 Lilli Leimi1*
Lilli Leimi1* Jessica R. Koski2,3,4Outi Kilpivaara2,3,4,5
Jessica R. Koski2,3,4Outi Kilpivaara2,3,4,5 Kim Vettenranta1
Kim Vettenranta1 A. Inkeri Lokki6†
A. Inkeri Lokki6† Seppo Meri5,6†
Seppo Meri5,6†- 1Pediatric Research Center, Children’s Hospital, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 2Applied Tumor Genomics Research Program, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 3Department of Medical and Clinical Genetics, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 4Medicum, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 5Diagnostic Center, Helsinki University Hospital, Helsinki, Finland
- 6Department of Bacteriology and Immunology and Translational Immunology Research Program, University of Helsinki, Helsinki, Finland
Introduction: Complement system has a postulated role in endothelial problems after hematopoietic stem cell transplantation (HSCT). In this retrospective, singlecenter study we studied genetic complement system variants in patients with documented endotheliopathy. In our previous study among pediatric patients with an allogeneic HSCT (2001-2013) at the Helsinki University Children´s Hospital, Finland, we identified a total of 19/122 (15.6%) patients with vascular complications, fulfilling the criteria of capillary leak syndrome (CLS), venoocclusive disease/sinusoidal obstruction syndrome (VOD/SOS) or thrombotic microangiopathy (TMA).
Methods: We performed whole exome sequencing (WES) on 109 patients having an adequate pre-transplantation DNA for the analysis to define possible variations and mutations potentially predisposing to functional abnormalities of the complement system. In our data analysis, we focused on 41 genes coding for complement components.
Results: 50 patients (45.9%) had one or several, nonsynonymous, rare germline variants in complement genes. 21/66 (31.8%) of the variants were in the terminal pathway. Patients with endotheliopathy had variants in different complement genes: in the terminal pathway (C6 and C9), lectin pathway (MASP1) and receptor ITGAM (CD11b, part of CR3). Four had the same rare missense variant (rs183125896; Thr279Ala) in the C9 gene. Two of these patients were diagnosed with endotheliopathy and one with capillary leak syndrome-like problems. The C9 variant Thr279Ala has no previously known disease associations and is classified by the ACMG guidelines as a variant of uncertain significance (VUS). We conducted a gene burden test with gnomAD Finnish (fin) as the reference population. Complement gene variants seen in our patient population were investigated and Total Frequency Testing (TFT) was used for execution of burden tests. The gene variants seen in our patients with endotheliopathy were all significantly (FDR < 0.05) enriched compared to gnomAD. Overall, 14/25 genes coding for components of the complement system had an increased burden of missense variants among the patients when compared to the gnomAD Finnish population (N=10 816).
Discussion: Injury to the vascular endothelium is relatively common after HSCT with different phenotypic appearances suggesting yet unidentified underlying mechanisms. Variants in complement components may be related to endotheliopathy and poor prognosis in these patients.
1 Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a well-established therapy for hematologic and lymphoid malignancies, but also for disorders of the immune or hematopoietic systems and metabolism incurable by other treatments. Despite the improvement in prognosis, toxicity and treatment-related mortality (TRM) still remain challenges (1, 2). In our recent study (3) nearly half (56/122, 45.9%) of the pediatric patients, who underwent allo-HSCT, had at least one severe adverse event (grade 3 or 4 by the Common Terminology Criteria for Adverse Events, CTCAE, 4.03 classification). Endothelial cell activation and dysfunction have recently been indicated in many severe complications of allo-HSCT (4). Thrombotic microangiopathy (TMA) is one of the best known endotheliopathy syndromes and a relatively common complication after HSCT affecting 10-20% of the patients (5–8). Previously, we identified a total of 19/122 (15.6%) patients with vascular complications, fulfilling the criteria of capillary leak syndrome (CLS), veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) or TMA. The patients with endotheliopathy had a poorer 5-year overall survival than those without it (77% versus 26%, p<0.001) (3).
Endothelial cells can be damaged and activated during the HSCT process by several mechanisms, including previous chemotherapy, conditioning (e.g. irradiation), drugs used in the transplantation process (such as G-CSF or calcineurin inhibitors), cytokines produced by activated immune cells or the injured tissues, infections, engraftment and alloreactivity (9–11). Injury and inflammation involve neutrophil activation leading to formation of neutrophil extracellular traps (NETs). NETs and other released intracellular materials are thought to activate the complement system (12, 13). On the other hand, overactive or activated complement system has been shown to disturb the normal function of the endothelium in many disease states (14–18). Previously, endotheliopathy in HSCT was considered to follow the “two-hit” –hypothesis. First, many risk factors in the conventional chemo or conditioning phase make the endothelium more vulnerable and procoagulant (Hit 1). This is followed by one or more factors (medications, infections, possible antibodies, or alloreactivity) in the post-HSCT recovery phase (Hit 2) promoting endothelial injury and related problems (19). Although not emphasized earlier, the second hit could, in addition to procoagulant activity, also involve “procomplement” activity with changes in the ability of endothelial cells to protect themselves against complement attack (5). Currently a “three hit” –hypothesis, where the first hit is the patients´ genetic predisposition to endothelial complications, is considered most relevant to the disease pathogenesis (5, 11, 20, 21).
Problems characterized by endothelial dysfunction vary in their clinical phenotype. Still, all of these share a common denominator: endothelial injury accompanied by proinflammatory, prothrombotic and proapoptotic processes likely related to complement activation (4, 8, 10). Here we focused on the following three entities: capillary leak syndrome (CLS), HSCT-related thrombotic microangiopathy (HSCT-TMA) and veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS). Other endotheliopathy syndromes, such as engraftment syndrome (ES), idiopathic pneumonia syndrome (IPS), posterior reversible encephalopathy syndrome (PRES), peri-engraftment respiratory distress syndrome (PERDS) and refractory acute GVHD will not be addressed here.
In this retrospective study, because of the postulated role of complement, we studied genetic complement system variants in patients with documented endotheliopathy. Thereby, our aim was to lay basis for understanding potential mechanisms behind the acute complications seen post-HSCT.
