
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 18 September 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1269015
This article is part of the Research Topic Innovations in Development, Translational Research and Manufacturing of CAR T cells View all 19 articles
 Sarah Staudt1†
Sarah Staudt1† Kai Ziegler-Martin1†Alexander Visekruna2John Slingerland3Roni Shouval4,5
Kai Ziegler-Martin1†Alexander Visekruna2John Slingerland3Roni Shouval4,5 Michael Hudecek1
Michael Hudecek1 Marcel van den Brink3
Marcel van den Brink3 Maik Luu1*
Maik Luu1*The opportunities genetic engineering has created in the field of adoptive cellular therapy for cancer are accelerating the development of novel treatment strategies using chimeric antigen receptor (CAR) and T cell receptor (TCR) T cells. The great success in the context of hematologic malignancies has made especially CAR T cell therapy a promising approach capable of achieving long-lasting remission. However, the causalities involved in mediating resistance to treatment or relapse are still barely investigated. Research on T cell exhaustion and dysfunction has drawn attention to host-derived factors that define both the immune and tumor microenvironment (TME) crucially influencing efficacy and toxicity of cellular immunotherapy. The microbiome, as one of the most complex host factors, has become a central topic of investigations due to its ability to impact on health and disease. Recent findings support the hypothesis that commensal bacteria and particularly microbiota-derived metabolites educate and modulate host immunity and TME, thereby contributing to the response to cancer immunotherapy. Hence, the composition of microbial strains as well as their soluble messengers are considered to have predictive value regarding CAR T cell efficacy and toxicity. The diversity of mechanisms underlying both beneficial and detrimental effects of microbiota comprise various epigenetic, metabolic and signaling-related pathways that have the potential to be exploited for the improvement of CAR T cell function. In this review, we will discuss the recent findings in the field of microbiome-cancer interaction, especially with respect to new trajectories that commensal factors can offer to advance cellular immunotherapy.
The intestinal microbiome belongs to one of the most complex communities of bacterial and fungal strains and has been shown to influence both host physiology and pathophysiology, as well as malignant and non-malignant disease development. The mutualistic interaction plays an essential role in educating immune cells and tolerogenic reactions. While the first studies in the field have demonstrated that intestinal microbial colonization is a key factor for the maintenance of gut homeostasis, work on dysbiosis showed that disease development beyond the gut, such as autoimmunity in the central nervous system and allergic reactions in the lung, is also dependent on commensal factors (1–3). The crucial impact of the microbiome in cancer was highlighted first by clinical data from patients receiving bone marrow transplantations (BMTs) who developed Graft-versus-Host-Disease (GVHD) in association with a loss of diversity of the intestinal microbiome (4). Moreover, changes in the patient microbiome composition and the production of microbial metabolites have been associated with different clinical responses in regard to immune checkpoint inhibition (ICI) and chimeric antigen receptors (CAR) T cell therapy (2, 5–9). Analyses of commensal influences on tumor cells have shown the potential to promote carcinogenesis and identified intratumoral colonization as an additional layer of the tumor microenvironment (TME) (10–12). These findings highlight the microbiome as a source for novel and therapeutically relevant strategies such as rational shaping of the commensal community in patients as preconditioning before or synergistically acting intervention combined with immunotherapies (e.g. ICI or adoptive T cell transfers).
CARs and transgenic T cell receptors (TCRs) are a result of the advances in synthetic biology and genetic engineering which have led to new strategies to redirect T cell specificity towards tumor-associated antigens (TAAs) (13, 14). The adoptive cell therapy (ACT) of engineered T cells has evolved into a therapeutic approach that has become a breakthrough in cancer immunotherapy (15), capable of demonstrating substantial response rates and efficacy in advanced malignancies (16–19).
TCRs composed of an α- and β-chain that form a heterodimer embedded in the CD3 signalling complex recognize their antigen in the context of a major histocompatibility complex (MHC) on specialized antigen-presenting cells (APCs) via MHC class II or virus infected and malignant cells via MHC class I (15, 20–22). Early approaches have isolated endogenous tumor-specific T cells from tumor lesions, blood or lymph nodes of patients which were expanded in vitro before adoptive transfer of mono- or oligoclonal repertoires into HLA-complementary recipients with encouraging results (23–26). The establishment of gene transfers to obtain T cells with transgenic TCR in combination with gene editing technologies has made ACT more accessible for the treatment of different disease entities (27, 28) However, the unique repertoire of MHC molecules among humans restricts the broad use of conventional TCRs for therapeutic applications to certain haplotypes (29). Furthermore, the identification of appropriate target and neoantigens is a laborious process supported by novel screening technologies (30).
In contrast, CARs are designed synthetically and consist in their basic embodiment of an extracellular binding domain that is derived from an antibody’s single chain fragment variable (scFv). The antigen-specific scFvs are fused to the intracellular signaling domains of the TCR, such as the CD3ζ chain, followed by one or more co-stimulatory domains (e.g. CD28 or 4-1BB) (31–33). Instead of scFvs, also extracellular portions of ligands or receptors for the target antigen can serve as CAR binding domain (34, 35). Consequently, activation of CARs upon antigen recognition occurs in a MHC-independent manner and does not cause mismatches with endogenous TCR chains (36). Modern gene transfer methods have enabled the generation of autologous and allogeneic CAR T cells from blood for a wide range of patients (19, 37, 38).
Although both TCR and CAR T cell approaches have shown impressive clinical outcomes, there is a necessity to investigate the hurdles and mechanisms that interfere with long-term fitness and response of engineered immune cells to broaden the range of hard-to-treat cancers. Host-specific factors such as the tumor microenvironment (TME) are crucially affecting the response towards immunotherapies.
In this review, we discuss the capacity of the intestinal microbiome to shape the TME, the host immune state, and the molecular mechanisms of microbiome-derived metabolites that modulate T cell responses to improve the efficacy of ACTs.
Over the last decades, insights into host-microbiome interaction have been facilitated by the establishment of gnotobiotic animals with defined colonization as model systems. Especially the dissection of the immune system in germ-free (GF) mice has highlighted the microbiome as a crucial factor for the education of T and B cells as well as for the generation of lymphoid organs. As compared to the standard wildtype strains kept under specific pathogen-free (SPF) conditions, GF mice show reduced antibody levels and repertoire diversity, underdeveloped lymphoid structures and impaired induction of T cell memory response (39–43).
The importance of microbiome-mediated imprinting of the host immune environment was also demonstrated by approaches that aimed for the generation of mouse models that closely resemble the natural mammalian metaorganism including coevolved commensals and pathogens. Laboratory mice are a pillar of biomedical research and many discoveries in immunology (44). It has become evident that the variable reproducibility of results originates from dissonant microbiota among laboratory animal facilities, which might therefore limit the capacity of these models to predict the complexity of the human immune environment. In order to mirror the physiology of free-living mammals in contrast to the sanitized environment in classic lab animals, Rosshart and colleagues developed strains by introducing C57BL/6 embryos into wild mice, such called “wildlings” (43). These wildlings have not only been shown to stably host the natural microbiota over multiple generations and antibiotic, under both dietary and microbial challenges, but also to reflect the clinical observations that could not be predicted in the according pre-clinical studies previously.
