
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 11 December 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1256133
 Berenice Fischer1
Berenice Fischer1 Tanja Kübelbeck1
Tanja Kübelbeck1 Antonia Kolb1
Antonia Kolb1 Julia Ringen2,3
Julia Ringen2,3 Ari Waisman4,5
Ari Waisman4,5 Miriam Wittmann1
Miriam Wittmann1 Susanne Karbach2,3,6
Susanne Karbach2,3,6 Stephan Marcus Kölsch7
Stephan Marcus Kölsch7 Daniela Kramer1,5*
Daniela Kramer1,5*Plaque psoriasis is an autoinflammatory and autoimmune skin disease, affecting 1-3% of the population worldwide. Previously, high levels of IL-36 family cytokines were found in psoriatic skin lesions, thereby contributing to keratinocyte hyperproliferation and infiltration of immune cells such as neutrophils. While treatment with anti-IL36 receptor (IL36R) antibodies was recently approved for generalized pustular psoriasis (GPP), it remains unclear, if targeting the IL36R might also inhibit plaque psoriasis. Here we show that antibody-mediated inhibition of IL36R is sufficient to suppress imiquimod-induced psoriasis-like skin inflammation and represses the disease’s development in a model that depends on IL-17A overexpression in the skin. Importantly, treatment with anti-IL36R antibodies inhibited skin inflammation and attenuated psoriasis-associated, systemic inflammation. This is possibly due to a widespread effect of IL36R inhibition, which not only suppresses pro-inflammatory gene expression in keratinocytes, but also the activation of other immune cells such as T-cells or dendritic cells. In conclusion, we propose that inhibition of the IL-36 signaling pathway might constitute an attractive, alternative approach for treating IL-17A-driven psoriasis and psoriasis-linked comorbidities.
There are five main types of psoriasis – plaque, guttate, inverse, pustular, and erythrodermic psoriasis. In general, psoriasis is an autoimmune and autoinflammatory skin disease characterized by parakeratosis, acanthosis, and a significant infiltration of various immune cells, especially IL-17-producing cells, neutrophils, and pro-inflammatory monocytes (1). Psoriasis vulgaris or plaque psoriasis is the most prevalent subtype of this skin disease (about 90% of the cases), characterized by an overactive immune response, leading to a chronic expression of IL-23 and IL-17A cytokines. While the causes of psoriasis are still not fully understood, it has become clear that psoriasis is not only a skin condition but also influences the development of several comorbidities such as psoriatic arthritis, metabolic syndrome, and cardiovascular disease (2). Why and which patients develop comorbidities and how these can be effectively treated, are the focus of ongoing investigations. Thus, there is still a medical need for new anti-psoriatic therapy approaches that also effectively inhibit psoriasis-associated comorbidities.
Besides IL-17A and IL-23, two members of the IL-1 superfamily, IL-36α and IL-36γ, were found to be overexpressed in the epidermal compartment of psoriasis vulgaris skin lesions (3, 4). Therefore, IL-36 signaling constitutes an attractive alternative pathway that could be targeted for the treatment of plaque psoriasis. IL-36 belongs to the IL-1 family, consisting of three activating isoforms (IL-36α, IL-36β, IL-36γ) and one antagonist (IL36Ra) (5). For activation or repression of IL-36 signaling, all IL-36 cytokines need to undergo N-terminally cleavage by endogenous cathepsins, which are neutrophil- or pathogen-derived proteases (6–9). Subsequently, activated IL-36 agonists bind to the IL-36 receptor leading to the recruitment of the IL-1RAcP adaptor molecule and subsequent downstream activation of MyD88, NF-κB, and STAT3, thereby triggering pro-inflammatory target gene expression (5, 10, 11). IL-36 cytokines, as well as the IL-36 receptor, are widely expressed in various murine and human cell types, such as keratinocytes, endothelial cells, dendritic cells (DCs), and CD4+ T-cells (12–14). IL-36 stimulation of keratinocytes, for example, induces the expression of chemokines such as Cxcl2, Cxcl5, Ccl2, and Ccl20, triggering a massive infiltration of neutrophils, monocytes, and T-cells into the skin (11). Moreover, in CD4+ T-cells, IL-36 treatment induces the proliferation of naïve T-cells and polarization to T helper 1 (Th1) cells, whereas IL-36-stimulated DCs promote T-cell priming (12, 15, 16). Further effects of IL-36 stimulation on the activation and function of endothelial cells, monocytes, CD8+ T-cells, and γδ T-cells have also been reported (16–18). Hence, IL-36 signaling seems to be an important signaling pathway that - depending on the initial trigger - can act on multiple cell types, thereby eliciting innate but also adaptive immune responses.
Originally, the prominent role of IL-36 cytokines in psoriasis was noted in pustular psoriasis, particularly in GPP. This rare and often severe dermatosis, marked by sterile pustules, is characterized by overactive IL-36 signaling due to loss-of-function mutations in the IL-36 receptor antagonist IL36RA (19). Confirming this pathomechanism, an antibody targeting the IL-36 receptor (IL36R) has recently been successfully developed and approved for the treatment of GPP (20). Whether inhibition of IL-36 signaling could also constitute an attractive approach for the treatment of plaque psoriasis, remains to be investigated though. Interestingly, global and keratinocyte-specific deletions of IL36R are sufficient to prevent experimentally induced psoriasis in mice (21–23). IL-36 has been shown to induce IL-23, and importantly IL-17 and IL-36 cytokines can induce each other, thereby amplifying pro-inflammatory gene expression in keratinocytes (24–26). Thus inhibition of the IL-36 signaling pathway might potentially also inhibit IL-17A-driven inflammation in plaque psoriasis. Accordingly, it was previously shown that the application of an anti-IL-36R antibody partially prevents imiquimod (IMQ)-, IL-23, or IL-36-mediated skin inflammation in mice. However, it is not clear if treatment with an anti-IL36R antibody can suppress psoriasis-like skin inflammation once it is already established.
To clarify, if inhibition of IL-36 signaling can block IL-17A-driven plaque psoriasis, especially when inflammation has already been induced, we investigated the effects of systemic anti-IL36R antibody application in two different mouse models. Effects of an anti-IL-36R antibody treatment were investigated either after establishment of IMQ-induced psoriasis or in keratinocyte-specific IL-17A overexpressing mice (K14-IL17Aind mice), harboring a heterogeneous deletion of the psoriasis-promoting factor IκBζ (Nfkbiz). Using both mouse models, we could show that inhibition of the IL-36 pathway effectively suppressed the recruitment of neutrophils, monocytes, T-cells, and dendritic cells to the affected skin. Moreover, keratinocyte hyperproliferation and psoriasis-associated, pro-inflammatory gene expression were effectively blocked in the skin of anti-IL36R-treated mice. We observed a widespread effect of anti-IL36R treatment, which affected local keratinocyte responses and the activation of other immune cells, leading to an inhibition of psoriasis-associated systemic inflammation. Thus, our data imply that inhibition of IL-36 signaling constitutes an attractive, alternative approach for the treatment of IL-17A-driven plaque psoriasis, which may also effectively inhibit psoriasis-associated comorbidities.
As a starting point of the project, we analyzed expression levels of the IL-36 receptor and IL-36 cytokines in human psoriasis vulgaris samples. As already reported before, IL-36 receptor expression (encoded by IL1RL2) (Figure 1A), as well as the expression of IL36A, IL36B, and IL36G are significantly upregulated in psoriasis vulgaris skin lesions, compared to healthy and non-lesional skin samples (Figure 1B) (27). Interestingly, when we re-analyzed the expression levels of known psoriasis-associated cytokines in previously published data sets, comparing gene expression of skin lesions to non-lesional controls derived from the same patient, IL36A, IL36B, and IL36G displayed the most significant increase in expression compared to conventional cytokines such as IL17A, IL23A or TNF (Figure 1B) (28, 29). Thus, IL-36 is a valid target for the treatment of plaque psoriasis, since IL-36 signaling is upregulated in plaque psoriasis, and its upregulation is even more pronounced than IL-17A and IL-22, which have already been successfully used as targets for the treatment of psoriasis vulgaris using neutralizing antibodies.
