
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 October 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1274654
This article is part of the Research TopicRegulated Cell Death Pathways in the Initiation and Reversal of Inflammation and Tissue RepairView all 8 articles
 Dan Ke1,2†
Dan Ke1,2† Zhen Zhang2,3†
Zhen Zhang2,3† Jieting Liu1,2†
Jieting Liu1,2† Peijian Chen1,2
Peijian Chen1,2 Yucen Dai1,2
Yucen Dai1,2 Xinhai Sun4
Xinhai Sun4 Yanhui Chu2*
Yanhui Chu2* Luxin Li1,2*
Luxin Li1,2*Diabetes mellitus is a metabolic disease that is characterized by chronic hyperglycemia due to a variety of etiological factors. Long-term metabolic stress induces harmful inflammation leading to chronic complications, mainly diabetic ophthalmopathy, diabetic cardiovascular complications and diabetic nephropathy. With diabetes complications being one of the leading causes of disability and death, the use of anti-inflammatories in combination therapy for diabetes is increasing. There has been increasing interest in targeting significant regulators of the inflammatory pathway, notably receptor-interacting serine/threonine-kinase-1 (RIPK1) and receptor-interacting serine/threonine-kinase-3 (RIPK3), as drug targets for managing inflammation in treating diabetes complications. In this review, we aim to provide an up-to-date summary of current research on the mechanism of action and drug development of RIPK1 and RIPK3, which are pivotal in chronic inflammation and immunity, in relation to diabetic complications which may be benefit for explicating the potential of selective RIPK1 and RIPK3 inhibitors as anti-inflammatory therapeutic agents for diabetic complications.
Diabetes mellitus (DM) is characterized by metabolic dysregulation resulting from impaired insulin secretion and/or insulin resistance (1). The consequential increased insulin resistance and disrupted glucose metabolism lead to chronic inflammation (2). Epidemiological findings imply that patients with type 2 diabetes showcase higher levels of acute phase reaction products and inflammatory mediators within their bloodstream (3). It has long been objectively recognized in clinical medicine that salicylic acid-based anti-inflammatory drugs can lower blood glucose levels (4). Basic medical research has found that inflammatory cytokines may impair insulin signaling due to “cross-talk” (5). Indeed, all the research findings till now has shown that pancreatic islets of T2DM patients exhibit detectable tissue inflammation which is accompanied by increased expression of inflammatory factors and immune cell infiltration. These are hallmark features of chronic inflammation, which leads to gradual progression of pathological fibrosis (6–9). The chronic low-grade inflammatory state is considered a crucial mechanism in the advancement of diabetic complications, including diabetic ophthalmopathy, diabetic cardiovascular complications, and diabetic nephropathy.
NF-κB pathway, which is the activation of inflammasome and cell death is thought to regulate the inflammation responsible for diabetic complications (10–12). NF-κB is a crucial moderator of the innate immune system and is instrumental in maintaining the normal physiological functions of the body. In response to inflammation, the body triggers a natural immune response, whereby immune-responsive cells release inflammatory agents whose transcription depends on NF-κB activation (13, 14). Rupture of necrotic or apoptotic cells causes the release of molecules of Damage-Associated Molecular Pattern (DAMPs). These substances are released into the interstitial space of cells or the blood circulation after rupture or necrosis of autologous cells, and are recognized as danger signals by receptors. This recognition leads to stimulation of the immune system, which results in inflammation (15, 16). Inflammasomes are crucial centers of innate immunity that facilitate the secretion of pro-inflammatory cytokines and regulate the inflammatory response via interactions with other cells (17). Both RIPK1 and RIPK3 are closely linked to these regulatory processes and are considered a highly promising target. Inhibiting their kinase activities has shown efficacy in treating several animal models of human diseases (18–21). The promising potential of RIPK1 inhibitors in treating autoimmune diseases, inflammation, acute illnesses (such as severe novel coronavirus pneumonia sepsis), and a range of other conditions has led to great anticipation for their development (22, 23).
While it is increasingly acknowledged that diabetes is an autoinflammatory condition and its complications stem from a prolonged low-grade inflammatory state, the development of drugs targeting RIPK1 and RIPK3 to alleviate diabetic complications remains inadequate. Since overactivation of RIPK1 and RIPK3 can lead to harmful inflammatory responses and tissue damage, understanding the precise mechanisms of RIPK1 and RIPK3 regulation of inflammation and the development of selective inhibitors are of utmost importance for the treatment of diabetes complications (19, 24). Therefore, this review firstly examines the primary inflammatory mechanisms of diabetic complications and highlights the intricate and crucial functions of RIPK1 and RIPK3 in these mechanisms. Subsequently, this paper provides a concise summary of the latest research on the relationship between diabetic complications and RIPK1 and RIPK3 and additionally explores potential issues and drug development targets.
Receptor-interacting protein kinases RIPK1 and RIPK3 are key protein kinases in the organism involved in a wide range of biological processes. Overexpression or aberrant function of RIPK1 and RIPK3 has been shown to correlate with the onset and progression of many diseases, ranging from inflammatory responses to neurodegenerative diseases and tumors.
Structure is the basis of a protein’s function, and different structural domains enabling different functions for RIPK1 and RIPK3. Three structural domains can be identified for RIPK1: the N-terminal kinase domain, the intermediate structural domain and the C-terminal death domain (DD) (25). The N-terminal kinase domain of RIPK1 plays a crucial role in RIPK1-dependent apoptosis and necroptosis, with its kinase activity being essential (26). Caspase-8, a crucial apoptotic protein, induces apoptosis through the cleavage of RIPK1 at D324 and the suppression of RIPK1 activation (25).
The intermediate structural domain of RIPK1 is involved in regulating NF-κB signaling (27, 28). RIPK1 can undergo ubiquitination on various residues within its structural domain, leading to the formation of complex I, which serves as a critical scaffold (29). Hai-Bing Zhang’s team discovered that the intermediate structural domain in RIPK1 holds a Lys377 polyubiquitination site, which is essential for building the NF-κB-activated IKK kinase complex (30). This complex can undergo ubiquitination by E3 ubiquitin ligases, including inhibitor of apoptosis 1 (cIAP1) or cIAP2 (31–33). The Lys377 residue facilitates transforming growth factor-β-activated kinase 1 (TAK1) activation by serving as a binding hub for downstream signals (e. g. NF-κB) through recruitment of the K63 ubiquitin-binding proteins TAK1-binding protein 2 (TAB2) or TAB3. This, in turn, promotes TAK1 activation (34, 35). Activated TAK1 mediates inhibitory phosphorylation of RIPK1 through activation of IκB kinase and MAPK. This leads to activation of the NF-κB pathway, promoting survival signaling (35, 36).
The C-terminal DD structural domain of RIPK1 heterodimerizes with the death structural domains of TNFR1, TRADD, and FADD. Additionally, it homodimerizes with itself, ultimately promoting the formation of death receptor signaling complex II. This leads to the activation of the N-terminal kinase domain and the promotion of necrotic apoptosis (29, 37, 38). DAMPs released through necrotic apoptosis establish feed-forward amplification loops via NF-κB-dependent production of inflammatory cytokines, thus further amplifying the inflammation and forming the cycle (39–41).
RIPK3 comprises an N-terminal structural domain of kinase and RHIM. The kinase activity of RIPK3 is linked with necroptotic apoptosis. Wang’s group demonstrated that RIPK3 undergoes autophosphorylation at residue S227 and binds to MLKL (42), forming a stabilizing complex that ultimately results in necrotic cell death through the creation of a cleavage pore in the plasma membrane. Another domain, the receptor-interacting protein kinase homotypic interaction motif (RHIM), is present in both RIPK1 and RIPK3 and serves a vital function. The RHIM enables interaction among RIPK1 and RIPK3 and facilitates transduction downstream signal for inflammation and cell death by allowing binding to various cellular junctions (43–45). For instance, as early as 2012 (46), research revealed that ZBP1 (Z-DNA binding protein 1; also known as DAI or DLM1) could trigger necrotic apoptosis via homotypic interaction between RHIM and RIPK3. As research progressed, the structure of ZBP1 was thoroughly detailed, revealing the presence of two RHIM structural domains; however, only one of the domains was found to be functional (47). The functional RHIM interacts with RIPK1 and RIPK3 to trigger the transduction of inflammatory signal. In this process, RIPK1 and RIPK3 act as scaffolds and are not associated with kinase activity or cell death (48). In both ZBP1 and TLR adaptors, RHIM is also the primary mode of TRIF signaling (26, 49). And following these receptors and associated adaptors, RIPK1 employs various mechanisms to regulate different modes (50), including activation of NF-κB in complex I bound to TNFR1, which is independent of kinase function, and maintaining balanced control of inappropriate activation of RIPK3 and caspase-8 (26, 50, 51). However, the full understanding of how RIPK1 balances the signaling of these various factors is still lacking. It is already known that the RHIM structural domain shared by RIPK1 and RIPK3 may serve as an integrator of the pro-death upstream signaling facilitated by TRIF and regulate the unnecessary activation of RIPK3 by ZBP1 (24, 25, 49).
The varied functions of RIPK1 and RIPK3 can be simply categorized into two roles: a scaffolding role and a kinase role. Interestingly, the scaffolding effect does not align completely with the kinase action of the process. In RIPK1, its scaffolding function controls pro-survival signaling and pro-inflammation, while the kinase function regulates cell death. RIPK3 exhibits similarity to RIPK1 in that the process of scaffolding is linked with pro-inflammatory signaling, while the state of kinase activity is linked with necrotic apoptosis. However, current research on RIPK1 inhibitors focuses primarily on its kinase activity, with even fewer investigations on RIPK3 inhibitors and drugs specifically targeting the role of the scaffold have yet to be developed. If using kinases like TAK1 and TBK1 as drug targets for the regulation of RIPK1 and RIPK3, it is important to consider their impact on RIPK1 and RIPK3. Therefore, further advancement of studies on the role of the RIPK1 and RIPK3 scaffolds is urgently needed. Coupled with the fact that the structure of RIPK lends itself to the development of pharmacologically specific small molecule inhibitors, more in-depth studies could help physicians tailor specific targeted inhibitors to the risk of progression of each patient, making precision medicine a reality.
RIPK1 and RIPK3 play a significant role in the transduction of repetitive signal within the body’s inflammatory response and are instrumental in regulating and activating multiple signaling pathways. RIPK1 and RIPK3 activate inflammation-related pathways like NF-κB, leading to the production of inflammatory factors and responses (52–54). Furthermore, both RIPK1 and RIPK3 have been linked to the activation of the NLRP3 inflammasome and are also implicated in the production of the pro-inflammatory cytokine IL-1β (55, 56). Recent studies have shown that both RIPK1 and RIPK3 play a role in the regulation of cell death pathway selection and balance. Moreover, RIPK1 has been found to activate numerous cell death pathways, like apoptosis and necroptosis, in response to external stimuli (25, 57).
NF-κB signaling pathway comprises receptor and receptor-proximal signaling interface proteins, IκB kinase complexes, IκB proteins, and NF-κB dimers (57). These proteins function as transcription factors that form dimers and regulate the expression of genes encoding acute phase response proteins, cytokines, immunomodulatory molecules, and cell adhesion molecules, among others (58–60). NF-κB is involved in various biological processes, including immune and inflammatory responses, as well as tumorigenesis, through the regulation of gene expression (61, 62). Numerous extracellular stimulatory signals, such as pro-inflammatory cytokines, physicochemical stimuli, LPS, and others, can activate the NF-κB signaling pathway (63, 64). Here we will use TNF-α, a classical pro-inflammatory cytokine, as an example.
Nutritional overload in peripheral metabolic tissues results in the production of TNF-α, which bind to TNFR1 and activate pre-assembled TNFR1 trimers, prompting the formation of complex I (65–69). The membrane complex I, also referred to as TNF receptor signaling complex (TNF-RSC), is composed of TNFR1 associated-death domain (TRADD), TNFR1 associated-factor 2 (TRAF2), RIPK1, cellular inhibitor of apoptosis proteins 1 (cIAP1), cIAP2, and linear ubiquitin chain assembly complex (LUBAC) (70). TRADD and RIPK1 are recruited to the intracellular death domain (DD) of activated TNFR1 through their respective DD structural domains to initiate formation of complex I (71, 72). In complex I, TRADD recruits the E3 ubiquitin ligase IAPs and then adds the K63 ubiquitin chain to RIPK1 (20).
RIPK1 serves as a crucial bridging protein in the classical NF-κB signaling pathway by enlisting the IKK complex, which comprises IKKα, IKKβ, and IKKγ, via its binding to IKKγ (also referred to as NEMO), an indispensable regulator of NF-κB signaling (31, 73, 74). Direct mediation of IKK complex activation occurs through an oligomerization- or ubiquitination-dependent mechanism of IKKγ (26, 75). The IKK complex, once activated, phosphorylates the IκB protein. As a result, phosphorylated IκB undergoes both ubiquitination and proteasomal degradation, thus releasing the NF-κB/Rel complexes (27, 76). The NF-κB/Rel complex is activated through post-translational modifications such as phosphorylation, acetylation, and glycosylation. Once activated, it is translocated into the nucleus and binds to target genes, thereby promoting transcription and increased expression of inflammatory mediators, including chemokines and cytokines (77–80) (Figure 1).
