 Denho Ravi1,2
Denho Ravi1,2 Erato Ntinopoulou1,2
Erato Ntinopoulou1,2 Nessim Guetta1,2
Nessim Guetta1,2 Manuela Weier1,2
Manuela Weier1,2 Verena Vogel3
Verena Vogel3 Barbara Spellerberg3†Parham Sendi4†
Barbara Spellerberg3†Parham Sendi4† Sandrine Gremlich1,2
Sandrine Gremlich1,2 Thierry Roger2†
Thierry Roger2† Eric Giannoni1,2*†
Eric Giannoni1,2*†- 1Clinic of Neonatology, Department Mother-Woman-Child, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 2Infectious Diseases Service, Department of Medicine, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 3Institute of Medical Microbiology and Hygiene, University of Ulm, Ulm, Germany
- 4Institute for Infectious Diseases, University of Bern, Bern, Switzerland
Introduction: Streptococcus agalactiae (Group B Streptococcus, GBS) is a leading pathogen of neonatal sepsis. The host-pathogen interactions underlying the progression to life-threatening infection in newborns are incompletely understood. Macrophages are first line in host defenses against GBS, contributing to the initiation, amplification, and termination of immune responses. The goal of this study was to compare the response of newborn and adult monocyte-derived macrophages (MDMs) to GBS.
Methods: Monocytes from umbilical cord blood of healthy term newborns and from peripheral blood of healthy adult subjects were cultured with M-CSF to induce MDMs. M-CSF-MDMs, GM-CSF- and IFNγ-activated MDMs were exposed to GBS COH1, a reference strain for neonatal sepsis.
Results: GBS induced a greater release of IL-1β, IL-6, IL-10, IL-12p70 and IL-23 in newborn compared to adult MDMs, while IL-18, IL-21, IL-22, TNF, RANTES/CCL5, MCP-1/CCL2 and IL-8/CXCL8 were released at similar levels. MDM responses to GBS were strongly influenced by conditions of activation and were distinct from those to synthetic bacterial lipopeptides and lipopolysaccharides. Under similar conditions of opsonization, newborn MDMs phagocytosed and killed GBS as efficiently as adult MDMs.
Discussion: Altogether, the production of excessive levels of Th1- (IL-12p70), Th17-related (IL-1β, IL-6, IL-23) and anti-inflammatory (IL-10) cytokines is consistent with a dysregulated response to GBS in newborns. The high responsiveness of newborn MDMs may play a role in the progression of GBS infection in newborns, possibly contributing to the development of life-threatening organ dysfunction.
Introduction
Every year, 2.5 million children aged less than five years, including over half a million newborns die from sepsis (1, 2). Despite improvements in perinatal care, Streptococcus agalactiae (Group B Streptococcus, GBS) remains a leading pathogen in early-life, and a major cause of neonatal sepsis (3–5). The mortality rate of GBS neonatal sepsis is 5% in high-income countries and 20% globally, and long-term disability occurs in 20% of survivors (4, 6, 7). It is believed that the developmental status of the immune system of neonates plays a key role in the susceptibility to GBS disease (8). Yet, our limited knowledge of the reasons underlying the transition from colonization by GBS to invasion and progression to life-threatening infection hampers the development of innovative strategies targeting GBS disease.
To ensure protection against infection, newborns rely mainly on their innate immune system and bioactive molecules (such as immunoglobulins and antimicrobial peptides) transmitted from the mother through the placenta in utero or through breastmilk postnatally (9, 10). Quantitative and qualitative differences between the neonatal and the adult innate immune systems have been described (9, 10). Concentrations of complement proteins and other antimicrobial peptides are reduced in newborns. In addition, recruitment of neutrophils to sites of infection and ability to phagocytose and kill pathogens are limited. Newborn dendritic cells and monocytes exposed to lipopolysaccharides (LPS) release lower amounts of the pro-inflammatory and Th1-polarizing cytokines tumor necrosis factor (TNF), IL-1β, IL-12p70, but similar or even higher levels of Th17-polarizing and anti-inflammatory cytokines IL-6, IL-23 and IL-10 compared to adult cells (11–16). However, the neonatal immune responses to GBS might differ from responses to purified microbial products (17–20). Moreover, newborns affected by GBS disease have strong systemic pro- and anti-inflammatory responses (21, 22), consistent with the observation that a dysregulated host response to infection contributes to the pathogenesis of sepsis (23).
Macrophages are distributed across all tissues, serving as resident innate immune cells (24). The differentiation, activation and function of macrophages are shaped by environmental cues, spanning from the induction of classically activated pro-inflammatory M1 macrophages to the promotion of alternatively activated pro-resolving/anti-inflammatory M2 macrophages (25). Macrophages play a crucial role to orchestrate host immunity and inflammatory response with the release of a large panel of cytokines (24). In murine models of pneumonia, newborn alveolar macrophages have a lower capacity to phagocytose and kill GBS compared to adult cells, suggesting that reduced macrophage responses might contribute to the vulnerability of newborns to this pathogen (26, 27). Yet, studies investigating the responses of human newborn macrophages to GBS are lacking.
Given the importance of macrophages in host defenses against bacteria and in the pathogenesis of sepsis (23), a deeper understanding of the interactions between GBS and newborn macrophages is needed. We developed a robust in vitro model to compare the responses of primary human newborn and adult monocyte-derived macrophages (MDMs) exposed to GBS. The phagocytosis, bacterial killing, production of cytokines and viability of MDMs was quantified under different conditions of macrophage activation. Here, we report a picture consistent with a dysregulated cytokine response with a conserved capacity to phagocytose and kill GBS in newborns.
Materials and methods
Ethical statement
The study protocol was approved by the ethics committee of the Canton de Vaud, Switzerland (CER-VD, project #2019-01772). Written informed consent was obtained from the mothers for the collection of umbilical cord blood and from adult volunteers for peripheral blood.
Blood samples
Umbilical cord blood was collected from 20 healthy term newborns by puncture of placental vessels, after delivery of the placenta. Blood was obtained from 18 healthy adult volunteers (age 20-59 years) by puncture of a peripheral vein. S-Monovettes® containing EDTA (Sarstedt AG & Co. KG) were used to collect blood.
Isolation, culture and activation of cells
Mononuclear cells were isolated from blood by gradient density centrifugation using Ficoll-Paque™ PLUS solution (GE Healthcare). CD14+ monocytes were purified from mononuclear cells by positive selection using magnetic microbeads coupled to anti-human CD14 antibodies (Miltenyi Biotec) (28–30). Cells were incubated with Pacific Blue™ anti-human CD14 antibody (BioLegend) to determine the purity of each monocyte preparation. Data were acquired using an Attune Nxt Flow Cytometer (ThermoFisher) and analysed using FlowJo (version 10.7, FlowJo LLC). For experiments, we only used monocyte preparations with a purity above 90%. The viability of mononuclear cells and monocytes was determined by trypan blue exclusion using a CountessTM Automated cell counter (Invitrogen). Monocytes (105 cells/well) were cultured in 96 well plates for seven days at 37°C, 5% CO2, in RPMI 1640 (ThermoFisher) supplemented with 10% native human serum (HS, from human male AB plasma, Sigma-Aldrich Corp.), 1% penicillin-streptomycin (ThermoFisher) and 50 ng/ml human macrophage colony-stimulating factor (M-CSF, PeproTech EC Ltd.) to induce their differentiation into monocyte-derived macrophages (MDMs). After 7 days, the medium was changed to RPMI containing 10% HS and different concentrations ranging from 10 to 100 ng/ml (50 ng/ml if not otherwise stated) of human M-CSF, granulocyte-macrophage colony-stimulating factor (GM-CSF, PeproTech EC Ltd.) or interferon gamma (IFNγ, PBL Assay Science) (30). Cells were incubated for 24 hours to induce GM-CSF- or IFNγ-activated MDMs (GM-CSF-MDMs, IFNγ-MDMs), or resting macrophages (M-CSF-MDMs) (31).
Preparation of GBS COH1
All experiments were performed with GBS COH1 (American Type Culture Collection BAA-1176), a serotype III sequence type 17 reference strain obtained from a newborn with sepsis (32). We incubated bacteria overnight at 37°C, with 5% CO2 in Brain Heart Infusion (BHI, ThermoFisher) broth. New tubes containing BHI medium were inoculated with overnight cultures (1:40) and incubated for two hours, to reach mid-log phase of growth, corresponding to 6 x 108 bacteria/ml. Bacteria were washed and resuspended in ice-cold phosphate-buffered saline solution (PBS) at 2 x 105 - 107 bacteria/100 µl. GBS was plated on Columbia III agar with 5% sheep blood (BD Biosciences) to determine the concentration of our bacterial culture. The minimum inhibitory concentration (MIC) of GBS COH1 to gentamicin was 64 µg/ml. MIC was determined by overnight BHI liquid cultures of GBS COH1 in 96 well plates (37°C, 5% CO2) with serial dilution of gentamicin (Sigma-Aldrich) ranging from 0.5 to 512 µg/ml. At the end of incubation, proliferation of bacteria was determined by optical density (O.D. 600 nm).
