
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 November 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1260470
This article is part of the Research TopicInnovations in Development, Translational Research and Manufacturing of CAR T cellsView all 19 articles
 Lorena Pérez-Amill1,2,3
Lorena Pérez-Amill1,2,3 Àlex Bataller4,5
Àlex Bataller4,5 Julio Delgado1,4,6
Julio Delgado1,4,6 Jordi Esteve1,4,6
Jordi Esteve1,4,6 Manel Juan1,6,3,7*
Manel Juan1,6,3,7* Nela Klein-González1,2,3*
Nela Klein-González1,2,3*Chimeric antigen receptor (CAR) T therapies are being developed for acute myeloid leukemia (AML) on the basis of the results obtained for other haematological malignancies and the need of new treatments for relapsed and refractory AML. The biggest challenge of CART therapy for AML is to identify a specific target antigen, since antigens expressed in AML cells are usually shared with healthy haematopoietic stem cells (HSC). The concomitant expression of the target antigen on both tumour and HSC may lead to on-target/off-tumour toxicity. In this review, we guide researchers to design, develop, and translate to the clinic CART therapies for the treatment of AML. Specifically, we describe what issues have to be considered to design these therapies; what in vitro and in vivo assays can be used to prove their efficacy and safety; and what expertise and facilities are needed to treat and manage patients at the hospital.
The beginning of cancer immunotherapy can be traced back to William Coley, often hailed as the pioneer of this field. In 1891, Coley embarked on a groundbreaking endeavour to stimulate the immune system as a means to treat sarcoma patients. This involved injecting heat-inactivated Streptococcus pyogenes and Serratia marcescens to these patients (1). However, it was not until the last century that several breakthroughs in immunotherapy, including the development of monoclonal antibodies, the utilization of cytokines, oncology vaccines, and the introduction of immune checkpoint inhibitors (such as CTLA-4 and PD-1) (2), along with the adoptive cell therapy, catapulted immunotherapy into becoming the most promising approach for cancer treatment.
The first foray into cell immunotherapy against cancer occurred with the introduction of allogeneic stem cell transplantation in 1957 by E. Donall Thomas. Leukaemia patients were treated with intravenous infusion of bone marrow from healthy donors, inducing the “graft-versus-leukemia” effect (3, 4). A significant stride in adoptive cell therapy (ACT) occurred in 1986 when Dr. Rosenberg and his team described the use of tumour infiltrating lymphocytes (TILS) from melanoma patients. These TILS were isolated from melanoma surgical specimens, expanded in vitro with IL-2 for several weeks, and subsequently reintroduced into melanoma patient, resulting substantial tumour regressions (5).
In subsequent years, two different groups (Kuwana Y, et al. from Japan and Gross G, et al. from Israel) described the concept of covalently linking the antibody’s variable domains (VL, VH) to the TCR constant regions (Cα, Cβ), thereby activating T cells in an HLA-independent manner (6, 7). Subsequently, Eshhar et al. and Brocker T, et al. independently designed a construct composed of a single-chain variable fragment (scFv) of an antibody linked to the signalling ζ or γ chain of the T-cell receptor (TCR), allowing the entire expression in one molecule, thus generating the first CAR molecule (8–11). Since this initial CAR design, numerous generations of CAR molecules have been developed (section 1.2).
Today, CAR molecules are synthetic chimeric receptors comprising an extracellular antigen-binding domain derived from an antibody and the intracellular signalling domain of the TCR. CART therapy involves modifying T cells to express CARs that recognize a specific antigen expressed on the surface of malignant cells and exert a cytotoxic effect towards them in an HLA-independent manner. Autologous T cells are isolated from the patient’s blood by leukapheresis, activated, genetically engineered ex vivo to express the CAR on their surface, and expanded in close-manufacturing bioreactors. After obtaining and characterizing the cell product, these cells are frequently cryopreserved. To ensure the engraftment of CART cells, patients usually undergo a lymphodepleting chemotherapy prior to CART infusion (Figure 1). Finally, they are infused into the patient to specifically kill cancer cells expressing the target antigen (Figure 1) (12, 13).
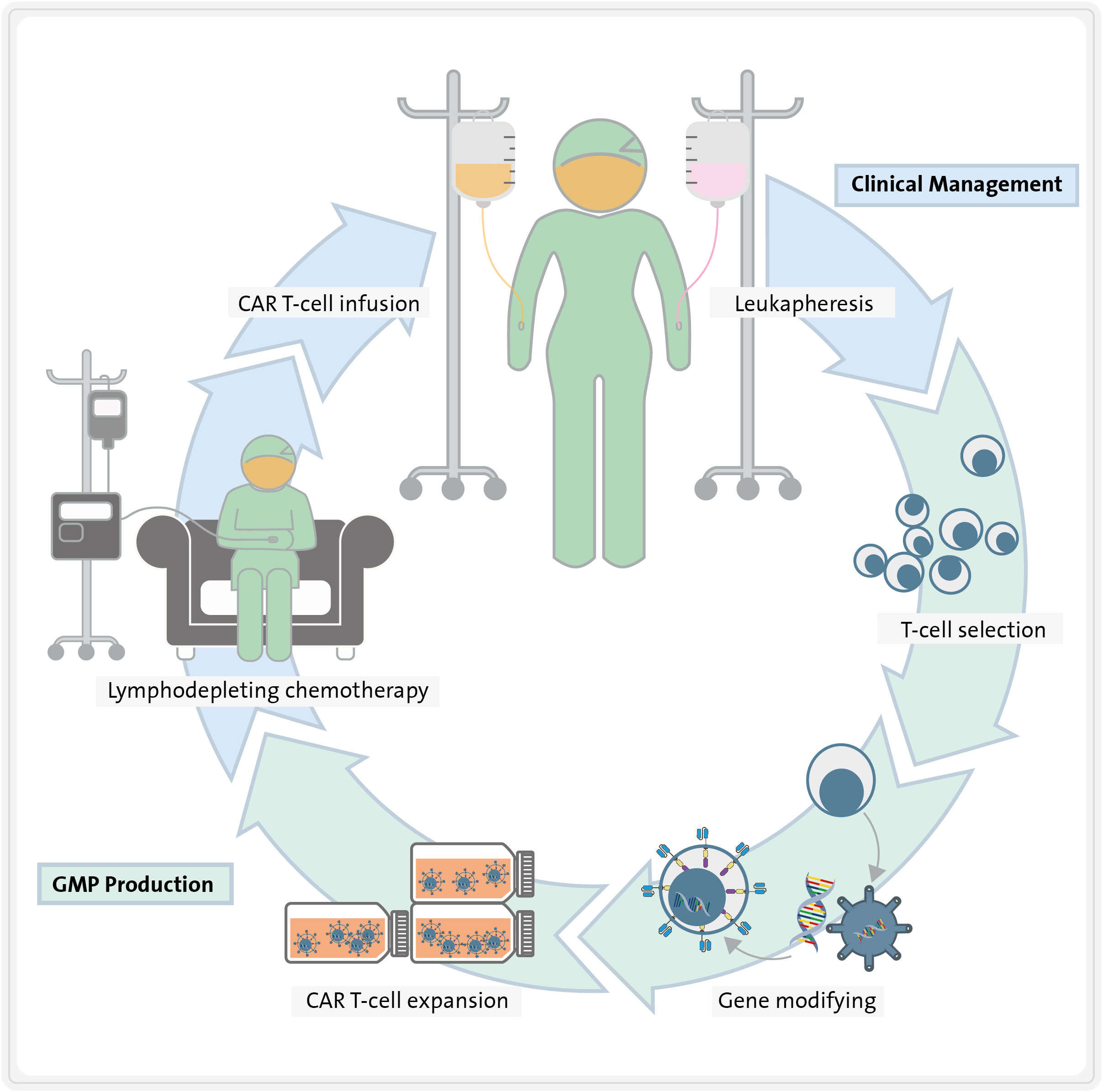
Figure 1 Steps required for CART therapy. First, T cells are isolated from the patient blood by leukapheresis. Second, they are activated and genetically engineered ex vivo to express the CARs on their surface. Third, they are expanded in close-manufacturing bioreactors, while patient undergoes lymphodepleting chemotherapy. Finally, CART cells are re-infused into the patient, where they exert specific cytotoxicity towards tumour cells.
CAR molecules are synthetic chimeric receptors characterized by three distinct domains: the extracellular antigen-binding domain, the transmembrane domain, and the intracellular CD3ζ signalling chain of the TCR (11, 14, 15) (Figure 2).
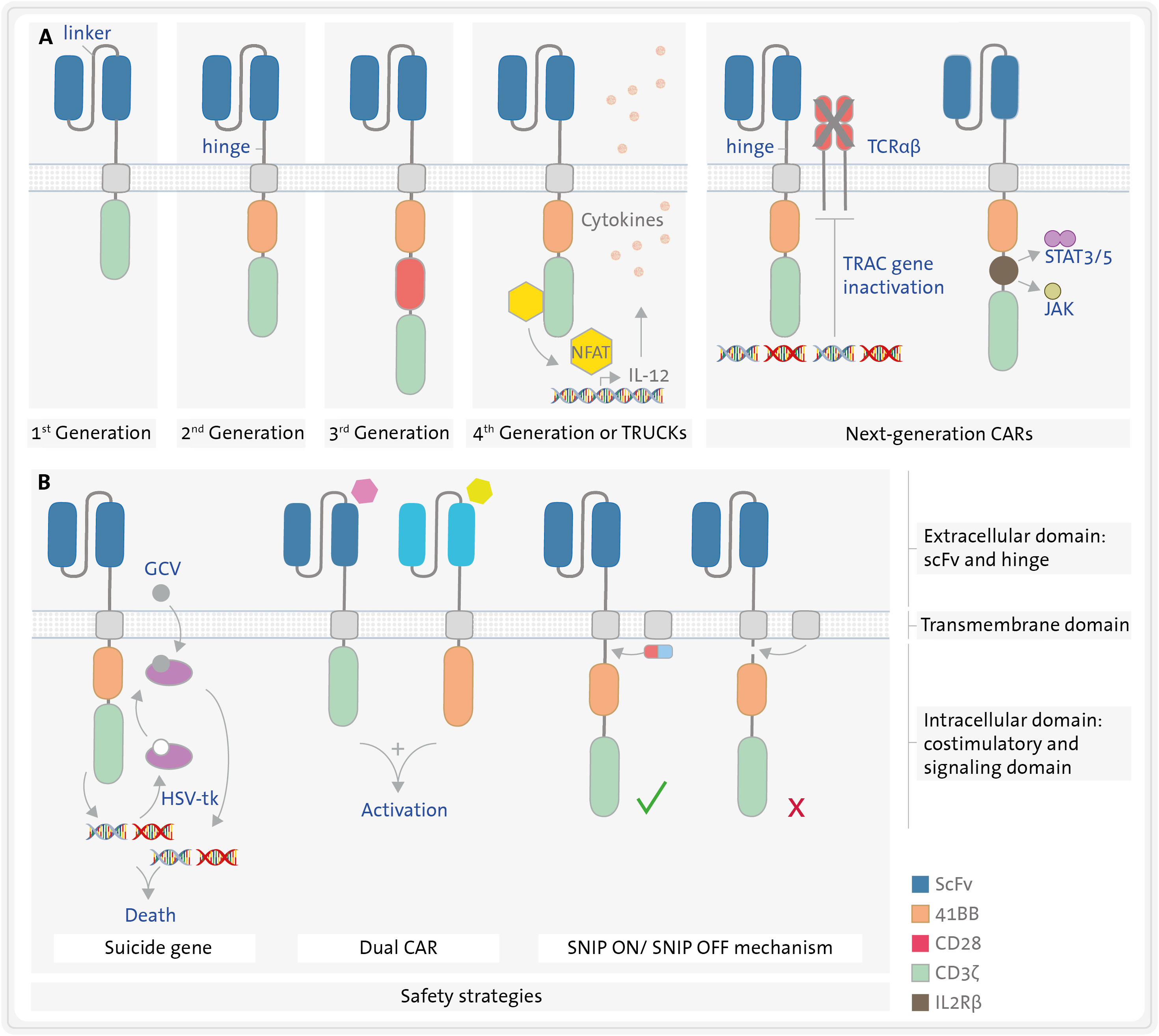
Figure 2 Structure of different CAR constructs. (A) CARs have three domains: an extracellular, a transmembrane, and an intracellular domain. The extracellular domain (blue) consists of a single chain variable fragment (scFv) formed by the variable region of a heavy chain (VH) and that of a light chain (VL) derived from an immunoglobulin, both connected by a linker. A hinge connects the extracellular domain to the transmembrane domain (grey). The intracellular domain consists of a signalling domain (green) and several possible costimulatory domains (orange, red, yellow, brown), such as 4-1BB or CD28. Five generations of CART cells varying in the number and type of costimulatory domains are represented in section (A, B). Safety strategies (B) include the use of suicide genes e.g., herpes virus thymidine kinase (HSV-tk) in combination with ganciclovir (GCV), dual CART cells, and inducible CART cells using SNIP mechanism. JAK/STAT3/5, janus kinase-signal transducer and activator of transcription pathway; NFAT, nuclear factor of activated T cells; SNIP, Signal Neutralization by an Inhibitable Protease; TRAC, T-cell receptor alpha constant; TRUCKs, T cells Redirected for Universal Cytokine Killing.
The extracellular domain is composed of a scFv. This is the domain that recognizes the antigen expressed on the surface of target cells. The scFv is a fusion protein that combined the variable region of a the heavy (VH) and the variable region of the light chain (VL) of an immunoglobulin, fused by a linker that confers its flexibility (16). Typically, it is derived from a murine antibody, although it can also be humanized or even fully human (e.g. originating from transgenic mice or a phage-display library) (17). The latter options may be preferred in certain circumstances over murine scFv as the latter can potentially lead to immunogenicity. However, disparate data have been reported about the immunogenicity induced by CART therapy (18–20). Some patients may develop a humoral anti-CAR response, the so-called HAMAs (human anti-mouse antibody) (21), which have been reported to potentially reduce the persistence and cytotoxic effect of the CART cells (22, 23). Nevertheless, other groups using the commercial CART anti-CD19 tisagenlecleucel did not observe any reduction in CART expansion, persistence or efficacy due to HAMAs (24).
The spacer or hinge connects the extracellular and the transmembrane domains, providing flexibility to the scFv and enhancing the interaction between the CAR and the target cell (25). Additionally, its length can influence the functionality of the immunological synapse (26). The transmembrane domain is embedded in the cellular membrane linking the extracellular and intracellular domains of the CAR. Both the hinge and the transmembrane domain are typically derived from either CD28, CD8 (T-cell surface glycoproteins) or Immunoglobulin G (IgG) (27).