2 Materials and methods
2.1 Patients
Patient selection was accomplished as previously described (3). We studied a cohort of 109 pediatric patients, who underwent allo-HSCT between 1/2001 and 12/2013 at the Helsinki University Children´s Hospital, Finland, and had an adequate pre-transplantation DNA available for whole exome sequencing (WES) and further bioinformatics and statistical analysis. The key demographic and clinical data of the endotheliopathy patients versus others are given in Table 1. All patients were of Finnish ancestry.
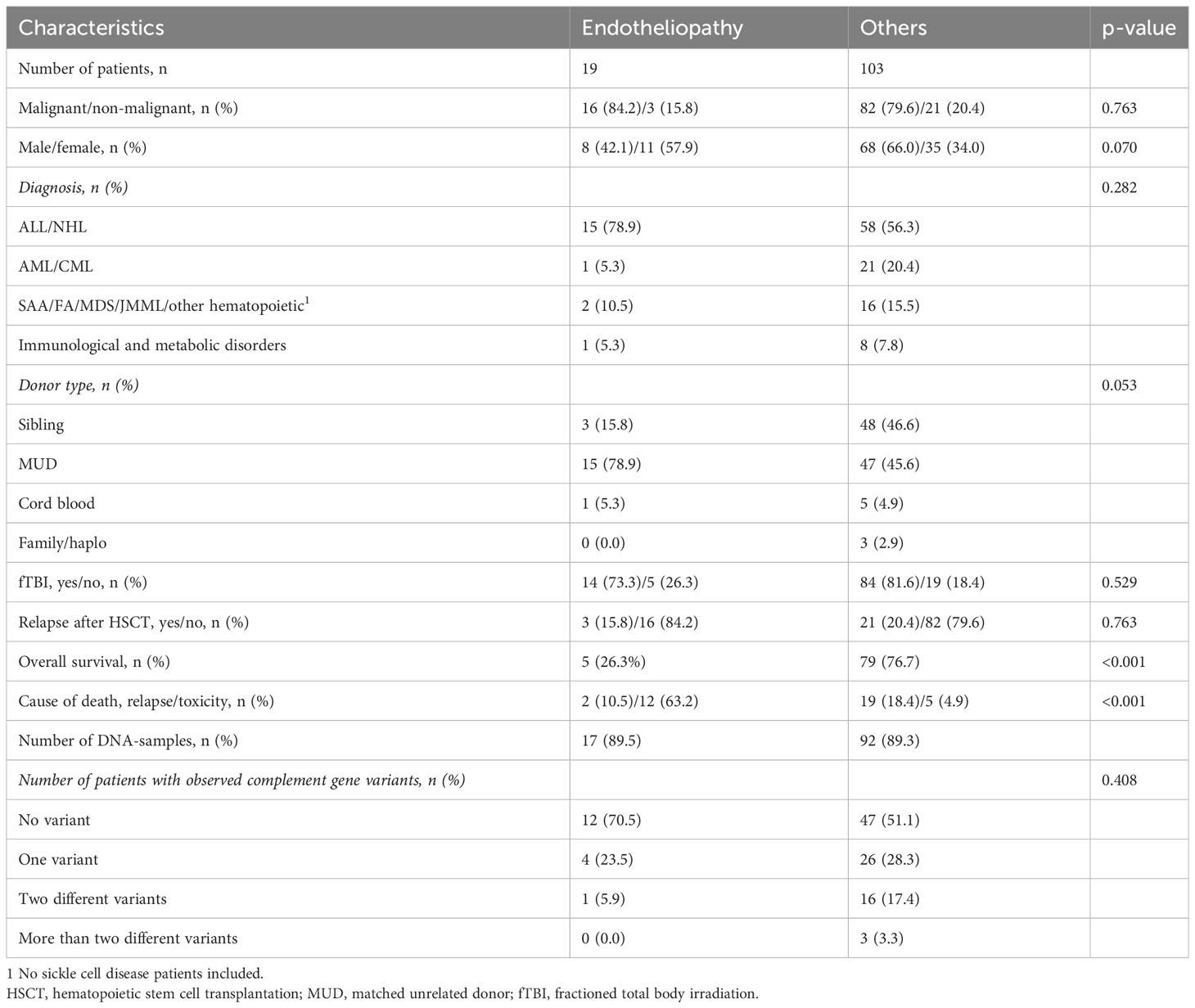
Table 1 The demographics of the patients with endotheliopathy and those without having received allo-HSCT at the Helsinki University Children´s Hospital during 2001-2013.
The study was approved by the Research Ethics Committee of the Helsinki University Hospital (79/13/03/03/2016, update 3082/2018). All samples and data were obtained following authorization by the ethics committee and the national supervisory authority Valvira. Written, informed consents were not attainable due to the retrospective nature of the study also including deceased patients.
2.2 Outcome measures
DNA analysis aimed to define possible variations and mutations in classical, lectin or terminal pathway factors or in the membrane-bound components of the complement system, potentially predisposing to activation or dysfunction of the complement cascade.
2.3 Data collection
Genomic DNA was isolated by the Finnish Red Cross Blood Service (HLA-lab), Helsinki, Finland, from pre-transplant peripheral blood samples. DNA-samples were collected from both the recipient and donor for HLA-matching. Clinical data was collected retrospectively from the medical records focusing on the first 100 days post-transplant as described previously (3).
2.4 Whole exome sequencing
WES was performed at Blueprint Genetics, Helsinki, Finland. The sample preparation and exome sequencing have been previously described (22). In data analysis, we focused on 41 genes coding for components of the classical, lectin and terminal pathways as well as for complement receptors and membrane-bound complement regulators. The list of studied genes is presented in the Supplementary Table S1.