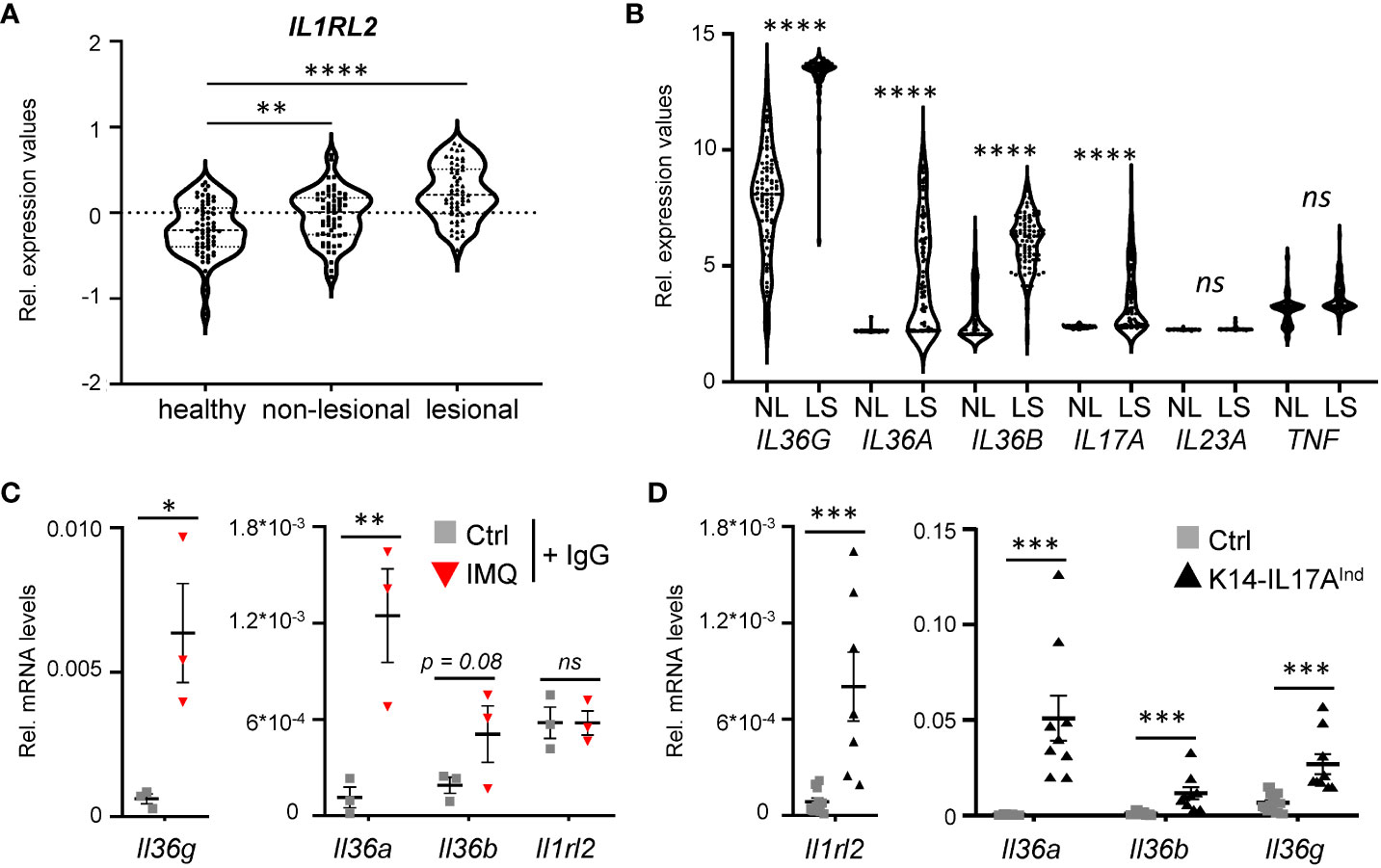
Figure 1 IL-36 signaling is upregulated in psoriatic skin lesions of mice and psoriasis patients. (A) mRNA expression levels of the IL36 receptor (IL36R/IL1RL2) in skin samples of 64 healthy individuals and in non-lesional and lesional samples from 58 psoriasis vulgaris patients (GDS4602). Significance was calculated using a 2-tailed Student’s t-test: ** p < 0.01**** < 0.0001. (B) mRNA expression levels of psoriasis-associated cytokines in lesional and non-lesional skin of 85 patients (GDS4600). Significance was calculated using a one-way ANOVA test. ns = not significant, **** p < 0.0001. (C, D) Gene expression of IL-36 cytokines and Il1rl2 in 7 days IMQ-treated skin (C) or lesions of K14-IL17Aind mice (D). Shown are the relative mRNA levels normalized to β-Actin. Significance was calculated using a 2-tailed Student’s t-test: * p < 0.05, ** p < 0.01, *** < 0.001, ns, not significant. Shown is the mean ± SEM of n = 3 (C) or n = 9-11 (D) animals.
Furthermore, we detected a similar upregulation of the IL-36 signaling pathway in psoriasis-like skin lesions of mice that either develop psoriasis upon IMQ treatment (Figure 1C), or in psoriatic mice that overexpress IL-17A in the epidermis (Figure 1D). Thus, IL-36 signaling is also upregulated in two mouse models of IL-17A-driven psoriasis, which can subsequently be utilized to investigate anti-IL36R antibody treatment in IL-17A-driven psoriasis.
Previously, it was shown that inhibition of IL-36 signaling by treatment with an anti-IL36R antibody or the IL-36RA antagonist partially prevents the development of IMQ- or IL-36-mediated psoriasis in mice (30–32). However, it remains unclear, if IMQ-induced psoriasis can be treated with an anti-IL36R antibody, once the skin inflammation has already developed. To test this, we pre-treated the ears of wildtype mice for 2 days with IMQ-containing Aldara® cream to induce psoriasis-like skin lesions. At this time point, keratinocyte hyperproliferation and gene expression of psoriasis-associated cytokines and chemokines were already detectable (Supplementary Figures 1A, B). Subsequently, from day 3 on, besides the topical IMQ treatment, mice received intraperitoneal (i.p.) injections of either IgG control or anti-IL-36R antibody (Figure 2A), which was able to inhibit IL-36-driven gene expression in keratinocytes in vitro (Supplementary Figure 1C). On day 7, mice were sacrificed and subsequently analyzed. Interestingly, systemic application of an anti-IL36R antibody was not only able to effectively inhibit ear swelling, but also significantly reduced keratinocyte hyperproliferation and infiltration of neutrophils, monocytes, dendritic cells, and T-cells into the ears of IMQ-treated mice (Figures 2B-D). Moreover, blockade of the IL-36 signaling pathway also suppressed systemic inflammation marked by elevated blood counts of neutrophils, monocytes, and dendritic cells in IMQ-treated mice (Supplementary Figure 1D).
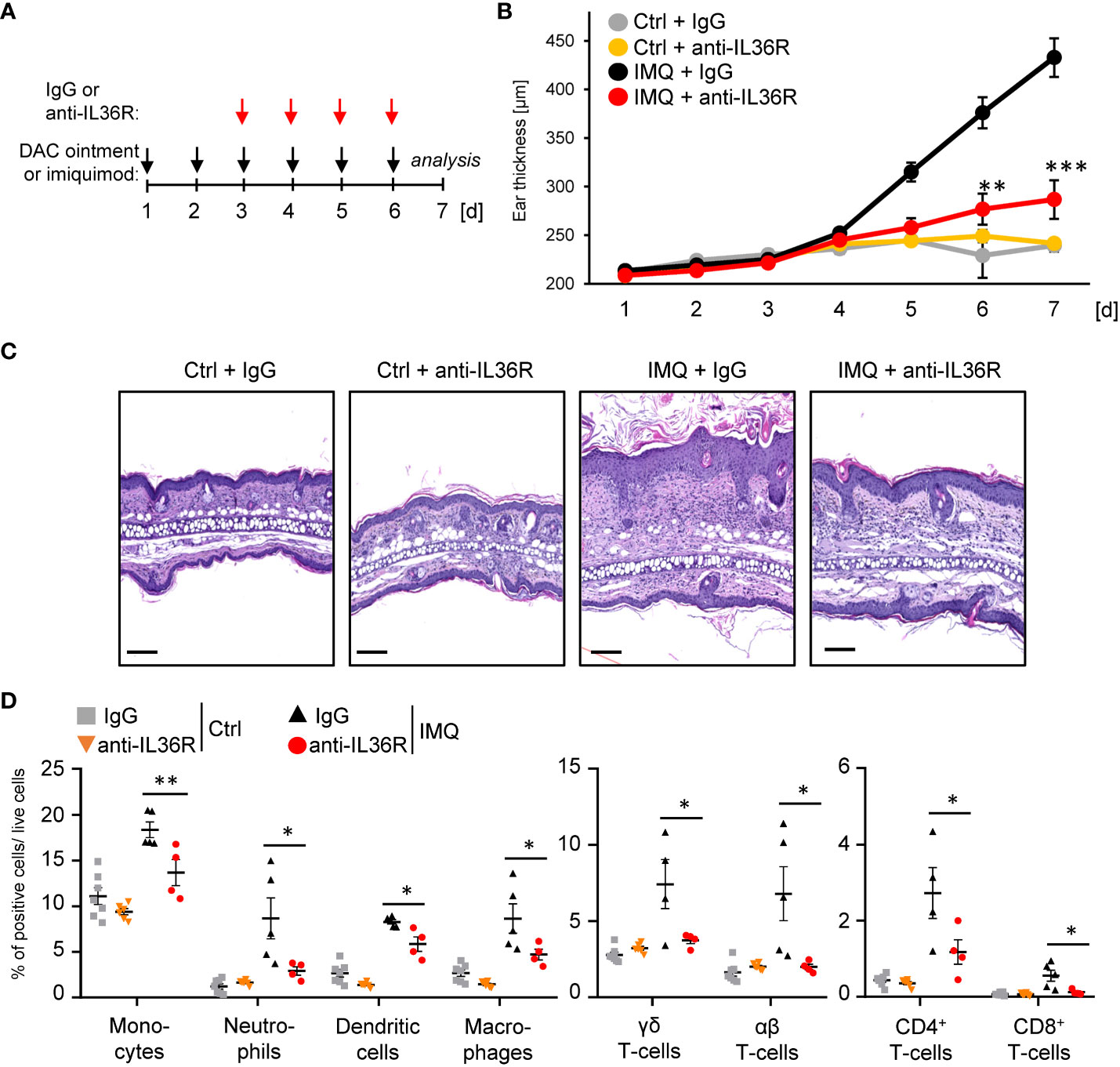
Figure 2 Anti-IL36R treatment suppresses keratinocyte hyperproliferation and infiltration of immune cells in IMQ-induced psoriasis-like skin inflammation. (A) Treatment scheme. Antibody treatment was started 2 days after IMQ treatment. (B) Ear thickness during the course of the experiment (n = 14, except for Ctrl + anti-IL36R n = 6). (C) H&E staining of ears from the treated mice at the endpoint at day 7. Scale: 100 µm. (D) Flow cytometry analysis of skin-infiltrating immune cells from treated animals at day 7 (n = 3 - 5 animals per group). Shown is the relative percentage of positive cells, after pre-gating on viable cells. Significance was calculated using a 2-tailed Student’s t-test: * p < 0.05, ** p < 0.01, *** < 0.001. Shown is the mean ± SEM.