Cytokine measurements
M-CSF-, GM-CSF- and IFNγ-MDMs were exposed to 107 GBS per well or 0.2 µg/ml lipopolysaccharide (LPS, Invivogen), or 2 µg/ml Pam3CysSerLys4 (Pam3CSK4, Invivogen). After one hour of incubation, we added gentamicin to reach a final concentration of 100 µg/ml. Cell-culture supernatants were collected after 18 hours to quantify cytokines by ELISA (BD Biosciences, for human TNF) and by ProcartaPlex immunoassays (Human Custom ProcartaPlex 16-plex, Invitrogen, for IL-1β, IL-1RA, IL-6, IL-8, IL-10, IL-12p70, IL-18, IL-20, IL-21, IL-22, IL-23, IL-27, MCP-1, MIF, RANTES, and IFNβ).
Gentamicin protection assay
The principle behind this method is to count bacteria (colony-forming units: CFU) inside macrophages after precise intervals of time in order to determine phagocytosis and intracellular killing of bacteria (33). Gentamicin is used to kill bacteria that are outside but not those that are inside macrophages, preventing extracellular growth of bacteria. M-CSF-, GM-CSF-, and IFNγ-MDMs in 96 well plates were exposed to 2 x 105 GBS/well to determine phagocytosis and intracellular killing. Plates were centrifuged 15 minutes at 50 RCF and 37°C to maximize contact between bacteria and MDMs, and incubated for 45 minutes. Cells were washed twice with warm PBS, and incubated 30 minutes in RPMI 10% HS containing 10-100 ng/ml M-CSF, GM-CSF or IFNγ and 100 µg/ml gentamicin to kill the remaining extracellular bacteria (33). Phagocytosis was determined after a washing step and lysis of MDMs with sterile water followed by plating of serial dilutions of cell lysates on Columbia III agar with 5% sheep blood plates (). A second and a third plate were incubated with RPMI 10% HS, 20 µg/ml gentamicin and 10-100 ng/ml M-CSF, GM-CSF or IFN-γ for 3 and 18 hours. Intracellular bacteria were quantified as described above, and data was presented relative to the number of phagocytosed bacteria ().
To validate the model, we quantified by plating the extracellular bacteria remaining after antibiotic treatment and washing of cell cultures. We also quantified the intracellular concentration of gentamicin by fluorescence polarization immunoassay (FPIA) using a Cobas Integra 400 plus (Roche). In additional validation steps, we inhibited phagocytosis by replacing HS with heat-inactivated fetal bovine serum (FBS, HyClone ThermoFisher), and by treating MDMs with 1, 5 and 10 µM cytochalasin D (Zigosporium mansonii, Merck & Cie), 1 and 10 µM oligomycin (Streptomyces diastatochromogenes, Sigma-Aldrich), and 10 and 50 mM 2-deoxy-D-glucose (2-DG, Roth AG) alone or in combination for 30 minutes (cytochalasin D and FBS) or one hour (oligomycin and 2-DG) before exposition to GBS.
Fluorescence microscopy
GBS was stained by incubating bacteria for 1 hour at 25°C with fluorescein isothiocyanate (FITC), followed by four washing steps (34). MDMs were exposed to FITC-labeled GBS and pictures of cells were taken at 1 hour with an EVOS M7000 Imaging System (ThermoFisher). Phagocytosis of FITC-labeled GBS was quantified in 100 macrophages. The viability of MDMs was assessed in cytokine (MOI 100) and inhibitor (MOI 2) plates, using the LIVE/DEAD™ Cell Imaging kit (Life-Technologies). The number of live and dead MDMs was quantified based on analysis of the pictures acquired with the EVOS M7000 Imaging System.
Statistical analysis
Analyses and graphs were performed using GraphPad Prism (GraphPad Software 9.5.1) and Microsoft Excel software. Data are expressed as means ± SEMs. Groups were compared by two-way ANOVA followed by Dunnett’s and Sidak’s multiple comparison tests, to compare each experimental condition with the control condition within the same donor group and to compare the newborns to the adults for a same experimental condition. Results were considered statistically significant when P < 0.05.
Results
Activation of newborn and adult MDMs by IFNγ increases GBS-induced TNF production
We compared the capacity of resting M-CSF-MDMs, GM-CSF-activated, and IFN-γ-activated newborn and adult MDMs to release TNF in response to live GBS COH1 (Figure 1). In comparison with resting M-CSF-MDMs, IFNγ-activated newborn and adult MDMs secreted 3.3- and 4.5-fold higher amounts of TNF in response to GBS. The trends towards more TNF production by GM-CSF-activated compared to resting MDMs and towards a greater production of TNF by newborn compared to adult MDMs did not reach statistical significance.
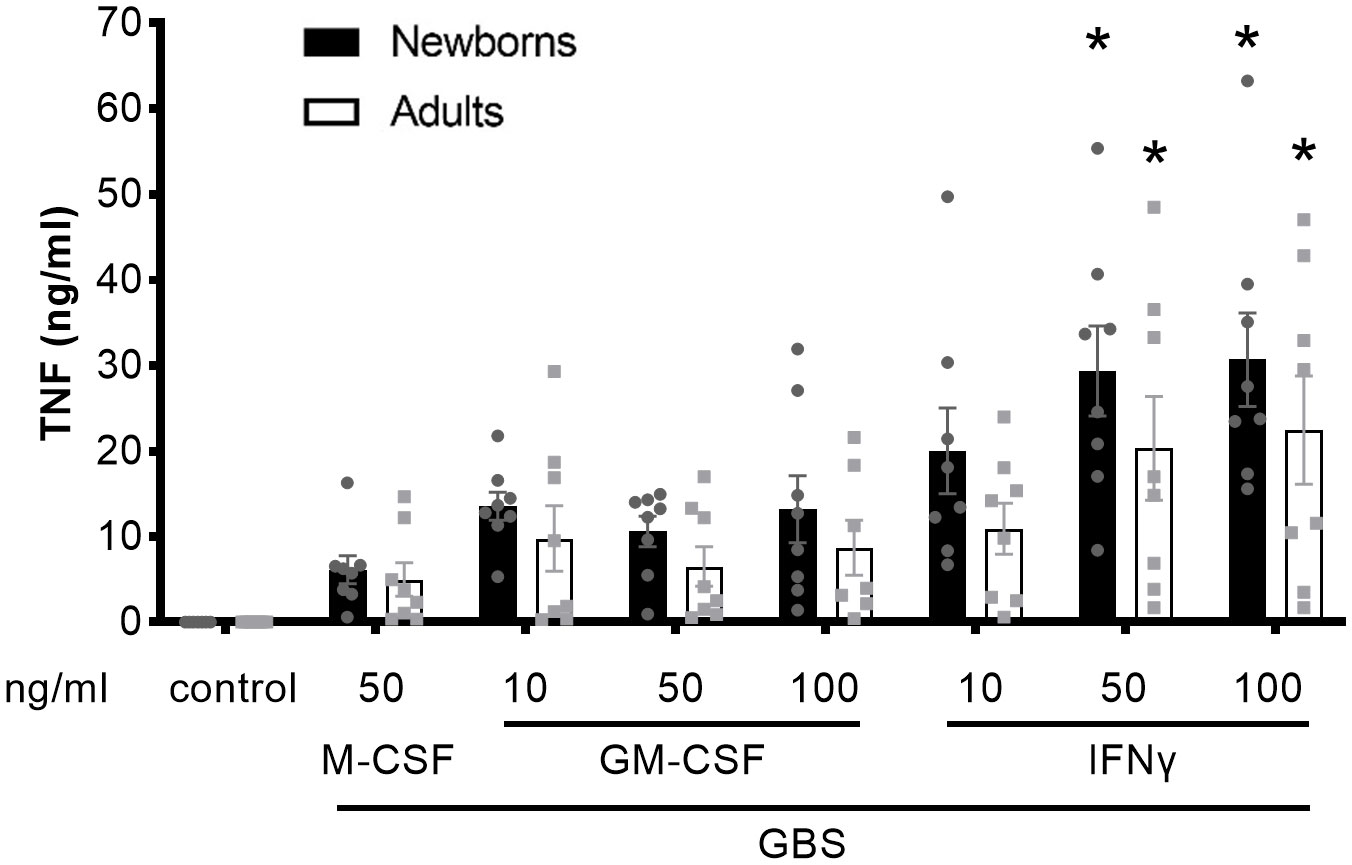
Figure 1 Activation of newborn and adult MDMs by IFNγ increases GBS-induced TNF production. Newborn and adult resting (M-CSF-) and GM-CSF- or IFNγ-activated MDMs were exposed to GBS (107 bacteria, MOI 100). The control condition corresponds to 50 ng/ml M-CSF-MDMs not exposed to GBS. Concentrations of TNF were measured in cell culture supernatants collected at 18 hours. Results are expressed as mean ± SEM of 8 newborns and 8 adults. Analysis by two-way ANOVA followed by Dunnett’s multiple comparison test to assess differences with the control condition of the same age group is presented. *P < 0.05 vs M-CSF-MDMs exposed to GBS.
