
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 20 November 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1273182
This article is part of the Research TopicKey Players of the Immune System in Skin Inflammation – Allergy and AutoimmunityView all 6 articles
 Zhao-Yuan Wang†
Zhao-Yuan Wang† Yu-Xin Zheng†
Yu-Xin Zheng† Fan Xu
Fan Xu Ying-Zhe Cui
Ying-Zhe Cui Xue-Yan Chen
Xue-Yan Chen Si-Qi Chen
Si-Qi Chen Bing-Xi Yan
Bing-Xi Yan Yuan Zhou
Yuan Zhou Min Zheng
Min Zheng Xiao-Yong Man*
Xiao-Yong Man*Atopic dermatitis (AD) is one of the most common inflammatory skin diseases with complex pathogenesis involving epidermal barrier dysfunction, skin microbiome abnormalities and type-2-skewed immune dysregulation. Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that plays critical roles in various biological processes. However, the role of STAT3 in epidermal keratinocytes in AD remains unclear. In this study, we generated an epidermal keratinocyte-specific Stat3-deficient mouse strain (termed Stat3 cKO mice). After topical 2,4-dinitrochlorobenzene (DNCB) treatment, Stat3 cKO mice developed worsened AD-like skin inflammation with increased Ki67+ cells, decreased filaggrin and loricrin expression, and downregulated S100A9 and LL37. The dominant microbial population in Stat3 cKO mice changed from Ralstonia to Staphylococcus. DNCB-treated Stat3 cKO mice displayed more infiltrating type-2 inflammatory cells, including mast cells, eosinophils, and CD4+T cells, accompanied by increased skin IL-4 and serum IgE levels. Moreover, thymic stromal lymphopoietin (TSLP), mainly produced by keratinocytes, was highly expressed in the ear skin of Stat3 cKO mice and chemoattracted more TSLPR+ cells. TSLP blockade significantly alleviated DNCB-induced AD-like skin inflammation in Stat3 cKO mice. Thus, epidermal keratinocyte-specific STAT3 deficiency can aggravate AD-like skin inflammation in mice, possibly through TSLP dysregulation.
Atopic dermatitis (AD) is one of the most common chronic inflammatory skin diseases (1), affecting 2.7% to 20.1% of children and 2.1% to 4.9% of adults across different countries (2, 3). AD is characterized by recurrent eczematous lesions and intense itching, and has a high heterogeneity in its natural course. The onset of AD occurs early in life in approximately 80% of cases, with the remainder developing AD during adulthood (4).
The causes of AD are complex and multifactorial, including epidermal barrier defects, skin microbiome abnormalities, and type-2-skewed immune dysregulation (5). These pathophysiological processes can promote and interact with others. Disruption of skin barrier function due to filaggrin deficiency and microbial dysbiosis promotes the release of epidermal alarmins, such as interleukin (IL)-33 and thymic stromal lymphopoietin (TSLP) (6, 7); these proinflammatory signals recruit and activate type 2 inflammatory cells (8, 9); local type 2 immune responses further impair the barrier function, thereby facilitating Staphylococcus aureus (S. aureus) colonization or infection (5).
Signal transducer and activator of transcription 3 (STAT3) is the downstream transcription factor of multiple cytokines and Janus kinases (JAKs), which is involved in the regulation of immune response, and cell growth, differentiation and apoptosis (10). Previous studies have focused on the role of STAT3 signaling in immune cells. In T cells, STAT3 acts as a critical transcription factor for Th17 cell differentiation (11). In B cells, impaired STAT3 signaling can reduce their cellular and molecular maturation, which results in the production of low-affinity IgE (12). However, given that epidermal keratinocytes are not only the first line of defense against external invaders but also essential for the initiation, progression, and persistence of AD (13), the contribution of keratinocyte STAT3 to AD remains unclear.
Therefore, in this study, we evaluated the role of STAT3 in keratinocytes by generating mice with inducible keratinocyte-specific deletion of STAT3 and treating them with 2,4-dinitrochlorobenzene (DNCB) to induce AD-like skin inflammation. Mice with keratinocyte-specific STAT3 deficiency (termed Stat3 cKO mice) developed more severe DNCB-induced AD-like skin inflammation accompanied by skin barrier impairment, Staphylococcus-dominant microbial dysbiosis, more infiltrating type-2 inflammatory cells, and increased IL-4 levels. Moreover, TSLP expression was increased in Stat3 cKO mice, which recruited more target cells to the affected skin. TSLP blockade attenuated DNCB-induced AD-like skin inflammation in Stat3 cKO mice, suggesting that keratinocyte-specific STAT3 dysfunction leads to TSLP-mediated skin inflammation in AD.
To generate keratinocyte-specific Stat3 cKO mice, Stat3f/f mice (B6.129S1-Stat3tm1Xyfu/J mice, Jackson Lab #016923) were crossed with K14-CreERT+ mice (STOCK Tg(KRT14-cre/ERT)20Efu/J mice, Jackson Lab # 005107), both of which were purchased from Jackson Laboratory. To confirm the deficiency of STAT3 in the mouse epidermis induced by intraperitoneal injection of tamoxifen, Western blotting and immunohistochemistry were performed using STAT3 and phospho-STAT3 specific antibodies. All mouse experiments were performed under a protocol approved by the Second Affiliated Hospital Zhejiang University School of Medicine Animal Care Committee.
AD-like skin lesions were induced by topical application of DNCB (Sigma-Aldrich, St. Louis, MO, USA), with minor modifications to the protocol. Briefly, the ear skin of mice was sensitized once on Day -7 by applying 20 μL of 1% DNCB dissolved in acetone: olive oil (3:1). Beginning on Day 0, mice were challenged by applying 20 μL of 0.5% DNCB to the ears every other day for up to 21 days. The thickness of ear skin was measured with a digital caliper once a week.
Ear tissues were fixed with 10% formalin and embedded in paraffin. Afterwards, paraffin-embedded sections (4 μm) were cut, deparaffined and stained with hematoxylin-eosin (H&E) or toluidine blue for light microscopic examinations.
Formalin-fixed, paraffin-embedded tissues were subjected to sectioning, deparaffinization, and rehydration. Then, heat-induced epitope retrieval was performed in a 0.01M sodium citrate buffer (pH 6.0), followed by blocking with normal goat serum (Boster, Beijing, China) for 1 hour at room temperature.
For IHC, tissue sections were incubated with p-STAT3 (Cell Signaling Technology, 9145, 1:250) and Ki67 (Abcam, ab264429, 1:500) antibodies respectively at 4°C overnight. Subsequently, HRP-labeled anti-rabbit secondary antibodies (Boster, SV0002, 1:500) were applied with DAB high-sensitivity substrate chromogen solution (Vector Laboratories, Burlingame, CA, USA).
For IF, slides were incubated with K14 (Abcam, ab181595, 1:500), CD4 (Abcam, ab183685, 1:500), CD8 (R&D, MAB116, 1:100), loricrin (Proteintech, 55439-1-AP, 1:100), filaggrin (Abcam, ab81468, 1:200), S100A9 (Abcam, ab63818, 1:250), LL37 (Abcam, ab180760, 1:250), TSLP (Abcam, ab188766, 1:250), CD68 (Santa Cruz, sc-17832, 1:100), CD11c (Abcam, ab11029, 1:200), TSLPR (BioLegend, 151802, 1:200), respectively at 4°C overnight. Then, Alexa 488 or Alexa 555 conjugated secondary antibodies (Thermo Fisher Scientific, 1:200) plus DAPI (Roche, Mannheim, Germany) were used for visualization.