Next, we wanted to understand, which molecular signaling pathways are affected by anti-IL36R treatment in IMQ-treated mice. As expected, suppression of IL 36 signaling did not only affect the expression of mostly keratinocyte-derived chemokines (Ccl20, Ccl2, Cxcl2) and cytokines (Il36g) (Figure 3A) but also effectively reduced the expression of psoriasis-associated cytokines produced by T-cells (Il17a, Il22, Tnf) and myeloid cells (Il1b) (Figure 3B, Supplementary Figures 2A-C). Importantly, the same chemokines and cytokines (e.g. CXCL2, CCL2, IL-1β, and IL-17) were also effectively downregulated on the protein level in IMQ- and anti-IL36R-treated animals compared to IMQ- and IgG-treated mice (Figure 3C, Supplementary Figure 2D). Thus, systemic anti-IL36R treatment was able to broadly inhibit psoriasis-like skin inflammation in IMQ-treated mice.
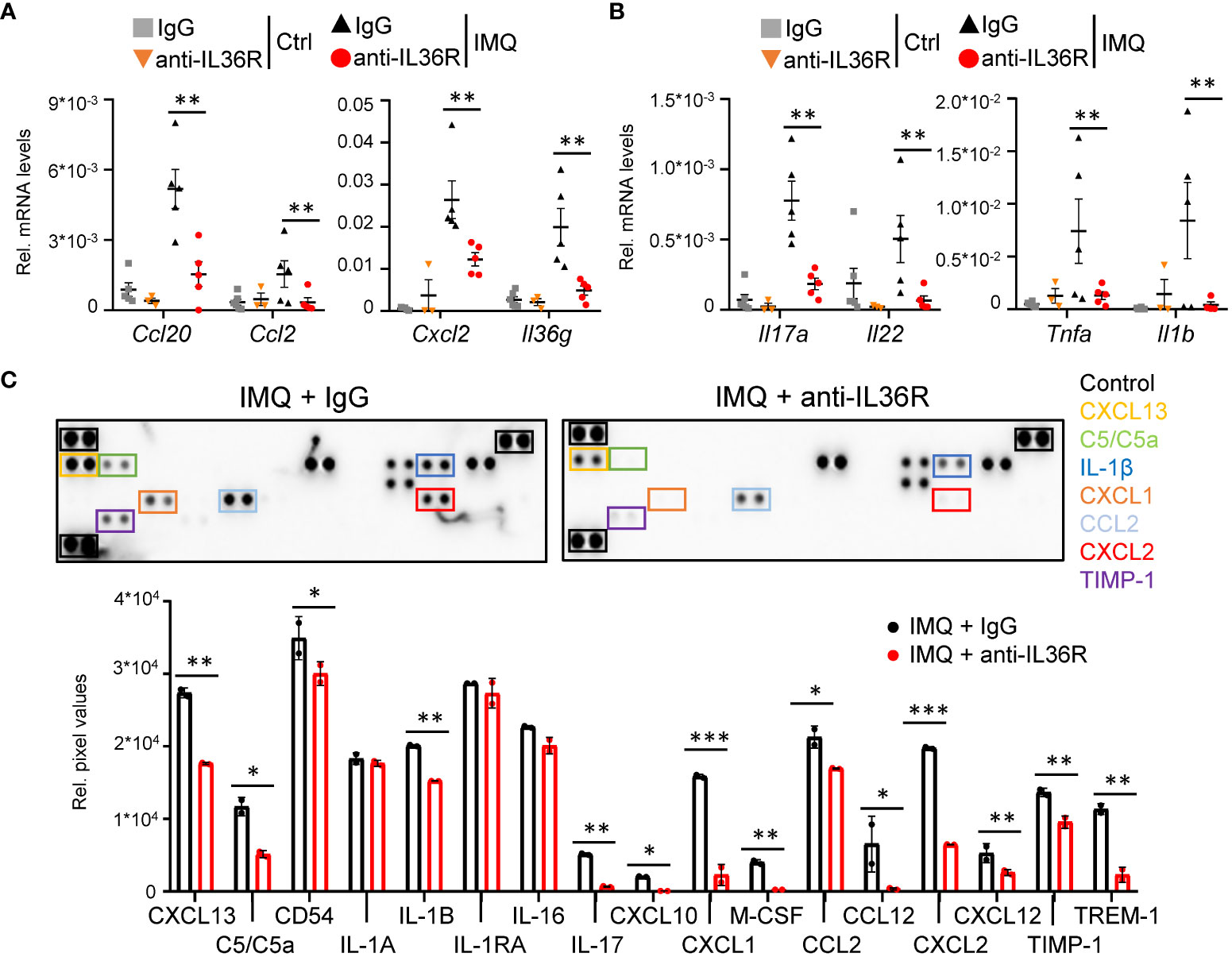
Figure 3 Anti-IL36R treatment suppresses pro-inflammatory cytokine and chemokine expression in IMQ-induced psoriasis-like skin inflammation. (A, B) Gene expression analysis of the skin from Ctrl or IMQ-treated animals that received IgG or anti-IL36R treatment. Shown is the relative gene expression of keratinocyte-derived cytokines and chemokines (A), as well as of psoriasis-associated cytokines deriving from T-cells and myeloid cells (B), normalized to β-Actin. n = 3 - 6. Shown is the mean ± SEM. (C) Protein levels of cytokines and chemokines from ears of IMQ + IgG and IMQ + anti-IL36R-treated animals. Shown are the immunoblot analysis and its evaluation normalized to the reference controls and depicted as relative pixel values. Samples were derived from 3 animals per group and pooled before analysis. Significance was calculated using a 2-tailed Student’s t-test: *p < 0.05, **p < 0.01, and ***p < 0.001.
Based on our findings using the IMQ mouse model, we next investigated the effects of anti-IL36R antibody treatment in a genetic mouse model of IL-17A-driven psoriasis. K14-IL17Aind mice develop psoriasis-like skin lesions due to constitutive, keratinocyte-derived overexpression of IL-17A (33). Of note, we utilized K14-IL17Aind mice harboring a heterozygous deletion of Nfkbiz, a well-known key factor of transcriptional responses in psoriasis (11, 34). Whereas K14-IL17Aind mice harboring wildtype Nfkbiz expression show severe systemic inflammation and skin lesions all over the whole body at young age (33, 35), keratinocyte-specific, heterozygous depletion of Nfkbiz in these mice renders the mice healthy until an age of 10–11 weeks, when the mice start to develop psoriasis-like skin lesions at the ears and back (34). Due to the temporal appearance and extent of psoriatic skin disease in these mice, we therefore decided to use this mouse model. Mice received i.p. injections with IgG control or anti-IL36R antibody once a week, starting at an age of 8 weeks (Figure 4A). Development of psoriasis was scored using a cumulative Psoriasis Area and Severity Index (PASI) score describing the lesion size and the severity of scaling and erythema at the sites of the lesions (34). At around weeks 15-20, when at least one mouse of a treatment group reached a PASI score of 3-4, all mice were sacrificed and analyzed. Although IgG- or anti-IL36R antibody-treated K14-IL17Aind mice overexpressed comparable levels of IL-17A (Supplementary Figure 3A) and showed comparable body weights (Figure 4B), blockade of the IL-36 signaling pathway was sufficient to partially inhibit the development of psoriatic lesions in these mice (Figures 4C, D). Likewise, keratinocyte hyperproliferation (Figure 4E) and the relative infiltration of neutrophils and monocytes into the skin of K14-IL17Aind mice were also reduced in mice that received anti-IL-36R antibody injections (Figure 4F).