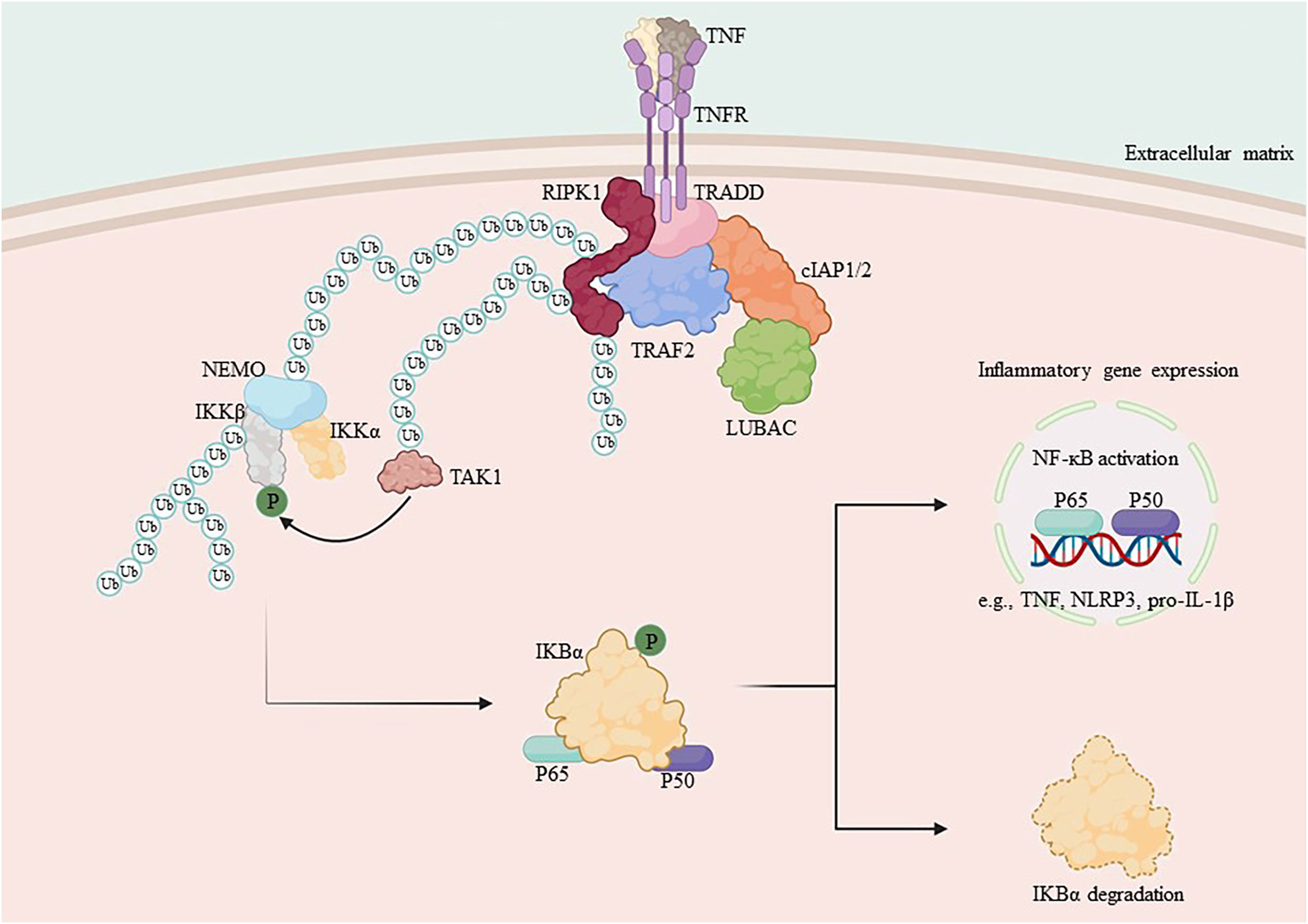
Figure 1 Mechanism of NF-κB activation by RIPK1. TNF-α binds to the membrane receptor TNFR1 and recruits TRADD, TRAF2, cIAP1/2 and RIPK1 to initiate the formation of complex I, which is composed of the ubiquitin chain assembly complex LUBAC and the E3 ubiquitin ligase. In complex I, LUBAC works with the E3 ubiquitin ligase to add the K63 ubiquitin chain to RIPK1, which binds to NEMO and recruits the IKK complex, activating the IκB protein and releasing NF-κB. Subsequent translocation of NF-κB to the nucleus promotes the transcription of pro-inflammatory genes and increases the expression of cytosolic inflammatory mediators.
Appropriate responses from NF-κB are vital for the survival of cells, thereby promoting immunity by regulating the expression of genes associated with inflammation and neutralizing the cytotoxic effects of TNF-α (51, 81). During chronic diabetes, however, persistent hyperglycemia causes elevated blood levels of advanced glycation end products (AGEs). Subsequently, AGEs induce activation of IκB kinase (IKKβ), facilitate phosphorylation- and ubiquitination-mediated proteasomal degradation of the Iκβ protein inhibitor by binding with RAGE, and consequently prompt the release of NF-κB (82–84). The activated master transcription factor NF-κB moves to the nucleus and enhances the expression of different inflammatory cytokines (IL-1β, IL-6, TNF-α), which can lead to insulin resistance. This is a critical process where the ubiquitination scaffolding of RIPK1 plays a crucial role. RIPK1-deficient mice experience systemic multiorgan inflammation and typically die soon after birth (82, 85, 86).
Early studies on RIPK3, which has the same origin with RIPK1, concentrated on inducing cell death and activating NF-κB, but the findings were perplexing, for RIPK3 hindered the activation of NF-κB by both Toll-like Receptor 3 (TLR3) and the TLR4 signaling adapters TRIF and TNFR1 (86, 87). However, other studies have shown increased NF-κB activation after RIPK3 enhancement, leading to speculation that RIPK3 may not significantly contribute to NF-κB activation (88). However, subsequent research indicated that phosphorylation and degradation of IκBα, induced by tumor necrosis factor, TLR2, and TLR4, remained unaltered in cells with double knockout of RIPK3 (88–91). The expression of tumor necrosis factor, IL-6, and IL-1β induced by endotoxin decreased in double knockout RIPK3 mice. Moreover, LPS-induced phosphorylation and degradation of IκBα remained normal in double knockout RIPK3 bone marrow-derived macrophages (BMDMs) (92). However, the RelB-p50 heterodimer’s nuclear translocation was significantly hindered, while that of other NF-κB subunits remained unaffected (93). The findings indicate that although RIPK1 promotes the early phosphorylation and degradation of IκBα, RIPK3 controls the activation of NF-κB in a cell type-dependent manner downstream of IκBκα. Thus, while RIPK1 and RIPK3 frequently work together to induce cell death, they regulate the activation of NF-κB separately through different mechanisms. Additional all-encompassing proteomic analyses investigating the molecular interactions linked to RIPK1 and RIPK3 are necessary to unveil further mechanistic details of these regulations.
Furthermore, prior research has demonstrated that signal transduction of RAGE/NF-κB additionally triggers the development of NLRP3 inflammasome. In human monocytes, lipopolysaccharide signaling through TRIF activates the NLRP3 inflammasome, which recruits RIPK1, FADD, and caspase-8 to trigger K+-non-efflux-dependent NLRP3 activation (18, 94). Notably, in mice bone marrow stromal cells, TLR4 activation alone directly stimulates NLRP3 inflammasome activity mediated by RIPK1, RIPK3 and caspase-8 and is independent of cell death (95, 96). NLRP3 inflammasome respond to cellular stress signals and participate in the maturation and secretion of the critical cytokines, IL-1β and IL-18, leading to inflammation (97–100). Given the complex regulatory mechanisms involved, it would be reasonable to take the view that inhibiting the kinases RIPK1 and RIPK3 could modulate NLRP3 inflammasome levels through the cell death pathway. Necrotic apoptosis releases DAMPs, which activate NLRP3, leading to intracellular potassium loss. This, in turn, results in NLRP3 cleavage, activating caspase-1 and IL-1β and triggers a complex network of cellular responses that cause local and systemic inflammation (9). The latest studies indicate that intravenous immunoglobulin (IVIg) is capable of regulating the expression and activation of NLRP3 inflammasomes by reducing RIPK1 levels. Therefore, the inhibition of RIPK1 can help to reduce inflammation by regulating inflammasomes. However, due to the complexity and variety of cell types found in the human body, further experiments are necessary to confirm the utility of RIPK inhibitors.
Cell death runs through the life activities of multicellular organisms, and countless diseases of living organisms arise from abnormal cell death, whether in excessive or inadequate amounts (101, 102). Regulatory cell death is a multifaceted process. With the development of the research, various modes of regulatory cell death have been identified, mainly categorized as apoptosis and necrotic apoptosis based on their effects. As previously mentioned, complex I activates NF-κB and supports cell survival. However, its short lifespan quickly transforms into the transition of the secondary cytoplasmic complex II, which regulates cell death. The different forms of complex II direct the cell towards different pathways of death (29, 37, 38).
Apoptosis is the-earliest-discovered regulated form of cell death that can be triggered by TNF-α through an exogenous pathway or by mitochondrial effectors via an endogenous pathway (103, 104). Caspases are crucial proteins that drive apoptosis. Complex IIa is the cytoplasmic complex consisting of FADD and caspase-8. The activated caspase-8 in complex IIa initiates the apoptotic program via a cascade reaction and cleaves necrotic mediators, including RIPK1 and RIPK3, among others (18, 25, 29). Caspase-8 cleavage of RIPK1’s C-terminal agonist domain at residue D324 is a crucial mechanism for inducing apoptosis and preventing both RIPK1 activation and necrotic apoptosis (105–107).
When caspase-8 activation is inhibited, RIPK1 is able to dimerize through its C-terminal DD, ultimately resulting in the activation of RIPK1 as well as the formation of complex IIb. This complex includes FADD, caspase-8, RIPK1, RIPK3, and MLKL (18, 29, 102). Subsequent ubiquitination of RIPK1 within complex IIb facilitates the activation of RIPK3 and MLKL. Additionally, RIPK3, which shares the same RHIM Domain as RIPK1, forms a necrosome with RIPK1 via a homotypic interaction, also referred to as complex IIc (18, 29). RIPK3 oligomers recruit MLKL to necrosomes, leading to its oligomerization (108). Oligomerized MLKL translocate to the plasma membrane through the Golgi-Microtubule-Actin pathway (109). and forms clusters of pores with tight junction proteins or modulates ion channel flux to induce necrotic apoptosis (Figure 2).
