 Roxana Zogorean
Roxana Zogorean Stefan Wirtz
Stefan Wirtz- 1Medizinische Klinik 1, Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 2Medical Immunology Campus Erlangen, FAU Erlangen-Nürnberg, Erlangen, Bavaria, Germany
Inflammatory bowel disease (IBD) is a chronic inflammatory disease of the gastrointestinal tract, defined by a clinical relapse-remitting course. Affecting people worldwide, the origin of IBD is still undefined, arising as a consequence of the interaction between genes, environment, and microbiota. Although the root cause is difficult to identify, data clearly indicate that dysbiosis and pathogenic microbial taxa are connected with the establishment and clinical course of IBD. The composition of the microbiota is shaped by plasma cell IgA secretion and binding, while cytokines such as IL10 or IFN-γ are important fine-tuners of the immune response in the gastrointestinal environment. B cells may also influence the course of inflammation by promoting either an anti-inflammatory or a pro-inflammatory milieu. Here, we discuss IgA-producing B regulatory cells as an anti-inflammatory factor in intestinal inflammation. Moreover, we specify the context of IgA and IgG as players that can potentially participate in mucosal inflammation. Finally, we discuss the role of B cells in mouse infection models where IL10, IgA, or IgG contribute to the outcome of the infection.
Introduction
Inflammatory Bowel diseases (IBD) are chronic inflammatory disorders of the gastrointestinal tract. Several factors may essentially contribute to the onset and progression of IBD: genetic factors, environmental factors, the host immune system and microbiota. The crosstalk between microbiota and the immune cells together with intestinal epithelial cells comprises the hallmark of intestinal homeostasis (1, 2). Although this interaction is essential for maintaining a balanced system and for initiating a protective response to luminal pathogens, it seems to be also critical in the pathogenesis of IBD. In response to intestinal barrier disruption, cytokines and chemokines are released in excess and are crucial contributors to acute intestinal inflammation (3). IBD patients are unable to resolve the inflammation which further develops into chronic intestinal inflammation due to sustained activation of the mucosal immune cells.
Importantly, inflammatory bowel diseases, Crohn’s disease and ulcerative colitis, are characterized as well by increased antibody responses at the mucosal site which participate in exacerbation of inflammation (4–8). Despite the fact that the pathogenic role of T cell subsets and soluble factors such as TNF and IL-23 have been intensively studied in intestinal inflammation (9–11), the role of B cells and antibodies still remains largely elusive. However, in recent years, studies have increasingly brought more light into the role of B cells in IBD. In particular the detection of autoantibodies in the serum of both ulcerative colitis (UC) and Crohn’s disease (CD) patients has led to a better understanding of possible therapeutic strategies (12). Different pathophysiology defines the two types of IBD in regards to autoantibodies: UC patients have higher IgG antibodies against tropomyosin 1 and 5 isoforms (13, 14) and anti-neutrophil cytoplasmic antibody (ANCA) (15). On the other hand, CD patients have much lower ANCA antibodies but increased levels of IgG and IgA against Saccharomyces cervisiae (16), flagellin (17) and E. coli (18). However, more studies are necessary for elucidating the pathogenic role and the relevance of autoantibodies in IBD (12).
Due to the disruption of the intestinal barrier in IBD, increased levels of plasma cells are found in the mucosa of UC patients and may be an indicator of severity or relapse (8, 19). In addition to the disruption of the intestinal barrier, IBD patients have altered microbiota composition compared to healthy controls (20–22). IgA, the most abundant antibody in the body, has been shown to have a role in targeting bacterial species to maintain homeostasis and control and elimination of pathogenic bacteria (23). The fitness of beneficial bacteria is an important parameter for health, and the secretion of IgA may have an advantageous role to play in this respect. While several commensal bacteria are known to play an immunosuppressive role in inflammation and the imbalance between beneficial and harmful bacteria is a feature of IBD, the precise function of gut bacteria in IBD still remains incompletely understood (24).
In homeostasis, B cells are involved in maintaining the intestinal barrier by producing antibodies such as IgA and IgM. In addition, they have immunosuppressive activity as regulatory B cells (Bregs) during inflammation, and their role has been described in a number of diseases, including IBD, in both humans and mice (25, 26). B cells have been suggested to be involved in the immunoregulatory response to intestinal inflammation through the production of the anti-inflammatory cytokine IL-10 in T cell-induced colitis or DSS colitis models (25, 26). B-cell depletion using anti-CD20 therapies also targets Bregs and IL-10, leading to increased CD4+ T-cell proliferation and proinflammatory cytokines and worsening UC (27, 28). In comparison to regulatory T cells, Bregs do not consist of a single subset of B cells (29) which raises the interest for their possible hindered functions.
However, in a murine model of IBD, B cells are found to be increased in the mesenteric lymph nodes and may increase the inflammation severity together with T cells (30). A recent study found that B cells with an IFN-I signature reduced the stromal-epithelial cell interaction required for mucosal healing. The study demonstrates the short-term beneficial effect of B cell depletion on wound healing in inflamed colonic tissue (31).
IgA is produced mainly in the gut-associated lymphoid tissues (GALT) such as Peyer’s patches, mesenteric lymph nodes (mLN), isolated lymphoid follicles (ILF), the cecal patch but also in the lamina propria. In mice, the germinal centers formed in the Peyer’s patches and mLN support the differentiation of IgA+ B cells and consequently the production of specific IgA, while ILF seem to contribute mostly to T-independent IgA synthesis (32, 33). Interestingly, human GALT ILFs have recently been classified into two subtypes, namely mucosal ILF (M-ILF) and submucosal ILF (SM-ILF). These, ILFs have been shown to contain germinal centers and memory B cells and are a major source of physiologically important IgA (34) indicating the existence of substantial species specific differences.
Although the function antibodies in the intestine environment may be better understood, acting in opsonization of pathogens, recruiting complement, binding Fc receptors (35–38), the function of IgA is still puzzling especially due to the dichotomic role of maintaining tolerance to commensals and elimination of pathogens.
Studies done on activation induced cytidine deaminase-deficient mice showed that IgA is important for modulating the bacterial communities in the intestine. Lack of IgA in the AID -/- mice determined the excessive growing of anaerobic bacteria in the small intestines. Colonization of germ-free mice with the same anaerobic bacteria showed strong induction of unspecific IgA (39). The changes in the microbial environment caused strong activation of the immune response with hypertrophy in the lymphoid tissues.
IgA modulates the microbiota-immune response cross talk by acting on bacterial epitopes and on bacterial metabolism (40, 41). For example IgA directed against the capsular polysaccharide CPS4 determines a less pro-inflammatory phenotype of Bacteroides thetaiotaomicron (42). IgA antibodies protect against colonizing mucosal pathogens and maintain a homeostatic environment for commensals using a variety of strategies that have been reviewed elsewhere (43, 44). An interesting observation was made by Bunker et al. when they tested the binding affinities of IgA antibodies to bacteria in vitro. The study shows that the antigen expression levels on bacteria that drives IgA binding are depending on the environment (45).
Herein, we review how pro-inflammatory cytokines may play an anti-inflammatory or pro-inflammatory role in the intestine and how microbiota is influenced by immune cells interaction and in return can influence the nature of immune response. In addition, we discuss regulatory B cells in relation to microbiota. We present newly described IgA producing Bregs as possible key players in shaping the microbiota to enhance suppression of inflammation.
Anti or pro-inflammatory cytokines: fine-tuners of the immune response
Cytokines and chemokines enable intercellular communication and participate in maintaining homeostasis of the intestine. In IBD patients, their functions seemed to be skewed to a continuous inflammatory state with negative effects on intestinal barrier integrity (3). However, the labels of pro- and anti-inflammatory describe a simplistic view of cytokines functions during an inflammatory process (46). When sensing microbial components through e.g. Toll like receptor (TLR) signaling, lamina propria macrophages and dendritic cells produce large amounts of IL-1β, IL-6, IL-18 and TNF (47). IL-6 is a pro-inflammatory cytokine which can activate antigen presenting cells and T cells from the intestine and may also have a function in the stimulation and expansion of intestinal epithelial cells (IEC) (48). However, blocking of IL-6 or IL-6 signaling has become an important direction especially in treatment of UC and CD patients with high production of IL-6 (49).
TNF is produced mostly by CD14+ macrophages, adipocytes, T cells, fibroblasts in patients with IBD (50–52) and has several effects in inducing intestinal inflammation in colitis: causing necroptosis in Paneth cells, activating macrophages and effector T cells, affecting the IEC and disrupting the intestinal barrier. IBD patients treated with anti-TNF antibodies show reduced inflammation (53), but the treatment with soluble TNF receptor has failed to improve the disease (54). It was thus hypothesized that soluble TNF is needed for self-renewal during intestinal inflammation while anti-TNF antibodies resolve inflammation by binding to membrane TNF on immune cells thereby causing apoptosis (55).
Innate lymphoid cells (ILCs) have been shown to produce high levels of mainly pro-inflammatory cytokines such as IFN-γ and IL23 which drive experimental innate immune mediated colitis (56). IL23 production has been found to be increased in several murine models of colitis, suggesting an essential role in intestinal inflammation (57–59). Furthermore, UC and CD patients have increased levels of IL23 in the serum and mucosa (60, 61). Several other mechanisms were described that render the balance to a pro-inflammatory state: the production of CC-chemokine ligand 3 by ILCs recruits CCR1+ inflammatory monocytes and intensify the inflammation in CD (62).
Moreover, in mice type 3 innate lymphoid cells are one of the primary producers of IL-22, a cytokine with pleiotropic effects mainly on non-hematopoietic cells (63). IL-22 production has protective roles in the context of mucosal inflammation by preserving intestinal stem cells after tissue damage (64) and by producing antimicrobial peptides which act on the intestinal barrier and microbiota (65, 66). Besides its beneficial role, studies however indicate that overexpression of IL-22 results in high antimicrobial peptides production with loss of bacterial diversity in IL10-/- mice, which develop spontaneous colitis compared to IL10-/-IL-22-/- mice (67).
Studies have revealed high numbers of T cells present in the intestine of IBD patients together with high levels of T cell-derived pro-inflammatory cytokines (68, 69). Interestingly, there are couples of differences in T cells subtypes present in CD compared to UC patients. Th1 infiltration and increased production of IFN-γ and IL-2 is detected mostly in the lamina propria of patients of CD (70), while NKT cells with Th2 type phenotype seem to be present in the lamina propria of UC patients (71, 72).
Another T cell subset, Th17 cells, has been shown to produce high levels of cytokines with pro-inflammatory activity in mouse models and potentially in both CD and UC (73, 74). Th17 cells, together with other types of immune cells such as γδ T cells and ILCs, are the major producers of IL-17 in response to stimulation by IL-1β and IL-23. During inflammation, IL-17 plays a role in recruiting neutrophils, antimicrobial peptide production and maintaining the intestinal barrier (75). Although IL-17 production protects against bacterial or fungal intestinal infections, it can also have a pathological role. High levels of IL-17 are found in the serum and in the mucosa of patients with UC and CD (61, 76). Whether its supports inflammation in IBD remains however unclear (77).
Integrity of the intestinal barrier is maintained also by γδ T cells, a cell type abundant in the intestinal epithelium. These cells were shown to produce antimicrobial peptides or cytokines such as IL-17, but can also contribute to intestinal immune reactions via molecules such as granzymes (78). Despite their described role in maintaining intestinal homeostasis in mice (79), their distribution in IBD patients is less clear (80). For instance, one study found increased expression of gut-homing chemokine receptor 9 on circulating γδ T cells in IBD patients (81). However, another recent study, using single cell sequencing, showed a decrease of CD8+ T cells and γδ T cells in the tissues of patients with severe CD (82). Nonetheless, treatment of CD patients with a neutralizing anti–IL-17 receptor monoclonal antibody has shown no efficacy and even worsened disease (83). Moreover, murine and human Th17 cells may produce a more diverse pro-inflammatory repertoire of cytokine such as IL-26 and IFN-γ (84–87) suggesting a strong role in inflammation and tissue damage in IBD.
