 Jerome Razanamahery
Jerome Razanamahery Amelie Godot2
Amelie Godot2 Bernard Bonnotte
Bernard Bonnotte- 1Department of Internal Medicine and Clinical Immunology, Dijon University Hospital, Dijon, France
- 2Department of Internal Medicine, Besancon University Hospital, Besancon, France
Histiocytoses encompass a wide spectrum of diseases, all characterized by tissue infiltration by CD68+ histiocytes. Most adult histiocytoses are considered clonal diseases because they highlight recurrent somatic mutations in the MAP-kinase pathway gene, primarily BRAF. The presence of BRAF mutation is associated with widespread disease in children with Langerhans cell histiocytosis (LCH) or cardiovascular/neurological involvement in Erdheim–Chester disease (ECD). Nevertheless, few data are available on adult clonal histiocytosis. This is why we have conducted a retrospective study of all patients with clonal histiocytosis in our institution and present the data according to the presence of BRAF mutation. Among 27 adult patients (10 ECD, 10 LCH, 5 Rosai–Dorfman disease (RDD), and 3 mixed ECD/LCH), 11 (39%) have BRAF mutation with gain of function (n = 9) and deletion (n = 2). Those patients had frequent multicentric disease with risk organ involvement, especially the brain and cardiovascular system. They had frequent associated myeloid neoplasms (mostly chronic myelomonocytic leukemia) and received more frequently targeted therapy as the front-line therapy. Nevertheless, its presence did not affect the overall survival or relapse-free survival probably due to the emergence of efficient therapies. To conclude, rapid and accurate molecular establishment in adult clonal histiocytoses is crucial because BRAFV600E mutation correlates with multicentric disease with organ involvement and incomplete metabolic response.
Introduction
Histiocytoses are clonal hematopoietic disorders characterized by CD68+ histiocyte tissue infiltration causing a wide clinical spectrum of diseases (1). The recent identification of somatic mutations in the Mitogen-Activated Protein Kinase (MAP-kinase) pathway gene, especially BRAFV600E in most tissue samples from adult histiocytoses [Langerhans cell histiocytosis (LCH) (2), Erdheim–Chester disease (ECD) (3), and Rosai–Dorfman disease (RDD) (4)], led to consider these diseases as myeloid neoplasm (5). Interestingly the genetic landscape analysis of various cancers, such as melanoma, colon cancer, lung cancer, and more recently histiocytic neoplasms, has highlighted the pivotal role of BRAFV600E gene, which is considered an important cornerstone in the development of human cancer. Its presence is associated with disseminated disease, neurodegeneration, and resistance to front-line therapy in pediatric LCH patients (6, 7). In adults, LCH is mainly restricted to the lung, making BRAFV600E testing difficult due to the risk of a pulmonary biopsy procedure and a low DNA quantity when available. Hence, few data are available regarding the impact of BRAFV600E mutation in adult extrapulmonary LCH. BRAFV600E mutation has also been described in ECD, a rare adult non-Langerhans cell histiocytosis. Its presence is significant because patients with cardiac and neurological locations are frequently BRAF-mutated (8). Because LCH, ECD, and RDD are characterized by clonal origins, we aimed to investigate the impact of BRAFV600E mutation on aggressiveness and outcomes in a cohort of adult histiocytoses. This study describes the distinct characteristics and outcomes of various adult histiocytoses separately, followed by an analysis according to BRAFV600E mutational status.
Method
This is a single-center retrospective study (local ethics board approved) for which informed consent was waived. Patients included were at least 18 years old with biopsy-confirmed histiocytosis diagnosed from 1990 to 2022. Tissue infiltration by histiocytes (CD68+) and hematoxylin and eosin staining for CD207, CD1a, and S100 analyses were mandatory. The diagnosis of histiocytoses was performed according to expert guidelines (9–11). LCH patients had compatible clinical manifestations (i.e., skin, liver, lung, colon, endocrine, or brain) and tissue biopsy showing infiltration by CD68+, CD1a+, and CD207+ histiocytes. Biopsy was mandatory except for isolated pulmonary LCH after exclusion for differential diagnosis. ECD patients had iconic features (long bone osteosclerosis, peri-renal infiltration, and vascular sheathing) along with histology showing CD68+ and CD1a− histocytes accompanied by varying degrees of fibrosis. RDD patients had typical histology features with CD68+ and CD1a− with emperipolesis.
