthalmol Case Rep (2022) 25:101305. doi: 10.1016/j.ajoc.2022.101305
95. Gambineri E, Mannurita SC, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol (2018) 9(NOV). doi: 10.3389/fimmu.2018.02411
96. Chida N, Kobayashi I, Takezaki S, Ueki M, Yamazaki Y, Garelli S, et al. Disease specificity of anti-tryptophan hydroxylase-1 and anti-AIE-75 autoantibodies in APECED and IPEX syndrome. Clin Immunol (2015) 156(1):36–42. doi: 10.1016/j.clim.2014.10.010
97. Lampasona V, Passerini L, Barzaghi F, Lombardoni C, Bazzigaluppi E, Brigatti C, et al. Autoantibodies to harmonin and villin are diagnostic markers in children with IPEX syndrome. PloS One (2013) 8(11):e78664. doi: 10.1371/journal.pone.0078664
98. Park JH, Lee KH, Jeon B, Ochs HD, Lee JS, Gee HY, et al. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review. Autoimmun Rev (2020) 19(6):102526. doi: 10.1016/j.autrev.2020.102526
99. Carneiro-Sampaio M, Moreira-Filho CA, Bando SY, Demengeot J, Coutinho A. Intrauterine IPEX. Front Pediatr (2020) 8:599283. doi: 10.3389/fped.2020.599283
100. Consonni F, Ciullini Mannurita S, Gambineri E. Atypical presentations of IPEX: expect the unexpected. Front Pediatr (2021) 9:643094. doi: 10.3389/fped.2021.643094
101. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
102. Van Coillie S, Wiernicki B, Xu J. Molecular and cellular functions of CTLA-4. Adv Exp Med Biol (2020) 1248:7–32. doi: 10.1007/978-981-15-3266-5_2
103. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for Malignancies in 131 affected CTLA4 mutation carriers. Front Immunol (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
104. Jamee M, Hosseinzadeh S, Sharifinejad N, Zaki-Dizaji M, Matloubi M, Hasani M, et al. Comprehensive comparison between 222 CTLA-4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review. Clin Exp Immunol (2021) 205(1):28–43. doi: 10.1111/cei.13600
105. Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U S A. (2015) 112(2):524–9. doi: 10.1073/pnas.1414576112
106. Maiarana J, Moncada-Velez M, Malbran E, Torre MG, Elonen C, Malbran A, et al. Deep immunophenotyping shows altered immune cell subsets in CTLA-4 haploinsufficiency. Pediatr Allergy Immunol (2023) 34(7):1–4. doi: 10.1111/pai.13994
107. Lougaris V, Malagola M, Baronio M, Morello E, Gazzurelli L, Benvenuto A, et al. Successful hematopoietic stem cell transplantation for complete CTLA-4 haploinsufficiency due to a de novo monoallelic 2q33.2-2q33.3 deletion. Clin Immunol (Orlando Fla.). United States; (2020) 220:108589. doi: 10.1016/j.clim.2020.108589
108. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
109. Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N, et al. Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis. Nat Commun (2019) 10(1):3106. doi: 10.1038/s41467-019-10812-x
110. Gámez-Díaz L, Seidel MG. Different apples, same tree: visualizing current biological and clinical insights into CTLA-4 insufficiency and LRBA and DEF6 deficiencies. Front Pediatr (2021) 9(April):1–9. doi: 10.3389/fped.2021.662645
111. Kiykim A, Ogulur I, Dursun E, Charbonnier LM, Nain E, Cekic S, et al. Abatacept as a long-term targeted therapy for LRBA deficiency. J Allergy Clin Immunol Pract (2019) 7(8):2790–2800.e15. doi: 10.1016/j.jaip.2019.06.011
112. Nakajima H, Takayama A, Ito T, Yoshikawa T. Acute encephalomyelitis with multiple herpes viral reactivations during abatacept therapy. BMJ Case Rep (2013) 2013. doi: 10.1136/bcr-2013-009731
113. Hoshino A, Tanita K, Kanda K, Imadome K-I, Shikama Y, Yasumi T, et al. High frequencies of asymptomatic Epstein-Barr virus viremia in affected and unaffected individuals with CTLA4 mutations. Clin Immunol (2018) 195:45–8. doi: 10.1016/j.clim.2018.07.012