The role of IL-26 in intestinal inflammation may depend on the disease state, with a protective effect in acute inflammation but damaging in the chronic inflammation of IBD patients (88). While IL-8 is known to have a role in immune cell recruitment to the site of infection or inflammation (89), it also has a role in differentiation of Caco-2 BBE monolayers to enterocytes (90). Although, IL-1β levels are elevated in IBD patients and are corelated with disease severity, macrophage derived IL-1β signals from sensing microbiota in steady state promote Treg differentiation and tolerance to dietary antigens (91).
Importantly, non-immune cells contribute to the production of pro-inflammatory cytokines in IBD (92). These cells may activate the immune cells by producing TNF and IL-6 or may respond to pro-inflammatory cytokines produced by lymphocytes or APC and thus participate in intestinal barrier destruction in IBD. On the contrary, IL-6 production by murine intestinal epithelial cells promoted crypt organoid proliferation and increased stem cell numbers (93). Moreover, anti-inflammatory cytokines such as IL-10 are induced in response to microbiota and participate in maintaining tolerance and homeostasis in the murine intestinal epithelium (94, 95).
In conclusion, the cytokine network involved in intestinal inflammation which then leads to IBD is regulated in a complex manner by genetic, microbial and immune factors. Therapeutic agents are used to block cytokines or cytokine signaling but have sometimes limited efficacy or different outcomes depending on patient subgroups (3, 96).
The role of microbiota in maintaining homeostasis
Homeostasis in the gut is characterized by immune tolerance to commensal bacteria while preserving an intact intestinal barrier and successfully fighting and clearing out pathogens. The intestinal microbiota is separated from the intestinal epithelium by a barrier of mucus (97, 98) enriched in antimicrobial peptides (AMPs). Not solely the mucus layer acts as shield, but is also the environment where bacteria are constrained by immune response (Figure 1).
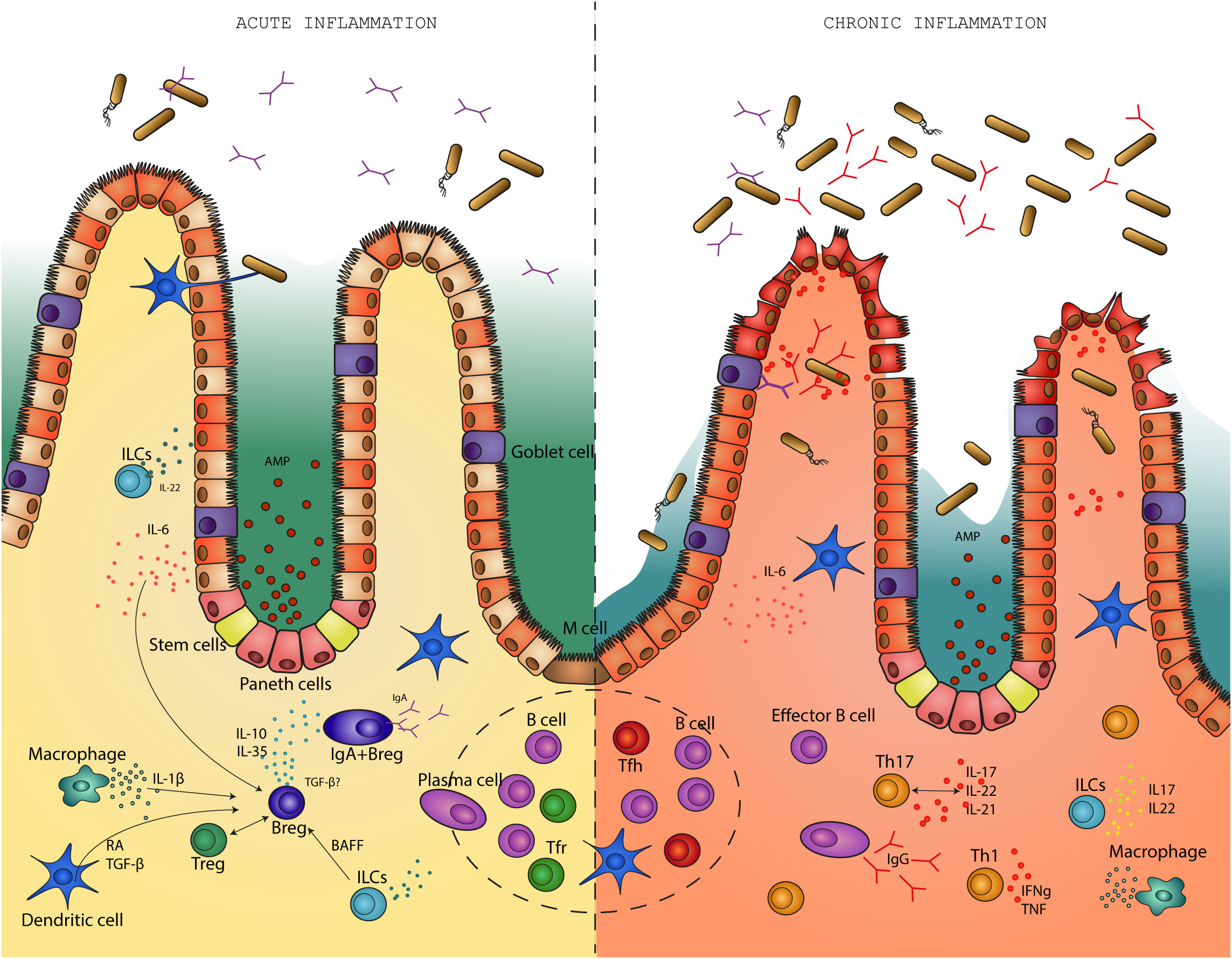
Figure 1 Regulatory B cells differentiate in response to pro-inflammatory stimuli during an acute infection. In a feedback loop with the microbiota, regulatory B cells suppress inflammation in the gut and help return the environment to homeostasis. On the other hand, in the absence of regulatory B cells, intestinal inflammation becomes chronic. This is associated with disruption of the intestinal barrier, bacterial translocation and high levels of pro-inflammatory cytokines. B effector cells produce high levels of IgG antibodies in response to disruption of the mucosal barrier.
Various intestinal dendritic cell (DC) subsets capture luminal antigens from commensal bacteria via several proposed mechanisms. One pathway refers to the delivery of luminal antigens and is dependent on M cells, neonatal Fc receptors or apoptosis. It was demonstrated recently that DC sampling of both soluble and particulate antigens via so called Goblet cell associated Passages (GAPs) could be of particular importance for mucosal immunity and tolerance (99, 100). The direct pathway involves intestinal dendrite extension into the lumen to sample antigens (101) followed by migration to the lymph nodes (102). Importantly, intestinal DC functions support IgA production by B cells (103). Commensal products such as polysaccharide A condition DCs to induce IL-10 producing T regs which help in preventing colonic inflammation in the mouse colitis models (104–106). Murine DCs can be conditioned by short-chain fatty acids: acetate, butyrate and propionate to further support IL-10 producing cells (107) or IgA production in B cells (108) (Figure 2).
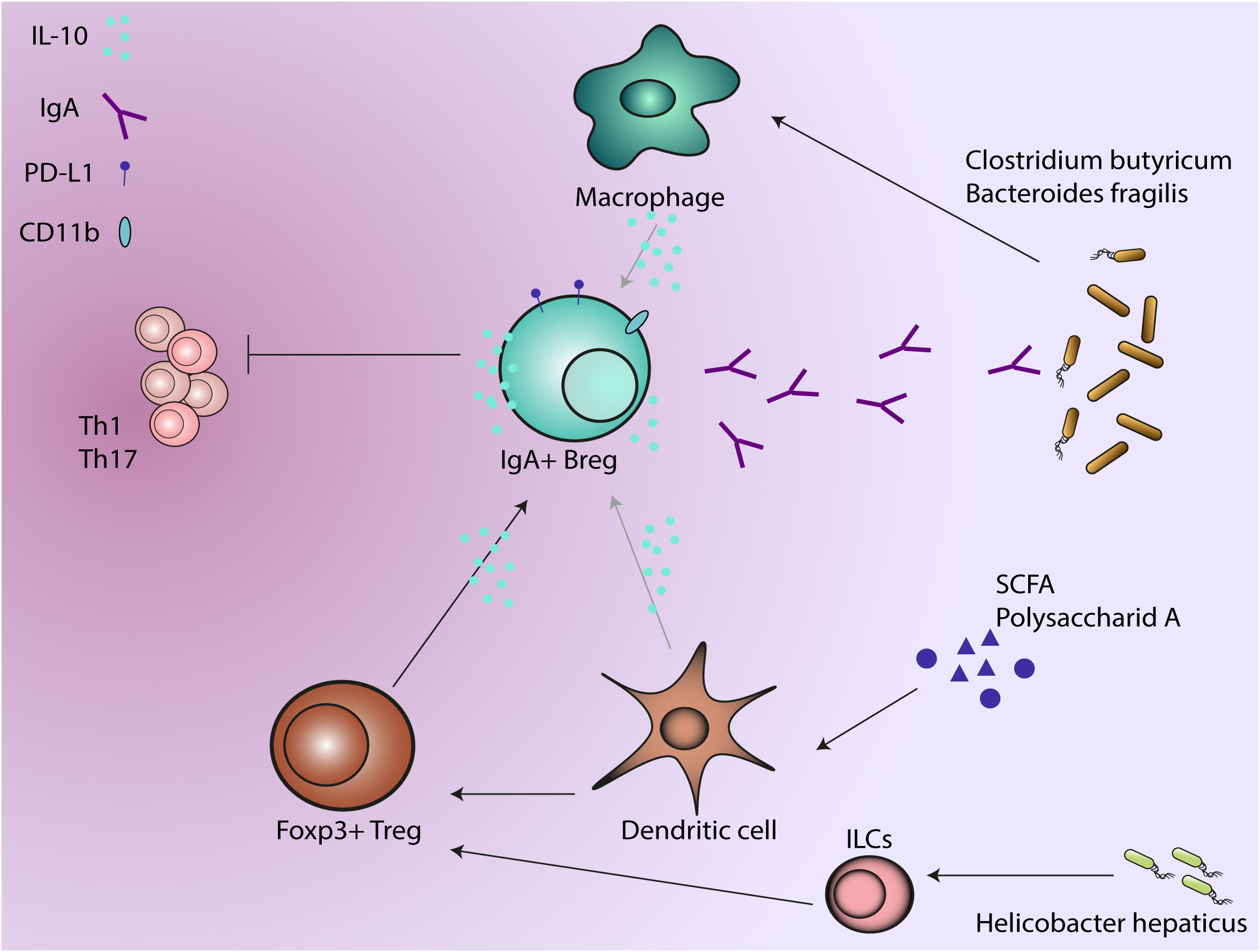
Figure 2 Regulatory B cells produce IgA in response to anti-inflammatory factors such as IL-10. As a dual protective phenotype, IgA production regulate the microbial composition while IL-10 production suppresses the activity of immune cells. Microbiota and microbial metabolites induce regulatory phenotype in other immune cells that further support IgA+ B regs, which will further regulate the microbiota and suppress the activity of T cells.
Mucosal tolerance is maintained by Tregs activated in response to Myd88-dependent microbial sensing which promote IgA production in mouse models and enforce commensal bacteria (109). Myd88 regulates the expression of antimicrobial protein RegIIIγ preventing the activation of the adaptive immune response to bacteria (110).
Nod-like receptors such as NOD2 participate in shaping the microbiota composition by limiting the proliferation of the certain bacterial taxa in NOD2-/- mice (111). In return, NOD2 can be stimulated by commensal bacteria to promote the survival of murine epithelial stem cells and epithelial renewal (112).
Commensal microbiota interacts with other innate immune effector cells like monocytes and macrophages to maintain homeostasis in the gut. Helicobacter hepaticus, a pathobiont, produces an immunomodulatory polysaccharide that induces IL-10 production in murine intestinal macrophages (113) and may further promote IL-10 producing Tregs (114)(Figure 2). Clostridium butyricum prevents acute experimental colitis in mice by inducing IL-10 production in intestinal macrophages (115). Anti-inflammatory response due to M2 polarization of macrophages is induced by Bacteroides fragilis (116) and Clostridia class, while Enterococcus faecalis induces a pro-inflammatory M1 phenotype (117, 118) (Figure 2).
