 Susan Yung
Susan Yung Tak Mao Chan
Tak Mao Chan- Department of Medicine, School of Clinical Medicine, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
Lupus nephritis (LN) is a common and severe manifestation of systemic lupus erythematosus and an important cause of acute and chronic kidney injury. Early diagnosis of LN and preventing relapses are key to preserving renal reserve. However, due to the complexity and heterogeneity of the disease, clinical management remains challenging. Kidney biopsy remains the gold standard for confirming the diagnosis of LN and subsequent assessment of kidney histopathology, but it is invasive and cannot be repeated frequently. Current clinical indicators of kidney function such as proteinuria and serum creatinine level are non-specific and do not accurately reflect histopathological changes, while anti-dsDNA antibody and C3 levels reflect immunological status but not kidney injury. Identification of novel and specific biomarkers for LN is prerequisite to improve management. Renal function deterioration is associated with changes in the endothelial glycocalyx, a delicate gel-like layer located at the interface between the endothelium and bloodstream. Inflammation induces endothelial cell activation and shedding of glycocalyx constituents into the circulation. This review discusses the potential role of soluble glycocalyx components as biomarkers of active LN, especially in patients in whom conventional serological and biochemical markers do not appear helpful.
1 Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by a breakdown of immune tolerance, autoantibody production and immune-mediated injury to multiple organs. SLE has a wide spectrum of clinical manifestations. Kidney involvement, termed lupus nephritis (LN), affects up to 60% of patients, and is an important cause of morbidity and mortality (1). Improvement in immunosuppressive treatment over the past two decades has improved short-term clinical outcomes. Cardiovascular disease (CVD) has emerged as an important cause of morbidity and mortality during long-term follow-up (2). Lupus patients have increased risk of developing CVD compared to age- and gender-matched controls, and the risk ratio is even greater in premenopausal women with LN compared with age-matched controls (3–5). Increased prevalence of sub-clinical atherosclerosis has also been reported, with up to 40% of patients showing evidence of carotid plaque or increased coronary calcification (6, 7). Factors that contribute to cardiovascular events in lupus patients include not only the traditional Framingham Study risk factors such as advanced age, duration of disease, hypertension, smoking and hypercholesterolemia, but also SLE-related factors such as immune complex-induced endothelial injury, antiphospholipid antibody-induced thrombosis, complement activation, and corticosteroid exposure (3).
Accumulating evidence suggests that vascular inflammation and endothelial cell activation in lupus patients precedes CVD (8). Increased secretion of pro-inflammatory cytokines and chemokines by immune and non-immune cells, and binding of anti-dsDNA antibodies to cell surface cross-reactive antigens on resident renal cells can promote the loss of the protective endothelial glycocalyx and exposes cell adhesion molecules on endothelial cells, which in turn, promotes leukocyte recruitment. Shedding of glycocalyx constituents such as syndecan-1 (also known as CD138), hyaluronan (HA), thrombomodulin and cell adhesion molecules into the circulation may serve as soluble biomarkers of endothelial cell activation and dysfunction in LN. More recently, results from our group and others showed that circulating levels of these glycocalyx components are associated with disease activity in LN patients and may contribute to the pathogenesis of LN as well as CVD. It is pertinent to note that severe forms of LN, namely Class 3 or 4 in the ISN-RPS Classification (9, 10), are characterized by inflammatory cell infiltration in the glomerular capillaries, and it is not uncommon to observe endothelial cell abnormalities at the ultrastructural level under electron microscopy. As a result, abnormalities affecting vascular endothelial cells affect patients with LN at both microvascular and macrovascular levels. This review aims to give a brief overview of the endothelium, mechanisms of endothelial injury in LN, and recent advances in the understanding of endothelial glycocalyx constituents in the pathogenesis of LN, and their potential role as biomarkers for disease activity or complications in clinical management.
2 Endothelium
The vascular endothelium comprises a monolayer of endothelial cells that lines the entire circulatory system and separates the blood from the interstitial compartments. The integrity of the endothelium is dependent on intercellular junction complexes including tight junctions, adherens junctions and gap junctions; and integrins that facilitate the adhesion of endothelial cells to their underlying substratum (11). The endothelium plays critical roles in the regulation of vascular tone, permselectivity, platelet aggregation, blood coagulation, leukocyte trafficking, antigen presentation and lipoprotein catabolism (12, 13). Endothelial cells release numerous vasodilatory and vasoconstrictive molecules to maintain vascular homeostasis (11). Nitric oxide (NO) is an important vasoactive compound synthesized by endothelial cells through the conversion of L-arginine by nitric oxide synthase (NOS). NO maintains blood flow by regulating vasodilation, inhibits platelet aggregation and leukocyte adhesion, and prevents smooth muscle cell proliferation. The luminal surface of endothelial cells is lined by a protective glycocalyx which regulates many functions of blood vessels (Table 1), for example, thrombomodulin and heparan sulphate proteoglycans are key constituents of the glycocalyx and both exhibit antithrombotic actions.
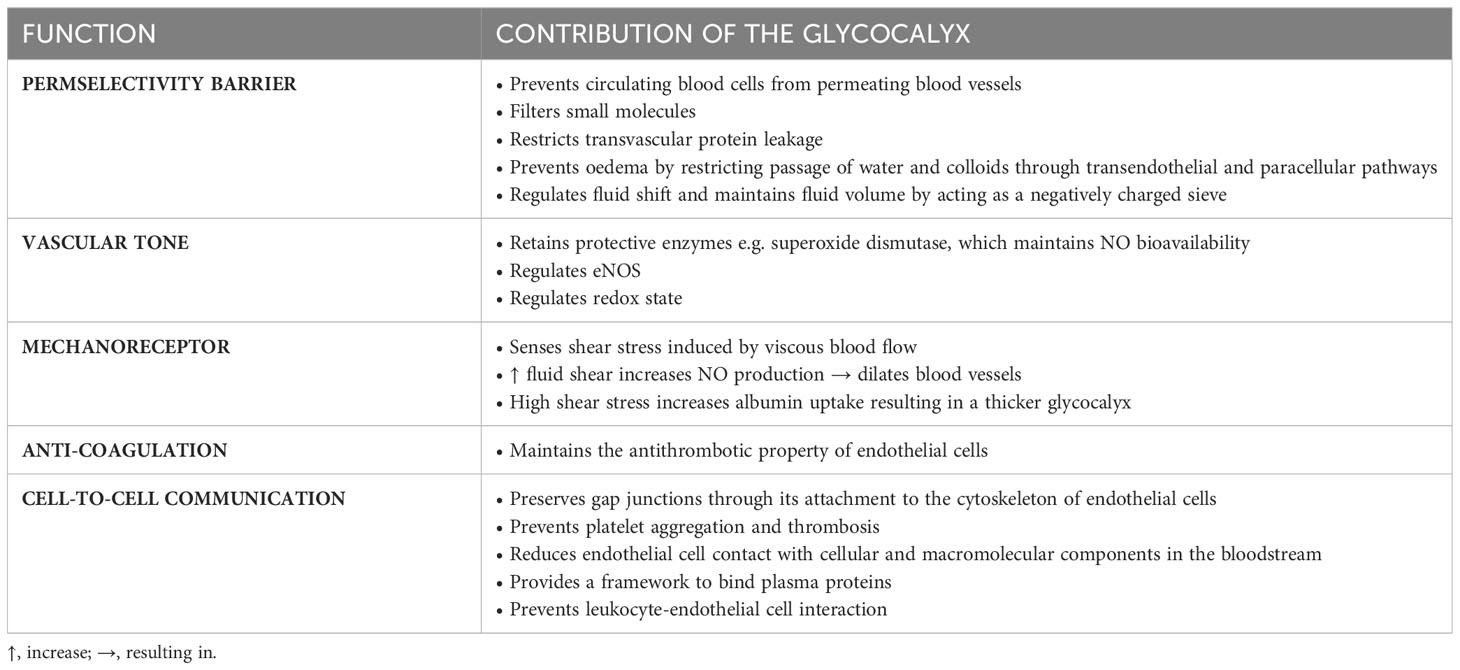
Table 1 Functions of the glycocalyx.
The endothelium also maintains the normal function of the kidney, a highly vascularized organ (14). The renal artery and its microvasculature provides blood to the kidney, delivers oxygen and nutrients to renal cells, and maintains the integrity of the endothelial monolayer through the expression of numerous proteins including interjunctional protein complexes, vascular endothelial cadherin, PECAM-1, VEGF/VEGFR complex, Tie2 and its ligands angiopoietin-1 (Ang1) and Ang2 (15). The microvasculature in the kidney cortex comprises afferent arterioles that direct blood into the glomerulus and efferent arterioles that transport blood from the glomerulus into peritubular capillaries surrounding tubules where they deliver blood into postcapillary venules. Renal endothelial cells are surrounded by support cells on their abluminal side, with smooth muscle cells supporting endothelial cells in arterioles and postcapillary venules, podocytes and mesangial cells supporting glomerular endothelial cells, and pericytes supporting peritubular capillaries (16). The microvasculature in the glomerulus serves as a selective permeability filter which permits small molecules such as water, sugars, electrolytes and small proteins to pass through while retaining high molecular weight proteins in the circulation (17). Peritubular microvessels remove waste molecules from the circulation into the urine and reabsorb molecules from pre-urine back into the blood. Compared to endothelial cells from other organs, fenestrated glomerular endothelial cells are endowed with a particularly thick glycocalyx that extends into the pores and also covers the surface of podocytes (18), and forms a critical component of the permeability barrier to solutes. Disturbances in the permeability of the glomerular capillary wall and loss of the glycocalyx contributes to albuminuria (19–21).
3 Endothelial glycocalyx
The luminal surface of endothelial cells is lined by a protective glycocalyx, also known as the pericellular matrix or endothelial surface layer. It is a delicate and heterogenous gel-like structure with a thickness ranging from 0.5 - 2 µm and it is composed of negatively-charged proteoglycans and glycosaminoglycans (GAGs) such as heparan sulphate, chondroitin sulphate and HA; glycoproteins bearing acidic oligosaccharides and terminal sialic acids; and plasma proteins including albumin and antithrombin. Glycoproteins such as selectins and cell adhesion molecules are located at the base of the glycocalyx and under physiological conditions, are obscured by a dense layer of proteoglycans covalently bound to the endothelial cell membrane. Proteoglycans such as syndecan-1, syndecan-4 and glypican-1, mask the binding sites of selectins and cell adhesion molecules thereby inhibiting leukocyte adhesion (22). On the luminal side, plasma proteins such as albumin are bound to GAG chains through electrostatic interactions between the negatively-charged sulphate and carboxylate groups on the GAG chains and positive arginine residues in albumin, and this interaction stabilizes the glycocalyx structure. The composition of the glycocalyx may vary depending on the blood vessel type, vascular bed and fluid shear stress applied to the endothelial cell surface (21). The glycocalyx synthesized by glomerular endothelial cells express more thrombomodulin compared to HUVEC but less than human brain microvascular endothelial cells, whereas expression of cell adhesion molecules are comparable between the aforementioned endothelial cells (23). The presence of proteoglycans and GAGs endows the glycocalyx with a high net negative charge that not only repels negatively charged molecules but prevents white blood cells and platelets from interacting with the endothelium. The glycocalyx serves to cushion and protect endothelial cells from injury and plays a vasculoprotective role by restricting transvascular protein leakage, leukocyte extravasation, thrombosis and complement activation; regulates redox state and NO release; and sequestration of antithrombotic factors, antioxidants, anti-inflammatory mediators, lipoproteins and protease inhibitors (24, 25) (Table 2). The glycocalyx also binds enzymes such as superoxide dismutase that protects the glycocalyx from oxidative stress. The integrity of the endothelial and glycocalyx barrier is regulated by endothelial stabilizing agents such as sphingosine-1-phosphate, which is transported to the endothelium by high density lipoprotein (HDL) and albumin, where it serves to stabilize the endothelial cortical actin cytoskeleton and inhibits secretion of matrix metalloproteinases (MMPs). These proteases degrade proteoglycans in the glycocalyx and matrix proteins which form the substratum on which endothelial cells adhere to (26).