Newborn MDMs release higher level of IL-1β, IL-6, IL-10, IL-12p70 and IL-23 than adult MDMs in response to GBS
To have a broad view on host responses, we quantified by multiplex immunoassay the secretion of 16 cytokines and chemokines by newborn and adult M-CSF-, GM-CSF- and IFNγ-MDMs exposed to live GBS COH1 (Figure 2). We presented the cytokines induced by GBS with a difference between newborn and adult MDMs in Figure 2A, the cytokines induced by GBS with no difference between newborn and adult MDMs in Figure 2B, and the cytokines not induced by GBS in Figure 2C. Exposure to GBS led to the secretion of IL-1β, IL-6, IL-10, IL-12p70, IL- 18, IL-21, IL-22, IL-23, and RANTES/CCL5, but not IL-1RA, IL-8/CXCL8, IL-20, IL-27, IFN-β, MCP-1/CCL2 and MIF in newborn and adult MDMs. GBS-induced secretion of IL-1β, IL-12p70 and IL-23 was more elevated in IFNγ- than in M-CSF-MDMs. Compared to adults, newborn M-CSF-MDMs exposed to GBS produced higher levels of IL-6 (3.8-fold) and IL-10 (3-fold), newborn GM-CSF-MDMs produced higher levels of IL-6 (4.5-fold), and newborn IFNγ-MDMs produced higher levels of IL-1β (4.9-fold), IL-12p70 (5.7-fold), IL-10 (3.8-fold) and IL-23 (1.8-fold). To evaluate the specificity of host responses to GBS, we exposed IFNγ-MDMs to the Toll-like receptor (TLR)-4 ligand LPS and the TLR1/2 ligand Pam3CSK4 (Supplementary Figure 1). LPS induced a higher production of IFNβ (2.5-fold) and IL-10 (10.6-fold) in newborn compared to adult IFNγ-MDMs. No difference in the secretion of the 16 cytokines and chemokines was observed between newborn and adult IFNγ-MDMs exposed to Pam3CSK4. We quantified the number of live and dead cells 18 hours after exposure to GBS to investigate a potential impact of cell survival on the differential responses of newborn and adult MDMs. Viability at 18 hours was 95 ± 2% and 92 ± 4% for newborn and adult M-CSF-MDMs, 94 ± 5% and 93 ± 6% for newborn and adult GM-CSF-MDMs, and 84 ± 5% and 87 ± 8% for newborn and adult IFNγ-MDMs (Figure 3). Normalizing cytokine levels according to the number of cells in each well (Supplementary Figure 2) did not modify our findings.
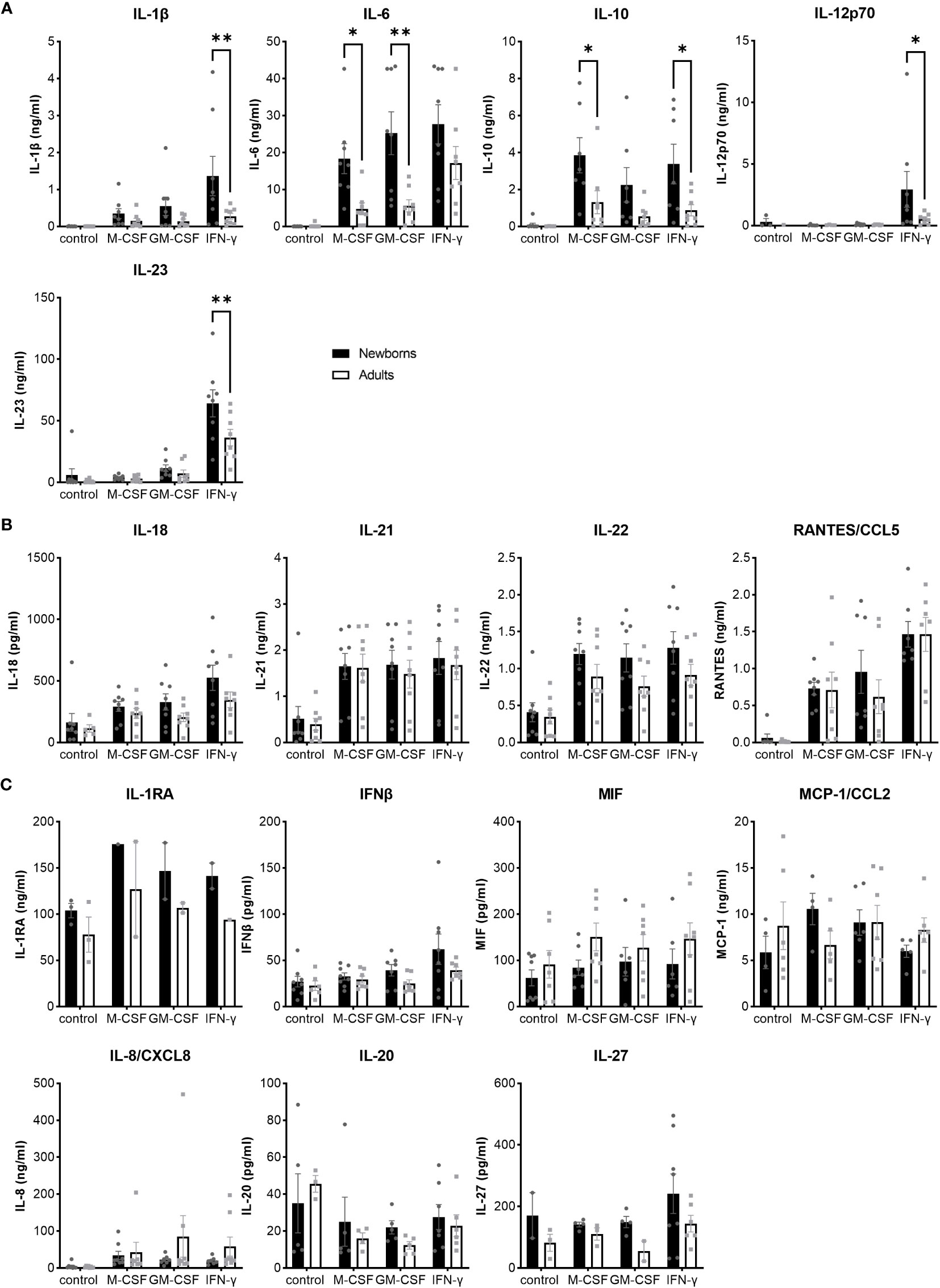
Figure 2 Distinct pattens of cytokine and chemokine release by newborn and adult MDMs in response to GBS. Newborn and adult resting (M-CSF-) and GM-CSF- or IFNγ-activated MDMs were exposed to GBS (107 bacteria, MOI 100). The control condition corresponds to M-CSF-MDMs not exposed to GBS. Concentrations of M-CSF, GM-CSF and IFNγ were 50 ng/ml. Concentrations of 16 cytokines/chemokines were measured in cell culture supernatants collected at 18 hours. (A) Cytokines differentially induced in newborn and adult MDMs: IL-1β, IL-6, IL-10, IL-12p70, IL-23. (B) Cytokines/chemokines induced at similar levels in newborn and adult MDMs: IL-18, IL-21, IL-22, RANTES/CCL5. (C) Cytokines/chemokines not induced by GBS, neither in newborn nor in adult MDMs. Results are expressed as mean ± SEM of 8 newborns and 8 adults (except for IL-1RA in panel C, n = 2-3). Analysis by two-way ANOVA followed by Sidak’s multiple comparison test to assess differences for the same condition between newborns and adults is presented. *P < 0.05, **P < 0.01.