2.5 Bioinformatics analysis
The germline variant analysis was performed using BasePlayer (v.1.0.2) (23). Raw variant calls were filtered to include only good quality variants in the set (1000 genomes mapability track) (24) and quality measures GQ>=20, QUAL>=20, coverage>6 reads, and AF>=30%). Only non-synonymous and splice variants in protein-coding transcripts with a minor allele frequency (MAF) of < 0.01 in population-specific controls gnomAD (v.2.1) non-cancer whole database and gnomAD non-cancer Finns (n=10 816) (25) were considered in the study. Variants were classified as pathogenic (P), likely pathogenic(LP), benign (B), likely benign (LB) or variants of uncertain significance (VUS) according to the American College of Medical Genetics and Genomics (ACMG) (26) guidelines using InterVar (27). Also ClinVar (version 23.01.2021) was used for the variant pathogenicity estimation (28).
Findings were validated visually with BasePlayer. Locations of amino acid variants in protein structure and potential effects on protein function were analyzed by UniProt (https://www.uniprot.org) with RCSB PDB (RCSB.org) (29) data models 5FMW (30) and 7AKK (31) and drawn with Biorender (https://www.biorender.com/).
2.6 Statistical analysis
To study the enrichment of rare variants in the genes studied, the frequency of variants in the patient set and among gnomAD non-cancer Finns were compared. The significances of differences were evaluated using 2-sided Fisher’s exact test. Burden test of variants was done using the Total Frequency Test (TFT) (32) by collapsing rare germline variants to the gene and pathway level. These were compared to the total allele counts of variants in gnomAD non-cancer Finns with the same filtering as in the patient set. Benjamini-Hochberg procedure was used to adjust the p-values for multiple testing. The analyses were performed in R (version 4.1.2, https://www.r-project.org/).
3 Results
The patient data and clinical findings of our patient cohort have been published before (3). In brief, the original patient cohort consisted of 122 patients with an allogeneic HSCT between 2001 and 2013 at the Helsinki University Children´s Hospital, Finland. We evaluated the acute adverse events emerging ≤ 100 days post-transplant and prevalence of key vascular complications. We identified a total of 19/122 (15.6%) patients with vascular complications, fulfilling the criteria of capillary leak syndrome (CLS), veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) or thrombotic microangiopathy (TMA). These patients had a poorer 5-year overall survival than those without vascular complications (26% versus 77%, p<0.001). These endotheliopathy-related adverse events appeared early on post-HSCT, varied in their clinical phenotype and predicted a poor outcome.
3.1 Gene analysis
Samples for genetic analyses were available from 109 patients, of whom 17 had endotheliopathy. Of the 109 patients 50 (45.9%) had one or several, nonsynonymous, rare (MAF <0.01) germline variants. These 109 patients had altogether 51 different and in total 66 variants in 25 complement genes of soluble classical, lectin or terminal pathway factors or in the membrane-bound components (receptors or regulators). As many as 21/66 (31.8%) of the variants were in the terminal pathway. The list of gene variants is presented in Supplementary Table S2.