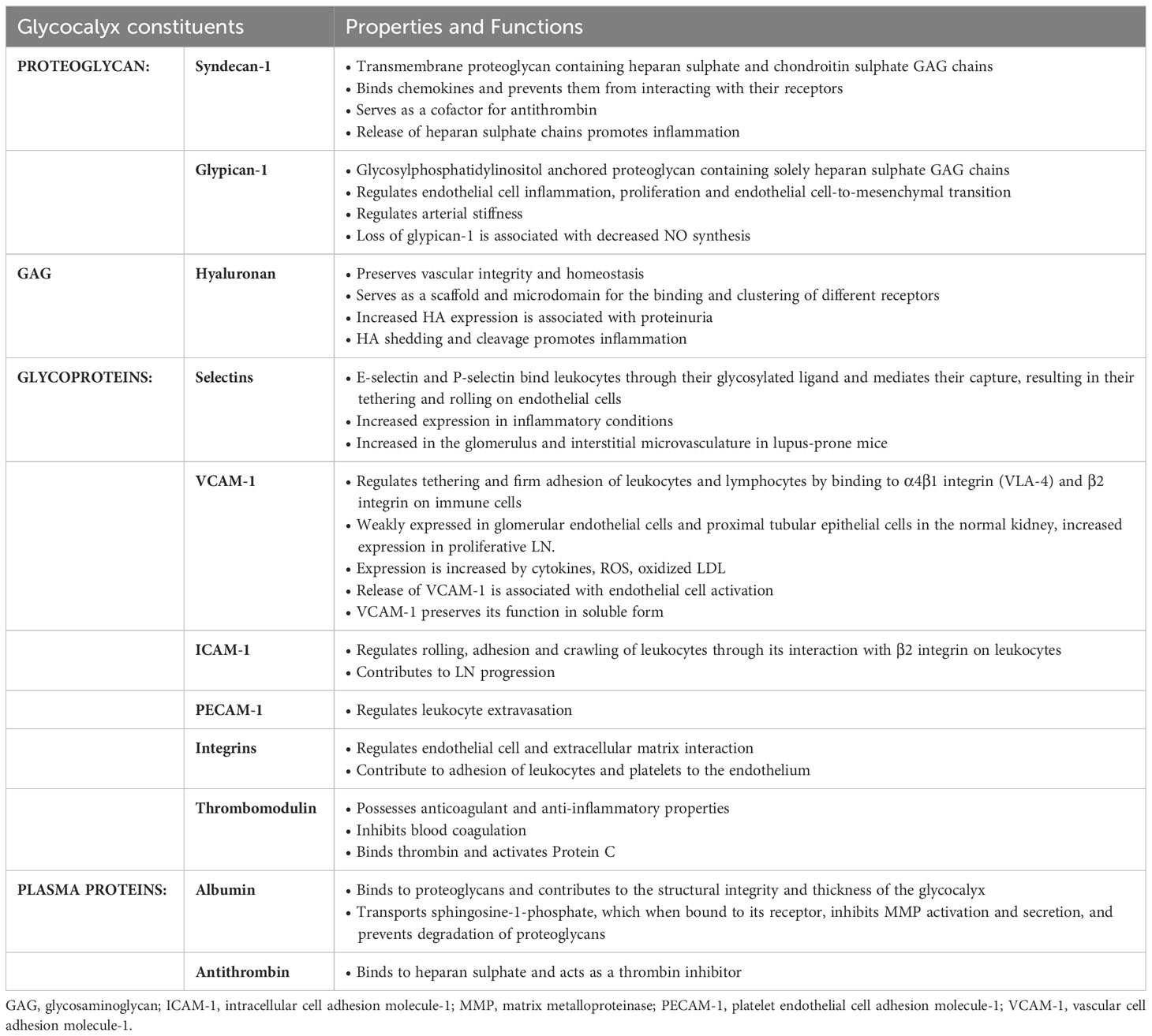
Table 2 Constituents of the endothelial glycocalyx and their functions.
Preserving the integrity of the glycocalyx is essential to maintain vascular homeostasis. Despite its important role in vasculoprotection, the glycocalyx is a fragile structure and can be easily damaged by changes in blood chemistry and flow patterns along the vessel walls, increased levels of hormones, neurotransmitters, vasoactive and atherogenic factors, TNF-α and MMPs, and a loss of plasma components especially albumin in the circulation (26–29). Inflammation or ischemia also induces endothelial cell activation, triggering downstream activation of proteases and shedding of the glycocalyx resulting in fluid extravasation, leukocyte and platelet adhesion, vascular inflammation, hypercoagulability, thrombosis, and loss of flow-responsive vasodilation (30). Shedding of the glycocalyx can reduce glycocalyx thickness by up to 65% and exacerbates systemic inflammatory processes by acting as danger associated molecular patterns (DAMPs) and initiate recruitment of neutrophils (30, 31). A loss of the glycocalyx leads to the acquisition of a non-adaptive phenotype characterized by a loss of the homeostatic mechanisms present in healthy endothelial cells. Alteration in the endothelial glycocalyx and composition is associated with CVD, hypertension and renal disease (19, 32–34) and precedes podocyte foot process effacement and proteinuria (35).
4 Endothelial injury in lupus nephritis
Endothelial cell activation, vascular inflammation and kidney injury can be induced through numerous effector mechanisms in LN and include the deposition of oxidized LDL (ox-LDL) in blood vessels, production of autoantibodies against endothelial cells and their subsequent binding to their cell surface antigens, induction of pro-inflammatory mediators including TNF-α, and formation of neutrophil extracellular traps (NETs).
Patients with Class 3 or 4 LN show glomerular endothelial injury, characterized by leukocyte accumulation and endocapillary proliferation associated with capillary wall destruction. These detrimental changes are initiated by immune complex deposition in the subendothelial space or thrombotic events in SLE-associated lupus anticoagulant syndrome (9, 10). The glomerular endothelium together with its negatively charged glycocalyx facilitates the deposition of positively charged autoantibodies (36). Deposition of immune complexes in the glomerular subendothelium or binding of anti-endothelial antibodies to cross-reactive antigens on the plasma membrane of endothelial cells results in the activation of procoagulation pathways, fibrin deposition and exudative lesions. Immune complex deposition, mediated in part through C5b-9 following complement activation, also increases secretion of cytokines and chemokines and expression of cell adhesion molecules leading to degradation of the glycocalyx, leukocyte recruitment, increased endothelial permeability, endothelial cell apoptosis and kidney injury (37–39). Inflammation is the body’s immediate response to tissue damage where circulating leukocytes adhere to activated endothelial cells and emigrate to the site of tissue damage with the aim of removing the injurious insult and start the healing process. This is a highly regulated, multi-step process comprising a series of coordinated interactions between leukocytes and endothelial cells. Neutrophils are the first-line responders to tissue injury and their adhesion to the vascular endothelium is essential to mount an inflammatory response. TNF-α released by macrophages at the site of injury facilitates the degradation of the endothelial glycocalyx constituents through activation of heparanase and MMP-9 in endothelial cells, and release of free radicals, proteases and heparanase from leukocytes and mast cells (40, 41). Shedding of heparan sulphate proteoglycans from the glycocalyx exposes selectins and cell adhesion molecules constitutively expressed on the endothelial cell surface, and engagement of P-selectin and E-selectin to sialylated, fucosylated mucin on leukocytes facilitate the initial attachment and rolling of leukocytes on the endothelium. Chemokines secreted by macrophages and stromal cells, which are immobilized by heparan sulphate proteoglycans in the glycocalyx, generate a chemotactic gradient that promotes activation of transmembrane G-protein coupled receptors on leukocytes, which initiates integrin clustering on leukocytes and their binding to VCAM-1, ICAM-1 and ICAM-2, leading to firm adhesion on the endothelium and subsequent extravasation or diapedesis (42–44). Secretion of pro-inflammatory cytokines such as IL-1, IFN-γ and TNF-α by tissue-resident macrophages and mast cells, and release of cytokines, chemokines and growth factors sequestered by heparan sulphate GAG chains following glycocalyx degradation, increases E-selectin, VCAM-1 and ICAM-1 expression and decreases thrombomodulin expression in endothelial cells, which augments neutrophil recruitment (23, 45). Increased expression of adhesion molecules on the luminal endothelial cell surface during inflammation is therefore associated with thinning of the glycocalyx. While glycocalyx degradation is crucial for neutrophilic response to danger signals, reconstitution of the glycocalyx is important for resolution of inflammatory processes. Elimination or suppression (as in the case of autoimmune diseases) of the injurious insult is essential to initiate both glycocalyx and tissue repair. In LN patients, a loss of self-tolerance, aberrant innate and adaptive immune responses and autoantibody production creates an inflammatory microenvironment in the kidney especially during relapse, and timely treatment is essential to prevent endothelial cell activation and vascular inflammation, and preserve patient and kidney survival (46).
Long-term use of immunosuppressive medication, impaired immune response, renal impairment and disease activity in LN patients is associated with an increased risk of infection (47). Neutrophils recruited to sites of infection release antimicrobial NETs, composed of chromatin and neutrophil components that trap and kill microbes in tissues and vasculature (48). Timely degradation and removal of NETs is crucial for tissue homeostasis and to prevent their presentation as autoantigens. DNase 1 is required to degrade NETs. LN patients have been reported to show impaired NET degradation consequent to increased serum level of DNase 1 inhibitors and production of anti-NET antibodies (49). Impairment of NET degradation correlates with disease activity in LN patients (49) and contribute to endothelial cell injury, vascular inflammation and thrombosis (48). Low density granulocytes (LDGs) are a distinct subset of pro-inflammatory neutrophils, which are increased in SLE patients and have been shown to promote endothelial cell injury in culture (50). Studies have shown that SLE-derived LGDs express high level of active MMP-9, which are externalized during NETosis and cluster on the plasma membrane of cultured endothelial cells and activates endothelial MMP-2 (51), which may contribute to shedding of the endothelial glycocalyx.
Chronic inflammation induces a plethora of inflammatory mediators including TNF-α, IL-1β and IL-6 and proteolytic enzymes, which augment shedding of endothelial glycocalyx constituents and their detection in the circulation and urine (52–56). Inflammation induces ROS and activation of MMPs, ADAMs and other proteases including elastase and cathepsin B, which induces cleavage of syndecan-1 and CD44 and releases their ectodomains from endothelial cells, while heparanase and hyaluronidase degrade heparan sulphate and HA GAG chains respectively. Although it was previously believed that MMPs were rapidly released following de novo synthesis, emerging evidence suggests that MMPs in their activated form are stored in secretory granules in the endothelial cells and can be rapidly released by inflammatory or angiogenic stimuli without prior upregulation of their mRNA (57). In experimental studies, considerable shedding of glycocalyx components in coronary vessels have been observed in isolated guinea pig hearts after 20 min following TNF-α perfusion (58), and this was associated with increased expression of cell adhesion molecules on the endothelium and reduced degradation of asymmetric dimethylarginine, the latter an endogenous NOS inhibitor (59). Serum TNF-α level is increase in LN patients with active disease and contribute to glycocalyx shedding and subsequent inflammatory processes in LN patients.
Endothelial injury leads to impaired vascular tone and permeability and reduced NO release resulting in diminished endothelium-dependent vasodilation and subsequent atherosclerotic lesion in SLE patients. Endothelial NOS (eNOS) expression inversely correlates with the degree of glomerular injury in LN patients, mediated in part through increased IFN-α signature (60, 61). Atherogenic risk and intimal-medial thickening have been reported to inversely correlate with the thickness of the glycocalyx (62). Aortic stiffness, defined as the reduced capability of an artery to expand and contract in response to changes in pressure (63), has been reported in SLE patients and predisposes patients to CVD. Artery stiffness associated with hypertension and ageing induces degradation of the endothelial glycocalyx (64, 65). HUVEC cultured on polyacrylamide gels with a substrate stiffness of 10 kPa, which mimicked subendothelial stiffness detected in aged or diseased arteries, showed a marked reduction in heparan sulphate GAG chains and glypican-1 core protein but not HA expression, and was accompanied by increased MCP-1 secretion and ICAM-1 expression when compared to cells cultured on gels with a subendothelial stiffness equivalent to that in healthy arteries (2.5 - 5.0 kPa) (66). To date, the clinico-association of glypican-1 with disease activity in SLE or LN patients has not been documented, and further studies are warranted to determine the association of glycocalyx-associated glypican-1 and vascular stiffness in LN patients.
Lipoprotein oxidation is an early event in atherogenesis resulting in the formation of various oxidation products that trigger local immune responses (67), and may play an important role in premature atherosclerosis in SLE patients. Increased levels of oxidized epitopes on LDL have been reported in SLE patients and is associated with coronary or peripheral arterial disease, carotid plaque, increased intima-medial thickness score, and kidney involvement (68). Whereas HDL possesses anti-oxidant properties and removes ROS from ox-LDL and inhibits the expression of adhesion molecules on endothelial cells, ox-LDL contributes to inflammatory processes through induction of inflammatory mediators and monocyte recruitment. In SLE patients, total HDL level is decreased whereas pro-inflammatory LDL level is increased (69). Increased inflammatory processes in atherogenesis results in the shedding of the glycocalyx constituents into the circulation leading to an impairment in its selective permeability barrier, which in turn, results in transendothelial leakage of atherogenic LDL at lesion-prone arterial sites (70). Areas of glycocalyx damage have been shown to correlate with ox-LDL cholesterol uptake. In apolipoprotein E-deficient mice, lipid accumulation and plaque formation increase in areas of the endothelium that showed reduced glycocalyx coverage, whereas plaque-free regions were observed in the endothelium with an intact glycocalyx (71, 72).
Endothelial cell apoptosis is a key feature of atherosclerosis and colocalizes with tissue factor activity in atherosclerotic plaques, possibly through activation of the redox-sensitive pathways and redistribution of phosphatidylserine residues (73, 74). Increased circulating apoptotic endothelial cells and decreased bone marrow-derived endothelial progenitor cells and myeloid angiogenic cells suggests an imbalance between endothelial cell injury and repair in SLE and LN patients (73). Stimulation of glomerular endothelial cells with serum from SLE and LN patients increases IL-1β and IL-6 transcripts and IL-8, IL-15 and PDGF-BB secretion and is associated with increased neutrophil recruitment (75, 76). These pro-inflammatory mediators also induce glycocalyx shedding.