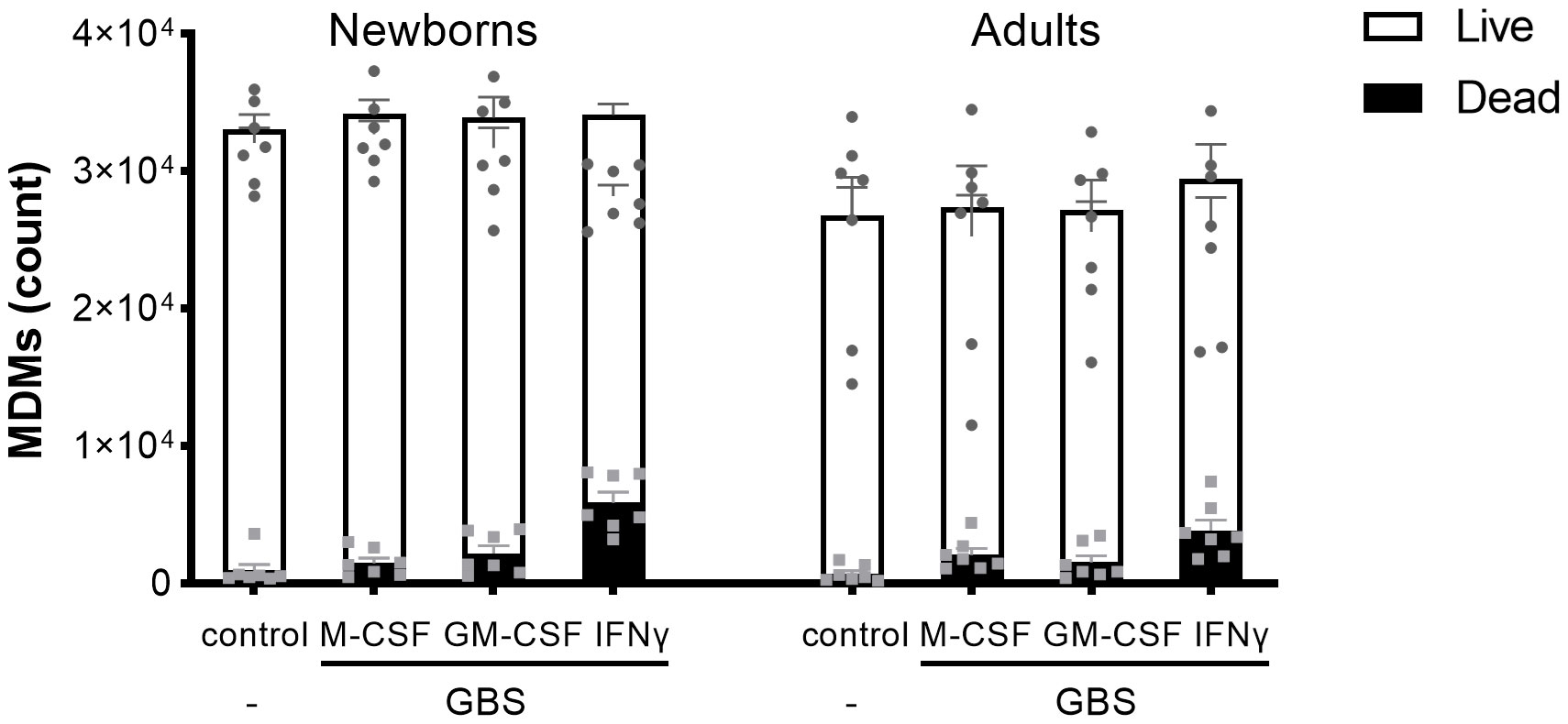
Figure 3 Newborn and adult MDMs maintain the same viability after exposure to GBS. Newborn and adult resting (M-CSF-) and GM-CSF- or IFNγ-activated MDMs were exposed to 107 GBS (MOI 100). The control condition corresponds to M-CSF-MDMs not exposed to GBS. Concentrations of M-CSF, GM-CSF and IFNγ were 50 ng/ml. The number of live and dead newborn and adult MDMs was quantified by fluorescence microscopy after 18 hours of incubation with GBS. Results are expressed as mean ± SEM of 7 newborns and 7 adults. Analysis by two-way ANOVA followed by Sidak’s multiple comparison test to assess differences for the same condition between newborns and adults is presented. *P < 0.05.
Newborn and adult MDMs have a similar rate of phagocytosis and killing of GBS
We analyzed the capacity of newborn and adult MDMs to phagocytose and kill GBS COH1 using a gentamicin protection assay. Newborn and adult M-CSF-MDMs phagocytosed 25 ± 5% and 27 ± 6% of the inoculum (Figure 4A). At 3 hours, 53 ± 12% and 44 ± 12% of phagocytosed bacteria survived in newborn and adult M-CSF-MDMs (Figure 4B). At 18 hours, survival of bacteria was drastically reduced to < 2% (Figure 4C), showing that both newborn and adult MDMs efficiently killed internalized bacteria. Activation of MDMs by GM-CSF or IFNγ at concentrations between 10 and 100 ng/ml did not influence phagocytosis or killing of GBS. We confirmed the absence of difference in phagocytosis between newborn and adult M-CSF-MDMs by quantifying 84 ± 13 and 100 ± 12 FITC-labeled GBS phagocytosed per 100 newborn and adult MDMs (Figure 4D).
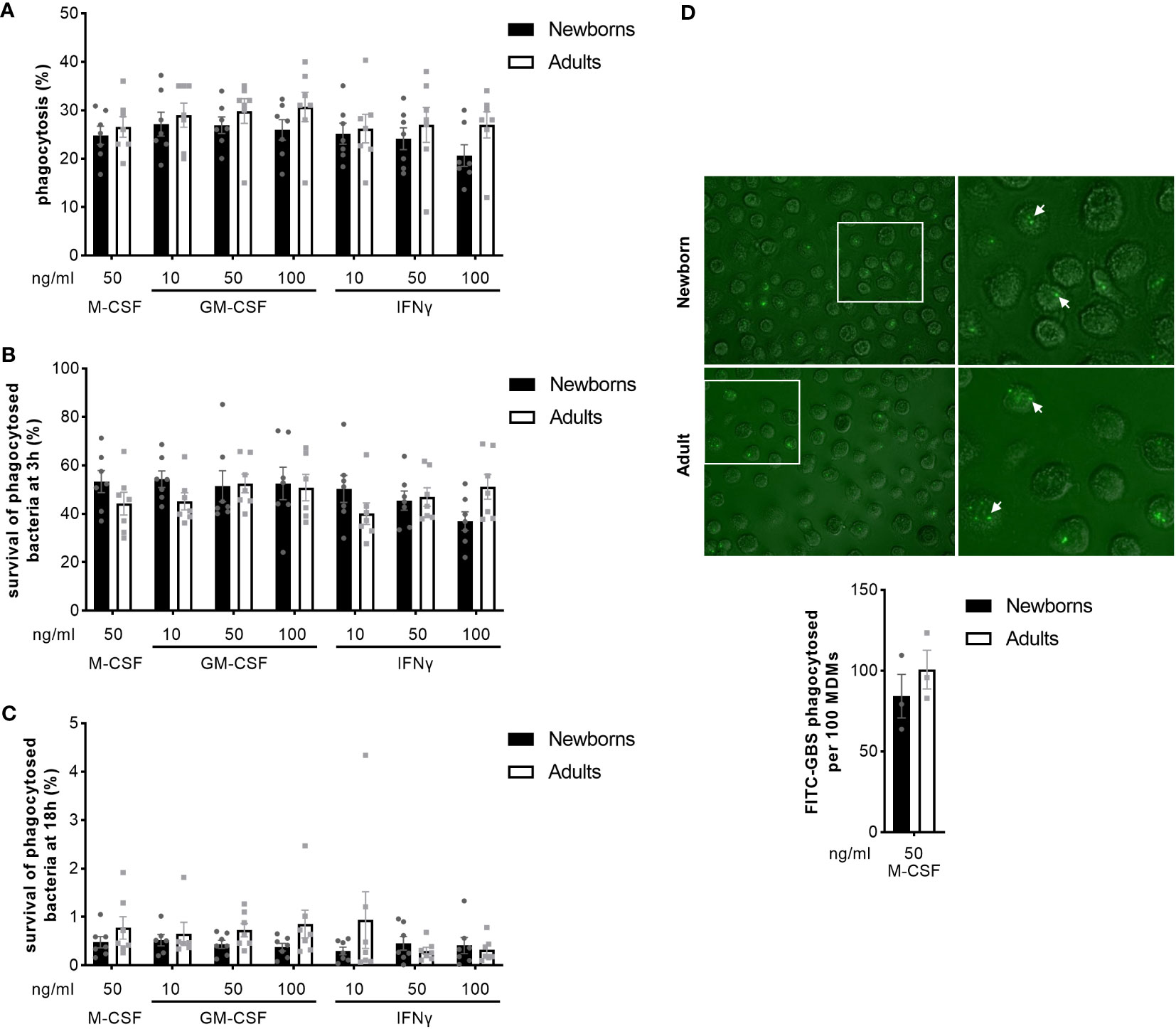
Figure 4 Newborn and adult MDMs phagocytose and kill GBS at a similar rate. Newborn and adult resting (M-CSF-) and GM-CSF- or IFNγ-activated MDMs were exposed to 2 x 105 GBS (MOI 2) during 1 hour. Phagocytosis of GBS was quantified by plating cell lysates (A). Intracellular survival of GBS was quantified by plating cell lysates at 3 (B) and 18 hours (C). Phagocytosis of GBS was visualized by fluorescence microscopy. Representative images and quantification of FITC-labelled GBS internalized by newborn and adult M-CSF-MDMs are shown, with a 100x magnification. The white arrows indicate phagocytosed bacteria. (D). Results are expressed as mean ± SEM of 7 newborns and 7 adults (A–C) and 3 newborns and 3 adults (D). Analysis by two-way ANOVA followed by Sidak’s multiple comparison test to assess differences for the same condition between newborns and adults is presented. *P < 0.05.