While administration of a CD28-superagonist caused an inflammatory cytokine response instead of the originally observed Treg expansion in wildlings, administration of anti-TNF-α treatment to wildlings during lethal endotoxemia did not lead to rescue as observed in conventional animals (45–47). Efforts to characterize wildlings and minimal microbiota consortia are critical for our understanding of how the microbiome is imprinting the host immune environment as a premise for immunotherapeutic interventions (43, 48).
Commensals contribute to gut homeostasis, a fine and dynamic balance of inflammatory and immunosuppressive mechanisms, by inducing the differentiation of regulatory T cells (Tregs) as gatekeepers of peripheral and mucosal tolerance. Simultaneously, members such as segmented filamentous bacteria (SFB) are required for the development of Th17 cells in the mucosa which both orchestrate the intestinal immune response against bacteria but were also identified as a prerequisite for the development of T cell-mediated autoimmunity. While GF mice show resistance to induction of experimental autoimmune encephalomyelitis (EAE), monocolonization with SFB is sufficient to generate mucosal Th17 cells as pathogenic drivers of central nervous system inflammation (49). Interestingly, soluble commensal metabolites have been shown to repress the SFB-mediated onset of EAE either by promoting the differentiation of Tregs or by inducing interleukin (IL)-10 as an anti-inflammatory regulator of the microenvironment (50, 51).
Also in other inflammatory diseases such as Graft-versus-Host-Disease (GVHD), expansion of certain microbiota strains was correlated with disease incidence (4). The lactose-dependent expansion of Enterococcus after allogeneic hematopoietic cell transplantation (allo-HCT) in the intestine of gnotobiotic animals enhanced GVHD severity which was attenuated by dietary depletion of the disaccharide. Similarly, allo-HCT patients with low capabilities to absorb lactose were dominantly colonized with Enterococcus post-antibiotically (4). Despite being involved in shaping an inflammatory environment, the microbiome is a crucial provider of nutrients promoting hematopoietic recovery after bone marrow transplantation (52). Commensal depletion reduced both dietary energy uptake and visceral fat storage and led to worse lymphocyte and neutrophil recovery as compared to WT hosts. The phenotype was restored by the administration of sucrose, compensating the caloric deficits. Interestingly, work on calorie restriction has reported the enrichment of Bifidobacterium bifidum which resulted in increased antitumor immunity by IFN-γ+ CD8 T cells infiltrating the TME (Figure 1) (53). These investigations have contributed to the hypothesis that an interplay between several commensal factors is involved in maintaining physiological homeostasis further strengthening the thought of dysbiosis as a detrimental cause of pathophysiology.
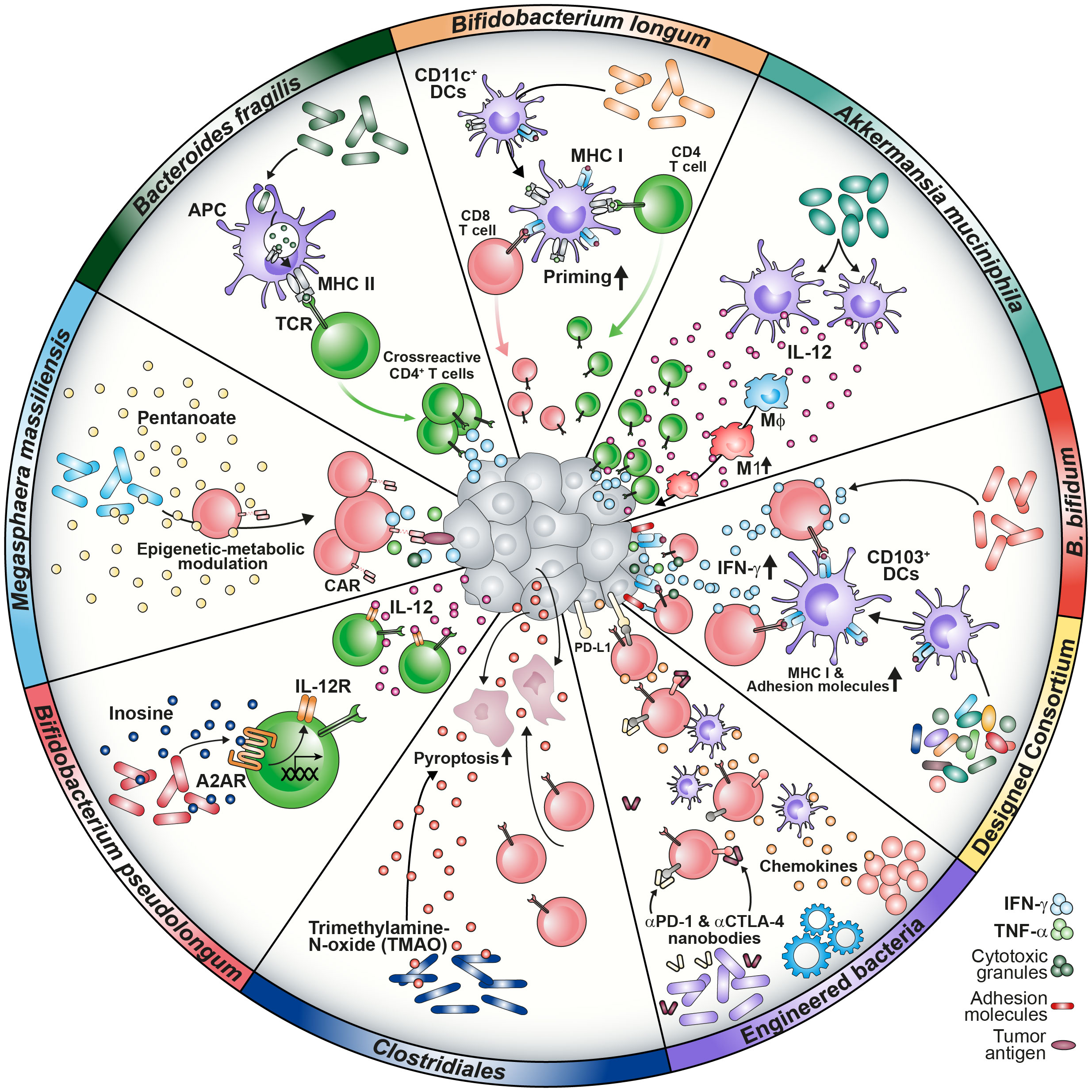
Figure 1 Graphical summary of microbiome-medicated mechanisms improving cancer immunotherapy approaches. Microbial composition shapes a responsive TME in several ways. Strains such as B. longum, B. fragilis, B bifidum and designed consortia have been found to improve T cell priming via increase of MHC class I and II molecules on DCs, tumor infiltration and IFN-γ secretion. Prevalence of A. muciniphila was associated with enhanced IL-12 secretion by DCs causing macrophage maturation towards the M1 phenotype. Microbial metabolites are capable of modulating T cells directly by epigenetic-metabolic reprogramming (M. massiliensis-derived pentanoate) or by inducing the IL-12 receptor on CD4 T cells via the inosine-A2AR axis (B. pseudolongum). TMAO triggers pyroptosis in tumor cells and increases CD8 T cell-mediated antitumor immunity (Clostridiales). Further, engineering of bacteria to produce ICI nanobodies or chemokines were reported to reprogram the TME favorably.