To detect the total serum IgE after DNCB modeling, blood samples were collected from the retroorbital plexus on Day 21 followed by centrifugation at 3000 rpm for 10 min at 4°C. The total serum IgE was measured by using the mouse ELISA kit (Mouse IgE ELISA MAX Deluxe, BioLegend).
After separating the epidermis and dermis of mouse ear skin, the epidermis was digested with 0.25% trypsin (Thermo Fisher Scientific, USA), whereas the dermis was digested with 1 mg/ml collagenase (Sigma-Aldrich, St. Louis, MO). After neutralization with fetal bovine serum, the mixture was centrifuged at 1000 rpm for 5 min, and the cells were resuspended in PBS to obtain a single-cell suspension. After Fc receptor blocking with Mouse TruStain FcX PLUS (1:100, BioLegend, San Diego, CA), the cells were incubated with Live/Dead Zombie dye (UV, BioLegend, 423106, 1:100) and antibodies specific for CD45 (BV510, BioLegend, 103138, 1:100) and FcϵRIα (PE/Cyanine7, BioLegend, 134318, 1:100) in the dark at room temperature for 30 min. After centrifugation, the cells were suspended in PBS and detected by the Beckman CytoFlex LX flow cytometer using CytExpert software (Beckman Coulter, Brea, CA).
After sacrifice, the mouse ear was removed and cut into pieces. Then, the tissues were grinded to powder in mortar and lysed with RIPA buffer containing phosphatase and protease inhibitors. The total protein concentration of the homogenate was measured by the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23227) and further diluted to 5 μg/μl. These samples were stored at -80°C for subsequent cytokine assays.
For the simultaneous detection of various cytokines in the mouse ear skin after DNCB induction, the R&D Luminex Assay (Catalog Number LXSAMSM) was used according to the manufacturer’s instructions. Here, IL-4, IL-10, IFN-γ, TNF-α, IL-1β, and IL-6 were measured, performed by the Novogene Bioinformatics Technology Co. (Beijing, China).
To profile the skin microbiota after DNCB treatment, the mouse ear skin was swabbed with sterile swabs which were further snap-frozen on dry ice and sent out for full-length 16s ribosomal RNA sequencing using Pacific Bioscience platform. After obtaining clean reads, the program Uparse V7.0.1001 was used to cluster these sequences into Operational Taxonomic Units with an average percent identity of 97%. Based on the SILVA database, the software Mothur was used for taxonomic classification. The above full-length 16s rRNA sequencing and bioinformatics analysis were performed by the Novogene Bioinformatics Technology Co. (Beijing, China).
RNA sequencing was performed on the total epidermal RNA from mouse ear skin after being treated with DNCB or vehicle on 21 days. Differentially expression analysis was conducted by the DESeq2 R package (1.20.0). The P-values were adjusted using the Benjamini & Hochberg method. Adjusted P < 0.05 and |log2 fold change| > 2 were set as the threshold to define statistical significance. Data processing and analysis are detailed in Supplementary Materials, which were performed by the Novogene Bioinformatics Technology Co. (Beijing, China).
For the detection of TSLP in the ear skin, the mouse ELISA kit (Mouse TSLP ELISA MAX Deluxe, BioLegend) was used according to the manufacturer’s protocol. To block TSLP activity in the skin, DNCB-treated Stat3 cKO mice and Stat3f/f mice on Day 23 (D0) were injected at ears with 20 μg of neutralizing TSLP antibody (BioLegend, 515202) or IgG2a control antibody (BioLegend, 407102). One hour after injection, both mice received topical DNCB treatment. The above procedures were carried out every other day until the 8th day, as well as the measurement of the ear thickness and dermatitis score.
The experimental data were analyzed by Prism 8.0 Software (GraphPad Software, La Jolla, CA, USA) and significance was assessed by the two-way ANOVA analysis or Mann-Whitney U test. For all experiments with error bars, standard deviations (SD) were calculated to indicate variations, and the data are represented as the Mean ± SD. Here, P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
To explore the role of epidermal keratinocyte STAT3 in the pathogenesis of AD, we developed tamoxifen-inducible K14-CreERT+Stat3flox/flox mice (termed K14-CreERT+-Stat3f/f mice). Intraperitoneal injection of tamoxifen into K14-CreERT+-Stat3f/f mice for 5 consecutive days lead to epidermal keratinocyte-specific STAT3 deficiency (termed Stat3 cKO) (Supplementary Figure S1A). Taking K14-CreER-Stat3flox/flox mice (termed Stat3f/f mice) as the control, the efficacy of STAT3 deficiency in the epidermis was confirmed by Western blotting and immunohistochemistry (IHC) (Supplementary Figures S1B, C).
After tamoxifen induction, DNCB was topically applied to the ears of the mice to simulate AD-like skin inflammation (Figure 1A). DNCB is a chemical hapten commonly used to induce allergic contact dermatitis and thought to evoke a Th1-dominated immune response. However, repeated challenges with DNCB over an extended period cause a chronic Th2-dominated inflammatory response similar to that in human AD (14). On day 21, the DNCB-treated ears of Stat3 cKO mice were red, thickened, and swollen, accompanied by exudation and crusting. Interestingly, these AD-like skin lesions were more severe than those in Stat3f/f mice (Figure 1B; Supplementary Figure S2A). The thickness of DNCB-treated ear in Stat3 cKO mice was significantly increased from Day 7, and the difference became greater over time (Figure 1C). Histologically, the DNCB-treated ear skin of Stat3 cKO mice showed marked acanthosis, increased granular layers, and thickened dermis along with dermal inflammatory cell infiltration (Figure 1D), which were similar to the histological changes observed in human AD. In addition, Stat3 cKO mice exhibited a significant increase in the number of Ki67+ cells among keratinocytes, which is a marker of epidermal hyperplasia (Figures 1E, F). After DNCB treatment, the phosphorylation of STAT3 was confirmed using IHC staining. As expected, keratinocyte-specific deficiency of phosphorylated STAT3 was observed in Stat3 cKO mice. Besides, there was increased STAT3 activation in Stat3f/f mice after DNCB treatment (Supplementary Figure S2B). Collectively, these results indicate that keratinocyte-specific STAT3 dysfunction exacerbated DNCB-induced AD-like skin inflammation.