Patients with endotheliopathy had variants in several complement pathways. The most notable ones were detected in the terminal pathway (C6 and C9), lectin pathway (MASP1) and in ITGAM encoding for CD11b, a part (the alpha-chain) of the β-integrin complement receptor type 3, CR3 (Figure 1). Examples of discovered variants are given in Figures 2, 3, where the locations of the variant in protein domain sequences (2A and 3A) and structures (2B and 3B) are indicated. Four patients had the same rare (gnomAD Finn MAF= 0.009608) missense variant (rs183125896; Thr279Ala) in the C9 gene. Two of these patients were diagnosed with endotheliopathy, CLS followed by thrombotic microangiopathy (TMA), and furthermore one with a capillary leak syndrome-like (CLS-like) phenotype. One of the patients with CLS and TMA had only this C9 variant, the other also a variant in C1s (rs149869489; Pro428Arg). This C9 variant has no previously known disease associations and is classified by the ACMG guidelines as a variant of uncertain significance (VUS). A schematic C9 protein structure and the location of the variant in both C9 and the MAC complex is shown in Figure 2. Two patients had a rare (gnomAD Finn MAF= 0.009841) variant (rs142896559; Gly452Glu) in the C6 gene. One of these patients was diagnosed with CLS, severe anal pain and diarrhea. This C6 variant is classified as likely benign.
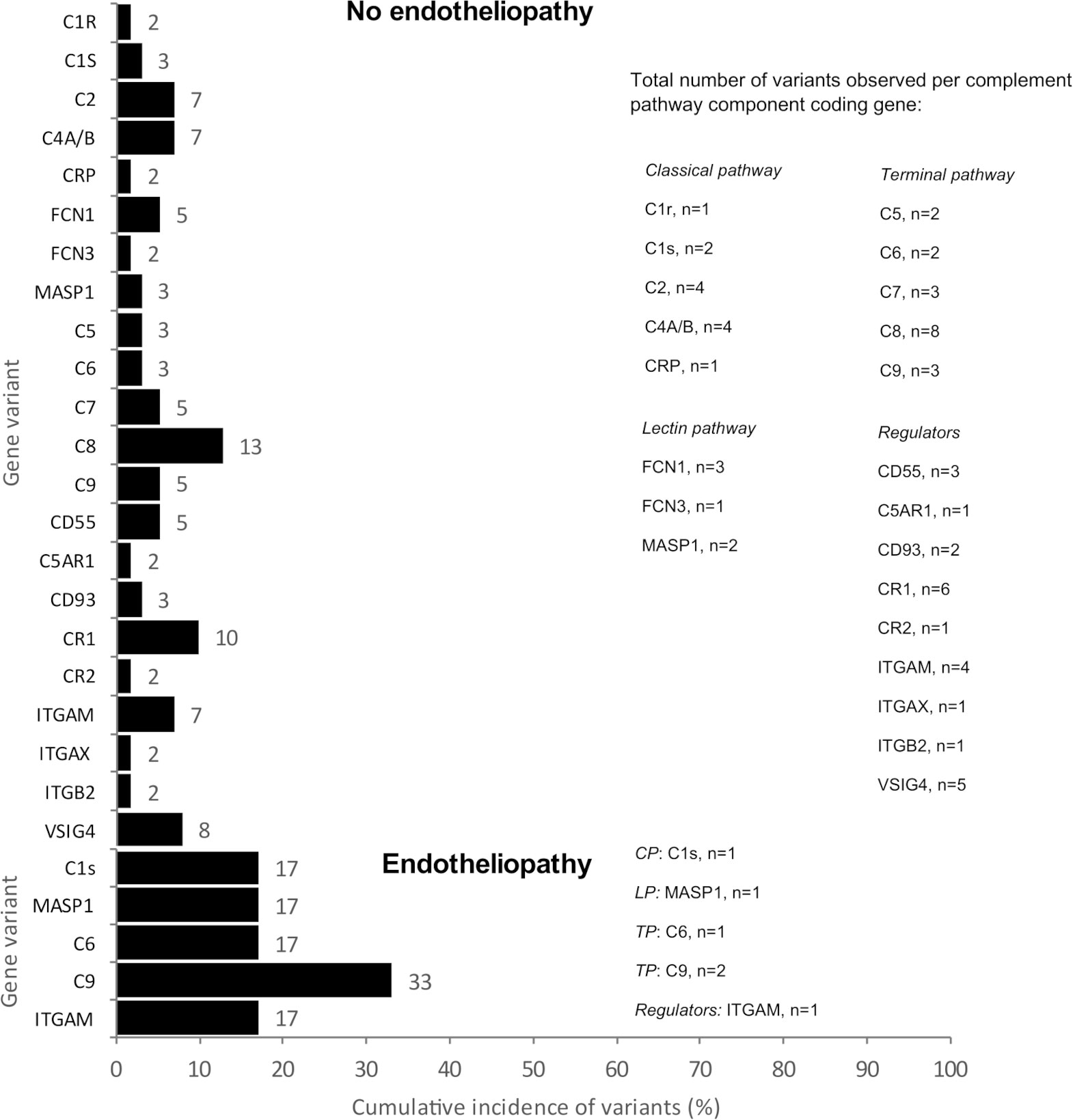
Figure 1 The cumulative incidence (%) and the total number (n) of variants observed per complement pathway component coding gene. The number of variants per gene is equal to the number of patients with a variant in that gene. Above are shown the variants of the patients without endotheliopathy (patient n=92) and below those with endotheliopathy (patient n=17). C1r, complement component C1r subcomponent; C1s, complement component C1s subcomponent; C2, complement component C2; C4A/B, complement components C4A and C4B; CRP, C-reactive protein; FCN1, Ficolin-1 (M-ficolin); FCN3, Ficolin-3; MASP1, Mannan-binding lectin serine protease 1; C5, complement component C5; C6, complement component C6; C7, complement component C7; C8, complement component C8; C9, complement component C9; CD55, DAF, Decay-accelerating factor; C5AR1, C5a anaphylatoxin chemotactic receptor 1; CD93, complement component C1q receptor; CR1, complement receptor type 1; CR2, complement receptor type 2; ITGAM, integrin subunit alpha M; complement receptor type 3, ITGAX, integrin subunit alpha X; ITGB2, integrin beta-2; VSIG4, V-set and immunoglobulin domain-containing protein 4 (also called complement and Ig receptor/CRIg).
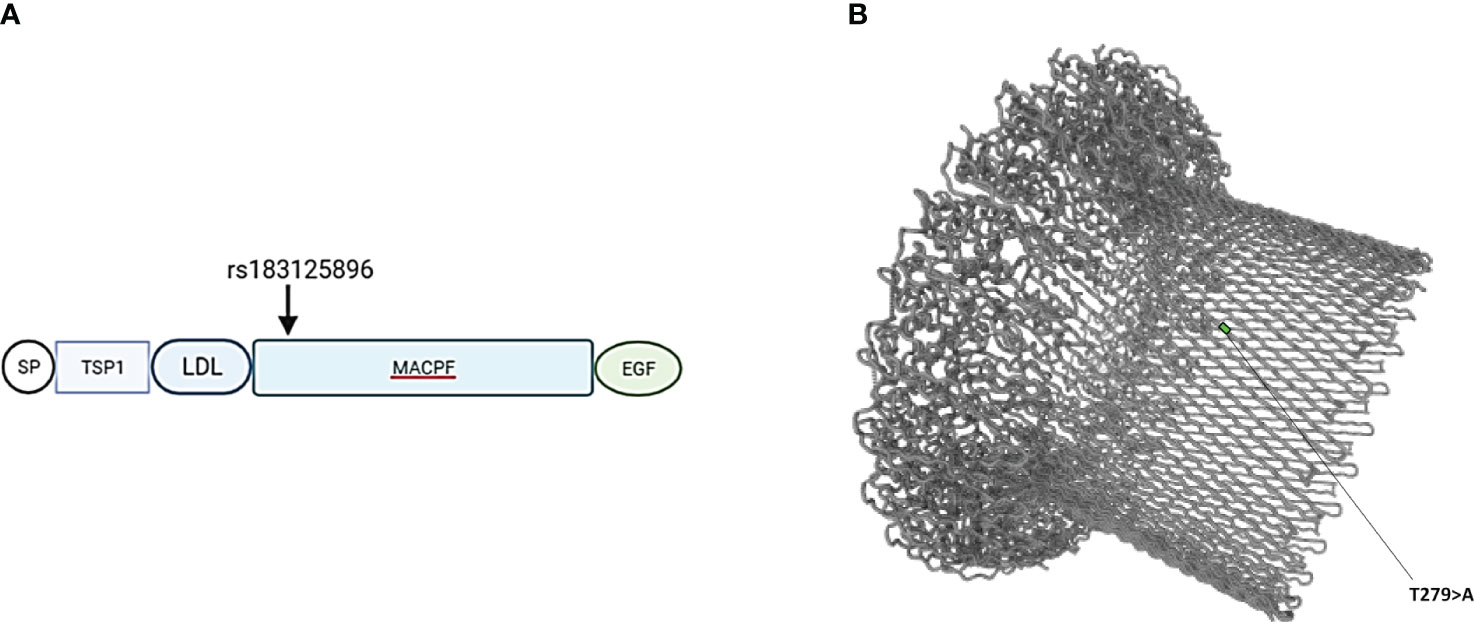
Figure 2 (A) Schematic location of variant rs183125896 encoded amino acid (Thr279Ala) in the domain structure of C9 protein. Variant is located in exon 6, which codes for the MACPF (membrane attack complex/perforin) domain. (B) Barrel structure of the membrane attack complex (https://www.uniprot.org). TSP1, thrombospondin type-1 repeat; LDL, low-density lipoprotein receptor class A repeat; EGF, epidermal growth factor- like domain.