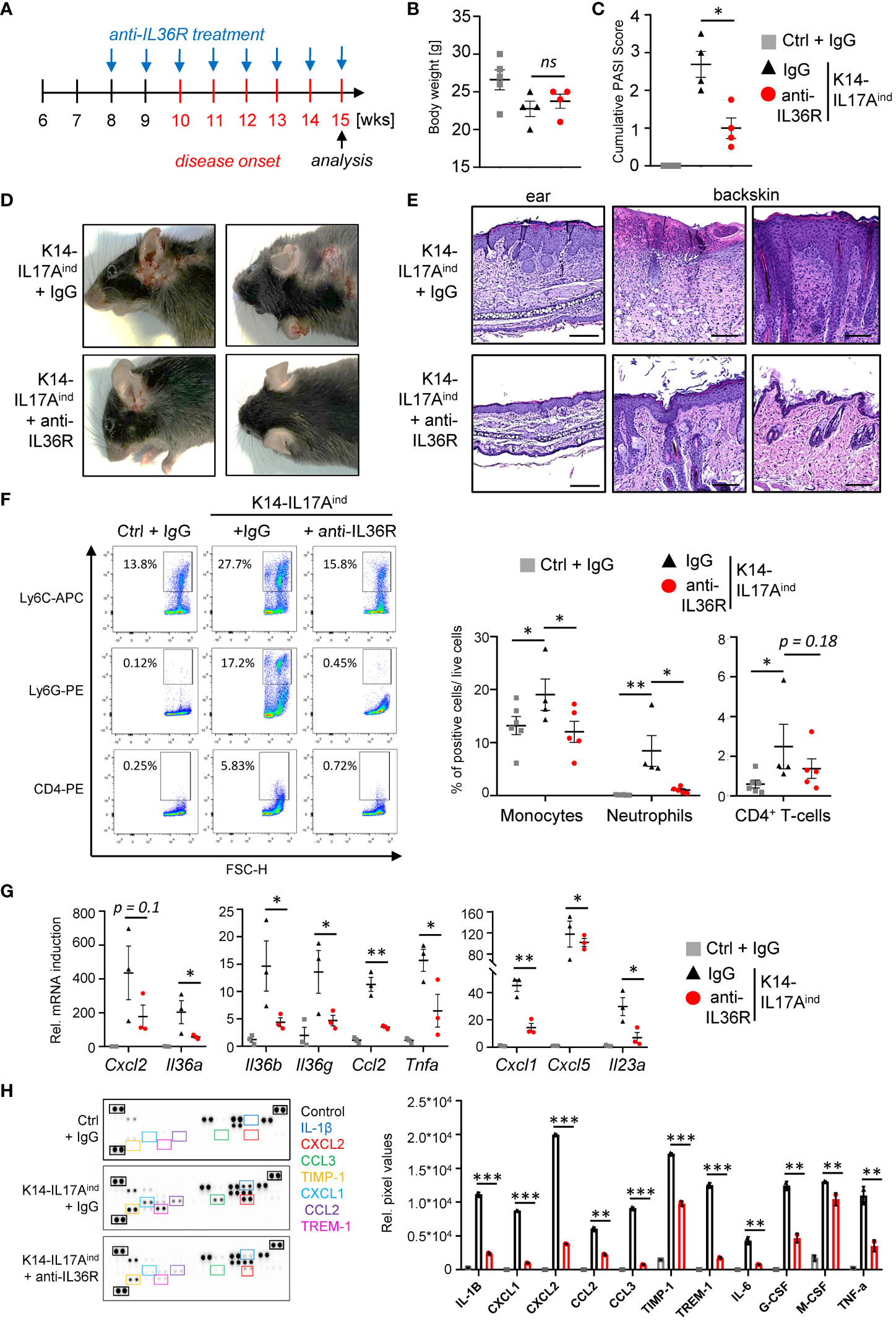
Figure 4 Anti-IL36R treatment suppresses skin lesion development and immune cell infiltration in IL-17A-induced psoriasis in vivo. (A) Treatment scheme. (A, C) Body Weight (B) and PASI Score (C) of the animals at the endpoint (15–20 weeks old). (D) Pictures of the animals at endpoint. (E) H&E staining of the ears and skin lesions at the endpoint. Scale: 100 µm (ears) and 200 µm (back skin). (F) Flow cytometry analysis of skin-infiltrating immune cells from treated animals at endpoint (n = 4 - 6 animals per group). Shown is the relative percentage of positive cells, after pre-gating on viable cells. (G) Gene expression analysis of the skin of IgG- or anti-IL36R-treated K14-IL17Aind mice. Relative gene expression was normalized to Rpl37A. Shown is the gene expression from one treatment group. (H) Protein levels from lesional skin of IgG- and anti-IL36R-treated K14-IL17Aind mice. n = 3 animals per group. Significance was calculated using a 2-tailed Student’s t-test: * p < 0.05, ** p < 0.01, *** < 0.001, ns, not significant. Shown is the mean ± SEM.
Molecular analysis of the skin of IgG- and anti-IL36R-treated K14-IL17Aind mice revealed effective suppression of psoriasis-associated cytokines (Il36a, Il36g, Il36b, Il23a, and Tnf), chemokines (Cxcl1, Cxcl5, and Ccl2) (Figure 4G) and antimicrobial-peptides (such as Defb4) on mRNA level (Supplementary Figure 3B). Moreover, several cytokines and chemokines, such as IL-1β, IL-6, TNF, CXCL2, and CCL2 were efficiently suppressed on protein level in the skin of anti-IL36R-treated K14-IL17Aind mice, compared to the IgG-treated control mice (Figure 4H). Thus, application of anti-IL36R antibodies effectively suppressed the development of IL-17A-driven psoriasis-like skin inflammation in these mice.
K14-IL17Aind mice do not only suffer from psoriasis-like skin lesions but also develop psoriasis-associated systemic inflammation, similar to psoriasis-related comorbidities in humans. Therefore, we investigated if the application of an anti-IL36R antibody was also able to inhibit the development of systemic inflammation in K14-IL17Aind mice. The first signs of systemic inflammation can be detected by increased granulopoiesis and myelopoiesis in the bone marrow. Next, these myeloid cells are released into the bloodstream, become overactive, and elicit more reactive oxygen species (ROS), which subsequently leads to vessel damage and the development of cardiovascular disease in the long run (35). Subsequently, systemic inflammation in several organs develops, which is marked by an increased expression of several cytokines and chemokines. This process may be due to, or triggered by, this systemic increase in myeloid cells and their activation and contributes to the development of psoriasis-associated comorbidities. Congruent with the less severe psoriatic skin lesions in anti-IL36R-treated K14-IL17Aind mice, we also detected significantly fewer monocytes and neutrophils being generated in the bone marrow (Figure 5A). Reactive oxygen species in the blood were found to be significantly increased in psoriatic mice as compared to healthy controls. Upon anti-IL36R treatment, the increase in ROS formation was still observed but was no longer significant (Figure 5B). Furthermore, whereas IgG-treated K14-IL17Aind mice showed signs of inflammation in the spleen and colon, as well as an increased expression of pro-inflammatory cytokines (such as Csf3, Il1b, Il6, and Tnf), treatment with an anti-IL36R antibody remarkedly inhibited systemic signs of inflammation in the organs of these mice (Figures 5C-E). This suggests, that in addition to suppression of the psoriatic skin disease, anti-IL36R antibodies could also inhibit psoriasis-associated systemic inflammation in K14-IL17Aind mice.
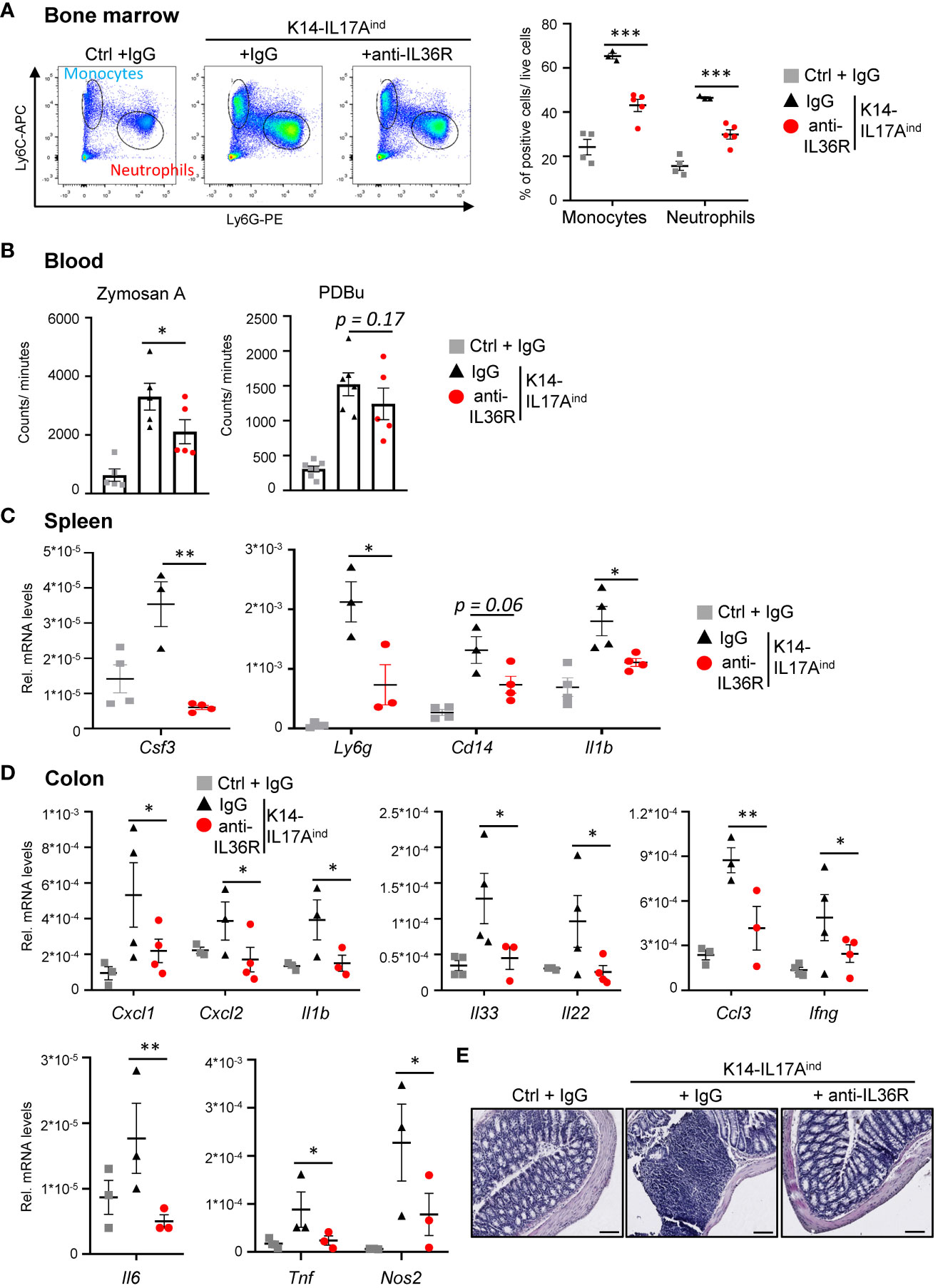
Figure 5 Anti-IL36R treatment inhibits systemic inflammation in K14-IL17Aind mice. (A) Flow cytometry analysis of the bone marrow from Ctrl mice or K14-IL17Aind mice that either received IgG or anti-IL36R treatment (n = 2-3 animals per group). The relative percentage of positive cells is shown after pre-gating on viable cells. (B) Oxidative burst was measured in venous blood of control or K14-IL17Aind mice by analyzing the formation of reactive oxygen and nitrogen species. Blood cells were either restimulated with phorbol-12,13-dibutyrate (PDBu) or with Zymosan A. After addition of the 8-amino-5-chloro-7-phenylpyridol-(3,4-d)pyridazine-1,4-(2H,3H)-dione sodium salt (L-012), the oxidative burst was detected by the created chemiluminescence using a SparkTM Multimode Microplate Reader. n = 4-5. C + D. Gene expression analysis of the spleen (C) and colon (D) from treated mice. Shown is the relative gene expression normalized to β-Actin (C) or Rpl37A (D) for n = 3-4 animals per group. Significance was calculated using a 2-tailed Student’s t-test: * p < 0.05, ** p < 0.01, *** p< 0.001. Shown is the mean ± SEM. (E) H&E staining of the colon at the endpoint. Scale: 100 µm.