Numerous physiological and pathological stimuli can alter endothelial permeability. Thrombin, histamine and pro-inflammatory cytokines can induce opening of adherens and tight junctional complexes through increased PKC phosphorylation, which in turn mediates cytoskeleton reorganization, altered expression of focal adhesion components including vinculin, α-actinin and talin, resulting in changes in cell-matrix interactions, a loss of cell-cell contact and increased paracellular transport (77). Although the PKC isoenzymes that mediated increased endothelial permeability was not determined, it is possible that PKC-α and PKC-β isoforms were increased since they are the predominant isoforms present in endothelial cells (78). We previously demonstrated that anti-dsDNA antibodies induced PKC-α, PKC-βI and PKC-βII phosphorylation in human mesangial cells and their expression were increased in the glomeruli in NZB/W F1 mice with active nephritis (79). Immune deposition in the subendothelial region is a characteristic feature in severe LN. It is possible that anti-dsDNA or other autoantibodies could bind to glomerular endothelial cells to induce endothelial injury through PKC activation or other signaling pathways. High molecular weight HA has been shown to reduce renal PKC activation in a murine model of diabetic nephropathy (80). Given that HA is a key component of the glycocalyx, its cleavage or shedding from the glycocalyx may induce PKC activation and subsequent acceleration of endothelial cell activation and dysfunction.
5 Clinical association of circulating glycocalyx constituents in patients with lupus nephritis
Early diagnosis of LN and monitoring of treatment efficacy is challenging, since conventional parameters used in clinical practice, such as proteinuria or deranged kidney function, are likely preceded by a stage of preclinical kidney injury. Kidney biopsy remains the gold standard in the diagnosis of LN and subsequent assessment of kidney histopathology but since it is invasive it is performed infrequently and cannot be repeated frequently. Conventional parameters for the assessment of kidney function such as proteinuria and serum creatinine are non-specific, and do not accurately reflect histopathological changes (54). Serial monitoring of anti-dsDNA and C3 levels is useful in some, but not all, patients (81, 82). There is currently no biomarker that shows a distinct advantage for flare prediction (83). Recently, we and others have demonstrated that measurement of glycocalyx constituents in serum and urine samples correlate with disease activity in LN.
Shedding of the glycocalyx constituents into the bloodstream is considered an early sign of endothelial activation and injury (34), and measurement of circulating glycocalyx components may serve as biomarkers of CVD and kidney injury in LN patients. Circulating fragments of the glycocalyx are emerging as diagnostic and prognostic tools for numerous pathological conditions including autoimmune diseases and sepsis. The next section will focus on circulating glycocalyx constituents that show association with disease parameters in SLE and LN patients.
5.1 Syndecan-1
Heparan sulphate comprises more than 50% of the total GAG content in the glycocalyx. It is synthesized in the Golgi apparatus and is composed of repeating disaccharide units of D-glucuronic acid and N-acetyl glucosamine residues. During biosynthesis, the GAG chain can be modified by N-deacetylation or N-sulphation of the glucosamine unit, O-sulphation of both monosaccharide units or C5 epimerization of glucuronic acid to iduronic acid (84). Given the different combination of modifications that can occur, the extent of sulphation and their location along the GAG chain, there is considerable structural diversity and 23 different disaccharides in heparan sulphate chains have so far been identified (85), which endow heparan sulphate GAG chains with the ability to bind to numerous ligands that mediate various biological functions (84). Heparan sulphate GAG chains are attached to a core protein and are present in the glycocalyx predominantly as syndecan-1.
Syndecan-1 is a transmembrane proteoglycan that has five potential attachment sites for GAG chains and may contain both heparan sulphate and chondroitin/dermatan sulphate GAG chains. Heparan sulphate GAG chains are restricted to the N terminus of the core protein whereas chondroitin/dermatan sulphate GAG chains are attached at the C terminus (86). Through its heparan sulphate GAG chains, syndecan-1 can interact with numerous extracellular matrix proteins such as collagen, laminin and fibronectin and it can serve as co-receptors for integrins and growth factors such as FGF, EGF and HGF, and inhibit IFN-γ and TNF-α activity (87, 88). Owing to these interactions, syndecan-1 plays key roles in cell proliferation, adhesion, signaling, wound healing and inflammation (88–90). Syndecan-1 can impede leukocyte adhesion to the endothelium by inhibiting the interaction of β2 integrin on leukocytes to ICAM-1 on endothelial cells. Transmigration of leukocytes from the circulation, across the endothelium and basement membrane to the site of injury is mediated through a chemotactic gradient. Syndecan-1 can immobilize chemokines such as CXCL1, 2 and 8 through electrostatic interactions between the negative charge of heparan sulphate GAG chains and the positive charge present in arginine and lysine residues in chemokines (85, 91), which alters the orientation and oligomerization of chemokines, which in turn, affects the interaction of chemokines with their receptors (91, 92). In patients with renal disease including LN, syndecan-1 present on tubular epithelial cells can bind L-selectin and MCP-1, and the degree of binding is associated with leukocyte infiltration (93). In addition to the glycocalyx, tubular epithelial cells may be another source of syndecan-1 in the kidney which can contribute to tubulo-interstitial fibrosis. In lupus-prone mice, syndecan-1 has been shown to interact with death receptor 6, an orphan immune regulator, which regulates expansion and activation of autoreactive follicular helper T cells and disease progression (94). A loss of syndecan-1 may exacerbate immunological responses in LN.
Endothelial cells are constantly exposed to the mechanical shearing forces of blood flow. Changes in the magnitude of shear stress in addition to temporal and spatial distribution have been reported to alter endothelial permeability and hydraulic conductivity and expression of cell adhesions. Fluid shear stress is a potent regulator of vascular homeostasis and induces AKT, Rho A and paxillin phosphorylation in endothelial cells, which play critical roles in NO production, regulation of oxidative stress and cell survival, cytoskeletal remodeling and adherence of endothelial cells to their underlying substratum (95–99). Heparan sulphate proteoglycans have been shown to act as mechanosensors (100). A loss of syndecan-1 in cultured endothelial cells through gene silencing inhibited AKT and Rho A activation and abolished the establishment of a paxillin phosphorylation gradient in response to flow, and induced a pro-atherosclerotic phenotype with decreased gene expression of Kruppel-like factor (KLF)-2 and KLF-4 and increased expression of KLF5 (101). KLF are a family of transcription factors and key regulators of vasomotor tone, inflammatory and thrombotic processes, and shear stress-induced phenotypic changes in endothelial cells. Whereas KLF-2 and KLF-4 can increase genes that are atheroprotective, KLF-5 induces gene expression of pro-inflammatory mediators in endothelial cells. These studies support a role for syndecan-1 in mechanosensing in cultured endothelial cells and suggests an atheroprotective role for syndecan-1 (101), and loss of syndecan-1 is associated with a pro-inflammatory phenotype. Pro-inflammatory mediators including TNF-α or IL-6 induces loss of syndecan-1 through increased MMP activation, which cleaves syndecan-1 ectodomain. Heparanase is an enzyme or sheddase that specifically cleaves heparan sulphate GAG chains from their protein core and is secreted by numerous cells including macrophages, podocytes and mast cells. Enzymatic removal of heparan sulphate GAG chains from the glycocalyx of cultured human glomerular endothelial cells resulted in an increase in albumin flux across the cell monolayer (102). Furthermore, increased glomerular heparanase expression is associated with proteinuria, increased BUN and glomerular injury (103, 104). The release of heparan sulphate GAG chains from syndecan-1 will in turn release a plethora of growth factors and cytokines sequestered on the GAG chains, triggering downstream inflammatory and fibrotic processes.
LN patients with active disease have increased serum syndecan-1 level compared to patients in remission and the level correlated with serological and clinical parameters of disease including anti-dsDNA antibody titre, proteinuria, serum creatinine level and both SLEDAI-2K and renal SLEDAI-2K scores (56, 105, 106). Proteinuria and renal SLEDAI-2K score are reliable markers of renal activity in LN and they reflect kidney injury. When compared to proteinuria and renal SLEDAI-2K score, serum syndecan-1 level showed comparable correlation with serological markers of active disease, and a better correlation with serum creatinine level and eGFR. Serum syndecan-1 level was also higher in LN patients with active disease compared to SLE patients with extrarenal manifestations (53, 56) and this may be attributed, at least in part, to a loss of syndecan-1 from the glomerular endothelial glycocalyx and downstream glomerular injury. Serum syndecan-1 level showed a higher sensitivity rate (85.91%) than anti-dsDNA antibody titre (75.00%) and C3 level (62.07%) in distinguishing patients with active LN from quiescent LN patients, while the specificity rate was comparable between all three markers (86.21%, 91.67% and 96.43% respectively) (53). Although anti-dsDNA antibody and C3 levels reflect serological activation, not all episodes of relapse are preceded by an increase in these clinical indicators. Serum syndecan-1 level also correlated with the severity of interstitial inflammation in renal biopsies from LN patients with active disease (53, 56). One possible mechanism through which soluble syndecan-1 contributes to interstitial inflammation may be through the release of chemokines after syndecan-1 shedding from tubular epithelial cells, which facilitates ligand-receptor interaction and recruitment of mononuclear cells (107). Serum syndecan-1 level has also been reported to correlate with circulating CD20-CD38+CD138+ plasma cells suggesting that syndecan-1 may also be derived from CD138+ plasma cells (105). Soluble syndecan-1 can activate B cell differentiation and autoantibody production in a murine model of SLE (108), whereas heparan sulphate fragments can induce secretion of pro-inflammatory cytokines and low molecular weight HA which exacerbate inflammatory processes through TLR-4 and highlights a pathogenic role of syndecan-1 in SLE and LN (109, 110). In a longitudinal study, increased serum syndecan-1 level preceded clinical flare by 3 to 4 months and decreased after treatment in parallel with clinical improvement suggesting that measurement of serum syndecan-1 level may be useful in monitoring impending disease flare (53). Whether syndecan-1 level might be related to cardiovascular complications in SLE patients remains to be investigated but it may be a predictor of endothelial cell activation. Despite an increase in serum syndecan-1 level, none of the LN patients in the longitudinal study developed CVD at the time of recruitment (53). Since endothelial cells play a key role in the early events of atherosclerosis, it is not surprising that endothelial cell activation and injury is observed in asymptomatic patients. Increased plasma syndecan-1 level and decreased capillary glycocalyx thickness is observed in patients with antiphospholipid syndrome and may contribute to endothelial injury and vascular thrombosis that are characteristic features in this condition (111). Increase plasma syndecan-1 level may serve as an independent risk factor for adverse cardiovascular events in patients with non-ischemic dilated cardiomyopathy (112).
5.2 Hyaluronan
HA is a non-sulphated GAG composed of repeating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine and is synthesized on the inner surface of the plasma membrane by one of three isoforms of HA synthase (HAS), namely HAS-1, HAS-2 and HAS-3. Nascent HA molecules are extruded through the plasma membrane onto the cell surface or into the extracellular matrix. Despite its simple chemical structure, HA has pleiotropic functions that depend on its molecular weight and tissue distribution. High molecular weight HA possesses anti-inflammatory and anti-angiogenic properties, whereas low molecular weight HA generated by either de novo synthesis or degradation of the parent molecule by ROS or hyaluronidase, possesses pro-inflammatory and angiogenic properties, and may exacerbate tissue inflammation, through its ability to induce chemokine secretion in macrophages (113). Its anionic properties are attributed to the presence of carboxyl groups. Unlike other GAGs, HA is not covalently attached to a core protein and is bound and tethered to endothelial cells by binding to CD44, its principal cell surface receptor. At the cell surface, HA polymerization generates macromolecules with molecular weight in excess of 1,000 kDa, with a length ranging from 2 - 25 µm. HA accounts for 5 - 20% of the total GAG content in the glycocalyx, and HA cables intertwines in the glycocalyx and functions as a scaffold and microdomain for the binding and clustering of different receptors and contributes to the permselectivity of vessels by repelling or preventing the passage of plasma proteins, thereby creating a small protein-free zone in the more compact inner layer of the glycocalyx. HA also contributes to the oncotic gradient directed towards the lumen of the blood vessel, which limits water efflux out of the bloodstream, and this gradient is reduced following the loss of HA from the glycocalyx. In animal studies, inactivation of HAS-2 in glomerular endothelial cells decreased HA expression in the endothelial glycocalyx by 80%, and this was associated with vascular destabilization characterized by capillary ballooning, loss of endothelial fenestrations, mesangiolysis, glomerulosclerosis and development of albuminuria (114). A loss of HA also resulted in a reduction of Ang1/Tie2 signaling, the latter a key regulator of endothelial quiescence and maintenance of endothelial cell barrier (114).