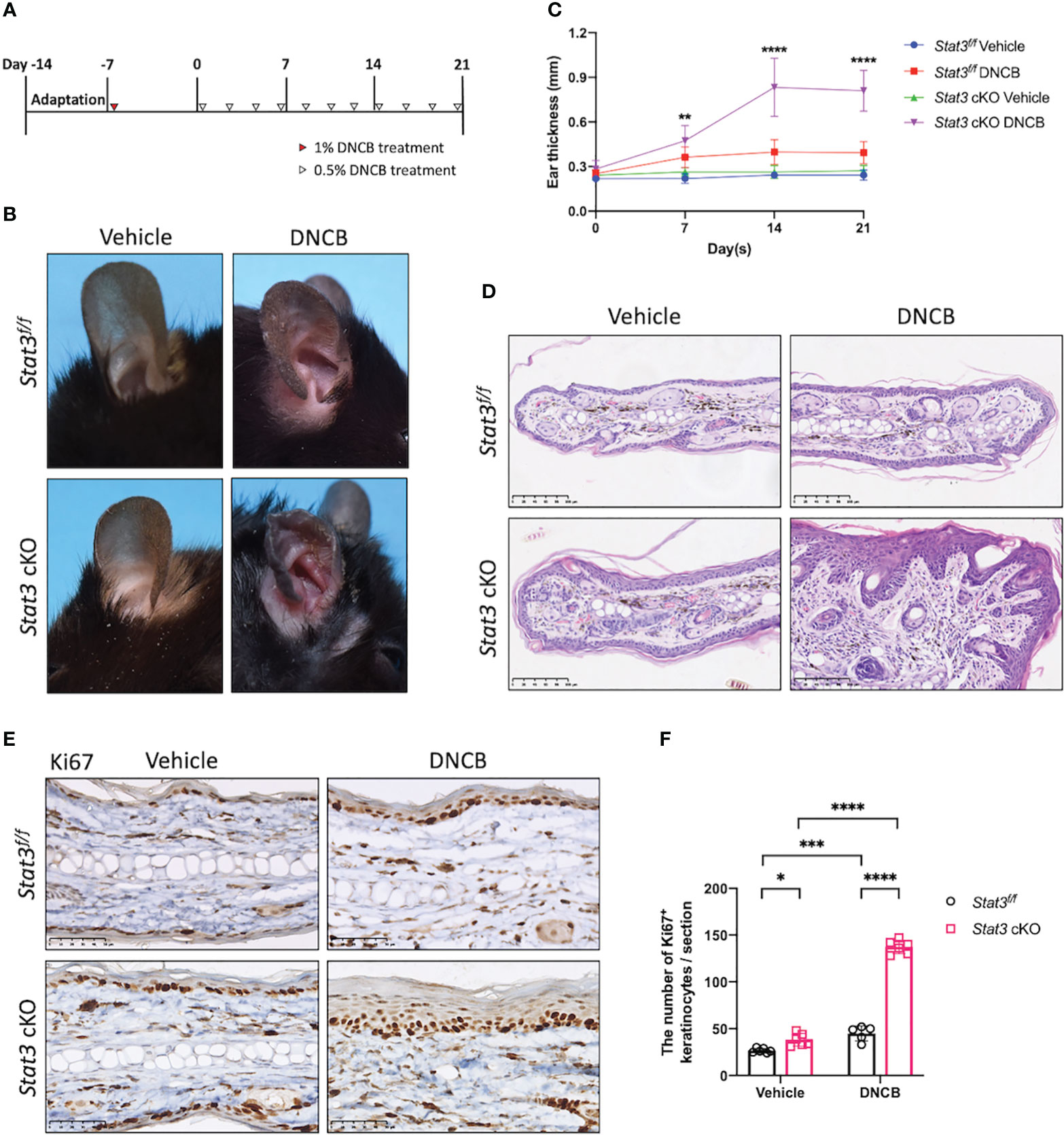
Figure 1 STAT3 deficiency in basal keratinocytes exacerbated DNCB-induced AD-like skin inflammation. (A) Schematic diagram of the experimental design of DNCB-induced AD-like skin inflammation. (B, D) Representative phenotypes and histological changes in Stat3f/f mice and Stat3 cKO mice after DNCB or vehicle treatment on Day 21. Bar = 250 μm. (C) During the induction period, ear thickness (mm) was measured weekly (n ≥ 10). (E) Representative IHC staining of Ki67 and (F) Ki67-positive keratinocyte counts (n = 5-6). Bar = 50 μm. The data are presented as the mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The epidermis is the first line of defense between the host and its environment. In AD, there usually exists a defective epidermal barrier, whose critical importance in the disease development has been apparent. To determine whether STAT3 dysfunction may impair skin barrier function, the expression of filaggrin and loricrin was studied by using immunofluorescence (IF). In vehicle-treated ear skin, there was relatively continuous expression of filaggrin and loricrin (Figure 2A). However, after repeated stimulation with DNCB, both were downregulated, especially in Stat3 cKO mice, in which they were barely expressed (Figure 2A).
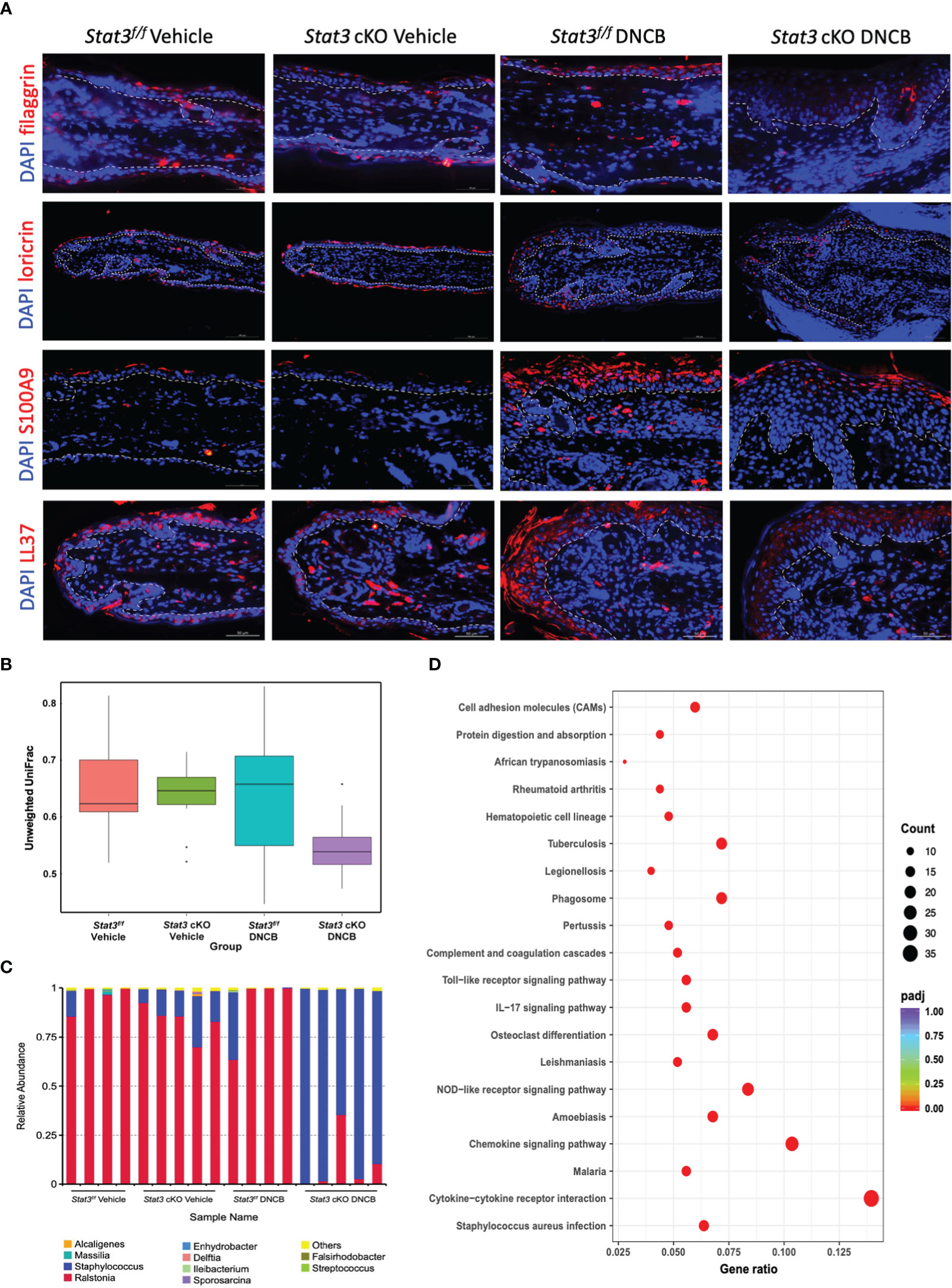
Figure 2 Epidermal barrier dysfunction was aggravated in Stat3 cKO mice with AD-like skin inflammation, accompanied by AMP insufficiency and microbial dysbiosis. (A) Representative IF staining of filaggrin, loricrin, S100A9, and LL37 (red) in the ear skin of Stat3f/f mice and Stat3 cKO mice after DNCB or vehicle treatment. Bar = 50 μm. (B, C) The microbial diversity on the surface of the ear skin and the relative abundance of different microbial genera determined by full-length 16s ribosomal RNA sequencing (n = 4-5). (D) KEGG pathway analysis of differentially expressed genes identified by RNA sequencing between the epidermis of Stat3f/f mice and Stat3 cKO mice after DNCB treatment (n = 3-4, adjusted P < 0.05, |log2-fold change| > 2).