Next, we wanted to investigate if anti-IL36R antibody treatment is also able to inhibit and resolve psoriasis in K14-IL17Aind mice once the psoriatic lesions already developed. For this purpose, we waited until approximately week 12, when the mice were scored with a PASI score of 1-2 and already displayed psoriatic lesions at the head and neck. Subsequently, we treated the mice two times a week for 2-3 weeks with either IgG as control or anti-IL36R antibody, until at least one animal of the control or experimental group reached a PASI score of approximately 4. At this time point (around 14-15 weeks of age), all mice were sacrificed and analyzed (Figure 6A). Interestingly, this short-term treatment with anti-IL36R antibody was sufficient to significantly diminish the lesion size, keratinocyte hyperproliferation, and infiltration of neutrophils, monocytes, and dendritic cells into the psoriatic skin lesions of K14-IL17Aind mice (Figures 6B-E). This was due to a significant repression of psoriasis-associated cytokine and chemokine expression in the previously affected lesional skin of anti-IL36R-treated animals, compared to IgG-treated control animals (Figure 6F), and to non-lesional skin samples of the same, anti-IL36R-treated mice (Supplementary Figure 4A). Furthermore, whereas IgG-treated K14-IL17Aind mice also displayed psoriasis-associated systemic inflammation, marked by granulopoiesis, myelopoiesis, and an elevated expression of inflammation markers in the spleen, mice that received a short-term treatment with anti-IL36R antibodies displayed a reduction in these markers of systemic inflammation (Supplementary Figures 4B, C).
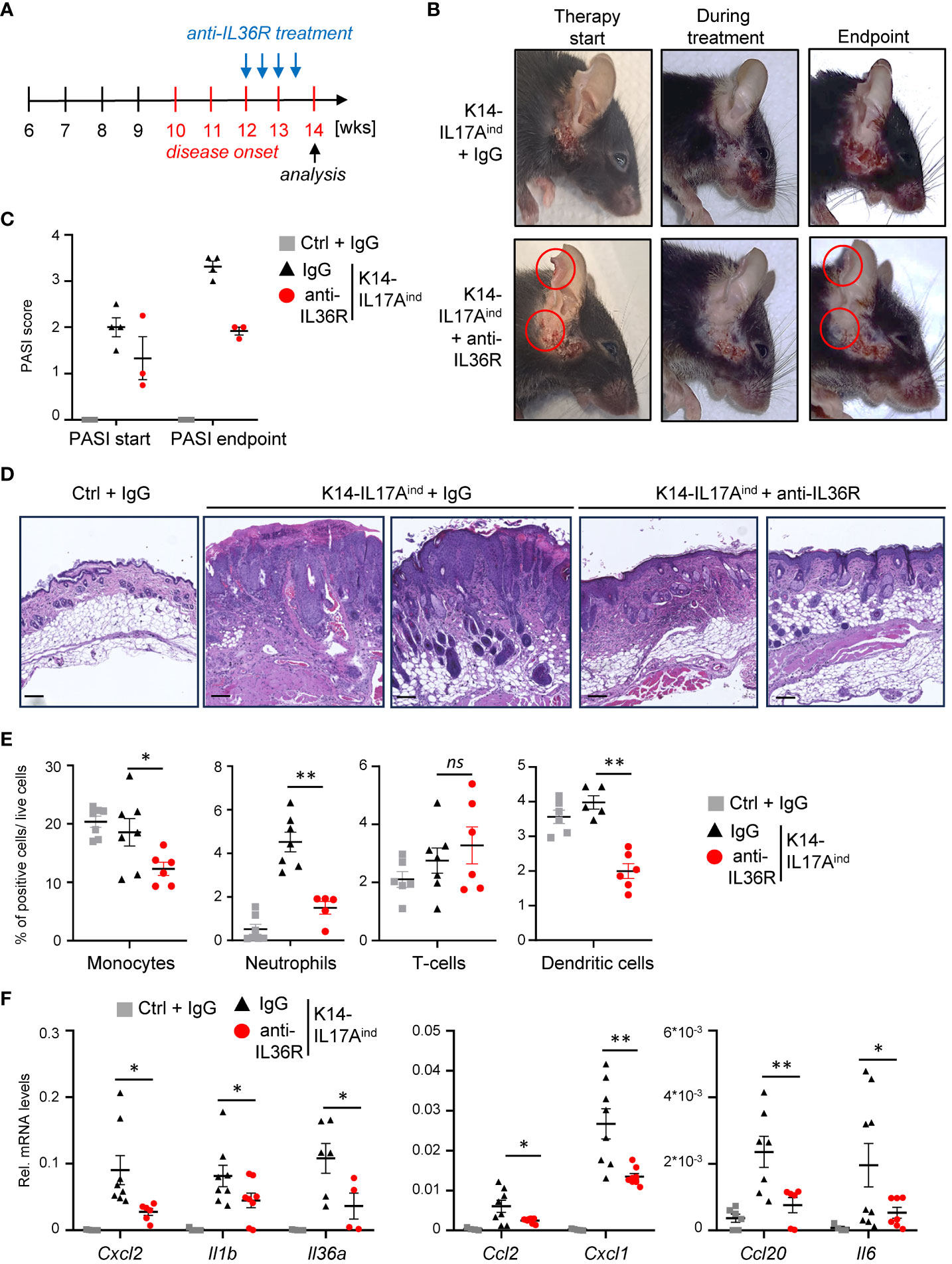
Figure 6 Acute suppression of psoriasis-associated inflammation in K14-IL17Aind mice upon anti-IL36R therapy. (A) Treatment scheme. (B) Pictures of an IgG or anti-IL36R-treated animal at the starting point of the treatment, during the treatment and at the endpoint. (C) PASI Score of the mice before treatment and at the endpoint. (D) H&E staining of the affected skin areas at the endpoint. Scale: 100 µm. (E) Flow cytometry analysis of skin-infiltrating immune cells from treated animals at endpoint (n = 3 - 5 animals per group). Shown is the relative percentage of positive cells, after pre-gating on viable cells. (F) Gene expression of the skin from IgG or anti-IL36R-treated K14-IL17Aind mice at the endpoint. Relative gene expression was normalized to Rpl37A. Significance was calculated using a 2-tailed Student’s t-test: * p < 0.05, ** p < 0.01, ns, not significant. Shown is the mean ± SEM.
In conclusion, anti-IL36R treatment is not only able to prevent the development of IL17A-driven psoriasis but also to inhibit and partially resolve psoriatic lesions and associated psoriasis-associated systemic inflammation.
An antibody targeting the IL-36 receptor has recently been approved for the treatment of GPP, a subtype of psoriasis (19). While this treatment is very effective in GPP, it remains an open question if an anti-IL36R antibody might also be beneficial for the treatment of IL-17A-dependent plaque psoriasis. Moreover, inhibition of IL-36 signaling in psoriasis might also be advantageous for the effective treatment of psoriasis-associated comorbidities. Here, we investigated the consequences of an anti-IL36R treatment in two mouse models of IL-17A-driven psoriasis: in IMQ-treated mice and in mice that overexpress IL-17A in the epidermis, thus developing IL-17A-depending psoriatic lesions. We found that systemic anti-IL36R treatment, given after the development of IMQ-induced psoriasis-like skin lesions, is able to inhibit keratinocyte hyperproliferation and immune cell infiltration into IMQ-treated skin. Furthermore, it was sufficient to repress skin-derived, pro-inflammatory cytokine and chemokine expression. Our findings are in agreement with previous studies, in which a global and keratinocyte-specific knockout of the IL-36 receptor (21, 23), or anti-IL-36R treatment (30, 36) was sufficient to effectively prevent IMQ-induced psoriasis-like skin inflammation. Limitations of the previous studies were, on the one hand, the experimental setup, as well as secondly, the IMQ model itself. In the previous studies, IL36R knockout or inhibition of IL-36 signaling was already present before the application of IMQ was started, thereby only showing that blockade of the IL-36 pathway can prevent IMQ-mediated skin inflammation (21, 23, 30, 36). We here add to these data by showing that anti-IL36R antibody treatment is also able to attenuate already fully established IMQ-mediated skin inflammation. Thus, anti-IL36R treatment efficiently blocks IMQ-mediated psoriasis-like skin disease in mice, similar to what has previously been shown for neutralizing antibodies against IL-17 or IL-23 (37, 38).