In a murine model of atherosclerosis, pharmacological inhibition of HA synthesis using 4-methylumbelliferone reduced the thickness of the endothelial glycocalyx and this was accompanied by endothelial dysfunction, increased thrombotic and inflammatory responses with macrophage retention in vascular lesions and a more rapid progression to atherosclerosis (115). Exposure of the glycocalyx to hyaluronidase also resulted in a marked decrease in glycocalyx thickness with a concomitant increase in capillary wall permeability and pericapillary oedema (116). In diabetic patients, the extent of glycocalyx shedding and release of HA into the circulation correlates with carotid artery intimal-medial thickness (117). A similar correlation may exist in LN patients although further studies are warranted to confirm this. A loss of glycocalyx HA, whether by enzymatic cleavage or pharmacological inhibition, promotes atherosclerotic lesion and highlights the importance of glycocalyx HA in maintaining homeostatic vascular function. Proteolytic degradation of CD44 by cathepsin B, elastase or thrombin can also release HA from the glycocalyx and may exert similar detrimental effects (118).
In the vasculature, HA is incorporated into the glycocalyx or extracellular matrix. Although glycocalyx HA plays a key role in preserving vascular integrity and homeostasis, recent studies have shown that it also contributes to progressive LN by acting as a ligand for CD44+ T cell binding (119). In healthy subjects, circulating HA has a half-life of 2 - 5 min and is rapidly removal from the circulation by the liver and kidney (120). Serum HA level is increased in LN patients with active disease compared to patients in remission, which may be attributed to either impaired clearance or increased synthesis, and it correlated with serological and clinical parameters of disease (53, 121). Whereas serum syndecan-1 level increased prior to clinical flare, HA level increase at the time of nephritic flare, decreased after treatment and returned to basal level after 9 months (53). Serum HA level showed similar sensitivity but lower specificity than anti-dsDNA antibody or C3 levels (sensitivity and specificity rates: 74.07% and 68.96% respectively for HA, 75.00% and 91.67% respectively for anti-dsDNA antibodies, and 62.07% and 96.43% respectively for C3), and lower sensitivity and specificity rates than proteinuria (sensitivity and specificity rates of 85.71% and 87.50% respectively) and renal SLEDAI-2K scores (sensitivity and specificity rates of 100.00% and 100.00% respectively) in distinguishing LN patients with active disease and remission (53). HA level was comparable in patients with LN and non-renal SLE, and given that HA plays an important role in inflammatory disorders and is expressed by both immune and non-immune cells (122), it is not surprising that HA level cannot distinguish between active LN and active non-renal SLE patients. Serum HA level correlates with tubular atrophy score, interstitial fibrosis score and overall chronicity index and showed no association with activity index or its components suggesting that HA may contribute to chronic kidney disease (53). In CKD patients, plasma syndecan-1 level was detected in patients with stage 4 CKD, whereas increased HA level was observed only in patients with stage 5 CKD suggesting that shedding of HA occurs later than that of syndecan-1 and may be indicative of more severe injury to the kidney (123).
HA has been reported to drive inflammatory and fibrotic processes in the lungs through activation of CD44 and TLR-4 (122). HA also contributes to kidney fibrosis in lupus-prone mice and this may explain at least in part, the association of HA with histologic chronicity index. HA expression is increased in the glomerulus of LN patients, mediated through anti-dsDNA antibody induction of HAS-2 and IL-1β and downstream synthesis of both high and low molecular weight HA (121). Low molecular weight HA may be generated by HAS-3 activation or through enzymatic degradation of high molecular weight HA by hyaluronidases. Given that anti-dsDNA antibodies had no effect on HAS-3 activation (121), an increase in low molecular weight HA may be due to enzymatic cleavage of the glomerular endothelial glycocalyx. Low molecular weight HA exacerbates inflammatory processes through its ability to expose cell adhesion molecules in the glycocalyx and subsequent macrophage binding and recruitment.
LN is characterized by aberrant influx of immune cells into the kidney. T cell homing in the glomerulus is mediated through the binding of CD44 on T cells to HA in the endothelial glycocalyx. The glycocalyx is thus the first point of contact between circulating immune cells and the local microenvironment and may contributes to pathogenesis of disease (119). In contrast to other experimental models of kidney disease where a reduction of endothelial glycocalyx is observed, murine models of LN have shown a 3-fold increase in the thickness of the endothelial glycocalyx, attributed to increased HA expression and this was accompanied by proteinuria, whereas removal of HA from the endothelial glycocalyx using hyaluronidase removed the number of activated T cells in the glomerulus and improved proteinuria (119). Whether an increase in glomerular endothelial glycocalyx thickness is also observed in LN patients has not been investigated. We and others have demonstrated that glomerular HA expression is increased in LN patients, mediated in part through anti-dsDNA antibodies and increased HAS-2 expression (121, 124). It is possible that HA plays a protective and pathogenic role in LN depending on its molecular weight.
5.3 Thrombomodulin
Thrombomodulin is a 74 kDa transmembrane protein that is predominantly expressed in the endothelial glycocalyx and to a lesser extent on mesangial cells, mesothelial cells, dendritic cells and monocytes (125, 126). Thrombomodulin acts as a membrane-bound, high-affinity receptor for thrombin and prevents its interaction with platelets and coagulation factors. Following its binding to thrombin, thrombomodulin activates protein C, which in turn degrades coagulation factors (127–129). Thrombomodulin may also contain chondroitin sulphate GAG chains which may serve as a weak ligand for thrombin (130). Thrombomodulin generates an anti-inflammatory and barrier-stabilizing microenvironment and has been shown to regulate NFκB signaling, IL-6 secretion and expression of cell adhesion molecules including ELAM-1, VCAM-1 and ICAM-1 (131). Decreased thrombomodulin expression in the endothelial glycocalyx is attributed to reduced transcription and translation induced by pro-inflammatory cytokines such as TNF-α (132), or through cleavage by neutrophil proteinases, with a concomitant increase in soluble thrombomodulin in serum, plasma and urine in patients with renal disease including LN (53, 133–135).
Increased soluble thrombomodulin level is associated with endothelial dysfunction and vascular risk, atherosclerosis, cardioembolic stroke and obesity (136). We and others have reported that serum thrombomodulin level is increased in patients with active LN compared to LN patients in remission, patients with non-renal SLE, CKD patients and healthy subjects (53, 133–135, 137–141). Serum thrombomodulin level correlated with serological and clinical parameters of disease including anti-dsDNA antibody level, renal SLEDAI-2K score, proteinuria and serum creatinine level, and inversely correlated with eGFR, serum albumin and C3 levels (53, 56, 135). In a longitudinal study, increased serum thrombomodulin level preceded clinical flare by almost 4 months and as with syndecan-1, may be a potential indicator of impending disease flare (53). Unlike syndecan-1 and HA levels which returned to baseline levels after approximately 9 months of treatment, serum thrombomodulin level persisted for a longer duration (53). Whether this suggests abnormality of endothelial cell function, ongoing low-grade immune-mediated inflammation or progressive kidney damage warrants further investigation. Serum thrombomodulin level can distinguish between patients with active LN and healthy subjects and patients with active non-renal SLE but did not show a high specificity in distinguishing LN patient and CKD patients suggesting that increased thrombomodulin level may be a potential biomarker of kidney damage (53). In a Multi-Ethnic Study of Atherosclerosis (MESA), serum thrombomodulin level showed a strong inverse correlation with glomerular filtration rate (142). When compared to conventional serological markers of active LN, thrombomodulin level was more sensitive (89.66%) than anti-dsDNA antibody and C3 levels in distinguishing active LN from remission and showed less specificity (68.97%) to conventional biomarkers (53). Serum thrombomodulin level correlated with the severity of interstitial inflammation in renal biopsies from patients with active LN and may reflect histopathological changes, for example, renal vascular lesions in LN is associated with increased plasma thrombomodulin levels (53, 134), and patients with end-stage kidney disease show a noticeable loss of their glycocalyx in the sublingual microvasculature, which is accompanied by an increase in plasma thrombomodulin levels compared with healthy subjects (125). Kidney transplantation replenishes the endothelial glycocalyx and can reduce serum thrombomodulin levels, except in patients who developed allograft CKD when thrombomodulin level remained elevated (125).
5.4 Cell adhesion molecules
Adhesion of circulating leukocytes to the endothelium and their transmigration to the site of injury is mediated through cell adhesion molecules such as E-selectin, VCAM-1 (also known as CD106) and ICAM-1. Under non-inflammatory conditions, the glycocalyx serves as the first line of defense against leukocyte adhesion and limits the interaction of leukocytes with cell adhesion molecules. Under inflammatory conditions, shedding of the endothelial glycocalyx exposes cell adhesion molecules, which permits circulating leukocytes to adhere to the endothelium followed by diapedesis. Diseases characterized by acute inflammation are often associated with increased E-selectin expression, whereas chronic inflammation is associated with increased expression of VCAM-1 and ICAM-1 (143). VCAM-1 and ICAM-1 expression is increased during systemic and local inflammation by TNF-α and IL-1β, and they initiate the strong adhesion of circulating leukocytes to the endothelium and their subsequent transmigration through endothelial cell junctions (144).
Adhesion molecules are shed from the activated endothelium following proteolytic cleavage and are detected in the circulation where they could potentially serve as soluble biomarkers of vascular endothelial dysfunction and cardiovascular disease (145, 146). Plasma VCAM-1 and E-selectin are associated with cardiovascular events, coronary calcium, and carotid plaques in SLE patients (147, 148). VCAM-1 and ICAM-1 expression have also been detected in the glomeruli and renal tubules in patients and mice with active LN, suggesting that resident renal cells may also contribute to circulating levels of adhesion molecules (149, 150).
Early studies that investigated serum VCAM-1 level in SLE and Class 3 or 4 LN patients were conflicting (133, 151–153). Independent researchers have shown an association between serum VCAM-1 level and proteinuria, but not with anti-dsDNA antibody or C3 levels, or SLEDAI score (134), while other studies demonstrated an increase in VCAM-1 level in SLE patients but no association with disease activity or organ involvement (154). Correlation between serum VCAM-1 level and coronary calcification/subclinical atherosclerosis has also been reported, irrespective of whether the patient had lupus (155). We recently demonstrated that serum VCAM-1 level was significantly higher in LN patients with nephritic flare compared to remission, and its level correlated with clinical and serological parameters of disease including anti-dsDNA antibody level, renal SLEDAI score and proteinuria and inversely correlated with C3 level. Serum VCAM-1 level also showed high sensitivity and specificity and could distinguish between patients with active LN from those in remission (sensitivity and specificity rates of 68.97% and 89.66% respectively), patients with active non-renal SLE (sensitivity and specificity rates of 90.91% and 86.21% respectively), CKD patients (sensitivity and specificity rates of 89.66% and 82.61% respectively) and healthy subjects (96.55% and 96.00% respectively) (52). When compared to conventional biomarkers of disease, serum VCAM-1 level showed comparable sensitivity and specificity as anti-dsDNA antibody and C3 levels in distinguishing patients with active LN and remission, and similar sensitivity and specificity as C3 (sensitivity and specificity rates of 90.00% and 89.29% respectively), but higher specificity than anti-dsDNA antibody titre (sensitivity and specificity rates of 100.00% and 42.86% respectively), in distinguishing active LN patients from patients with active non-renal SLE. The measurement of a panel of biomarkers rather than a single biomarker, may be more useful to predict nephritis flares (156, 157) and VCAM-1 in combination with C3, proteinuria or serum levels of syndecan-1, HA and thrombomodulin was superior to C3, anti-dsDNA antibody titre or serum creatinine level in distinguishing active LN from quiescent disease (52). In a longitudinal study, serum VCAM-1 level increased 4.5 months before nephritic flare was evident clinically as shown by an increase of serum creatinine level and/or proteinuria and VCAM-1 level returned to baseline after one year, suggesting that serum VCAM-1 level may serve as an early indicator of impending nephritic flare. Serum VCAM-1 level correlated with serum levels of syndecan-1, HA and thrombomodulin suggesting a close association between components of the endothelial glycocalyx. At the time of flare, serum VCAM-1 level correlates with leukocyte infiltration score and fibrinoid necrosis/karyorrhexis score in renal biopsies from LN patients highlighting its role in leukocyte infiltration into the kidney (52). Increased urine VCAM-1 level has also been reported in patients with active LN and strongly correlated with renal biopsy activity score. In this respect, urine VCAM-1 level has been proposed to serve as a noninvasive marker in assessing histopathologic changes in the kidney and is associated with inferior long-term renal outcome (54, 158–161). In childhood-onset SLE, urinary VCAM-1 outperformed anti-dsDNA antibody titre and C3 level as biomarkers to predict nephritic flare (55). Using aptamer-based screening followed by ELISA validation, urinary VCAM-1 was identified as a strong candidate to distinguish between active LN and quiescent disease irrespective of ethnicity (162). More recently, the assessment of urine: serum fractional excretion ratios of cell adhesion molecules outperformed corresponding urine and serum levels in identifying active LN from remission (163).