114. Parker L, Kapadia M, Chellapandian D, Whangbo J, Grace R, Prince C, et al. Limitations of Bu/Flu-based preparative regimen in CTLA-4 haploinsufficiency to achieving disease phenotype correction. Clin Immunol (2023) 250:109396. doi: 10.1016/j.clim.2023.109396
115. Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol (2016) 138(2):615–619.e1. doi: 10.1016/j.jaci.2016.01.045
116. Margarit-Soler A, Deyà-Martínez À, Canizales JT, Vlagea A, García-García A, Marsal J, et al. Case report: Challenges in immune reconstitution following hematopoietic stem cell transplantation for CTLA-4 insufficiency-like primary immune regulatory disorders. Front Immunol (2022) 13(December):1–10. doi: 10.3389/fimmu.2022.1070068
117. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med (2007) 357(16):1608–19. doi: 10.1056/NEJMoa073687
118. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096
119. Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr Opin Immunol (2015) 34:75–82. doi: 10.1016/j.coi.2015.02.008
120. Villarino AV, Kanno Y, Shea JJO. Mechanism of JAK/STAT signaling in immunity disease". J Immunol (2016) 194(1):21–7. doi: 10.4049/jimmunol.1401867
121. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi: 10.1038/ng.3040
122. Lodi L, Faletti LE, Maccari ME, Consonni F, Groß M, Pagnini I, et al. STAT3-confusion-of-function: beyond the loss and gain dualism. J Allergy Clin Immunol (2022) 150(5):1237–41. doi: 10.1016/j.jaci.2022.06.007
123. Wang W, Liu L, Hui X, Wang Y, Ying W, Zhou Q, et al. Efficacy of tocilizumab therapy in a patient with severe pancytopenia associated with a STAT3 gain-of-function mutation. BMC Immunol (2021) 22(1):19. doi: 10.1186/s12865-021-00411-1
124. Wegehaupt O, Muckenhaupt T, Johnson MB, Schwab KO, Speckmann C. Ruxolitinib controls lymphoproliferation and diabetes in a STAT3-GOF patient. J Clin Immunol (2020) 40(8):1207–10. doi: 10.1007/s10875-020-00864-w
125. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol (2018) 15(4):234–48. doi: 10.1038/nrclinonc.2018.8
126. Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich P-S, et al. Clinical aspects of STAT3 gain-of-function germline mutations: A systematic review. J Allergy Clin Immunol Pract (2019) 7(6):1958–1969.e9. doi: 10.1016/j.jaip.2019.02.018
127. Puck JM, Deschênes SM, Porter JC, Dutra AS, Brown CJ, Willard HF, et al. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet (1993) 2(8):1099–104. doi: 10.1093/hmg/2.8.1099
128. Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell (1993) 73(1):147–57. doi: 10.1016/0092-8674(93)90167-O
129. Hernandez JD, Hsieh EWYY. A great disturbance in the force: IL-2 receptor defects disrupt immune homeostasis. Curr Opin Pediatr (2022) 34(6):580–8. doi: 10.1097/MOP.0000000000001181
130. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi: 10.1182/blood-2006-02-004747
131. Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol (2006) 6(8):595–601. doi: 10.1038/nri1901
132. Vignoli M, Ciullini Mannurita S, Fioravanti A, Tumino M, Grassi A, Guariso G, et al. CD25 deficiency: A new conformational mutation prevents the receptor expression on cell surface. Clin Immunol (2019) 201(February):15–9. doi: 10.1016/j.clim.2019.02.003
133. Lai N, Liu L, Lin L, Cui C, Wang Y, Min Q, et al. Effective and safe treatment of a novel IL2RA deficiency with rapamycin. J Allergy Clin Immunol Pract (2020) 8(3):1132–1135.e4. doi: 10.1016/j.jaip.2019.09.027