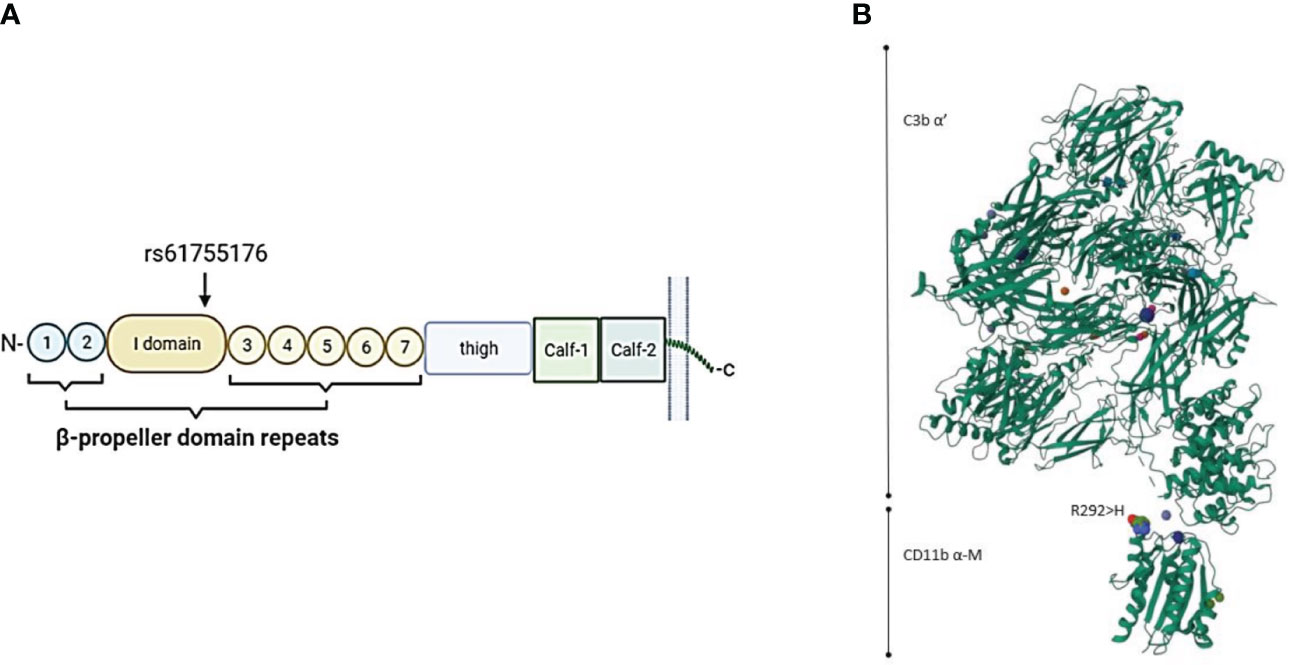
Figure 3 (A) Schematic location of the ITGAM variant rs61755176 encoded amino acid (Arg292His) in the CR3 alpha-chain (CD11b). The variant is located in exon 9 coding the edge of the I-domain surrounded by the beta-propeller domain repeat domains in the CD11b part (coded by ITGAM) of the CR3 receptor. (B) 3-dimensional structure of the CD11b-iC3b complex (https://www.uniprot.org). The variant amino acid is located close to the binding site of CR3 to iC3b.
We found four different integrin alpha-M (ITGAM) variants in five patients, all classified as VUS. One of our patients with a missense variant in the ITGAM gene (rs61755176; Arg292His: gnomAD Finn MAF= 0.0005198) was diagnosed with CLS and TMA, but also showing signs of VOD. The other patient with a missense variant in the ITGAM gene (rs775141495; Ile13Ser: gnomAD Finn MAF<0.0001) had a similar gastrointestinal bleeding and bilirubin increase suggesting endotheliopathy, but not fulfilling the diagnostic criteria for TMA or VOD. This patient also developed hypertension and acute respiratory distress syndrome (ARDS) and died D+65 post-transplant. No infection etiology was found for the patient´s increasing oxygen requirement and pulmonary problems. The schematic structure of the α-M domain of the CR3 protein in complex with C3b and the location of variant rs61755176 on the CR3 is shown in Figure 3. According to this structure, the location of Arg292His is close to the attachment site to iC3b. On the other hand, and although the published protein structure of CR3 includes the integrin α-M-chain, it does not include the Ile13Ser variant (rs775141495) located in the first blade of the β-propeller domain of CD11b. Thus, no predictions can be made to its expected effect on CR3 function.
One patient with severe CLS and TMA/VOD with rapid weight gain, ascites, pleural effusion, elevated bilirubin, hepatomegaly, low levels of coagulation factors and GI-bleeding, had an ultrarare missense variant in the MASP1 gene (rs1712360640; Ala544Thr). This variant was absent in gnomAD but found in ALFA project Europe with an allele frequency <0.0001 and is classified as VUS. The variant has not been reported in ClinVar. This variant is located in the serine protease domain, but its possible effect on the protease function is not known.