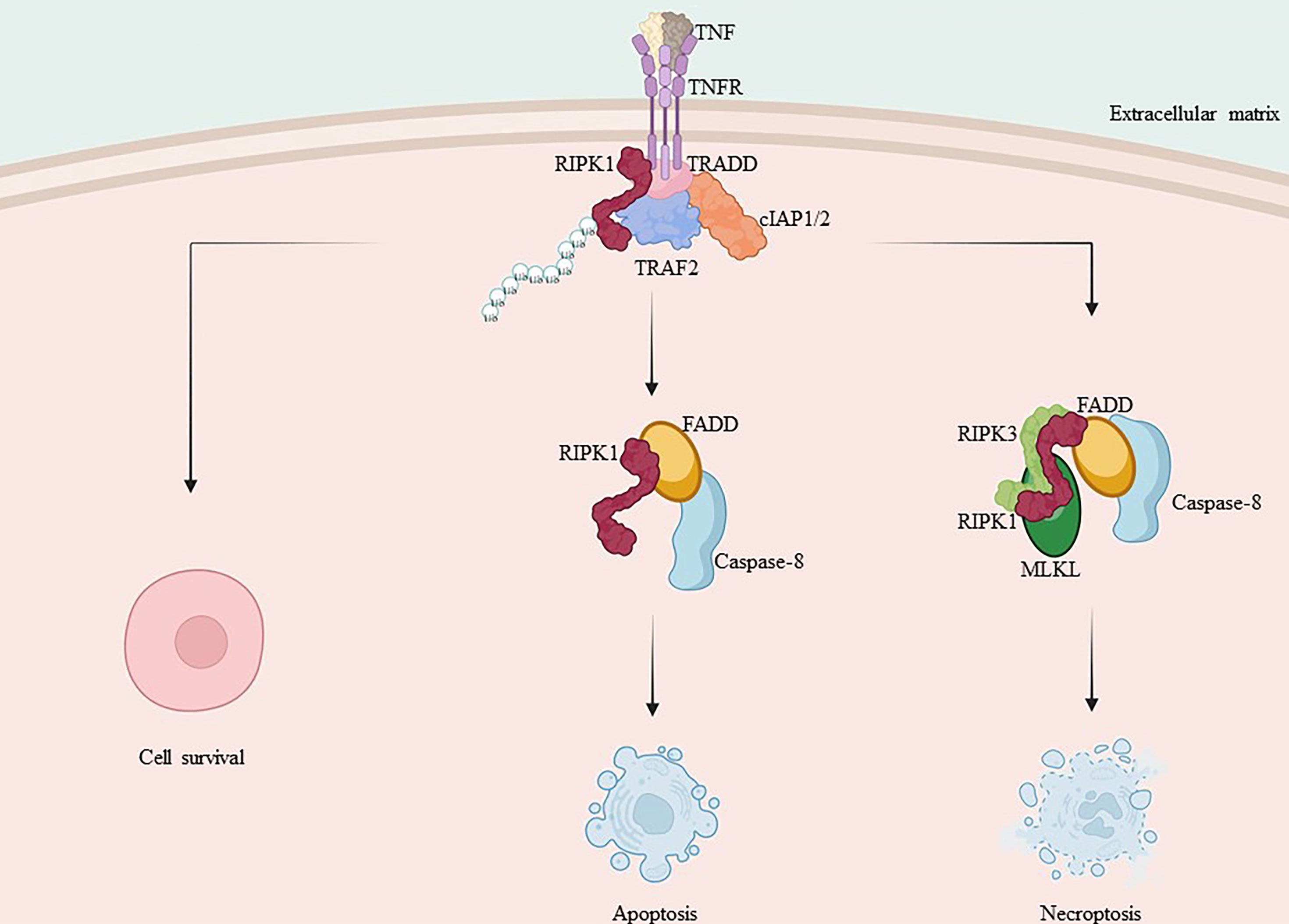
Figure 2 Mechanism of RIPK1 and RIPK3 in apoptosis and necroptosis. TNF-α binds to the membrane receptor TNFR1, recruiting TRADD, TRAF2, cIAP1/2 and RIPK1, initiating the formation of pro-survival complex I. Caspase-8 is activated and RIPK1 and TRADD in complex I bind to FADD, forming complex II a with activated caspase-8, promoting apoptosis. When caspase-8 activation is inhibited, RIPK1 and RIPK3 interact via RHIM isoforms, recruiting MLKL and forming complex II b, leading to necroptosis.
It should be noted that while RIPK1 in most cases is generally accepted as the upstream kinase of RIPK3, some research has demonstrated that RIPK3 can trigger necrotic apoptosis without reliance on RIPK1. Furthermore, this effect has been observed in cases where there is an overexpression of RIPK3 (15, 110–112).
Necrotic apoptosis is generally considered as a significant contributor to cell death and inflammation in numerous pathological contexts (112–115). For example, the important role of cell death in driving inflammation is provided by mutation of the proline-serine-threonine phosphatase-interacting protein 2 gene in mice (Pstpip2cmo), which causes inflammatory lesions in the bones and various degrees of skin and paw inflammation that closely resemble chronic recurrent multifocal osteomyelitis in humans (116). Necrotic and apoptotic cells display enlarged and swollen organelles along with early plasma membrane damage, leading eventually to rupture (115). The rapid loss of integrity of the plasma membrane leads to the release of DAMPs, which include IL-1α, HMGB1, and uric acid (117, 118). Since this type of inflammation is not associated with pathogen infection, it is known as aseptic inflammation. Like pathogen-induced inflammation, danger-associated molecular patterns (DAMPs) have the ability to activate both immune cells (such as neutrophils, macrophages, and dendritic cells) and nonimmune cells (like endothelial cells and fibroblasts) (119). These activated cells release numerous cytokines and chemokines, resulting in the recruitment of inflammatory cells and the activation of an adaptive immune response (120).
Although we have separately discussed regulatory cell death and the NF-κB pathway, but in fact these two processes they often work together. Infection, injury, and stress can lead to cytokine production to prime response by directly inducing regulatory cell death and triggering immune cell activation, leading to cytokine production (121). Dead cells initiate adaptive immunity by providing antigenic and inflammatory stimuli to dendritic cells (DCs). The DCs then activate CD8 T cells through a process called antigenic cross-stimulation (121).
However, Malek’s team discovered that the release of inflammatory mediators, including DAMPs, from dead cells was inadequate for initiating CD8 T cell crossover, and that successful crossover initiation required RIPK1 signaling and NF-κB to induce transcription within the dead cells (122). To orchestrate the adaptive immune response, inflammatory and cell death signaling pathways work together (123, 124). Receptors that elicit necrotic apoptosis also effectively induce NF-κB (51, 125), The effector molecule RIPK1 has been identified as a crucial mediator in regulating cellular activity and inflammation through the formation of common modules, complex I and complex II.
Overall, complex I promotes survival, complex IIa triggers apoptosis, and complex IIb induces necrosis. The type of complex II (either a or b) and caspase-8 status (either activated or inactivated) determine the mode of cell death, which can either be apoptosis or necrotic apoptosis. As a shared constituent of individual complexes, RIPK1 integrates different signals from external sources, including cytokines, growth factors, and pathogens, and regulates cellular behavior. Inflammatory cytokine production involves signaling through the TNF receptor (TNFR) superfamily and innate immune signal transduction through pattern recognition receptors (PRRs). Depending on the stimulus and environment, IAPs may ubiquitinate RIPK1, leading to inflammatory signal transduction via NF-κB, inhibiting RIPK3 necrosis-inducing complex formation, or inhibiting RIPK1/inflammatory pathways triggered by RIPK3 complex activation (57, 126, 127). Simultaneously activated immune cells produce cytokines that stimulate inflammation by activating pro-inflammatory genes. Additionally, they initiate regulated cell death, which closes a loop and amplifies the inflammatory response (Figure 3).
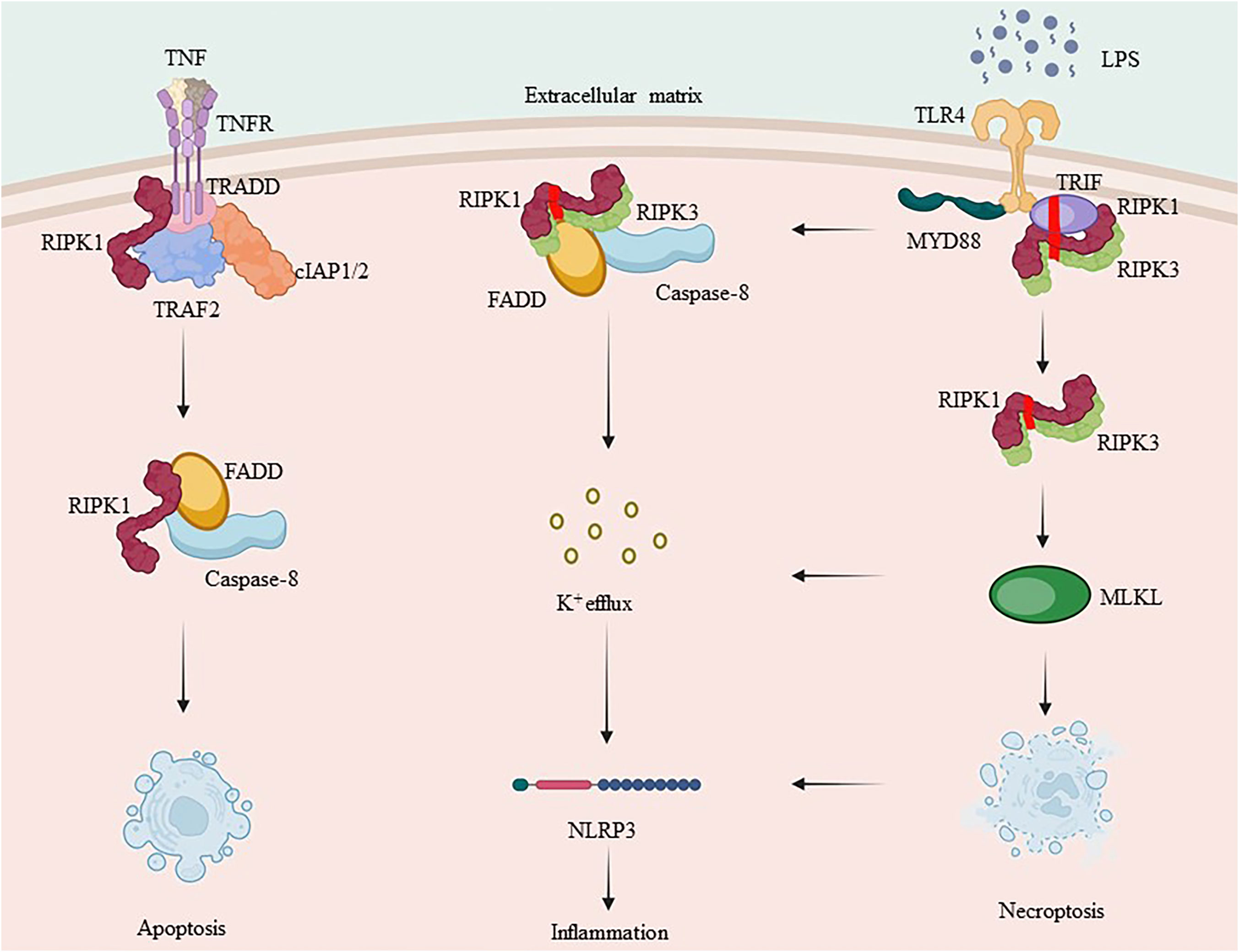
Figure 3 RIPK1 and RIPK3’s complex crosstalk in inflammation. TNF-α binds to the membrane receptor TNFR1 and recruits TRADD, TRAF2, cIAP1/2 and RIPK1 to initiate complex I formation. RIPK1, which is involved in complex I formation, is a key protein in both complex IIa and complex IIb. Ubiquitination of RIPK1 plays an essential role in NF-κB signaling. Caspase-8, when activated, forms complex IIa with RIPK1, leading to apoptosis, whereas inhibition of caspase-8 promotes necrotic apoptosis by forming complex IIb. RIPK3 can also independently induce the formation of inflammasome.
As far back as 1876, researchers discovered that anti-inflammatory treatments provided benefits for diabetes, including the improvement of blood sugar in diabetic patients with the use of sodium salicylate (128). More than a century later, Shoelson and colleagues demonstrated that inhibition of the NF-κB pathway mediates this antidiabetic effect (129). NF-κB functions as a primary transcription factor in the upregulation of RAGE, inflammatory cytokines, and associated insulin resistance genes, and inhibition of NF-κB may be an effective treatment for diabetic complications (82). Blocking pro-inflammatory cytokines has been employed to treat other cytokine storm-related diseases, though with varied success. This underscores the limited comprehension of the underlying mechanisms (130). Currently, the primary objective of inflammation-targeted therapy against NF-κB is to hinder its activity. This can be achieved by inhibiting the phosphorylation of the IκBα protein to prevent its ubiquitination and degradation, or by hindering the DNA-binding activity of NF-κB (130). Similar to the mixed success of drugs blocking proinflammatory cytokines like TNF-α (131). Considering that NF-κB plays a crucial role in mediating normal immune response, inhibiting its function comes with the risk of inhibiting immune response while also reducing inflammation, how to balance this relationship is a major challenge in drug development. Thus, targeting the development of specific small molecules meticulously is necessary to ensure the vision of effective therapy.
Recent researches indicate that the upstream activating proteins of NF-κB, namely RIPK1 and RIPK3, have significant roles in immunity and inflammation. RIPK inhibitors have exhibited anti-inflammatory potential by obstructing inflammatory signal transduction. Developing highly selective inhibitors that target RIPK1 may offer innovative solutions for treating a broad range of diseases, including neurological disorders, inflammatory diseases, tumors, and sepsis (132–136). Therefore, a combination of inhibitors targeting RIPK1 and RIPK3 and hypoglycemic drugs may increase effectiveness in treating diabetes mellitus (137–139).
It has been proved in experiments that Nec-1, Nec-1s, and GSK872 are inhibitors that target RIPK1 and RIPK3 for treating inflammatory diseases, such as inflammatory response syndrome, diabetic ophthalmopathy, atherosclerosis, pancreatitis, nonalcoholic fatty liver disease, and ischemia-reperfusion injury of the brain, heart, and kidneys. These essential proof-of-concept studies have steered the development of RIPKs in pharmaceutical companies towards antagonists for clinical trials. Harris et al. disclosed in early 2016 that a superb benzoxazinone-like lead compound, which inhibits RIPK1, was discovered in a DNA-encoded compound library (GSK481) (140). After one year, they published another article on the optimization of GSK481 for acquiring a clinical candidate compound (GSK2982772) and presented its outstanding features (141). GSK2982772 specifically binds to RIPK1 and efficiently blocks TNF-dependent cellular pathways, including NF-κB and necrotic apoptosis-inducing inflammatory pathways. As a new anti-inflammatory compound, it reduces the spontaneous production of cytokines in ulcerative colitis tissues. Due to its favorable physicochemical properties and ADMET, it is anticipated to be a low-dose oral drug. The compound is undergoing Phase II clinical trials for the treatment of plaque psoriasis, rheumatoid arthritis, and ulcerative colitis. Phase I clinical trials for inflammatory bowel disease have been completed. Pharmaceutical companies, including GlaxoSmithKline, Sanofi, and Denali, are currently prioritizing the development of drugs that inhibit RIPK1. Table 1 presents the pertinent developments in this area.