Depending on the intracellular domain employed, in accordance with their evolutionary trajectory, five generations of CARs can be discerned, (Figure 2). The intracellular CD3ζ chain from the TCR complex (signalling domain) is a common feature across all CAR versions. It initiates the first signal for T-cell activation (22, 28). First-generation CARs rely only on this domain to promote the activation of CART cells, which does not ensure sustained cytotoxic activity in vivo. Consequently, second and third generations of CARs were engineered with one or two additional costimulatory domains, respectively, incorporating the second costimulatory signal requisite for T lymphocyte activation. These augment activation levels, efficacy against target cells and persistence of the CART cells in vivo (29, 30) (Figure 2). The most frequently employed costimulatory domains are CD28 (31) and 4-1BB (CD137) (32), although others like ICOS, OX40, CD27 or DAP-12 have also been utilized (15). Clinical findings indicate that CARs with a CD28 domain lead to greater T-cell activation, while those with a 4-1BB domain result in enhanced persistence in vivo (33–35).
Fourth-generation CARs also produce and secrete transgenic proteins, such us cytokines like interleukin 12 (IL-12) under the control of the nuclear factor of activated T cells (NFAT) (15, 36). Cytokine secretion can either improve the activation of CART cells or diminish the immunosuppressive tumour microenvironment, thereby increasing their survival and persistence. This, in turn, translates into a higher cytotoxicity over an extended period in preclinical models (37). Fourth-generation CARs are also referred to TRUCKs: T cells Redirected for Universal Cytokine Killing (37).
Finally, consensus is lacking regarding what constitutes a “fifth-generation”. In fact, next-generation CART cells come in at least two flavours. The first option involves a signal from a truncated cytoplasmatic cytokine receptor, such as IL2Rβ, which activates the JAK-STAT3/5 pathway. These CART cells have three immune activation signals – TCR activation by the CD3ζ, the costimulatory signal (typically 4-1BB or CD28) and the cytokine signal through IL2Rβ (38). The second option entails gene-edited CART cells, for instance, with inactivation of the T-cell receptor alpha constant (TRAC) gene via CRISPR-Cas9, to prevent TCR expression for donor-derived allogeneic CART cells (39) (Figure 2A).
CAR construct modifications focusing on safety will be discussed in more detail later. Briefly, there are several strategies that employ a suicide gene or express an antigen on the CART cells. This allows for the targeting of CART cells by approved antibodies, inducing antibody-dependent cell-mediated cytotoxicity (ADCC) and enabling the elimination of CART cells in cases of severe toxicity (40) (Figure 2B). Other strategies to enhance CART safety involve engineering them to exert a cytotoxic effect only when more than one target antigen is detected (e.g., dual CART cells) or conditionally expressing CAR molecules, such as Signal Neutralization by an Inhibitable Protease (SNIP) (41) (Figure 2B).
The initial successful clinical outcomes of CART cells were simultaneously reported by three distinct institutions: 1) the National Cancer Institute (NCI) (42), 2) the Memorial Sloan-Kettering Cancer Center (MSKCC) (43) and 3) the University of Pennsylvania (UPenn) (44). Various second-generation CART therapies targeting the CD19 antigen were developed and assessed in patients. The NCI group treated a patient with advanced follicular lymphoma, resulting in a partial response and B-cell aplasia after CART treatment (42). At MSKCC Hospital, nine patients diagnosed with refractory chronic lymphocytic leukemia (CLL) or relapsed B-cell acute lymphoblastic leukemia (B-ALL) were treated with CART therapy, demonstrating both safety and promising therapeutic potential (43). Meanwhile, UPenn reported the initial complete response of patient with refractory CLL treated with anti-CD19 CART therapy (44).
Since then, six CART therapies have received approval from the U.S. Food and Drug Administration (FDA) (Figure 3). Among them, four of them are CART cells targeting the B-cell antigen CD19: tisagenlecleucel (Kymriah) (45), axicabtagene ciloleucel (Yescarta) (46), brexucabtagene autoleucel (Tecartus) (47), and lisocabtagene maraleucel (Breyanzi) (48). The remaining two target the B-cell maturation antigen (BCMA): idecabtagene vicleucel (Abecma) (49) and ciltacabtagene autoleucel (Carvykti) (50). CD19-directed CART therapies are approved for treatment of patients with relapsed or refractory (R/R) B-ALL and B-cell lymphomas; while BCMA-directed CART therapies are indicated for R/R multiple myeloma (MM). All approved CART products express second-generation CARs, with either a 4-1BB or CD28 costimulatory domain. The majority of approved products employ a murine scFv with the exception of ciltacabtagene autoleucel, which utilizes a 2-epitope binding camelid. The different domains utilized in each CAR, including the hinge and TM domains, are illustrated in Figure 3. Additionally, there is the option of clinical application via a clause known as Hospital Exemption, as exemplified by varnimcabtagene autoleucel for the treatment of adult patients with R/R ALL in Spain (51).
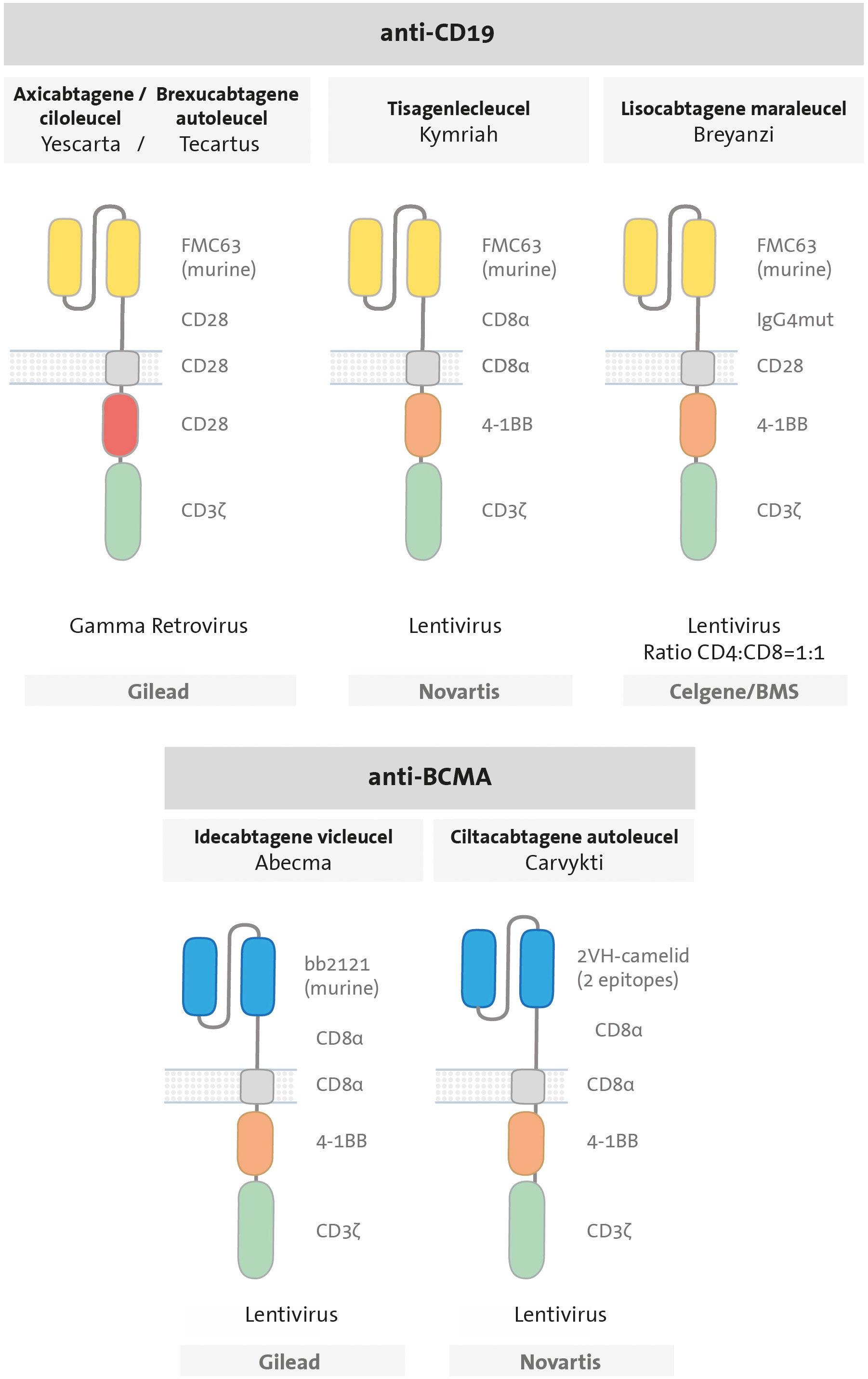
Figure 3 Design of CAR T cells approved by the FDA. All are second-generation CARs, composed by an scFv (anti-CD19 in yellow or anti-BCMA in blue); a hinge (CD28, CD8a or IgG4mut); a transmembrane domain (CD28 or CD8 in grey), one costimulatory domain (CD28 in red or 4-1BB in orange) and CD3ζ signalling domain in green.
Acute myeloid leukemia (AML) is a heterogeneous neoplasm characterized by uncontrolled clonal expansion of transformed immature haematopoietic precursors, often associated with recurrent genetic alterations (52, 53). Its incidence is approximately 4.2 cases per 100,000 habitants, with a median age of presentation of 68 years, although it can be manifest at any age (54, 55).
For patients eligible for intensive treatments, chemotherapeutic agents remain the therapy of choice, typically including cytarabine and anthracyclines in most cases. However, new drugs are progressively being introduced. For patients who are not eligible for intensive treatments, novel active low-intensity regimens have been emerged and provide meaningful antileukemic activity. These include the combination of the bcl-2 inhibitor, venetoclax, with hypomethylating agents or low-dose cytarabine, as well as the isocitrate dehydrogenases 1 (IDH1) inhibitor, ivosidenib, for AML cases harbouring an activating mutation in this metabolic gene (56, 57). Nonetheless, none of these therapeutic approaches lead to complete leukemia eradication, and most patients will experience a clinical progression after several cycles.
Allogeneic haematopoietic stem-cell transplantation (allo-HSCT) has been a pivotal procedure for non-favourable risk AML, due to its potential to eliminate residual leukemic cells through the graft-versus-leukemia (GvL) effect (58, 59). Relapsed or refractory (R/R) AML presents a frequent and challenging scenario, occurring in 40-50% of younger patients and even more frequently in older individuals. A standard treatment protocol for these patients is still lacking and survival is poor, with an overall survival rate of 10% at 5 years (60, 61). Therefore, there is a clear need for novel therapeutic approaches in this scenario.
The therapeutic landscape of AML has undergone recent transformation with the introduction of monoclonal antibodies (mAb), such as CD33 gemtuzumab ozogamicin, (GO) and targeted therapies like the aforementioned venetoclax, as well as IDH1/2 and FLT3 inhibitors (56, 57, 62). Novel immunotherapeutic approaches are also being explored. For instance, ongoing clinical trials are assessing the efficacy of CD3-engaging bispecific antibodies, such as the CD123xCD3-targeting flotetuzumab. Additional targets being explored include CD33xCD3 and WT1xCD3 (63, 64).
Another interesting approach involves utilizing drugs relieving the blockage imposed by various immune checkpoints expressed by leukemic cells. Whereas CTLA-4 and PD-1 inhibitors, which are efficacious in several solid tumours and specific lymphoma subtypes, have not exhibited significant antileukemic activity in AML, promising initial results have been reported targeting other innate immune checkpoints. For example, the anti-TIM3 monoclonal antibody sabatolimab or the antibodies magrolimab and lemzoparlimab targeting CD47 (“do not eat me” signal), may restore macrophage phagocytosis of AML cells, and are currently under investigation in several clinical trials (65–67).
While several CART products targeting CD19 or BCMA have yielded outstanding clinical outcomes and some have been gained regulatory approval for treating B-cell malignancies, none has yet received regulatory approval for AML. In the subsequent sections, we describe the CART approaches that have been tested or are currently under investigation in AML. Finally, we integrate published data with the expertise of our group in the development CART products, offering guidance to researchers on how to design, develop, and translate these therapies for the treatment of AML.
The foremost challenge in CART therapy for AML lies in identifying a specific target antigen that is expressed on the surface of malignant cells but not on healthy cells, since the concomitant expression of the target antigen on both leukemic and healthy cells could result in on-target/off-tumour toxicity of varying severity. For instance, in B-cell malignancies, CD19-directed CART cells eradicate both malignant and healthy B cells/B-cell progenitors. However, B-cell aplasia and consequent hypogammaglobulinemia can be effectively managed with intravenous immunoglobulin reposition (68). In contrast, in some cases, on-target/off-tumour toxicity can be fatal, as was the case of a patient who died five days after receiving ERBB2-directed CART therapy for metastatic colon cancer. In this instance, CART cells targeted pulmonary epithelial cells expressing the ERBB2 antigen leading to severe respiratory distress followed by cardiac arrest (69). Therefore, choosing an appropriate target is the pivotal initial step in designing a CART cell (70).
The majority of surface antigens identified to date in AML cells are either shared with healthy haematopoietic stem cells (HSC) or not universally expressed in all AML cells. The on-target/off-tumour effect of CART cells on HSC could result in prolonged cytopenia, putting the patient at risk of infections or bleeding. This challenge can only be circumvented if targets exclusively expressed in AML cells are identified. This can be achieved, for example, through whole-genome sequencing (71), scrutinizing the surfaceome of AML (72) or carrying out proteomic and transcriptomic studies to compare antigen expression in leukemic stem cells and healthy stem cells (70). The ideal target antigen should possess the following characteristics: 1) restricted expression to malignant cells, i.e. minimally or not expressed at all on their healthy counterparts to minimize hematologic toxicity; 2) restricted expression to malignant cells without being expressed on other healthy tissues to avoid further on-target/off-tumour toxicity; 3) prevalent expression in most AML cases, enduring over time and under selective pressure; 4) expression in both malignant myeloid mature blasts and leukemic stem cells (LSC) to prevent the escape of the latter, which could lead to relapse (73).
In this section, we will discuss the most frequently targeted antigens by CART therapies (Figure 4), and later, we will provide a summary of the pertinent clinical data associated with these products (Table 1).