As with VCAM-1, conflicting results have also been reported for serum ICAM-1 and E-selectin levels in SLE and LN patients and their association with clinical and serological parameters of disease (52, 152, 162, 164–171). The discrepancies may be related to differences in the characteristics of patients, ethnicity and management, the timing of sample collection in relation to disease activity, and differences in the sensitivity of assays used (52, 134, 172). In our study, whereas VCAM-1 showed a high seropositivity rate (93%) in patients with active LN, a lower seropositivity rate was observed for ICAM-1 at 38%. Although the longitudinal profile of ICAM-1 followed a similar trend to that of disease activity and response to treatment, serum ICAM-1 level showed no association with anti-dsDNA antibody or C3 levels. In addition to its expression on endothelial cells, VCAM-1 and ICAM-1 expression has also been observed on monocytes, mesangial cells, proximal tubular epithelial cells and smooth muscle cells. Increased VCAM-1 expression is observed in the kidney of lupus-prone mice with active disease when compared to non-autoimmune mice, suggesting a role in mediating leukocyte infiltration in the inflamed kidney parenchyma (149). The mechanisms though which renal VCAM-1 or ICAM-1 expression is induced in LN remain to be fully defined although their induction by anti-dsDNA antibodies has been proposed (173, 174).
6 Conclusions
Early diagnosis of LN, accurate assessment of disease activity and timely monitoring of treatment efficacy is essential for patient and kidney survival, but this remains challenging due to the heterogeneity and complexity of LN. A renal biopsy remains the gold standard for the diagnosis and prognosis of LN but since it is invasive, it is not feasible to perform frequent kidney biopsies for routine monitoring of histopathologic changes and disease activity. Conventional clinical and serological parameters to assess disease activity rely on non-specific indicators of kidney injury or function such as proteinuria and serum creatinine level, which could present late in the course of disease activation and do not reflect histopathologic changes. Serological tests for anti-dsDNA antibody and C3 levels reflect immunological status but not kidney injury and not all patients show an association of anti-dsDNA antibody titre with disease activity. Identification of novel biomarkers for LN should facilitate early diagnosis of nephritic flare and monitoring of treatment response in order to preserve residual kidney function. Biomarkers for LN should accurately reflect disease activity, be reliable and useful in clinical practice, easily measured routinely, have biological and pathophysiological relevance across ethnicities, and show superiority to current conventional markers (175). Furthermore, biomarkers for LN should be able to distinguish between LN and non-renal SLE patients. Given the heterogeneity and complexity of disease, it is unlikely that one molecule can serve as a biomarker for LN. Over the past few decades, biomarker discovery in LN has progressed from individual candidates to unbiased high-throughput platforms mass spectrometry, proteomics and aptamer-based screening. Urinary biomarkers have yielded promising results in predicting the activity of LN, but their clinical utility requires further assessment. The current challenge in the development of novel biomarkers following their discovery for LN is their validation in a large population with ethnic diversity. A biomarker panel may provide superior information regarding LN compared to a single marker, without overlap with other types of glomerulonephritis (176).
We and others have shown that constituents of the glycocalyx correlate with disease activity and may have potential diagnostic and prognostic value in LN, endothelial cell activation and possible cardiovascular complications. Table 3 compares the clinico-pathological association of serum syndecan-1, HA, thrombomodulin and VCAM-1 with conventional indicators of kidney injury and immunologic activity in LN patients. Further studies are warranted in a larger cohort to validate these findings especially in longitudinal studies and different ethnic groups to ascertain whether certain biomarker panels may be used for specific ethnicity and genetic composition. A schematic diagram detailing changes in the glycocalyx in LN patients is presented in Figure 1.
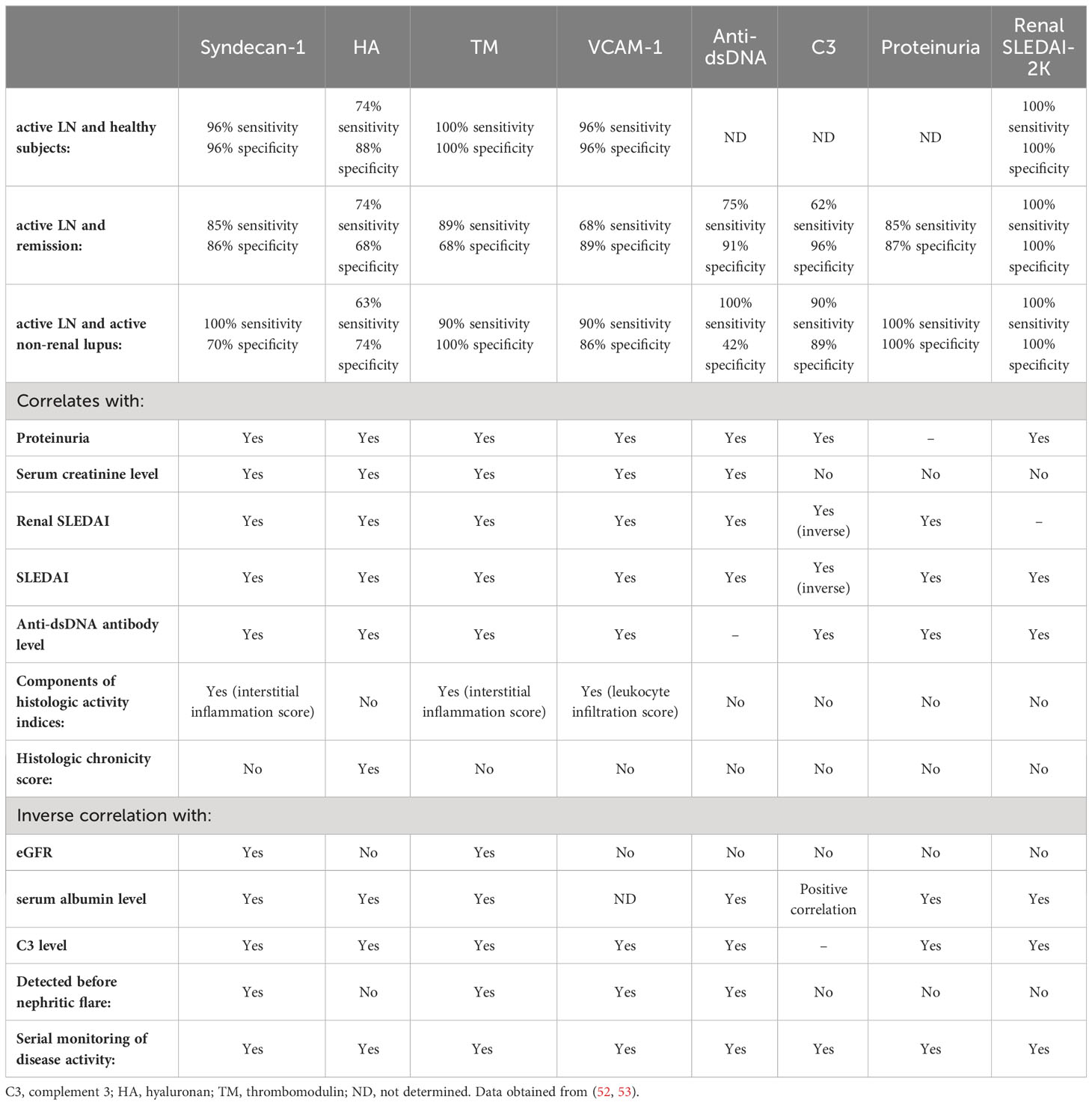
Table 3 Comparison of syndecan-1, hyaluronan, thrombomodulin and VCAM-1 as putative biomarkers for lupus nephritis compared to conventional serological and clinical markers of disease.
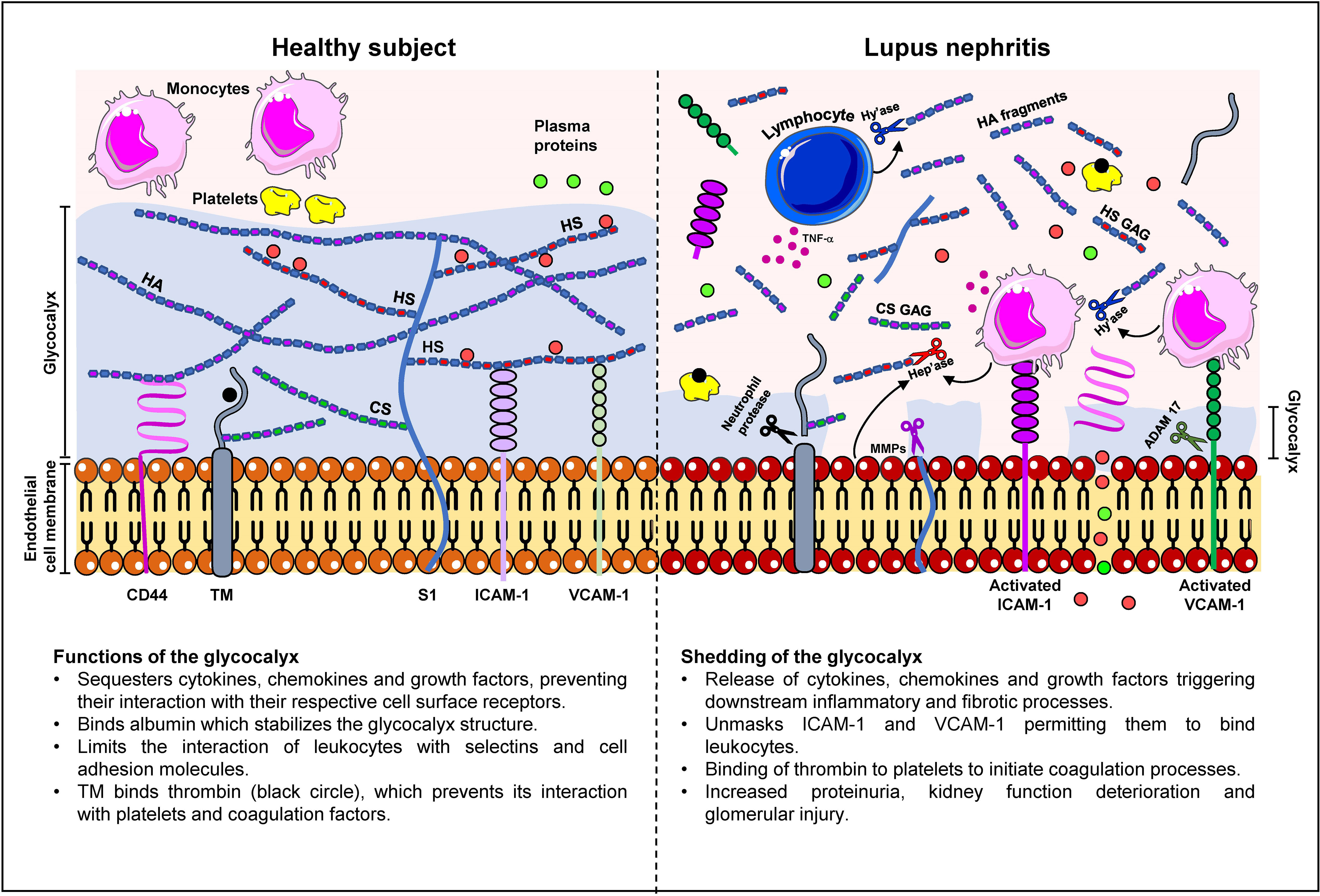
Figure 1 Changes in the glycocalyx composition in lupus nephritis patients. In a healthy subject, the glycocalyx ranges from 0.5 - 2.0 μm in thickness and comprises negatively-charged proteoglycans, glycosaminoglycans (GAG) and glycoproteins such as syndecan-1 (S1), CD44, HA, thrombomodulin (TM), and cell adhesion molecules. The glycocalyx serves to cushion and protect endothelial cells and restrict transvascular protein leakage. The interaction of albumin (red circle) with heparan sulphate GAG chains (HS GAG) stabilizes the glycocalyx structure. S1 and TM may also contain chondroitin sulphate (CS) GAG chains which contribute to the electronegative charge of the glycocalyx. The high net negative charge of the glycocalyx prevents leukocytes and platelets from interacting with the endothelium and contributes to the maintenance of endothelial permeability to plasma proteins (green circles). Proteoglycans also mask the binding sites of selectins and cell adhesion molecules, which inhibits leukocyte adhesion. In LN, chronic inflammation is accompanied by increased cytokine, chemokine and growth factor secretion in immune and non-immune cells, which in turn, increases synthesis of enzymes that cleave glycocalyx constituents leading to the destruction of the glycocalyx. These enzymes include hyaluronidase (Hy’ase) that cleaves HA, heparanase (Hep’ase) that cleaves HS GAG chains, MMPs that cleave syndecan-1 ectodomain and CD44, neutrophil protease that cleaves TM, and ADAM-17 that cleaves cell adhesion molecules. Shedding of the glycocalyx releases pro-inflammatory mediators that were bound to glycocalyx constituents and unmasks the binding sites in VCAM-1 and ICAM-1 thereby permitting the binding of leukocytes and exacerbation of inflammation. Shedding of the glycocalyx also contributes to transvascular protein leakage and proteinuria.
Author contributions
Drafting the article: SY and TC. Approval of the final version for submission: SY and TC. All authors contributed to the article.