BRAFV600E status was established using droplet PCR (12) and next-generation sequencing (NGS) technology if the DNA quantity was sufficient (13). The liver, spleen, and bone marrow were risk organs for LCH patients, and the cardiovascular system was at risk for ECD patients. The brain was a risk organ for all patients.
The treatment approach was based on symptoms and organ involvement according to guidelines and expert consensus. For patients with multisystemic Langerhans cell histiocytosis (LCH), chemotherapy was the first-line therapy, while immunosuppressive agents were considered for cases of isolated skin LCH. All patients with LCH were advised to stop smoking.
For the ECD patients, treatment options included interferon, biologic agents (such as IL-1 or TNF-alpha inhibitors), or targeted therapy for severe cases (i.e., cardiovascular or neurological involvement). Patients with RDD received treatment when presenting symptomatic lymphadenopathy or autoimmune-associated conditions. Non-symptomatic patients without risk of organ infiltration were closely monitored. For all patients, targeted therapies were proposed following relapse or failure of conventional treatments. Specifically, patients with the BRAFV600E mutation received a BRAF inhibitor (vemurafenib) at a dose of 420 mg twice daily, while those without the mutation received a MEK inhibitor (cobimetinib) at a dose of 400 mg for 21 days within a 28-day cycle. All treatment decisions were validated with the reference center for histiocytosis at Pitié-Salpétrière Hospital.
Disease activity was assessed using 18fluorodeoxyglucose positron tomography (according to Positron Emission Tomography Response Criteria In Solid Tumors (PERCIST) criteria). Complete metabolic response was defined by the normalization of all lesions to at or below the standardized uptake value (SUV) of the liver background. Partial metabolic response was defined by a ≥50% decrease from the baseline sum of all target lesions’ SUV. Progressive metabolic disease was defined by a ≥50% increase in the nadir sum of all target or new evaluable lesions’ SUV. Stable metabolic disease was defined if the patient did not meet the previous criteria. To evaluate the efficacy response following front-line therapy, patients with either a complete metabolic response or a partial metabolic response were classified as responders. In contrast, patients with stable or progressive metabolic disease were categorized as non-responders. The complete diagnosis procedures are reported in Supplemental Data. Comparison between groups was performed using the Mann–Whitney test, chi2, or Fisher’s test. For multiple comparison groups, an analysis of variance (ANOVA) test with the Kruskal–Wallis procedure was used followed by Dunn’s multiple comparison. Multiple logistic regression analysis for factors associated with BRAF mutation was performed with all variables with a p-value <0.2 in simple logistic regression. Statistical significance was set at p < 0.05 (two-sided). Statistical analyses were performed using GraphPad Prism software V.10 (GraphPad, San Diego, CA, USA).
Results
Twenty-eight adult patients were included in the study. Ten patients had ECD, 10 LCH, 5 RDD, and 3 mixed “ECD/LCH”. Comparison between the four groups showed differences in terms of phenotype and disease course. ECD patients had a more frequent retroperitoneal involvement than LCH patients (80% vs. 0%; p = 0.024) and a more frequent digestive localization than RDD patients (70% vs. 0%; p = 0.046). Their mesentery biopsies revealed histiocyte infiltration more frequently than in LCH patients (50% vs. 0%; p = 0.0249). However, ECD patients had limited cutaneous histiocytic infiltration as compared with mixed “ECD/LCH” (10% vs. 100%; p = 0.024). Patients with LCH had more frequent endocrine system involvement (“arginine vasopressin deficiency”) than ECD patients (60% vs. 10%; p = 0.048). In addition, RDD patients had more lymph node involvement than ECD (40% vs. 0%; p = 0.03) and LCH patients (40% vs. 0%; p = 0.03) as confirmed by histological samples. They also had a lower rate of risk organ involvement compared to “mixed ECD/LCH” patients (0% vs. 100%; p = 0.042). At the last follow-up, RDD patients more often achieved a complete metabolic response compared with ECD patients (100% vs. 10%; p = 0.0050), while ECD patients often had more stable metabolic disease compared with LCH patients (60% vs. 0%; p = 0.03). Regarding the mutational landscape, 11 patients (39%) had BRAFV600E mutation (5 LCH, 4 ECD, and 2 “ECD/LCH”) with gain-of-function mutation on exon 15 (n = 9) and deletion in exon 12 (n = 2). BRAF-mutated patients also had mutations in KRAS, TET2, DNMT3A, ASXL1, and SRSF2 within biopsy tissue. BRAF wild-type patients had MAP2K1 gene mutation in exon 2 [RDD (n = 2) and ECD (n = 1)] (Table 1, Supplementary Table 1). There is no statically relevant correlation between the frequency of BRAFV600E mutation and the type of histiocytosis. All patients had overexpression of phosphorylated extracellular signal-regulated kinase (phospho-Erk) in tissue samples. One BRAF wild-type patient had overexpression of both the “Colony stimulating factor 1 receptor” (CSF1R) and “Programmed death ligand 1” (PDL1). Three patients could not undergo NGS analysis due to insufficient DNA quantity.