Signals from microbiota can shape the phenotypic diversity and function of innate lymphoid cells ILCs. For example, microbial metabolite sensor Ffar2 regulates the proliferation and function of mouse type 3 ILCs (119). A study done by Kedmi et al. (120) showed that RORγt type 3 ILCs are necessary in inducing Tregs by the Helicobacter hepaticus showing that microbiota target multiple antigen presenting cells. Type 3 ILCs are important as immune surveillance players of the microbiota, participating in controlling the colonization resistance to pathogens by production of IL22 (121).
Although the role of the Th17 subset is controversial in the context of intestinal inflammation (122), their inflammatory activity may be determined by specific bacterial taxa inducing their differentiation. Segmented filamentous bacteria (SFB) induce non-inflammatory Th17 while Citrobacter rodentium induced production of pro-inflammatory cytokines from Th17 (123). Xu et al. demonstrated that TGF-β signaling drives the production of anti-inflammatory cytokine IL-10 in Th17 cells in the ileum and Peyers patches of the mice showing that plasticity of Th17 cells depends on environmental cues (124).
Another subset of T cells, follicular helper T cells (Tfh), can also play a role in microbiota composition. One study showed that deficiency of PD-1 co-receptor determines an altered phenotype of murine Tfh that affects IgA binding capacity to bacteria and results in an increase of the Enterobacteriaceae family (125). Tfh cells are crucial for GC formation in both mice and humans, assisting B cells in the generation of high affinity antibody production (126). Consequently, impairment in Tfh cells caused by lack of receptors, for example ATP-gated ionotropic P2RX7, translates to low IgA binding to commensals (127). On the other hand, microbiota can also have an impact on Tfh cells. Germ free mice have impairment in Tfh development, due to the loss of MYD88 signaling through TLR2 (128). Tfh cells are continuously exposed to microbial byproducts that infer in the GC reaction and the IgA production, participating in the regulation of the microbiota-host interaction (129, 130).
Microbiota-T cells-IgA crosstalk in inflammation processes
Alterations in the gut microbiota due to multiple factors, such as environment or genetic susceptibility, contribute to development of intestinal inflammation and IBD. Multiple IBD loci have been linked to sensing and defense against bacteria, supporting the evidence of aberrant immune responses to intestinal microbiota (131–133). One of the first line of defense between luminal antigens and intestinal epithelium is represented by IgA production at mucosal surfaces (134). Although commensal bacteria induce homeostatic IgA production which in turn binds to bacterial communities to regulate their abundance and diversity (43, 135), higher levels of IgA were found on pathogenic and colitogenic bacteria (4).
In patients with IBD, bacteria taxa coated with IgA were shown to participate in the pathogenesis of inflammatory processes in mouse models (4, 6). Nevertheless, IgA coating plays a role in restricting pathogenic strains from disrupting the mucosal barrier (136, 137). Moreover, IgA coated bacteria from healthy individuals have immunosuppressive roles in the gut in comparison with taxa identified from IBD patients or from mice with dysbiosis. The outcome on intestinal inflammation from the two situations with IgA coated strains may be caused by different mechanism of T cell dependent IgA production. The inflammatory pathway is induced by Th17 cells while the immunoregulatory pathway depends on regulatory T cells (138).
Cong et al. (139) focused specifically on T dependent IgA production by Foxp3+Tregs in CBir1 Tg flagellin transgenic mice. The study showed that Foxp3+ Tregs when stimulated by a specific antigen only, in this case flagellin, promote the production of IgA in B cells. Upon depletion of Foxp3+ Tregs, IgA levels and IgA+ B cells were reduced in the lamina propria suggesting that Foxp3+ Tregs may also be important for the survival of intestinal IgA+ B cells. Foxp3+ T cells suppress inflammation and sustain a balanced and diverse microbiota, which in turn, stimulates the proliferation of Foxp3+ T cells and IgA production in a feedback regulatory loop (140). Foxp3+ T cells differentiate to T follicular regulatory (Tfr) cells and induce IgA production in B cells. When Tfr are absent, T follicular helper cells increase and affect the response of GC reaction towards more poly or self-reactive B cells (141, 142) (Figure 1). Furthermore, the production of a low specific IgA may determine changes in the microbiota and their microbial products that are necessary for the induction of regulatory T cells (143, 144). Th17 cells maintain homeostasis of the intestinal barrier in the steady state but can also act as a two-edged sword by promoting pathogenic inflammation in a genetic susceptible host (145, 146). IL-10 deficient mice have severe inflammation in the gut due to inflammatory and destructive processes mediated by Th17 responses (6). The study done by Britton et al. (138) offers a hypothesis regarding the immunoregulatory capability of microbiota in the etiology of IBD: the anti- or pro-inflammatory arm of the balance is influenced by the microbial communities in the gut which can either enrich a tolerogenic RORγt+Tregs cells or enrich proinflammatory Th17 cells (Figure 1).
IgA-seq helped to identify bacterial taxa coated with high levels of IgA from the gut of IBD patients (147) which were shown to worsen the development of DSS-induced colitis in gnotobiotic mice (4). However, the highly IgA coated bacteria are part of commensals and may be insufficient to drive the inflammation alone without having a specific susceptible environment or genetic conditions. For example, Prevotellaceae, one of the taxa identified by Palm et al. (4) aggravated chemically induced colitis in mice (148) and absence of IL-10 predisposed to colitis in the presence of Helicobacter species (149, 150).
Regulatory B cells
B cells are involved in the generation of an effective immune response through antigen presentation, antibody and cytokine production. Interestingly, B cells can also become Bregs, which play a role in the suppression of the inflammatory milieu. The suppressive capacity has mainly been described by the production of IL-10. However, other immunoregulatory mechanisms are also employed (151, 152).
The phenotypic characterization of Bregs is challenging because of high heterogeneity in markers and lack of prototypic transcription factors which precisely identify the population. Consequently, Bregs are defined based upon their immunoregulatory function. B cells can become Bregs at different stages of development or differentiation, suggesting that the regulatory program may depend on environmental conditions and not on predisposed fate-decisions (153).
B cells involved in the first line of defense are represented by marginal zone B cells and B1 cells. To become mature, B cells have to travel from the bone marrow to the secondary lymphoid organs. As they progress to their maturation they go through 3 stages identified, in mice, as transitional B cells T1, T2 and T3. Transitional 2-marginal zone precursors (T2-MZP) were identified to produce IL-10 and have regulatory function in vitro and in vivo (154–156). Additionally, marginal zone CD23-CD1dhigh B cells produce high amount of IL-10 and have suppressive activity (157–159). B10 cells are another population of Bregs, defined as CD1dhi CD5+, and can be found in the spleen or peritoneal cavity (160–162). B10 produce solely IL-10 and originate either in the bone marrow or fetal liver (163). Antibody secreting cells (ASC), such as plasmablasts and plasma cells, can also become Bregs with important suppressive functions in experimental autoimmune encephalomyelitis (EAE) and a Salmonella infection model (164, 165). Other Breg subsets were described in mouse and humans with overlapping markers and suppressive function (166).
Bregs differentiate in response to inflammatory signals by expressing inhibitory markers and anti-inflammatory cytokines (167). Pro-inflammatory cytokines such as IL-6 or IL-1β are induced during inflammation and drive the differentiation of T2-MZP IL-10+ Bregs cells in a model of antigen induced arthritis. Interestingly, the production of IL-6 and IL-1β is regulated by the microbiota of mice manifesting arthritis (168). Nevertheless, IL10+ B cells can co-express IL10 together with pro-inflammatory cytokines including IL-6 and TNF-α, which may suggest a possible role of IL-6 and TNF-α for enhancing IL-10 production (169).
Factors such as B-cell activating factor (BAFF) and A proliferation-inducing ligand (APRIL) are involved in IgA B cell induction and survival (128, 170, 171), as well as in the induction of IL-10+ and IL-35+ Bregs during inflammation (172–176). Moreover, it was shown that human CD40+ILC3s engage in a feedback loop with B cells in which they induce the differentiation of Bregs through BAFF production (177) (Figure 1). The cytokine IL-21 is produced by Th17 and Tfh cells. Together with TGF-β or retinoic acid, IL-21 strongly promotes IgA class switching and production in mice (178). Furthermore, IL-21, IL-15 and granulocyte macrophage colony-stimulating factor (GM-CSF) are essential for the induction of Bregs (179, 180).
Although inflammatory stimuli induce the activation of Bregs, BCR recognition plays a critical role as well. By deleting two calcium sensors STIM1 and STIM2 in mice, causing a BCR impairment, Matsumoto et al. have shown that B cells produce less IL-10 due to defective BCR stimulation (181). IL-10 production by IL10+ plasmablasts requires engaging of TLR and BCR which further determine the transcription of IRF4 that binds to the IL-10 genomic locus (165). Another study found that the differentiation of B10 cells and IL-10 production can be induced by stimulating the B cells with LPS, PMA (Phorbol-12-myristat-13-acetat) and ionomycin (182). Moreover, Bregs are dominant in lymphoid tissues and the peritoneal cavity but their role can be hindered depending on the murine model of choice due to the presence of myeloid cells producing IL-10 in liver and blood. While IL10+ B cells play no essential role in endotoxemia, IL10+ B cells decrease the numbers of CD8+ T cells during infection with murine cytomegalovirus and control the immune activation when the mice were challenged with anti-IgD antibodies (183).
Bregs differentiation and IL-10 production depends also on CD40 stimulation and signaling and can be enhanced by IL-21 (169, 184). Follicular T helper cells express both IL-21 and CD40L in the GC, driving the class-switching of B cells to become memory B cells and plasma cells. Hence, the GC reactions might be an environment where effector and suppressive memory and plasma cells are produced (153).
An efficient immune response is comprised also of effective antigen presentation by B cells, which can further induce pro-inflammatory or anti-inflammatory reaction. For example, the lack of co-stimulatory ligands such as CD80 and CD86 on B cells induce T cell anergy or Tregs (153, 185). In return, Tregs can deplete CD80 and CD86 ligands from antigen presenting cells such as B cells and dendritic cells to further suppress the activation of conventional T cells (186).
Stimulation of B cells with IL-35, an anti-inflammatory cytokine, stimulates the conversion of B cells to IL35+ Bregs which further act on suppression of T cells (187). It was shown that in a model of experimental autoimmune uveitis both IL-10 and IL-35 signaling might be essential for the suppression function of IL35+Bregs (188). The same research group discovered that the IL-12p35 subunit has immune-regulatory functions and is responsible for inducing Bregs and Tregs expansion (189).
Several remaining questions still yet have to be answered regarding Bregs activation and differentiation. Is the fate of a Bregs written in their ontogeny or there are several factors that contribute to the conversion of B cells to Breg? Given the missing link of a transcription factor or the high diversity of markers that describe Bregs, is their state a stable one or it is transitory to a pro-inflammatory B cell? Moreover, looking at similar inducible cues for both Bregs and effector B cells, does it depend on a certain concentration or combination of factors? (153).
New studies addressing intracellular and extracellular metabolic signals in Breg offered some light in understanding the immunoregulatory balance of B cells. Although the microenvironmental signals seem to be similar for both regulatory and effector B cells, several pathways were identified as regulators of different outcomes. For example, gut and microbial metabolites (short-chain fatty acids, 5-hydroxyindoleacetic acid, fatty acids) particularly supportes regulatory B cell differentiation (166, 190, 191) (Figure 2).
How does the microbiota interact with Bregs?
Microbiota promotes the differentiation of Bregs which in response suppress inflammatory signals. The regulatory function of Bregs has been described in several studies in autoimmune disease models of multiple sclerosis, rheumatoid arthritis and IBD (192–195). Bregs regulate the Th1/Th17 to Tregs balance in both mice and humans. Mice that have a conditional IL-10 deletion in B cells have reduced numbers of Tregs and an increase in pro-inflammatory Th1 and Th17 cells (156, 196) (Figure 1).