The other limitation of the previous studies derives from the IMQ mouse model itself. While in patients, triggers for the induction of psoriatic lesions are multifactorial but seem to derive from “stressed” keratinocytes (39), IMQ constitutes a TLR7/8 agonist, that activates skin-resident immune cells in the first instance. Thus, although IMQ is still the standard model for testing anti-psoriatic drugs, it remains to be seen if it is the best pre-clinical model available (40). Furthermore, it has recently been shown that IMQ fails to induce a pure psoriasis response in humans but instead causes an IL-17-dominated contact dermatitis (41). Therefore, we decided to additionally investigate the consequences of an anti-IL36R treatment in a genetically driven psoriasis mouse model. K14-IL17Aind Nfkbiz het mice (referred to as K14-IL17Aind) which chronically overexpress IL-17A in the epidermal compartment, in combination with a heterozygous depletion of Nfkbiz in keratinocytes. While these mice look completely healthy at birth, they develop psoriatic skin lesions at the head and back in adulthood (34). The disease slowly progresses, and subsequently also triggers the development of psoriasis-associated comorbidities, thus much better reflecting the human psoriasis pathology (34, 35). Interestingly, while IgG control-treated K14-IL17Aind mice displayed psoriasis-like skin lesions starting at an age of 11 weeks, mice that were treated with anti-IL36R antibodies in parallel developed significantly fewer and less severe lesions, although exhibiting comparable levels of IL-17A overexpression. Moreover, inhibition of IL-36 signaling was able to effectively inhibit particularly the infiltration of monocytes and neutrophils into the skin lesions, as well as the expression of known psoriasis-associated cytokines and chemokines. Importantly, using this mouse model, we could also show that short term treatment using anti-IL36R antibodies was sufficient to successfully inhibit psoriasis-associated inflammation once it has already been established.
Previously, it was shown that IL-23, IL-17, and IL-36 expression are closely interconnected with each other in keratinocytes and that IL-36, downstream of IL-23 and IL-17, constitutes a critical effector molecule of IL-23 and IL-17 responses in psoriasis (42–44). Thus, IL-17A secretion by T-cells does not only induce the expression of IL-36α and IL-36γ in various cell types but also synergistically induce the expression of various cytokines and chemokines driving the progression of psoriasis (14, 24). Vice versa, IL-36γ expression in psoriatic lesions is one of the earliest genes which become repressed upon treatment with anti-IL17 antibodies (14). In agreement with these findings, we detected a strong downregulation of several IL-23 and IL-17 target genes (such as Il36g, Defb4, or S100a9) in the skin of anti-IL36R-treated mice. These genes have previously been shown to be important markers for effective therapeutic responses in human psoriasis vulgaris patients, treated with anti-IL17 or anti-IL23 antibodies (45, 46). Thus, since anti-IL36R treatment downregulates similar genes as anti-IL17 or anti-IL23 therapy does, blockade of the IL-36 signaling pathway might be also similar effective as other biologicals for the treatment of psoriasis vulgaris patients.
IgG-control-treated K14-IL17Aind mice also suffered from psoriasis-associated systemic inflammation, which is characterized by an elevated granulopoiesis, myelopoiesis, inflamed spleen, and colon. There were no signs of systemic inflammation detectable in the simultaneously aged and anti-IL36R-treated K14-IL17Aind mice. We suggest that these effects derive from the widespread anti-inflammatory effect of anti-IL-36 antibody treatment, as previous studies revealed that not only keratinocytes but also endothelial cells, fibroblasts, T-cells, and dendritic cells respond to IL-36 (13, 16, 47). Our findings have major therapeutic implications, since antibodies against IL36R might not only be important for the direct treatment of the psoriatic skin but also for psoriasis-associated comorbidities, which impact life expectancy. Previous studies showed that IL-36 overexpression contributes to the development of arthritis (4), metabolic disorders (48), cardiovascular diseases (49), and inflammatory bowel disease (50). Future approaches should therefore investigate to what extent IL-36 overactivation contributes to the development of psoriasis-associated comorbidities, and if anti-IL36R antibodies, alone or in combination with other anti-psoriatic drugs, are helpful in the treatment of these comorbidities.
In summary, our pre-clinical study showed that inhibition of the IL-36 signaling pathway constitutes an attractive approach for the treatment of IL-17A-driven psoriasis vulgaris. As it is still not clear which class of neutralizing antibodies are the most efficient therapy for the whole psoriatic disease spectrum and with regard to long-term disease trajectories, anti-IL36R antibodies might constitute an attractive alternative with unique properties targeting both skin and systemic symptoms.
All animal experiments have been approved by the local animal ethics committee (Landesuntersuchungsamt Rheinland-Pfalz). All experiments were conducted in accordance with German law and guidelines for animal care. For the anti-IL36R treatment of IMQ-induced psoriasis, both ears of male C57BL/6NCrl wild-type mice (8-10 weeks old, purchased from Charles River Laboratories) were treated for 6 consecutive days with Aldara® cream (5% IMQ, 3M Pharmaceuticals), while control mice received diluted basis cream (DAC). After 2 days of treatment with DAC or IMQ, mice additionally received intraperitoneal (i.p.) injections of either 750 µg IgG or anti-IL36R antibodies (both from Boehringer Ingelheim) from the third day of IMQ treatment onwards. Ear thickness was daily measured using a precise caliper (IP67/C110T, Kroeplin). On day 7, mice were sacrificed and analyzed.
To study the effect of anti-IL36R treatment in IL-17A-driven psoriasis, both female and male K14-IL17Aind Nfkbizfl/+ mice were utilized (referred to as K14-IL17Aind), which develop skin lesions starting at an age around 11 weeks old. These mice were generated by crossing homozygous KRT14-Cre mice (B6N.Cg-Tg(KRT14-cre)1Amc/J) from Jackson Laboratories (Stock 018964) to homozygous IL17Aind Nfkbizfl/fl (B6.Gt(ROSA)26Sortm3(CAG-Il17a)Awai; Nfkbiztm1.1Muta/Tarc) mice. Starting at an age of 8 weeks, K14-IL17Aind mice were intraperitoneally injected with 500 µg IgG or anti-IL36R antibody once a week. As a control, IL17Aind Nfkbizfl/+ mice (referred to as Ctrl) of the same age and gender were treated with IgG antibodies. Mice were scored with the cumulative PASI Score describing the degree of scaling, erythema, and percentage of the affected area (0 = no lesions, 1 = very mild, 2 = mild, 3 = intermediate, 4 = severe, and 5 = very severe). When at least one animal of the treatment group reached a PASI score of 3 - 5, at an age of 15 - 20 weeks, all mice of the same treatment group were sacrificed and analyzed in parallel. For the therapeutic approach, control and K14-IL17Aind mice were treated with 500 µg IgG or anti-IL36R antibody twice a week for 3 to 4 weeks, starting as soon as a PASI score of 1-2 was reached. Sacrification and analysis were carried out after reaching the endpoint as described above.
8- to 12-weeks-old wildtype mice were sacrificed and tails were wiped with 70% isopropanol. In order to isolate mKC, the skin was separated from the muscle and incubated overnight at 4°C in equal amounts of basal keratinocyte-SFM medium (Cat.17005042 Gibco, Thermo Fisher Scientific) and 5 U/mL dispase I solution (Cat.07913, StemCell). The next day, the epidermis was separated from the dermis and incubated for 15 min with 0.05% trypsin-EDTA solution (Cat.25300-054, Thermo Fisher Scientific) at room temperature (RT). Stopping the reaction with DMEM medium supplemented with 10% FCS, cells were carefully washed out and transferred to a collection tube using a 100 µm cell strainer. After centrifugation (1000 rpm, 5 min), the pellet was resuspended in complete keratinocyte-SFM medium (containing BPE and EGF), supplemented with 0.05 M CaCl2. Cells were seeded on collagen type I-coated 6-well plates (Cat.A1064401, Thermo Fisher Scientific). When the plates reached a confluency of 70-80% confluency, cells were starved overnight (SFM Medium without CaCl2 and supplements) and subsequently treated with 100 ng/mL IL-36α (Cat.555902, Biolegend) for 1.5 h, in the presence of either 50 µg/mL IgG or anti-IL36R antibody.
Total RNA isolation and qPCR analysis were performed as described before (51). Briefly, 1 or 4 µg total RNA (from cell culture samples or tissue) was reverse transcribed into cDNA. Gene expression was quantified by real-time PCR (CFX384, BioRad) using self-designed primers (Supplementary Table 1) and GreenMasterMix (Cat.M3023, Genaxxon) using the following PCR protocol: 15 min initial denaturation at 95°C, 40 cycles of 95°C for 15 s and 60°C for 45 s. Relative mRNA levels were normalized to the reference gene ActB (keratinocytes, skin, spleen), Rpl37a (colon) using the 2-ΔCt method.
Quantitative levels of murine IL-17A were measured in skin and serum samples using the commercial ELISA MAX™ Deluxe Set Mouse IL-17A kit (Cat.432504, Biolegend). Measurement was performed using a microplate reader (Hidex Sense, Hidex) and concentration was determined based on four parameter logistic (4PL) regression.