Table 1 Ongoing and completed clinical trials for RIPK inhibitors.
In conclusion, NF-κB and its associated kinases RIPK1 and RIPK3 hold potential as specific targets for chronic immune-related illnesses. However, due to the multifaceted role of NF-κB, targeting it may result in unintended repercussions, ultimately leading to a high incidence of side effects. Instead, utilizing the more nuanced RIPK1 inhibitors may be a better approach to targeting inflammation-related signals. Additionally, RIPK1, comprised of multiple structural domains (including the RHIM structural domain, DD structural domain, and kinase activity domain), coordinates cell death and innate immunity signals to orchestrate adaptive immunity. Currently, there are no effective inhibitors of RIPK3 in clinical trials. However, as noted previously, RIPK3 functions independently of RIPK1. Therefore, targeting other elements, such as specific downstream effector RIPK3, in the development of RIPK1 inhibitor medications could potentially enhance the drug’s therapeutic effectiveness in certain cases. Currently, the kinase activity of RIPK3 remains poorly understood, and development of drugs targeting RIPK1 holds more promise.
The development of RIPK1 inhibitors has primarily targeted its kinase active site, including the ATP binding site and hydrophobic pocket, with limited attention given to its scaffolding role. As a result, exploring and targeting scaffolding proteins at the intersection of cell death and host defense pathways may present novel therapeutic avenues for immunotherapy research.
The development of diabetic complications, including ophthalmopathy, cardiovascular complications, and nephropathy, is closely linked to the chronic low-grade inflammatory state caused by diabetes mellitus (142–144). Chronic hyperglycemia and insulin resistance result in the release of inflammatory catalysts and activation of immune cells, subsequently contributing to the onset and sustainability of the inflammatory response (145). Controlling the inflammatory response is crucial for preventing and treating diabetes-associated inflammatory complications.
Diabetic retinopathy (DR) is a specific microvascular complication of diabetes mellitus that occurs in about one third of patients with DM, and with the incidence of DM increasing every year, DR has become a major cause of vision loss in the general population, along with blindness in patients with DM (146).
Microglia are resident macrophages in the retina that can monitor their surroundings and maintain tissue homeostasis by dynamically changing their morphology and function in response to changing cellular microenvironment (147). In DR, microglia proliferate and migrate from the inside of retina to the outside, secreting a variety of inflammatory cytokines, leading to increased vascular permeability, and causing intraretinal fluid accumulation and disruption of the blood-retinal barrier (147). Indeed, elevated chemokines and cytokines can be detected in the serum, vitreous, retina and atrial fluid of patients with DR, and inhibition of these inflammation-associated factors may be beneficial in halting angiogenesis and neurodegeneration in DR (148).
Upon recognition of hyperglycemia-generated DAMPs, microglia recruit RIPK1 as a cell death complex (26). RIPK1 mediates both its own activation and necroptosis. TNF-α released by activated microglia further activates the TNF-α-RIPK1-NF-κB signaling pathway to promote its transcription, leading to increased expression of IL-6 and IL-8 and activation of caspase-1 (149). In diabetic mouse model, activated caspase-1 promotes neovascularization with new vessels sprouting from the optic nerve head and small veins in the mid-peripheral retina (150).These vessels eventually bleed into the vitreous cavity due to fibrotic/glial cell vitreoretinal adhesion and traction, ultimately leading to retinal detachment, making RIPK1 a very important source target (151). In addition, in the vitro studies have shown that endothelial cell apoptosis and necroptosis increase in response to inflammatory cytokine stimulation, further leading to the release of DAMPs and promoting the inflammatory cycle (18, 152, 153). Necroptosis is cell death dependent on the activity of RIPK1 and RIPK3 kinases, and inhibition of RIPK1 and RIPK3 effectively reduces necroptosis. Inhibition of RIPK1 and RIPK3 affects the developmental progression of retinopathy in animal models of diabetes. In addition, Kate E. Lawlor et al. proved that RIPK3 directly regulates NLRP3 inflammasome activation in macrophages, but the exact mechanism involved is still unclear and further studies are needed to explore the possibility of RIPK3 as a therapeutic target for DR (154).
Inhibition of pro-inflammatory molecules has been confirmed to attenuate diabetes-induced vascular and neurodegenerative disease in animal models of DR (155), and studies using anti-inflammatory agents such as salicylates or minocycline in patients with DR have further demonstrated that modulation of the inflammatory response may be beneficial in preventing long-term irreversible vascular and neuronal damage (156–158). Rosenbaum et al. demonstrated that Nec-1 treatment prevents retinal cell death in a rat model of retinal ischemia, suggesting that RIPK1 is a promising target for DR (159). However, we have discussed that inflammation responds differently in different immune cells, so more research is needed to determine the precise molecular mechanisms and functions of each type of inflammatory cell in the pathogenesis and development of DR. It may pave the way for targeted drugs.
It is widely accepted that hyperglycemia is not inherently frightening for patients with diabetes; rather, what causes concern is the multitude of morbidity and mortality complications associated with the disease. Among these complications, cardiovascular complications, to which atherosclerosis (AS) is identified as the primary pathological mechanism contributing, are the primary cause of death in diabetic patients (160, 161).
AS initiates with inflammatory effects on endothelial cells that uphold the integrity and regular function of the arterial wall. The elevated NF-κB pathway stimulates endothelial cells, which leads to endothelial cell activation by inflammatory factors, such as TNF-α. When the fine balance of endothelial cells is compromised, monocytes adhere, and foam cells accumulate lipids. Low-density lipoprotein (LDL) deposition oxidation is closely linked to AS. Oxidized-LDL deposited in the endothelium induces RIPK3 expression in macrophages, promotes the phosphorylation of both RIPK3 and MLKL, and causes necrotic apoptosis in macrophages (108, 115, 162, 163). while the death of macrophages significantly affects the formation of necrotic cores and destabilization of plaques in advanced AS lesions (164, 165).
The necrotic core of the plaque is associated not only with macrophages but also with vascular smooth muscle cells. These cells play a role in the formation of vascular plaques during atherosclerotic lesions. Adhesion of these cells to the subendothelial space is associated with various cytokines. It is well known that neutrophils in diabetic patients secrete higher levels of cytokines and growth factors, such as TNF-α, IL-1β, and IL-8, than in healthy individuals (166, 167). The harmful and inflammatory impacts of TNF-α are primarily executed by controlling the crucial transcription factor NF-κB. Endothelial cells, triggered by TNF-α, display elevated RIPK1 levels to promote inflammation that relies on NF-κB (168–170).
Inflammasome also contributes to initiating this process. For instance, TNF-α can stimulate the synthesis of inflammasome, including the pivotal NLRP3, by upregulating NLRP3 expression in human vascular smooth muscle cells (171). Additionally enhancing neutrophil migration to the inflammation site, phagocytosis, cytosolic protease release, reactive oxygen species (ROS) production, and apoptosis are associated with increased NF-κB activation and inflammatory cytokine production due to ROS overproduction. Furthermore, inhibiting NF-κB may help mitigate endocardial inflammation as well as the reorganization of cell-substrate interactions (168–170). Indeed, NF-κB has a direct impact on cell death, calcium management, cell attachment, and the production of inflammatory proteins such as COX2 and iNOS. Suppressing NF-κB prevents hypertrophy and heart failure in a mouse model of pressure overload as well as after coronary artery ligation (27, 172). Furthermore, RIPK3 has the capability to produce mature IL-1β through its activation of the NLRP3 inflammasome, subsequently leading to the generation of an inflammatory response (173).
The formation and expansion of necrotic cores within plaques are significant factors in the pathology of unstable atherosclerotic plaques (174, 175). Heng Chen et al.’s team demonstrated that Arctiin protects the rat heart by reducing necrotic apoptosis through scavenging ROS and restoring mitochondrial functions (176). Based on bioinformatics data, it is suggested that Arctiin can exert its anti-necrotic and apoptotic effects by directly targeting RIPK1 and/or MLKL. Shizuka Koshinuma et al.’s experiments perfusing guinea pig hearts demonstrate that concurrent inhibition of necroptosis and apoptosis improves cardioprotection (177). Therefore, the therapy of combining RIPK inhibitors with standard statin presents an appealing option for preventing cardiovascular complications associated with diabetes. Additionally, RIPK inhibitors show the possibility in replacing aspirin for preventing cardiac malignancies, contingent on addressing the issue of hepatoxicity.
Diabetic nephropathy (DN) is a frequent and severe complication of DM, marked by the reduction of GFR and the elevation of urinary protein excretion. As DN advances, it may result in serious complications like renal insufficiency, hypertension, and cardiovascular disease, ultimately leading to heightened all-cause mortality risk in patients. Additionally, common symptoms of DN like edema, anemia, and fatigue significantly impact patients’ daily lives. The disease’s progression may require long-term dialysis or kidney transplantation, resulting in repeated treatments, extended care, hefty financial burden, and both physical and mental damage to patients.
Chronic inflammation in the kidney is a crucial pathological mechanism that triggers DN. Although there is ongoing debate regarding whether DN can be classified as an inflammatory condition, mounting evidence suggests that persistent inflammation of both circulatory system and renal tissue is a significant physiological component of microangiopathy in DN (178). In the prediabetic stage, it has been observed that there is a significant increase in glomerular filtration rate along with increased microproteinuria and C-reactive protein levels, which indicates that the inflammation has appeared at this stage. The work by Bohle’s team and subsequent research have confirmed macrophage infiltration in the renal tissues of diabetic patients. A considerable number of recent studies further reveal the role of inflammation and immune response in the development of DN.
The inflammation of DN is inherently linked with NF-κB, a crucial and pervasive transcription factor that can be rapidly activated by numerous inflammatory mediators found in DN (179). including hyperglycemia, advanced glycation end products (AGE), mechanical stress, ROS, inflammatory cytokines, angiotensin II (Ang-II), and albuminuria (180). activated NF-κB stimulates the transcription of pro-inflammatory cytokines, chemokines, and adhesion molecules (181). The development of drugs targeting the NF-κB pathway encounters many difficulties. This is primarily due to the pathway’s complexity, which makes drugs capable of easily causing harmful effects on other cells or tissues, thereby posing greater risks and side effects. Nevertheless, RIPK inhibitors, with their larger therapeutic window and fewer side effects, exhibit potential for development. Man Guo et al. created an in vitro cellular model to investigate the intervention of high glucose levels by using normal rat renal tubular cells (NRK-52E). Additionally, they utilized an in vivo mouse model of DN and found that Nec-1 and N-acetylcysteine (NAC) could improve renal function by inhibiting RIPK1 (182). Therefore, RIPK1 and targeted antioxidants may be a potential therapeutic target for DN.
This conclusion is also corroborated by alternative studies that identify necrotic apoptosis as a strongly inflammatory type of cell death, which plays a significant role in the advancement of DN (183). Yuyin Xu found that necroptosis might significantly contribute to foot cell injury and depletion in diabetic neuropathy by activating the RIPK1-RIPK3-MLKL signaling pathway. This was observed through measuring necroptosis, apoptosis, and apoptosis in foot cells in vivo and in vitro (184). Xian Wang and colleagues’ research on Paeoniflorin (PF) demonstrates that PF directly binds and facilitates degradation of TNFR1 in podocytes (185), which, in turn, prevents injury to foot cells in DN by regulating necrotic apoptosis through the RIPK1-RIPK3 signaling pathway.
Many recent studies have reported that RIPK3 can independently promote cytokine release and the formation of inflammasome containing NLRP3 (186–188). Upon stimulation by danger-associated molecular patterns, the Pyrin structural domain of NLRP3 binds to apoptosis-associated speck-like proteins that contain cysteine-rich domain recruitment structural domains. These domains then bind to pro-caspase-1 via CARD-CARD interactions, converting pro-caspase-1 to active caspase-1. Meanwhile, pro-IL-1β is cleaved by inflammasome, resulting in the maturation of IL-1β and subsequent inflammatory responses (189, 190). Thus, it is conceivable that RIPK3 could function as an innovative target for inflammatory therapy in DN. Sadly, there are no on-going initiatives for developing inhibitors that target RIPK3 precisely, which exhibits potential as a research hotspot for future immune drug and inflammatory therapy development.