The suppressive function of Bregs is not only supported by their IL-10 production (166). Bregs can regulate the immune response through other mechanisms such as: TGF-β (197), EBI3/IL-35 (Epstein-Barr virus induced gene 3) participates in inducing Tregs (164, 189), GITRL (glucocorticoid-induced tumor necrosis factor receptor-related protein ligand) (194), FasL (Fas ligand) induced apoptosis by binding to its receptor (198), PD-L1 (programmed death ligand 1) participate in restricting T cell differentiation (173), CD73 has an immunosuppressive effect by converting adenosine monophosphate to adenosine (199, 200). B cells cooperate with Tregs in feedback loops to attenuate inflammatory processes in the gut. In DSS-colitis, B cell deficient mice have lower number of Tregs in GALT and display more severe colitis. Interestingly, B cells attenuated colitis in an IL10-independent pathway and induced the differentiation of Tregs which supported the functions of IgA-plasma cells (25).
The immunogenicity of bacteria seems to play an important role in B cell activation and differentiation during the interaction of B cells with host microbiota. If the encountered bacteria have a strong immunogenic potential then Bregs will respond with a strong immunoregulatory response to maintain immune tolerance (201). Although, a more intense suppressive response by Bregs can regulate pro-inflammatory immune responses caused by the same bacteria, in the absence of Bregs the strong immunogenic bacteria will aggravate the disease progression (202–204).
Defects in PI3Kγ signaling translate to reduced numbers of IL-10 producing Bregs in response to resident bacteria. B cells lacking PI3Kγ were unable to resolve the intestinal inflammation in T cell mediated-colitis (205). The human IBD7 susceptibility locus harbors the PIK3cd gene (206, 207) and genetic deletion of PI3Kγ drives spontaneous colitis in the presence of commensals (208, 209). These findings might explain the roles of specific pathways in the activation of immune cells by microbiota, since the PI3Kγ pathway seems to contribute to the immunoregulation mostly when intestinal microbiota is the main activator of inflammation (205). Another study done by Mishima et al. supports the role of PI3Kp110d signaling for IL-10 production by bacteria murine activated-B cells and its importance in maintaining mucosal tolerance (210). They also observed that continuous bacterial stimulation is essential for mounting a sustained activation of Bregs in vitro.
Breg cells are characterized by their ability to suppress the immune system, but their high diversity in phenotypic marker and transcription factor expression makes specific phenotyping difficult (152). The transcription factor AhR (aryl-hydrocarbon receptor) contributes to the interaction of immune cells and microbiota by binding diverse ligands from dietary components or from microbiota (211). Recently, several studies revealed that Ahr drives and maintains the immunoregulatory function of splenic Breg (212, 213). Importantly, Piper et al. shows that, in mice, Ahr responds to inflammatory stimuli and promotes the differentiation of Bregs by regulating the production of IL-10 and silencing the transcription of pro-inflammatory cytokines (212). Moreover, Rosser et al. shows that presence of gut microbiota derived metabolites such as butyrate increases the Ahr ligand availability which results in supporting Breg function (190).
In addition to IL-10; other cytokines such as IL-35 are essential in the outcome of several diseases (194). IL35+ B cells interact with microbiota through their microbial metabolites and maintain intestinal homeostasis. A microbial metabolite, indoleacetic acid, supports the expansion of IL35+ Bregs which are the main source of IL-35 in the intestine during DSS-induced colitis. In turn, the presence of IL-35 has immunosuppressive function and shapes the intestinal microbiota (214). Moreover, IL-35 production plays an important role in reducing the inflammation in models of T cell dependent colitis (215) and DSS-induced colitis (216). Although the IL-35 secretion is decreased in patients with UC compared to controls, higher IL-35 in the mucosa of UC was correlated with higher IL-10 secretion in the gut (217). Moreover, B cells from CD patients have a higher expression of IL-35 but lower protein secretion compared to controls (218).
Several bacterial pathogens can induce immunosuppressive response to infection as an immunological escape. For example, Helicobacter pylori infection can induce differentiation of Foxp3+ Tregs and CD19+ IL10+Bregs (219). When mice are infected with H. pylori and treated with DSS-colitis, the colitis scores were significantly reduced with less pro-inflammatory cytokines in the colonic mucosa (220, 221). The studies suggest the role of H. pylori in attenuation of acute and chronic colitis. Salmonella sp. infection helped to identify a subset of IL-35 plasma cells which is induced during infection together with IL-10 plasma cells and highlighted the role of IL-35 Bregs as essential regulators of the immune system (164, 222).
IgA Bregs
Although IgA plasma cells modulate the immune response in particular at the mucosal sites through production of IgA, their role as immunosuppressive players is not yet fully understood.
In vitro studies showed that IL-10 stimulation of human but not mouse B cells contributes to proliferation of activated B cells and may drive IgA production (223, 224). Kunisawa et al. (225) found that IL-10 induced CD11b+, IgA+ plasma cells and determine IgA production on those cells. Although it is not clear if IL-10 alone is necessary for IgA production or some other factors are participating in maintaining CD11b+ IgA+ plasma cells, the study identified the CD150 surface marker as potential mechanism through which IL-10 can mediate B cell proliferation. CD11b+ B cells are present in the GALT in DSS-induced colitis and in UC patients. CD11b+ B cells play an immunosuppressive role by inhibiting colitis in mice and produce increased levels of IgA (226) (Figure 2).
As described before, another immunosuppressive mechanism of Bregs is through expression of PD-L1 upon sensing inflammatory signals (227, 228). IgA plasma cells in the lamina propria of mice were found to express PD-L1 and to induce FOXP3+ Tregs cells but not pro-inflammatory IFN-γ+ Th1 cells (229) (Figure 2). Despite the fact that IgA production is not the main focus, one study by Serrán et al. (230) found expression of high levels of PD-L1 on plasmablasts during infection with Trypanosoma cruzi with a suppressive effect on T cells.
Fehres et al. identified a new subset of human IL-10 producing B regs that express IgA (173). In this study, they describe that APRIL stimulation determines differentiation to IgA+ B regs, while other stimulations such as TGF-β and BAFF does not induce IL-10 production in IgA+ Bregs. APRIL-induced IgA+ Bregs significantly, suppressed CD4+ proliferation and induced Foxp3+ Tregs. Furthermore, APRIL-induced IgA+ Bregs also inhibited TNF production by macrophages through IL-10 and PD-L1. Albeit this role of APRIL for production of IL-10 by IgA+ Bregs, some other studies identified TGF-β as potent stimulatory factor for differentiation of immunosuppressive plasma cells which express IgA, IL-10 and PD-L1 in a mouse model of prostate cancer (231).
Non-resolving inflammatory responses in IBD are associated with the development of colitis-associated colorectal cancer (CAC) (232, 233). Even though IL-10 deficiency in B cells aggravates the outcome of DSS-induced chronic colitis, it does not contribute to the development of tumorigenesis in CAC, which suggests the presence of other immunosuppressive mechanisms. Melcher et al. found that Bregs can exhibit dual protective phenotypes: IL-10 production for the suppression on inflammatory environment and on IgA production for shaping a protective microbiota (234)(Figure 2). Interestingly, in this study the IL-10 producing Bregs exert suppressive effects on Th1/Th17 cells in mouse chronic colitis and then differentiate into IgA+ plasma cells in response to TLR activation. After the differentiation into IgA+ plasma cells, the Bregs lost the capacity for IL-10 production.
Homeostasis in the colon may also depend on the interaction of IgA antibody secreting cells (IgA-ASC) and Tregs. IgA-ASC that express the gut homing chemokine receptor CCR10+ form conjugates with Tregs to direct their migration into the colon to support a homeostatic environment (235). Moreover, CCR10+ IgG1-ASC have immunoregulatory role and can compensate for the loss of CCR10+ IgA-ASC in IgA-ko mice. Furthermore, IgA-ASC originate in the gut in response to commensals and can migrate to central nervous system during experimental autoimmune encephalomyelitis (EAE) where they play a critical role in regulation of inflammation (236).
Bregs have heterogeneity in phenotypic markers and express different transcription factors in both mice and human, giving rise to the hypothesis that Bregs do not comprise a distinct cellular lineage but any B cells can differentiate to Breg cells in an appropriate environment (167). Latest research show that IgA production by Bregs may be a new mechanism to regulate the immune response and may bring new light in understanding the anti-inflammatory and pro-inflammatory balance.
Pro-inflammatory IgA and IgG
IBD patients have increased numbers of B cells in the inflamed intestine compared to healthy individuals. Interestingly, UC patients described as ulcerative colitis type 1, have increased expression of the B cell activation factor BAFF, suggesting a possible role for B cells in IBD. Further studies have also confirmed the expansion of B cells in UC patients. In a study done by Rubin et al. they observed more gut trophic CD45RO+ B cells in the inflamed tissue of CD patients compared to UC patients (237). These CD45+RO B cells are considered a biomarker for CD activity index and permeability of the gut. Moreover, a lower expression of pSTAT3 levels in CD25+ B cells in inflamed tissue of CD patients was detected compared to uninflamed tissue which can be associated with lower levels of regulatory B cells (237). The study of Pararasa et al. suggests that the increase in CD27-IgD- memory B cells in GALT of IBD patients is rather determined by the proliferation at the site of inflammation and less by the recruitment of cells from the blood. However, is interesting to notice the same immune response for both UC and CD, supporting the notion that the response to intestinal challenge determine the expansion of CD27- B cells (238). The inflamed gut of UC patients has increased IgA+ and IgG+ plasma cell populations compared with healthy controls. Uzzan et al. suggests that IgG+ plasma cells could participate in enhancing the inflammatory milieu (8). The increased number of B cells during chronic inflammation in the intestine develops in lymphoid structures recently described as tertiary lymphoid structures (TLSs). However, TLS are present more in biopsies from CD patients and still remain enigmatic in their role during the disease (239, 240).
IgA produced at mucosal sites is considered to have anti-inflammatory proprieties by keeping tolerance to commensals and protecting against pathogens. Despite its high abundance in the blood, the role of IgA in the serum is less understood (241). In recent years the role of IgA binding to FcαRI (CD89) gained increasing attention. FcαRI expression is observed in neutrophils, eosinophils, macrophages, monocytes and Kuppfer cells (242). Hansen et al. show that serum IgA forms immune complexes, binds to FcαRI and induces pro-inflammatory cytokine production in different human myeloid cells (243). High levels of IgA immune complexes, IgA autoantibodies or increased IgA levels in the serum are found in diseases such as celiac disease (244), IgA nephropathy (245); and IBD (246).
Furthermore, patients with IBD have increased levels of commensal-targeting IgG in the serum which contributes to inflammation of the intestinal mucosa (247). Uzzan et al. found reduced VDJ gene mutation in IgG+ plasma cells in UC which might suggest an impairment of GC for differentiation of IgG+ and IgA+ plasma cells due to hyperstimulation of B cells in an inflammatory environment or might be explained by higher accumulation of autoreactive cells due to inflammation (8). A variant of FCGR2A, which alters the binding affinity of the antibody receptor it encodes, FcγRIIA, for IgG, has been found in genome-wide association studies (GWAS) in UC. The study of Castro-Dopico et al. shows that colonic mucosa of UC patients with FCGR2A genotype have anti-commensal IgG which forms complexes with macrophages and induces NLRP3 and reactive oxygen species with the production of pro-inflammatory IL-1β and neutrophil-recruiting chemokines (248).
Conclusion
In conclusion, the cytokine network in intestinal inflammation leading to IBD is complex and highly regulated by genetic, microbial, and immune factors. So far therapeutic agents targeting cytokines have at least in some patients limited efficacy and varied outcomes. Targeting all B cells with Rituximab has proven to be unsuccessful and rather detrimental to IBD patients, most likely particularly because potential functions of Bregs have been overlooked (12). Better understanding the proinflammatory role of B cells and the factors that induce intestinal Bregs could help to design better treatments that do not interfere with the activity of beneficial Bregs and provide new insights into stimulating Bregs to tilt the balance towards suppression of inflammation. The emergence of IgA-producing Bregs adds another mechanism for regulating inflammation in close interaction with the microbiota. Further research is needed to fully comprehend the intricate mechanisms and interactions within this complex network and to develop more effective treatments for IBD.
Author contributions
RZ: Writing – original draft. SW: Writing – review & editing.