In addition to the above structural proteins, keratinocytes also synthesize various host defense molecules such as antimicrobial peptides (AMPs), which exert broad-spectrum effects against bacteria, fungi, and viruses (15). Thus, we examined the expression of S100A9 and cathelicidin (LL37) in the ear skin by IF. As shown in Figure 2A, S100A9 was barely expressed in Stat3f/f mice that received topical application of vehicle, while LL37 was slightly expressed in the cytoplasm of keratinocytes in the upper epidermis. After topical DNCB treatment, the expression of S100A9 and LL37 in Stat3f/f and Stat3 cKO mice was upregulated, and these proteins were mainly distributed in the upper layers of the ear epidermis. However, compared with Stat3f/f mice, Stat3 cKO mice showed a reduced expression level of both proteins (Figure 2A).
AD flares are generally associated with a disordered microbiome, with S. aureus being a dominant colonizer and pathogen (16). Dysfunction of the epidermal barrier, together with an insufficient upregulation of specific AMPs, might further enhance S. aureus colonization (1). Therefore, we performed full-length 16s ribosomal RNA sequencing to identify microbial populations on the ear skin surface. The microbial diversity of DNCB-treated Stat3 cKO mice was decreased compared with that of the other groups (Figure 2B). Moreover, the relative abundance of genera was different among the 4 groups (Figure 2C). On the ear skin of Stat3f/f mice, the dominant genus was Ralstonia regardless of topical vehicle or DNCB treatment. However, in Stat3 cKO mice, the Staphylococcus genus with a small proportion appeared on the vehicle-treated ear skin, and became dominant after DNCB treatment (Figure 2C). To further explore the effects of Staphylococcus dominance, we analyzed the epidermal transcriptome using RNA sequencing, which identified 651 upregulated genes and 139 downregulated genes (adjusted P < 0.05, |log2-fold change| > 2, Supplementary Figure S3A). KEGG pathway analysis showed that the upregulated genes were most significantly enriched in the S. aureus infection pathway, suggesting that Staphylococcus dominance influenced the expression of related genes in Stat3 cKO mice (Figure 2D; Supplementary Figure S3B).
Taken together, these data suggest that epidermal barrier impairment was aggravated in Stat3 cKO mice with AD-like skin inflammation, accompanied by specific AMP insufficiency and Staphylococcus-dominant microbial dysbiosis.
Since the cutaneous inflammation of AD is characterized by sequential and progressive patterns of inflammatory cell infiltration, predominantly involving type-2-skewed immune dysregulation, we studied several type-2 inflammatory cells and cytokines. Serum IgE levels were significantly elevated in Stat3 cKO mice compared with Stat3f/f mice after DNCB treatment (Figure 3A). As a high-affinity Fc receptor for IgE, FcϵRIα is abundantly expressed on mast cells and basophils (17). Using flow cytometry, we found that the proportion of FcϵRIα+ cells in the ear dermis of Stat3 cKO mice was significantly increased (Figure 3B). Among these FcϵRIα+ cells, mast cells were detected by toluidine blue staining. Stat3 cKO mice treated with DNCB exhibited more infiltrating mast cells in the dermis (Figures 3C, E). The presence of eosinophils in AD inflammation has long been established (18). Consistently, there was a significant increase in eosinophil number in Stat3 cKO mice with DNCB-induced AD-like skin inflammation (Figures 3C, E).