Similarly, there are reports on the role of RIPK in necrotic apoptosis, but few studies have directly explored the role of RIPK1 and RIPK3 in DN, and the relevant areas remain highly investigable. Therefore, further research is necessary to investigate the precise role of RIPK1 and RIPK3 in DN. Most known studies utilize acute injury models with increased necrotic apoptosis. For instance, the RIPK1 inhibitor Nec-1 can reduce renal ischemia and reperfusion injury as well as sepsis-related acute kidney injury (191, 192). Lack of RIPK3 can protect against renal tubular injury in acute kidney injury induced by sepsis (193, 194). Deletion of RIPK3 or MLKL can hinder oxalate crystal-induced acute kidney injury and renal damage in a mouse model of renal ischemia-reperfusion injury, among other effects (195–197). However, further experiments with chronic injury models are necessary to provide ample evidence supporting the role of RIPK inhibitors for DN. Nonetheless, given the inextricable link between necrotic apoptosis and the onset of inflammation, it is reasonable to hypothesize that inhibitors of RIPK1 and RIPK3 could aid in controlling DN and may prove more effective when used in conjunction with antihyperglycemic agents.
With the surge in the number of people suffering from the disease, diabetes mellitus has become a major challenge in the field of medicine and health. The current treatment model of type 2 diabetes mellitus is mainly based on lowering blood glucose, such as interfering with intestinal glucose absorption and increasing glucose utilization conversion in peripheral tissues, etc (198, 199). However, the incidence of chronic complications of T2DM is still high, and the effective effects of traditional drugs and the huge side effects of new drugs have also been a difficult problem for clinical application, so there is an urgent need of a new target drug based on the molecular mechanisms of diabetic inflammation in an attempt to radically reduce the harm of diabetic complications.
The pathological mechanism of diabetic complications cannot be separated from inflammation, and the extensive roles of RIPK1 and RIPK3 in NF-κB, necro-apoptotic pro-inflammatory pathways, and inflammatory crosstalk demonstrate the potential of targeting RIPK1 and RIPK3 as a therapeutic for diabetic complications. With the current enthusiasm of major pharmaceutical companies for RIPK1 inhibitors, there is an urgent need to develop inflammation-targeting agents for DM complications as soon as possible, based on existed clinical use.
However, it is important to point out that despite the anti-inflammatory effects of RIPK inhibitors, the problems of RIPK1 and RIPK3 being dependent on cell type has arisen in previous experiments (198, 200–202). Therefore, further development and refinement of the studies on human primary cells (especially human cells important for inflammation) is necessary, and we need to conduct further studies on the effects of RIPK inhibitors in general models of inflammation as well. Diabetic complications are not confined to a single site, but often involve the whole body, and more detailed studies on different cell types will further reveal the complex interactions of these small molecule antagonists in the inflammatory pathways associated with diabetic complications, including TNFR1, MAPK and NF-κB signaling. In addition, whether the role of drug development falls on the kinase binding process of RIPK1 and RIPK3 or the scaffolding structure of RIPK1 and RIPK3 is also worth discussing.
RIPKs form a convoluted and interconnected network of signaling pathways. However, RIPK1 and RIPK3 have distinct and interdependent inflammatory roles, thereby necessitating an investigation into both pathways when signaling from small molecules or small interfering RNA to identify specific scenarios where targeting either or both pathways could be advantageous. Are these two inflammatory pathways of disparate significance in distinct inflammatory scenarios? Do these pathways undergo dissimilar alterations in diverse anatomical structures and cell types, and what is the ideal dosage to attain an optimal outcome? Furthermore, which pathway plays the leading role in influencing biological outcomes? Do these pathways undergo dissimilar alterations in diverse anatomical structures and cell types, and what is the ideal dosage to attain an optimal outcome?
In conclusion, RIPK1/3 seems to be promising anti-inflammatory targets for diabetic complications. Although many RIPK1 inhibitors remain in early clinical phases, their toxicity and off-target issues impede development, likely due to drug absorption in the liver and RIPK1’s regulation of hepatocytes as a housekeeping factor for homeostasis and inflammation (203). For this reason, RIPK1 inhibitors are prone to hepatotoxicity. Learning from the experience of terminated RIPK1 inhibitors, the key challenges for future clinical development include tracking real-time activation or necrosis biomarkers of RIPK1 in vivo and developing highly specific, efficient, and safe small-molecule inhibitors of RIPK1. It is hypothesized that RIPK3 signaling inhibitors may be more suitable for specifically targeting inflammation-related signals, which requires further demonstration through relevant studies for drug development. It is hoped that these inhibitors will undergo testing in patients with T2DM complications and be implemented in clinical practices in the near future.
DK: Conceptualization, Writing – original draft. ZZ: Funding acquisition, Writing – review & editing. JL: Funding acquisition, Writing – review & editing. PC: Writing – review & editing. YD: Writing – review & editing. XS: Writing – review & editing. YC: Funding acquisition, Supervision, Writing – review & editing. LL: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Local Colleges and Universities Talent Development Funding from Heilongjiang Provincial Department of Finance (No. 2020GSP09), the Natural Science Foundation of Heilongjiang Province (No. LH2022H099), the Basic Scientific Research Project of University belongs to Heilongjiang (No. 2021-KYYWF-0519), the Heilongjiang Provincial Key Research and Development Program (No. GZ20210121), the Torch Program of Mudanjiang Medical University (No. 2022-MYHJ-003) and the Project of Young Innovative Talents Training Program of Regular Undergraduate Colleges and Universities in Heilongjiang Province (No. UNPYSCT-2020064).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet (2005) 365(9468):1415–28. doi: 10.1016/s0140-6736(05)66378-7
2. Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, et al. Insulin resistance and altered systemic glucose metabolism in mice lacking nur77. Diabetes (2009) 58(12):2788–96. doi: 10.2337/db09-0763
3. Laino AS, Woods D, Vassallo M, Qian X, Tang H, Wind-Rotolo M, et al. Serum interleukin-6 and C-reactive protein are associated with survival in melanoma patients receiving immune checkpoint inhibition. J Immunother Cancer (2020) 8(1):e000842. doi: 10.1136/jitc-2020-000842
4. Leijte GP, Kiers D, van der Heijden W, Jansen A, Gerretsen J, Boerrigter V, et al. Treatment with acetylsalicylic acid reverses endotoxin tolerance in humans in vivo: A randomized placebo-controlled study. Crit Care Med (2019) 47(4):508–16. doi: 10.1097/ccm.0000000000003630
5. Garcia-Martinez I, Alen R, Pereira L, Povo-Retana A, Astudillo AM, Hitos AB, et al. Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance. JHEP Rep (2023) 5(8):100756. doi: 10.1016/j.jhepr.2023.100756
6. Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci U.S.A. (2020) 117(6):2751–60. doi: 10.1073/pnas.1920004117
7. Ikeno Y, Ohara D, Takeuchi Y, Watanabe H, Kondoh G, Taura K, et al. Foxp3+ Regulatory T cells inhibit ccl(4)-induced liver inflammation and fibrosis by regulating tissue cellular immunity. Front Immunol (2020) 11:584048. doi: 10.3389/fimmu.2020.584048
8. Guo Y, Zhang H, Zhao Z, Luo X, Zhang M, Bu J, et al. Hyperglycemia induces meibomian gland dysfunction. Invest Ophthalmol Vis Sci (2022) 63(1):30. doi: 10.1167/iovs.63.1.30
9. Sun SC. The non-canonical Nf-Kb pathway in immunity and inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
10. Wilson RB. Hypoxia, cytokines and stromal recruitment: parallels between pathophysiology of encapsulating peritoneal sclerosis, endometriosis and peritoneal metastasis. Pleura Peritoneum (2018) 3(1):20180103. doi: 10.1515/pp-2018-0103
11. Yuan Q, Tang B, Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct Target Ther (2022) 7(1):182. doi: 10.1038/s41392-022-01036-5
12. Wang YL, Sun GY, Zhang Y, He JJ, Zheng S, Lin JN. Tormentic acid inhibits H2o2-induced oxidative stress and inflammation in rat vascular smooth muscle cells via inhibition of the Nf-Kb signaling pathway. Mol Med Rep (2016) 14(4):3559–64. doi: 10.3892/mmr.2016.5690
13. Dong T, Li M, Gao F, Wei P, Wang J. Construction and imaging of a neurovascular unit model. Neural Regener Res (2022) 17(8):1685–94. doi: 10.4103/1673-5374.332131
14. Liu X, Wang Z, Qian H, Tao W, Zhang Y, Hu C, et al. Natural medicines of targeted rheumatoid arthritis and its action mechanism. Front Immunol (2022) 13:945129. doi: 10.3389/fimmu.2022.945129
15. Zindel J, Kubes P. Damps, pamps, and lamps in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
16. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol (2023) 23(3):159–73. doi: 10.1038/s41577-022-00760-x
17. Chen C, Wang J, Chen J, Zhou L, Wang H, Chen J, et al. Morusin alleviates mycoplasma pneumonia via the inhibition of wnt/B-catenin and Nf-Kb signaling. Biosci Rep (2019) 39(6):bsr20190190. doi: 10.1042/bsr20190190
18. Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev (2017) 277(1):76–89. doi: 10.1111/imr.12541
19. Qiao S, Hong L, Zhu Y, Zha J, Wang A, Qiu J, et al. RIPK1-RIPK3 mediates myocardial fibrosis in type 2 diabetes mellitus by impairing autophagic flux of cardiac fibroblasts. Cell Death Dis (2022) 13(2):147. doi: 10.1038/s41419-022-04587-1
20. Meng X, Dang T, Chai J. From apoptosis to necroptosis: the death wishes to cancer. Cancer Control (2021) 28:10732748211066311. doi: 10.1177/10732748211066311
21. Muendlein HI, Sarhan J, Liu BC, Connolly WM, Schworer SA, Smirnova I, et al. Constitutive interferon attenuates RIPK1/3-mediated cytokine translation. Cell Rep (2020) 30(3):699–713.e4. doi: 10.1016/j.celrep.2019.12.073
22. Yang Q, Ren GL, Wei B, Jin J, Huang XR, Shao W, et al. Conditional knockout of tgf-Brii/smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics (2019) 9(26):8277–93. doi: 10.7150/thno.35686
23. Schnause AC, Komlosi K, Herr B, Neesen J, Dremsek P, Schwarz T, et al. Marfan syndrome caused by disruption of the fbn1 gene due to a reciprocal chromosome translocation. Genes (Basel) (2021) 12(11):1836. doi: 10.3390/genes12111836
24. Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, et al. Zbp1/dai ubiquitination and sensing of influenza vrnps activate programmed cell death. J Exp Med (2017) 214(8):2217–29. doi: 10.1084/jem.20170550
25. Shan B, Pan H, Najafov A, Yuan J. Necroptosis in development and diseases. Genes Dev (2018) 32(5-6):327–40. doi: 10.1101/gad.312561.118
26. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U.S.A. (2019) 116(20):9714–22. doi: 10.1073/pnas.1901179116
27. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. Nrf2 and Nf-Kb interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol (2019) 21:101059. doi: 10.1016/j.redox.2018.11.017
28. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