To validate our observations, we addressed potential methodological drawbacks: non-specific killing of intracellular bacteria by internalized gentamicin, and non-exhaustive killing of extracellular and membrane-bound bacteria. Intracellular gentamicin concentrations measured in newborn and adult M-CSF-MDMs at the end of the killing assay were below 0.1 µg/ml (Supplementary Table I), in line with the notion that gentamicin has a slow rate of entry into eukaryotic cells (35). Since we measured the minimal inhibitory concentration (MIC) of gentamicin for GBS COH1 to be 64 µg/ml, intracellular bacteria could not be affected by gentamicin. Exhaustive killing of extracellular and membrane-bound bacteria was verified by plating the cell culture supernatant 3 and 18 hours after gentamicin treatment. For newborn and adult MDMs, the proportion of live extracellular bacteria was < 0.5% of the inoculum used for infection (Supplementary Table II).
Opsonization enhances while inhibition of actin polymerization or energy metabolism prevents phagocytosis of GBS
The coating of pathogens with opsonins, followed by reorganization of the actin cytoskeleton, and metabolic reprogramming of macrophages are key steps to ensure optimal phagocytosis of bacteria (36). Given that previous research has focused on murine macrophages and cell lines, we investigated the impact of opsonization, actin polymerization, glycolysis and oxidative phosphorylation on the capacity of newborn and adult MDMs to phagocytose GBS. We quantified GBS uptake in the presence of heat-inactivated FBS, which contains very low concentrations of opsonins (37). Replacement of native HS by heat-inactivated FBS decreased GBS phagocytosis from 25 ± 7% to 8 ± 6% in newborn and from 26 ± 6% to 5 ± 5% in adult M-CSF-MDMs (Figure 5A). Pre-incubation of M-CSF-MDMs with 1, 5 and 10 µM cytochalasin D, an inhibitor of actin polymerization, hindered phagocytosis in a dose-dependent manner. Cytochalasin D at 10 µM reduced phagocytosis by newborn and adult MDMs from 26 ± 6% to 10 ± 5% and from 28 ± 6% to 9 ± 2% (Figure 5B), without significantly affecting cell viability (Figure 5C). Inhibition of glycolysis with 2-DG or inhibition of oxidative phosphorylation by oligomycin did not affect GBS phagocytosis (Figure 5D). However, the combination of both inhibitors elicited a large and dose-dependent reduction of phagocytosis (Figure 5D), with a 39% ± 18% and 39% ± 19% decrease in phagocytosis at 10 mM 2-DG and 1-10 µM oligomycin in adult MDMs, and a 98 ± 0.5% and 98% ± 1%, 96% ± 2% and 97% ± 1% decrease at 50 mM 2-DG and 1-10 µM oligomycin for newborn and adult M-CSF-MDMs. Both inhibitors, either alone or in combinations, at concentrations up to 50 mM for 2-DG and 10 µM for oligomycin had no impact on cell viability (Figure 5E).
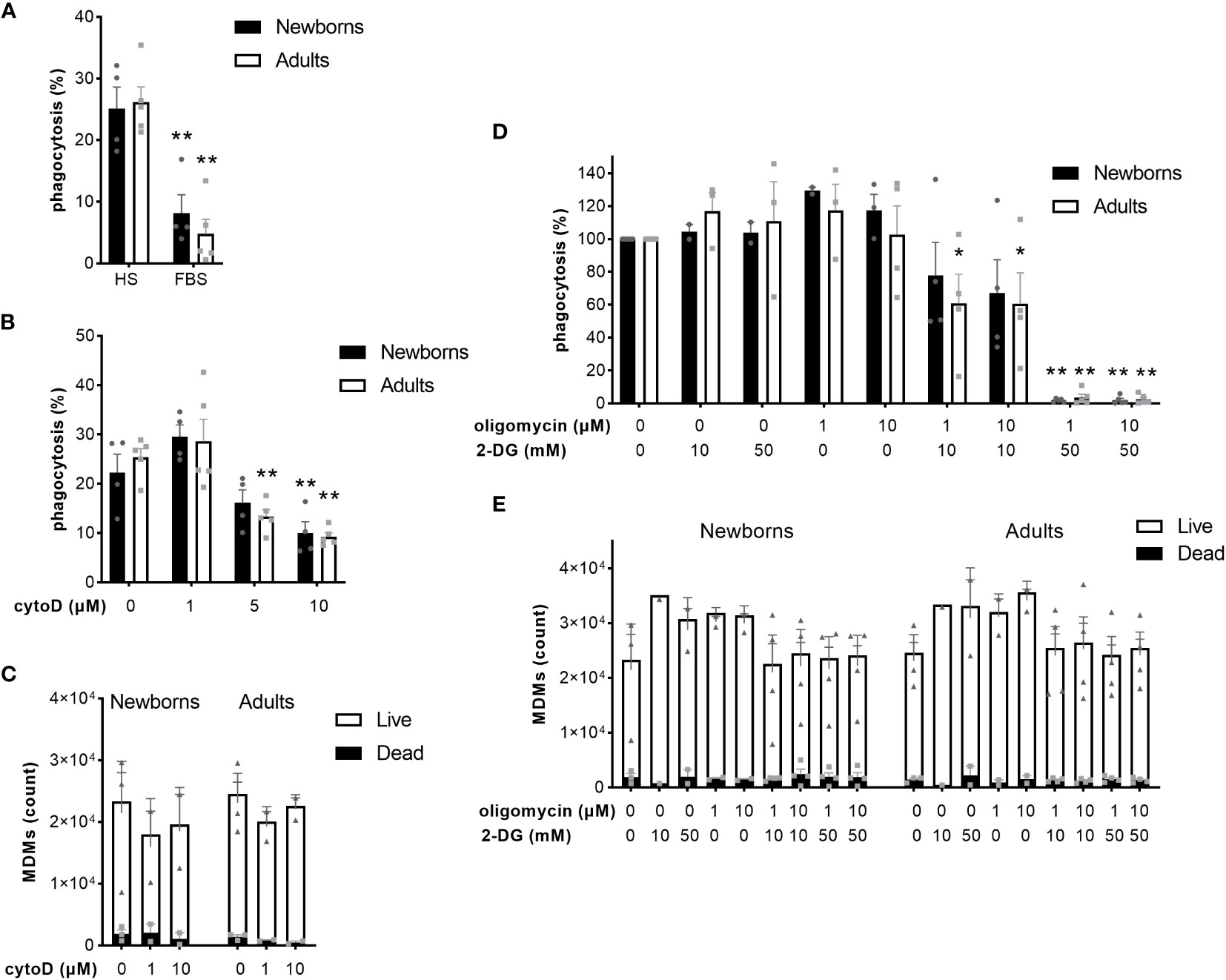
Figure 5 Opsonization, actin polymerization and energy metabolism contribute to phagocytosis of GBS by newborn and adult MDMs. Newborn and adult M-CSF-MDMs were pre-incubated with 10% native human serum (HS) (A-E) or 10% heat-inactivated fetal bovine serum (FBS) (A), with increasing concentrations of cytochalasin D (cytoD) for 30 minutes (B, C), oligomycin (D, E) and 2 deoxy-glucose (2-DG) (D, E) for one hour, or vehicle (DMSO for oligomycin or cytoD) and then exposed to 2 x 105 GBS (MOI 2) during 1 hour. Phagocytosis of GBS was quantified by plating cell lysates (A, B, D). The number of live and dead MDMs was quantified by fluorescence microscopy (C, E). Results are expressed as mean ± SEM (A–C, E) or normalized to controls (D) of 5 newborns and 5 adults. Analysis by two-way ANOVA followed by Dunnett’s multiple comparison test to assess differences with the control condition within the same age group is presented. *P < 0.05, **P < 0.01 vs controls.