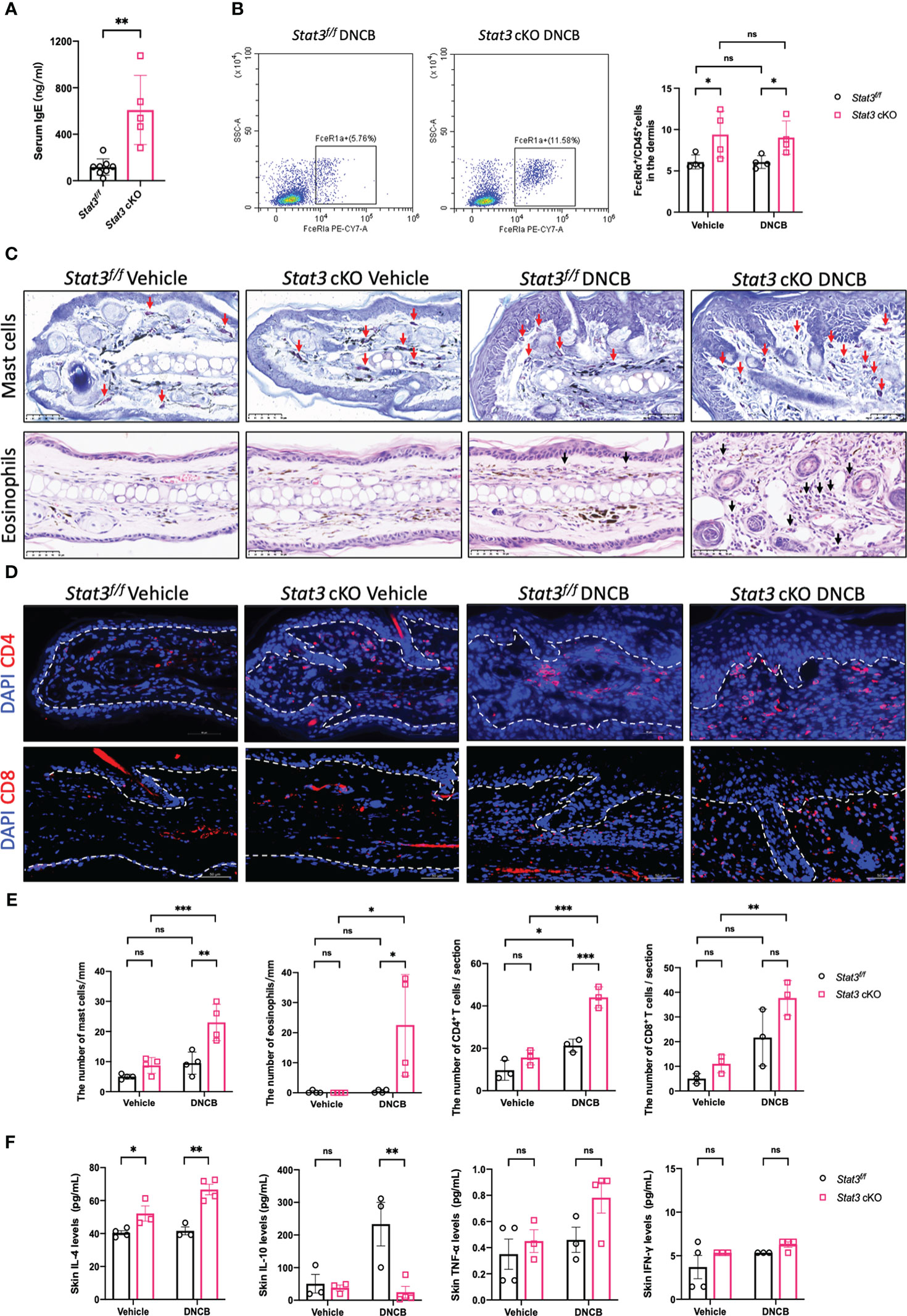
Figure 3 Keratinocyte-specific STAT3 deficiency predominantly increased the infiltration of type-2 inflammatory cells and the secretion of related cytokines. (A) Analysis of the serum IgE levels (ng/ml) in Stat3f/f mice (n = 8) and Stat3 cKO mice (n = 5) after DNCB treatment using ELISA. (B) Flow cytometry analysis of FcϵRIα+ cells in the dermis of Stat3f/f mice and Stat3 cKO mice (n = 4). (C) Toluidine blue staining of mast cells (red arrows) and H&E staining of eosinophils (black arrows) in mouse ear skin of each group. Bar = 50 μm. (D) Representative IF staining of CD4+ or CD8+ cells (red) in the ear skin of Stat3f/f mice and Stat3 cKO mice. Bar = 50 μm. (E) Statistical analysis results of the number of mast cells, eosinophils, and CD4+ or CD8+ cells (n = 3-4). (F) The expression levels of Il-4, IL-10, TNF-α, and IFN-γ (pg/ml) in the ear skin of Stat3f/f mice and Stat3 cKO mice (n = 3-4). The data are presented as the mean ± SD; P > 0.05 denotes not significant (ns); *P < 0.05, **P < 0.01, and ***P < 0.001.
In addition to the above inflammatory cells, T-cell infiltration is crucial for the development of AD skin inflammation. Using IF, we found that the number of CD4+ T cells was significantly increased in the DNCB-treated ear skin of both mice, but Stat3 cKO mice exhibited more CD4+ T cells, especially in the dermal-epidermal interface (Figures 3D, E). Unlike CD4+ T cells, there were no significant changes in the number of CD8+ T cells (Figures 3D, E).
We further measured the expression levels of cytokines in the ear skin using a Luminex Assay. Compared with Stat3f/f mice, the type-2 cytokine IL-4 was significantly upregulated in Stat3 cKO mice, regardless of vehicle or DNCB treatment (Figure 3F). In contrast, the inhibitory cytokine IL-10 expression was significantly decreased in the DNCB-treated ear skin of Stat3 cKO mice (Figure 3F). In addition, the expression of the proinflammatory cytokine TNF-α and type-1 cytokine IFN-γ showed no significant changes (Figure 3F).
Collectively, these results indicate that STAT3 dysfunction in basal keratinocytes predominantly promoted the infiltration of type-2 inflammatory cells and the secretion of related cytokines.
In barrier-impaired skin, keratinocytes send proinflammatory signals through alarmins including thymic stromal lymphopoietin (TSLP) (5). TSLP serves as a key “bridge”, which exerts its biological effects by binding to a high-affinity heteromeric complex composed of a TSLP receptor (TSLPR) subunit and a IL-7 receptor (IL-7R) α chain (19). This functional TSLPR is expressed by a variety of immune cell populations, including dendritic cells (DCs), monocytes/macrophages, mast cells, eosinophils, basophils, and T and B cells (20, 21). Our ELISA data showed that the expression level of TSLP was significantly increased in the ear skin of Stat3 cKO mice, regardless of vehicle or DNCB induction (Figure 4A). IF staining revealed that TSLP was primarily expressed in keratinocytes, and the expression differences among these groups were consistent with those revealed by ELISA (Figure 4B). Moreover, there exhibited a significantly increased infiltration of TSLPR+ cells in the ear dermis of DNCB-treated Stat3 cKO mice compared with that of DNCB-treated Stat3f/f mice (Figures 4C, D). Specifically, there was a significant increase in the number of infiltrating CD11c+ DCs and CD68+ macrophages into the skin (Figures 4C, D). These data reveal that keratinocyte-specific STAT3 deficiency promoted the expression and secretion of TSLP by keratinocytes, which recruited specific target cells in AD-like skin inflammation.