Funding
This work has received funding from the German Research Foundation (DFG) (GRK2599: P3) and (DFG TRR241: A03).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol (2008) 8(6):411–20. doi: 10.1038/nri2316
2. Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol (2010) 10(3):159–69. doi: 10.1038/nri2710
3. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi: 10.1038/nri3661
4. Palm NW, Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell (2014) 158(5):1000–10. doi: 10.1016/j.cell.2014.08.006
5. van der Waaij LA, Kroese FG, Visser A, Nelis GF, Westerveld BD, Jansen PL, et al. Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur J Gastroenterol Hepatol (2004) 16(7):669–74. doi: 10.1097/01.meg.0000108346.41221.19
6. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn's disease spondyloarthritis promote T(H)17-dependent inflammation. Sci Transl Med (2017) 9(376):669–74. doi: 10.1126/scitranslmed.aaf9655
7. Alexander KL, Zhao Q, Reif M, Rosenberg AF, Mannon PJ, Duck LW, et al. Human microbiota flagellins drive adaptive immune responses in Crohn’s disease. Gastroenterology (2021) 161(2):522–535.e6. doi: 10.1053/j.gastro.2021.03.064
8. Uzzan M, Martin JC, Mesin L, Livanos AE, Castro-Dopico T, Huang R, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat Med (2022) 28(4):766–79. doi: 10.1038/s41591-022-01680-y
9. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
10. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314(5804):1461–3. doi: 10.1126/science.1135245
11. Giuffrida P, Di Sabatino A. Targeting T cells in inflammatory bowel disease. Pharmacol Res (2020) 159:105040. doi: 10.1016/j.phrs.2020.105040
12. Castro-Dopico T, Colombel JF, Mehandru S. Targeting B cells for inflammatory bowel disease treatment: back to the future. Curr Opin Pharmacol (2020) 55:90–8. doi: 10.1016/j.coph.2020.10.002
13. Geng X, Biancone L, Dai HH, Lin JJ, Yoshizaki N, Dasgupta A, et al. Tropomyosin isoforms in intestinal mucosa: production of autoantibodies to tropomyosin isoforms in ulcerative colitis. Gastroenterology (1998) 114(5):912–22. doi: 10.1016/S0016-5085(98)70310-5
14. Taniguchi M, Geng X, Glazier KD, Dasgupta A, and Lin JJC, Das KM. Cellular immune response against tropomyosin isoform 5 in ulcerative colitis. Clin Immunol (2001) 101(3):289–95. doi: 10.1006/clim.2001.5133
15. Roozendaal C, Kallenberg CG. Are anti-neutrophil cytoplasmic antibodies (ANCA) clinically useful in inflammatory bowel disease (IBD)? Clin Exp Immunol (1999) 116(2):206–13. doi: 10.1046/j.1365-2249.1999.00905.x
16. Walker LJ, Aldhous MC, Drummond HE, Smith BR, Nimmo ER, Arnott ID, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) in Crohn's disease are associated with disease severity but not NOD2/CARD15 mutations. Clin Exp Immunol (2004) 135(3):490–6. doi: 10.1111/j.1365-2249.2003.02392.x
17. Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest (2004) 113(9):1296–306. doi: 10.1172/JCI200420295
18. Zhang S, Morgan X, Dogan B, Martin F-P, Strickler S, Oka A, et al. Mucosal metabolites fuel the growth and virulence of E. coli linked to Crohn’s disease. JCI Insight (2023) 7(10). doi: 10.1172/jci.insight.157013
19. Kevans D, Kirsch R, Dargavel C, Kabakchiev B, Riddell R, Silverberg MS, et al. Histological markers of clinical relapse in endoscopically quiescent ulcerative colitis. Inflammation Bowel Dis (2020) 26(11):1722–9. doi: 10.1093/ibd/izz308
20. Andoh A, Sakata S, Koizumi Y, Mitsuyama K, Fujiyama Y, Benno Y. Terminal restriction fragment length polymorphism analysis of the diversity of fecal microbiota in patients with ulcerative colitis. Inflammation Bowel Dis (2007) 13(8):955–62. doi: 10.1002/ibd.20151
21. Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol (2018) 53(1):95–106. doi: 10.1007/s00535-017-1384-4
22. Vila AV, Hu S, Andreu-Sánchez S, Collij V, Jansen BH, Augustijn HE, et al. Faecal metabolome and its determinants in inflammatory bowel disease. Gut (2023) 72(8):1472–85. doi: 10.1136/gutjnl-2022-328048
23. Pabst O, Slack E. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol (2020) 13(1):12–21. doi: 10.1038/s41385-019-0227-4
24. Chen Y, Cui W, Li X, Yang H. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front Immunol (2021) 12:761981. doi: 10.3389/fimmu.2021.761981
25. Wang L, Ray A, Jiang X, Wang JY, Basu S, Liu X, et al. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol (2015) 8(6):1297–312. doi: 10.1038/mi.2015.20
26. Schmidt EG, Larsen HL, Kristensen NN, Poulsen SS, Claesson MH, Pedersen AE, et al. B cells exposed to enterobacterial components suppress development of experimental colitis. Inflammation Bowel Dis (2012) 18(2):284–93. doi: 10.1002/ibd.21769
27. Leiper K, Martin K, Ellis A, Subramanian S, Watson AJ, Christmas SE, et al. Randomised placebo-controlled trial of rituximab (anti-CD20) in active ulcerative colitis. Gut (2011) 60(11):1520–6. doi: 10.1136/gut.2010.225482
28. Goetz M, Atreya R, Ghalibafian M, Galle PR, Neurath MF. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflammation Bowel Dis (2007) 13(11):1365–8. doi: 10.1002/ibd.20215
29. Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M, Veen van W. Regulatory B cells, A to Z. Allergy (2021) 76(9):2699–715. doi: 10.1111/all.14763
30. Olson TS, Reuter BK, Scott KG-E, Morris MA, Wang X-M, Hancock LN, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med (2006) 203(3):541–52. doi: 10.1084/jem.20050407
31. Frede A, Czarnewski P, Monasterio G, Tripathi KP, Bejarano DA, Flores Ramirez RO, et al. B cell expansion hinders the stroma-epithelium regenerative cross talk during mucosal healing. Immunity (2022) 55(12):2336–2351.e12. doi: 10.1016/j.immuni.2022.11.002
32. Suzuki K, Fagarasan S. How host-bacterial interactions lead to IgA synthesis in the gut. Trends Immunol (2008) 29(11):523–31. doi: 10.1016/j.it.2008.08.001
33. Tsuji M, Suzuki K, Kitamura H, Maruya M, Ivanov Kinoshita K, II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity (2008) 29(2):261–71. doi: 10.1016/j.immuni.2008.05.014
34. Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune profiling of human gut-associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region-specific immunity. Immunity (2020) 52(3):557–570.e6. doi: 10.1016/j.immuni.2020.02.001
35. Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol (1991) 9:457–92. doi: 10.1146/annurev.iy.09.040191.002325
36. den Dunnen J, Vogelpoel LT, Wypych T, Muller FJ, Boer L, Kuijpers TW, et al. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcγRIIa in human dendritic cells. Blood (2012) 120(1):112–21. doi: 10.1182/blood-2011-12-399931
37. Magri G, Comerma L, Pybus M, Sintes J, Lligé D, Segura-Garzón D, et al. Human secretory IgM emerges from plasma cells clonally related to gut memory B cells and targets highly diverse commensals. Immunity (2017) 47(1):118–134.e8. doi: 10.1016/j.immuni.2017.06.013
38. Liu J, Wang Y, Xiong E, Hong R, Lu Q, Ohno H, et al. Role of the IgM Fc receptor in immunity and tolerance. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00529
39. Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr (1999) 69(5):1046s–51s. doi: 10.1093/ajcn/69.5.1046s
40. Joglekar P, Ding H, Canales-Herrerias P, Pasrich PJ, Sonnenburg JL, Peterson DA. Intestinal IgA regulates expression of a fructan polysaccharide utilization locus in colonizing gut commensal Bacteroides thetaiotaomicron. mBio (2019) 10(6). doi: 10.1128/mBio.02324-19
41. Porter NT, Canales P, Peterson DA, Martens EC. A subset of polysaccharide capsules in the human symbiont bacteroides thetaiotaomicron promote increased competitive fitness in the mouse gut. Cell Host Microbe (2017) 22(4):494–506.e8. doi: 10.1016/j.chom.2017.08.020
42. Peterson DA, Canales P, Peterson DA, Martens EC. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe (2007) 2(5):328–39. doi: 10.1016/j.chom.2007.09.013
43. Bunker JJ, Bendelac A. IgA responses to microbiota. Immunity (2018) 49(2):211–24. doi: 10.1016/j.immuni.2018.08.011
44. Takeuchi T, Ohno H. IgA in human health and diseases: Potential regulator of commensal microbiota. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1024330
45. Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science (2017) 358(6361). doi: 10.1126/science.aan6619
46. Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol (2018) 9:1270. doi: 10.3389/fimmu.2018.01270
47. Ng SC, Benjamin JL, McCarthy NE, Hedin CRH, Koutsoumpas A, Plamondon S, et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in Crohn's disease. Inflammatory Bowel Dis (2010) 17(10):2027–37. doi: 10.1002/ibd.21590.
48. Kuhn KA, Manieri NA, Liu T-C, Stappenbeck TS. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PloS One (2014) 9(12):e114195. doi: 10.1371/journal.pone.0114195
49. Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell (2009) 15(2):79–80. doi: 10.1016/j.ccr.2009.01.009
50. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118(6):2269–80. doi: 10.1172/JCI34610
51. Atreya R, Zimmer M, Bartsch B, Waldner MJ, Atreya I, Neumann H, et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology (2011) 141(6):2026–38. doi: 10.1053/j.gastro.2011.08.032
52. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology (2013) 145(2):407–15. doi: 10.1053/j.gastro.2013.04.011
53. Waetzig GH, Rosenstiel P, Arlt A, Till A, Bräutigam K, Schäfer H, et al. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J (2005) 19(1):91–3. doi: 10.1096/fj.04-2073fje
54. Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, et al. Etanercept for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology (2001) 121(5):1088–94. doi: 10.1053/gast.2001.28674
55. Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P, et al. Epithelial TNF receptor signaling promotes mucosal repair in inflammatory bowel disease. J Immunol (2017) 199(5):1886–97. doi: 10.4049/jimmunol.1601066
56. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi: 10.1038/nature08949
57. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest (2006) 116(5):1310–6. doi: 10.1172/JCI21404
58. Arnold IC, Mathisen S, Schulthess J, Danne C, Hegazy AN, Powrie F. CD11c(+) monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol (2016) 9(2):352–63. doi: 10.1038/mi.2015.65
59. Becker C, Dornhoff H, Neufert C, Fantini MC, Wirtz S, Huebner S, et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol (2006) 177(5):2760–4. doi: 10.4049/jimmunol.177.5.2760
60. Noviello D, Mager R, Roda G, Borroni RG, Fiorino G, Vetrano S. The IL23-IL17 immune axis in the treatment of ulcerative colitis: successes, defeats, and ongoing challenges. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.611256
61. Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in Crohn's disease. Front Immunol (2021) 12:622934. doi: 10.3389/fimmu.2021.622934
62. Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Bègue B, et al. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity (2012) 37(1):108–21. doi: 10.1016/j.immuni.2012.05.013
63. Arshad T, Mansur F, Palek R, Manzoor S, Liska V. A double edged sword role of interleukin-22 in wound healing and tissue regeneration. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.02148
64. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med (2015) 212(11):1783–91. doi: 10.1084/jem.20150318
65. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med (2013) 210(6):1117–24. doi: 10.1084/jem.20121588
66. Bergstrom KSB, Morampudi V, Chan JM, Bhinder G, Lau J, Yang H, et al. Goblet cell derived RELM-β Recruits CD4+ T cells during infectious colitis to promote protective intestinal epithelial cell proliferation. PloS Pathog (2015) 11(8):e1005108. doi: 10.1371/journal.ppat.1005108
67. Gunasekera DC, Ma J, Vacharathit V, Shah P, Ramakrishnan A, Uprety P, et al. The development of colitis in Il10–/– mice is dependent on IL-22. Mucosal Immunol (2020) 13(3):493–506. doi: 10.1038/s41385-019-0252-3
68. Baumgart DC, Sandborn WJ. Crohn's disease. Lancet (2012) 380(9853):1590–605. doi: 10.1016/S0140-6736(12)60026-9
69. Imam T, Park S, Kaplan MH, Olson MR. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol (2018) 9:1212. doi: 10.3389/fimmu.2018.01212
70. Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut (2009) 58(8):1152–67. doi: 10.1136/gut.2008.163667
71. Nakase H, Sato N, Mizuno N, Ikawa Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun Rev (2022) 21(3):103017. doi: 10.1016/j.autrev.2021.103017
72. Fuss IJ, Neurath M, Boirivant M, Klein JS, Motte C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol (1996) 157(3):1261–70.
73. Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut (2008) 57(12):1682–9. doi: 10.1136/gut.2007.135053
74. Zhao J, Lu Q, Liu Y, Shi Z, Hu L, Zeng Z, et al. Th17 cells in inflammatory bowel disease: cytokines, plasticity, and therapies. J Immunol Res (2021) 2021:8816041. doi: 10.1155/2021/8816041
75. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol (2023) 23(1):38–54. doi: 10.1038/s41577-022-00746-9
76. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52(1):65–70. doi: 10.1136/gut.52.1.65
77. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61(12):1693–700. doi: 10.1136/gutjnl-2011-301668
78. Ribot JC, Lopes N, Silva-Santos B. γδ T cells in tissue physiology and surveillance. Nat Rev Immunol (2021) 21(4):221–32. doi: 10.1038/s41577-020-00452-4
79. Siegmund B, Sennello JA, Lehr HA, Senaldi G, Dinarello CA, Fantuzzi G, et al. Frontline: Interferon regulatory factor-1 as a protective gene in intestinal inflammation: role of TCR γ δ T cells and interleukin-18-binding protein. Eur J Immunol (2004) 34(9):2356–64. doi: 10.1002/eji.200425124
80. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol (2011) 11(7):445–56. doi: 10.1038/nri3007
81. Mann ER, McCarthy NE, Peake STC, Milestone AN, Al-Hassi HO, Bernardo D, et al. Skin- and gut-homing molecules on human circulating γδ T cells and their dysregulation in inflammatory bowel disease. Clin Exp Immunol (2012) 170(2):122–30. doi: 10.1111/j.1365-2249.2012.04649.x
82. Jaeger N, Gamini R, Cella M, Schettini JL, Bugatti M, Zhao S, et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat Commun (2021) 12(1):1921. doi: 10.1038/s41467-021-22164-6
83. Targan SR, Sheikh F, Dickensheets H, Savan R, Young HA, Walter MR, et al. Mo2083 A randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and efficacy of AMG 827 in subjects with moderate to severe Crohn's disease. Gastroenterology (2012) 143(3):e26. doi: 10.1053/j.gastro.2012.07.084
84. Donnelly RP, Sheikh F, Dickensheets H, Savan R, Young HA, Walter MR, et al. Interleukin-26: an IL-10-related cytokine produced by Th17 cells. Cytokine Growth Factor Rev (2010) 21(5):393–401. doi: 10.1016/j.cytogfr.2010.09.001
85. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci (2015) 112(22):7061–6. doi: 10.1073/pnas.1415675112
86. He Y, Lin LJ, Zheng CQ, Jin Y, Lin Y. Cytokine expression and the role of Th17 cells in a mouse model of colitis. Mol Med Rep (2012) 6(6):1438–42. doi: 10.3892/mmr.2012.1111
87. Dambacher J, Beigel F, Zitzmann K, Toni De EN, Göke B, Diepolder HM, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut (2009) 58(9):1207–17. doi: 10.1136/gut.2007.130112
88. Corridoni D, Antanaviciute A, Gupta T, Fawkner-Corbett D, Aulicino A, Jagielowicz M, et al. Single-cell atlas of colonic CD8+ T cells in ulcerative colitis. Nat Med (2020) 26(9):1480–90. doi: 10.1038/s41591-020-1003-4
89. Arnott ID, Drummond HE, Ghosh S. Gut mucosal secretion of interleukin 1β and interleukin-8 predicts relapse in clinically inactive Crohn's disease. Digestive Dis Sci (2001) 46:402–9. doi: 10.1023/A:1005617302718
90. Rossi O, Karczewski J, Stolte EH, Brummer RJ, Nieuwenhoven van MA, Meijerink M, et al. Vectorial secretion of interleukin-8 mediates autocrine signalling in intestinal epithelial cells via apically located CXCR1. BMC Res Notes (2013) 6:431. doi: 10.1186/1756-0500-6-431
91. Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature (2019) 568(7752):405–9. doi: 10.1038/s41586-019-1082-x
92. Curciarello R, Canziani KE, Docena GH, Muglia CI. Contribution of non-immune cells to activation and modulation of the intestinal inflammation. Front Immunol (2019) 10:647. doi: 10.3389/fimmu.2019.00647
93. Jeffery V, Goldson AJ, Dainty JR, Chieppa M, Sobolewski A. IL-6 signaling regulates small intestinal crypt homeostasis. J Immunol (2017) 199(1):304–11. doi: 10.4049/jimmunol.1600960
94. Kominsky DJ, Campbell EL, Ehrentraut SF, Wilson KE, Kelly CJ, Glover LE, et al. IFN-γ-mediated induction of an apical IL-10 receptor on polarized intestinal epithelia. J Immunol (2014) 192(3):1267–76. doi: 10.4049/jimmunol.1301757
95. Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, et al. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J Immunol (2017) 199(8):2976–84. doi: 10.4049/jimmunol.1700105
96. Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity (2019) 50(4):992–1006. doi: 10.1016/j.immuni.2019.03.017
97. Konrad A, Cong Y, Duck W, Borlaza R, Elson CO. Tight mucosal compartmentation of the murine immune response to antigens of the enteric microbiota. Gastroenterology (2006) 130(7):2050–9. doi: 10.1053/j.gastro.2006.02.055
98. Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol (2013) 14(7):646–53. doi: 10.1038/ni.2604
99. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483(7389):345–9. doi: 10.1038/nature10863
100. Kulkarni DH, Gustafsson JK, Knoop KA, McDonald KG, Bidani SS, Davis JE, et al. Goblet cell associated antigen passages support the induction and maintenance of oral tolerance. Mucosal Immunol (2020) 13(2):271–82. doi: 10.1038/s41385-019-0240-7
101. Chang S-Y, Ko H-J, Kweon M-N. Mucosal dendritic cells shape mucosal immunity. Exp Mol Med (2014) 46(3):e84–4. doi: 10.1038/emm.2014.16
102. Esterházy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat Immunol (2016) 17(5):545–55. doi: 10.1038/ni.3408
103. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science (2004) 303(5664):1662–5. doi: 10.1126/science.1091334
104. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe (2014) 15(4):413–23. doi: 10.1016/j.chom.2014.03.006
105. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature (2008) 453(7195):620–5. doi: 10.1038/nature07008
106. Wiechers C, Zou M, Galvez E, Beckstette M, Ebel M, Strowig T, et al. The microbiota is dispensable for the early stages of peripheral regulatory T cell induction within mesenteric lymph nodes. Cell Mol Immunol (2021) 18(5):1211–21. doi: 10.1038/s41423-021-00647-2
107. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40(1):128–39. doi: 10.1016/j.immuni.2013.12.007
108. Wu W, Sun M, Chen F, Cao AT, Liu H, Zhao Y, et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol (2017) 10(4):946–56. doi: 10.1038/mi.2016.114
109. Wang S, Charbonnier LM, Rivas Noval M, Georgiev P, Li N, Gerber G, et al. MyD88 adaptor-dependent microbial sensing by regulatory T cells promotes mucosal tolerance and enforces commensalism. Immunity (2015) 43(2):289–303. doi: 10.1016/j.immuni.2015.06.014
110. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi: 10.1126/science.1209791
111. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity (2014) 41(2):311–24. doi: 10.1016/j.immuni.2014.06.015
112. Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe (2014) 15(6):792–8. doi: 10.1016/j.chom.2014.05.003
113. Danne C, Ryzhakov G, Martínez-López M, Ilott NE, Franchini F, Cuskin F, et al. A large polysaccharide produced by helicobacter hepaticus induces an anti-inflammatory gene signature in macrophages. Cell Host Microbe (2017) 22(6):733–745.e5. doi: 10.1016/j.chom.2017.11.002
114. Kullberg MC, Jankovic D, Gorelick PL, Caspar P, Letterio JJ, Cheever AW, et al. Bacteria-triggered CD4(+) T regulatory cells suppress Helicobacter hepaticus-induced colitis. J Exp Med (2002) 196(4):505–15. doi: 10.1084/jem.20020556
115. Hayashi A, Sato T, Kamada N, Mikami Y, Matsuoka K, Hisamatsu T, et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe (2013) 13(6):711–22. doi: 10.1016/j.chom.2013.05.013
116. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U.S.A. (2010) 107(27):12204–9. doi: 10.1073/pnas.0909122107
117. Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe (2012) 12(4):496–508. doi: 10.1016/j.chom.2012.09.009
118. Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM, et al. Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Trans Oncol (2013) 6(5):596–IN8. doi: 10.1593/tlo.13412
119. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-sensing receptor ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity (2019) 51(5):871–884.e6. doi: 10.1016/j.immuni.2019.09.014
120. Kedmi R, Najar TA, Mesa KR, Grayson A, Kroehling L, Hao Y, et al. A RORγt(+) cell instructs gut microbiota-specific T(reg) cell differentiation. Nature (2022) 610(7933):737–43. doi: 10.1038/s41586-022-05089-y
121. Guo X, Liang Y, Zhang Y, Lasorella A, Kee BL, Fu YX. Innate Lymphoid Cells Control Early Colonization Resistance against Intestinal Pathogens through ID2-Dependent Regulation of the Microbiota. Immunity (2015) 42(4):731–43. doi: 10.1016/j.immuni.2015.03.012
122. Martinez GJ, Nurieva RI, Yang XO, Dong C. Regulation and function of proinflammatory TH17 cells. Ann N Y Acad Sci 2008 (1143) p:188–211. doi: 10.1196/annals.1443.021
123. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity (2019) 51(1):77–89.e6. doi: 10.1016/j.immuni.2019.05.004
124. Xu H, Agalioti T, Zhao J, Steglich B, Wahib R, Vesely MCA, et al. The induction and function of the anti-inflammatory fate of T(H)17 cells. Nat Commun (2020) 11(1):3334. doi: 10.1038/s41467-020-17097-5
125. Kim YI, Song JH, Ko HJ, Kweon MN, Kang CY, Reinecker HC, et al. CX(3)CR1(+) macrophages and CD8(+) T cells control intestinal IgA production. J Immunol (2018) 201(4):1287–94. doi: 10.4049/jimmunol.1701459
126. Zhao Z, Xu B, Wang S, Zhou M, Huang Y, Guo C, et al. Tfh cells with NLRP3 inflammasome activation are essential for high-affinity antibody generation, germinal centre formation and autoimmunity. Ann Rheum Dis (2022) 81(7):1006–12. doi: 10.1136/annrheumdis-2021-221985
127. Proietti M, Cornacchione V, Jost Rezzonico T, Romagnani A, Faliti CE, Perruzza L, et al. ATP-gated ionotropic P2X7 receptor controls follicular T helper cell numbers in Peyer's patches to promote host-microbiota mutualism. Immunity (2014) 41(5):789–801. doi: 10.1016/j.immuni.2014.10.010
128. Castigli E, Scott S, Dedeoglu F, Bryce P, Jabara H, Bhan AK, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U.S.A. (2004) 101(11):3903–8. doi: 10.1073/pnas.0307348101
129. Jones L, Ho WQ, Ying S, Ramakrishna L, Srinivasan KG, Yurieva M, et al. A subpopulation of high IL-21-producing CD4+ T cells in Peyer’s Patches is induced by the microbiota and regulates germinal centers. Sci Rep (2016) 6(1):1–17. doi: 10.1038/srep30784
130. Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev (2014) 260(1):76–85. doi: 10.1111/imr.12189
131. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature (2001) 411(6837):603–6. doi: 10.1038/35079114
132. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature (2001) 411(6837):599–603. doi: 10.1038/35079107
133. Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol (2019) 17(8):497–511. doi: 10.1038/s41579-019-0213-6
134. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol (2010) 28:243–73. doi: 10.1146/annurev-immunol-030409-101314
135. Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity (2015) 43(3):541–53. doi: 10.1016/j.immuni.2015.08.007
136. Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe (2013) 14(5):571–81. doi: 10.1016/j.chom.2013.10.009
137. Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O'Connell RM, et al. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe (2015) 17(2):153–63. doi: 10.1016/j.chom.2014.12.009
138. Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R, et al. Microbiotas from humans with inflammatory bowel disease alter the balance of gut th17 and RORγt(+) regulatory T cells and exacerbate colitis in mice. Immunity (2019) 50(1):212–224.e4. doi: 10.1016/j.immuni.2018.12.015
139. Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U.S.A. (2009) 106(46):19256–61. doi: 10.1073/pnas.0812681106
140. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity (2014) 41(1):152–65. doi: 10.1016/j.immuni.2014.05.016
141. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38(3):596–605. doi: 10.1016/j.immuni.2012.11.020
142. Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsuic Y, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science (2012) 336(6080):485–9. doi: 10.1126/science.1217718
143. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331(6015):337–41. doi: 10.1126/science.1198469
144. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500(7461):232–6. doi: 10.1038/nature12331
145. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32(6):815–27. doi: 10.1016/j.immuni.2010.06.001
146. Longman RS, Yang Y, Diehl GE, Kim SV, Littman DR. Microbiota: host interactions in mucosal homeostasis and systemic autoimmunity. Cold Spring Harb Symp Quant Biol (2013) 78:193–201. doi: 10.1101/sqb.2013.78.020081
147. Shapiro JM, Zoete MR, Palm NW, Laenen Y, Bright R, Mallette M, et al. Immunoglobulin A targets a unique subset of the microbiota in inflammatory bowel disease. Cell Host Microbe (2021) 29(1):83–93.e3. doi: 10.1016/j.chom.2020.12.003
148. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi: 10.1016/j.cell.2011.04.022
149. Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun (1998) 66(11):5157–66. doi: 10.1128/IAI.66.11.5157-5166.1998
150. Keubler LM, Buettner M, Häger C, Bleich A. A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflammation Bowel Dis (2015) 21(8):1967–75. doi: 10.1097/MIB.0000000000000468
151. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
152. Fillatreau S. Regulatory functions of B cells and regulatory plasma cells. BioMed J (2019) 42(4):233–42. doi: 10.1016/j.bj.2019.05.008
153. Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
154. Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol (2007) 178(12):7868–78. doi: 10.4049/jimmunol.178.12.7868
155. Blair PA, Chavez-Rueda KA, Evans JG, Shlomchik MJ, Eddaoudi A, Isenberg DA, et al. Selective targeting of B cells with agonistic anti-CD40 is an efficacious strategy for the generation of induced regulatory T2-like B cells and for the suppression of lupus in MRL/lpr mice. J Immunol (2009) 182(6):3492–502. doi: 10.4049/jimmunol.0803052
156. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186(10):5569–79. doi: 10.4049/jimmunol.1100284
157. Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol (2005) 25(1):29–40. doi: 10.1007/s10875-005-0355-6
158. Miles K, Heaney J, Sibinska Z, Salter D, Savill J, Gray D, et al. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci U.S.A. (2012) 109(3):887–92. doi: 10.1073/pnas.1109173109
159. Bankoti R, Gupta K, Levchenko A, Stäger S. Marginal zone B cells regulate antigen-specific T cell responses during infection. J Immunol (2012) 188(8):3961–71. doi: 10.4049/jimmunol.1102880
160. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Tedder TF, Sato S, et al. IL-10-producing regulatory B10 cells inhibit intestinal injury in a mouse model. Am J Pathol (2011) 178(2):735–43. doi: 10.1016/j.ajpath.2010.10.022
161. Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol (2009) 182(12):7459–72. doi: 10.4049/jimmunol.0900270
162. Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol (2015) 194(4):1395–401. doi: 10.4049/jimmunol.1401329
163. Maseda D, Candando KM, Smith SH, Kalampokis I, Weaver CT, Plevy SE, et al. Peritoneal cavity regulatory B cells (B10 cells) modulate IFN-γ+CD4+ T cell numbers during colitis development in mice. J Immunol (2013) 191(5):2780–95. doi: 10.4049/jimmunol.1300649
164. Shen P, Roch T, Lampropoulou V, R A, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507(7492):366–70. doi: 10.1038/nature12979
165. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41(6):1040–51. doi: 10.1016/j.immuni.2014.10.016
166. Rosser EC, Mauri C. The emerging field of regulatory B cell immunometabolism. Cell Metab (2021) 33(6):1088–97. doi: 10.1016/j.cmet.2021.05.008
167. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
168. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat Med (2014) 20(11):1334–9. doi: 10.1038/nm.3680
169. Glass MC, Glass DR, Oliveria JP, Mbiribindi B, Esquivel CO, Krams SM, et al. Human IL-10-producing B cells have diverse states that are induced from multiple B cell subsets. Cell Rep (2022) 39(3):110728. doi: 10.1016/j.celrep.2022.110728
170. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med (2005) 201(1):35–9. doi: 10.1084/jem.20032000
171. Rickert RC, Jellusova J, Miletic AV. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev (2011) 244(1):115–33. doi: 10.1111/j.1600-065X.2011.01067.x
172. den Hartog G, Osch van TLJ, Vos M, Meijer B, Savelkoul HFJ, Neerven van RJJ, et al. BAFF augments IgA2 and IL-10 production by TLR7/8 stimulated total peripheral blood B cells. Eur J Immunol (2018) 48(2):283–92. doi: 10.1002/eji.201646861
173. Fehres CM, Uden van NO, Yeremenko NG, Fernandez L, Salinas Franco G, Duivenvoorde van LM, et al. APRIL induces a novel subset of IgA(+) regulatory B cells that suppress inflammation via expression of IL-10 and PD-L1. Front Immunol (2019) 10:1368. doi: 10.3389/fimmu.2019.01368
174. Hua C, Audo R, Yeremenko N, Baeten D, Hahne M, Combe B, et al. A proliferation inducing ligand (APRIL) promotes IL-10 production and regulatory functions of human B cells. J Autoimmun (2016) 73:64–72. doi: 10.1016/j.jaut.2016.06.002
175. Yang M, Sun L, Wang S, Ko KH, Xu H, Zheng BJ, et al. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J Immunol (2010) 184(7):3321–5. doi: 10.4049/jimmunol.0902551
176. Zhang Y, li J, Zhou N, Zhang Y, Wu M, Xu J, et al. The Unknown Aspect of BAFF: Inducing IL-35 Production by a CD5 + CD1d hi FcγRIIb hi Regulatory B Cell Subset in lupus. J Invest Dermatol (2017) 137:2532–43. doi: 10.1016/j.jid.2017.07.843
177. Komlósi ZI, Kovács N, Veen van W, Kirsch AI, Fahrner HB, Wawrzyniak M, et al. Human CD40 ligand-expressing type 3 innate lymphoid cells induce IL-10-producing immature transitional regulatory B cells. J Allergy Clin Immunol (2018) 142(1):178–194.e11. doi: 10.1016/j.jaci.2017.07.046
178. Cao AT, Yao S, Gong B, Nurieva RI, Elson CO, Cong Y, et al. Interleukin (IL)-21 promotes intestinal IgA response to microbiota. Mucosal Immunol (2015) 8(5):1072–82. doi: 10.1038/mi.2014.134
179. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi: 10.1038/nature11501
180. Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E, et al. A granulocyte-macrophage colony-stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med (2009) 15(9):1038–45. doi: 10.1038/nm.2003
181. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y, et al. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity (2011) 34(5):703–14. doi: 10.1016/j.immuni.2011.03.016
182. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28(5):639–50. doi: 10.1016/j.immuni.2008.03.017
183. Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol (2009) 183(4):2312–20. doi: 10.4049/jimmunol.0900185
184. Bankó Z, Pozsgay J, Szili D, Tóth M, Gáti T, Nagy G, et al. Induction and differentiation of IL-10-producing regulatory B cells from healthy blood donors and rheumatoid arthritis patients. J Immunol (2017) 198(4):1512–20. doi: 10.4049/jimmunol.1600218
185. Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun (2019) 101:145–52. doi: 10.1016/j.jaut.2019.04.016
186. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118(30). doi: 10.1073/pnas.2023739118
187. Zhang Y, Wei S, Wu Q, Shen X, Dai W, Zhang Z, et al. Interleukin-35 promotes Breg expansion and interleukin-10 production in CD19+ B cells in patients with ankylosing spondylitis. Clin Rheumatol (2022) 41(8):2403–16. doi: 10.1007/s10067-022-06137-8
188. Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med (2014) 20(6):633–41. doi: 10.1038/nm.3554
189. Dambuza IM, He C, Choi JK, Yu C-R, Wang R, Mattapallil MJ, et al. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat Commun (2017) 8(1):719. doi: 10.1038/s41467-017-00838-4
190. Rosser EC, Piper CJM, Matei DE, Blair PA, Rendeiro AF, Orford M, et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab (2020) 31(4):837–851.e10. doi: 10.1016/j.cmet.2020.03.003
191. Luu M, Pautz S, Kohl V, Singh R, Romero R, Lucas S, et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun (2019) 10(1):760. doi: 10.1038/s41467-019-08711-2
192. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest (2008) 118(10):3420–30. doi: 10.1172/JCI36030
193. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med (2003) 197(4):489–501. doi: 10.1084/jem.20021293
194. Ray A, Wang L, Dittel BN. IL-10-independent regulatory B-cell subsets and mechanisms of action. Int Immunol (2015) 27(10):531–6. doi: 10.1093/intimm/dxv033
195. Zhu Q, Rui K, Wang S, Tian J. Advances of regulatory B cells in autoimmune diseases. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.592914
196. Carter NA, Rosser EC, Mauri C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther (2012) 14(1):R32. doi: 10.1186/ar3736
197. Huai G, Markmann JF, Deng S, Rickert CG TGF-β-secreting regulatory B cells: unsung players in immune regulation. Clin Transl Immunol (2021) 10(4):e1270. doi: 10.1002/cti2.1270
198. Loxton AG, van Rensburg IC. FasL regulatory B-cells during Mycobacterium tuberculosis infection and TB disease. J Mol Biol (2021) 433(13):166984. doi: 10.1016/j.jmb.2021.166984
199. Kaku H, Cheng KF, and Al-Abed Y, Rothstein TL. A novel mechanism of B cell–mediated immune suppression through CD73 expression and adenosine production. J Immunol (2014) 193(12):5904–13. doi: 10.4049/jimmunol.1400336
200. Saze Z, Schuler PJ, Hong C-S, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell–mediated suppression of activated T cells. Blood (2013) 122(1):9–18. doi: 10.1182/blood-2013-02-482406
201. Maerz JK, Trostel C, Lange A, Parusel R, Michaelis L, Schäfer A, et al. Bacterial immunogenicity is critical for the induction of regulatory B cells in suppressing inflammatory immune responses. Front Immunol (2019) 10:3093. doi: 10.3389/fimmu.2019.03093