Table 1 Characteristics of patients with histiocytoses according to BRAFV600E status.
BRAFV600E-mutated patients had multicentric disease (91% vs. 48%; p = 0.041) with “risk organ” involvement (81% vs. 23%; p = 0.005), particularly the cardiovascular system (45% vs. 5.8%; p = 0.022) and the brain (45% vs. 5.8%; p = 0.022). They also had frequent endocrine involvement (54% vs. 11%; p = 0.029). Biological tests at diagnosis were similar (data not shown).
The occurrence of “risk organ” involvement remained higher in BRAFV600E-mutated patients after the exclusion of RDD patients (81% vs. 33%; p = 0.036). However, there was only a trend toward cardiovascular and brain involvement (45% vs. 8%; p = 0.06 for both). The BRAFV600E-mutated patients also had myeloproliferative neoplasm (three chronic myelomonocytic leukemia and one JAK2-mutated essential thrombocythemia) compared to the BRAF wild-type group (36% vs. 0%; p = 0.046). The frequency of clonal hematopoiesis (characterized by the presence of myeloid gene mutation on bone marrow without morphologic evidence for myelodysplastic or myeloproliferative disease) was similar in both groups. Patients in the study received several lines of treatment ranging from none to six depending on relapse rate. All BRAF-mutated patients had symptoms requiring treatment ranging from non-steroidal inflammatory agents (n = 1) for LCH and from conventional therapies (n = 6) to targeted therapies (n = 4) for severe cases. Some BRAF wild-type patients received no treatment, such as those with non-symptomatic RDD (n = 4), while others received surgery (n = 1), conventional agents including chemotherapy (n = 7), or biological agents (n = 3). Among BRAFV600E patients receiving first-line targeted therapy were multisystemic ECD (n = 3), LCH (n = 1), and mixed ECD/LCH (n = 1) with risk organ involvement. One BRAFwt patient received first-line treatment with cobimetinib due to contraindications to other ECD therapies. Hence, the use of targeted therapy as first-line treatment tends to be more frequent in BRAFV600E-mutated patients (36% vs. 6%; p = 0.062). Efficacy of first-line therapy was similar between groups (55% vs. 82%; p = 0.1998). Some patients received targeted therapies during the disease course [ECD (n = 4) and LCH (n = 1)] after a relapse. Almost all patients (90%) treated with targeted therapies had a partial metabolic response after treatment initiation, but no patient achieved a complete metabolic response. Furthermore, the use of BRAF inhibitor was responsible for CMML worsening in three patients requiring treatment discontinuation but no specific treatment such as azacytidine or hydroxycarbamide. Subsequent administration of a MEK inhibitor successfully restored the metabolic response associated with the normalization of the monocyte count.
The median [IQR] follow-up was 113 [78–180] months. BRAF wild-type patients had a more complete metabolic response (48% vs. 0%; p = 0.007), but the relapse rate was not influenced by BRAFV600E status. Death tends to be more frequent in BRAF patients (36% vs. 5.8%; p = 0.061). The cause of death was infections, except for two patients (one in both groups) who died from disease progression. The rate of complete metabolic response in BRAF wild-type patients remained higher after the exclusion of RDD patients (41% vs. 0%; p = 0.004).
Univariate logistic regression analysis showed that BRAFV600E was associated with “risk-organ” involvement (OR = 14.63; 95% CI [2.555–128]; p = 0.005), especially brain involvement (OR = 12.50; 95% CI [1.589–268]; p = 0.035), multicentric disease (OR = 11.25; 95% CI [1.610–230.7]; p = 0.036), and endocrine involvement (OR = 8.40; 95% CI [1.422–72.31]; p = 0.028) but not in multivariate analysis (Supplementary Table 2). Neither overall survival (HR: 5.574; 95% CI [0.90–33.5]; p = 0.064) nor progression-free survival (HR: 1.613; 95% CI [0.3847–6.760]; p = 0.51) was impacted by BRAFV600E status (Figure 1).