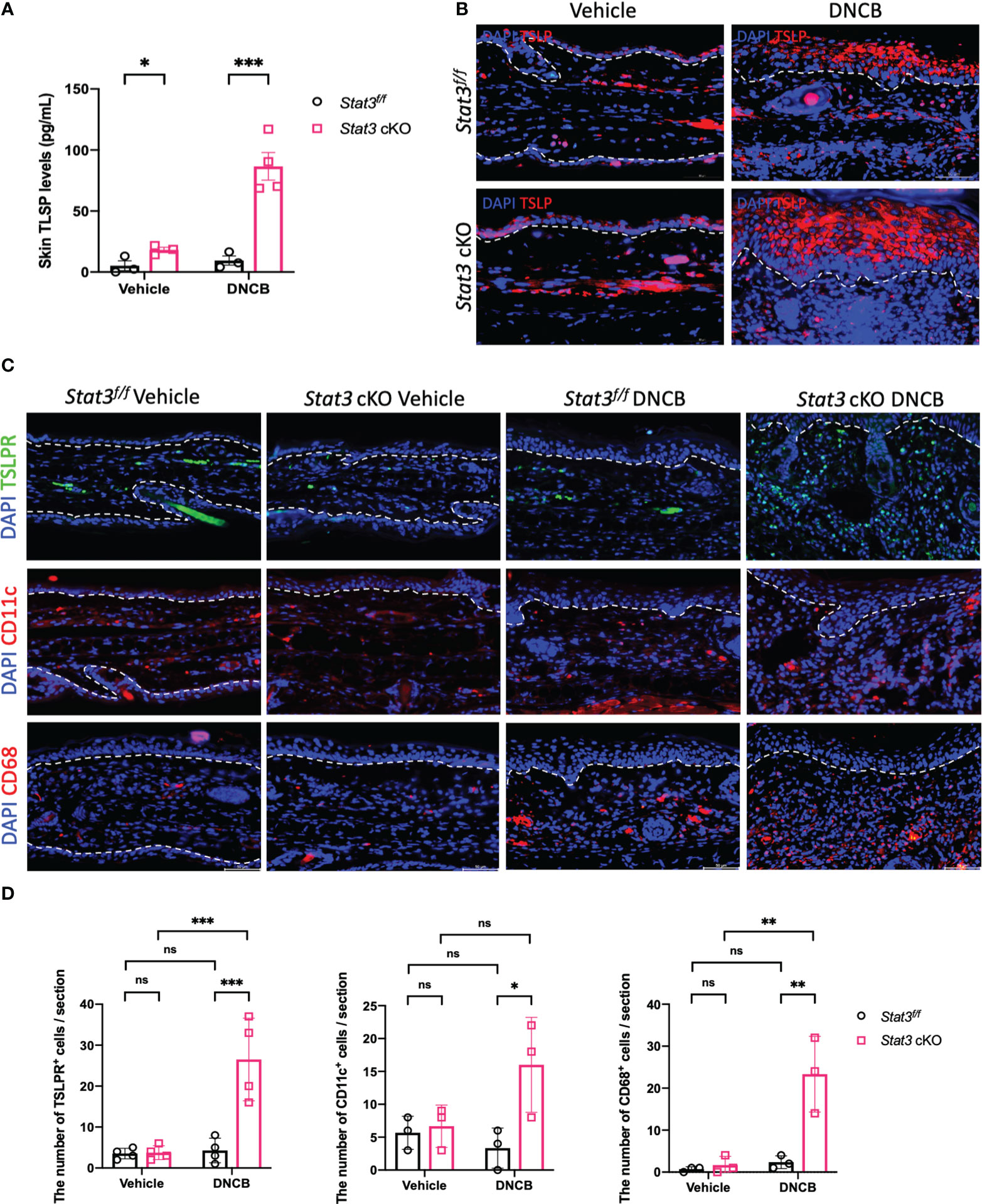
Figure 4 The production of TSLP by keratinocytes was increased in Stat3 cKO mice, which recruited more target cells in AD-like skin inflammation. (A) Analysis of TSLP expression level (pg/ml) in the ear skin of Stat3f/f mice and Stat3 cKO mice after DNCB or vehicle induction by ELISA (n = 3-4). (B) IF staining showed that TSLP was primarily expressed in keratinocytes (red). Bar = 50 μm. (C) IF staining revealed that the infiltration of TSLPR+ cells (green), CD11c+ cells (red), and CD68+ cells (red) was increased in the dermis of DNCB-treated Stat3 cKO mice compared with Stat3f/f mice. (D) Statistical analysis of the abovementioned cells (n = 3-4). The data are presented as the mean ± SD; P > 0.05 denotes ns; *P < 0.05, **P < 0.01, and ***P < 0.001.
Several studies have reported that keratinocyte-derived cytokine TSLP acts as a master switch of AD skin inflammation (22, 23). To determine whether the exacerbation of AD-like skin inflammation was triggered by TSLP, the ear skin of Stat3 cKO mice was injected with an anti-TSLP neutralizing antibody during DNCB challenge. We found that anti-TSLP antibody treatment attenuated DNCB-induced AD-like skin inflammation in Stat3 cKO mice (Figure 5A), as the ear thickness and dermatitis score were significantly decreased (Figure 5B). Histologically, the ear skin of Stat3 cKO mice injected with the anti-TSLP antibody showed decreased epidermal and dermal thickness, reduced exudation and crusting, and fewer infiltrating dermal inflammatory cells, including mast cells (Figure 5C). Moreover, there was a significant decrease in the number of TSLPR+ cells in the dermis of Stat3 cKO mice after anti-TSLP antibody treatment (Figure 5C). These results suggest that TSLP was the critical factor underlying severe AD-like skin inflammation in Stat3 cKO mice.
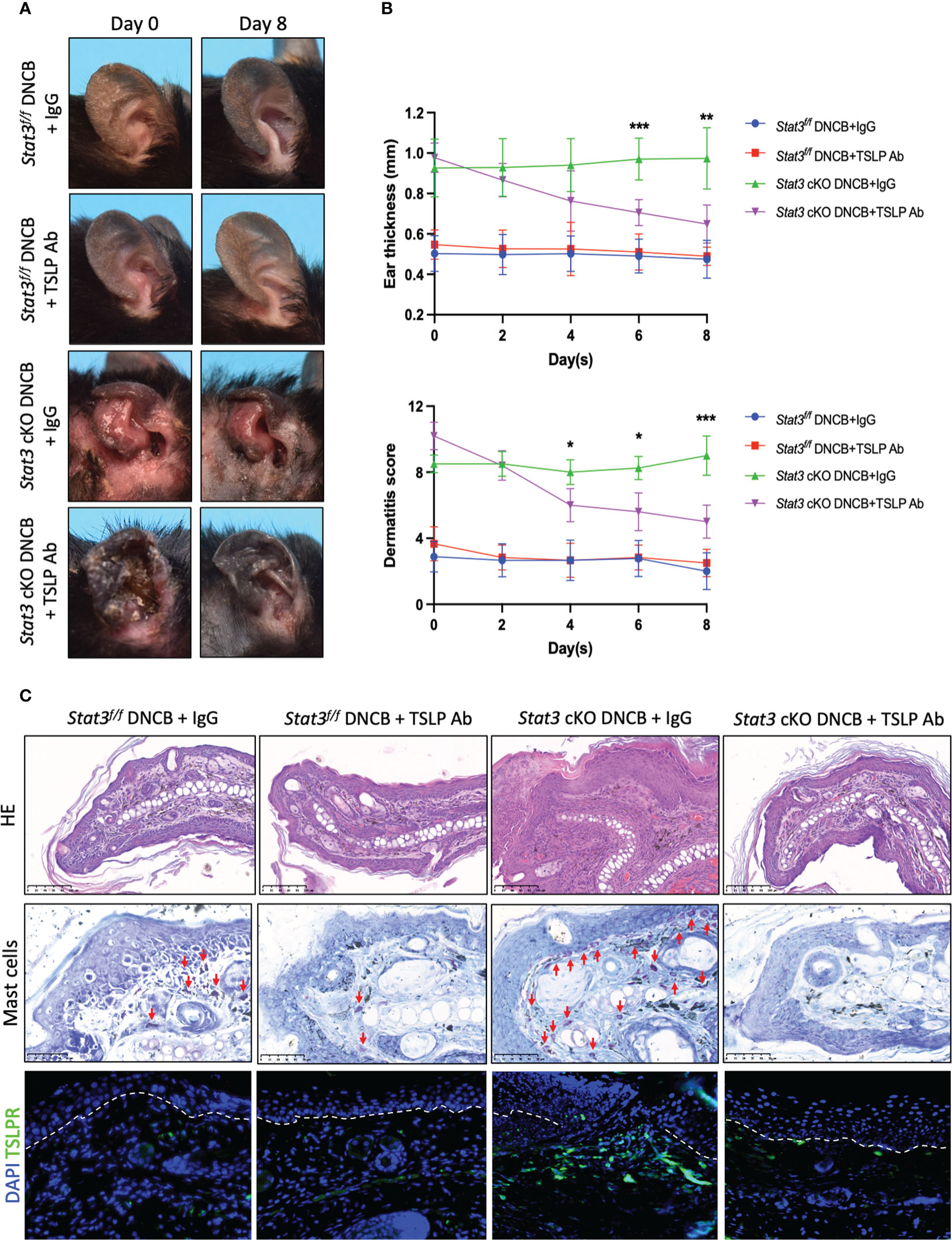
Figure 5 DNCB-induced AD-like skin inflammation of Stat3 cKO mice was alleviated by an anti-TSLP neutralizing antibody. (A) Representative photographs of DNCB-treated Stat3f/f mice and Stat3 cKO mice after anti-TSLP antibody or isotype IgG antibody injection on Day 8. (B) The ear thickness (mm) and dermatitis score were measured every other day (n = 5-8). (C) Analysis of the histological changes in ear skin by HE staining (Bar = 100 μm), mast cells by toluidine blue staining (red arrows, Bar = 50 μm), and TSLPR-positive cells by IF staining (green, Bar = 50 μm). The data are presented as the mean ± SD; P > 0.05 denotes ns; *P < 0.05, **P < 0.01, and ***P < 0.001.