202. Steimle A, Michaelis L, Lorenzo Di F, Kliem T, Münzner T, Maerz JK, et al. Weak agonistic LPS restores intestinal immune homeostasis. Mol Ther (2019) 27(11):1974–91. doi: 10.1016/j.ymthe.2019.07.007
203. Waidmann M, Bechtold O, Frick JS, Lehr HA, Schubert S, Dobrindt U, et al. Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology (2003) 125(1):162–77. doi: 10.1016/S0016-5085(03)00672-3
204. Gronbach K, Flade I, Holst O, Lindner B, Ruscheweyh HJ, Wittmann A, et al. Endotoxicity of lipopolysaccharide as a determinant of T-cell–mediated colitis induction in mice. Gastroenterology (2014) 146(3):765–75. doi: 10.1053/j.gastro.2013.11.033
205. Oka A, Mishima Y, Liu B, Herzog JW, Steinbach EC, Kobayashi T, et al. Phosphoinositide 3-kinase P110δ-signaling is critical for microbiota-activated IL-10 production by B cells that regulate intestinal inflammation. Cells (2019) 8(10). doi: 10.3390/cells8101121
206. Cho JH, Nicolae DL, Ramos R, Fields CT, Rabenau K, Corradino S, et al. Linkage and linkage disequilibrium in chromosome band 1p36 in American Chaldeans with inflammatory bowel disease. Hum Mol Genet (2000) 9(9):1425–32. doi: 10.1093/hmg/9.9.1425
207. Seki N, Nimura Y, Ohira M, Saito T, Ichimiya S, Nomura N, et al. Identification and chromosome assignment of a human gene encoding a novel phosphatidylinositol-3 kinase. DNA Res (1997) 4(5):355–8. doi: 10.1093/dnares/4.5.355
208. Steinbach EC, Kobayashi T, Russo SM, Sheikh SZ, Gipson GR, Kennedy ST, et al. Innate PI3K p110δ regulates Th1/Th17 development and microbiota-dependent colitis. J Immunol (2014) 192(8):3958–68. doi: 10.4049/jimmunol.1301533
209. Uno JK, Rao KN, Matsuoka K, Sheikh SZ, Kobayashi T, Li F, et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110δ. Gastroenterology (2010) 139(5):1642–53, 1653.e1-6. doi: 10.1053/j.gastro.2010.07.008
210. Mishima Y, Oka A, Liu B, Herzog JW, Eun CS, Fan TJ, et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. J Clin Invest (2019) 129(9):3702–16. doi: 10.1172/JCI93820
211. Zhou L. AHR function in lymphocytes: emerging concepts. Trends Immunol (2016) 37(1):17–31. doi: 10.1016/j.it.2015.11.007
212. Piper CJM, Rosser EC, Oleinika K, Nistala K, Krausgruber T, Rendeiro AF, et al. Aryl hydrocarbon receptor contributes to the transcriptional program of IL-10-producing regulatory B cells. Cell Rep (2019) 29(7):1878–1892.e7. doi: 10.1016/j.celrep.2019.10.018
213. Sherr DH, Monti S. The role of the aryl hydrocarbon receptor in normal and Malignant B cell development. Semin Immunopathology (2013) 35(6):705–16. doi: 10.1007/s00281-013-0390-8
214. Xie M, Zhu Y, Zhou Y, Wang Q, Gu E, Chu Y, et al. Interleukin-35 -producing B cells rescues inflammatory bowel disease in a mouse model via STAT3 phosphorylation and intestinal microbiota modification. Cell Death Discovery (2023) 9(1):67. doi: 10.1038/s41420-023-01366-5
215. Wirtz S, Billmeier U, McHedlidze T, Blumberg RS, Neurath MF. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology (2011) 141(5):1875–86. doi: 10.1053/j.gastro.2011.07.040
216. Wang Y, Mao Y, Zhang J, Shi G, Cheng L, Lin Y, et al. IL-35 recombinant protein reverses inflammatory bowel disease and psoriasis through regulation of inflammatory cytokines and immune cells. J Cell Mol Med (2018) 22(2):1014–25. doi: 10.1111/jcmm.13428
217. Wang S, Qin C. Interleukin 35 rescues regulatory B cell function, but the effect is dysregulated in ulcerative colitis. DNA Cell Biol (2017) 36(5):413–21. doi: 10.1089/dna.2016.3570
218. Zhao M, Gu J, Wang Z. B cells in Crohn’s patients presented reduced IL-35 expression capacity. Mol Immunol (2020) 118:124–31. doi: 10.1016/j.molimm.2019.12.005
219. Wei L, Wang J, Liu Y. Prior to Foxp3+ regulatory T-cell induction, interleukin-10-producing B cells expand after Helicobacter pylori infection. Pathog Dis (2014) 72(1):45–54. doi: 10.1111/2049-632X.12182
220. Li X, Tan J, Zhang F, Xue Q, Wang N, Cong X, et al. H.pylori infection alleviates acute and chronic colitis with the expansion of regulatory B cells in mice. Inflammation (2019) 42(5):1611–21. doi: 10.1007/s10753-019-01022-0
221. Zhang H, Dai Y, Liu Y, Wu T, Li J, Wang X, et al. Helicobacter pylori colonization protects against chronic experimental colitis by regulating Th17/Treg balance. Inflammatory Bowel Dis (2018) 24(7):1481–92. doi: 10.1093/ibd/izy107
222. Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, et al. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity (2010) 33(5):777–90. doi: 10.1016/j.immuni.2010.10.016
223. Brière F, Bridon JM, Chevet D, Souillet G, Bienvenu F, Guret C, et al. Interleukin 10 induces B lymphocytes from IgA-deficient patients to secrete IgA. J Clin Invest (1994) 94(1):97–104. doi: 10.1172/JCI117354
224. DeFrance T, Vanbervliet B, Brière F, Durand I, Rousset F, Banchereau J, et al. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med (1992) 175(3):671–82. doi: 10.1084/jem.175.3.671
225. Kunisawa J, Gohda M, Hashimoto E, Ishikawa I, Higuchi M, Suzuki Y, et al. Microbe-dependent CD11b+ IgA+ plasma cells mediate robust early-phase intestinal IgA responses in mice. Nat Commun (2013) 4:1772. doi: 10.1038/ncomms2718
226. Fu Y, Wang Z, Yu B, Lin Y, Huang E, Liu R, et al. Intestinal CD11b(+) B cells ameliorate colitis by secreting immunoglobulin A. Front Immunol (2021) 12:697725. doi: 10.3389/fimmu.2021.697725
227. Zacca ER, Onofrio LI, Acosta CDV, Ferrero PV, Alonso SM, Ramello MC, et al. PD-L1+ Regulatory B cells are significantly decreased in rheumatoid arthritis patients and increase after successful treatment. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02241
228. Sun X, Zhang T, Li M, Yin L, Xue J. Immunosuppressive B cells expressing PD-1/PD-L1 in solid tumors: A mini review. Qjm (2019) 115(8):507–12,. doi: 10.1093/qjmed/hcz162
229. Doi T, Kanai T, Mikami Y, Sujino T, Jun L, Ono Y, et al. IgA plasma cells express the negative regulatory co-stimulatory molecule programmed cell death 1 ligand and have a potential tolerogenic role in the intestine. Biochem Biophys Res Commun (2012) 425(4):918–23. doi: 10.1016/j.bbrc.2012.08.010
230. Serrán MG, Vernengo FF, Almada L, Beccaria CG, Gazzoni Y, Canete PF, et al. Extrafollicular plasmablasts present in the acute phase of infections express high levels of PD-L1 and are able to limit T cell response. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.828734
231. Shalapour S, Font-Burgada J, Caro Di G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature (2015) 521(7550):94–8. doi: 10.1038/nature14395
232. Birch RJ, Burr N, Subramanian V, Tiernan JP, Hull MA, Finan P, et al. Inflammatory bowel disease-associated colorectal cancer epidemiology and outcomes: an english population-based study. Am J Gastroenterol (2022) 117(11):1858–70. doi: 10.14309/ajg.0000000000001941
233. Maryńczak K, Włodarczyk J, Sabatowska Z, Dziki A, Dziki Ł, Włodarczyk M, et al. Colitis-associated colorectal cancer in patients with inflammatory bowel diseases in a tertiary referral center: A propensity score matching analysis. J Clin Med (2022) 11(3). doi: 10.3390/jcm11030866
234. Melcher C, Yu J, Duong VHH, Westphal K, Farimany Siasi Helmi N, Shaverskyi A, et al. B cell-mediated regulatory mechanisms control tumor-promoting intestinal inflammation. Cell Rep (2022) 40(2):111051. doi: 10.1016/j.celrep.2022.111051
235. Zhao L, Hu S, Davila ML, Yang J, Lin YD, Albanese JM, et al. Coordinated co-migration of CCR10(+) antibody-producing B cells with helper T cells for colonic homeostatic regulation. Mucosal Immunol (2021) 14(2):420–30. doi: 10.1038/s41385-020-0333-3
236. Rojas OL, Pröbstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, et al. Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell (2019) 176(3):610–624.e18. doi: 10.1016/j.cell.2018.11.035
237. Rubin SJS, Bai L, Haileselassie Y, Garay G, Yun C, Becker L, et al. Mass cytometry reveals systemic and local immune signatures that distinguish inflammatory bowel diseases. Nat Commun (2019) 10(1):2686. doi: 10.1038/s41467-019-10387-7
238. Pararasa C, Zhang N, Tull TJ, Chong MHA, Siu JHY, Guesdon W, et al. Reduced CD27(-)IgD(-) B cells in blood and raised CD27(-)IgD(-) B cells in gut-associated lymphoid tissue in inflammatory bowel disease. Front Immunol (2019) 10:361. doi: 10.3389/fimmu.2019.00361
239. McNamee EN, Rivera-Nieves J. Ectopic tertiary lymphoid tissue in inflammatory bowel disease: protective or provocateur? Front Immunol (2016) 7:308. doi: 10.3389/fimmu.2016.00308
240. Sura R, Colombel J-F, Van Kruiningen HJ. Lymphatics, tertiary lymphoid organs and the granulomas of Crohn’s disease: an immunohistochemical study. Alimentary Pharmacol Ther (2011) 33(8):930–9. doi: 10.1111/j.1365-2036.2011.04605.x
241. Breedveld A, van Egmond M. IgA and FcαRI: pathological roles and therapeutic opportunities. Front Immunol (2019) 10:553. doi: 10.3389/fimmu.2019.00553
242. Bakema JE, van Egmond M. The human immunoglobulin A Fc receptor FcαRI: a multifaceted regulator of mucosal immunity. Mucosal Immunol (2011) 4(6):612–24. doi: 10.1038/mi.2011.36
243. Hansen IS, Hoepel W, Zaat SAJ, Baeten DLP, Dunnen den J. Serum IgA immune complexes promote proinflammatory cytokine production by human macrophages, monocytes, and kupffer cells through FcαRI-TLR cross-talk. J Immunol (2017) 199(12):4124–31. doi: 10.4049/jimmunol.1700883
244. Lindfors K, Ciacci C, Kurppa K, Lundin KEA, Makharia GK, Mearin ML, et al. Coeliac disease. Nat Rev Dis Primers (2019) 5(1):3. doi: 10.1038/s41572-018-0054-z
245. Habas E, Ali E, Farfar K, Errayes M, Alfitori J, Habas E, et al. IgA nephropathy pathogenesis and therapy: Review & updates. Med (Baltimore) (2022) 101(48):e31219. doi: 10.1097/MD.0000000000031219
246. Kazemi-Shirazi L, Gasche CH, Natter S, Gangl A, Smolen J, Spitzauer S, et al. IgA autoreactivity: a feature common to inflammatory bowel and connective tissue diseases. Clin Exp Immunol (2002) 128(1):102–9. doi: 10.1046/j.1365-2249.2002.01804.x
247. Rengarajan S, Vivio EE, Parkes M, Peterson DA, Roberson EDO, Newberry RD, et al. Dynamic immunoglobulin responses to gut bacteria during inflammatory bowel disease. Gut Microbes (2020) 11(3):405–20. doi: 10.1080/19490976.2019.1626683
Keywords: regulatory B cells (Bregs), IgA, microbiota, IBD, intestinal inflammation, IgG, plasma cells
Citation: Zogorean R and Wirtz S (2023) The yin and yang of B cells in a constant state of battle: intestinal inflammation and inflammatory bowel disease. Front. Immunol. 14:1260266. doi: 10.3389/fimmu.2023.1260266
Received: 17 July 2023; Accepted: 18 September 2023;
Published: 02 October 2023.
Edited by:
Saurabh Mehandru, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Andrea Cerutti, Catalan Institution for Research and Advanced Studies (ICREA), SpainEvelyn Tsantikos, Monash University, Australia
Copyright © 2023 Zogorean and Wirtz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Wirtz, U3RlZmFuLldpcnR6QHVrLWVybGFuZ2VuLmRl