134. Gars E, Purington N, Scott G, Chisholm K, Gratzinger D, Martin BA, et al. Bone marrow histomorphological criteria can accurately diagnose hemophagocytic lymphohistiocytosis. Haematologica (2018) 103(10):1635–41. doi: 10.3324/haematol.2017.186627
135. Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
136. Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne AC, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract (2018) 6(5):1508–17. doi: 10.1016/j.jaip.2018.05.031
137. Moshous D, Briand C, Castelle M, Dupic L, Morelle G, Abou Chahla W, et al. Alemtuzumab as first line treatment in children with familial lymphohistiocytosis. Blood (2019) 134(Supplement_1):80. doi: 10.1182/blood-2019-124477
138. Marsh RA, Kim M-O, Liu C, Bellman D, Hart L, Grimley M, et al. An intermediate alemtuzumab schedule reduces the incidence of mixed chimerism following reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant. (2013) 19(11):1625–31. doi: 10.1016/j.bbmt.2013.09.001
139. Broglie L, Pommert L, Rao S, Thakar M, Phelan R, Margolis D, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv (2017) 1(19):1533–6. doi: 10.1182/bloodadvances.2017007526
140. Weitzman S. Approach to hemophagocytic syndromes. Hematol Am Soc Hematol Educ Progr. (2011) 2011:178–83. doi: 10.1182/asheducation-2011.1.178
141. Bergsten E, Horne A, Hed Myrberg I, Aricó M, Astigarraga I, Ishii E, et al. Stem cell transplantation for children with hemophagocytic lymphohistiocytosis: results from the HLH-2004 study. Blood Adv (2020) 4(15):3754–66. doi: 10.1182/bloodadvances.2020002101
142. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet (2000) 25(2):173–6. doi: 10.1038/76024
143. Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol (1995) 154(11):6122–31. doi: 10.4049/jimmunol.154.11.6122
144. Jessen B, Bode SFN, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood (2013) 121(15):2943–51. doi: 10.1182/blood-2012-10-463166
145. Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood (2005) 106(1):40–2. doi: 10.1182/blood-2005-01-0319
146. Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A. (1998) 95(23):13765–70. doi: 10.1073/pnas.95.23.13765
147. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2022) 40(1):24–64. doi: 10.1007/s10875-022-01289-3
148. Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. (2012) 122(3):821–32. doi: 10.1172/JCI61014
149. Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest. (2012) 122(3):814–20. doi: 10.1172/JCI60224
150. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. J Clin Immunol (2015) 35(4):331–8. doi: 10.1007/s10875-015-0141-9
151. Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol (2015) 39:115–23. doi: 10.1016/j.semcdb.2015.01.015
152. Mudde ACA, Booth C, Marsh RA. Evolution of our understanding of XIAP deficiency. Front Pediatr (2021) 9:660520. doi: 10.3389/fped.2021.660520
153. Yang L, Booth C, Speckmann C, Seidel MG, Worth AJJ, Kindle G, et al. Phenotype, genotype, treatment, and survival outcomes in patients with X-linked inhibitor of apoptosis deficiency. J Allergy Clin Immunol (2022) 150(2):456–66. doi: 10.1016/j.jaci.2021.10.037
154. Ono S, Okano T, Hoshino A, Yanagimachi M, Hamamoto K, Nakazawa Y, et al. Hematopoietic stem cell transplantation for XIAP deficiency in Japan. J Clin Immunol (2017) 37(1):85–91. doi: 10.1007/s10875-016-0348-4
155. Krei JM, Møller HJ, Larsen JB. The role of interleukin-18 in the diagnosis and monitoring of hemophagocytic lymphohistiocytosis/macrophage activation syndrome - a systematic review. Clin Exp Immunol (2021) 203(2):174–82. doi: 10.1111/cei.13543