Discussion
GBS causes a major burden of disease in early-life, including stillbirths, neonatal and infant mortality, and long-term disability (3–6). Here, we show that GBS induces a distinct host response in newborn and adult MDMs, with differences depending on the conditions of macrophage activation. Overall, newborn MDMs exposed to GBS release higher amounts of Th1, Th17 and anti-inflammatory cytokines and phagocytose and kill the bacteria to the same extent as adult MDMs.
Depending on the state of macrophage activation, live GBS triggers a greater release of IL-1β, IL-6, IL-10, IL-12p70, and IL-23 in newborn compared to adult MDMs. IL-12p70 is necessary to drive the differentiation of Th1 cells, while IL-1β, IL-6 and IL-23 are required to induce and maintain Th17 cell differentiation (38). IL-10 dampens inflammatory responses, thereby limiting tissue damage. Therefore, our results point towards greater Th1, Th17, and anti-inflammatory responses in newborn MDMs. These findings contrast with previous studies indicating that neonatal monocytes and dendritic cells exposed to TLR 1/2, 3, 4, 7/8, 8 and 9 agonists release lower amounts pro-inflammatory and Th1-polarizing cytokines than adult cells (11–16). The most consistent observation reported in the literature is the reduced capacity of neonatal innate immune cells to release IL-12p70 in response to TLR agonists (12, 15, 16, 39). Innate immune cells detect live bacteria through multiple pattern recognition receptors. Studies conducted in animal models and immortalized cell lines indicate that innate immune cells sense GBS through TLR2/6, TLR7, TLR8, TLR9, cyclic GMP-AMP synthase (cGAS) and NOD-like receptor family, pyrin domain containing 3 (NLRP3) (40–46). Mixed populations of newborn mononuclear cells (MNCs) produce lower levels of IL-10, IL-18 and IFNγ but similar levels of TNF and IL-6 compared to adult cells in response to heat killed GBS (47–50). Our results clearly show that cytokine responses of MDMs to live GBS are different from those to purified TLR2 or TLR4 agonists or heat inactivated bacteria, emphasizing the importance of using live bacteria in experimental models of infection.
Murine newborn macrophages have reduced capacities to phagocytose and kill bacteria, including GBS (26, 27, 51, 52). Conversely, human newborn and adult MNCs (19, 53) and MDMs (our data) internalize and eliminate GBS to the same extent. Opsonization enhances phagocytosis of GBS by murine macrophages and human MNCs from adults (54, 55). Our results suggest that opsonization may play an important role in promoting the uptake of GBS by primary human MDMs. In addition, newborn MDMs exhibit comparable efficiency as adult MDMs in internalizing GBS across various opsonization conditions. This suggests that newborn MDMs do not have an intrinsically reduced capacity to phagocytose GBS. Phagocytosis is a high energy-requiring process. During bacterial infection, a metabolic shift reprograms macrophages towards aerobic glycolysis (56). While high glycolytic activity is associated with efficient phagocytosis in murine macrophages (57–59), the impact of oxidative phosphorylation on phagocytosis has not been investigated. We report that inhibitors of both glycolysis and oxidative phosphorylation are required to reduce phagocytosis by newborn and adult MDMs, in line with the importance of metabolic pathways to supply the energy required for the uptake of bacteria. Previous studies have shown that GBS can survive in macrophage cell lines (60–62) and can induce macrophage cell death (63, 64). Our data indicate that primary human MDMs from newborns and adults are able to fully eliminate phagocytosed GBS without an impact on the viability of MDMs. Therefore, the vulnerability of neonates to GBS disease may not be related to a reduced capacity of macrophages to phagocytose and kill the bacteria.
During infection, Th1, Th17 and anti-inflammatory responses are required to initiate, amplify, and terminate the responses required to clear pathogens while minimizing tissue damage. Th1 CD4+ T cells provide protection against intracellular pathogens through the production of IFNγ and IL-2. IFNγ promotes bacterial clearance of GBS in newborn mice by increasing the bactericidal capacity of whole blood and peritoneal macrophages (62, 65). Given that activation by IFNγ does not increase the bactericidal capacity of human MDMs, the strong production of Th1 cytokines by newborn MDMs in vitro may not translate into enhanced protective responses during GBS infection. Th17 CD4+ T cells confer protection against bacteria through early recruitment of neutrophils and other inflammatory cells (66). However, naïve human neonatal CD4+ T cells exposed to IL-1β, IL-6 and IL-23 preferentially adopt an immunoregulatory Th22 phenotype, rather than a stereotypic Th17 phenotype (67). The higher production of Th1, Th17 and anti-inflammatory cytokines in newborns compared to adult MDMs in response to GBS challenges the concept that the neonatal immune system is poorly responsive, in line with recent literature (29, 68, 69). This may represent an age-adapted protective response to a dangerous pathogen. Alternatively, our findings are consistent with a dysregulated neonatal immune response to GBS. Indeed, a dysregulated host response, characterized by excessive activation of the immune system, concomitantly with features of immune suppression, plays a central role in the pathogenesis of sepsis, and sepsis-related adverse outcomes in adults (23). In accordance with this concept, GBS infection is associated with strong systemic pro- and anti-inflammatory responses in newborns (21, 22, 70).
Previous studies in humans have investigated mixed populations of umbilical cord blood MNCs or monocyte-derived cells, without a phenotypic characterization of differentiated cells or validation of phagocytosis and killing assays. Several validation steps ensured the robustness of our model. The viability of MDMs obtained from highly purified preparations of CD14+ monocytes was confirmed in each experiment. The production of a large number of cytokines by MDMs was quantified in several conditions of activation. We addressed the potential pitfalls of the gentamicin protection assay by multiple validation experiments.
Our study has several limitations. An in vitro system does not reflect the tissue-specific environment of macrophages in vivo. For example, newborns have low circulating levels of opsonins (71). In our experiments, the use of human adult serum or heat inactivated FBS in the culture medium provided the same amount of opsonins for newborn and adult MDMs, allowing to analyze their intrinsic phagocytic capacities. However, we did not test the impact of specific opsonins in the phagocytosis of GBS. Although birth is the time of life when the risk of developing GBS infection is highest, studying children could inform on changes reflecting the transition from a newborn GBS-sensitive to an adult GBS-resistant immune system. While GBS COH1 is a reference strain for neonatal studies, it may not be representative of the strains that are causing GBS infection nowadays. Moreover, we did not assess the mechanisms underlying the differences in cytokine production observed between newborn and adult MDMs.
In summary, we provide an extensive description of a robust methodology aimed at evaluating the cytokine response and the capacity of newborn and adult MDMs to phagocytose and kill GBS. We show that newborn MDMs have an increased Th1- and Th17-related cytokine response, together with enhanced release of IL-10 in response to GBS. Newborn MDMs do not have an intrinsically reduced capacity to phagocytose or kill GBS. These findings indicate that the neonatal innate immune system is highly responsive to GBS, and point towards a dysregulated macrophage response to this bacterium in newborns. This may have strong implications for the understanding of the mechanisms underlying the progression towards life-threatening organ dysfunction during GBS infection. Additionally, our results could be relevant for the development of immune modulating therapies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Ethics Committee on human research of the Canton de Vaud (CER-VD). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
DR: Writing – original draft, Data curation, Methodology, Writing – review & editing, Conceptualization, Formal Analysis, Investigation. EN: Writing – review & editing, Formal Analysis, Investigation. NG: Formal Analysis, Investigation, Writing – review & editing. MW: Investigation, Writing – review & editing. VV: Writing – review & editing, Formal Analysis. BS: Formal Analysis, Writing – review & editing, Conceptualization, Methodology. PS: Conceptualization, Formal Analysis, Methodology, Writing – review & editing. SG: Formal Analysis, Methodology, Writing – review & editing, Investigation, Supervision, Writing – original draft. TR: Conceptualization, Formal Analysis, Methodology, Supervision, Writing – review & editing. EG: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Swiss National Science Foundation (grant #320030_197618 to EG, grant #310030_207418 to TR), the WEGH Foundation (to EG), and by a grant from the European Society for Pediatric Research (to EG). EN was supported by a Swiss Government Excellence Research Scholarship.