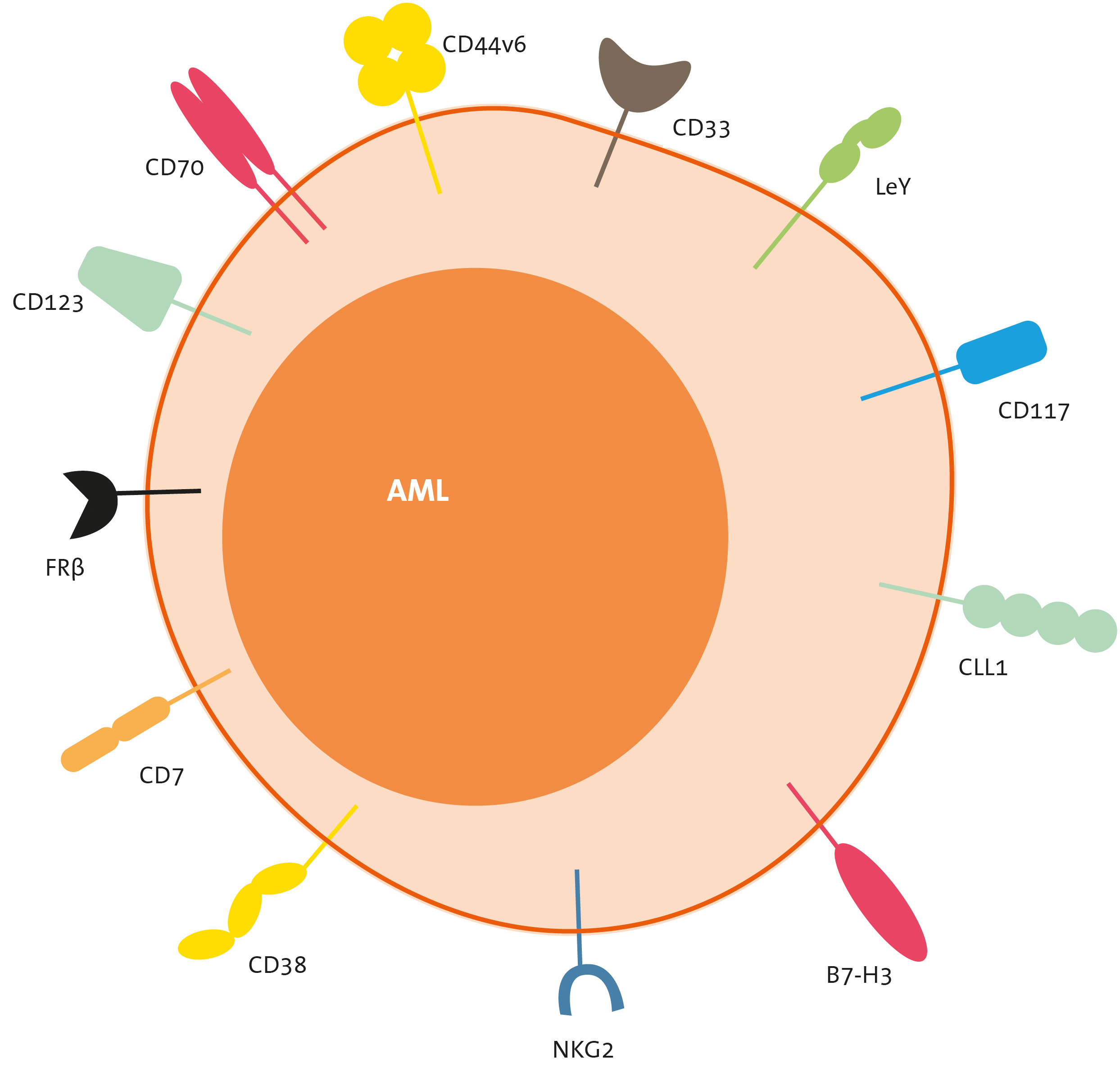
Figure 4 Target antigens of CART therapies for AML. CLL-1, C-type lectin-like molecule-1; FLT-3, FMS-like tyrosine kinase 3; FRb, folate receptor b; LeY, Lewis Y; NKG2, natural killer group 2 member.

Table 1 Published clinical trials using CART in AML: CART design.
CD33 is a transmembrane receptor expressed in the majority of AML cases (approximately 80%) and has therefore been extensively studied as a target for CART therapy in AML (89, 90). However, it is also expressed on myeloid cells, both mature and progenitors, as well as on certain cells of lymphoid lineage (91, 92). The humanized anti-CD33 antibody drug-conjugated GO (GO, Mylotarg® Pfizer) has gained approval for frontline therapy, in combination with intensive chemotherapy, especially in cases with favourable-risk cytogenetics AML (93). The main toxicities associated with GO include myelotoxity and the risk of inducing sinusoidal obstruction syndrome (SOS). Nonetheless, this latter side effect appears to be target-independent damage, probably related to the conjugated moiety of the drug, calicheamicin. This is evident as a similar toxicity is also observed with inotuzumab ozogamicin, an anti-CD22 antibody conjugated with the same molecule (94, 95).
CD123 or alpha chain of the interleukin 3 receptor (IL-3Rα) is expressed in 80% to 90% of AML cases. Moreover, it is prevalent not only in the bulk of AML blasts but also in LSC. Preclinical studies have demonstrated robust anti-leukemia effects with CD123-directed CART cells (96–98). Several CART therapies targeting this antigen are currently in clinical development for AML. However, this antigen is also expressed on healthy myeloid lineage cells, and there is conflicting data regarding the myelotoxic effect of CD123-directed CART cells. Some studies report low expression of CD123 on HSC (99, 100) while others describe a myeloablative effect of CD123-directed CART cells in various humanized mouse models (101). Consequently, in most clinical trials, CD123-directed CART therapy is utilized as a bridge therapy to allo-HSCT (see section on Clinical trials in AML) (76).
Additionally, CD123 is expressed on healthy endothelial cells of small-calibre blood vessels (102, 103) and this introduces another potential on-target/off-tumour effect of CD123-directed CART cells, namely capillary leak syndrome. CD123 has been clinically validated as a target for the treatment of blastic plasmacytoid dendritic-cell neoplasm (BPDCN) with the use of tagraxofusp (SL-401), a CD123-directed cytotoxin containing a truncated diphtheria toxin (70). Notably, capillary leak syndrome was a frequent adverse effect experienced by approximately 25% of patients.
CLL-1 or C-type lectin-like molecule 1 (also known as C-type lectin domain family 12 member A, CLEC12A) is expressed in over 80% of AML cases, on both blasts and LSC (104, 105). It is also expressed on healthy differentiated myeloid cells, but notably not on HSC or other non-haematologic human tissues (104, 106). Preclinical studies have demonstrated robust anti-leukemia activity of CLL-1-directed CART cells, both in vitro and in in vivo, without causing myelosuppression (107–109). This makes CLL-1 a promising target for CART therapy in AML.
In addition to the previously mentioned targets, which are common myeloid antigens, several others are currently under exploration. For instance, NKG2D exhibits an up-regulation in AML and while maintaining limited expression in healthy tissues. Nevertheless, it is important to note that inflammation and stressful events can lead to an up-regulation of this antigen in healthy tissues (110). CD7 is expressed in approximately 30% of adult AML cases and is associated with a more aggressive course of disease. However, it is also expressed on normal activated T cells, NK cells, and some progenitor cells. Consequently, CD7-directed CART therapies may lead to CART fratricide, and knocking out CD7 in autologous cells might be a necessary step in the CART generation process.
Other potential targets include Lewis Y (LeY), which was the initial targeted in CART therapy for AML patients (69, 70); FLT3 (111, 112); folate receptor β (113, 114); CD38 (115); CD44v6 (116, 117); CD117 or c-kit (118); CD276 (119); and B7-H3 (120). All these targets have shown promising preclinical results.
While various antigens have been explored in preclinical and clinical settings for CART in AML, none has yet demonstrated results comparable to those achieved with CD19 antigens in the context of B-cell malignancies, both in terms of efficacy and safety. However, there are emerging strategies that hold the potential to broaden the therapeutic window of CART therapy in AML.
The choice of various domains in CART cells is pivotal in increasing their efficacy and persistence. The scFv derived from different murine antibodies, humanized antibodies or fully human scFv exhibit varying binding affinities to antigens (121). Hence, it proves beneficial to assess multiple scFv directly against the same antigen and select the one that best suits the chosen strategy. Additionally, studies have noted that CART cells containing CD28 as the costimulatory domain display greater sensitivity to low levels of antigen compared to CART with 4-1BB. On the other hand, CART cells containing the 4-1BB costimulatory domain have demonstrated increased persistence in vitro, in vivo and in clinical trials (35, 122, 123).
TRUCKs, the fourth generation of CART, offer two distinct avenues for enhancement. They can heighten the cytotoxicity of CART cells by releasing cytokines like IL-12, or alternatively, promote their expansion and persistence through the release of cytokines such as IL-15. Atilla PA and collaborators found that CLL1-CART cells co-expressing transgenic IL-15 displayed a less terminally differentiated state and demonstrated superior expansion compared to CART cells lacking IL-15 (124). Moving forward, the fifth generation of CART cells further heightens the persistence of CART cells through the provision of a third immune activation signal. In addition, gene editing of the CAR gene in TRAC locus using CRISPR-Cas9 technology presents another option to enhance the potency of CART cells (39).
The combination of CART therapy with other immunotherapies, such as immune checkpoints inhibitors, holds the potential for a synergistic effect, ultimately increasing its efficacy. Ongoing clinical trials are investigating whether the combination of CART-19 and PD-1 or PD-L1 blockade can enhance CART persistence and clinical responses (125, 126). Given the heterogeneity of AML, selecting a single antigen present in all tumoral cells can be challenging. Some identified antigens for AML exhibit a regulatable expression. For instance, CD70 is widely expressed on AML cells but not on normal HSC and its expression on malignant cells can be heightened by the use of azacytidine (127). Similarly, the folate receptor B is typically expressed on 70% of AML cells, but its expression can be upregulated when AML cells are treated with all-trans retinoic acid (ATRA) (113).
An alternative approach involves engineering dual CART cells to modulate antigen recognition on tumour cells. There exists a wide range of dual CAR options, employing Boolean logic to determine when CART cells will be activated. Three primary logic gates can be distinguished: 1) AND: both antigens must be recognized for CART activation; 2) OR: either one of the two antigens must be recognized for CART activation, and 3) NOT: only antigen 1 must be present; the presence of antigen 2 inhibits CART activation (128). In cases where the antigen is downregulated or expressed at low levels, a dual CAR with an “OR logic-gate” strategy may prevent tumour escape. Here, CART is fully activated when targeting either one of the two target antigens (e.g. tandem, bicistronic or co-infusion CARTs).
Taking this concept further, a combination of a CAR with an HLA-Independent TCR (HIT) can increase sensitivity and decrease tumour escape associated with low target antigen expression. Theoretically, this strategy allows for a more precise selection of both target antigens compared to the dual strategy. Mansilla-soto and collaborators proposed choosing a lower-density target for the HIT (increasing sensitivity to this target and avoiding antigen escape) and a higher-density target for the CAR (129).
Currently, the majority of procedures in clinical trials involving CART cells in AML are followed by an allo-HSCT to overcome their potential myelotoxic effect. One strategy to improve safety involves engineering dual CART cells that calibrate their affinity to antigens present on both malignant and healthy cells, thereby reducing the on-target/off-tumour effect.
One approach is to design dual CART cells that only eliminate cells that express both target antigens, utilizing an “AND logic-gate” strategy, as previously described (Figure 2). In this scenario, two CAR molecules are expressed in the same T cell. One CAR molecule is a first-generation CAR that recognizes antigen 1 with the intracellular CD3ζ signalling domain and the second CAR molecule is a chimeric costimulatory receptor (CCR) that recognizes antigen 2 with an intracellular costimulatory domain (130). With this AND-gate strategy, CART cells are fully activated only when encountering both antigens (131–133). For instance, dual CART cells can be engineered to target a specific leukemia antigen (e.g., CD7) and an antigen both expressed on the leukemic blasts and on HSC (e.g., CD33). These bispecific CART cells would exert a cytotoxic effect only when encountering leukemic cells expressing both target antigens (i.e., CD33+ CD7+), but not when encountering HSC (CD33+ CD7-) or mature T cells (CD33-, CD7+) (134).
Another innovative approach is the new “IF-better gate” strategy, which modulates the detection and activity of CART cells based on the density of antigens expressed on both leukemic cells and healthy cells. In this scenario, the CART cell targets malignant cells with a high density of target 1 through the CAR (first construct). Cells with a low density of target 1 are only eliminated when they also exhibit a high density of target 2, recognized simultaneously by the concomitant CCR recognition (135, 136).
Another approach is the “NOT logic-gate” strategy. In this scenario, the first CAR molecule recognizes an antigen present on tumour cells, while the second CAR recognizes an antigen present only on healthy cells. This second recognition inhibits the activation of the first CAR in a reversible manner (130).
Utilizing mRNA CART cells is a different strategy to enhance safety; mRNA CART are “biodegradable” CARs since RNA is not integrated into the genome. This means they will not persist in the patient for more than a few days, aiming to mitigate myelotoxicity. However, this approach carries the potential risk of disease relapse due to low persistence of CART cells (137, 138). This mRNA strategy has been studied both in vitro and in vivo with CD33 (139) and CD123 (140) antigens for AML. Nevertheless, initial clinical trials with CD123 “biodegradable” CART cells did not yield clinical responses (75).
To mitigate myelotoxicity, one approach is to generate CART cells targeting an antigen on leukemic cells that has been deliberately knocked out from the donor HSC for the subsequent allo-HSCT (141). Kim and co-workers successfully deleted CD33 from HSC, leading to a long-term and functional engraftment with an immune system resistant to the anti-CD33 CART cells in a xenograft mouse model (141). Similarly, Nils and colleagues published a CART therapy targeting the pan-haematologic CD45 antigen. They base-edited the targeted CD45 epitope in human HSC (CD45edited) to prevent their killing by the anti-CD45 CART cells (142, 143).
Another alternative to mitigate both myelosuppression and cytokine release syndrome (CRS) induced by CART cells is the implementation of switch-off strategies (Figure 2) (40, 133), such as the expression of suicide genes. For instance, the inducible Caspase-9 suicide gene (iCasp9) can be expressed in CART cells, where the administration of the AP1903 molecule induces the Caspase3 apoptosis pathway in this cell. Another well-studied suicide gene is the Herpes Simplex virus thymidine kinase (HSV-tk); with the addition of ganciclovir (GCV); the HSV-tk phosphorylates GCV developing a toxic triphosphate which competes with triphosphate triggering to DNA synthesis inhibition and cell apoptosis (144). The other switch-off strategy, as previously described, is to express an antigen on the CART cells that can be targeted by antibodies to induce ADCC. For instance, the expression of EGFR or CD20 on the surface of CART cells, that could be targeted with the monoclonal antibodies cetuximab or rituximab, respectively, to induce ADCC and eliminate the CART cells if needed (145). Moreover, recently, SNIP CART cells have been successfully engineered to be non-constitutively active, but instead switched between an ON- and an OFF-status by a protease, thus enabling tuning of CAR activity to improve safety (41, 146).