156. Chiang SCC, Bleesing JJ, Marsh RA. Current flow cytometric assays for the screening and diagnosis of primary HLH. Front Immunol (2019) 10(July):1740. doi: 10.3389/fimmu.2019.01740
157. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology (2014) 147(5):990–1007. doi: 10.1053/j.gastro.2014.07.023
158. Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361(21):2033–45. doi: 10.1056/NEJMoa0907206
159. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
160. Murugan D, Albert MH, Langemeier J, Bohne J, Puchalka J, Järvinen PM, et al. Very early onset inflammatory bowel disease associated with aberrant trafficking of IL-10R1 and cure by T cell replete haploidentical bone marrow transplantation. J Clin Immunol (2014) 34(3):331–9. doi: 10.1007/s10875-014-9992-8
161. Meena S, Varla H, Swaminathan VV, Chandar R, Jayakumar I, Ramakrishnan B, et al. Hematopoietic stem cell Transplantation in Children with very Early Onset Inflammatory Bowel Disease Secondary to Monogenic Disorders of immune-dysregulation. Indian J Hematol Blood Transfus an Off J Indian Soc Hematol Blood Transfus. (2023) 39(2):183–90. doi: 10.1007/s12288-022-01586-2
162. Lien R, Lin YF, Lai MW, Weng HY, Wu RC, Jaing TH, et al. novel Mutations of the Tetratricopeptide Repeat Domain 7A gene and Phenotype/genotype comparison. Front Immunol (2017) 8(SEP):1–10. doi: 10.3389/fimmu.2017.01066
163. Fabre A, Bourgeois P, Coste M-E, Roman C, Barlogis V, Badens C. Management of syndromic diarrhea/tricho-hepato-enteric syndrome: A review of the literature. Intractable rare Dis Res (2017) 6(3):152–7. doi: 10.5582/irdr.2017.01040
164. Cuchet-Lourenço D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science (2018) 361(6404):810–3. doi: 10.1126/science.aar2641
165. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi: 10.1146/annurev-immunol-032712-095946
166. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342(6160):866–71. doi: 10.1126/science.1243292
167. Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, Jadidi-Niaragh F, et al. Clinical, immunological, and genetic features in patients with activated PI3Kδ Syndrome (APDS): a systematic review. Clin Rev Allergy Immunol (2020) 59(3):323–33. doi: 10.1007/s12016-019-08738-9
168. Maccari ME, Wolkewitz M, Schwab C, Lorenzini T, Leiding JW, Aladjdi N, et al. Activated phosphoinositide 3-kinase δ syndrome: Update from the ESID Registry and comparison with other autoimmune-lymphoproliferative inborn errors of immunity. J Allergy Clin Immunol (2023) 152(4):984–96. doi: 10.1016/j.jaci.2023.06.015
169. Tessarin G, Rossi S, Baronio M, Gazzurelli L, Colpani M, Benvenuto A, et al. Activated phosphoinositide 3-kinase delta syndrome 1: clinical and immunological data from an Italian cohort of patients. J Clin Med (2020) 9(10):3335. doi: 10.3390/jcm9103335
170. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi: 10.1038/ni.2771
171. Duggan S, Al-Salama ZT. Leniolisib: first approval. Drugs (2023) 83(10):943–8. doi: 10.1007/s40265-023-01895-4
173. Martinez-Valle F, Balada E, Ordi-Ros J, Bujan-Rivas S, Sellas-Fernandez A, Vilardell-Tarres M. DNase1 activity in systemic lupus erythematosus patients with and without nephropathy. Rheumatol Int (2010) 30(12):1601–4. doi: 10.1007/s00296-009-1199-6
174. Gatselis NK, Vakrakou AG, Zachou K, Androutsakos T, Azariadis K, Hatzis G, et al. Decreased serum DNase1-activity in patients with autoimmune liver diseases. Autoimmunity (2017) 50(2):125–32. doi: 10.1080/08916934.2017.1279610
175. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet (2001) 28(4):313–4. doi: 10.1038/91070