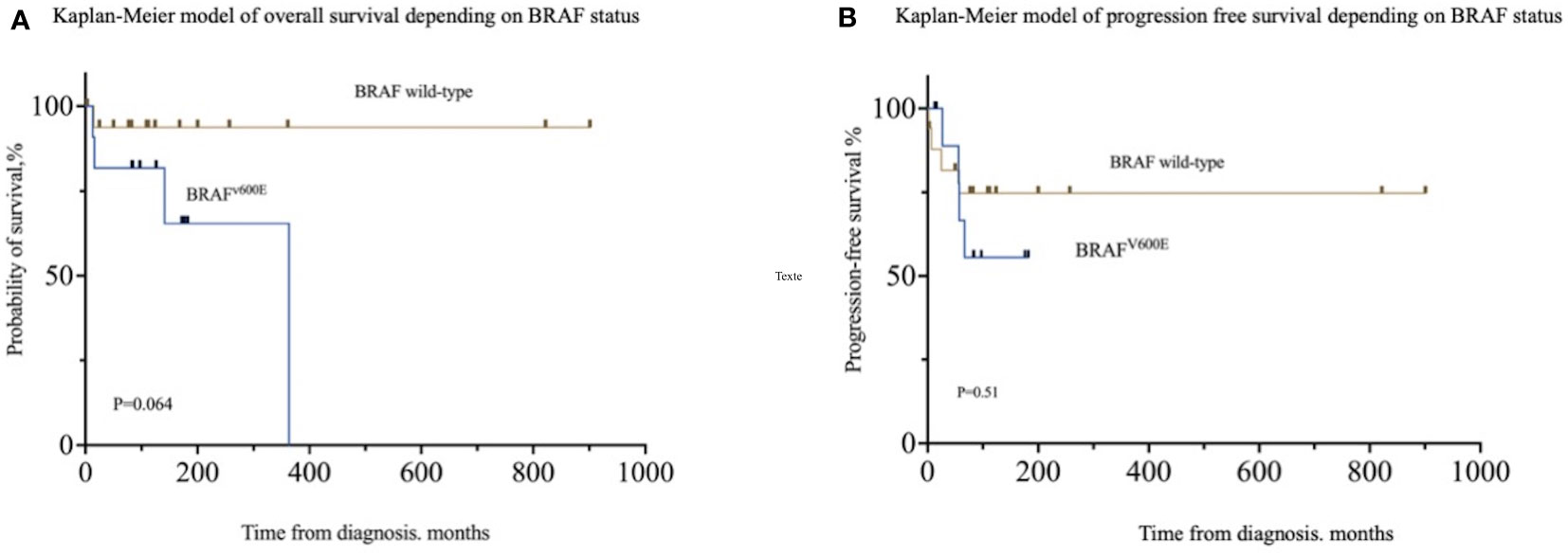
Figure 1 Patient outcomes according to BRAFV600E status. (A) Overall survival in patients depending on BRAFV600E status. (B) Progression-free survival depending on BRAFV600E status.
Discussion
In this study, we present the distinctive features of 28 adults with various forms of clonal histiocytosis, combined with an exploration of the impact of the presence of BRAFV600E mutation on presentation and outcome.
In the first part of the analysis, we observed that all cases of clonal histiocytosis exhibited a unique phenotype consistent with the literature (1). Patients diagnosed with Rosai–Dorfman disease had mutations in genes related to the MAP-kinase pathway, excluding BRAFv600E. Notably, their disease course appeared distinct compared to “L-group histiocytosis”. Our data showed that BRAFV600E mutation was associated with multicentric disease and risk organ involvement. This phenotype is in line with pediatric LCH (6) (7) studies and ECD cohort (8). Excluding RDD patients, the presence of BRAFV600E correlated with risk organ involvement irrespective of the type of “L-group histiocytosis”.
Our low frequency of BRAFV600E mutation can be attributed to the presence of RDD patients, in which BRAFV600E mutation has only been reported in two cases. (14). The mutational landscape regarding BRAFV600E and other MAP-kinase pathway genes is similar to the literature. In addition, the expression of phospho-Erk in all patients confirms the activation of the MAP-kinase pathway cascade (15) and confirms the clonal origins of those histiocytoses. In this series, the NGS analysis identified additional mutations of clonal hematopoiesis genes on biopsy samples, confirming a continuum between myeloid hematopoiesis and histiocytoses, especially in BRAF-mutated patients (1). This finding is reinforced by the occurrence of authentic myeloid neoplasms in BRAF-mutated patients and the efficacy of targeted therapy on both diseases in these patients. The recent advances in ontogeny understanding of histiocytic disorders partly explain the clinical presentation. In LCH, BRAFV600E mutation in bone marrow progenitors gives rise to mutated clones infiltrating myeloid dendritic cells, which infiltrate virtually all tissues (16). The same mechanism is proposed in ECD with a predominant involvement of CD14+ monocytes (17) rather than dendritic cells and also an abnormal activation of “trained immunity” mediated by BRAF (18). The understanding of the brain involvement mechanism has yet to be elucidated. It may result from a continuum in the blood–brain barrier between BRAFV600E monocytes deriving from yolk sack (19) progenitors and bone marrow (20).