In conclusion, a range of strategies have been devised to enhance the safety of CART therapy in AML. These include the precise and sophisticated engineering of dual CART cells, the utilization of biodegradable mRNA CART cells, and the implementation of switch-off mechanisms. Additionally, gene editing of donor HSCs offer promising avenues to mitigate potential toxicities.
Creating effective CART therapies involves adjusting various components like the scFv’s affinity and recognized epitope. This includes the scFv itself, the order of its VH/VL domains, the length of the linker in between, as well as the hinge, TM domain, costimulatory domain/s, and implementing a safety strategy if wanted. By modifying one or more of these factors, different CARTs can be designed with distinct efficacy and safety profiles (147, 148). These CARTs are then assessed through a series of lab tests to select the most promising candidate for further clinical development.
The majority of CART therapies currently in development utilize T cells, which are potent killers and relatively easy to manipulate. These T cells can be obtained either from the patient (autologous) or a healthy donor (allogeneic). By using autologous CART cells, the risk of graft-versus-host disease (GvHD) is avoided. In some cases, patients have relapsed after an allo-HSCT before receiving the CART treatment. In these cases, CART cells are engineered from autologous T cells, which technically come from the patient’s rebuilt donor immune system. Surprisingly, even in that case, significant GvHD has not been observed (149, 150). Alongside T cells, other immune cells like natural killer cells (NK), cytokine-induced killer cells (CIK), macrophages and regulatory or γδ T cells are being explored (151). NK or γδ T cells are advantageous as they do not cause GvHD, allowing them to be used in “off-the-shelf” allogeneic therapies (152–154). Another approach to prevent GvHD involves knocking out (KO) the endogenous T-cell receptor (TCR by TRAC gene KO) using genome editing tools like zing-finger nuclease or CRISPR/Cas9 (98, 154, 155).
From this point forward, we will focus on the steps needed to generate autologous CART cells. First, T cells are separated from peripheral blood mononuclear cells (PBMC) by isolating the cells that are positive for CD3 antigen. The ratio between the subpopulations of CD4+ helper T cells and CD8+ cytotoxic T cells can vary from person to person. Some studies suggest using products with a specific ratio (e.g., 1:1), while others do not adjust this parameter (48, 156). Currently, among the six FDA-approved CART products, only one has a defined 1:1 CD4:CD8 ratio (157). Further research is required to determine if a particular CD4:CD8 ratio is necessary to achieve better therapeutic results.
After isolating the T cells, they need to be activated with antibodies or antibody-coated beads (CD3-CD28) and cytokines (IL-2/IL-7/IL-15). Then, they are expanded using a culture medium supplemented with the cytokines (IL-2 or IL-7/IL-15) (158). Traditionally, IL-2 is used for in vitro expansion. However, it has been reported that using IL-15 leads to less T-cell senescence, which could improve their efficacy and persistence in the body (159).
Gene editing can be achieved in two ways: permanently using through viral means (such as γ-retroviruses, lentiviruses, adenoviruses or adeno-associated viruses) or temporarily through non-viral means (like transposons or mRNA).
The retroviridae family includes γ-retroviruses and lentiviruses. They facilitate the integration of the new genetic information (transgene) into the genome of T cells, allowing for stable, long-term gene expression (112). While γ-retroviruses can only infect dividing-cells, lentiviruses can infect both dividing and non-dividing cells. Among the lentiviruses, the second and third generations lentiviruses (LV) are commonly used in this process. The former has a higher transduction rate, while the latter is considered safer (160). It is important to note that when using these methods, the transgene is inserted randomly into the genome of T cells (29).
On the other hand, transposons are a more cost-effective option as they do not need the extensive viral manufacturing process. While they theoretically allow for transgene integration in less critical areas of the genome compared to viruses, potentially making them safer, they still insert genes randomly (161–163). In 2016, the first clinical trial using a non-viral Sleepy Beauty system to generate anti-CD19 CART cells was published (164). This method is not as established as the viral technology and requires further research (165).
Finally, mRNA transfer through electroporation is highly efficient in terms of cost, time, and achieving desired expression levels. However, CAR expression will decrease and eventually fade away in days or weeks as T cells divide, since RNA is not integrated into the genome (139, 166). A CART product that used mRNA electroporation to transiently express the CAR was found to be safe but did not show efficacy in AML patients (75).
To date, most CART cells used to treat AML patients have been genetically engineered using retroviral (108, 151, 167) or lentiviral methods (83, 101, 154). This approach allows for efficient and reproducible genetic modification of T cells that can be produced at scale following good manufacturing practices (GMP).
Typically, confirmation of CAR expression on T cells is determined using either by flow cytometry or real-time PCR (168). Additionally, the impact of CAR expression on T-cell subsets distribution and the expression of exhaustion and senescence markers can be assessed using flow cytometry. Subsequently, in vitro efficacy is evaluated by testing: 1) cytotoxicity towards tumour cells, 2) proliferation and 3) cytokine production (169).
Cytotoxicity is studied by co-culturing CART cells with AML target cells at various effector: target ratios (ranging from 10:1 to 1:8) for different durations (from 4 to 96 hours). The commonly utilized AML models include the MOLM-13/14, THP-1, Kasumi-1 and K562 cell lines. The latter is derived from a chronic myeloid leukemia at a blast crisis. It is crucial to determine the level of expression of the target antigen in the chosen cell line(s). Using one or more cell lines with varying expression levels (high/low) can be advantageous. Target cells can be easily distinguished from CART cells through flow cytometry by transfecting them with a reporter like green fluorescent protein (GFP) or a monomeric cherry red fluorescent protein (mCherry) (170). Common cytotoxicity assays are the 4-hour Chromium-51 (51Cr) release assay (108, 171); the luciferase killing assay (172); and assays where the surviving target cells are quantified by flow cytometry at the end of the co-culture (e.g., with 7-AAD or GFP/mCherry as preciously explained) (101). Based on the existing literature, it is anticipated that effective CART cells can eliminate the majority of target AML cells after 24 or 48 hours at low effector: target ratios (101).
Efficient CART cells demonstrate the ability to multiply when they engage with their specific antigen. This can be evaluated by co-culturing previously labelled CART cells (using carboxyfluorescein succinimidyl ester (CFSE)) with one or more target cells, and then tracking their proliferation by measuring CFSE dilution over several generations using flow cytometry (112). In this assay, IL-2 can be used as a positive, non-specific stimulus.
Additionally, when in contact with target cells, CART cells rapidly produce and release various cytokines. In most studies, this is determined by co-culturing CART cells with target cells that either express or do not express the specific antigen (e.g., CD123-directed CART cells are co-cultured with CD123+ and CD123- AML cells). The levels of these cytokines in the culture medium are typically measured using enzyme-linked immunosorbent assay (e.g. ELISA) (171). Specifically, IL-2 is measured to assess the activation status of CART cells, while interferon γ (IFN-γ) and granzyme B are checked to evaluate their cytotoxic activity. Both aspects can also be evaluated using CD107a degranulation assay; CD107a expression indicates the activation and cytotoxic degranulation state of immune cells (173). CART cells also release numerous pro-inflammatory cytokines, such as tumour necrosis factor a (TNF-α), interleukin 6 (IL-6), and interleukin 1b (IL-1b), which play a key role in activating the immune system. However, an exacerbated secretion of these molecules may lead to a strong cytokine release syndrome (CRS), although this can be managed with treatments like anti-IL-6 antibodies (174).
Considering the characteristics of most AML antigens, it is crucial to investigate whether CART cells can recognize the target antigen on HSC and assess if they might eliminate or hinder the proliferation of these cells, potentially causing haematologic toxicity.
To carry out these tests, HSC may be obtained from a cord blood unit or from the bone marrow (BM) of a healthy donor for allo-HSCT (151). These samples can then be used to measure the expression of the target antigen using flow cytometry. If the target antigen is present, cytotoxicity assays and proliferation colony-formation unit assays can be performed after co-culturing the HSC with CART cells. This approach has been employed to establish that both CD123- and CD33-directed CART cells reduced the capacity of HSC to form colonies (101, 167), suggesting that targeting these antigens may lead to myelosuppression. Indeed, in a clinical setting, it is essential to have an available allogeneic donor in case patients require an allo-HSCT after CD123- or CD33- directed CART treatment.
To assess the efficacy of a novel CART cell in a living organism in vivo, it is recommended to initially study their ability to eliminate grafts of AML cell lines first, followed by assessments on AML patient-derived xenograft (PDXs). Nod-scid-gamma (NSG) mice are typically the preferred model for in vivo experiments (175) due to their immunodeficiency, which allows human AML cells to engraft in their bone marrow. To improve cell engraftment of cells, especially that of PDX cells, mice are often sub-lethally irradiated 4 to 24 hours prior to the injection of AML cells. The majority of published in vivo studies employ AML cell lines like MOLM-13 (101), MOLM-14 (166), THP-1 (176), or Kasumi-1 (175). In these cases, leukemic cells are usually modified with a plasmid expressing the luciferase enzyme, allowing their growth to be periodically monitored through bioluminescence imaging after injecting luciferin intraperitoneally in live mice. Additionally, cells can be transfected with a reporter, such as GFP, to assess their presence in the peripheral blood during the experiment and in other tissues at the end of the study using flow cytometry (139, 167).
At the beginning of the experiment, AML cells (typically 0.1-10x106) are intravenously (i.v.) injected into mice. Once the AML is established, it can be detected through bioluminescence. Following this, which can take from one to several days, CART cells can be i.v. injected into the mice. The dosage of AML cells and CART cells will be determined by the AML proliferation rate and the specific objectives of the study. The most important measurement in these experiments is to quantify the presence of malignant cells.
For investigating potential myelotoxicity induced by CART therapy in vivo, utilizing a humanized mouse model, which involves mice with a “human” immune system, is the most effective approach. In this model, a human immune system is established by i.v. injecting human HSC or PBMC (175, 176) into previously irradiated NSG or NSG-SGM3 mice (6-12 weeks old). The NSG-SGM3 strain, while more immunosuppressed than NSG, allows for superior engraftment of human cells, leading to in vivo expansion of a greater number of human cells from both lymphoid and myeloid lineages. It is worth noting that this strain is more expensive and delicate.
Once the haematopoietic system is established (approximately 6 to 12 weeks after HSC or PBMC injection), animals are treated with either autologous human CART cells (derived from the humanized mice) (Figure 5 option 1) or heterologous CART cells (derived from a human donor) (Figure 5 option 2). In the latter case, it is important to consider a potential allogeneic effect if the HSC/PBMC and T cells are sourced from different donors (and are not HLA-matched). To evaluate the myelotoxicity induced by CART cells, the condition of the animals and the persistence of HSC will be regularly monitored. The persistence of HSC can be assessed during the experiment through bone marrow aspirates and in peripheral blood, or at the end of the experiment through flow cytometry quantification of HSC in the bone marrow (typically 4 to 12 weeks after CART injection) (120, 177).
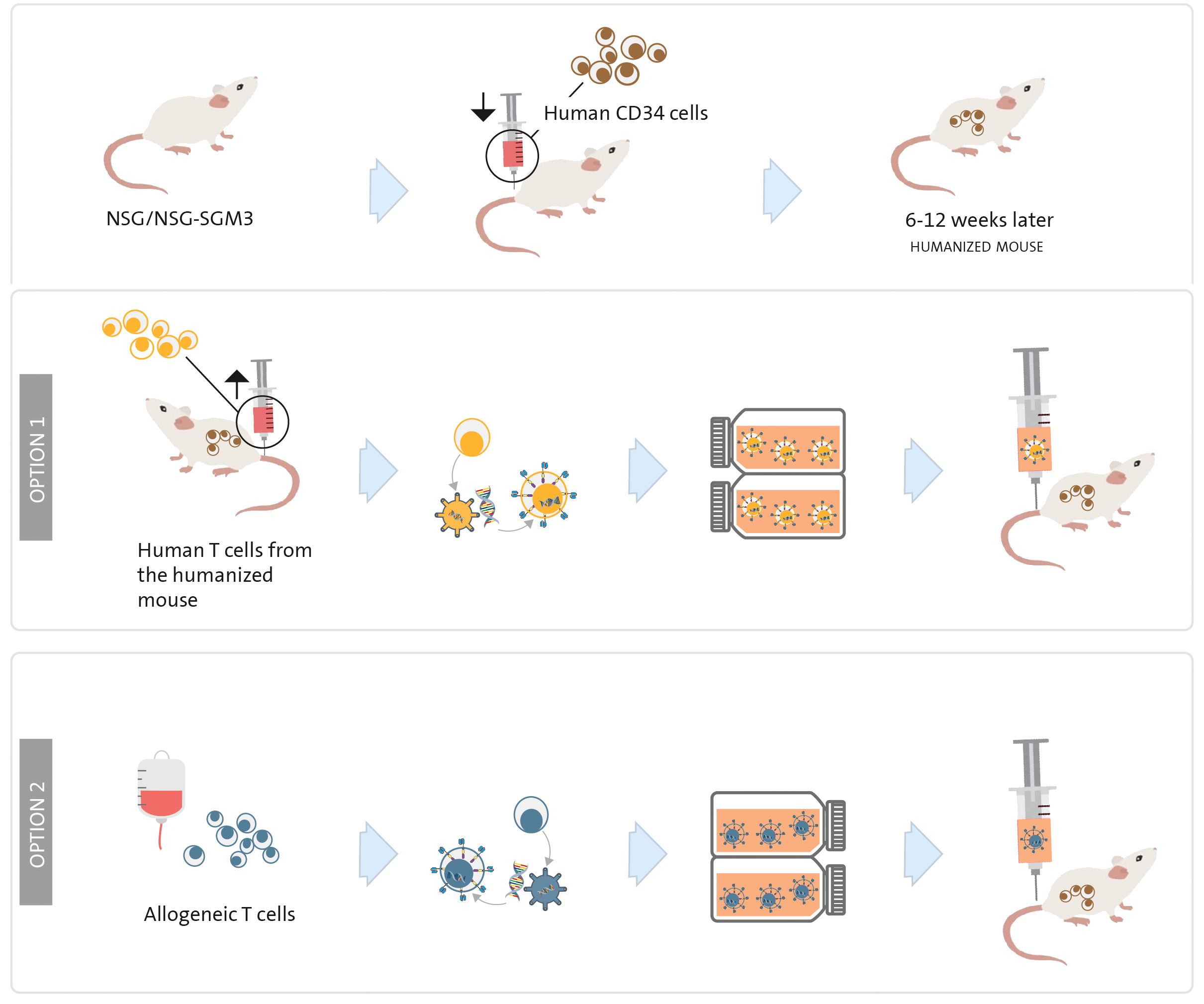
Figure 5 Humanized mouse model to study myelosuppression. NSG or NSG-SGM3 mice are irradiated on day 0 and a human immune system is established by human CD34+ haematopoietic stem cells or PBMC transplantation. Once the animals present a “human” immune system (between 6 and 12 weeks later), human CART cells are injected; they can be engineered with T cells obtained from the same humanized mouse (option 1) or from a human T-cell donor (option 2).