STAT3 is well documented as an important regulator of complex biological functions. Increasing evidence suggests that STAT3 is involved in the development of allergic inflammation, but the role of STAT3 in epidermal keratinocytes in AD has not been directly evaluated. In this study, we found that Stat3 cKO mice exhibited worsened AD-like skin inflammation characterized by a defective skin barrier, Staphylococcus-dominant microbial dysbiosis, more infiltrating immune cells, and an increased concentration of keratinocyte-derived TSLP.
Previous studies have found that the transcription of the STAT3 gene is increased in lesional skin versus nonlesional skin in AD patients (24). Furthermore, as a downstream transcription factor of IL-4, IL-13, and IL-22, activated STAT3 in keratinocytes acts as a negative regulator of keratinocyte differentiation by suppressing the expression of filaggrin and loricrin, leading to epidermal barrier dysfunction (25, 26). On the other hand, dominant-negative mutations of the STAT3 gene result in the classical autosomal dominant hyper-IgE syndrome (HIES) (27), which is characterized by AD, recurrent infections, and elevated serum IgE levels and eosinophil counts (28). AD and HIES have overlapping clinical features, but the changes in STAT3 expression and function are the opposite in these conditions, suggesting that STAT3 might play bidirectional roles in the development of AD skin lesions. Here, by using mice with keratinocyte-specific deletion of STAT3, we provide relatively direct evidence for the potential inhibitory role of keratinocyte STAT3 in the pathogenesis of AD-like skin inflammation.
TSLP is a cytokine expressed by epithelial cells, including keratinocytes (21). Recent work has highlighted the role of TSLP in various inflammatory diseases, which is an IL-7-like cytokine initiating and promoting type-2 inflammation, including AD, allergic rhinitis, and asthma (21). TSLP was found to be highly expressed in the lesional skin of AD patients and to potently activated DCs to secrete Th2-recruiting chemokines. These TSLP-activated DCs induce Th2 cell differentiation to produce IL-4, IL-5, IL-13, and TNF-α, while downregulating IL-10 and IFN-γ (29). Interestingly, these upregulated cytokines can synergistically promote the production of TSLP by keratinocytes (30), suggesting the existence of a positive feedback loop that amplifies skin inflammation. In addition, keratinocyte TSLP can trigger the atopic march in mice (8), and mice overexpressing TSLP in keratinocytes showed a spontaneous AD-like phenotype, with the development of eczematous lesions, increased circulating Th2 cells, and higher serum IgE levels (31). In our study, the expression level of TSLP was significantly increased in the skin of Stat3 cKO mice and was further elevated after DNCB induction. More importantly, blockade of TSLP activity by TSLP neutralizing antibody markedly reduced AD-like skin inflammation in Stat3 cKO mice. Thus, TSLP may be an important determinant for promoting AD-like skin inflammation in the absence of keratinocyte STAT3.
At barrier interfaces, TSLP expression can be triggered by environmental stimuli, including allergens, bacterial and fungal products, viruses, mechanical injury, and cigarette smoke extracts (20). In addition, several endogenous factors, such as proinflammatory cytokines, Th2-related cytokines and IgE, have positive effects on TSLP production (32). However, it is still unclear how STAT3 dysfunction promotes the expression of TSLP in keratinocytes. It has been indicated that S. aureus-derived ligands of TLRs can induce TSLP expression in keratinocytes, leading to Th2-skewed sensitization to environmental allergens and S. aureus-derived allergens, or both (33, 34). In this study, Staphylococcus dominance and corresponding transcriptomic alterations in the skin were found in Stat3 cKO mice with AD-like skin inflammation, suggesting that Staphylococcus-dominant microbial colonization may be trigger TSLP expression in Stat3 cKO mice. Future studies should address this important issue and clarify the molecular interactions or pathways involved in the process.
Atopic skin has been characterized by a relatively impaired induction of AMPs (35). Ong et al. firstly found that the expression of AMPs, including LL37 and human β-defensin 2 (hBD2), was significantly decreased in atopic lesions versus psoriatic lesions (35). Some studies have attributed this impairment to elevated Th2-related cytokines in AD, such as IL-4, IL-13, and TSLP (15, 36, 37). Among them, IL-4 and IL-13 negatively regulate LL-37 and hBD expression through STAT6 in keratinocytes (36, 37), while TSLP downregulates the production of S100A7 and hBD2 by keratinocytes via the JAK2/STAT3-dependent mechanism (15). Here, keratinocyte-specific STAT3 dysfunction led to increased TSLP and IL-4 levels accompanied by decreased S100A9 and LL37 levels. The detailed mechanisms regulating the expression of AMPs in keratinocytes need further investigation.
In summary, our findings provide insights into the role of keratinocyte STAT3 in regulating AD skin inflammation. Future studies elucidating the regulatory mechanisms among STAT3, TSLP, and AMPs should help identify new therapeutic strategies for AD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SRP451094 (SRA).