Preclinical studies have contributed to our knowledge about the microbiome-cancer axis and the outcome of immunotherapy. Several aspects, such as the priming and activation of immune cells or the attraction to the tumor site, have been described (54). The investigation of correlations between bacterial strains and immunotherapy outcome was driven by the initial observation that the different microbial composition of laboratory mice derived from either TAC of JAX resulted in different tumor growth kinetics and had an impact on ICI response. Sivan and colleagues identified Bifidobacterium longum as commensal that enhanced T cell priming by dendritic cells (DCs) and their accumulation in tumors, which improved tumor treatment. Administration of the strain further acted synergistically with α-PD-L1 therapy (55). Moreover, Bacteroides fragilis was associated with an improved response to anti-cytotoxic T-lymphocyte-associated protein-4 (α-CTLA-4) blockade due to the development of B. fragilis-specific T cells combating the tumor (Figure 1) (6). This implies that antigen mimicry can be mechanistically involved in inducing antitumor immunity (56).
Analysis of clinical data correlated antibiotics treatment with a decreased survival of NSCLC patients and indicated a positive link between Akkermansia muciniphila abundance and response to α-PD-1 ICI. Of note, fecal microbiota transplantation (FMT) from α-PD-1 responders into either GF or microbiome-depleted animals reflected the clinical outcome in preclinical mouse models in an IL-12-dependent manner (5) (Figure 1). In accordance, first FMT studies have promoted the ICI response in immunotherapy-refractory melanoma patients (57).
The importance of IL-12 as a TME-modulating factor was recently demonstrated by CAR T cells engineered to release the pro-inflammatory cytokine upon CAR engagement (58, 59). Recruitment and enhancement of macrophage function were accompanied by improved killing of tumor cells with antigen-loss in a TNF-α-dependent manner (59). Integration of IL-12 into the CAR exodomain conferred NK-like killing properties to CD8 T cells (58). Of note, intratumoral delivery of the cytokine both supported the antitumor activity of CAR T cells and the repolarization of the TME by attracting infiltrating CD4 T cells, further highlighting that microbiome-induced IL-12 could boost engineered T cell function (60).
Similarly, the synergism between CAR T cells and α-PD-1 blockade was shown in HER2- and GD2-dependent preclinical models (61, 62). Engineering strategies mediating T cell-intrinsic PD-1 pathway interference were tested to improve the potency of CAR T cells (63, 64). However, although the first results from clinical trials combining ACT and ICI have obtained encouraging results as well, no cases with significant increase in CAR T expansion and persistence were reported upon checkpoint blockade (65–67). Based on these insights, future studies might include the microbiome modulation to enhance the response towards ICI in combination with CAR T cell therapy.
The promising results from ICI studies have encouraged research teams to design bacterial consortia capable of shaping the TME favorably to improve T cell response. In accordance with that concept, Tanoue and colleagues generated a defined consortium of 11 strains derived from the human microbiome capable of increasing the frequency of IFN-γ+ CD8 T cells. Colonization with the selected strains improved resistance against Listeria monocytogenes infection as well as ICI in a CD103+ DC-dependent manner (68) (Figure 1). Besides the importance of IFN-γ for T cell differentiation towards anti-viral and –tumoral responses, this cytokine has been shown to act as a crucial modulator of the TME (69–71). Kantari-Mimoun and colleagues described an IFN-γ-dependent two-step process that involves the ICAM-1/LFA-A1 axis, allowing CAR T cells to traffic from the periphery into the tumor islets (72). Similarly, Larsson and colleagues showed the importance of the IFN-γ pathway, highlighting that enhancing CAR T cell binding affinity and adhesion to target cells might increase solid tumor responses (73).
Hence, the design of microbial consortia could be considered as a strategy to prime the TME for efficient CAR T infiltration and response in tumor islets (Figures 1, 2).
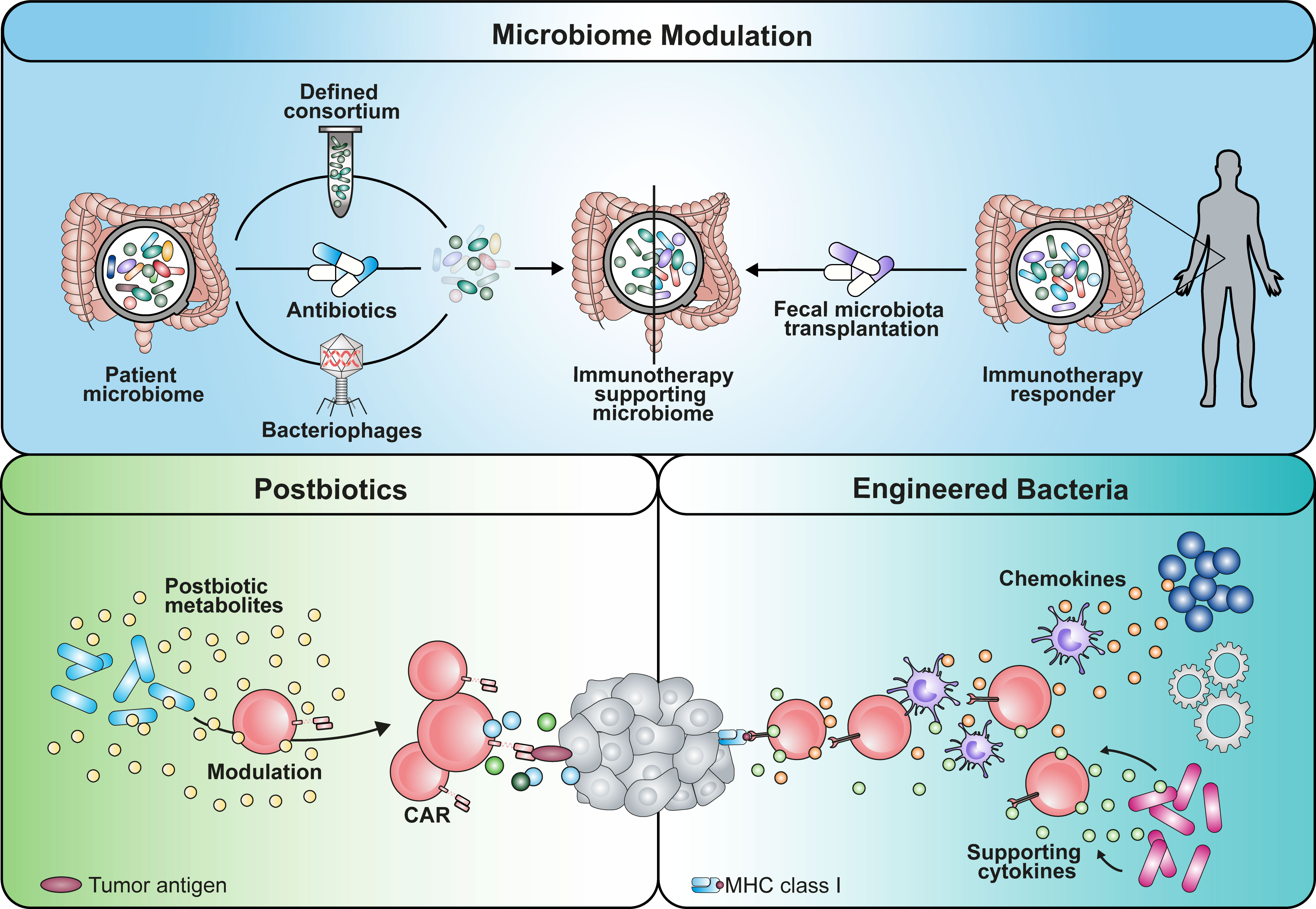
Figure 2 Graphical summary of potential implementation strategies that can be used to apply microbiome-derived mechanisms in cancer immunotherapy. An immunotherapy-favoring microbiome modulation could be achieved by either depleting strains using antibiotics or phages with certain selectivity. Alternatively, establishment of beneficial commensals could be enabled by transplantation of a defined consortium or fecal microbiota of responding patients. Similarly, engineered bacteria reprogramming the TME with soluble mediators is a potential avenue. Additionally, the use of microbial metabolites as postbiotic drugs has the potential to boost immunotherapy.