Funding
This work was supported by the University of Hong Kong Small Project Funding [grant number: 201409176201], the Department of Medicine Academic Activities Fund, UGC Matching Grant Scheme and kind donations from Mr. C. S. Yung, Mr. S. Ho, and Hui Hoy & Chow Sin Lan Charity Fund and the Family of Mr. Hui Ming. SY is supported by the Endowment Fund established for the ‘Yu Chiu Kwong Professorship in Medicine’ awarded to TC, the Wai Hung Charitable Foundation Limited and Mr. and Mrs. Tam Wing Fan Edmund Renal Research Fund. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Chan TM. Treatment of severe lupus nephritis: the new horizon. Nat Rev Nephrol (2015) 11(1):46–61. doi: 10.1038/nrneph.2014.215
2. Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med (1976) 60(2):221–5. doi: 10.1016/0002-9343(76)90431-9
3. Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du Berger R, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheumatol (2001) 44(10):2331–7. doi: 10.1002/1529-0131(200110)44:10<2331::AID-ART395>3.0.CO;2-I
4. Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA Jr., Jansen-McWilliams L, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol (1997) 145(5):408–15. doi: 10.1093/oxfordjournals.aje.a009122
5. Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med (2003) 349(25):2407–15. doi: 10.1056/NEJMoa035611
6. Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med (2003) 349(25):2399–406. doi: 10.1056/NEJMoa035471
7. Bruce IN, Burns RJ, Gladman DD, Urowitz MB. Single photon emission computed tomography dual isotope myocardial perfusion imaging in women with systemic lupus erythematosus. I. Prevalence and distribution of abnormalities. J Rheumatol (2000) 27(10):2372–7.
8. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol (2015) 161(1):59–63. doi: 10.1016/j.clim.2015.05.024
9. Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol (2004) 15(2):241–50. doi: 10.1097/01.ASN.0000108969.21691.5D
10. Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int (2004) 65(2):521–30. doi: 10.1111/j.1523-1755.2004.00443.x
11. Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol (2012) 9(8):439–53. doi: 10.1038/nrcardio.2012.64
12. Bordy R, Totoson P, Prati C, Marie C, Wendling D, Demougeot C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat Rev Rheumatol (2018) 14(7):404–20. doi: 10.1038/s41584-018-0022-8
13. Cahill PA, Redmond EM. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis (2016) 248:97–109. doi: 10.1016/j.atherosclerosis.2016.03.007
14. Yuen DA, Gilbert RE, Marsden PA. Bone marrow cell therapies for endothelial repair and their relevance to kidney disease. Semin Nephrol (2012) 32(2):215–23. doi: 10.1016/j.semnephrol.2012.02.008
15. Yu X, Seegar TC, Dalton AC, Tzvetkova-Robev D, Goldgur Y, Rajashankar KR, et al. Structural basis for angiopoietin-1-mediated signaling initiation. Proc Natl Acad Sci U S A (2013) 110(18):7205–10. doi: 10.1073/pnas.1216890110
16. Lemos DR, Marsh G, Huang A, Campanholle G, Aburatani T, Dang L, et al. Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am J Physiol Renal Physiol (2016) 311(6):F1230–f42. doi: 10.1152/ajprenal.00030.2016
17. Quaggin SE, Kreidberg JA. Development of the renal glomerulus: good neighbors and good fences. Development (2008) 135(4):609–20. doi: 10.1242/dev.001081
18. Okada H, Takemura G, Suzuki K, Oda K, Takada C, Hotta Y, et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit Care (2017) 21(1):261. doi: 10.1186/s13054-017-1841-8
19. Rabelink TJ, de Zeeuw D. The glycocalyx - linking albuminuria with renal and cardiovascular disease. Nat Rev Nephrol (2015) 11(11):667–76. doi: 10.1038/nrneph.2015.162
20. Salmon AH, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol (2012) 23(8):1339–50. doi: 10.1681/asn.2012010017
21. Salmon AH, Satchell SC. Endothelial glycocalyx dysfunction in disease: albuminuria and increased microvascular permeability. J Pathol (2012) 226(4):562–74. doi: 10.1002/path.3964
22. Butler MJ, Down CJ, Foster RR, Satchell SC. The pathological relevance of increased endothelial glycocalyx permeability. Am J Pathol (2020) 190(4):742–51. doi: 10.1016/j.ajpath.2019.11.015
23. Sartain SE, Turner NA, Moake JL. TNF regulates essential alternative complement pathway components and impairs activation of protein C in human glomerular endothelial cells. J Immunol (2016) 196(2):832–45. doi: 10.4049/jimmunol.1500960
24. Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev (2008) 88(2):451–87. doi: 10.1152/physrev.00055.2006
25. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev BioMed Eng (2007) 9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959
26. Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol (2014) 306(3):H363–72. doi: 10.1152/ajpheart.00687.2013
27. Lipowsky HH. Microvascular rheology and hemodynamics. Microcirculation (2005) 12(1):5–15. doi: 10.1080/10739680590894966
28. Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann BioMed Eng (2012) 40(4):840–8. doi: 10.1007/s10439-011-0427-x
29. Tarbell JM. Shear stress and the endothelial transport barrier. Cardiovasc Res (2010) 87(2):320–30. doi: 10.1093/cvr/cvq146
30. Barry M, Pati S. Targeting repair of the vascular endothelium and glycocalyx after traumatic injury with plasma and platelet resuscitation. Matrix Biol Plus (2022) 14:100107. doi: 10.1016/j.mbplus.2022.100107
31. Wiesinger A, Peters W, Chappell D, Kentrup D, Reuter S, Pavenstadt H, et al. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PloS One (2013) 8(11):e80905. doi: 10.1371/journal.pone.0080905
32. Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int (2007) 72(2):151–6. doi: 10.1038/sj.ki.5002312
33. Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL, et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol (2001) 12(7):1434–47. doi: 10.1681/asn.V1271434
34. Rabelink TJ, de Boer HC, van Zonneveld AJ. Endothelial activation and circulating markers of endothelial activation in kidney disease. Nat Rev Nephrol (2010) 6(7):404–14. doi: 10.1038/nrneph.2010.65
35. Ebefors K, Wiener RJ, Yu L, Azeloglu EU, Yi Z, Jia F, et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int (2019) 96(4):957–70. doi: 10.1016/j.kint.2019.05.007
36. Nangaku M, Couser WG. Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin Exp Nephrol (2005) 9(3):183–91. doi: 10.1007/s10157-005-0357-8
37. van Paassen P, Duijvestijn A, Debrus-Palmans L, Damoiseaux J, Vroomen M, Tervaert JW. Induction of endothelial cell apoptosis by IgG antibodies from SLE patients with nephropathy: a potential role for anti-endothelial cell antibodies. Ann N Y Acad Sci (2007) 1108:147–56. doi: 10.1196/annals.1422.017
38. Moscato S, Pratesi F, Bongiorni F, Scavuzzo MC, Chimenti D, Bombardieri S, et al. Endothelial cell binding by systemic lupus antibodies: functional properties and relationship with anti-DNA activity. J Autoimmun (2002) 18(3):231–8.doi: 10.1006/jaut.2002.0583
39. Dieudé M, Senécal JL, Raymond Y. Induction of endothelial cell apoptosis by heat-shock protein 60-reactive antibodies from anti-endothelial cell autoantibody-positive systemic lupus erythematosus patients. Arthritis Rheumatol (2004) 50(10):3221–31. doi: 10.1002/art.20564
40. Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, et al. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: a contributor to endothelial cell glycocalyx dysfunction. FASEB J (2014) 28(11):4686–99. doi: 10.1096/fj.14-252221
41. Dogné S, Flamion B. Endothelial glycocalyx impairment in disease: focus on hyaluronan shedding. Am J Pathol (2020) 190(4):768–80. doi: 10.1016/j.ajpath.2019.11.016
42. Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol (2014) 14(4):232–46. doi: 10.1038/nri3641
43. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
44. Romagnani P, De Paulis A, Beltrame C, Marone G, Romagnani S. Chapter 36 - chemokine receptors on human mast cells. In: Marone G, Lichtenstein LM, Galli SJ, editors. Mast cells and basophils. London: Academic Press (2000). p. 579–96.
45. Glennon-Alty L, Hackett AP, Chapman EA, Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic Biol Med (2018) 125:25–35. doi: 10.1016/j.freeradbiomed.2018.03.049
46. Yap DY, Tang CS, Ma MK, Lam MF, Chan TM. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol Dial Transplant (2012) 27(8):3248–54. doi: 10.1093/ndt/gfs073
47. Thong KM, Chan TM. Infectious complications in lupus nephritis treatment: a systematic review and meta-analysis. LUPUS (2019) 28(3):334–46. doi: 10.1177/0961203319829817
48. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
49. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi: 10.1073/pnas.0909927107
50. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol (2010) 184(6):3284–97. doi: 10.4049/jimmunol.0902199
51. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis (2015) 74(7):1417–24. doi: 10.1136/annrheumdis-2013-204837
52. Yu KY, Yung S, Chau MK, Tang CS, Yap DY, Tang AH, et al. Clinico-pathological associations of serum VCAM-1 and ICAM-1 levels in patients with lupus nephritis. LUPUS (2021) 30(7):1039–50. doi: 10.1177/09612033211004727
53. Yu KYC, Yung S, Chau MKM, Tang CSO, Yap DYH, Tang AHN, et al. Serum syndecan-1, hyaluronan and thrombomodulin levels in patients with lupus nephritis. Rheumatol (Oxford) (2021) 60(2):737–50. doi: 10.1093/rheumatology/keaa370
54. Singh S, Wu T, Xie C, Vanarsa K, Han J, Mahajan T, et al. Urine VCAM-1 as a marker of renal pathology activity index in lupus nephritis. Arthritis Res Ther (2012) 14(4):R164. doi: 10.1186/ar3912
55. Soliman SA, Haque A, Vanarsa K, Zhang T, Ismail F, Lee KH, et al. PF4 and VCAM-1 surpass conventional metrics in identifying nephritis disease activity in childhood-onset systemic lupus erythematosus. Front Immunol (2022) 13:885307. doi: 10.3389/fimmu.2022.885307
56. Kim KJ, Kim JY, Baek IW, Kim WU, Cho CS. Elevated serum levels of syndecan-1 are associated with renal involvement in patients with systemic lupus erythematosus. J Rheumatol (2015) 42(2):202–9. doi: 10.3899/jrheum.140568
57. Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol (2002) 160(2):673–80. doi: 10.1016/s0002-9440(10)64887-0
58. Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, et al. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol (2009) 104(1):78–89. doi: 10.1007/s00395-008-0749-5
59. Ito A, Tsao PS, Adimoolam S, Kimoto M, Ogawa T, Cooke JP. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation (1999) 99(24):3092–5. doi: 10.1161/01.cir.99.24.3092
60. Buie JJ, Renaud LL, Muise-Helmericks R, Oates JC. IFN-α Negatively regulates the expression of endothelial nitric oxide synthase and nitric oxide production: implications for systemic lupus erythematosus. J Immunol (2017) 199(6):1979–88. doi: 10.4049/jimmunol.1600108
61. Jones Buie JN, Pleasant Jenkins D, Muise-Helmericks R, Oates JC. L-sepiapterin restores SLE serum-induced markers of endothelial function in endothelial cells. Lupus Sci Med (2019) 6(1):e000294. doi: 10.1136/lupus-2018-000294
62. van den Berg BM, Spaan JA, Rolf TM, Vink H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am J Physiol Heart Circ Physiol (2006) 290(2):H915–20. doi: 10.1152/ajpheart.00051.2005
63. Gavish B, Izzo JL Jr. Arterial stiffness: going a step beyond. Am J Hypertens (2016) 29(11):1223–33. doi: 10.1093/ajh/hpw061
64. Weinbaum S, Cancel LM, Fu BM, Tarbell JM. The glycocalyx and its role in vascular physiology and vascular related diseases. Cardiovasc Eng Technol (2021) 12(1):37–71. doi: 10.1007/s13239-020-00485-9
65. Ikonomidis I, Voumvourakis A, Makavos G, Triantafyllidi H, Pavlidis G, Katogiannis K, et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J Clin Hypertens (Greenwich) (2018) 20(4):672–79. doi: 10.1111/jch.13236
66. Mahmoud M, Mayer M, Cancel LM, Bartosch AM, Mathews R, Tarbell JM. The glycocalyx core protein Glypican 1 protects vessel wall endothelial cells from stiffness-mediated dysfunction and disease. Cardiovasc Res (2021) 117(6):1592–605. doi: 10.1093/cvr/cvaa201
67. Karvonen J, Päivänsalo M, Kesäniemi YA, Hörkkö S. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation (2003) 108(17):2107–12. doi: 10.1161/01.Cir.0000092891.55157.A7
68. Frostegård J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, et al. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheumatol (2005) 52(1):192–200. doi: 10.1002/art.20780
69. McMahon M, Grossman J, Skaggs B, Fitzgerald J, Sahakian L, Ragavendra N, et al. Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheumatol (2009) 60(8):2428–37. doi: 10.1002/art.24677
70. van den Berg BM, Spaan JA, Vink H. Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Arch (2009) 457(6):1199–206. doi: 10.1007/s00424-008-0590-6
71. Mitra R, O’Neil GL, Harding IC, Cheng MJ, Mensah SA, Ebong EE. Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr Atheroscler Rep (2017) 19(12):63. doi: 10.1007/s11883-017-0691-9
72. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis (2016) 252:136–46. doi: 10.1016/j.atherosclerosis.2016.07.930
73. Rajagopalan S, Somers EC, Brook RD, Kehrer C, Pfenninger D, Lewis E, et al. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood (2004) 103(10):3677–83. doi: 10.1182/blood-2003-09-3198
74. Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood (1997) 89(7):2429–42. doi: 10.1182/blood.V89.7.2429
75. Dimou P, Wright RD, Budge KL, Midgley A, Satchell SC, Peak M, et al. The human glomerular endothelial cells are potent pro-inflammatory contributors in an in vitro model of lupus nephritis. Sci Rep (2019) 9(1):8348. doi: 10.1038/s41598-019-44868-y
76. Russell DA, Markiewicz M, Oates JC. Lupus serum induces inflammatory interaction with neutrophils in human glomerular endothelial cells. Lupus Sci Med (2020) 7(1): e000418. doi: 10.1136/lupus-2020-000418
77. Lum H, Malik AB. Mechanisms of increased endothelial permeability. Can J Physiol Pharmacol (1996) 74(7):787–800. doi: 10.1139/y96-081
78. Bussolino F, Silvagno F, Garbarino G, Costamagna C, Sanavio F, Arese M, et al. Human endothelial cells are targets for platelet-activating factor (PAF). Activation of alpha and beta protein kinase C isozymes in endothelial cells stimulated by PAF. J Biol Chem (1994) 269(4):2877–86.