In terms of outcome, the complete metabolic response achievement is influenced by the absence of the BRAFV600E mutation. Nevertheless, it has no impact on overall survival probably due to the efficacy of the drugs. Hence, targeted therapies have revolutionized the prognosis of the disease (21–24). In our cohort, these treatments were efficient in almost all patients, although they can cause various side effects, prompting consideration for a transition from BRAF inhibitors to MEK inhibitors in order to regain efficacy. These treatments may be of interest as a front-line therapy to avoid several lines of chemotherapy regimens in elderly patients who die mainly from infections. Furthermore, these treatments reduce the mutational BRAFV600E load and thus the risk of brain invasion by mutated clones causing delayed neurodegeneration. However, it is important to notice that these treatments are only suspensive and not curative for these diseases. The absence of complete metabolic disease in this cohort confirms this postulate and is in line with the literature (22). Other signaling pathways for which we have specific inhibitors (AKT/mTOR, ALK, and CSF1R) (25, 26) now pave the way for targeted therapies rather than conventional chemotherapy regimens in adult clonal histiocytosis.
This study has certain limitations. The retrospective nature and the data collection bias the analysis. 18FDG-PET or MRI was not available for the patients with the earliest diagnosis. The choice of therapeutic approach was influenced by the recent upgrade of targeted therapies, which were only considered as salvage therapy previously.
Nevertheless, the main strength of this study is the thorough molecular (including bone marrow) evaluation in most adult clonal histiocytoses mixing adult extrapulmonary LCH, ECD, and RDD, followed by a morphologic and metabolic response to assess the impact of BRAFV600E mutation.
Conclusion
Rapid and accurate molecular establishment in adult clonal histiocytoses is crucial because BRAFV600E mutation correlates with multicentric disease with organ involvement and incomplete metabolic response.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Dijon university hospital local ethics committe. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
JR: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. AG: Data curation, Writing – review & editing. VL-S: Data curation, Writing – review & editing. MS: Data curation, Writing – review & editing. SA: Data curation, Writing – review & editing. BB: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank Prof. Julien Haroche and Prof. Fleur Cohen-Aubart for their expertise on the patients at the Pitié-Salpétrière Hospital. They also thank Prof. Jean-Francois Emile for his review of difficult cases and NGS technology on BRAFwt patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1260193/full#supplementary-material
References
1. Emile J-F, Cohen-Aubart F, Collin M, Fraitag S, Idbaih A, Abdel-Wahab O, et al. Histiocytosis. Lancet (2021) 398(10295):157–170. doi: 10.1016/S0140-6736(21)00311-1
2. Badalian-Very G, Vergilio J-A, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood (2010) 116:1919–23. doi: 10.1182/blood-2010-04-279083
3. Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood (2012) 120:2700–3. doi: 10.1182/blood-2012-05-430140
4. Garces S, Medeiros LJ, Patel KP, Li S, Pina-Oviedo S, Li J, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol (2017) 30:1367–77. doi: 10.1038/modpathol.2017.55
5. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
6. Héritier S, Emile J-F, Barkaoui M-A, Thomas C, Fraitag S, Boudjemaa S, et al. BRAF mutation correlates with high-risk langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol (2016) 34:3023–30. doi: 10.1200/JCO.2015.65.9508
7. Hélias-Rodzewicz Z, Donadieu J, Terrones N, Barkaoui M-A, Lambilliotte A, Moshous D, et al. Molecular and clinicopathologic characterization of pediatric histiocytoses. Am J Hematol (2023) 98:1058–69. doi: 10.1002/ajh.26938
8. Cohen-Aubart F, Emile J-F, Carrat F, Helias-Rodzewicz Z, Taly V, Charlotte F, et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol (2018) 93:E114–7. doi: 10.1002/ajh.25055