A study investigating the human cancer microbiome uncovered that intratumoral bacteria are present in various solid malignancies, such as breast and ovarian cancer, lung and pancreatic tumor tissues. Surprisingly, these bacteria could be detected even in those that have no direct communication with the external environment (e.g., glioblastoma or bone tumors) (74). This discovery gave rise to the term oncobiome (74, 75). The presence of tumor-associated bacteria in immunosuppressive micro niches points to a highly organized colonization of transformed tissues that affects the behavior of tumor and immune cells (12). Intriguingly, it was postulated that the cell-associated members of the intratumoral microbiota could drive the migration of cancer cells and impact on the cellular heterogeneity of the TME. Intestinal bacteria and some oral bacteria have been found in colorectal carcinoma (CRC) samples. The orally found commensal Fusobacterium nucleatum was shown to translocate to the colon. Enrichment of the bacterium in the tumor tissue has led to worse radiotherapy outcome and promoted colorectal carcinogenesis (10, 76, 77). Intratumoral colonization was shown to directly modulate the efficacy of chemotherapeutic agents due to metabolization of pharmacologic compounds, highlighting the microbiome as a TME factor affecting several layers of tumor-host interaction (78). Recently, Bender and fellows showed that the as probiotic considered strain Lactobacillus reuteri can translocate to the tumor tissue in a preclinical melanoma model where it is driving the effector function of CTLs depending on a tryptophan-enriched diet (79). Additionally, colonization of the cancerous mammary gland with Clostridiales genera was associated with an activated immune microenvironment due to the presence of bacterial-derived trimethylamine N-oxide (TMAO) that improved immunotherapy response (80) (Figure 1). These results underline not only the involvement of nutrition and diet in shaping the oncobiome, but also a potential for intratumoral bacteria to modulate TME favorably for immune cells (79, 81).
In support of this concept, the engineering of microbes capable of secreting α-PD-L1 or α-CTLA-4 nanobodies to mediate ICI or chemokines such as CXCL16 and CCL20 to recruit CTLs and DCs to the malignancy site has been shown to elicit antitumor effects (82, 83) (Figures 1, 2). Two recently published studies focusing on CD19 CAR T cell intervention for B cell malignancy patients found that antibiotic treatment prior to CAR T cell infusion was correlated with adverse outcomes (8, 9). Especially the administration of piperacillin/tazobactam, imipenem/cilastatin and meropenem (P-I-M) within a 4 week window pre-CAR T cell treatment was associated with worse survival and increased neurotoxicity (9). About 80% of patients receiving CD19 CAR T cell therapy experience cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS). While the analysis of a prospective cohort revealed an association between Clostridia species and day 100 complete response, the results highlighted that both CAR T efficacy and CRS/ICANS are influenced by the microbiome composition which might be responsible for the immune cell activation state and environment. As Clostridia are well known to produce metabolites such as short-chain fatty acids (SCFAs), the lack of bacterial factors in antibiotics-treated patients might contribute to the described adverse outcomes (50, 84). Further, Stein-Thoeringer and colleagues correlated exposure to wide-spectrum antibiotics with decreased survival and CAR T responsiveness with Bacteroides, Ruminococcus, Eubacterium and Akkermansia (8).
An immunostimulating effect of the commensals, which is dampened by antibiotic treatment, could also be attributed to other factors like Toll-like receptor (TLR) ligands. Work from Paulos et al. has demonstrated that adoptive transfer of tumor-specific CD8 T cells into lymphodepleted host show reduced antitumor activity upon reduction of the microbiota via antibiotics or neutralization of serum LPS (85). Similarly, irradiated TLR4-deficient mice showed worse tumor repression compared to WT controls. Notably, the administration of ultrapure LPS to lymphodepleted hosts enhanced the potency of ACT in a syngeneic model. In line with these findings, we have investigated an association between the abundance of the bacterial groups and relapse/progression of disease after allogeneic hematopoitic-cell transplantation (allo-HCT). Analysis of stool samples from 541 patients admitted for allo-HCT was performed via 16S sequencing during a two year-follow up after the treatment. Interestingly, the analysis revealed an association between abundance of Eubacterium limosum and less relapse/disease progression post allo-HCT (86).
A study performed by Hu and colleagues explored the role of the microbiome on CAR T cell-mediated CRS in relapsed/refractory multiple myeloma (87). By collecting microbiota samples before CAR-T infusion, during infusion prior to cytokine storm development, during active cytokine storm, and up to fourteen days after CAR-T treatment, the group could link severe CRS to the reduction of Bifidobacteria abundance and observed a decline in microbiota diversity after CAR T cell administration. Interestingly, the decline was accompanied by higher abundance of Enterococcus and Actinomyces. Complete response in patients was associated with enrichment of Bifidobacterium and Prevotella species. The study is another example of how commensal composition is connected to ACT outcome.