Compared to gnomAD, the gene variants seen in our patients with endotheliopathy were all significantly (FDR < 0.05) enriched. The associations between the genetic findings and endotheliopathy diagnoses are shown in Figure 4. Overall, 14/25 genes coding for components of the complement system had an increased burden of missense variants among our patients in comparison to the gnomAD Finnish population. In our pathway burden analysis, variants of the classical pathway of complement activation were found to be enriched in the patients in comparison to gnomAD controls. All complement gene variants enriched in our patient population are shown in Table 2. Although a fair comparison can be made only to the gnomAD Finnish population, we also made a comparison to gnomAD Europe (Supplementary Table S2). It showed features that distinguish the Finnish population from the rest of Europeans.
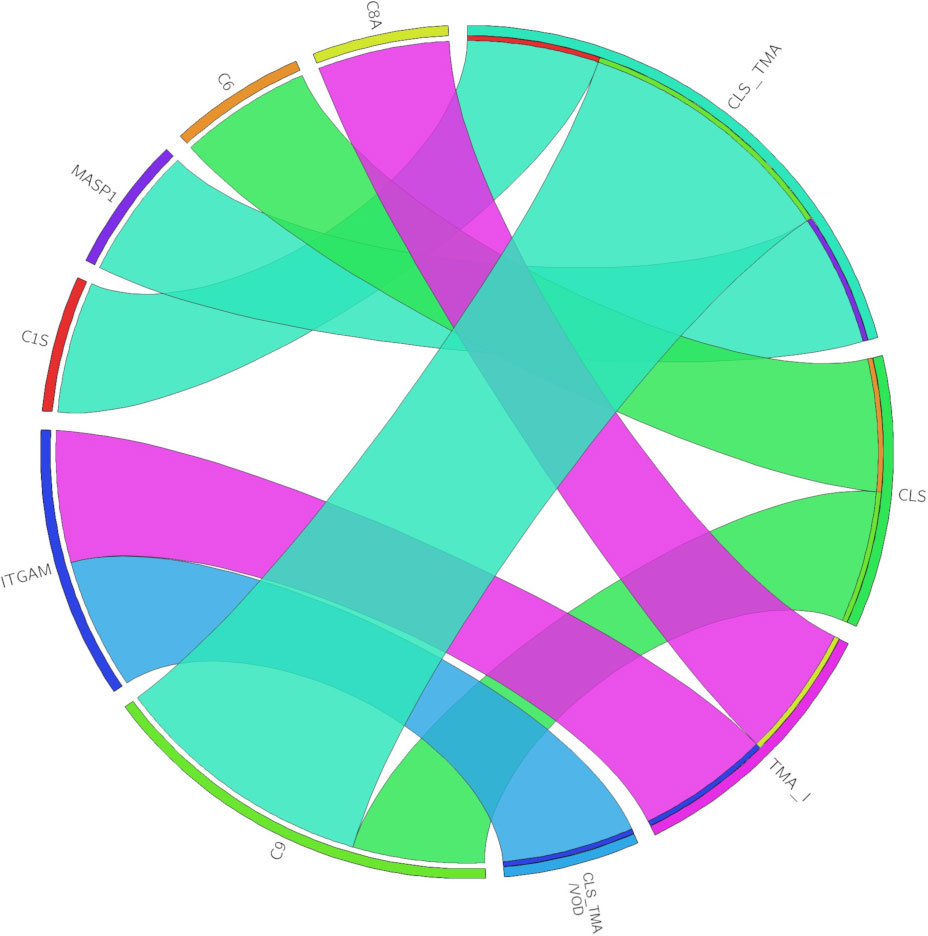
Figure 4 The chord diagram depicts the associations between genetic findings (left) and diagnoses (right). The width of each chord is determined by the number of patients in the diagnosis group with variants in the gene. CLS, capillary leak syndrome; TMA, thrombotic microangiopathy; TMA_I, TMA-like; CLS_TMA, CLS+TMA; CLS_TMA/VOD, CLS+TMA/VOD.
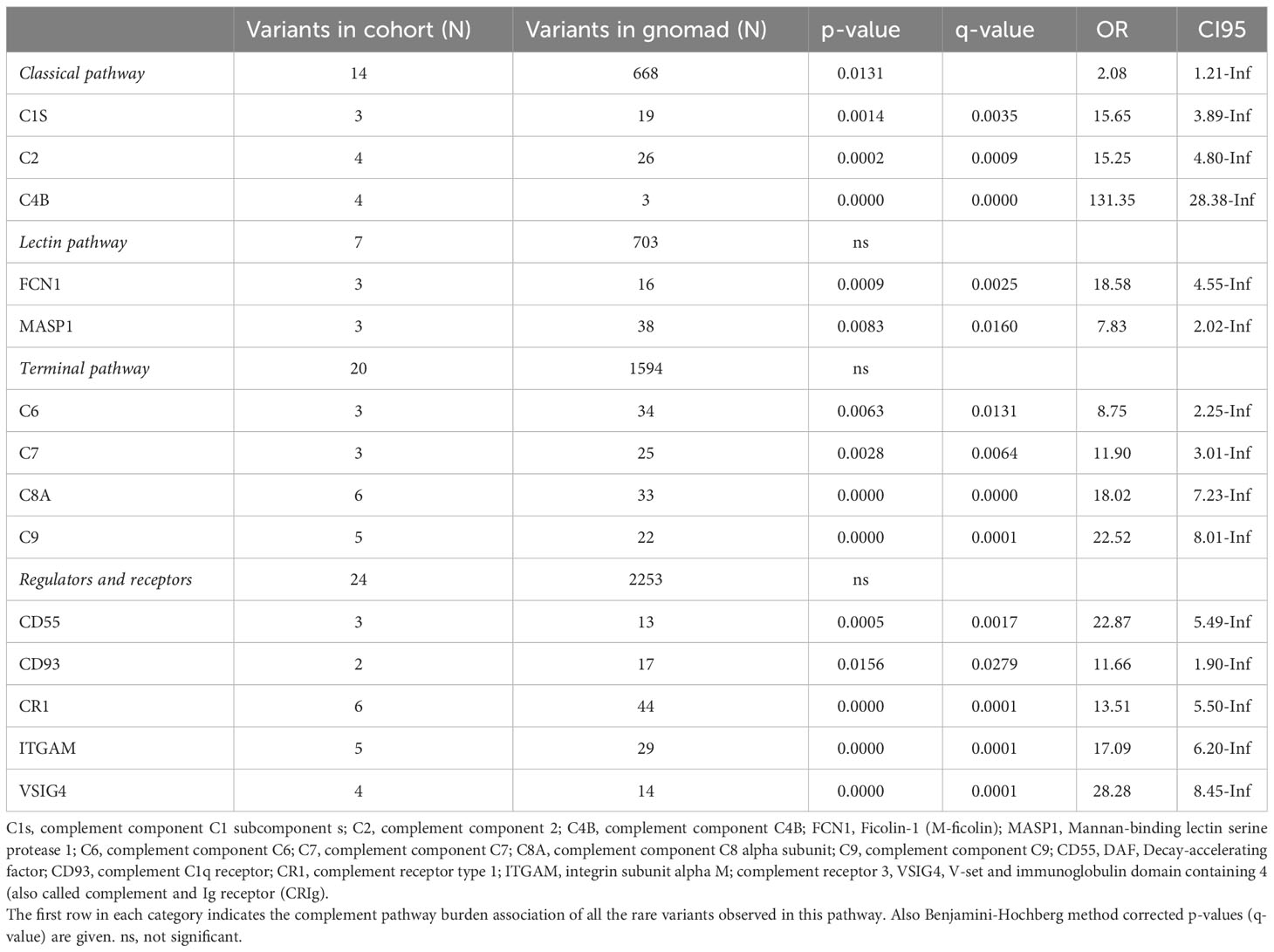
Table 2 Complement gene variants enriched in the patient population (N=109) compared to gnomAD non-cancer Finn population (N=10 816).