29. Li S, Ning LG, Lou XH, Xu GQ. Necroptosis in inflammatory bowel disease and other intestinal diseases. World J Clin cases (2018) 6(14):745–52. doi: 10.12998/wjcc.v6.i14.745
30. Zhang X, Zhang H, Xu C, Li X, Li M, Wu X, et al. Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nat Commun (2019) 10(1):4158. doi: 10.1038/s41467-019-11839-w
31. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res (2016) 26(4):457–83. doi: 10.1038/cr.2016.40
32. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer (2018) 18(2):69–88. doi: 10.1038/nrc.2017.105
33. He S, Wang X. Rip kinases as modulators of inflammation and immunity. Nat Immunol (2018) 19(9):912–22. doi: 10.1038/s41590-018-0188-x
34. Xu YR, Lei CQ. Tak1-tabs complex: A central signalosome in inflammatory responses. Front Immunol (2020) 11:608976. doi: 10.3389/fimmu.2020.608976
35. Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of tak1 and ikk. Oncogene (2007) 26(22):3214–26. doi: 10.1038/sj.onc.1210413
36. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by tak1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun (2017) 8(1):359. doi: 10.1038/s41467-017-00406-w
37. Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. Rip1/rip3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med (2019) 44(3):771–86. doi: 10.3892/ijmm.2019.4244
38. Zhang T, Ding C, Chen H, Zhao J, Chen Z, Chen B, et al. M(6)a mrna modification maintains colonic epithelial cell homeostasis via Nf-Kb-mediated antiapoptotic pathway. Sci Adv (2022) 8(12):eabl5723. doi: 10.1126/sciadv.abl5723
39. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest (2017) 127(1):55–64. doi: 10.1172/jci88881
40. Zera KA, Buckwalter MS. The local and peripheral immune responses to stroke: implications for therapeutic development. Neurotherapeutics (2020) 17(2):414–35. doi: 10.1007/s13311-020-00844-3
41. Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discovery (2021) 20(5):384–405. doi: 10.1038/s41573-021-00154-z
42. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of rip3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
43. Liu T, Zhang L, Joo D, Sun SC. Nf-Kb signaling in inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
44. Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, et al. Autophagy regulates inflammatory programmed cell death via turnover of rhim-domain proteins. Elife (2019) 8:e44452. doi: 10.7554/eLife.44452
45. Zhang H, Wu X, Li X, Li M, Li F, Wang L, et al. Crucial roles of the rip homotypic interaction motifs of RIPK3 in RIPK1-dependent cell death and lymphoproliferative disease. Cell Rep (2020) 31(7):107650. doi: 10.1016/j.celrep.2020.107650
46. Upton JW, Kaiser WJ, Mocarski ES. Dai/zbp1/dlm-1 complexes with rip3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vira. Cell Host Microbe (2012) 11(3):290–7. doi: 10.1016/j.chom.2012.01.016
47. Karki R, Kanneganti TD. Adar1 and zbp1 in innate immunity, cell death, and disease. Trends Immunol (2023) 44(3):201–16. doi: 10.1016/j.it.2023.01.001
48. Muendlein HI, Connolly WM, Magri Z, Smirnova I, Ilyukha V, Gautam A, et al. Zbp1 promotes lps-induced cell death and il-1β Release via rhim-mediated interactions with RIPK1. Nat Commun (2021) 12(1):86. doi: 10.1038/s41467-020-20357-z
49. Kuriakose T, Kanneganti TD. Zbp1: innate sensor regulating cell death and inflammation. Trends Immunol (2018) 39(2):123–34. doi: 10.1016/j.it.2017.11.002
50. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
51. Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol (2015) 33:79–106. doi: 10.1146/annurev-immunol-032414-112248
52. Wong J, Garcia-Carbonell R, Zelic M, Ho SB, Boland BS, Yao SJ, et al. RIPK1 mediates tnf-induced intestinal crypt apoptosis during chronic nf-Kb activation. Cell Mol Gastroenterol Hepatol (2020) 9(2):295–312. doi: 10.1016/j.jcmgh.2019.10.002
53. Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, et al. Tnf can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis (2013) 4(1):e465. doi: 10.1038/cddis.2012.201
54. Huang X, Tan S, Li Y, Cao S, Li X, Pan H, et al. Caspase inhibition prolongs inflammation by promoting a signaling complex with activated RIPK1. J Cell Biol (2021) 220(6):e202007127. doi: 10.1083/jcb.202007127
55. Speir M, Lawlor KE. Rip-roaring inflammation: RIPK1 and RIPK3 driven nlrp3 inflammasome activation and autoinflammatory disease. Semin Cell Dev Biol (2021) 109:114–24. doi: 10.1016/j.semcdb.2020.07.011
56. Wu YH, Mo ST, Chen IT, Hsieh FY, Hsieh SL, Zhang J, et al. Caspase-8 inactivation drives autophagy-dependent inflammasome activation in myeloid cells. Sci Adv (2022) 8(45):eabn9912. doi: 10.1126/sciadv.abn9912
57. Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of tak1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med (2020) 217(3):jem.20191644. doi: 10.1084/jem.20191644
58. Hettmann T, DiDonato J, Karin M, Leiden JM. An essential role for nuclear factor kappab in promoting double positive thymocyte apoptosis. J Exp Med (1999) 189(1):145–58. doi: 10.1084/jem.189.1.145
59. Yurtseven E, Ural D, Baysal K, Tokgözoğlu L. An update on the role of pcsk9 in atherosclerosis. J Atheroscler Thromb (2020) 27(9):909–18. doi: 10.5551/jat.55400
60. Tantry US, Schror K, Navarese EP, Jeong YH, Kubica J, Bliden KP, et al. Aspirin as an adjunctive pharmacologic therapy option for covid-19: anti-inflammatory, antithrombotic, and antiviral effects all in one agent. J Exp Pharmacol (2021) 13:957–70. doi: 10.2147/jep.S330776
61. Li WL, Yu SP, Chen D, Yu SS, Jiang YJ, Genetta T, et al. The regulatory role of nf-Kb in autophagy-like cell death after focal cerebral ischemia in mice. Neuroscience (2013) 244:16–30. doi: 10.1016/j.neuroscience.2013.03.045
62. Liu Z, Huo JH, Dong WT, Sun GD, Li FJ, Zhang YN, et al. A study based on metabolomics, network pharmacology, and experimental verification to explore the mechanism of qinbaiqingfei concentrated pills in the treatment of mycoplasma pneumonia. Front Pharmacol (2021) 12:761883. doi: 10.3389/fphar.2021.761883
63. Kaltschmidt C, Greiner JFW, Kaltschmidt B. The transcription factor nf-Kb in stem cells and development. Cells (2021) 10(8):2042. doi: 10.3390/cells10082042
64. Liang Y, Feng G, Wu L, Zhong S, Gao X, Tong Y, et al. Caffeic acid phenethyl ester suppressed growth and metastasis of nasopharyngeal carcinoma cells by inactivating the nf-Kb pathway. Drug Des Devel Ther (2019) 13:1335–45. doi: 10.2147/dddt.S199182
65. Seo HY, Kim MK, Lee SH, Hwang JS, Park KG, Jang BK. Kahweol ameliorates the liver inflammation through the inhibition of nf-Kb and stat3 activation in primary kupffer cells and primary hepatocytes. Nutrients (2018) 10(7):863. doi: 10.3390/nu10070863
66. Ren B, Tan L, Song Y, Li D, Xue B, Lai X, et al. Cerebral small vessel disease: neuroimaging features, biochemical markers, influencing factors, pathological mechanism and treatment. Front Neurol (2022) 13:843953. doi: 10.3389/fneur.2022.843953
67. Li N, Hua J. Immune cells in liver regeneration. Oncotarget (2017) 8(2):3628–39. doi: 10.18632/oncotarget.12275
68. Kong YH, Xu SP. Salidroside prevents skin carcinogenesis induced by dmba/tpa in a mouse model through suppression of inflammation and promotion of apoptosis. Oncol Rep (2018) 39(6):2513–26. doi: 10.3892/or.2018.6381
69. He M, Hu C, Chen M, Gao Q, Li L, Tian W. Effects of gentiopicroside on activation of nlrp3 inflammasome in acute gouty arthritis mice induced by msu. J Nat Med (2022) 76(1):178–87. doi: 10.1007/s11418-021-01571-5
70. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery (2016) 15(5):348–66. doi: 10.1038/nrd.2015.6
71. Rodríguez-Gómez JA, Kavanagh E, Engskog-Vlachos P, Engskog MKR, Herrera AJ, Espinosa-Oliva AM, et al. Microglia: agents of the cns pro-inflammatory response. Cells (2020) 9(7):1717. doi: 10.3390/cells9071717
72. Han JH, Park J, Kang TB, Lee KH. Regulation of caspase-8 activity at the crossroads of pro-inflammation and anti-inflammation. Int J Mol Sci (2021) 22(7):3318. doi: 10.3390/ijms22073318
73. Cho JA, Park E. Curcumin utilizes the anti-inflammatory response pathway to protect the intestine against bacterial invasion. Nutr Res Pract (2015) 9(2):117–22. doi: 10.4162/nrp.2015.9.2.117
74. Liu M, Xiao CQ, Sun MW, Tan MJ, Hu LH, Yu Q. Xanthatin inhibits stat3 and nf-Kb signalling by covalently binding to jak and Ikk kinases. J Cell Mol Med (2019) 23(6):4301–12. doi: 10.1111/jcmm.14322
75. Xie F, Xu L, Zhu H, Li Y, Nong L, Chen Y, et al. Serum metabolomics based on gc-ms reveals the antipyretic mechanism of ellagic acid in a rat model. Metabolites (2022) 12(6):479. doi: 10.3390/metabo12060479
76. Gao F, Mu X, Wu H, Chen L, Liu J, Zhao Y. Calreticulin (Calr)-induced activation of Nf-ĸb signaling pathway boosts lung cancer cell proliferation. Bioengineered (2022) 13(3):6856–65. doi: 10.1080/21655979.2022.2040874
77. Truong VL, Jeong WS. Cellular defensive mechanisms of tea polyphenols: structure-activity relationship. Int J Mol Sci (2021) 22(17):9109. doi: 10.3390/ijms22179109
78. Yang RQ, Guo PF, Ma Z, Chang C, Meng QN, Gao Y, et al. Effects of simvastatin on inos and caspase−3 levels and oxidative stress following smoke inhalation injury. Mol Med Rep (2020) 22(4):3405–17. doi: 10.3892/mmr.2020.11413
79. Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol (2018) 15(2):111–28. doi: 10.1038/nrgastro.2017.119
80. Jayasinghe AMK, Han EJ, Kirindage K, Fernando IPS, Kim EA, Kim J, et al. 3-bromo-4,5-dihydroxybenzaldehyde isolated from polysiphonia morrowii suppresses tnf-A/ifn-Γ-stimulated inflammation and deterioration of skin barrier in hacat keratinocytes. Mar Drugs (2022) 20(9):563. doi: 10.3390/md20090563
81. Yuan Y, Liu Q, Zhao F, Cao J, Shen X, Li C. Holothuria leucospilota polysaccharides ameliorate hyperlipidemia in high-fat diet-induced rats via short-chain fatty acids production and lipid metabolism regulation. Int J Mol Sci (2019) 20(19):4738. doi: 10.3390/ijms20194738
82. Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules (2022) 12(4):542. doi: 10.3390/biom12040542
83. Van Quickelberghe E, De Sutter D, van Loo G, Eyckerman S, Gevaert K. A protein-protein interaction map of the tnf-induced Nf-Kb signal transduction pathway. Sci Data (2018) 5:180289. doi: 10.1038/sdata.2018.289
84. Mazhar M, Yang G, Mao L, Liang P, Tan R, Wang L, et al. Zhilong huoxue tongyu capsules ameliorate early brain inflammatory injury induced by intracerebral hemorrhage via inhibition of canonical Nfкβ Signalling pathway. Front Pharmacol (2022) 13:850060. doi: 10.3389/fphar.2022.850060
85. Zhang K, Zhang X, Lv A, Fan S, Zhang J. Saccharomyces boulardii modulates necrotizing enterocolitis in neonatal mice by regulating the sirtuin 1/nf−Kb pathway and the intestinal microbiota. Mol Med Rep (2020) 22(2):671–80. doi: 10.3892/mmr.2020.11138
86. Ye Y, Wang P, Zhou F. Mir-489-3p inhibits tlr4/Nf-Kb signaling to prevent inflammation in psoriasis. Exp Ther Med (2021) 22(1):744. doi: 10.3892/etm.2021.10176
87. Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci (2020) 41(3):209–24. doi: 10.1016/j.tips.2020.01.002
88. Wei J, Chen L, Wang D, Tang L, Xie Z, Chen W, et al. Upregulation of rip3 promotes necroptosis via a ros−Dependent Nf−Kb pathway to induce chronic inflammation in hk−2 cells. Mol Med Rep (2021) 24(5):783. doi: 10.3892/mmr.2021.12423
89. Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. Rip3, a novel apoptosis-inducing kinase. J Biol Chem (1999) 274(24):16871–5. doi: 10.1074/jbc.274.24.16871
90. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. Dai/zbp1 recruits rip1 and rip3 through rip homotypic interaction motifs to activate nf-kappab. EMBO Rep (2009) 10(8):916–22. doi: 10.1038/embor.2009.109
91. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of nf-kappa B activation via the DNA-dependent activator of ifn regulatory factors. J Immunol (2008) 181(9):6427–34. doi: 10.4049/jimmunol.181.9.6427
92. Balic JJ, Albargy H, Luu K, Kirby FJ, Jayasekara WSN, Mansell F, et al. Stat3 serine phosphorylation is required for tlr4 metabolic reprogramming and il-1β Expression. Nat Commun (2020) 11(1):3816. doi: 10.1038/s41467-020-17669-5
93. Florio TJ, Lokareddy RK, Yeggoni DP, Sankhala RS, Ott CA, Gillilan RE, et al. Differential recognition of canonical Nf-Kb dimers by importin A3. Nat Commun (2022) 13(1):1207. doi: 10.1038/s41467-022-28846-z
94. Swanson KV, Deng M, Ting JP. The nlrp3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
95. Tonnus W, Belavgeni A, Beuschlein F, Eisenhofer G, Fassnacht M, Kroiss M, et al. The role of regulated necrosis in endocrine diseases. Nat Rev Endocrinol (2021) 17(8):497–510. doi: 10.1038/s41574-021-00499-w
96. Li X, Li F, Zhang X, Zhang H, Zhao Q, Li M, et al. Caspase-8 auto-cleavage regulates programmed cell death and collaborates with RIPK3/mlkl to prevent lymphopenia. Cell Death Differ (2022) 29(8):1500–12. doi: 10.1038/s41418-022-00938-9
97. Li X, Shang X, Sun L. Tacrolimus reduces atherosclerotic plaque formation in apoe(-/-) mice by inhibiting nlrp3 inflammatory corpuscles. Exp Ther Med (2020) 19(2):1393–9. doi: 10.3892/etm.2019.8340
98. Zhang YZ, Sui XL, Xu YP, Gu FJ, Zhang AS, Chen JH. Nlrp3 inflammasome and lipid metabolism analysis based on uplc-Q-tof-ms in gouty nephropathy. Int J Mol Med (2019) 44(1):172–84. doi: 10.3892/ijmm.2019.4176
99. Sakaguchi N, Sasai M, Bando H, Lee Y, Pradipta A, Ma JS, et al. Role of gate-16 and gabarap in prevention of caspase-11-dependent excess inflammation and lethal endotoxic shock. Front Immunol (2020) 11:561948. doi: 10.3389/fimmu.2020.561948
100. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, et al. Concerted activation of the aim2 and nlrp3 inflammasomes orchestrates host protection against aspergillus infection. Cell Host Microbe (2015) 17(3):357–68. doi: 10.1016/j.chom.2015.01.006
101. Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S, et al. Ampk: potential therapeutic target for ischemic stroke. Theranostics (2018) 8(16):4535–51. doi: 10.7150/thno.25674
102. Rothlin CV, Hille TD, Ghosh S. Determining the effector response to cell death. Nat Rev Immunol (2021) 21(5):292–304. doi: 10.1038/s41577-020-00456-0
103. Fogg VC, Lanning NJ, Mackeigan JP. Mitochondria in cancer: at the crossroads of life and death. Chin J Cancer (2011) 30(8):526–39. doi: 10.5732/cjc.011.10018
104. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol (2020) 21(7):398–414. doi: 10.1038/s41580-020-0232-1
105. Nguyen LN, Kanneganti TD. Panoptosis in viral infection: the missing puzzle piece in the cell death field. J Mol Biol (2022) 434(4):167249. doi: 10.1016/j.jmb.2021.167249
106. Simpson DS, Pang J, Weir A, Kong IY, Fritsch M, Rashidi M, et al. Interferon-Γ Primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity (2022) 55(3):423–41.e9. doi: 10.1016/j.immuni.2022.01.003
107. Green DR. The coming decade of cell death research: five riddles. Cell (2019) 177(5):1094–107. doi: 10.1016/j.cell.2019.04.024
108. Koch A, Jeiler B, Roedig J, van Wijk SJL, Dolgikh N, Fulda S. Smac mimetics and trail cooperate to induce mlkl-dependent necroptosis in burkitt’s lymphoma cell lines. Neoplasia (2021) 23(5):539–50. doi: 10.1016/j.neo.2021.03.003
109. Meng Y, Horne CR, Samson AL, Dagley LF, Young SN, Sandow JJ, et al. Human RIPK3 C-lobe phosphorylation is essential for necroptotic signaling. Cell Death Dis (2022) 13(6):565. doi: 10.1038/s41419-022-05009-y
110. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
111. Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, et al. RIPK3 promotes kidney fibrosis via akt-dependent atp citrate lyase. JCI Insight (2018) 3(3):e94979. doi: 10.1172/jci.insight.94979
112. Liu C, Li P, Zheng J, Wang Y, Wu W, Liu X. Role of necroptosis in airflow limitation in chronic obstructive pulmonary disease: focus on small-airway disease and emphysema. Cell Death Discovery (2022) 8(1):363. doi: 10.1038/s41420-022-01154-7
113. Meng X, Guo R, Fan C, Li Y, Liu X, Chen X, et al. RIPK1 downregulation enhances neutrophil extracellular traps in psoriasis. Postepy Dermatol Alergol (2022) 39(1):72–80. doi: 10.5114/ada.2022.113803
114. Hu X, Xu Y, Zhang H, Li Y, Wang X, Xu C, et al. Role of necroptosis in traumatic brain and spinal cord injuries. J Adv Res (2022) 40:125–34. doi: 10.1016/j.jare.2021.12.002
115. Paudel S, Jeyaseelan S. Kill two birds with one stone: role of the ripk-3 in necroptosis and inflammasome activation. Am J Respir Cell Mol Biol (2021) 64(5):525–7. doi: 10.1165/rcmb.2021-0085ED
116. Karki R, Kanneganti TD. The ‘Cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol (2021) 42(8):681–705. doi: 10.1016/j.it.2021.06.001
117. Mahaling B, Low SWY, Beck M, Kumar D, Ahmed S, Connor TB, et al. Damage-associated molecular patterns (Damps) in retinal disorders. Int J Mol Sci (2022) 23(5):2591. doi: 10.3390/ijms23052591
118. Zhang Y, Karki R, Igwe OJ. Toll-like receptor 4 signaling: A common pathway for interactions between prooxidants and extracellular disulfide high mobility group box 1 (Hmgb1) protein-coupled activation. Biochem Pharmacol (2015) 98(1):132–43. doi: 10.1016/j.bcp.2015.08.109
119. Chen L, Huang Q, Zhao T, Sui L, Wang S, Xiao Z, et al. Nanotherapies for sepsis by regulating inflammatory signals and reactive oxygen and nitrogen species: new insight for treating covid-19. Redox Biol (2021) 45:102046. doi: 10.1016/j.redox.2021.102046
120. McCulloch L, Alfieri A, McColl BW. Experimental stroke differentially affects discrete subpopulations of splenic macrophages. Front Immunol (2018) 9:1108. doi: 10.3389/fimmu.2018.01108
121. Sastre B, García-García ML, Cañas JA, Calvo C, Rodrigo-Muñoz JM, Casas I, et al. Bronchiolitis and recurrent wheezing are distinguished by type 2 innate lymphoid cells and immune response. Pediatr Allergy Immunol (2021) 32(1):51–9. doi: 10.1111/pai.13317
122. Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, et al. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity (2016) 45(1):46–59. doi: 10.1016/j.immuni.2016.06.007
123. Gao F, Hu X, Liu W, Wu H, Mu Y, Zhao Y. Calcium-activated nucleotides 1 (Cant1)-driven nuclear factor-K-gene binding (Nf-ĸb) signaling pathway facilitates the lung cancer progression. Bioengineered (2022) 13(2):3183–93. doi: 10.1080/21655979.2021.2003131
124. Yurchenko M, Skjesol A, Ryan L, Richard GM, Kandasamy RK, Wang N, et al. Slamf1 is required for tlr4-mediated tram-trif-dependent signaling in human macrophages. J Cell Biol (2018) 217(4):1411–29. doi: 10.1083/jcb.201707027
125. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B (2022) 12(1):1–17. doi: 10.1016/j.apsb.2021.08.026
126. Liu Q, Qiu J, Liang M, Golinski J, van Leyen K, Jung JE, et al. Akt and mtor mediate programmed necrosis in neurons. Cell Death Dis (2014) 5(2):e1084. doi: 10.1038/cddis.2014.69
127. Takezaki A, Tsukumo SI, Setoguchi Y, Ledford JG, Goto H, Hosomichi K, et al. A homozygous sftpa1 mutation drives necroptosis of type ii alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J Exp Med (2019) 216(12):2724–35. doi: 10.1084/jem.20182351
128. Rumore MM, Kim KS. Potential role of salicylates in type 2 diabetes. Ann Pharmacother (2010) 44(7-8):1207–21. doi: 10.1345/aph.1M483
129. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest (2006) 116(7):1793–801. doi: 10.1172/jci29069
130. Mathes E, O’Dea EL, Hoffmann A, Ghosh G. Nf-κb dictates the degradation pathway of ikappabalpha. EMBO J (2008) 27(9):1357–67. doi: 10.1038/emboj.2008.73
131. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke (2018) 13(6):612–32. doi: 10.1177/1747493018778713
132. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol (2015) 25(6):347–53. doi: 10.1016/j.tcb.2015.01.001
133. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in cns diseases. Nat Rev Neurosci (2019) 20(1):19–33. doi: 10.1038/s41583-018-0093-1
134. Karunakaran D, Nguyen MA, Geoffrion M, Vreeken D, Lister Z, Cheng HS, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce Nf-Kb activation and atherogenesis in mice. Circulation (2021) 143(2):163–77. doi: 10.1161/circulationaha.118.038379
135. Yan L, Zhang T, Wang K, Chen Z, Yang Y, Shan B, et al. Senp1 prevents steatohepatitis by suppressing RIPK1-driven apoptosis and inflammation. Nat Commun (2022) 13(1):7153. doi: 10.1038/s41467-022-34993-0
136. Van Eeckhoutte HP, Donovan C, Kim RY, Conlon TM, Ansari M, Khan H, et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of copd. Eur Respir J (2023) 61(4):2201506. doi: 10.1183/13993003.01506-2022
137. Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, et al. Tak1 kinase switches cell fate from apoptosis to necrosis following tnf stimulation. J Cell Biol (2014) 204(4):607–23. doi: 10.1083/jcb.201305070
138. Jia Y, Wang F, Guo Q, Li M, Wang L, Zhang Z, et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via jnk1/2-ros signaling in hepatic stellate cells. Redox Biol (2018) 19:375–87. doi: 10.1016/j.redox.2018.09.007
139. Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Gonçalves A, et al. Caspase-1 engagement and tlr-induced C-flip expression suppress asc/caspase-8-dependent apoptosis by inflammasome sensors nlrp1b and nlrc4. Cell Rep (2017) 21(12):3427–44. doi: 10.1016/j.celrep.2017.11.088
140. Harris PA, King BW, Bandyopadhyay D, Berger SB, Campobasso N, Capriotti CA, et al. DNA-encoded library screening identifies benzo[B][1,4]Oxazepin-4-ones as highly potent and monoselective receptor interacting protein 1 kinase inhibitors. J Med Chem (2016) 59(5):2163–78. doi: 10.1021/acs.jmedchem.5b01898
141. Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a first-in-class receptor interacting protein 1 (Rip1) kinase specific clinical candidate (Gsk2982772) for the treatment of inflammatory diseases. J Med Chem (2017) 60(4):1247–61. doi: 10.1021/acs.jmedchem.6b01751
142. Zhou Z, Sun B, Yu D, Zhu C. Gut microbiota: an important player in type 2 diabetes mellitus. Front Cell Infect Microbiol (2022) 12:834485. doi: 10.3389/fcimb.2022.834485
143. Wu T, Qiao S, Shi C, Wang S, Ji G. Metabolomics window into diabetic complications. J Diabetes Investig (2018) 9(2):244–55. doi: 10.1111/jdi.12723
144. Swärd K, Stenkula KG, Rippe C, Alajbegovic A, Gomez MF, Albinsson S. Emerging roles of the myocardin family of proteins in lipid and glucose metabolism. J Physiol (2016) 594(17):4741–52. doi: 10.1113/jp271913
145. Koenen M, Hill MA, Cohen P, Sowers JR. Obesity, adipose tissue and vascular dysfunction. Circ Res (2021) 128(7):951–68. doi: 10.1161/circresaha.121.318093
146. Ye L, Guo H, Wang Y, Peng Y, Zhang Y, Li S, et al. Exosomal circehmt1 released from hypoxia-pretreated pericytes regulates high glucose-induced microvascular dysfunction via the nfia/nlrp3 pathway. Oxid Med Cell Longev (2021) 2021:8833098. doi: 10.1155/2021/8833098
147. Guo L, Choi S, Bikkannavar P, Cordeiro MF. Microglia: key players in retinal ageing and neurodegeneration. Front Cell Neurosci (2022) 16:804782. doi: 10.3389/fncel.2022.804782
148. Sato T, Enoki T, Karasawa Y, Someya H, Taguchi M, Harimoto K, et al. Inflammatory factors of macular atrophy in eyes with neovascular age-related macular degeneration treated with aflibercept. Front Immunol (2021) 12:738521. doi: 10.3389/fimmu.2021.738521
149. Brás JP, Bravo J, Freitas J, Barbosa MA, Santos SG, Summavielle T, et al. Tnf-alpha-induced microglia activation requires mir-342: impact on nf-kb signaling and neurotoxicity. Cell Death Dis (2020) 11(6):415. doi: 10.1038/s41419-020-2626-6