Based on these data, the understanding of TME-modifying probiotic bacteria can contribute to new combinatorial cancer therapies using engineered microbes or defined consortia in synergy with adoptively transferred T cells that benefit from improved function and response.
The microbiome harbors an enormous repertoire of genes encoding for enzymatic pathways which enable the production of small molecules that can serve as postbiotics (Figure 2). These commensal metabolites serve as second messengers that, in contrast to most gut-resident bacteria, can cross the epithelium and diffuse through the lamina propria reaching the systemic circulation (88). Thus, soluble microbial metabolites can modulate immune and non-immune cells in both proximity and distance to the gut bridging the gap between host and microbiome. Initially, the mechanistic investigation of short-chain fatty acids (SCFAs), a dominant class of commensal products, as physiological inducers of Tregs in the intestine has drawn attention to their therapeutic potential.
We have demonstrated that the treatment of CD8 T cells with the SCFA pentanoate enhances the expression of CTL-associated genes such as IFN-γ and TNF-α via epigenetic-metabolic reprogramming. Pentanoate acts as a specific inhibitor of histone deacetylase (HDAC) class I and impacts the mTOR pathway as one of the key metabolic regulators (84, 89). In in vivo experiments, pentanoate-treated TCR-transgenic T cells showed increased antitumor activity and persistence in syngeneic solid tumor models. These features were conferred to CAR T cells which elicited superior tumor control in contrast to the untreated control group in a pancreatic TME. Moreover, acetate, which is abundantly produced by commensals and tumor cells, is capable of fueling the T cell metabolism in glucose-restricted CD8 T cells. The SCFA enhances the IFN-γ secretion in an acetyl-CoA synthetase (ACSS)-dependent manner (90). Although the modulation of histones is one of the intensively studied mechanisms in the SCFA field, their impact on DNA methylation has remained rather understudied. An effect of butyrate has been described as an inductor of ERK phosporylation which in turn down-regulates the DNA methyltransferase DNMT1 (91). Consequently, demethylation of tumor suppressor genes was observed. However, more in-depth research in immune cells needs to be done to assess the mechanisms involved in microbiome-mediated modulation of DNA methylation which bears a strong potential for ACTs. Recent work has highlighted DNA methylation as reprogramming mechanism capable of determining CAR T cell fate (92).
Remarkably, although tumor-derived lactate has been identified as a glycolysis-derived metabolite with immunosuppressive characteristics in the TME that induces M2-like polarization in TAMs, a new study has reported an increase in CD8 T cell stemness and antitumor response following lactate treatment (93, 94). Mechanistically, lactate mediated these effects by suppressing HDAC activity, which caused hyperacetylation at H3K27 of the Tcf7 super enhancer locus. The subsequent Tcf7 gene expression was associated with a stem-like phenotype (95). These results raise a question about the involvement of the dominant commensal-derived lactate and Lactobacillus strains in tuning T cell function and immune cell activation (4, 94).
Denk and colleagues identified that urolithin A (UA), a metabolite derived from the conversion of ellagitannins by the gut microbiome, is improving mitochondrial health. By engaging the Pink1-Pgam5 axis, UA triggered mitophagy and compensatory mitochondrial biogenesis causing the differentiation of T memory stem cells (TSCMs) with superior CD8 antitumor immunity (96).
Not only CD8 T cells, but also CD4 T cells appear to be modulated by microbial metabolites. The analysis of Bifidobacterium pseudolongum in the context of immunotherapy revealed that the production of inosine induced the expression of the IL-12 receptor on CD4 T cells via the adenosine A2A receptor (Figure 1). Translocation of inosine through a leaky gut barrier into the systemic circulation improved antitumor response. The crucial role of CD4 CAR T cells was recently pointed out by Melenhorst and colleagues, who observed that CD4+ CAR T cells dominated the CAR T cell population of patients with decade-long leukemia remission at later monitoring time points. Those cells maintained cytotoxic characteristics and proliferation (97, 98).
These insights suggest that commensal metabolites with the capacity to reprogram host immune cells can be utilized as postbiotic physiologic drugs to potentiate effector function and memory features of ACT products. Furthermore, commensal-based adjuvants might provide new tools to achieve long-term function and antitumor reactivity of CAR T cells.
The microbiome is a complex network of microorganisms that directly and indirectly influence host physiology and immunological homeostasis. The education of the host immune system is a prerequisite not only for a functional defense against infectious diseases and malignant transformation but also for promoting tolerance. Studies have demonstrated that response and resistance to cancer immunotherapy is dependent on the immune cell activity within the TME, which can be modulated by the intestinal or translocated microbiome. On one hand, these findings allow us to think about the microbial composition as a predictive indicator that could guide the choice of the immunotherapeutic approach to achieve long-lasting remission (9, 87, 99).
On the other hand, understanding how commensals prime the TME can be an important asset to shape the surrounding for endogenous and engineered T cells beneficially. More detailed knowledge about the affected cell populations in the TME, such as myeloid-derived suppressor cells and tumor-associated macrophages, is necessary. Based on these insights, different strategies have the potential to be implemented in clinical setting (Figure 2). The first approach might include the administration of defined microbial consortia as probiotics or of personalized antibiotic treatment regimens that shape the microbiome favorably (9, 68). Using strain-specific phages could be an alternative technology with higher precision compared to antibiotic treatment (100). In line with this concept, fecal microbiota transplantation (FMT) from donors that have shown response to immunotherapy might be capable of overcoming resistance mechanisms. As a second approach, engineering of microbes for intratumoral colonization can be used synergistically with ACTs (82, 83). These are able to reprogram the TME by either secreting modulatory factors themselves (e.g. antibodies, cytokines) or recruiting endogenous immune cells using chemokines (101, 102). However, we need to keep in mind that the inter-individual diversity of the microbiome remains a challenge in the process of creating the optimal formulation for a designer consortium. Further research needs to be performed to clarify whether a “one-size fits all” drug is more suitable rather than a personalized medicine approach.
While TME reprogramming is a potential avenue to obtain a less suppressive milieu, rewiring immune cells themselves to overcome the latter is an additional course. Commensal-derived metabolites that bridge the host-microbiome interaction are a powerful source of physiological molecules with drug-like properties, also considered as postbiotics (e.g. SCFAs). While their production within the host can be modulated and utilized by diet, metabolite libraries might be valuable to screen and identify novel tools to improve ACTs and immune cell engineering (7, 79, 84, 96, 103).