The humanized mouse model can also be utilized to evaluate efficacy. This can be achieved by injecting HSC into the liver of sub-lethally irradiated newborn NSG-SGM3 mice (176) or by following the method described in the previous paragraph (141); waiting until the animals develop a human immune system; injecting AML cells as previously outlined; and finally, administering CART cells engineered from T cells obtained from the humanized mice. This model enables simultaneous study of both myelotoxicity and efficacy (elimination of AML cells).
Additionally, a humanized mouse model proves valuable in to assessing CRS potentially caused by the CART cells. These animals contain human monocytes, T cells, and other cells implicated in this syndrome (176).
At the Hospital Clínic of Barcelona, we have developed several academic CART products, including varnimcabtagene autoleucel (ARI-0001) for patients with R/R B-ALL, B-cell lymphoma and CLL, as well as cesnicabtagene autoleucel (ARI-0002h) for R/R MM patients (21, 169, 170). Drawing from our experience in conducting clinical trials with CART cells and insights from existing literature, we will now outline the requirements for GMP production of CART therapies.
As previously mentioned, in the majority of clinical trials involving CART therapies for AML, autologous T cells are preferred over allogeneic T cells. Autologous T cells are obtained via leukapheresis from the patient, followed by a T-cell selection process (Figure 1). After 24 h, retroviral or lentiviral vectors that were previously manufactured are introduced to the cell culture in order to transduce T cells in accordance with GMP standards. Subsequently, the CART cells need to be expanded for a period ranging from 6 to 12 days (depending on required dosage for each clinical trial. This is done in specialized, close-automated bioreactors such as the CliniMacs Prodigy® (178), Wave® Bioreactor (154), G-Rex® or Cocoon® (179).
Of note, Milone’s group achieved highly effective lentiviral transduction of non-activated T cells, managing to produce CART cells within 24 hours (180). Both the CliniMacs Prodigy® and the Cocoon® offer complete automation of the entire production process, ensuring sterility and enhancing reproducibility under GMP conditions. In our view, these bioreactors currently represent the best options for point-of-care production of CART cells (181). CART cells are typically cryopreserved while undergoing sterility and microbiological safety assessments. They are thawed just prior to infusion into the patient. In certain clinical trials (e.g., Atalanta-1/CP0201-NHL), fresh CART cells are administered to patients with a vein-to-vein time of only 7 days. In such cases, a sample is rigorously assessed for safety beforehand.
CART cells must meet specific criteria during the final stages of the manufacturing process. These criteria encompass aspects such as visual appearance, viability, quantity, potency, sterility and safety. Only after meeting these standards can the CART cells be approved for release.
To gain regulatory approval for a clinical trial, the validation of the CART production process under GMP must be submitted to the national regulatory agency. This validation process meticulously covers every aspect of the CART therapy production, ensuring compliance with GMP and conducting through quality controls to ensure safety and evaluate efficacy (168).
The emergence of CART therapies for haematologic malignancies requires specialized training for healthcare professionals, including physicians and nurses. Additionally, it entails the establishment of specific units dedicated to the administration and management of these treatments.
Clinical trials involving CART therapies for AML entail the collaboration of various hospital departments. These include: 1. Haemato-Oncology Department: responsible for treating the patient and, if necessary, overseeing the allo-HSCT; 2. Apheresis Unit: where T cells are collected from the patient; 3. Immunotherapy Unit: this Unit is responsible for generating or receiving, processing and analysing the CART product; and 4. Intensive Care Unit (ICU) that may also be involved in patient care (21).
Serious side effects, such as CRS and neurological toxicity, have been reported in up to one third of AML patients undergoing CART therapies (182, 183). Other reported adverse effects include infections, persistent cytopenia, macrophage activation syndrome, as well as various on-target/off-tumour effects.
Cytokine-release syndrome results from the massive release of inflammatory cytokines (e.g., IL-1 and IL-6) following target recognition by the CART cells. This leads to the activation of other immune cell, like tissue macrophages, which can induce changes or damage in extra tumoral tissues. Neurotoxicity, also known as immune effector cell-associated neurotoxicity syndrome (ICANS), shares a similar underlying pathophysiology with CRS. In this case, the inflammatory cytokines produced by CART cells and the tumour microenvironment diffuse into the central nervous system. This, in turn, can trigger activation of microglial cells, leading to neurological symptoms (184).
As previously mentioned, the majority of the antigens found on AML cells are also present on healthy HSC. CART cells targeting these antigens could potentially result in prolonged cytopenia. Specialized care is required to manage these conditions. Therefore, utilizing risk-stratification tools like the CAR-HEMATOTOX score may prove beneficial in assessing the risk of hematotoxicity and anticipating patient needs. This tool can correlate the duration of severe neutropenia based on predictive biomarkers of hematotoxicity for R/R B-cell lymphoma (185). In 2023, a comprehensive survey conducted by EHA-EBMT groups outlined grading and management guidelines for hematoxicity following CART therapy. The results emphasized the widespread use of CTCAE (Common Terminology Criteria for Adverse Events) criteria for grading post-CART cytopenias and CIBMTR for evaluating hematopoieitic recovery by clinicians (6).
Given the anticipated side effects, AML patients who are considered for CART therapy must exhibit a satisfactory performance status and lack major comorbidities that could potentially worsen their prognosis. Eligibility assessments should be conducted by a multidisciplinary team experienced in this form of therapy, and equipped with the necessary resources to manage or treat potential complications.
The inaugural Phase I trial evaluating CART cells in AML patients was documented in 2013. This trial utilized a second-generation CAR featuring a CD28 costimulatory domain, targeting the LeY antigen. Four patients with R/R AML participated (74), and this trial yielded the initial indication of CART activity against AML in humans. Notably, three patients exhibited either disease stability or a reduction in blast count. Moreover, one patient with skin infiltration (known as leukemia cutis) experienced a transient improvement, as confirmed by a lymphocytic infiltration in the skin biopsy. This finding suggested that CART cells may be effective in eliminating AML in cases of extramedullary disease.
Currently, CD123 is the most frequently targeted antigen by CART therapies in AML patients (see Figure 6). As mentioned earlier, CD123 is expressed in the majority of AML cases and its expression has also been identified on LSC (177, 186). However, it is also present in endothelial cells, which means that on-target/off-tumour toxicity could lead to life-threatening consequences, such as capillary-leak syndrome (103). Moreover, since CD123 is found in HSC, CD123-targeted CART cells may impair normal haematopoiesis and potentially cause irreversible myeloablation, though this is still a subject of debate (100, 101, 187). The first patient treated with an allogeneic “universal” CD123-directed CART therapy (UCART123; NCT03203369) was a 78-year-old male diagnosed with BPDCN who died due to a combination of CRS and capillary-leak syndrome. Consequently, the trial was initially discontinued (188) and later resumed with a dose reduction and the introduction of an upper age limit (78).
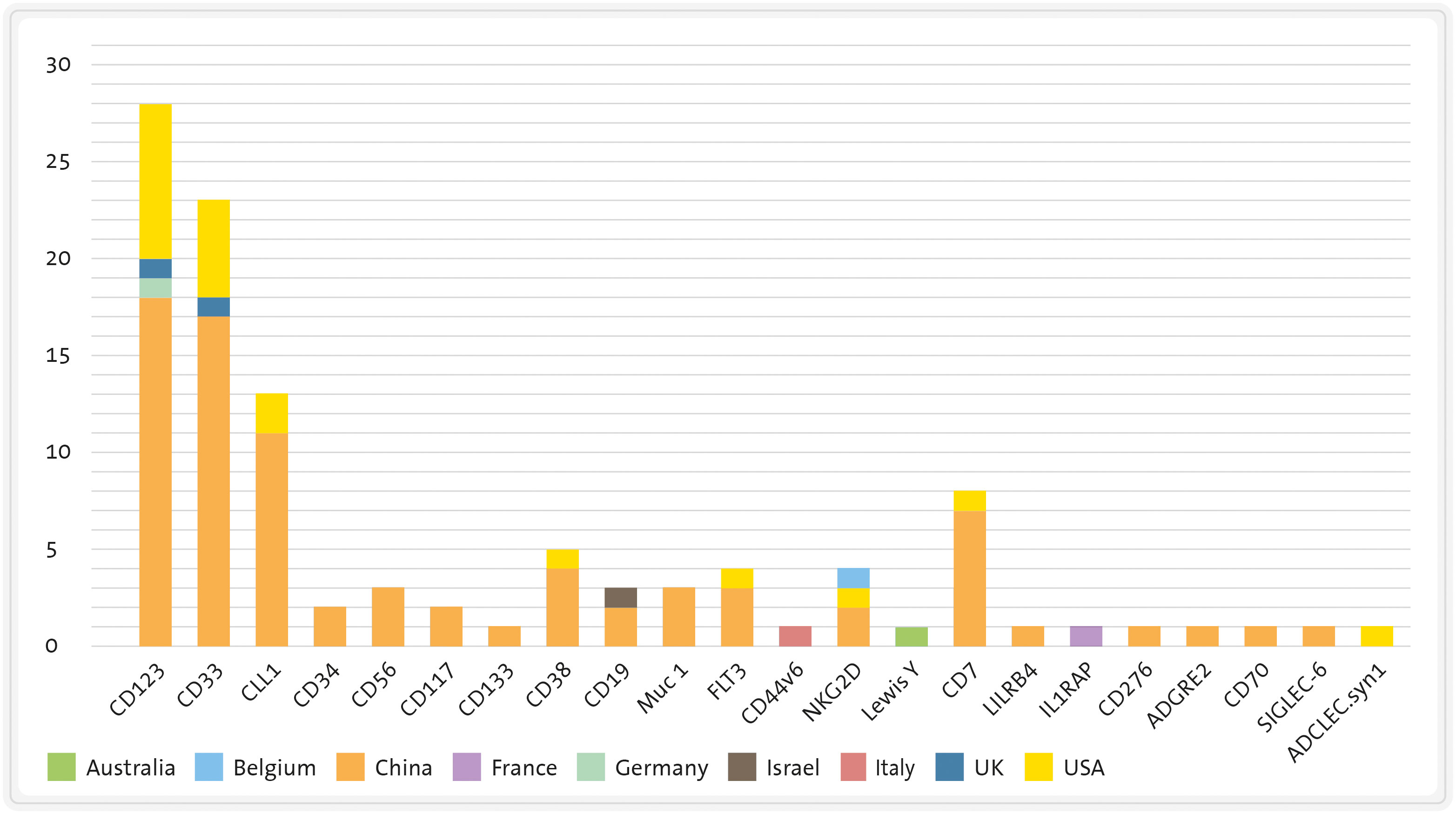
Figure 6 Number of clinical trials of CART therapies for AML divided by target antigen and country. CLL1, C-type lectin-like molecule-1 FLT3: FMS-like tyrosine kinase 3 LILRB4, leukocyte immunoglobulin like receptor B4. Source: www.clinicaltrials.gov (26/06/2023).
Another trial employed a distinct strategy to decrease toxicity: a “biodegradable” CD123-directed CAR through mRNA electroporation (75). Published data revealed that all treated patients experienced fever, and four out of five suffered from CRS; the trial was discontinued due to lack of antitumoral efficacy.
Finally, ongoing trials in the USA and China (NCT02159495, NCT04318678, and NCT03114670) involve a CD123 CAR construct that also expresses EGFR or CD20 on the T-cell surface, providing a safety switch off upon treatment with EGFR/CD20-specific antibodies (cetuximab and rituximab, respectively) (77, 145).
CD33 is another target currently being explored in clinical trials for AML patients (80, 167, 186). However, there have been limited published results from registered trials involving CART (NCT01864902, NCT0186902, and NCT03126864) or CAR-NK cell therapies targeting CD33 (NCT02944162) in AML. Preliminary findings from a trial conducted in China (NCT01864902) indicated that one patient experienced an initial reduction in blasts and systemic inflammatory symptoms; however, after two weeks, the disease progressed (79). Notably, this patient did not exhibit clinical or analytical signs of sinusoidal obstruction syndrome, a complication associated with GO, as previously described (189).
A CART therapy targeting two antigens, either CD33 or CLL-1, is currently undergoing phase I trial testing (NCT03795779). It has been reported that a patient enrolled in this study was able to undergo allo-HSCT after successful eradication of leukemia and myeloablation (81).
The most recent clinical trial utilizing CART therapy directed specifically towards CLL-1, either alone or in combination with CD33 as previously mentioned (81), obtained promising outcomes in both adult patients (84) and paediatric R/R AML patients (82, 85), (Table 1). Among the eight paediatric patients treated, four achieved a state of morphologic leukemia-free state (MLFS) along minimal residual disease (MRD) negativity (85).
A CART therapy targeting NKG2D has also undergone evaluation in a phase I trial encompassing AML and other haematological neoplasms (NCT02203825) (86). In this study, CART cells exhibited biological activity, as evidenced by an increase in inflammation parameters post-injection. Nevertheless, their anti-AML effect was limited (190). The potential improvement of clinical efficacy through combination with therapies that upregulate NKG2D expression, such as decitabine (191), warrants further investigation.
Furthermore, CART therapies targeting additional antigens are currently undergoing patient trials, albeit with limited published data, as seen in trials NCT03473457, NCT03291444, NCT04351022, etc. See Tables 1 and 2 for detailed results of published clinical trials for AML (87, 88).