The animal study was approved by the Second Affiliated Hospital Zhejiang University School of Medicine Animal Care Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Z-YW: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Visualization, Data curation, Investigation, Validation, Writing – original draft. Y-XZ: Conceptualization, Data curation, Investigation, Methodology, Validation, Writing – original draft. FX: Writing – original draft, Conceptualization, Data curation, Investigation. Y-ZC: Investigation, Writing – original draft, Validation. X-YC: Investigation, Writing – original draft. S-QC: Methodology, Writing – review & editing. B-XY: Writing – review & editing, Supervision. YZ: Supervision, Writing – review & editing. MZ: Supervision, Writing – review & editing. X-YM: Writing – review & editing, Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Visualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (No. 82203905, 82230104, 81930089).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1273182/full#supplementary-material
1. Weidinger S, Novak N. Atopic dermatitis. Lancet (2016) 387(10023):1109–22. doi: 10.1016/S0140-6736(15)00149-X
2. Silverberg JI, Barbarot S, Gadkari A, Simpson EL, Weidinger S, Mina-Osorio P, et al. Atopic dermatitis in the pediatric population: A cross-sectional, international epidemiologic study. Ann Allergy Asthma Immunol (2021) 126(4):417–28 e2. doi: 10.1016/j.anai.2020.12.020
3. Barbarot S, Auziere S, Gadkari A, Girolomoni G, Puig L, Simpson EL, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy (2018) 73(6):1284–93. doi: 10.1111/all.13401
4. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov (2022) 21(1):21–40. doi: 10.1038/s41573-021-00266-6
5. Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet (2020) 396(10247):345–60. doi: 10.1016/S0140-6736(20)31286-1
6. Palmer CNA, Irvine AD, Terron-Kwiatkowski A, Zhao YW, Liao HH, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet (2006) 38(4):441–6. doi: 10.1038/ng1767
7. Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus aureus and atopic dermatitis: A complex and evolving relationship. Trends Microbiol (2018) 26(6):484–97. doi: 10.1016/j.tim.2017.11.008
8. Leyva-Castillo JM, Hener P, Jiang H, Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol (2013) 133(1):154–63. doi: 10.1038/jid.2012.239
9. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA (2013) 110(34):13921–6. doi: 10.1073/pnas.1307321110
10. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
11. Chen Z, Laurence A, O'Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol (2007) 19(6):400–8. doi: 10.1016/j.smim.2007.10.015
12. van de Veen W, Kratz CE, McKenzie CI, Aui PM, Neumann J, van Noesel CJM, et al. Impaired memory B-cell development and antibody maturation with a skewing toward IgE in patients with STAT3 hyper-IgE syndrome. Allergy (2019) 74(12):2394–405. doi: 10.1111/all.13969
13. Chieosilapatham P, Kiatsurayanon C, Umehara Y, Trujillo-Paez JV, Peng G, Yue H, et al. Keratinocytes: innate immune cells in atopic dermatitis. Clin Exp Immunol (2021) 204(3):296–309. doi: 10.1111/cei.13575
14. Jin HL, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol (2009) 129(1):31–40. doi: 10.1038/jid.2008.106
15. Lee H, Ryu WI, Kim HJ, Bae HC, Ryu HJ, Shin JJ, et al. TSLP down-regulates S100A7 and ss-defensin 2 via the JAK2/STAT3-dependent mechanism. J Invest Dermatol (2016) 136(12):2427–35. doi: 10.1016/j.jid.2016.07.027
16. Totte JE, van der Feltz WT, Hennekam M, van Belkum A, van Zuuren EJ, Pasmans SG. Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: a systematic review and meta-analysis. Br J Dermatol (2016) 175(4):687–95. doi: 10.1111/bjd.14566
17. Kraft S, Kinet JP. New developments in FcepsilonRI regulation, function and inhibition. Nat Rev Immunol (2007) 7(5):365–78. doi: 10.1038/nri2072
18. Simon D, Braathen LR, Simon HU. Eosinophils and atopic dermatitis. Allergy (2004) 59(6):561–70. doi: 10.1111/j.1398-9995.2004.00476.x
19. Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol (2000) 1(1):59–64. doi: 10.1038/76923
20. Varricchi G, Pecoraro A, Marone G, Criscuolo G, Spadaro G, Genovese A, et al. Thymic stromal lymphopoietin isoforms, inflammatory disorders, and cancer. Front Immunol (2018) 9:1595. doi: 10.3389/fimmu.2018.01595
21. Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol (2012) 130(4):845–52. doi: 10.1016/j.jaci.2012.07.010
22. Wang SH, Zuo YG. Thymic stromal lymphopoietin in cutaneous immune-mediated diseases. Front Immunol (2021) 12:698522. doi: 10.3389/fimmu.2021.698522
23. Ebina-Shibuya R, Leonard WJ. Role of thymic stromal lymphopoietin in allergy and beyond. Nat Rev Immunol (2023) 23(1):24–37. doi: 10.1038/s41577-022-00735-y
24. Mobus L, Rodriguez E, Harder I, Stolzl D, Boraczynski N, Gerdes S, et al. Atopic dermatitis displays stable and dynamic skin transcriptome signatures. J Allergy Clin Immunol (2021) 147(1):213–23. doi: 10.1016/j.jaci.2020.06.012
25. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol (2015) 136(3):667–77 e7. doi: 10.1016/j.jaci.2015.03.051
26. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol (2005) 174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695
27. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096
28. Al-Shaikhly T, Ochs HD. Hyper IgE syndromes: clinical and molecular characteristics. Immunol Cell Biol (2019) 97(4):368–79. doi: 10.1111/imcb.12209
29. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol (2002) 3(7):673–80. doi: 10.1038/ni805
30. Bogiatzi SI, Fernandez I, Bichet JC, Marloie-Provost MA, Volpe E, Sastre X, et al. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J Immunol (2007) 178(6):3373–7. doi: 10.4049/jimmunol.178.6.3373
31. Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med (2005) 202(4):541–9. doi: 10.1084/jem.20041503
32. Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int (2012) 61(1):3–17. doi: 10.2332/allergolint.11-RAI-0395
33. Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol (2010) 126(5):985–93, 93 e1-3. doi: 10.1016/j.jaci.2010.09.002
34. Brauweiler AM, Goleva E, Leung DYM. Staphylococcus aureus lipoteichoic acid initiates a TSLP-basophil-IL4 axis in the skin. J Invest Dermatol (2020) 140(4):915–7 e2. doi: 10.1016/j.jid.2019.09.004
35. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med (2002) 347(15):1151–60. doi: 10.1056/NEJMoa021481
36. Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity (2006) 24(3):341–8. doi: 10.1016/j.immuni.2006.02.006
37. Albanesi C, Fairchild HR, Madonna S, Scarponi C, De Pita O, Leung DY, et al. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol (2007) 179(2):984–92. doi: 10.4049/jimmunol.179.2.984
Keywords: atopic dermatitis, DNCB, keratinocytes, stat3, TSLP
Citation: Wang Z-Y, Zheng Y-X, Xu F, Cui Y-Z, Chen X-Y, Chen S-Q, Yan B-X, Zhou Y, Zheng M and Man X-Y (2023) Epidermal keratinocyte-specific STAT3 deficiency aggravated atopic dermatitis-like skin inflammation in mice through TSLP upregulation. Front. Immunol. 14:1273182. doi: 10.3389/fimmu.2023.1273182
Received: 05 August 2023; Accepted: 07 November 2023;
Published: 20 November 2023.
Edited by:
Roopesh Singh, University of Virginia, United StatesCopyright © 2023 Wang, Zheng, Xu, Cui, Chen, Chen, Yan, Zhou, Zheng and Man. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Yong Man, bWFueHlAemp1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.