A crucial prerequisite to obtain the knowledge required might be the availability of preclinical models that reflect the human microbiome and its influences on host immunity as well as immunotherapy. Previous studies have pointed out that the transfer of human microbiota into gnotobiotic mice was capable to reflect the ICI responses in patients (5, 6). Also the establishment of wilding colonies had a strong predictive value with regards to the outcome of clinical studies (43). These revolutionary steps are still in need of standardization to assure reproducibility and accessibility of “humanized” gnotobiotic animals for a broad community of researchers (43, 104).
For a better understanding of the host-microbiome interaction, an interdisciplinary approach is needed that synergizes bioinformatics and systems biology, molecular microbiology, immunology and biotechnology in order to facilitate bench-to-bedside and back translation. As research labs are highly specialized and have limited capacities to cover both extensive clinical data collection from international cohorts and molecular studies of the microbiome-mediated effects, innovative ecosystems are required to move the field forward. Within the EU-funded research consortium T2EVOLVE, a public-private partnership of CAR and TCR T cell key opinion leaders, we aim to accelerate the development and improve access to engineered T cell therapy (105, 106). Synergies with the microbiome-CAR T consortium CARTOMICS will establish an infrastructure that boosts collaborative multidisciplinary work between partners, stakeholders, cell engineering approaches and microbiome-associated facets that guide the next-generation of cellular immunotherapy.
SS: Writing – original draft, Writing – review & editing. KZ-M: Writing – original draft, Writing – review & editing. AV: Writing – original draft, Writing – review & editing, Funding acquisition. JS: Writing – original draft, Writing – review & editing. RS: Writing – original draft, Writing – review & editing. MH: Writing – original draft, Writing – review & editing. MvdB: Writing – original draft, Writing – review & editing. ML: Writing – original draft, Conceptualization, Funding acquisition, Visualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No. 116026 (T2EVOLVE; MH and ML). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA. Moreover, funding was received from the Wilhelm-Sander-Stiftung (Grant No. 2022.134.1, AV and ML), ERA-NET TRANSCAN-3 (EC co-funded call 2021, SmartCAR-T; KZM, MH and ML), the Paula & Rodger Riney Foundation (MvdB, MH and ML), IZKF Würzburg (S-511; SS), the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, TRR 221, subproject A03; and TRR338, subproject A02; MH and ML), the Bavarian Center for Cancer Research (Bayerisches Zentrum für Krebsforschung, BZKF; MH and ML), the National Cancer Institute award numbers, R35-CA284024, P01-CA023766, R01-CA228308, and P30 CA008748 MSK Cancer Center Support Grant/Core Grant; National Heart, Lung, and Blood Institute (NHLBI) award number R01-HL164902; National Institute on Aging award number P01-AG052359, and TriInstitutional Stem Cell Initiative. Additional funding was received from The Lymphoma Foundation, The Susan and Peter Solomon Family Fund, The Solomon Microbiome Nutrition and Cancer Program, Cycle for Survival, Parker Institute for Cancer Immunotherapy, Research Initiative, Starr Cancer Consortium, and Seres Therapeutics (RS, JS and MvdB). This publication was supported by the Open Access Publication Fund of the University of Würzburg.
ML, MH, and AV are inventors on a patent application related to the use of pentanoate that has been filed by Philipps-University Marburg and Julius-Maximilians University Würzburg WO2021/058811A1. MH is inventor on patent applications related to CAR T cell manufacturing. MH is listed as an inventor on patent applications and granted patents related to CAR-T technologies that have been filed by the Fred Hutchinson Cancer Research Center, Seattle, WA and by the University of Würzburg, Würzburg, Germany. MH is a co-founder and equity owner of T-CURX GmbH, Würzburg, Germany. MH received honoraria from Celgene/BMS, Janssen, Kite/Gilead. MvdB has received research support and stock options from Seres Therapeutics and stock options from Notch Therapeutics and Pluto Therapeutics; he has received royalties from Wolters Kluwer; has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, Rheos Medicines, Ceramedix, Pluto Therapeutics, Thymofox, Garuda, Novartis (Spouse), Synthekine (Spouse), Beigene (Spouse), Kite (Spouse); he has IP Licensing with Seres Therapeutics and Juno Therapeutics; and holds a fiduciary role on the Foundation Board of DKMS (a nonprofit organization).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66. doi: 10.1038/nm.3444
2. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
3. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity (2016) 44:951–3. doi: 10.1016/j.immuni.2016.04.006
4. Stein-Thoeringer CK, Nichols KB, Lazrak A, Docampo MD, Slingerland AE, Slingerland JB, et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science (2019) 366:1143–9. doi: 10.1126/science.aax3760
5. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359:91–7. doi: 10.1126/science.aan3706
6. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350:1079–84. doi: 10.1126/science.aad1329
7. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369:1481–9. doi: 10.1126/science.abc3421
8. Stein-Thoeringer CK, Saini NY, Zamir E, Blumenberg V, Schubert M-L, Mor U, et al. A non-antibiotic-disrupted gut microbiome is associated with clinical responses to CD19-CAR-T cell cancer immunotherapy. Nat Med (2023) 1–11. doi: 10.1038/s41591-023-02234-6
9. Smith M, Dai A, Ghilardi G, Amelsberg KV, Devlin SM, Pajarillo R, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med (2022) 28:713–23. doi: 10.1038/s41591-022-01702-9
10. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
11. McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PloS One (2013) 8:e53653. doi: 10.1371/journal.pone.0053653
12. Galeano Niño JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature (2022) 611:810–7. doi: 10.1038/s41586-022-05435-0
13. June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Trans Med (2015) 7:280ps7. doi: 10.1126/scitranslmed.aaa3643
14. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
15. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
16. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. New Engl J Med (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
17. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
18. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
19. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New Engl J Med (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
20. Birnbaum ME, Berry R, Hsiao Y-S, Chen Z, Shingu-Vazquez MA, Yu X, et al. Molecular architecture of the αβ T cell receptor-CD3 complex. Proc Natl Acad Sci (2014) 111:17576–81. doi: 10.1073/pnas.1420936111
21. Szeto C, Lobos CA, Nguyen AT, Gras S. TCR recognition of peptide-MHC-I: rule makers and breakers. Int J Mol Sci (2020) 22. doi: 10.3390/ijms22010068
22. van Laethem F, Tikhonova AN, Singer A. MHC restriction is imposed on a diverse T cell receptor repertoire by CD4 and CD8 co-receptors during thymic selection. Trends Immunol (2012) 33:437–41. doi: 10.1016/j.it.2012.05.006
23. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Trans Med (2013) 5:174ra27. doi: 10.1126/scitranslmed.3004916
24. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A Preliminary Rep N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
25. Yoshizawa H, Chang AE, Shu S. Specific adoptive immunotherapy mediated by tumor-draining lymph node cells sequentially activated with anti-CD3 and IL-2. J Immunol (1991) 147:729–37.