79. Yung S, Zhang Q, Zhang CZ, Chan KW, Lui SL, Chan TM. Anti-DNA antibody induction of protein kinase C phosphorylation and fibronectin synthesis in human and murine lupus and the effect of mycophenolic acid. Arthritis Rheumatol (2009) 60(7):2071–82. doi: 10.1002/art.24573
80. Campo GM, Avenoso A, Micali A, Nastasi G, Squadrito F, Altavilla D, et al. High-molecular weight hyaluronan reduced renal PKC activation in genetically diabetic mice. Biochim Biophys Acta (2010) 1802(11):1118–30. doi: 10.1016/j.bbadis.2010.08.004
81. Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatol (Oxford) (2007) 46(7):1052–6. doi: 10.1093/rheumatology/kem112
82. Yap DYH, Kwan LPY, Ma MKM, Mok MMY, Chan GCW, Chan TM. Preemptive immunosuppressive treatment for asymptomatic serological reactivation may reduce renal flares in patients with lupus nephritis: a cohort study. Nephrol Dial Transplant (2019) 34(3):467–73. doi: 10.1093/ndt/gfy024
83. Gensous N, Marti A, Barnetche T, Blanco P, Lazaro E, Seneschal J, et al. Predictive biological markers of systemic lupus erythematosus flares: a systematic literature review. Arthritis Res Ther (2017) 19(1):238. doi: 10.1186/s13075-017-1442-6
84. Ledin J, Staatz W, Li JP, Götte M, Selleck S, Kjellén L, et al. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem (2004) 279(41):42732–41. doi: 10.1074/jbc.M405382200
85. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem (2002) 71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458
86. Kokenyesi R, Bernfield M. Core protein structure and sequence determine the site and presence of heparan sulfate and chondroitin sulfate on syndecan-1. J Biol Chem (1994) 269(16):12304–9. doi: 10.1016/S0021-9258(17)32716-3
87. Sarrazin S, Bonnaffé D, Lubineau A, Lortat-Jacob H. Heparan sulfate mimicry: a synthetic glycoconjugate that recognizes the heparin binding domain of interferon-gamma inhibits the cytokine activity. J Biol Chem (2005) 280(45):37558–64. doi: 10.1074/jbc.M507729200
88. Teng YH, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol (2012) 31(1):3–16. doi: 10.1016/j.matbio.2011.10.001
89. Couchman JR. Transmembrane signaling proteoglycans. Annu Rev Cell Dev Biol (2010) 26:89–114. doi: 10.1146/annurev-cellbio-100109-104126
90. Alexander CM, Reichsman F, Hinkes MT, Lincecum J, Becker KA, Cumberledge S, et al. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet (2000) 25(3):329–32. doi: 10.1038/77108
91. Proudfoot AE. The biological relevance of chemokine-proteoglycan interactions. Biochem Soc Trans (2006) 34(Pt 3):422–6. doi: 10.1042/BST0340422
92. Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood (2002) 100(12):3853–60. doi: 10.1182/blood.V100.12.3853
93. Celie JW, Reijmers RM, Slot EM, Beelen RH, Spaargaren M, Ter Wee PM, et al. Tubulointerstitial heparan sulfate proteoglycan changes in human renal diseases correlate with leukocyte influx and proteinuria. Am J Physiol Renal Physiol (2008) 294(1):F253–63. doi: 10.1152/ajprenal.00429.2007
94. Fujikura D, Ikesue M, Endo T, Chiba S, Higashi H, Uede T. Death receptor 6 contributes to autoimmunity in lupus-prone mice. Nat Commun (2017) 8:13957. doi: 10.1038/ncomms13957
95. Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA Jr. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res (2007) 101(7):723–33. doi: 10.1161/circresaha.107.152942
96. Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res (1998) 83(3):334–41. doi: 10.1161/01.res.83.3.334
97. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature (1999) 399(6736):601–5. doi: 10.1038/21224
98. Cicha I, Goppelt-Struebe M, Muehlich S, Yilmaz A, Raaz D, Daniel WG, et al. Pharmacological inhibition of RhoA signaling prevents connective tissue growth factor induction in endothelial cells exposed to non-uniform shear stress. Atherosclerosis (2008) 196(1):136–45. doi: 10.1016/j.atherosclerosis.2007.03.016
99. Zaidel-Bar R, Kam Z, Geiger B. Polarized downregulation of the paxillin-p130CAS-Rac1 pathway induced by shear flow. J Cell Sci (2005) 118(Pt 17):3997–4007. doi: 10.1242/jcs.02523
100. Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res (2003) 93(10):e136–42. doi: 10.1161/01.Res.0000101744.47866.D5
101. Voyvodic PL, Min D, Liu R, Williams E, Chitalia V, Dunn AK, et al. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem (2014) 289(14):9547–59. doi: 10.1074/jbc.M113.541573
102. Singh A, Satchell SC, Neal CR, McKenzie EA, Tooke JE, Mathieson PW. Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J Am Soc Nephrol (2007) 18(11):2885–93. doi: 10.1681/asn.2007010119
103. Garsen M, Benner M, Dijkman HB, van Kuppevelt TH, Li JP, Rabelink TJ, et al. Heparanase is essential for the development of acute experimental glomerulonephritis. Am J Pathol (2016) 186(4):805–15. doi: 10.1016/j.ajpath.2015.12.008
104. Gil N, Goldberg R, Neuman T, Garsen M, Zcharia E, Rubinstein AM, et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes (2012) 61(1):208–16. doi: 10.2337/db11-1024
105. Minowa K, Amano H, Nakano S, Ando S, Watanabe T, Nakiri Y, et al. Elevated serum level of circulating syndecan-1 (CD138) in active systemic lupus erythematosus. Autoimmunity (2011) 44(5):357–62. doi: 10.3109/08916934.2010.545846
106. Mosaad NA, Lotfy HM, Farag YM, Mahfouz RH, Shahin RM. Study of serum syndecan-1 levels in a group of Egyptian juvenile systemic lupus erythematosus patients. Immunol Lett (2017) 181:16–9. doi: 10.1016/j.imlet.2016.11.005
107. Celie JW, Katta KK, Adepu S, Melenhorst WB, Reijmers RM, Slot EM, et al. Tubular epithelial syndecan-1 maintains renal function in murine ischemia/reperfusion and human transplantation. Kidney Int (2012) 81(7):651–61. doi: 10.1038/ki.2011.425
108. Liu L, Akkoyunlu M. Circulating CD138 enhances disease progression by augmenting autoreactive antibody production in a mouse model of systemic lupus erythematosus. J Biol Chem (2021) 297(3):101053. doi: 10.1016/j.jbc.2021.101053
109. Noble PW. Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol (2002) 21(1):25–9. doi: 10.1016/s0945-053x(01)00184-6
110. Goodall KJ, Poon IK, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PloS One (2014) 9(10):e109596. doi: 10.1371/journal.pone.0109596
111. Miranda S, Billoir P, Le Besnerais M, Joannides R, Richard V, Levesque H, et al. New insights into antiphospholipid-related endothelial dysfunction by assessment of vascular glycocalyx layer: results from a preliminary cross-sectional study. LUPUS (2020) 29(2):157–64. doi: 10.1177/0961203319897958
112. Liu W, Wang Y, Zheng J, Song D, Zheng S, Ren L, et al. Syndecan-1 as an independent risk factor for the incidence of adverse cardiovascular events in patients having stage C and D heart failure with non-ischemic dilated cardiomyopathy. Clin Chim Acta (2019) 490:63–8. doi: 10.1016/j.cca.2018.12.022
113. Horton MR, McKee CM, Bao C, Liao F, Farber JM, Hodge-DuFour J, et al. Hyaluronan fragments synergize with interferon-gamma to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. J Biol Chem (1998) 273(52):35088–94. doi: 10.1074/jbc.273.52.35088
114. van den Berg BM, Wang G, Boels MGS, Avramut MC, Jansen E, Sol W, et al. Glomerular function and structural integrity depend on hyaluronan synthesis by glomerular endothelium. J Am Soc Nephrol (2019) 30(10):1886–97. doi: 10.1681/asn.2019020192
115. Nagy N, Freudenberger T, Melchior-Becker A, Röck K, Ter Braak M, Jastrow H, et al. Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: novel insights into the role of hyaluronan synthesis. Circulation (2010) 122(22):2313–22. doi: 10.1161/circulationaha.110.972653
116. Henry CB, Duling BR. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol (1999) 277(2):H508–14. doi: 10.1152/ajpheart.1999.277.2.H508
117. Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JA, Ince C, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes (2006) 55(4):1127–32. doi: 10.2337/diabetes.55.04.06.db05-1619
118. Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol (2015) 80(3):389–402. doi: 10.1111/bcp.12629
119. Kadoya H, Yu N, Schiessl IM, Riquier-Brison A, Gyarmati G, Desposito D, et al. Essential role and therapeutic targeting of the glomerular endothelial glycocalyx in lupus nephritis. JCI Insight (2020) 5(19): e131252. doi: 10.1172/jci.insight.131252
120. Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med (1997) 242(1):27–33. doi: 10.1046/j.1365-2796.1997.00170.x
121. Yung S, Tsang RC, Leung JK, Chan TM. Increased mesangial cell hyaluronan expression in lupus nephritis is mediated by anti-DNA antibody-induced IL-1beta. Kidney Int (2006) 69(2):272–80. doi: 10.1038/sj.ki.5000042
122. Jiang DH, Liang JR, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev (2011) 91(1):221–64. doi: 10.1152/physrev.00052.2009
123. Padberg JS, Wiesinger A, di Marco GS, Reuter S, Grabner A, Kentrup D, et al. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis (2014) 234(2):335–43. doi: 10.1016/j.atherosclerosis.2014.03.016
124. Feusi E, Sun L, Sibalic A, Beck-Schimmer B, Oertli B, Wuthrich RP. Enhanced hyaluronan synthesis in the MRL-Fas(lpr) kidney: role of cytokines. Nephron (1999) 83(1):66–73. doi: 10.1159/000045475
125. Dane MJ, Khairoun M, Lee DH, van den Berg BM, Eskens BJ, Boels MG, et al. Association of kidney function with changes in the endothelial surface layer. Clin J Am Soc Nephrol (2014) 9(4):698–704. doi: 10.2215/cjn.08160813
126. Pruna A, Peyri N, Berard M, Boffa MC. Thrombomodulin is synthesized by human mesangial cells. Kidney Int (1997) 51(3):687–93. doi: 10.1038/ki.1997.99
127. Lay AJ, Donahue D, Tsai MJ, Castellino FJ. Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood (2007) 109(5):1984–91. doi: 10.1182/blood-2006-07-037945
128. Nozaki Y, Ri J, Sakai K, Niki K, Funauchi M, Matsumura I. Protective effects of recombinant human soluble thrombomodulin on lipopolysaccharide-induced acute kidney injury. Int J Mol Sci (2020) 21(7): 2519. doi: 10.3390/ijms21072519
129. Nesheim M, Wang W, Boffa M, Nagashima M, Morser J, Bajzar L. Thrombin, thrombomodulin and TAFI in the molecular link between coagulation and fibrinolysis. Thromb Haemost (1997) 78(1):386–91. doi: 10.1055/s-0038-1657557
130. Hashiguchi T, Mizumoto S, Nishimura Y, Tamura J, Yamada S, Sugahara K. Involvement of human natural killer-1 (HNK-1) sulfotransferase in the biosynthesis of the GlcUA(3-O-sulfate)-Gal-Gal-Xyl tetrasaccharide found in alpha-thrombomodulin from human urine. J Biol Chem (2011) 286(38):33003–11. doi: 10.1074/jbc.M111.279174
131. Nara H, Okamoto H, Minota S, Yoshio T. Mouse monoclonal anti-human thrombomodulin antibodies bind to and activate endothelial cells through NF-kappaB activation. vitro Arthritis Rheumatol (2006) 54(5):1629–37. doi: 10.1002/art.21797
132. Boehme MW, Deng Y, Raeth U, Bierhaus A, Ziegler R, Stremmel W, et al. Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: in vivo and in vitro studies. Immunology (1996) 87(1):134–40.