176. Al-Mayouf SM, Sunker A, Abdwani R, Al Abrawi S, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet (2011) 43(12):1186–8. doi: 10.1038/ng.975
177. Englert H, Göbel J, Khong D, Omidi M, Wolska N, Konrath S, et al. Targeting NETs using dual-active DNase1 variants. Front Immunol (2023) 14:1181761. doi: 10.3389/fimmu.2023.1181761
178. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet (2016) 48(1):67–73. doi: 10.1038/ng.3459
179. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis (2018) 77(5):728–35. doi: 10.1136/annrheumdis-2017-212403
180. Henderson C, Goldbach-Mansky R. Monogenic IL-1 mediated autoinflammatory and immunodeficiency syndromes: Finding the right balance in response to danger signals. Clin Immunol (2010) 135(2):210–22. doi: 10.1016/j.clim.2010.02.013
181. Picard C, Puel A, Bonnet M, Ku C-L, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science (2003) 299(5615):2076–9. doi: 10.1126/science.1081902
182. Bal E, Lim AC, Shen M, Douangpanya J, Madrange M, Gazah R, et al. Mutation in IL 36 RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome. Exp Dermatol (2019) 28(10):1114–7. doi: 10.1111/exd.13387
183. Staus P, Rusch S, El-Helou S, Müller G, Krausz M, Geisen U, et al. The GAIN registry — a new prospective study for patients with multi-organ autoimmunity and autoinflammation. J Clin Immunol (2023) 43(6):1289–301. doi: 10.1007/s10875-023-01472-0
184. Cole T, McKendrick F, Titman P, Cant AJ, Pearce MS, Cale CM, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: A comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol (2013) 33(1):8–13. doi: 10.1007/s10875-012-9758-0
185. Shah AJ, Sokolic R, Logan B, Yin Z, Iyengar S, Scalchunes C, et al. Quality of life of patients with Wiskott Aldrich syndrome and X-linked thrombocytopenia: a study of the primary immune deficiency consortium (PIDTC), immune deficiency foundation, and the Wiskott-Aldrich foundation. J Clin Immunol (2019) 39(8):786–94. doi: 10.1007/s10875-019-00689-2
186. Abd Hamid IJ, Slatter MA, McKendrick F, Pearce MS, Gennery AR. Long-term health outcome and quality of life post-HSCT for IL7Rα-, artemis-, RAG1- and RAG2-deficient severe combined immunodeficiency: a single center report. J Clin Immunol (2018) 38(6):727–32. doi: 10.1007/s10875-018-0540-9
187. Rider NL, Kutac C, Hajjar J, Scalchunes C, Seeborg FO, Boyle M, et al. Health-related quality of life in adult patients with common variable immunodeficiency disorders and impact of treatment. J Clin Immunol (2017) 37(5):461–75. doi: 10.1007/s10875-017-0404-8
188. Kluger N, Jokinen M, Krohn K, Ranki A. What is the burden of living with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) in 2012? A health-related quality-of-life assessment in Finnish patients. Clin Endocrinol (Oxf). (2013) 79(1):134–41. doi: 10.1111/cen.12087
Keywords: hemophagocytic lymphohistiocytosis, immunodeficiency, immune dysregulation, HSCT, targeted therapies
Citation: Tsilifis C, Slatter MA and Gennery AR (2023) Too much of a good thing: a review of primary immune regulatory disorders. Front. Immunol. 14:1279201. doi: 10.3389/fimmu.2023.1279201
Received: 17 August 2023; Accepted: 16 October 2023;
Published: 31 October 2023.
Edited by:
Francisco Javier Espinosa-Rosales, Fundación Mexicana para Niñas y Niños con Inmunodeficiencias (FUMENI), MexicoReviewed by:
Aziz Bousfiha, Hassan II University Casablanca, MorroccoCopyright © 2023 Tsilifis, Slatter and Gennery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christo Tsilifis, Yy50c2lsaWZpc0BuaHMubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.