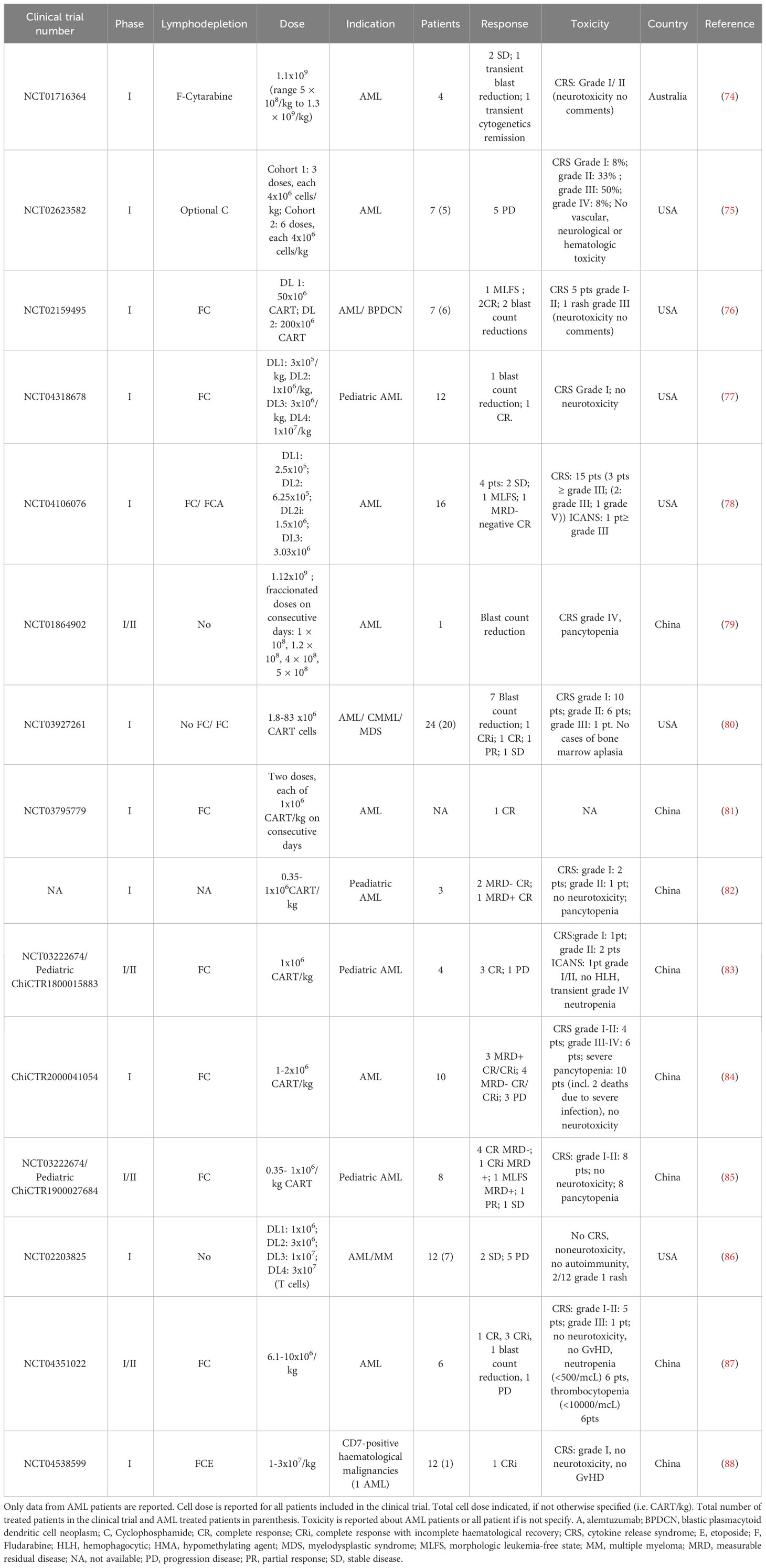
Table 2 Published clinical trials using CART in AML: Clinical results.
Allogeneic hematopoietic stem cells transplantation remains a cornerstone in the therapeutic plan for eligible AML patients, serving both as a consolidation approach for high-risk AML and as part of a salvage option for R/R AML (141). Therefore, integrating CART therapy into the treatment of these patients needs a comprehensive plan that includes allo-HSCT (192–194).
In the case of R/R AML patients who have not previously undergone allo-HSCT, the administration of a CART therapy followed by allo-HSCT represents a rational strategy. This approach aims to combine the potential benefits of CART infusion, which is critical for achieving AML cytoreduction, with the advantages of allo-HSCT. The latter can provide both additional GvL effects and a haematopoietic rescue after cytopenia probably caused by the CART cells. Executing this combined strategy is nonetheless logistically and clinically challenging, particularly managing the short interval between CART infusion and allo-HSCT (see Figure 7). It requires precise coordination with the haematopoietic cell donation process to minimize the period of CART-induced myelotoxicity and maximize the effectiveness of the CART cells. Moreover, the removal of CART cells to avoid interference with engraftment requires the lymphodepleting effects of a conditioning allo-HSCT regimen. Finally, patients may face frequent and sometimes severe complications associated with this sequential tandem procedure. These include immune-mediated complications post-CART infusion (e.g., CRS, ICANS, …) and the complications typically linked with allo-HSCT, such as GvHD and severe immunosuppression.
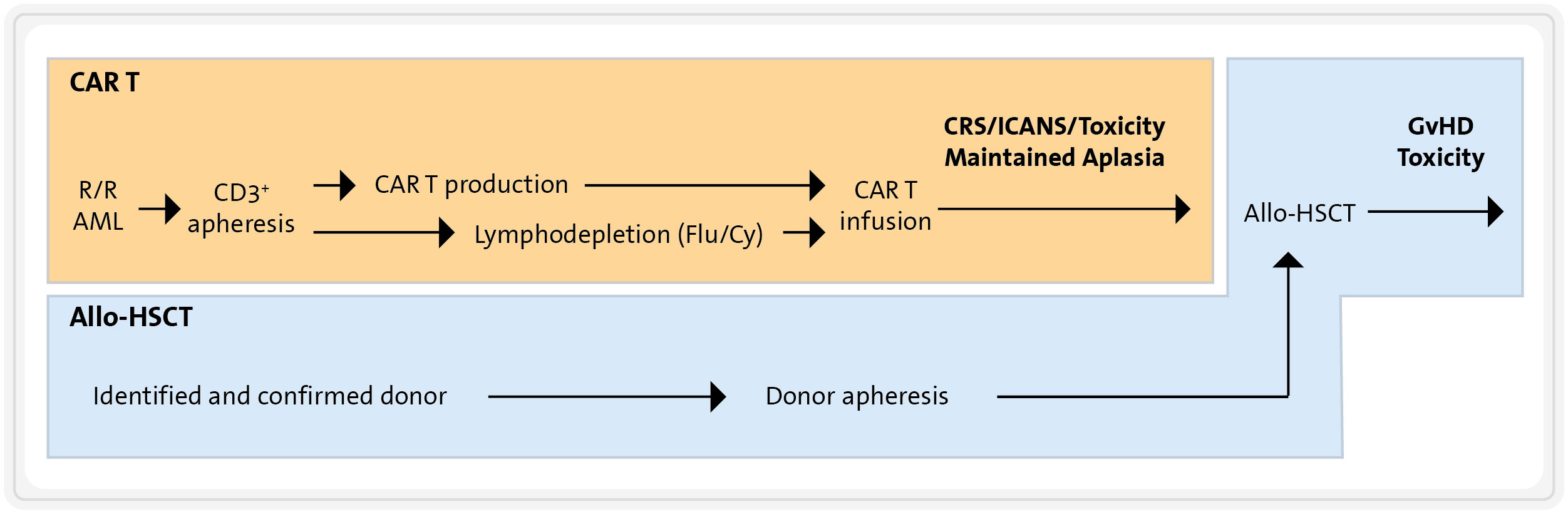
Figure 7 Combination of CART therapy and allo-HSCT. Flu, fludarabine; Cy, cyclophosphamide; CRS, cytokine release syndrome; ICANS, immune effector cell-associated neurotoxicity syndrome; GVHD, graft-versus-host disease.
Furthermore, CART cells hold promise for treating AML who experience a relapse after undergoing allo-HSCT. In this setting, any severe cytopenia following CART infusion could potentially be rescued using the same graft source utilized in the previous allo-HSCT.
While CART therapies have shown remarkable success in treating haematological diseases such as B-ALL, B-cell lymphoma and MM, their application in AML patients has faced greater challenges. The primary hurdle is the lack of a specific antigen exclusively expressed in AML cells. Despite this, clinical trials have predominantly focused on targets like CD123, CD33 and CLL-1. While published clinical data offer glimpses of the potential of CART therapies in AML treatment, they also underscore the limitations due to on-target/off-tumour toxicities. Therefore, in the majority of clinical trials involving CART therapies for AML, the availability of an allogeneic HSC donor is imperative to eventually rescue patients from life-threatening cytopenia. There are safety strategies, including switch-off mechanisms or the exploration of various dual CAR strategies, that can be employed to mitigate the potential toxicity induced by CART cells in AML (21, 195). Finally, refining clinical protocols, like combining CART cells with other therapies or integrating allo-HSCT, may help unlock the full potential of this therapeutic modality in AML patients, rendering it both safer and more effective (196).
LP-A: Writing – original draft, Writing – review & editing. ÀB: Writing – original draft. JD: Supervision, Writing – review & editing. JE: Writing – review & editing. MJ: Writing – review & editing, Funding acquisition. NK-G: Writing – original draft, Writing – review & editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported by Secretaria d’Universitat i Recerca del Departament d’Empresa i Coneixement, Generalitat de Catalunya, project 2020PANDE00079. LP-A is funded by “Programa Torres Quevedo” grant PTQ2020-011012 funded by MCIN/AEI/10.13039/501100011033; Gyala Therapeutics and FCRB-IDIBAPS are funded by project CPP2021-008553 funded by MCIN/AEI/10. 13039/501100011033 and by the European Union ‘NextGenerationEU/PRTR; Gyala Therapeutics is funded by a project from CDTI with the collaboration of the Ministry of Science and Innovation and co-financed by the European Union Next Generation EU with the file number SNEO-2021111060 (subsidized by CDTI). The project that gave rise to these results has received funding from ”la Caixa” Foundation*under the grant agreement LCF/PR/SP23/52950004.
LP-A: Gyala Therapeutics S.L. current employment. NK-G: honoraria of Gyala Therapeutics S.L. MJ: research funding of Gyala Therapeutics S.L.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Coley WB. The treatment of Malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Clin Ortho Relat Res (1893) 262:3–11.
2. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Sci (80-. ). (1996) 271(5256):1734–6. doi: 10.1126/SCIENCE.271.5256.1734
3. Thomas ED, Lochte HLJ, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med (2010) 257(11):491–6. doi: 10.1056/NEJM195709122571102
4. Thomas ED, Buckner CD, Clift RA, Fefer A, Johnson FL, Neiman PE, et al. Marrow transplantation for acute nonlymphoblastic leukemia in first remission. N Engl J Med (1979) 41(3):597–9. doi: 10.1056/NEJM197909133011109
5. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med (1988) 319(25):1676–80. doi: 10.1056/NEJM198812223192527
6. Rejeski K, Greco R, Onida F, Sánchez-Ortega I, Bonini C, Sureda A, et al. An international survey on grading, diagnosis, and management of immune effector cell-associated hematotoxicity (ICAHT) following CAR T-cell therapy on behalf of the EBMT and EHA. HemaSphere (2023) 7(5):e889. doi: 10.1097/HS9.0000000000000889
7. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U. S. A. (1989) 86(24,):10024. doi: 10.1073/PNAS.86.24.10024
8. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA (1993) 90(2,):720. doi: 10.1073/PNAS.90.2.720
9. Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. Eur J Immunol (1993) 23(7):1435–9. doi: 10.1002/EJI.1830230705
10. Dobosz P, Dzieciątkowski T. The intriguing history of cancer immunotherapy. Front In Immunol (2019) 10:2965. doi: 10.3389/fimmu.2019.02965
11. Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: Chimeric antigen receptor-redirected T cells. Front Immunol (2013) 4:371. doi: 10.3389/fimmu.2013.00371
12. Davila ML, Brentjens R, Wang X, Rivière I, Sadelain M. How do cars work? Early insights from recent clinical studies targeting CD19. OncoImmunology (2012) 1(9):1577–83. doi: 10.4161/onci.22524
13. Cummins KD, Gill S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin Hematol (2019) 56:155–163. doi: 10.1053/j.seminhematol.2018.08.008
14. Perez-Amill L, Marzal B, Urbano-Ispizua A, Juan M, Martín-Antonio B. CAR-T cell therapy: A door is open to find innumerable possibilities of treatments for cancer patients. Turkish J Hematol (2018) 35(4):217–28. doi: 10.4274/tjh.2018.0196
15. Zhang Q, Ping J, Huang Z, Zhang X, Zhou J, Wang G, et al. CAR-T cell therapy in cancer: tribulations and road ahead. J Immunol Res (2020) 2020:2020. doi: 10.1155/2020/1924379
16. Chen X, Zaro J, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev (2012) 65(10):1357–69. doi: 10.1016/j.addr.2012.09.039
17. Smith EL, Staehr M, Masakayan R, Tatake IJ, Purdon TJ, Wang X, et al. Development and evaluation of an optimal human single-chain variable fragment-derived BCMA-targeted CAR T cell vector. Mol Ther (2018) 26(6):1447–56. doi: 10.1016/j.ymthe.2018.03.016
18. Potthoff B, McBlane F, Spindeldreher S, Sickert D. A cell-based immunogenicity assay to detect antibodies against chimeric antigen receptor expressed by tisagenlecleucel. J Immunol Methods (2020) 476:112692. doi: 10.1016/J.JIM.2019.112692
19. Khan AN, Chowdhury A, Karulkar A, Jaiswal AK, Banik A, Asija S, et al. Immunogenicity of CAR-T cell therapeutics: evidence, mechanism and mitigation. Front Immunol (2022) 13:886546/BIBTEX. doi: 10.3389/FIMMU.2022.886546/BIBTEX
20. Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol (2021) 18(6):379. doi: 10.1038/S41571-021-00476-2
21. Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez-Ribas D, Caballero-Baños M, et al. CART19-BE-01: A multicenter trial of ARI-0001 cell therapy in patients with CD19+ Relapsed/refractory Malignancies. Mol Ther (2021) 29(2):636–44. doi: 10.1016/J.YMTHE.2020.09.027
22. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20):6106. doi: 10.1158/1078-0432.CCR-06-1183
23. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer (2017) 5:1. doi: 10.1186/S40425-017-0222-9
24. Mueller KT, Grupp SA, Maude SL, Levine JE, Pulsipher MA, Boyer MW, et al. Tisagenlecleucel immunogenicity in relapsed/refractory acute lymphoblastic leukemia and diffuse large B-cell lymphoma. Blood Adv (2021) 5(23):4980. doi: 10.1182/BLOODADVANCES.2020003844
25. Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol (2017) 10(1). doi: 10.1186/s13045-017-0437-8
26. Oda SK, Daman AW, Garcia NM, Wagener F, Schmitt TM, Tan X, et al. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood (2017) 130(22):2410. doi: 10.1182/BLOOD-2017-04-777052
27. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and Development of Therapies using Chimeric Antigen Receptor-Expressing T cells. Immunological Rev (2014) 257(1):107–26. doi: 10.1111/imr.12131
28. Janeway CA Jr, Travers P, Walport M. Immunobiology: the immune system in health and disease. 5th Edition. New York: Garland Science (2001).
29. Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells (2019) 8(5):472. doi: 10.3390/cells8050472
30. Abken H. Building on synthetic immunology and T cell engineering: A brief journey through the history of chimeric antigen receptors. Hum Gene Ther (2021) 32(19–20):1011–28. doi: 10.1089/HUM.2021.165
31. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121(5):1822–6. doi: 10.1172/JCI46110
32. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol (2021) 18(11):715–27. doi: 10.1038/S41571-021-00530-Z
33. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI 3 kinase/AKT/Bcl-X L activation and CD8 T cell-mediated tumor eradication. Mol Ther (2010) 18(2):413–20. doi: 10.1038/MT.2009.210/ATTACHMENT/C5A72333-F7D9-48A8-BFF1-0CD8CDAA817C/MMC2.ZIP
34. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell (2015) 28(4):415–28. doi: 10.1016/j.ccell.2015.09.004
35. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
36. Yeku OO, Brentjens RJ. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans (2016) 44:412–8:2. doi: 10.1042/BST20150291
37. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv Cell Gene Ther (2020) 3(3):e84. doi: 10.1002/ACG2.84
38. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer 2018 1201 (2018) 120(1):26–37. doi: 10.1038/s41416-018-0325-1
39. Eyquem J, Mansilla-Soto J, Giavridis T, Van Der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113. doi: 10.1038/NATURE21405
40. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol Cancer (2019) 18(1):1–13. doi: 10.1186/S12943-019-1057-4/FIGURES/1
41. Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell (2022) 185(10):1745–1763.e22. doi: 10.1016/J.CELL.2022.03.041
42. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116(20):4099–102. doi: 10.1182/blood-2010-04-281931
43. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118(18):4817–28. doi: 10.1182/blood-2011-04-348540
44. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
45. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
46. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
47. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. www.thelancet.com (2021) 398:2021. doi: 10.1016/S0140-6736(21)01222-8
48. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (2020) 396(10254,):839–52. doi: 10.1016/S0140-6736(20)31366-0
49. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMOA2024850/SUPPL_FILE/NEJMOA2024850_DATA-SHARING.PDF
50. Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy (2020) 22:57–69, 2020. doi: 10.1016/j.jcyt.2019.12.004
51. Martinez-Cibrian N, Español-Rego M, Pascal M, Delgado J, Ortiz-Maldonado V. Practical aspects of chimeric antigen receptor T-cell administration: From commercial to point-of-care manufacturing. Front Immunol (2022) 13:1005457. doi: 10.3389/fimmu.2022.1005457
52. Longo DL, Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373(12):1136–52. doi: 10.1056/NEJMRA1406184
53. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMOA1516192/SUPPL_FILE/NEJMOA1516192_DISCLOSURES.PDF
54. Grimwade D, Ivey A, Huntly BJP. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood (2016) 127(1):29. doi: 10.1182/BLOOD-2015-07-604496
55. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood (2022) 140(11,):1200–28. doi: 10.1182/BLOOD.2022015850
56. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med (2020) 383(7):617–29. doi: 10.1056/NEJMOA2012971
57. Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med (2022) 386(16):1519–31. doi: 10.1056/NEJMOA2117344
58. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424. doi: 10.1182/BLOOD-2016-08-733196
59. Heuser M, Ofran Y, Boissel N, Brunet Mauri S, Craddock C, Janssen J, et al. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol (2020) 31(6):697–712. doi: 10.1016/J.ANNONC.2020.02.018/ATTACHMENT/8FD9C8AD-B48C-472C-8302-DCF911D402B8/MMC1.DOCX
60. Thol F, Ganser A. Treatment of relapsed acute myeloid leukemia. Curr Treat Options Oncol (2020) 21(8,):1–11. doi: 10.1007/s11864-020-00765-5
61. DeWolf S, Tallman MS. How I treat relapsed or refractory AML. Blood (2020) 136(9):1023. doi: 10.1182/BLOOD.2019001982
62. DiNardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood (2020) 135(2):85–96. doi: 10.1182/BLOOD.2019001239
63. Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood (2021) 137(6):751–62. doi: 10.1182/BLOOD.2020007732
64. Clark MC, Stein A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Pract Res Clin Haematol (2020) 33(4):101224. doi: 10.1016/J.BEHA.2020.101224
65. Sallman DA, Asch AS, Al Malki MM, Lee DJ, Donnellan WB, Marcucci G, et al. The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. Blood (2019) 134(Supplement_1):569–9. doi: 10.1182/BLOOD-2019-126271
66. Brunner AM, Esteve J, Porkka K, Knapper S, Vey N, Scholl S, et al. Efficacy and safety of sabatolimab (MBG453) in combination with hypomethylating agents (HMAs) in patients with acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS): updated results from a phase 1b study. Blood (2020) 136(Supplement 1):1–2. doi: 10.1182/BLOOD-2020-136855
67. Abaza Y, Zeidan AM. Immune checkpoint inhibition in acute myeloid leukemia and myelodysplastic syndromes. Cells (2022) 11:14. doi: 10.3390/CELLS11142249
68. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
69. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
70. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell (2017) 32(4):506. doi: 10.1016/J.CCELL.2017.09.004
71. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia, N. Engl J Med (2013) 368(22,):2059–74. doi: 10.1056/nejmoa1301689
72. Köhnke T, Liu X, Haubner S, Bücklein V, Hänel G, Krupka C, et al. Integrated multiomic approach for identification of novel immunotherapeutic targets in AML. biomark Res (2022) 10(1,):1–13. doi: 10.1186/s40364-022-00390-4
73. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007 2511 (2007) 25(11):1315–21. doi: 10.1038/NBT1350
74. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the leY antigen in acute myeloid leukemia. Mol Ther (2013) 21(11,):2122. doi: 10.1038/MT.2013.154
75. Cummins K, Frey N, Nelson A, Schmidt AH, Luger S, Isaacs R, et al. Treating Relapsed / Refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood (2017) 130:1359. doi: 10.1182/BLOOD.V130.SUPPL_1.1359.1359
76. Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: A first-in-human clinical trial. Blood (2017) 130(Suppl_1):811–1. doi: 10.1182/BLOOD.V130.SUPPL_1.811.811
77. Naik S, Madden RM, Lipsitt A, Lockey T, Bran J, Rubnitz JE, et al. Safety and anti-leukemic activity of CD123-CAR T cells in pediatric patients with AML: preliminary results from a phase 1 trial. Blood (2022) 140(Supplement 1):4584–5. doi: 10.1182/blood-2022-170201
78. Sallman DA, DeAngelo DJ, Pemmaraju N, Dinner S, Gill S, Olin RL, et al. Ameli-01: A phase I trial of UCART123v1.2, an anti-CD123 allogeneic CAR-T cell product, in adult patients with relapsed or refractory (R/R) CD123+ Acute myeloid leukemia (AML). Blood (2022) 140(Supplement 1):2371–3. doi: 10.1182/BLOOD-2022-169928
79. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther (2015) 23(1):184. doi: 10.1038/MT.2014.164
80. Sallman DA, Elmariah H, Sweet K, Mishra A, Cox CA, Chakaith M, et al. Phase 1/1b safety study of prgn-3006 ultracar-T in patients with relapsed or refractory CD33-positive acute myeloid leukemia and higher risk myelodysplastic syndromes. Blood (2022) 140(Suppl 1):10313–5. doi: 10.1182/blood-2022-169142
81. Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood (2018) 132(Supplement 1):901–1. doi: 10.1182/BLOOD-2018-99-110579
82. Bu C. Phase I clinical trial of autologous CLL1 CAR-T therapy for pediatric patients with relapsed and refractory acute myeloid leukemia. Blood Am Soc Hematol (2020).
83. Zhang H, Wang P, Li Z, He Y, Gan W, Jiang H. Anti-CLL1 chimeric antigen receptor T-cell therapy in children with relapsed/refractory acute myeloid leukemia. Clin Cancer Res (2021) 27(13):3549–55. doi: 10.1158/1078-0432.CCR-20-4543
84. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol (2022) 15(1):1–5. doi: 10.1186/s13045-022-01308-1
85. Zhang H, Bu C, Peng Z, Li G, Zhou Z, Ding W, et al. Characteristics of anti-CLL1 based CAR-T therapy for children with relapsed or refractory acute myeloid leukemia: the multi-center efficacy and safety interim analysis. Leukemia (2022) 36(11):2596–604. doi: 10.1038/s41375-022-01703-0
86. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML / MDS and multiple myeloma. Cancer Res Immunology (2019) 7:100–12. doi: 10.1158/2326-6066.CIR-18-0307
87. Hematol J, Cui Q, Qian C, Xu N, Kang L, Dai H, et al. CD38 − directed CAR − T cell therapy : a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol (2021) 4–8. doi: 10.1186/s13045-021-01092-4
88. Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge W, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed / refractory CD7-positive hematological Malignancies : a phase I clinical study. Cell Res (2022) 32(11):995–1007. doi: 10.1038/s41422-022-00721-y
89. Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev (2014) 28(4,):143–53. doi: 10.1016/J.BLRE.2014.04.001
90. Gill SI. How close are we to CAR T-cell therapy for AML? Best Pract Res Clin Haematol (2019) 32(4):101104. doi: 10.1016/J.BEHA.2019.101104
91. Garnache-Ottou F, Chaperot L, Biichle S, Ferrand C, Remy-Martin JP, Deconinck E, et al. Expression of the myeloid-associated marker CD33 is not an exclusive factor for leukemic plasmacytoid dendritic cells. Blood (2005) 105(3):1256–64. doi: 10.1182/BLOOD-2004-06-2416
92. Hernández-Caselles T, Martínez-Esparza M, Pérez-Oliva AB, Quintanilla-Cecconi AM, García-Alonso A, Alvarez-López DMR, et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. J Leukoc Biol (2006) 79(1):46–58. doi: 10.1189/JLB.0205096
93. Hofmann S, Schubert ML, Wang L, He B, Neuber B, Dreger P, et al. Chimeric antigen receptor (CAR) T cell therapy in acute myeloid leukemia (AML). J Clin Med (2019) 8(2). doi: 10.3390/JCM8020200
94. Guffroy M, Falahatpisheh H, Biddle K, Kreeger J, Obert L, Walters K, et al. Liver microvascular injury and thrombocytopenia of antibody-calicheamicin conjugates in cynomolgus monkeys-mechanism and monitoring. Clin Cancer Res (2017) 23(7):1760–70. doi: 10.1158/1078-0432.CCR-16-0939
95. Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol (2014) 15(9):986–96. doi: 10.1016/S1470-2045(14)70281-5
96. Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun (2021) 12(1):1–20. doi: 10.1038/s41467-021-26683-0
97. Mardiros A, Forman SJ, Budde LE. T cells expressing CD123 chimeric antigen receptors for treatment of acute myeloid leukemia. Curr Opin Hematol (2015) 22(6,):484. doi: 10.1097/MOH.0000000000000190
98. Sugita M, Galetto R, Zong H, Ewing-Crystal N, Trujillo-Alonso V, Mencia-Trinchant N, et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat Commun (2022) 131:1–11. doi: 10.1038/s41467-022-29668-9
99. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leuk 2000 1410 (2000) 14(10):1777–84. doi: 10.1038/sj.leu.2401903
100. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α Chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell (2009) 5(1):31–42. doi: 10.1016/J.STEM.2009.04.018
101. Baroni ML, Sanchez Martinez D, Gutierrez Aguera F, Roca Ho H, Castella M, Zanetti S, et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J Immunother Cancer (2020) 8(1):e000845. doi: 10.1136/jitc-2020-000845
102. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, et al. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther (2017) 25(8):1933–45. doi: 10.1016/J.YMTHE.2017.04.017
103. Pemmaraju N, Lane AA, Sweet KL, Stein AS, Vasu S, Blum W, et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N Engl J Med (2019) 380(17):1628–37. doi: 10.1056/NEJMOA1815105/SUPPL_FILE/NEJMOA1815105_DATA-SHARING.PDF
104. Bakker ABH, Van Den Oudenrijn S, Bakker AQ, Feller N, Van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res (2004) 64(22):8443–50. doi: 10.1158/0008-5472.CAN-04-1659
105. Bill M, Bvan Kooten Niekerk P, SWoll P, Laine Herborg L, Stidsholt Roug A, Hokland P, et al. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC12A-related cancer stem cell biology. J Cell Mol Med (2018) 22(4):2311–8. doi: 10.1111/JCMM.13519
106. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol Oncol (2019) 12(1):41. doi: 10.1186/S13045-019-0726-5
107. Laborda E, Mazagova M, Shao S, Wang X, Quirino H, Woods AK, et al. Development of A chimeric antigen receptor targeting C-type lectin-like molecule-1 for human acute myeloid leukemia. Int J Mol Sci (2017) 18:2259. doi: 10.3390/IJMS18112259
108. Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol Ther (2017) 25(9):2202–13. doi: 10.1016/J.YMTHE.2017.05.024
109. Wang J, Chen S, Xiao W, Li W, Wang L, Yang S, et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J Hematol Oncol (2018) 11(1):2018. doi: 10.1186/S13045-017-0553-5
110. Hilpert J, Grosse-Hovest L, Grünebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol (2012) 189(3):1360–71. doi: 10.4049/JIMMUNOL.1200796
111. Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol Oncol (2018) 11(1). doi: 10.1186/S13045-018-0603-7
112. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-cells targeting FLT3 have potent activity against FLT3–ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leuk (2018) 325:1168–79. doi: 10.1038/S41375-018-0009-0
113. Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor–expressing T cells. Blood (2015) 125(22):3466–76. doi: 10.1182/BLOOD-2014-11-612721
114. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia (2016) 30(6):1355–64. doi: 10.1038/LEU.2016.35
115. Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica (2022) 107(2):437–45. doi: 10.3324/haematol.2020.271908
116. Ciceri F, Bonini C, Stanghellini MTL, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I–II study. Lancet Oncol (2009) 10(5):489–500. doi: 10.1016/S1470-2045(09)70074-9
117. Stasi A, Tey S.-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi: 10.1056/NEJMOA1106152
118. Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and Malignant CD117-expressing hematopoietic cells. Leukemia (2020) 34(10):2688–703. doi: 10.1038/S41375-020-0818-9
119. Zhang Z, Jiang C, Liu Z, Yang M, Tang X, Wang Y, et al. B7-H3-targeted CAR-T cells exhibit potent antitumor effects on hematologic and solid tumors. Mol Ther - Oncolytics (2020) 17:180–9. doi: 10.1016/J.OMTO.2020.03.019