Acknowledgments
We thank all participants who donated cord and adult blood samples for this study, and the midwives of the University Hospital of Lausanne for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1268804/full#supplementary-material
References
1. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7
2. Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al. Global, regional, and national causes of under-5 mortality in 2000-15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet (2016) 388(10063):3027–35. doi: 10.1016/S0140-6736(16)31593-8
3. Giannoni E, Agyeman PKA, Stocker M, Posfay-Barbe KM, Heininger U, Spycher BD, et al. Neonatal sepsis of early onset, and hospital-acquired and community-acquired late onset: A prospective population-based cohort study. J Pediatr (2018) 201:106–14 e4. doi: 10.1016/j.jpeds.2018.05.048
4. Goncalves BP, Procter SR, Paul P, Chandna J, Lewin A, Seedat F, et al. Group B streptococcus infection during pregnancy and infancy: estimates of regional and global burden. Lancet Glob Health (2022) 10(6):e807–e19. doi: 10.1016/S2214-109X(22)00093-6
5. Collaborators GBDAR Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet (2022) 400(10369):2221–48. doi: 10.1016/S0140-6736(22)02185-7
6. Mynarek M, Vik T, Andersen GL, Brigtsen AK, Hollung SJ, Larose TL, et al. Mortality and neurodevelopmental outcome after invasive group B streptococcal infection in infants. Dev Med Child Neurol (2023). doi: 10.1111/dmcn.15643
7. Libster R, Edwards KM, Levent F, Edwards MS, Rench MA, Castagnini LA, et al. Long-term outcomes of group B streptococcal meningitis. Pediatrics (2012) 130(1):e8–15. doi: 10.1542/peds.2011-3453
8. Korir ML, Manning SD, Davies HD. Intrinsic maturational neonatal immune deficiencies and susceptibility to group B streptococcus infection. Clin Microbiol Rev (2017) 30(4):973–89. doi: 10.1128/CMR.00019-17
9. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity (2017) 46(3):350–63. doi: 10.1016/j.immuni.2017.03.009
10. Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol (2017) 17(8):495–507. doi: 10.1038/nri.2017.54
11. Sharma AA, Jen R, Kan B, Sharma A, Marchant E, Tang A, et al. Impaired NLRP3 inflammasome activity during fetal development regulates IL-1beta production in human monocytes. Eur J Immunol (2015) 45(1):238–49. doi: 10.1002/eji.201444707
12. Aksoy E, Albarani V, Nguyen M, Laes JF, Ruelle JL, De Wit D, et al. Interferon regulatory factor 3-dependent responses to lipopolysaccharide are selectively blunted in cord blood cells. Blood (2007) 109(7):2887–93. doi: 10.1182/blood-2006-06-027862
13. Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol (2017) 18(6):622–32. doi: 10.1038/ni.3745
14. Belderbos ME, van Bleek GM, Levy O, Blanken MO, Houben ML, Schuijff L, et al. Skewed pattern of Toll-like receptor 4-mediated cytokine production in human neonatal blood: low LPS-induced IL-12p70 and high IL-10 persist throughout the first month of life. Clin Immunol (2009) 133(2):228–37. doi: 10.1016/j.clim.2009.07.003
15. Belderbos ME, Levy O, Stalpers F, Kimpen JL, Meyaard L, Bont L. Neonatal plasma polarizes TLR4-mediated cytokine responses towards low IL-12p70 and high IL-10 production via distinct factors. PloS One (2012) 7(3):e33419. doi: 10.1371/journal.pone.0033419
16. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183(11):7150–60. doi: 10.4049/jimmunol.0901481
17. Levy O, Jean-Jacques RM, Cywes C, Sisson RB, Zarember KA, Godowski PJ, et al. Critical role of the complement system in group B streptococcus-induced tumor necrosis factor alpha release. Infect Immun (2003) 71(11):6344–53. doi: 10.1128/IAI.71.11.6344-6353.2003
18. Mohamed MA, Cunningham-Rundles S, Dean CR, Hammad TA, Nesin M. Levels of pro-inflammatory cytokines produced from cord blood in-vitro are pathogen dependent and increased in comparison to adult controls. Cytokine (2007) 39(3):171–7. doi: 10.1016/j.cyto.2007.07.004
19. Gille C, Leiber A, Mundle I, Spring B, Abele H, Spellerberg B, et al. Phagocytosis and postphagocytic reaction of cord blood and adult blood monocyte after infection with green fluorescent protein-labeled Escherichia coli and group B Streptococci. Cytometry B Clin Cytom (2009) 76(4):271–84. doi: 10.1002/cyto.b.20474
20. Kolter J, Henneke P. Codevelopment of microbiota and innate immunity and the risk for group B streptococcal disease. Front Immunol (2017) 8:1497. doi: 10.3389/fimmu.2017.01497
21. Andersen J, Christensen R, Hertel J. Clinical features and epidemiology of septicaemia and meningitis in neonates due to Streptococcus agalactiae in Copenhagen County, Denmark: a 10 year survey from 1992 to 2001. Acta Paediatr (2004) 93(10):1334–9. doi: 10.1111/j.1651-2227.2004.tb02933.x
22. Bender L, Thaarup J, Varming K, Krarup H, Ellermann-Eriksen S, Ebbesen F. Early and late markers for the detection of early-onset neonatal sepsis. Dan Med Bull (2008) 55(4):219–23.
23. van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity (2021) 54(11):2450–64. doi: 10.1016/j.immuni.2021.10.012
24. Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol (2023) 23:563–579. doi: 10.1038/s41577-023-00848-y
25. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
26. Martin TR, Ruzinski JT, Rubens CE, Chi EY, Wilson CB. The effect of type-specific polysaccharide capsule on the clearance of group B streptococci from the lungs of infant and adult rats. J Infect Dis (1992) 165(2):306–14. doi: 10.1093/infdis/165.2.306
27. Lund SJ, Patras KA, Kimmey JM, Yamamura A, Butcher LD, Del Rosario PGB, et al. Developmental immaturity of siglec receptor expression on neonatal alveolar macrophages predisposes to severe group B streptococcal infection. iScience (2020) 23(6):101207. doi: 10.1016/j.isci.2020.101207
28. Giannoni E, Guignard L, Knaup Reymond M, Perreau M, Roth-Kleiner M, Calandra T, et al. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect Immun (2011) 79(7):2690–8. doi: 10.1128/IAI.00076-11
29. Roger T, Schneider A, Weier M, Sweep FC, Le Roy D, Bernhagen J, et al. High expression levels of macrophage migration inhibitory factor sustain the innate immune responses of neonates. Proc Natl Acad Sci U S A (2016) 113(8):E997–1005. doi: 10.1073/pnas.1514018113
30. Schneider A, Weier M, Herderschee J, Perreau M, Calandra T, Roger T, et al. IRF5 is a key regulator of macrophage response to lipopolysaccharide in newborns. Front Immunol (2018) 9:1597. doi: 10.3389/fimmu.2018.01597
31. Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol (2013) 281(1):51–61. doi: 10.1016/j.cellimm.2013.01.010
32. Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci U S A (2005) 102(39):13950–5. doi: 10.1073/pnas.0506758102
33. Kaneko M, Emoto Y, Emoto M. A simple, reproducible, inexpensive, yet old-fashioned method for determining phagocytic and bactericidal activities of macrophages. Yonsei Med J (2016) 57(2):283–90. doi: 10.3349/ymj.2016.57.2.283
34. Coligan JE. Current Protocols in Immunology Vol. 5 14.6.4. Coligan JE, Bierer BE, Marguiles DH, Shevach EM, Strober W, editors. Hoboken, New Jersey: Wiley (1993). Current Protocols.
35. Van Bambeke F, Barcia-Macay M, Lemaire S, Tulkens PM. Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives. Curr Opin Drug Discov Devel (2006) 9(2):218–30.
36. Gordon S. Phagocytosis: an immunobiologic process. Immunity (2016) 44(3):463–75. doi: 10.1016/j.immuni.2016.02.026
37. Pryzwansky KB, Lambris JD, MacRae EK, Schwab JH. Opsonized streptococcal cell walls cross-link human leukocytes and erythrocytes by complement receptors. Infect Immun (1985) 49(3):550–6. doi: 10.1128/iai.49.3.550-556.1985
38. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9(6):641–9. doi: 10.1038/ni.1610
39. Goriely S, Van Lint C, Dadkhah R, Libin M, De Wit D, Demonte D, et al. A defect in nucleosome remodeling prevents IL-12(p35) gene transcription in neonatal dendritic cells. J Exp Med (2004) 199(7):1011–6. doi: 10.1084/jem.20031272
40. Talati AJ, Kim HJ, Kim YI, Yi AK, English BK. Role of bacterial DNA in macrophage activation by group B streptococci. Microbes Infect (2008) 10(10-11):1106–13. doi: 10.1016/j.micinf.2008.06.001
41. Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe (2008) 4(6):543–54. doi: 10.1016/j.chom.2008.11.002
42. Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group B streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe (2016) 20(1):49–59. doi: 10.1016/j.chom.2016.06.003
43. Deshmukh SD, Kremer B, Freudenberg M, Bauer S, Golenbock DT, Henneke P. Macrophages recognize streptococci through bacterial single-stranded RNA. EMBO Rep (2011) 12(1):71–6. doi: 10.1038/embor.2010.189
44. Ehrnstrom B, Beckwith KS, Yurchenko M, Moen SH, Kojen JF, Lentini G, et al. Toll-like receptor 8 is a major sensor of group B streptococcus but not escherichia coli in human primary monocytes and macrophages. Front Immunol (2017) 8:1243. doi: 10.3389/fimmu.2017.01243
45. Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, et al. Activation of the NLRP3 inflammasome by group B streptococci. J Immunol (2012) 188(4):1953–60. doi: 10.4049/jimmunol.1102543
46. Gupta R, Ghosh S, Monks B, DeOliveira RB, Tzeng TC, Kalantari P, et al. RNA and beta-hemolysin of group B Streptococcus induce interleukin-1beta (IL-1beta) by activating NLRP3 inflammasomes in mouse macrophages. J Biol Chem (2014) 289(20):13701–5. doi: 10.1074/jbc.C114.548982
47. Joyner JL, Augustine NH, Taylor KA, La Pine TR, Hill HR. Effects of group B streptococci on cord and adult mononuclear cell interleukin-12 and interferon-gamma mRNA accumulation and protein secretion. J Infect Dis (2000) 182(3):974–7.