4 Discussion
The present study found a higher-than-expected frequency of rare complement gene variants possibly related to vascular complications following HSCT in children. Particularly interesting associations were found to the terminal pathway complement components C6 and C9 as well as to the main phagocytic complement receptor type 3 (CR3, CD11b/CD18). While pathway-burden analysis indicated that rare missense variants in genes coding for classical pathway are enriched in patients in general, from the point of view of endotheliopathy, main links were found to the terminal pathway complement components.
We found five patients with mutations in the ITGAM gene encoding for CD11b (integrin alpha-M), a central part of the key complement receptor CR3. CD11b alpha chain forms the CR3 receptor together with the integrin beta 2 chain (CD18 encoded by ITGB2). CR3 binds structures coated with iC3b, which is an inactivated form of C3b. CR3 regulates the adherence of neutrophils and monocytes to stimulated endothelium. Importantly, CR3 mediates the phagocytosis of complement iC3b-coated microbes and other particles. CR3 functions also as a receptor for the coagulation system proteins fibrinogen and factor X (FX). Mutated ITGAM may thus result in diminished neutrophil adhesion and migration, reduced phagocytosis, increased cytokine production and elevated anti-dsDNA antibody formation, which may further trigger the complement cascade (33). Genetic variants in the ITGAM gene are strongly associated with systemic lupus erythematosus (SLE) and its complications (lupus nephritis, etc) (33), as well as with pre-eclampsia (34). CR3 and CR4 are both β2-integrins that specifically recognize iC3b. It has been previously found that variant rs2230424 in CR4, located in the first blade of the β-propeller domain, results in a decreased binding capacity of CR4 to iC3b (34). We speculate, that ITGAM variant (rs775141495), also in the first blade of the β-propeller domain, may also interfere with the binding of CR3 to iC3b.
Acute respiratory distress syndrome (ARDS) is characterized by an increased permeability of endothelium and epithelium leading to an inflammatory reaction with accumulation of neutrophils and a protein-rich edema of the lungs. Later, also macrophages, strongly expressing CR3, promote the inflammatory response. Biomarkers of the endothelial damage have been linked to increased morbidity and mortality (35–37). One of our patients with the ITGAM variant (rs775141495; Arg292His) had an endotheliopathy-like clinical phenotype with ARDS. The patient´s ARDS symptoms evolved from day 30 post-transplant, thus matching the timeline of the post-engraftment acute phase (30-100 days post-transplant) pulmonary complications. An impaired cellular immunity plays a central role in this phase in both the infectious and non-infectious complications with the latter including many entities related to endothelial damage (38, 39). Our patient with the ITGAM variant Arg292His with CLS followed by TMA/VOD was treated with defibrotide (6.25mg/kg iv q. 6 hours) and survived. The other one with ARDS was not treated with defibrotide and died. Defibrotide is found to diminish the endothelial activation and enhance the local antithrombotic effects, while having also anti-inflammatory effects by interacting with the endothelium (40). In the clinical trial by Frame et al. (41) defibrotide was found safe, and even with the study not being designed to address the efficacy of defibrotide, the survival rates of the patients treated with defibrotide were promising, when compared to other patients with SARS-CoV-2 ARDS.
Mannan-binding lectin-associated serine protease -1 (MASP-1) is an initial part of the lectin pathway and can promote activation of both complement and coagulation. It is a procoagulant by inducing fibrin formation with thrombin, but it also directly activates prothrombin, FXIII and thrombin-activatable fibrinolysis inhibitor (TAFI) (42, 43). Thrombin, on the other hand, has been reported to directly activate the complement cascade by cleaving C3 and C5 (44). The lectin pathway was suggested to mediate complement activation in SARS-CoV-2 infection, and the MASP2-inhibitor narsoplimab had a promising effect in critically ill COVID-19 patients (45). The lectin pathway may also play a role in HSCT-related TMA. Adult patients with HSCT-TMA treated in a phase II study with narsoplimab showed significant improvement in TMA-related laboratory values, and the treatment resulted in a good clinical response with improved overall survival (46). Our patient with the MASP-1 variant Ala544Thr had severe and persistent diffuse gastrointestinal bleeding leading to ICU-admission twice and ending up being lethal. Massive haemorrhage is thought to be a possible trigger of complement activation (47, 48) and activated complement could contribute to tissue damage seen at the site of the haemorrhage (49, 50). We speculate that the MASP-1 variant rs1712360640 with severe GI-bleeding could lead to a more vigorous activation of the complement and coagulation cascade, leading to endothelium-related complications.