Secretion of cytokines was analyzed using the Proteome Profiler Mouse Cytokine Array Kit Panel A (Cat.ARY006, R&D systems) according to the manufacturer′s instructions. Skin samples were homogenized with C-tubes (Miltenyi) using the gentleMACS™ dissociator with protein lysis buffer containing 20 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM Na2EDTA, 1 mM EGTA, 1 mM β-glycerophosphate, 2 M urea and 1x protease inhibitor cocktail (Cat.4693124001, Sigma). After 10 min incubation on ice, samples were briefly sonicated (Bioruptor, Diagenode), centrifuged (1000 rpm, 5 min, 4°C), and the supernatant transferred into a new tube. Subsequently, the protein concentration was determined using the Qubit Protein Assay (Cat.Q33212, Thermo Fisher Scientific). Prior to analysis, input lysates were normalized to equal β-Actin levels by immunoblot analysis using an anti-β-Actin antibody (Cat.3700, Cell signaling). For each treatment group, equal amounts of samples deriving from 3 different animals were pooled and analyzed together. Membranes were developed using the Fusion FX imager (Peqlab) and relative cytokine expression levels were calculated as relative pixel values, normalized to the reference spots, using the dot blot analyzer (ImageJ).
The venous blood of the mice was taken by heart puncture and directly anti-coagulated with sodium citrate (0.1% v/v) to analyze the formation of reactive oxygen and nitrogen species (RONS) as previously described (52, 53). The blood was either incubated for 15 min with phorbol-12,13-dibutyrate (PDBu) or over 40 min with Zymosan A to induce oxidative burst of the whole blood. After the addition of the chemiluminescence dye 8-amino-5-chloro-7-phenylpyridol-(3,4-d)pyridazine-1,4-(2H,3H)-dione sodium salt (L-012), the created chemiluminescence directly correlating with the underlying ROS formation was measured, using a Spark™ Multimode Microplate Reader (Tecan Trading AG, Männedorf, Switzerland).
For analysis of infiltrating immune cell populations in skin samples, back skin or whole ears of approx. 1.5 x 1.5 cm was chopped and digested for 1.5 h at 37°C while shaking using 1 mL digestion mix containing 0.25 mg/mL Liberase TM (Cat.5401127001 Sigma), 100 µg/mL DNase I (Cat.11284932001 diluted in HBSS, Sigma), 0.5 mM CaCl2 and basal RPMI medium without supplements. Afterwards, skin samples were homogenized with C-tubes (Miltenyi) using the gentleMACS™ dissociator. The suspension was transferred to a 50 mL centrifuge tube using a 100 µm strainer to obtain a single-cell suspension and the digestion was stopped by adding 20 mL DMEM medium, supplemented with 10% FCS. After centrifugation (10 min at 450 x g) the cell pellet was washed in a FACS buffer containing 5% FCS and 1 mM EDTA. For preparation of bone marrow samples, cells were extracted from femur and tibia and washed in FACS buffer. Subsequently, prepared single-cell suspensions of skin and bone marrow were treated with TruStain FcX anti-mouse (Cat.101320, Biolegend) for 10 min on ice, washed and further surface-stained for 30 min on ice in the dark using the following antibodies: anti-CD3-APC (Cat.100236, clone 17A2, BioLegend), anti-CD4-PE (Cat.100408, clone GK1.5, BioLegend), anti-CD8a-APC (Cat.100712, clone 53-6.7, BioLegend), anti-Ly6G-PE (Cat.127607, clone 1A8, BioLegend), anti-Ly6C-APC (Cat.128016, clone HK1.4, BioLegend), anti-CD11c-APC (Cat.117310, clone N418, BioLegend), anti-F4/80-PE (Cat.123110, clone BM8, BioLegend), anti-αβTCR-PE (Cat.22155214, clone H57-597, Immunotools) and anti-γδ-APC (Cat.118115, clone GL3, BioLegend). Data was acquired on a LSR II flow cytometer (Becton Dickson) and gates were set based on the respective isotype controls (Cat.400612, APC Rat IgG2b, κ, BioLegend; Cat.400655, Brilliant Violet 421 Rat IgG2b, κ, BioLegend; Cat.400363, PE Rat IgG2b, κ, BioLegend; Cat.550085, PE Hamster IgG2, κ, BD). For live/dead staining the samples were co-stained with DAPI (Cat.422801, BioLegend) or Zombie Green fixable viability kit (Cat.423111, Biolegend). Cell populations were gated on living, single cells using FlowJo (Tree Star Inc.) software.
Tissues (ear skin, back skin, colon) were fixed overnight in 4% formaldehyde solution and washed in PBS. After fixation and paraffin-embedding, 5 μm (skin) or 7 μm (colon) sections were prepared. H&E staining and detection of phospho-RB (Cat.8516, Cell Signaling) was done as previously described (51).
Obtained results are either represented as the mean ± standard deviation (SD; in vitro stimulation) or mean ± SEM (standard error of the mean; n= animal numbers). Significance is depicted as asterisks (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P< 0.0001) and was calculated based on 2-tailed Student’s t-test. All data plots were generated using GraphPad Prism software.
Publicly available datasets were analyzed in this study. This data can be found here: GDS4602 and GDS4600 (27).
The animal study was approved by Landesuntersuchungsamt Rheinland-Pfalz, Germany. The study was conducted in accordance with the local legislation and institutional requirements.
BF: Formal analysis, Investigation, Project administration, Validation, Visualization, Writing – review & editing. TK: Formal analysis, Investigation, Writing – review & editing. AK: Formal analysis, Investigation, Writing – review & editing. JR: Formal analysis, Investigation, Visualization, Writing – review & editing. AW: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. MW: Investigation, Writing – review & editing. SK: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. SMK: Writing – review & editing. DK: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from Boehringer Ingelheim (BI). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. The study was further supported by grants from the Else-Kröner-Fresenius-Stiftung (DK), the Peter-Hans Hofschneider Foundation (DK), and the Deutsche Forschungsgemeinschaft TRR156/3 (project number 246807620) (DK, SK, AW) and TRR355/1 (project number 490846870) (DK, AW). Moreover SK received funding by the Federal Ministry of Education and Research (BMBF 01EO1503) and by the Deutsche Herzstiftung and by the DZHK Excellence Program.
We thank Boehringer Ingelheim for donating us anti-IL36R and IgG antibodies. Moreover, we thank Katharina Perius, Annika Lehmann, Dagmar Löck, Claudia Braun and Bonny Adami for their experimental support.
SMK is employed by Boehringer Ingelheim Pharma GmbH & Co. KG. SK declares having received consultancy honorary from Almirall and lecture honoraria from Janssen-Cilag, which both was not associated with this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1256133/full#supplementary-material
1. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
2. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. JAMA (2020) 323(19):1945–60. doi: 10.1001/jama.2020.4006
3. D'Erme AM, Wilsmann-Theis D, Wagenpfeil J, Holzel M, Ferring-Schmitt S, Sternberg S, et al. IL-36 gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol (2015) 135(4):1025–32. doi: 10.1038/jid.2014.532
4. Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, et al. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and crohn's disease. Clin Exp Immunol (2016) 184(2):159–73. doi: 10.1111/cei.12761
5. Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem (2004) 279(14):13677–88. doi: 10.1074/jbc.M400117200
6. Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem (2011) 286(49):42594–602. doi: 10.1074/jbc.M111.267922
7. Berekmeri A, Latzko A, Alase A, Macleod T, Ainscough JS, Laws P, et al. Detection of IL-36gamma through noninvasive tape stripping reliably discriminates psoriasis from atopic eczema. J Allergy Clin Immunol (2018) 142(3):988–91 e4. doi: 10.1016/j.jaci.2018.04.031
8. Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep (2016) 14(4):708–22. doi: 10.1016/j.celrep.2015.12.072
9. Macleod T, Ainscough JS, Hesse C, Konzok S, Braun A, Buhl AL, et al. The proinflammatory cytokine IL-36gamma is a global discriminator of harmless microbes and invasive pathogens within epithelial tissues. Cell Rep (2020) 33(11):108515. doi: 10.1016/j.celrep.2020.108515
10. Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev (2018) 281(1):169–78. doi: 10.1111/imr.12610
11. Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, et al. IkappaBzeta is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci USA (2018) 115(40):10088–93. doi: 10.1073/pnas.1801377115
12. Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, et al. IL-36R ligands are potent regulators of dendritic and t cells. Blood (2011) 118(22):5813–23. doi: 10.1182/blood-2011-05-356873
13. Bridgewood C, Stacey M, Alase A, Lagos D, Graham A, Wittmann M. IL-36gamma has proinflammatory effects on human endothelial cells. Exp Dermatol (2017) 26(5):402–8. doi: 10.1111/exd.13228