48. La Pine TR, Joyner JL, Augustine NH, Kwak SD, Hill HR. Defective production of IL-18 and IL-12 by cord blood mononuclear cells influences the T helper-1 interferon gamma response to group B Streptococci. Pediatr Res (2003) 54(2):276–81. doi: 10.1086/315796
49. Currie AJ, Curtis S, Strunk T, Riley K, Liyanage K, Prescott S, et al. Preterm infants have deficient monocyte and lymphocyte cytokine responses to group B streptococcus. Infect Immun (2011) 79(4):1588–96. doi: 10.1128/IAI.00535-10
50. Berner R, Welter P, Brandis M. Cytokine expression of cord and adult blood mononuclear cells in response to Streptococcus agalactiae. Pediatr Res (2002) 51(3):304–9.10.1203/00006450-200203000-00007
51. Bakker JM, Broug-Holub E, Kroes H, van Rees EP, Kraal G, van Iwaarden JF. Functional immaturity of rat alveolar macrophages during postnatal development. Immunology (1998) 94(3):304–9. doi: 10.1046/j.1365-2567.1998.00518.x
52. Zhang Q, Coveney AP, Yu S, Liu JH, Li Y, Blankson S, et al. Inefficient antimicrobial functions of innate phagocytes render infant mice more susceptible to bacterial infection. Eur J Immunol (2013) 43(5):1322–32. doi: 10.1002/eji.201243077
53. Marodi L, Kaposzta R, Nemes E. Survival of group B streptococcus type III in mononuclear phagocytes: differential regulation of bacterial killing in cord macrophages by human recombinant gamma interferon and granulocyte-macrophage colony-stimulating factor. Infect Immun (2000) 68(4):2167–70. doi: 10.1128/IAI.68.4.2167-2170.2000
54. Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, et al. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol (2002) 169(7):3970–7. doi: 10.4049/jimmunol.169.7.3970
55. Anthony BF, Concepcion NF. Opsonic activity of human IgG and IgM antibody for type III group B streptococci. Pediatr Res (1989) 26(4):383–7. doi: 10.1203/00006450-198910000-00021
56. Ramond E, Jamet A, Coureuil M, Charbit A. Pivotal role of mitochondria in macrophage response to bacterial pathogens. Front Immunol (2019) 10:2461. doi: 10.3389/fimmu.2019.02461
57. Michl J, Ohlbaum DJ, Silverstein SC. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages II. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and ATP generation. J Exp Med (1976) 144(6):1484–93. doi: 10.1084/jem.144.6.1484
58. Pavlou S, Wang L, Xu H, Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflammation (Lond) (2017) 14:4. doi: 10.1186/s12950-017-0151-x
59. Ciarlo E, Heinonen T, Theroude C, Herderschee J, Mombelli M, Lugrin J, et al. Sirtuin 2 deficiency increases bacterial phagocytosis by macrophages and protects from chronic staphylococcal infection. Front Immunol (2017) 8:1037. doi: 10.3389/fimmu.2017.01037
60. Valentin-Weigand P, Benkel P, Rohde M, Chhatwal GS. Entry and intracellular survival of group B streptococci in J774 macrophages. Infect Immun (1996) 64(7):2467–73. doi: 10.1128/iai.64.7.2467-2473.1996
61. Korir ML, Laut C, Rogers LM, Plemmons JA, Aronoff DM, Manning SD. Differing mechanisms of surviving phagosomal stress among group B Streptococcus strains of varying genotypes. Virulence (2017) 8(6):924–37. doi: 10.1080/21505594.2016.1252016
62. Cornacchione P, Scaringi L, Fettucciari K, Rosati E, Sabatini R, Orefici G, et al. Group B streptococci persist inside macrophages. Immunology (1998) 93(1):86–95. doi: 10.1046/j.1365-2567.1998.00402.x
63. Oliveira L, Madureira P, Andrade EB, Bouaboud A, Morello E, Ferreira P, et al. Group B streptococcus GAPDH is released upon cell lysis, associates with bacterial surface, and induces apoptosis in murine macrophages. PloS One (2012) 7(1):e29963. doi: 10.1371/journal.pone.0029963
64. Flaherty RA, Aronoff DM, Gaddy JA, Petroff MG, Manning SD. Distinct group B streptococcus sequence and capsule types differentially impact macrophage stress and inflammatory signaling responses. Infect Immun (2021) 89(5). doi: 10.1128/IAI.00647-20
65. Cusumano V, Mancuso G, Genovese F, Delfino D, Beninati C, Losi E, et al. Role of gamma interferon in a neonatal mouse model of group B streptococcal disease. Infect Immun (1996) 64(8):2941–4. doi: 10.1128/iai.64.8.2941-2944.1996
66. Chen J, Yang S, Li W, Yu W, Fan Z, Wang M, et al. IL-17A Secreted by Th17 Cells Is Essential for the Host against Streptococcus agalactiae Infections. J Microbiol Biotechnol (2021) 31(5):667–75. doi: 10.4014/jmb.2103.03053
67. Razzaghian HR, Sharafian Z, Sharma AA, Boyce GK, Lee K, Da Silva R, et al. Neonatal T helper 17 responses are skewed towards an immunoregulatory interleukin-22 phenotype. Front Immunol (2021) 12:655027. doi: 10.3389/fimmu.2021.655027
68. Wynn JL, Wilson CS, Hawiger J, Scumpia PO, Marshall AF, Liu JH, et al. Targeting IL-17A attenuates neonatal sepsis mortality induced by IL-18. Proc Natl Acad Sci U S A (2016) 113(19):E2627–35. doi: 10.1073/pnas.1515793113
69. Brook B, Harbeson DJ, Shannon CP, Cai B, He D, Ben-Othman R, et al. BCG vaccination-induced emergency granulopoiesis provides rapid protection from neonatal sepsis. Sci Transl Med (2020) 12(542). doi: 10.1126/scitranslmed.aax4517
70. Chishiki M, Go H, Endo K, Ueda NK, Takehara H, Namai Y. Cytokine profiles before and after exchange transfusions in severe late-onset neonatal group B streptococcus meningitis: A case report. Tohoku J Exp Med (2021) 253(4):269–73. doi: 10.1620/tjem.253.269
Keywords: newborn, group B streptococcus, phagocytosis, cytokine, macrophage, innate immunity, streptococcus agalactiae
Citation: Ravi D, Ntinopoulou E, Guetta N, Weier M, Vogel V, Spellerberg B, Sendi P, Gremlich S, Roger T and Giannoni E (2023) Dysregulated monocyte-derived macrophage response to Group B Streptococcus in newborns. Front. Immunol. 14:1268804. doi: 10.3389/fimmu.2023.1268804
Received: 28 July 2023; Accepted: 16 October 2023;
Published: 14 November 2023.
Edited by:
Joseph Alex Duncan, University of North Carolina at Chapel Hill, United StatesReviewed by:
Wendy W. J. Unger, Erasmus Medical Center, NetherlandsAsmaa Tazi, INSERM U1016 Institut Cochin, France
Pascal M. Lavoie, University of British Columbia, Canada
Copyright © 2023 Ravi, Ntinopoulou, Guetta, Weier, Vogel, Spellerberg, Sendi, Gremlich, Roger and Giannoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Giannoni, ZXJpYy5naWFubm9uaUBjaHV2LmNo
†ORCID: Barbara Spellerberg, orcid.org/0000-0001-7552-8764
Parham Sendi, orcid.org/0000-0002-7347-6312
Thierry Roger, orcid.org/0000-0002-9358-0109
Eric Giannoni, orcid.org/0000-0003-0897-6529