9. Goyal G, Heaney ML, Collin M, Cohen Aubart F, Vaglio A, Durham BH, et al. Erdheim-Chester disease: Consensus recommendations for the evaluation, diagnosis, and treatment in the molecular era. Blood (2020) 135(22)1929–1945. doi: 10.1182/blood.2019003507
10. Goyal G, Tazi A, Go RS, Rech KL, Picarsic J, Vassallo R, et al. Expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood (2022) 139(17)2601–2621, 2021014343. doi: 10.1182/blood.2021014343
11. Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile J-F, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood (2018) 131:2877–90. doi: 10.1182/blood-2018-03-839753
12. Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discovery (2016) 6:154–65. doi: 10.1158/2159-8290.CD-15-0913
13. Melloul S, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Fraitag S, Terrones N, et al. Highly sensitive methods are required to detect mutations in histiocytoses. Haematologica (2019) 104:e97–9. doi: 10.3324/haematol.2018.201194
14. Fatobene G, Haroche J, Hélias-Rodzwicz Z, Charlotte F, Taly V, Ferreira AM, et al. BRAF V600E mutation detected in a case of Rosai-Dorfman disease. Haematologica (2018) 103:e377–9. doi: 10.3324/haematol.2018.190934
15. Chow S, Patel H, Hedley DW. Measurement of MAP kinase activation by flow cytometry using phospho-specific antibodies to MEK and ERK: potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry (2001) 46:72–8. doi: 10.1002/cyto.1067
16. Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood (2020) 135:1319–31. doi: 10.1182/blood.2019000934
17. Durham BH, Roos-Weil D, Baillou C, Cohen-Aubart F, Yoshimi A, Miyara M, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood (2017) 130:176–80. doi: 10.1182/blood-2016-12-757377
18. Biavasco R, Molteni R, Stefanoni D, Nemkov T, Netea MG, Domínguez-Andrés J, et al. Oncogene-induced maladaptive activation of trained immunity in the pathogenesis and treatment of Erdheim-Chester disease. Blood (2021) 138(17):1554–1569. doi: 10.1182/blood.2020009594
19. Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, Ozkaya N, et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature (2017) 549:389–93. doi: 10.1038/nature23672
20. Amann L, Masuda T, Prinz M. Mechanisms of myeloid cell entry to the healthy and diseased central nervous system. Nat Immunol (2023) 24:393–407. doi: 10.1038/s41590-022-01415-8
21. Cohen Aubart F, Emile J-F, Carrat F, Charlotte F, Benameur N, Donadieu J, et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood (2017) 130:1377–80. doi: 10.1182/blood-2017-03-771873
22. Diamond EL, Subbiah V, Lockhart AC, Blay J-Y, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600-mutant erdheim-chester disease and langerhans cell histiocytosis: analysis of data from the histology-independent, phase 2, open-label VE-BASKET study. JAMA Oncol (2018) 4:384–8. doi: 10.1001/jamaoncol.2017.5029
23. Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature (2019) 567:521–4. doi: 10.1038/s41586-019-1012-y
24. Abeykoon JP, Rech KL, Young JR, Ravindran A, Ruan GJ, Dasari S, et al. Outcomes after treatment with cobimetinib in patients with rosai-dorfman disease based on KRAS and MEK alteration status. JAMA Oncol (2022) 8:1816–20. doi: 10.1001/jamaoncol.2022.4432
25. Abeykoon JP, Lasho TL, Dasari S, Rech KL, Ranatunga WK, Manske MK, et al. Sustained, complete response to pexidartinib in a patient with CSF1R-mutated Erdheim-Chester disease. Am J Hematol (2022) 97:293–302. doi: 10.1002/ajh.26441
Keywords: histiocytosis, Erdheim-Chester disease, Langerhans cell histiocytosis (LCH), Rosai Dorfman disease, BRAF
Citation: Razanamahery J, Godot A, Leguy-Seguin V, Samson M, Audia S and Bonnotte B (2023) Impact of BRAFV600E mutation on aggressiveness and outcomes in adult clonal histiocytosis. Front. Immunol. 14:1260193. doi: 10.3389/fimmu.2023.1260193
Received: 17 July 2023; Accepted: 04 September 2023;
Published: 22 September 2023.
Edited by:
Masaki Yasukawa, Ehime Prefectural University of Health Sciences, JapanCopyright © 2023 Razanamahery, Godot, Leguy-Seguin, Samson, Audia and Bonnotte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome Razanamahery, cmF6YW5hbWFoZXJ5Lmplcm9tZUBob3RtYWlsLmZy