14. Mercurio L, Failla CM, Capriotti L, Scarponi C, Facchiano F, Morelli M, et al. Interleukin (IL)-17/IL-36 axis participates to the crosstalk between endothelial cells and keratinocytes during inflammatory skin responses. PloS One (2020) 15(4):e0222969. doi: 10.1371/journal.pone.0222969
15. Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E, et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ t cells. Blood (2012) 120(17):3478–87. doi: 10.1182/blood-2012-06-439026
16. Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol (2014) 192(12):6053–61. doi: 10.4049/jimmunol.1301481
17. Cho WJ, Elbasiony E, Singh A, Mittal SK, Chauhan SK. IL-36gamma augments ocular angiogenesis by promoting the vascular endothelial growth factor-vascular endothelial growth factor receptor axis. Am J Pathol (2023) 193(11):1740–9. doi: 10.1016/j.ajpath.2023.01.003
18. Wang X, Zhao X, Feng C, Weinstein A, Xia R, Wen W, et al. IL-36gamma transforms the tumor microenvironment and promotes type 1 lymphocyte-mediated antitumor immune responses. Cancer Cell (2015) 28(3):296–306. doi: 10.1016/j.ccell.2015.07.014
19. Baum P, Visvanathan S, Garcet S, Roy J, Schmid R, Bossert S, et al. Pustular psoriasis: Molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol (2022) 149(4):1402–12. doi: 10.1016/j.jaci.2021.09.035
20. Blair HA. Spesolimab: First approval. Drugs (2022) 82(17):1681–6. doi: 10.1007/s40265-022-01801-4
21. Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. (2012) 122(11):3965–76. doi: 10.1172/JCI63451
22. Hernandez-Santana YE, Leon G, St Leger D, Fallon PG, Walsh PT. Keratinocyte interleukin-36 receptor expression orchestrates psoriasiform inflammation in mice. Life Sci Alliance. (2020) 3(4):e201900586. doi: 10.26508/lsa.201900586
23. Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, et al. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci Alliance. (2020) 3(6):e202000688. doi: 10.26508/lsa.202000688
24. Pfaff CM, Marquardt Y, Fietkau K, Baron JM, Luscher B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci Rep (2017) 7(1):15631. doi: 10.1038/s41598-017-15892-7
25. Miura S, Garcet S, Salud-Gnilo C, Gonzalez J, Li X, Murai-Yamamura M, et al. IL-36 and IL-17A cooperatively induce a psoriasis-like gene expression response in human keratinocytes. J Invest Dermatol (2021) 141(8):2086–90. doi: 10.1016/j.jid.2021.01.019
26. Bridgewood C, Fearnley GW, Berekmeri A, Laws P, Macleod T, Ponnambalam S, et al. IL-36gamma is a strong inducer of IL-23 in psoriatic cells and activates angiogenesis. Front Immunol (2018) 9:200. doi: 10.3389/fimmu.2018.00200
27. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet (2009) 41(2):199–204. doi: 10.1038/ng.311
28. Correa da Rosa J, Kim J, Tian S, Tomalin LE, Krueger JG, Suarez-Farinas M. Shrinking the psoriasis assessment gap: Early gene-expression profiling accurately predicts response to long-term treatment. J Invest Dermatol (2017) 137(2):305–12. doi: 10.1016/j.jid.2016.09.015
29. Suarez-Farinas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol (2012) 132(11):2552–64. doi: 10.1038/jid.2012.184
30. Ganesan R, Raymond EL, Mennerich D, Woska JR Jr., Caviness G, Grimaldi C, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs (2017) 9(7):1143–54. doi: 10.1080/19420862.2017.1353853
31. Hovhannisyan Z, Liu N, Khalil-Aguero S, Panea C, VanValkenburgh J, Zhang R, et al. Enhanced IL-36R signaling promotes barrier impairment and inflammation in skin and intestine. Sci Immunol (2020) 5(54):eaax1686. doi: 10.1126/sciimmunol.aax1686
32. Todorovic V, Su Z, Putman CB, Kakavas SJ, Salte KM, McDonald HA, et al. Small molecule IL-36gamma antagonist as a novel therapeutic approach for plaque psoriasis. Sci Rep (2019) 9(1):9089. doi: 10.1038/s41598-019-45626-w
33. Croxford AL, Karbach S, Kurschus FC, Wortge S, Nikolaev A, Yogev N, et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J Invest Dermatol (2014) 134(3):728–35. doi: 10.1038/jid.2013.404
34. Lorscheid S, Muller A, Loffler J, Resch C, Bucher P, Kurschus FC, et al. Keratinocyte-derived IkappaBzeta drives psoriasis and associated systemic inflammation. JCI Insight (2019) 4(22):e130835. doi: 10.1172/jci.insight.130835
35. Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol (2014) 34(12):2658–68. doi: 10.1161/ATVBAHA.114.304108
36. Su Z, Paulsboe S, Wetter J, Salte K, Kannan A, Mathew S, et al. IL-36 receptor antagonistic antibodies inhibit inflammatory responses in preclinical models of psoriasiform dermatitis. Exp Dermatol (2019) 28(2):113–20. doi: 10.1111/exd.13841
37. Li Q, Liu W, Gao S, Mao Y, Xin Y. Application of imiquimod-induced murine psoriasis model in evaluating interleukin-17A antagonist. BMC Immunol (2021) 22(1):11. doi: 10.1186/s12865-021-00401-3
38. Takahashi T, Koga Y, Kainoh M. Anti-IL-12/IL-23p40 antibody ameliorates dermatitis and skin barrier dysfunction in mice with imiquimod-induced psoriasis-like dermatitis. Eur J Pharmacol (2018) 828:26–30. doi: 10.1016/j.ejphar.2018.03.018
39. Zhou X, Chen Y, Cui L, Shi Y, Guo C. Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis (2022) 13(1):81. doi: 10.1038/s41419-022-04523-3
40. Hawkes JE, Gudjonsson JE, Ward NL. The snowballing literature on imiquimod-induced skin inflammation in mice: A critical appraisal. J Invest Dermatol (2017) 137(3):546–9. doi: 10.1016/j.jid.2016.10.024
41. Garzorz-Stark N, Lauffer F, Krause L, Thomas J, Atenhan A, Franz R, et al. Toll-like receptor 7/8 agonists stimulate plasmacytoid dendritic cells to initiate T(H)17-deviated acute contact dermatitis in human subjects. J Allergy Clin Immunol (2018) 141(4):1320–33 e11. doi: 10.1016/j.jaci.2017.07.045
42. Miura S, Garcet S, Li X, Cueto I, Salud-Gnilo C, Kunjravia N, et al. Cathelicidin antimicrobial peptide LL37 induces toll-like receptor 8 and amplifies IL-36gamma and IL-17C in human keratinocytes. J Invest Dermatol (2023) 143(5):832–41 e4. doi: 10.1016/j.jid.2022.10.017
43. Park YJ, Kim YH, Lee ES, Kim YC. Comparative analysis of single-cell transcriptome data reveals a novel role of keratinocyte-derived IL-23 in psoriasis. Front Immunol (2022) 13:905239. doi: 10.3389/fimmu.2022.905239
44. Ma F, Plazyo O, Billi AC, Tsoi LC, Xing X, Wasikowski R, et al. Single cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat Commun (2023) 14(1):3455. doi: 10.1038/s41467-023-39020-4
45. Krueger JG, Wharton KA Jr., Schlitt T, Suprun M, Torene RI, Jiang X, et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol (2019) 144(3):750–63. doi: 10.1016/j.jaci.2019.04.029
46. Visvanathan S, Baum P, Vinisko R, Schmid R, Flack M, Lalovic B, et al. Psoriatic skin molecular and histopathologic profiles after treatment with risankizumab versus ustekinumab. J Allergy Clin Immunol (2019) 143(6):2158–69. doi: 10.1016/j.jaci.2018.11.042
47. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut (2017) 66(5):823–38. doi: 10.1136/gutjnl-2015-310374
48. Giannoudaki E, Hernandez-Santana YE, Mulfaul K, Doyle SL, Hams E, Fallon PG, et al. Interleukin-36 cytokines alter the intestinal microbiome and can protect against obesity and metabolic dysfunction. Nat Commun (2019) 10(1):4003. doi: 10.1038/s41467-019-11944-w
49. El-Awaisi J, Kavanagh DP, Rink MR, Weston CJ, Drury NE, Kalia N. Targeting IL-36 improves age-related coronary microcirculatory dysfunction and attenuates myocardial ischemia/reperfusion injury in mice. JCI Insight (2022) 7(5):e155236. doi: 10.1172/jci.insight.155236
50. Leon G, Hernandez Santana YE, Irwin N, Giannoudaki E, O'Neill S, Csizmadia I, et al. IL-36 cytokines imprint a colitogenic phenotype on CD4(+) t helper cells. Mucosal Immunol (2022) 15(3):491–503. doi: 10.1038/s41385-022-00488-w
51. Muller A, Dickmanns A, Resch C, Schakel K, Hailfinger S, Dobbelstein M, et al. The CDK4/6-EZH2 pathway is a potential therapeutic target for psoriasis. J Clin Invest. (2020) 130(11):5765–81. doi: 10.1172/JCI134217
52. Daiber A, August M, Baldus S, Wendt M, Oelze M, Sydow K, et al. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue l-012. Free Radic Biol Med (2004) 36(1):101–11. doi: 10.1016/j.freeradbiomed.2003.10.012
Keywords: anti-IL36R, psoriasis, IL-17A, keratinocytes, imiquimod, systemic inflammation, spesolimab
Citation: Fischer B, Kübelbeck T, Kolb A, Ringen J, Waisman A, Wittmann M, Karbach S, Kölsch SM and Kramer D (2023) IL-17A-driven psoriasis is critically dependent on IL-36 signaling. Front. Immunol. 14:1256133. doi: 10.3389/fimmu.2023.1256133
Received: 10 July 2023; Accepted: 20 November 2023;
Published: 11 December 2023.
Edited by:
Roberto Paganelli, Institute for Advanced Biologic Therapies, ItalyReviewed by:
Cem Gabay, University Hospitals Geneva Medical Center, United StatesCopyright © 2023 Fischer, Kübelbeck, Kolb, Ringen, Waisman, Wittmann, Karbach, Kölsch and Kramer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Kramer, a3JhbWVyZGFAdW5pLW1haW56LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.