120. Lichtman EI, Du H, Shou P, Song F, Suzuki K, Ahn S, et al. Preclinical evaluation of B7-H3-specific chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Clin Cancer Res (2021) 27(11):3141. doi: 10.1158/1078-0432.CCR-20-2540
121. Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia (2017) 31(10):2191–9. doi: 10.1038/leu.2017.57
122. Shah TJ, Nirali N. Fry, Mechanisms of resistance to CAR T cell therapy. Nat Re (2019) 16:6, 372–385, 2019. doi: 10.4049/jimmunol.1801473.The
123. Zhao X, Yang J, Zhang X, Lu XA, Xiong M, Zhang J, et al. Efficacy and safety of CD28- or 4-1BB-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia. Mol Ther - Oncolytics (2020) 18(11,):272–81. doi: 10.1016/j.omto.2020.06.016
124. Atilla PA, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating tnfα activity allows transgenic il15-expressing cll-1 car t cells to safely eliminate acute myeloid leukemia. J Immunother Cancer (2020) 8(2):1–11. doi: 10.1136/jitc-2020-001229
125. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-ts): combination or built-in CAR-T. Int J Mol Sci (2018) 19(2). doi: 10.3390/IJMS19020340
126. Maude SL, Hucks GE, Seif AE, Talekar MK, Teachey DT, Baniewicz D, et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). N Engl J Med (2017) 35(15_suppl):103–3. doi: 10.1200/JCO.2017.35.15_SUPPL.103
127. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood (2021) 138(4):318–30. doi: 10.1182/BLOOD.2020008221
128. Hamieh M, Mansilla-Soto J, Rivière I, Sadelain M. Programming CAR T cell tumor recognition: tuned antigen sensing and logic gating. Cancer Discovery (2023) 13(4,):829–43. doi: 10.1158/2159-8290.CD-23-0101
129. Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med (2022) 28(2):345–52. doi: 10.1038/S41591-021-01621-1
130. Savanur MA, Weinstein-Marom H, Gross G. Implementing logic gates for safer immunotherapy of cancer. Front Immunol (2021) 12:780399/BIBTEX. doi: 10.3389/FIMMU.2021.780399/BIBTEX
131. Vishwasrao P, Li G, Boucher JC, Smith DL, Hui SK. Emerging CAR T cell strategies for the treatment of AML. Cancers (Basel) (2022) 14(5,):1–26. doi: 10.3390/cancers14051241
132. Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol (2019) 12(1,):1–10. doi: 10.1186/s13045-019-0813-7
133. Andrea AE, Chiron A, Bessoles S, Hacein-Bey-abina S. Engineering next-generation car-t cells for better toxicity management. Int J Mol Sci (2020) 21(22):1–25. doi: 10.3390/ijms21228620
134. Lanitis E, Coukos G, Irving M. All systems go: converging synthetic biology and combinatorial treatment for CAR-T cell therapy. Curr Opin Biotechnol (2020) 65:75–87. doi: 10.1016/j.copbio.2020.01.009
135. Katsarou A, Sjöstrand M, Naik J, Mansilla-Soto J, Kefala D, Kladis G, et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Transl Med (2021) 13:623. doi: 10.1126/SCITRANSLMED.ABH1962/SUPPL_FILE/SCITRANSLMED.ABH1962_DATA_FILE_S1.ZIP
136. Haubner S, Mansilla-Soto J, Nataraj S, He X, Park JH, Wang X, et al. ‘IF-better’ Gating: combinatorial targeting and synergistic signaling for enhanced CAR T cell efficacy. Blood (2021) 138(Supplement 1):2774–4. doi: 10.1182/BLOOD-2021-149263
137. Rajan TS, Gugliandolo A, Bramanti P, Mazzon E. In vitro-transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) therapy in hematologic and solid tumor management: a preclinical update. Int J Mol Sci (2020) 21(18):1–13. doi: 10.3390/IJMS21186514
138. Foster JB, Barrett DM, Karikó K. The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol Ther (2019) 27(4):747. doi: 10.1016/J.YMTHE.2019.01.018
139. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leuk 2015 298 (2015) 29(8):1637–47. doi: 10.1038/LEU.2015.52
140. Tasian SK, Kenderian SS, Shen F, Li Y, Ruella M, Fix WC, et al. Efficient termination of CD123-redirected chimeric antigen receptor T cells for acute myeloid leukemia to mitigate toxicity. Blood (2015) 126(23). doi: 10.1182/BLOOD.V126.23.565.565
141. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell (2018) 173(6):1439. doi: 10.1016/J.CELL.2018.05.013
142. Wellhausen N, Rennels AK, Lesch S, Agarwal S, Charria B, Choi G, et al. Epitope editing in hematopoietic cells enables CD45-directed immune therapy. Blood (2022) 140(Supplement 1):862–4. doi: 10.1182/BLOOD-2022-158684
143. Wellhausen N, O’Connell RP, Lesch S, Engel NW, Rennels AK, Gonzales D, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Transl Med (2023) 15(714):eadi1145. doi: 10.1126/SCITRANSLMED.ADI1145
144. Casucci M, Falcone L, Camisa B, Norelli M, Porcellini S, Stornaiuolo A, et al. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T cells co-expressing a suicide gene. Front Immunol (2018) 9:507. doi: 10.3389/FIMMU.2018.00507
145. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest (2016) 126(11):4262. doi: 10.1172/JCI84813
146. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science (2015) 350(6258):aab4077. doi: 10.1126/SCIENCE.AAB4077
147. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nat (1975) 256(5517):495–7. doi: 10.1038/256495A0
148. Roth KDR, Wenzel EV, Ruschig M, Steinke S, Langreder N, Heine PA, et al. Developing recombinant antibodies by phage display against infectious diseases and toxins for diagnostics and therapy. Front Cell Infect Microbiol (2021) 11:697876. doi: 10.3389/FCIMB.2021.697876
149. Bartoló-Ibars A, Uribe-Herranz M, Muñoz-Sánchez G, Arnaldos-Pérez C, Ortiz-Maldonado V, Urbano-Ispizua Á, et al. CAR-T after stem cell transplantation in B-cell lymphoproliferative disorders: are they really autologous or allogenic cell therapies? Cancers (2021) 13(18):4664. doi: 10.3390/CANCERS13184664
150. Brudno JN, Somerville RPT, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell Malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol (2016) 34(10):1112–21. doi: 10.1200/JCO.2015.64.5929
151. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leuk 2014 288 (2014) 28(8):1596–605. doi: 10.1038/LEU.2014.62
152. Christodoulou I, Ho J, Marple A, Ravich JW, Tam A, Rahnama R, et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J Immunother Cancer (2021) 9:3894. doi: 10.1136/jitc-2021-003894
153. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res (2018) 8(6):1083. doi: 10.1126/scitranslmed.aaj2013
154. Cai T, Gouble A, Black KL, Skwarska A, Naqvi AS, Taylor D, et al. Targeting CD123 in blastic plasmacytoid dendritic cell neoplasm using allogeneic anti-CD123 CAR T cells. Nat Commun (2022) 13(1):1–11. doi: 10.1038/s41467-022-29669-8
155. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9:374. doi: 10.1126/scitranslmed.aaj2013
156. Berdeja JG, Alsina M, Shah ND, Siegel DS, Jagannath S, Madduri D, et al. Updated results from an ongoing phase 1 clinical study of bb21217 anti-bcma CAR T cell therapy. Blood (2019) 134(Supplement_1):927. doi: 10.1182/blood-2019-126660
157. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med (2016) 8:355. doi: 10.1126/SCITRANSLMED.AAF8621/SUPPL_FILE/8-355RA116_SM.PDF
158. Watanabe N, Mo F, McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol (2022) 13:876339. doi: 10.3389/FIMMU.2022.876339
159. Battram AM, Bachiller M, Lopez V, de Larrea CF, Urbano-Ispizua A, Martín-Antonio B. Il-15 enhances the persistence and function of bcma-targeting car-t cells compared to il-2 or il-15/il-7 by limiting car-t cell dysfunction and differentiation. Cancers (Basel) (2021) 13(14):3534. doi: 10.3390/CANCERS13143534/S1
160. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol (1998) 72(11):8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998
161. Aronovich EL, McIvor RS, Hackett PB. The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet (2011) 20(R1):R14. doi: 10.1093/HMG/DDR140
162. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leuk (2017) 311:186–94. doi: 10.1038/LEU.2016.180
163. Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther (2022) 30(10):3155–75. doi: 10.1016/J.YMTHE.2022.06.006
164. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate. J Clin Invest (2016) 126(9):3363–76. doi: 10.1172/JCI86721DS1
165. Prommersberger S, Reiser M, Beckmann J, Danhof S, Amberger M, Quade-Lyssy P, et al. CARAMBA: a first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther (2021) 28(9):560. doi: 10.1038/S41434-021-00254-W
166. Tasian SK, Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood (2017) 129(17):2395–407. doi: 10.1182/BLOOD-2016-08-736041
167. O’Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica (2015) 100(3):336–44. doi: 10.3324/HAEMATOL.2014.112748
168. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-of-care CAR T-cell production (ARI-0001) using a closed semi-automatic bioreactor: Experience from an academic phase i clinical trial. Front Immunol (2020) 11:482/FULL. doi: 10.3389/FIMMU.2020.00482/FULL
169. Castella M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a novel anti-CD19 chimeric antigen receptor: A paradigm for an affordable CAR T cell production at academic institutions. Mol Ther - Methods Clin Dev (2019) 12:134–44. doi: 10.1016/j.omtm.2018.11.010
170. Perez-Amill L, Suñe G, Antoñana-Vildosola A, Castella M, Najjar A, Bonet J, et al. Preclinical development of a humanized chimeric antigen receptor against B cell maturation antigen for multiple myeloma. Haematologica (2020) 106(1):228577. doi: 10.3324/haematol.2019.228577
171. Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood (2013) 122(18):3138–48. doi: 10.1182/BLOOD-2012-12-474056
172. Bachiller M, Perez-Amill L, Battram AM, Carné SC, Najjar A, Verhoeyen E, et al. Original research: NK cells enhance CAR-T cell antitumor efficacy by enhancing immune/tumor cells cluster formation and improving CAR-T cell fitness. J Immunother Cancer (2021) 9(8):2866. doi: 10.1136/JITC-2021-002866
173. Lorenzo-Herrero S, Sordo-Bahamonde C, Gonzalez S, López-Soto A. CD107a degranulation assay to evaluate immune cell antitumor activity. Methods Mol Biol (2019) 1884:119–30. doi: 10.1007/978-1-4939-8885-3_7
174. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol (2018) 11:1. doi: 10.1186/s13045-018-0571-y
175. Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rγ null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. Int J Cancer (2008) 123(9):2222–7. doi: 10.1002/IJC.23772
176. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018 246 (2018) 24(6):739–48. doi: 10.1038/S41591-018-0036-4
177. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood (2014) 123(15):2343. doi: 10.1182/BLOOD-2013-09-529537
178. Loff S, Dietrich J, Meyer JE, Riewaldt J, Spehr J, von Bonin M, et al. Rapidly switchable universal CAR-T cells for treatment of CD123-positive leukemia. Mol Ther - Oncolytics (2020) 17:408–20. doi: 10.1016/J.OMTO.2020.04.009
179. Vucinic V, Quaiser A, Lückemeier P, Fricke S, Platzbecker U, Koehl U. Production and application of CAR T cells: current and future role of europe. Front Med (2021) 8:713401. doi: 10.3389/fmed.2021.713401
180. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng (2022) 6(2):118–28. doi: 10.1038/s41551-021-00842-6
181. Garcia-Aponte OF, Herwig C, Kozma B. Lymphocyte expansion in bioreactors: upgrading adoptive cell therapy. J Biol Eng (2021) 15(1):1–34. doi: 10.1186/S13036-021-00264-7
182. Penack O, Koenecke C. Complications after CD19+ CAR T-cell therapy. Cancers (Basel) (2020) 12(11):1–17. doi: 10.3390/CANCERS12113445
183. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMRA1706169
184. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol (2021) 22(2):85–96. doi: 10.1038/s41577-021-00547-6
185. Rejeski K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, et al. CAR-HEMATOTOX: a model for CAR T-cell–related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood (2021) 138(24):2499–513. doi: 10.1182/blood.2020010543
186. Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leuk (2018) 33(1):64–74. doi: 10.1038/s41375-018-0180-3
187. Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood (2005) 106(13):4086. doi: 10.1182/BLOOD-2005-03-1072
188. Cellectis reports clinical hold of UCART123 studies | Cellectis. Available at: https://cellectis.com/en/press/cellectis-reports-clinical-hold-of-ucart123-studies (Accessed Jun. 08, 2022).
189. Rajvanshi P, Shulman HM, Sievers EL, McDonald GB. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood (2002) 99(7):2310–4. doi: 10.1182/BLOOD.V99.7.2310
190. Murad JM, Baumeister SH, Werner L, Daley H, Trébéden-Negre H, Reder J, et al. Manufacturing development and clinical production of NKG2D Chimeric Antigen Receptor-expressing T cells for autologous adoptive cell therapy. Cytotherapy (2018) 20(7):952. doi: 10.1016/J.JCYT.2018.05.001
191. Raneros AB, Minguela A, Rodriguez RM, Colado E, Bernal T, Anguita E, et al. Increasing TIMP3 expression by hypomethylating agents diminishes soluble MICA, MICB and ULBP2 shedding in acute myeloid leukemia, facilitating NK cell-mediated immune recognition. Oncotarget (2017) 8(19):31959. doi: 10.18632/ONCOTARGET.16657
192. Jiang H, Hu Y, Mei H. Consolidative allogeneic hematopoietic stem cell transplantation after chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia: who? When? Why? Biomark Res (2020) 8(1):66. doi: 10.1186/S40364-020-00247-8
193. Liu J, Zhang X, Zhong JF, Zhang C. CAR-T cells and allogeneic hematopoietic stem cell transplantation for relapsed/refractory B-cell acute lymphoblastic leukemia. Immunotherapy (2017) 9(13):1115–25. doi: 10.2217/IMT-2017-0072/ASSET/IMAGES/LARGE/FIGURE1.JPEG
194. Jacoby E. The role of allogeneic HSCT after CAR T cells for acute lymphoblastic leukemia. Bone Marrow Transplant (2019) 54(2):810–4. doi: 10.1038/s41409-019-0604-3
195. Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol (2019) 38:415–22. doi: 10.1200/JCO.19
Keywords: acute myeloid leukemia, chimeric antigen receptor, myelotoxicity, hematologic toxicity, cytopenia, on-target/off-tumor toxicity
Citation: Pérez-Amill L, Bataller À, Delgado J, Esteve J, Juan M and Klein-González N (2023) Advancing CART therapy for acute myeloid leukemia: recent breakthroughs and strategies for future development. Front. Immunol. 14:1260470. doi: 10.3389/fimmu.2023.1260470
Received: 17 July 2023; Accepted: 30 October 2023;
Published: 30 November 2023.
Edited by:
Stephen Goldrick, University College London, United KingdomCopyright © 2023 Pérez-Amill, Bataller, Delgado, Esteve, Juan and Klein-González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manel Juan, bWp1YW5AY2xpbmljLmNhdA==; Nela Klein-González, a2xlaW5AcmVjZXJjYS5jbGluaWMuY2F0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.