150. Mohr S, Xi X, Tang J, Kern TS. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes (2002) 51(4):1172–9. doi: 10.2337/diabetes.51.4.1172
151. Forrester JV, Kuffova L, Delibegovic M. The role of inflammation in diabetic retinopathy. Front Immunol (2020) 11:583687. doi: 10.3389/fimmu.2020.583687
152. Wafa IA, Pratama NR, Sofia NF, Anastasia ES, Konstantin T, Wijaya MA, et al. Impact of covid-19 lockdown on the metabolic control parameters in patients with diabetes mellitus: A systematic review and meta-analysis. Diabetes Metab J (2022) 46(2):260–72. doi: 10.4093/dmj.2021.0125
153. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science (2020) 369(6511):1633–7. doi: 10.1126/science.abb9818
154. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and nlrp3 inflammasome activation in the absence of mlkl. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
155. Yan C, Zhou Y, Chen Q, Luo Y, Zhang JH, Huang H, et al. Dysfunction of the neurovascular unit in diabetes-related neurodegeneration. BioMed Pharmacother (2020) 131:110656. doi: 10.1016/j.biopha.2020.110656
156. Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol (2013) 169(2):337–52. doi: 10.1111/bph.12139
157. Early Treatment Diabetic Retinopathy Study Research Group. Early treatment diabetic retinopathy study design and baseline patient characteristics. Etdrs report number 7. Ophthalmology (1991) 98(5 Suppl):741–56. doi: 10.1016/s0161-6420(13)38009-9
158. Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, Barton J, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med (2018) 379(16):1529–39. doi: 10.1056/NEJMoa1804988
159. Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res (2010) 88(7):1569–76. doi: 10.1002/jnr.22314
160. Pérez-García A, Torrecilla-Parra M, Fernández-de Frutos M, Martín-Martín Y, Pardo-Marqués V, Ramírez CM. Posttranscriptional regulation of insulin resistance: implications for metabolic diseases. Biomolecules (2022) 12(2):208. doi: 10.3390/biom12020208
161. Boarescu PM, Boarescu I, Pop RM, Roşian ŞH, Bocșan IC, Rus V, et al. Evaluation of oxidative stress biomarkers, pro-inflammatory cytokines, and histological changes in experimental hypertension, dyslipidemia, and type 1 diabetes mellitus. Int J Mol Sci (2022) 23(3):1438. doi: 10.3390/ijms23031438
162. Zhang Q, Lin W, Tian L, Di B, Yu J, Niu X, et al. Oxidized low-density lipoprotein activates extracellular signal-regulated kinase signaling to downregulate sortilin expression in liver sinusoidal endothelial cells. J Gastroenterol Hepatol (2021) 36(9):2610–8. doi: 10.1111/jgh.15486
163. Gong P, Wang P, Pi S, Guo Y, Pei S, Yang W, et al. Proanthocyanidins Protect against Cadmium-Induced Diabetic Nephropathy through P38 Mapk and Keap1/Nrf2 Signaling Pathways. Front Pharmacol (2021) 12:801048. doi: 10.3389/fphar.2021.801048
164. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The at04a vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in apoe*3leiden.Cetp mice. Eur Heart J (2017) 38(32):2499–507. doi: 10.1093/eurheartj/ehx260
165. Seijkens TTP, Poels K, Meiler S, van Tiel CM, Kusters PJH, Reiche M, et al. Deficiency of the T cell regulator casitas B-cell lymphoma-B aggravates atherosclerosis by inducing cd8+ T cell-mediated macrophage death. Eur Heart J (2019) 40(4):372–82. doi: 10.1093/eurheartj/ehy714
166. Franz S, Ertel A, Engel KM, Simon JC, Saalbach A. Overexpression of S100a9 in obesity impairs macrophage differentiation via tlr4-nfkb-signaling worsening inflammation and wound healing. Theranostics (2022) 12(4):1659–82. doi: 10.7150/thno.67174
167. Diaz-Ruiz A, Nader-Kawachi J, Calderón-Estrella F, Mata-Bermudez A, Alvarez-Mejia L, Ríos C. Dapsone, more than an effective neuro and cytoprotective drug. Curr Neuropharmacol (2022) 20(1):194–210. doi: 10.2174/1570159x19666210617143108
168. Rochette L, Ghibu S. Mechanics insights of alpha-lipoic acid against cardiovascular diseases during covid-19 infection. Int J Mol Sci (2021) 22(15):7979. doi: 10.3390/ijms22157979
169. Wang Z, Dong H, Wang J, Huang Y, Zhang X, Tang Y, et al. Pro-survival and anti-inflammatory roles of Nf-Kb C-rel in the parkinson’s disease models. Redox Biol (2020) 30:101427. doi: 10.1016/j.redox.2020.101427
170. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
171. Wang Z, Xu G, Li Z, Xiao X, Tang J, Bai Z. Nlrp3 inflammasome pharmacological inhibitors in glycyrrhiza for nlrp3-driven diseases treatment: extinguishing the fire of inflammation. J Inflammation Res (2022) 15:409–22. doi: 10.2147/jir.S344071
172. Vaiciuleviciute R, Bironaite D, Uzieliene I, Mobasheri A, Bernotiene E. Cardiovascular drugs and osteoarthritis: effects of targeting ion channels. Cells (2021) 10(10):2572. doi: 10.3390/cells10102572
173. Ji N, Qi Z, Wang Y, Yang X, Yan Z, Li M, et al. Pyroptosis: A new regulating mechanism in cardiovascular disease. J Inflammation Res (2021) 14:2647–66. doi: 10.2147/jir.S308177
174. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif (2019) 52(2):e12563. doi: 10.1111/cpr.12563
175. Mehrotra P, Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discovery (2022) 21(8):601–20. doi: 10.1038/s41573-022-00470-y
176. Chen H, Tang LJ, Tu H, Zhou YJ, Li NS, Luo XJ, et al. Arctiin protects rat heart against ischemia/reperfusion injury via a mechanism involving reduction of necroptosis. Eur J Pharmacol (2020) 875:173053. doi: 10.1016/j.ejphar.2020.173053
177. Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J Anesth (2014) 28(2):235–41. doi: 10.1007/s00540-013-1716-3
178. Samsu N. Diabetic nephropathy: challenges in pathogenesis, diagnosis, and treatment. BioMed Res Int (2021) 2021:1497449. doi: 10.1155/2021/1497449
179. Foresto-Neto O, Albino AH, Arias SCA, Faustino VD, Zambom FFF, Cenedeze MA, et al. Nf-Kb system is chronically activated and promotes glomerular injury in experimental type 1 diabetic kidney disease. Front Physiol (2020) 11:84. doi: 10.3389/fphys.2020.00084
180. Koolaji N, Shammugasamy B, Schindeler A, Dong Q, Dehghani F, Valtchev P. Citrus peel flavonoids as potential cancer prevention agents. Curr Dev Nutr (2020) 4(5):nzaa025. doi: 10.1093/cdn/nzaa025
181. Zhang H, Sun SC. Nf-Kb in inflammation and renal diseases. Cell Biosci (2015) 5:63. doi: 10.1186/s13578-015-0056-4
182. Guo M, Chen Q, Huang Y, Wu Q, Zeng Y, Tan X, et al. High glucose-induced kidney injury via activation of necroptosis in diabetic kidney disease. Oxid Med Cell Longev (2023) 2023:2713864. doi: 10.1155/2023/2713864
183. Zhang J, Liu D, Zhang M, Zhang Y. Programmed necrosis in cardiomyocytes: mitochondria, death receptors and beyond. Br J Pharmacol (2019) 176(22):4319–39. doi: 10.1111/bph.14363
184. Xu Y, Gao H, Hu Y, Fang Y, Qi C, Huang J, et al. High glucose-induced apoptosis and necroptosis in podocytes is regulated by uchl1 via RIPK1/RIPK3 pathway. Exp Cell Res (2019) 382(2):111463. doi: 10.1016/j.yexcr.2019.06.008
185. Wang X, Liu XQ, Jiang L, Huang YB, Zeng HX, Zhu QJ, et al. Paeoniflorin directly binds to tnfr1 to regulate podocyte necroptosis in diabetic kidney disease. Front Pharmacol (2022) 13:966645. doi: 10.3389/fphar.2022.966645
186. Chakraborty C, Sharma AR, Bhattacharya M, Lee SS. A detailed overview of immune escape, antibody escape, partial vaccine escape of sars-cov-2 and their emerging variants with escape mutations. Front Immunol (2022) 13:801522. doi: 10.3389/fimmu.2022.801522
187. Tian G, Shi Y, Cao X, Chen W, Gu Y, Li N, et al. Preparation of the RIPK3 polyclonal antibody and its application in immunoassays of nephropathogenic infectious bronchitis virus-infected chickens. Viruses (2022) 14(8):1747. doi: 10.3390/v14081747
188. Rajesh Y, Kanneganti TD. Innate immune cell death in neuroinflammation and alzheimer’s disease. Cells (2022) 11(12):1885. doi: 10.3390/cells11121885
189. Navarro-Pando JM, Alcocer-Gómez E, Castejón-Vega B, Navarro-Villarán E, Condés-Hervás M, Mundi-Roldan M, et al. Inhibition of the nlrp3 inflammasome prevents ovarian aging. Sci Adv (2021) 7(1):eabc7409. doi: 10.1126/sciadv.abc7409
190. Zielinski MR, Gerashchenko D, Karpova SA, Konanki V, McCarley RW, Sutterwala FS, et al. The nlrp3 inflammasome modulates sleep and nrem sleep delta power induced by spontaneous wakefulness, sleep deprivation and lipopolysaccharide. Brain Behav Immun (2017) 62:137–50. doi: 10.1016/j.bbi.2017.01.012
191. Spaide RF, Fujimoto JG, Waheed NK, Sadda SR, Staurenghi G. Optical coherence tomography angiography. Prog Retin Eye Res (2018) 64:1–55. doi: 10.1016/j.preteyeres.2017.11.003
192. Liu XQ, Jiang L, Li YY, Huang YB, Hu XR, Zhu W, et al. Wogonin protects glomerular podocytes by targeting bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta Pharmacol Sin (2022) 43(1):96–110. doi: 10.1038/s41401-021-00721-5
193. Tobeiha M, Jafari A, Fadaei S, Mirazimi SMA, Dashti F, Amiri A, et al. Evidence for the benefits of melatonin in cardiovascular disease. Front Cardiovasc Med (2022) 9:888319. doi: 10.3389/fcvm.2022.888319
194. Yu H, Liu D, Shu G, Jin F, Du Y. Recent advances in nanotherapeutics for the treatment and prevention of acute kidney injury. Asian J Pharm Sci (2021) 16(4):432–43. doi: 10.1016/j.ajps.2020.11.001
195. Lau A, Rahn JJ, Chappellaz M, Chung H, Benediktsson H, Bihan D, et al. Dipeptidase-1 governs renal inflammation during ischemia reperfusion injury. Sci Adv (2022) 8(5):eabm0142. doi: 10.1126/sciadv.abm0142
196. Gao Y, Xu W, Guo C, Huang T. Gata1 regulates the microrna−328−3p/pim1 axis via circular rna itgb1 to promote renal ischemia/reperfusion injury in hk−2 cells. Int J Mol Med (2022) 50(2):100. doi: 10.3892/ijmm.2022.5156
197. Chen Q, Yu J, Rush BM, Stocker SD, Tan RJ, Kim K. Ultrasound super-resolution imaging provides A noninvasive assessment of renal microvasculature changes during mouse acute kidney injury. Kidney Int (2020) 98(2):355–65. doi: 10.1016/j.kint.2020.02.011
198. Drucker DJ. Diabetes, obesity, metabolism, and sars-cov-2 infection: the end of the beginning. Cell Metab (2021) 33(3):479–98. doi: 10.1016/j.cmet.2021.01.016
199. Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap–bile acids in metabolic control. Nat Rev Endocrinol (2014) 10(8):488–98. doi: 10.1038/nrendo.2014.60
200. Kist M, Komuves LG, Goncharov T, Dugger DL, Yu C, Roose-Girma M, et al. Impaired RIPK1 ubiquitination sensitizes mice to tnf toxicity and inflammatory cell death. Cell Death differentiation (2021) 28(3):985–1000. doi: 10.1038/s41418-020-00629-3
201. Khan HA, Ahmad MZ, Khan JA, Arshad MI. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat Dis Int (2017) 16(3):245–56. doi: 10.1016/s1499-3872(17)60014-6
202. Orning P, Lien E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J Leukoc Biol (2021) 109(1):121–41. doi: 10.1002/jlb.3mr0420-305r
Keywords: diabetes, diabetic complications, inflammation, regulatory cell death, receptor interacting protein kinase
Citation: Ke D, Zhang Z, Liu J, Chen P, Dai Y, Sun X, Chu Y and Li L (2023) RIPK1 and RIPK3 inhibitors: potential weapons against inflammation to treat diabetic complications. Front. Immunol. 14:1274654. doi: 10.3389/fimmu.2023.1274654
Received: 08 August 2023; Accepted: 05 October 2023;
Published: 26 October 2023.
Edited by:
Rajendra Karki, St. Jude Children’s Research Hospital, United StatesReviewed by:
Rksubbarao Malireddi, St. Jude Children’s Research Hospital, United StatesCopyright © 2023 Ke, Zhang, Liu, Chen, Dai, Sun, Chu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luxin Li, bGlsdXhpbkBtZGptdS5lZHUuY24=; Yanhui Chu, eWFuaHVpX2NodUBzaW5hLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.