133. Constans J, Dupuy R, Blann AD, Resplandy F, Seigneur M, Renard M, et al. Anti-endothelial cell autoantibodies and soluble markers of endothelial cell dysfunction in systemic lupus erythematosus. J Rheumatol (2003) 30(9):1963–6.
134. Yao GH, Liu ZH, Zhang X, Zheng CX, Chen HP, Zeng CH, et al. Circulating thrombomodulin and vascular cell adhesion molecule-1 and renal vascular lesion in patients with lupus nephritis. LUPUS (2008) 17(8):720–6. doi: 10.1177/0961203308089441
135. Boehme MW, Raeth U, Galle PR, Stremmel W, Scherbaum WA. Serum thrombomodulin-a reliable marker of disease activity in systemic lupus erythematosus (SLE): advantage over established serological parameters to indicate disease activity. Clin Exp Immunol (2000) 119(1):189–95. doi: 10.1046/j.1365-2249.2000.01107.x
136. Ohdama S, Yoshizawa Y, Kubota T, Aoki N. Plasma thrombomodulin as an indicator of thromboembolic disease in systemic lupus erythematosus. Int J Cardiol (1994) 47(1 Suppl):S1–6. doi: 10.1016/0167-5273(94)90319-0
137. Boehme MW, Nawroth PP, Kling E, Lin J, Amiral J, Riedesel J, et al. Serum thrombomodulin. A novel marker of disease activity in systemic lupus erythematosus. Arthritis Rheumatol (1994) 37(4):572–7. doi: 10.1002/art.1780370419
138. el-Gamal YM, Heshmat NM, el-Kerdany TH, Fawzy AF. Serum thrombomodulin in systemic lupus erythematosus and juvenile idiopathic arthritis. Pediatr Allergy Immunol (2004) 15(3):270–7. doi: 10.1111/j.1399-3038.2004.00085.x
139. Ho CY, Wong CK, Li EK, Tam LS, Lam CW. Elevated plasma concentrations of nitric oxide, soluble thrombomodulin and soluble vascular cell adhesion molecule-1 in patients with systemic lupus erythematosus. Rheumatol (Oxford) (2003) 42(1):117–22. doi: 10.1093/rheumatology/keg045
140. Hu YQ, Wang ZX, Xiang K, He YS, Feng YT, Shuai ZW, et al. Elevated circulating thrombomodulin levels in systemic lupus erythematosus: A systematic review and meta-analysis. Curr Pharm Des (2022) 28(4):306–12. doi: 10.2174/1381612827666211111152319
141. Kotajima L, Aotsuka S, Sato T. Clinical significance of serum thrombomodulin levels in patients with systemic rheumatic diseases. Clin Exp Rheumatol (1997) 15(1):59–65.
142. Dubin R, Cushman M, Folsom AR, Fried LF, Palmas W, Peralta CA, et al. Kidney function and multiple hemostatic markers: cross sectional associations in the multi-ethnic study of atherosclerosis. BMC Nephrol (2011) 12:3. doi: 10.1186/1471-2369-12-3
143. Adams DH, Shaw S. Leucocyte-endothelial interactions and regulation of leucocyte migration. Lancet (1994) 343(8901):831–6. doi: 10.1016/S0140-6736(94)92029-X
144. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi: 10.1038/nri3908
145. Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, Gotto AM Jr., et al. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation (1997) 96(12):4219–25. doi: 10.1161/01.CIR.96.12.4219
146. Nakai K, Itoh C, Kawazoe K, Miura Y, Sotoyanagi H, Hotta K, et al. Concentration of soluble vascular cell adhesion molecule-1 (VCAM-1) correlated with expression of VCAM-1 mRNA in the human atherosclerotic aorta. Coron Artery Dis (1995) 6(6):497–502.
147. Reynolds HR, Buyon J, Kim M, Rivera TL, Izmirly P, Tunick P, et al. Association of plasma soluble E-selectin and adiponectin with carotid plaque in patients with systemic lupus erythematosus. Atherosclerosis (2010) 210(2):569–74. doi: 10.1016/j.atherosclerosis.2009.12.007
148. Gustafsson J, Gunnarsson I, Borjesson O, Pettersson S, Moller S, Fei GZ, et al. Predictors of the first cardiovascular event in patients with systemic lupus erythematosus - a prospective cohort study. Arthritis Res Ther (2009) 11(6):R186. doi: 10.1186/ar2878
149. Wuthrich RP. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int (1992) 42(4):903–14. doi: 10.1038/ki.1992.367
150. Daniel L, Sichez H, Giorgi R, Dussol B, Figarella-Branger D, Pellissier JF, et al. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney Int (2001) 60(6):2215–21. doi: 10.1046/j.1523-1755.2001.00055.x
151. Wellicome SM, Kapahi P, Mason JC, Lebranchu Y, Yarwood H, Haskard DO. Detection of a circulating form of vascular cell adhesion molecule-1: raised levels in rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Immunol (1993) 92(3):412–18. doi: 10.1111/j.1365-2249.1993.tb03413.x
152. Spronk PE, Bootsma H, Huitema MG, Limburg PC, Kallenberg CG. Levels of soluble VCAM-1, soluble ICAM-1, and soluble E-selectin during disease exacerbations in patients with systemic lupus erythematosus (SLE); a long term prospective study. Clin Exp Immunol (1994) 97(3):439–44. doi: 10.1111/j.1365-2249.1994.tb06107.x
153. Ikeda Y, Fujimoto T, Ameno M, Shiiki H, Dohi K. Relationship between lupus nephritis activity and the serum level of soluble VCAM-1. LUPUS (1998) 7(5):347–54. doi: 10.1191/096120398678920172
154. Hajialilo M, Tayari P, Ghorbanihaghjo A, Khabbazi A, Malek Mahdavi A, Rashtchizadeh N. Relationship between serum vascular cell adhesion molecule-1 and endothelin-1 levels with organ involvement and disease activity in systemic lupus erythematosus patients. LUPUS (2018) 27(12):1918–25. doi: 10.1177/0961203318796285
155. Rho YH, Chung CP, Oeser A, Solus J, Raggi P, Gebretsadik T, et al. Novel cardiovascular risk factors in premature coronary atherosclerosis associated with systemic lupus erythematosus. J Rheumatol (2008) 35(9):1789–94.
156. Fasano S, Pierro L, Borgia A, Coscia MA, Formica R, Bucci L, et al. Biomarker panels may be superior over single molecules in prediction of renal flares in systemic lupus erythematosus: an exploratory study. Rheumatol (Oxford) (2020) 59(11):3193–200. doi: 10.1093/rheumatology/keaa074
157. Wolf BJ, Spainhour JC, Arthur JM, Janech MG, Petri M, Oates JC. Development of biomarker models to predict outcomes in lupus nephritis. Arthritis Rheumatol (2016) 68(8):1955–63. doi: 10.1002/art.39623
158. Soliman S, Mohamed FA, Ismail FM, Stanley S, Saxena R, Mohan C. Urine angiostatin and VCAM-1 surpass conventional metrics in predicting elevated renal pathology activity indices in lupus nephritis. Int J Rheum Dis (2017) 20(11):1714–27. doi: 10.1111/1756-185x.13197
159. Parodis I, Gokaraju S, Zickert A, Vanarsa K, Zhang T, Habazi D, et al. ALCAM and VCAM-1 as urine biomarkers of activity and long-term renal outcome in systemic lupus erythematosus. Rheumatol (Oxford) (2020) 59(9):2237–49. doi: 10.1093/rheumatology/kez528
160. Mok CC, Soliman S, Ho LY, Mohamed FA, Mohamed FI, Mohan C. Urinary angiostatin, CXCL4 and VCAM-1 as biomarkers of lupus nephritis. Arthritis Res Ther (2018) 20(1):6. doi: 10.1186/s13075-017-1498-3
161. Kiani AN, Wu T, Fang H, Zhou XJ, Ahn CW, Magder LS, et al. Urinary vascular cell adhesion molecule, but not neutrophil gelatinase-associated lipocalin, is associated with lupus nephritis. J Rheumatol (2012) 39(6):1231–7. doi: 10.3899/jrheum.111470
162. Stanley S, Vanarsa K, Soliman S, Habazi D, Pedroza C, Gidley G, et al. Comprehensive aptamer-based screening identifies a spectrum of urinary biomarkers of lupus nephritis across ethnicities. Nat Commun (2020) 11(1):2197. doi: 10.1038/s41467-020-15986-3
163. Soliman SA, Stanley S, Vanarsa K, Ismail F, Mok CC, Mohan C. Exploring urine:serum fractional excretion ratios as potential biomarkers for lupus nephritis. Front Immunol (2022) 13:910993. doi: 10.3389/fimmu.2022.910993
164. Egerer K, Feist E, Rohr U, Pruss A, Burmester GR, Dorner T. Increased serum soluble CD14, ICAM-1 and E-selectin correlate with disease activity and prognosis in systemic lupus erythematosus. LUPUS (2000) 9(8):614–21. doi: 10.1191/096120300678828749
165. Sfikakis PP, Charalambopoulos D, Vayiopoulos G, Oglesby R, Sfikakis P, Tsokos GC. Increased levels of intercellular adhesion molecule-1 in the serum of patients with systemic lupus erythematosus. Clin Exp Rheumatol (1994) 12(1):5–9.
166. Kling E, Bieg S, Boehme M, Scherbaum WA. Circulating intercellular adhesion molecule 1 as a new activity marker in patients with systemic lupus erythematosus. Clin Investig (1993) 71(4):299–304. doi: 10.1007/BF00184731
167. Sari RA, Taysi S, Erdem F, Yilmaz O, Keles S, Kiziltunc A, et al. Correlation of serum levels of soluble intercellular adhesion molecule-1 with disease activity in systemic lupus erythematosus. Rheumatol Int (2002) 21(4):149–52. doi: 10.1007/s00296-001-0159-6
168. Tulek N, Aydintug O, Ozoran K, Tutkak H, Duzgun N, Duman M, et al. Soluble intercellular adhesion molecule-1 (sICAM-1) in patients with systemic lupus erythematosus. Clin Rheumatol (1996) 15(1):47–50. doi: 10.1007/BF02231684
169. Wais T, Fierz W, Stoll T, Villiger PM. Subclinical disease activity in systemic lupus erythematosus: immunoinflammatory markers do not normalize in clinical remission. J Rheumatol (2003) 30(10):2133–9.
170. Mason JC, Kapahi P, Haskard DO. Detection of increased levels of circulating intercellular adhesion molecule 1 in some patients with rheumatoid arthritis but not in patients with systemic lupus erythematosus. Lack of correlation with levels of circulating vascular cell adhesion molecule 1. Arthritis Rheumatol (1993) 36(4):519–27. doi: 10.1002/art.1780360412
171. Lhotta K, Schlogl A, Kronenberg F, Joannidis M, Konig P. Soluble intercellular adhesion molecule-1 (ICAM-1) in serum and urine: correlation with renal expression of ICAM-1 in patients with kidney disease. Clin Nephrol (1997) 48(2):85–91.
172. Skeoch S, Haque S, Pemberton P, Bruce IN. Cell adhesion molecules as potential biomarkers of nephritis, damage and accelerated atherosclerosis in patients with SLE. LUPUS (2014) 23(8):819–24. doi: 10.1177/0961203314528061
173. Chan TM, Yu PM, Tsang KL, Cheng IK. Endothelial cell binding by human polyclonal anti-DNA antibodies: relationship to disease activity and endothelial functional alterations. Clin Exp Immunol (1995) 100(3):506–13. doi: 10.1111/j.1365-2249.1995.tb03730.x
174. Wuthrich RP, Jevnikar AM, Takei F, Glimcher LH, Kelley VE. Intercellular adhesion molecule-1 (ICAM-1) expression is upregulated in autoimmune murine lupus nephritis. Am J Pathol (1990) 136(2):441–50.
175. Monroy Trujillo JM, Fine DM. Lupus nephritis in the era of biomarkers. Clin J Am Soc Nephrol (2016) 11(1):4–5. doi: 10.2215/cjn.12371115
Keywords: lupus nephritis, glycocalyx, syndecan-1, thrombomodulin, hyaluronan, cell adhesion molecules
Citation: Yung S and Chan TM (2023) Endothelial cell activation and glycocalyx shedding - potential as biomarkers in patients with lupus nephritis. Front. Immunol. 14:1251876. doi: 10.3389/fimmu.2023.1251876
Received: 02 July 2023; Accepted: 18 September 2023;
Published: 03 October 2023.
Edited by:
Jian Gao, Shanghai Children’s Medical Center, ChinaCopyright © 2023 Yung and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Yung, c3N5eXVuZ0Boa3UuaGs=; Tak Mao Chan, ZHRtY2hhbkBoa3UuaGs=