26. Stevens EJ, Jacknin L, Robbins PF, Kawakami Y, el Gamil M, Rosenberg SA, et al. Generation of tumor-specific CTLs from melanoma patients by using peripheral blood stimulated with allogeneic melanoma tumor cell lines. Fine specificity and MART-1 melanoma antigen recognition. J Immunol (1995) 154:762–71.
27. Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, et al. A pilot trial of the combination of transgenic NY-ESO-1-reactive adoptive cellular therapy with dendritic cell vaccination with or without ipilimumab. Clin Cancer Res (2019) 25:2096–108. doi: 10.1158/1078-0432.CCR-18-3496
28. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18:807–15. doi: 10.1038/nm.2700
29. Song S, Han M, Zhang H, Wang Y, Jiang H. Full screening and accurate subtyping of HLA-A*02 alleles through group-specific amplification and mono-allelic sequencing. Cell Mol Immunol (2013) 10:490–6. doi: 10.1038/cmi.2013.33
30. Tretter C, de Andrade Krätzig N, Pecoraro M, Lange S, Seifert P, Frankenberg C, et al. Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification. Nat Commun (2023) 14:4632. doi: 10.1038/s41467-023-39570-7
31. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
32. Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Therapy: J Am Soc Gene Ther (2010) 18:413–20. doi: 10.1038/mt.2009.210
33. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U.S.A. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
34. Seif M, Kakoschke TK, Ebel F, Bellet MM, Trinks N, Renga G, et al. CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci Trans Med (2022) 14:eabh1209. doi: 10.1126/scitranslmed.abh1209
35. Schmidts A, Ormhøj M, Choi BD, Taylor AO, Bouffard AA, Scarfò I, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv (2019) 3:3248–60. doi: 10.1182/bloodadvances.2019000703
36. Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun (1987) 149:960–8. doi: 10.1016/0006-291x(87)90502-x
37. Jetani H, Navarro-Bailón A, Maucher M, Frenz S, Verbruggen C, Yeguas A, et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood (2021) 138:1830–42. doi: 10.1182/blood.2020009192
38. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
39. Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, Wu W-JH, Saldana-Morales FB, et al. Thymic development of gut-microbiota-specific T cells. Nature (2021) 594:413–7. doi: 10.1038/s41586-021-03531-1
40. Ohwaki M, Yasutake N, Yasui H, Ogura R. A comparative study on the humoral immune responses in germ-free and conventional mice. Immunology (1977) 32:43–8.
41. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
42. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity (2011) 34:794–806. doi: 10.1016/j.immuni.2011.03.021
43. Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science (2019) 365. doi: 10.1126/science.aaw4361
44. Vandamme TF. Use of rodents as models of human diseases. J Pharm Bioallied Sci (2014) 6:2–9. doi: 10.4103/0975-7406.124301
45. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl J Med (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
46. Lin C-H, Hünig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol (2003) 33:626–38. doi: 10.1002/eji.200323570
47. Ashkenazi A, Marsters SA, Capon DJ, Chamow SM, Figari IS, Pennica D, et al. Protection against endotoxic shock by a tumor necrosis factor receptor immunoadhesin. Proc Natl Acad Sci U.S.A. (1991) 88:10535–9. doi: 10.1073/pnas.88.23.10535
48. Fischer F, Romero R, Hellhund A, Linne U, Bertrams W, Pinkenburg O, et al. Dietary cellulose induces anti-inflammatory immunity and transcriptional programs via maturation of the intestinal microbiota. Gut Microbes (2020) 12:1–17. doi: 10.1080/19490976.2020.1829962
49. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
50. Luu M, Pautz S, Kohl V, Singh R, Romero R, Lucas S, et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun (2019) 10:760. doi: 10.1038/s41467-019-08711-2
51. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci (2011) 108 Suppl 1:4615–22. doi: 10.1073/pnas.1000082107
52. Staffas A, Da Burgos Silva M, Slingerland AE, Lazrak A, Bare CJ, Holman CD, et al. Nutritional support from the intestinal microbiota improves hematopoietic reconstitution after bone marrow transplantation in mice. Cell Host Microbe (2018) 23:447–457.e4. doi: 10.1016/j.chom.2018.03.002
53. Mao Y-Q, Huang J-T, Zhang S-L, Kong C, Li Z-M, Jing H, et al. The antitumour effects of caloric restriction are mediated by the gut microbiome. Nat Metab (2023) 5:96–110. doi: 10.1038/s42255-022-00716-4
54. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell (2018) 33:570–80. doi: 10.1016/j.ccell.2018.03.015
55. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350:1084–9. doi: 10.1126/science.aac4255
56. Boesch M, Baty F, Rothschild SI, Tamm M, Joerger M, Früh M, et al. Tumour neoantigen mimicry by microbial species in cancer immunotherapy. Br J Cancer (2021) 125:313–23. doi: 10.1038/s41416-021-01365-2
57. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science (2021) 371:602–9. doi: 10.1126/science.abb5920
58. Hombach A, Barden M, Hannappel L, Chmielewski M, Rappl G, Sachinidis A, et al. IL12 integrated into the CAR exodomain converts CD8+ T cells to poly-functional NK-like cells with superior killing of antigen-loss tumors. Mol Therapy: J Am Soc Gene Ther (2022) 30:593–605. doi: 10.1016/j.ymthe.2021.10.011
59. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
60. Agliardi G, Liuzzi AR, Hotblack A, Feo D, Núñez N, Stowe CL, et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun (2021) 12:444. doi: 10.1038/s41467-020-20599-x
61. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther (2016) 24:1135–49. doi: 10.1038/mt.2016.63
62. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
63. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/JCI83092
64. Lesch S, Nottebrock A, Rataj F, Heise C, Endres S, Kobold S. PD-1-CD28 fusion protein strengthens mesothelin-specific TRuC T cells in preclinical solid tumor models. Cell Oncol (Dordr) (2023) 46:227–35. doi: 10.1007/s13402-022-00747-9
65. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
66. Hill BT, Roberts ZJ, Xue A, Rossi JM, Smith MR. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transplant (2020) 55:1184–7. doi: 10.1038/s41409-019-0657-3
67. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
68. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
69. Song M, Ping Y, Zhang K, Yang L, Li F, Zhang C, et al. Low-dose IFNγ Induces tumor cell stemness in tumor microenvironment of non-small cell lung cancer. Cancer Res (2019) 79:3737–48. doi: 10.1158/0008-5472.CAN-19-0596
70. Ni C, Wu P, Zhu X, Ye J, Zhang Z, Chen Z, et al. IFN-γ selectively exerts pro-apoptotic effects on tumor-initiating label-retaining colon cancer cells. Cancer Lett (2013) 336:174–84. doi: 10.1016/j.canlet.2013.04.029
71. Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer (2011) 128:371–8. doi: 10.1002/ijc.25350
72. Kantari-Mimoun C, Barrin S, Vimeux L, Haghiri S, Gervais C, Joaquina S, et al. CAR T-cell entry into tumor islets is a two-step process dependent on IFNγ and ICAM-1. Cancer Immunol Res (2021) 9:1425–38. doi: 10.1158/2326-6066.CIR-20-0837
73. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature (2022) 604:563–70. doi: 10.1038/s41586-022-04585-5
74. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (2020) 368:973–80. doi: 10.1126/science.aay9189
75. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The microbiome and human cancer. Science (2021) 371. doi: 10.1126/science.abc4552
76. Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, et al. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep (2019) 20. doi: 10.15252/embr.201847638
77. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell (2017) 170:548–563.e16. doi: 10.1016/j.cell.2017.07.008
78. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (2017) 357:1156–60. doi: 10.1126/science.aah5043
79. Bender MJ, McPherson AC, Phelps CM, Laughlin CR, Shapira JH, Medina Sanchez L, et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell (2023) 186:1846–1862.e26. doi: 10.1016/j.cell.2023.03.011
80. Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab (2022) 34:581–594.e8. doi: 10.1016/j.cmet.2022.02.010
81. Simpson RC, Shanahan ER, Batten M, Reijers ILM, Read M, Silva IP, et al. Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiome. Nat Med (2022) 28:2344–52. doi: 10.1038/s41591-022-01965-2
82. Gurbatri CR, Lia I, Vincent R, Coker C, Castro S, Treuting PM, et al. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci Trans Med (2020) 12. doi: 10.1126/scitranslmed.aax0876
83. Savage TM, Vincent RL, Rae SS, Huang LH, Ahn A, Pu K, et al. Chemokines expressed by engineered bacteria recruit and orchestrate antitumor immunity. Sci Adv (2023) 9:eadc9436. doi: 10.1126/sciadv.adc9436
84. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat Commun (2021) 12:4077. doi: 10.1038/s41467-021-24331-1
85. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest (2007) 117:2197–204. doi: 10.1172/JCI32205
86. Peled JU, Devlin SM, Staffas A, Lumish M, Khanin R, Littmann ER, et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J Clin Oncol (2017) 35:1650–9. doi: 10.1200/JCO.2016.70.3348
87. Hu Y, Li J, Ni F, Yang Z, Gui X, Bao Z, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic Malignancies. Nat Commun (2022) 13:5313. doi: 10.1038/s41467-022-32960-3
88. Chen L, Zhernakova DV, Kurilshikov A, Andreu-Sánchez S, Wang D, Augustijn HE, et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat Med (2022) 28:2333–43. doi: 10.1038/s41591-022-02014-8
89. Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep (2018) 8:14430. doi: 10.1038/s41598-018-32860-x
90. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27:2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022
91. Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res (2011) 31:2723–32.
92. Zebley CC, Brown C, Mi T, Fan Y, Alli S, Boi S, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep (2021) 37:110079. doi: 10.1016/j.celrep.2021.110079
93. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513:559–63. doi: 10.1038/nature13490
94. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun (2022) 13:4981. doi: 10.1038/s41467-022-32521-8
95. Pais Ferreira D, Silva JG, Wyss T, Fuertes Marraco SA, Scarpellino L, Charmoy M, et al. Central memory CD8+ T cells derive from stem-like Tcf7hi effector cells in the absence of cytotoxic differentiation. Immunity (2020) 53:985–1000.e11. doi: 10.1016/j.immuni.2020.09.005
96. Denk D, Petrocelli V, Conche C, Drachsler M, Ziegler PK, Braun A, et al. Expansion of T memory stem cells with superior anti-tumor immunity by Urolithin A-induced mitophagy. Immunity (2022) 55:2059–2073.e8. doi: 10.1016/j.immuni.2022.09.014
97. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
98. Bove C, Arcangeli S, Falcone L, Camisa B, El Khoury R, Greco B, et al. CD4 CAR-T cells targeting CD19 play a key role in exacerbating cytokine release syndrome, while maintaining long-term responses. J ImmunoTherapy Cancer (2023) 11. doi: 10.1136/jitc-2022-005878
99. Pianko MJ, Devlin SM, Littmann ER, Chansakul A, Mastey D, Salcedo M, et al. Minimal residual disease negativity in multiple myeloma is associated with intestinal microbiota composition. Blood Adv (2019) 3:2040–4. doi: 10.1182/bloodadvances.2019032276
100. Li Y, Zhu F, Li Y, Pan S, Wang H, Yang Z, et al. Bacteriophages allow selective depletion of gut bacteria to produce a targeted-bacterium-depleted mouse model. Cell Rep Methods (2022) 2:100324. doi: 10.1016/j.crmeth.2022.100324
101. Saltzman DA, Heise CP, Hasz DE, Zebede M, Kelly SM, Curtiss R, et al. Attenuated Salmonella typhimurium containing interleukin-2 decreases MC-38 hepatic metastases: a novel anti-tumor agent. Cancer Biother Radiopharm (1996) 11:145–53. doi: 10.1089/cbr.1996.11.145
102. Loeffler M, Le’Negrate G, Krajewska M, Reed JC. IL-18-producing Salmonella inhibit tumor growth. Cancer Gene Ther (2008) 15:787–94. doi: 10.1038/cgt.2008.48
103. Dekkers KF, Sayols-Baixeras S, Baldanzi G, Nowak C, Hammar U, Nguyen D, et al. An online atlas of human plasma metabolite signatures of gut microbiome composition. Nat Commun (2022) 13:5370. doi: 10.1038/s41467-022-33050-0
104. Park JC, Im S-H. Of men in mice: the development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp Mol Med (2020) 52:1383–96. doi: 10.1038/s12276-020-0473-2
105. Donnadieu E, Luu M, Alb M, Anliker B, Arcangeli S, Bonini C, et al. Time to evolve: predicting engineered T cell-associated toxicity with next-generation models. J ImmunoTherapy Cancer (2022) 10. doi: 10.1136/jitc-2021-003486
Keywords: microbiome, immunotherapy, immunology, cancer immune cell therapy, CAR T cell
Citation: Staudt S, Ziegler-Martin K, Visekruna A, Slingerland J, Shouval R, Hudecek M, van den Brink M and Luu M (2023) Learning from the microbes: exploiting the microbiome to enforce T cell immunotherapy. Front. Immunol. 14:1269015. doi: 10.3389/fimmu.2023.1269015
Received: 28 July 2023; Accepted: 30 August 2023;
Published: 18 September 2023.
Edited by:
Marco Orecchioni, Augusta University, United StatesReviewed by:
Hossam Abdelsamed, University of Pittsburgh Medical Center, United StatesCopyright © 2023 Staudt, Ziegler-Martin, Visekruna, Slingerland, Shouval, Hudecek, van den Brink and Luu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maik Luu, THV1X21AdWt3LmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.