C6 is an essential part of the membrane attack complex (MAC). The variant (rs142896559; Gly452Glu) seen in our patient with CLS in located in the MAC perforin (MACPF) domain of the C6 protein (51). The variants seen in C6 and C9 are both located in the MACPF domain. The functional consequences of this variant in C6 are currently not known. However, the essential role of C6 in the generation of the complement membrane attack complex (MAC) and location of the variant in one of the key domains suggests that it could be involved in abnormal assembly of MAC.
While the functional consequences of the C9 variant (rs183125896; Thr279Ala) observed in four patients are also unclear, interestingly, reports on other C9 variants in connection to TMA have recently been published (52, 53). It is therefore possible that, in the HSCT patients, this mutation, normally tolerable, could contribute to triggering TMA.
Many variants were overrepresented in the whole HSCT population (Table 2). Interestingly, also transplant patients without vascular problems showed enrichment of variants in many complement genes. This could be related to the underlying reasons for the HSCT (usually malignancy, but also hematopoietic syndromes, immunological or metabolic disorders; Table 1). In this scenario, complement may have functioned insufficiently to prevent the disorder. Furthermore, the variants observed in the endotheliopathy patients were enriched in the patient population, although the small sample size of endotheliopathy patients did not warrant direct statistical comparison. A reason for this could be an incompatibility in the complement system between the recipient and the donor. Components from the donor and the recipient (e.g. between an activating and a regulating factor) may have a mismatch with each other and lead to events that harm the vascular endothelium. This hypothesis is supported by the fact that patients with endotheliopathy had more matched unrelated donors than those without endotheliopathy (p=0.031).
The limitation of our study is the size of our cohort. Gene burden analysis may be influenced by the relatively small N and the findings should be evaluated using a larger validation cohort. The strength of our study is the uniform Finnish ethnicity of the patients. Because of the centralization of more than 95% of the paediatric HSCTs in Finland to one, single centre, the cohort covers the whole Finland and represents the national population well. Thus, the frequency of variants in the patient set and gnomAD non-cancer Finns was comparable. Comparison to gnomAD Europeans (Supplementary Table S2) showed differences in complement variant distribution between Finns and the rest of Europeans. This is understandable, because it is well known that Finns are a genetically relatively uniform but distinct population in Europe (54).
Treatment-related toxicity is still a significant clinical problem in paediatric allo-HSCT. Injury to the vascular endothelium is relatively common after HSCT with varying phenotypic appearances suggesting hitherto unidentified underlying mechanisms. Rare and ultrarare variants in complement genes are overexpressed in patients with injury to the vascular endothelium. These nonsynonymous variants in complement genes may be related to endotheliopathy and poor prognosis seen in these patients. Our hypothesis is that in addition to these variants, also genetic incompatibility between the donor and the recipient predisposes to endothelial damage and problems seen post-HSCT. Studies to address this hypothesis are underway.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by The Research Ethics Committee of the Helsinki University Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because all samples and data were obtained following authorization by the ethics committee and the national supervisory authority Valvira. Written, informed consents were not attainable due to the retrospective nature of the study also including deceased patients.
Author contributions
LL performed research and collected data; JK analyzed and interpreted data; KV and OK supervised the students and interpreted data; IL and SM designed research, supervised the students, and interpreted data. LL, JK, KV, IL, and SM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Aamu Pediatric Cancer Foundation, Finnish Pediatric Research Foundation, Nona and Kullervo Väre Foundation, the State Subsidy to Hospitals (TYH2019311) and Sigrid Jusélius Foundation.
Acknowledgments
We thank the LYHTY group Professor Kirsi Jahnukainen, MD, PhD, Docent Minna Koskenvuo, MD, PhD, Docent Ulla Wartiovaara-Kautto, MD, PhD, Docent Eliisa Kekäläinen, MD, PhD and PhD student Adam Alexandersson, MD, for the valuable and inspiring discussions on the subject.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1249958/full#supplementary-material
References
1. Mateos MK, O'Brien TA, Oswald C, Gabriel M, Ziegler DS, Cohn RJ, et al. Transplant-related mortality following allogeneic hematopoeitic stem cell transplantation for pediatric acute lymphoblastic leukemia: 25-year retrospective review. Pediatr Blood Bancer. (2013) 60(9):1520–7. doi: 10.1002/pbc.24559
2. Brissot E, Rialland F, Cahu X, Strullu M, Corradini N, Thomas C, et al. Improvement of overall survival after allogeneic hematopoietic stem cell transplantation for children and adolescents: a three-decade experience of a single institution. Bone Marrow Transplantation. (2016) 51(2):267–72. doi: 10.1038/bmt.2015.250
3. Leimi L, Jahnukainen K, Olkinuora H, Meri S, Vettenranta K. Early vascular toxicity after pediatric allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplantation. (2022) 57(5):705–11. doi: 10.1038/s41409-022-01607-8
4. Luft T, Dreger P, Radujkovic A. Endothelial cell dysfunction: a key determinant for the outcome of allogeneic stem cell transplantation. Bone Marrow Transplantation. (2021) 56(10):2326–35. doi: 10.1038/s41409-021-01390-y
5. Meri S, Bunjes D, Cofiell R, Jodele S. The role of complement in HSCT-TMA: basic science to clinical practice. Adv Ther (2022) 39(9):3896–915. doi: 10.1007/s12325-022-02184-4
6. Dandoy CE, Rotz S, Alonso PB, Klunk A, Desmond C, Huber J, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Advances. (2021) 5(1):1–11. doi: 10.1182/bloodadvances.2020003455
7. Jodele S, Laskin BL, Dandoy CE, Myers KC, El-Bietar J, Davies SM, et al. A new paradigm: Diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev (2015) 29(3):191–204. doi: 10.1016/j.blre.2014.11.001
8. Young JA, Pallas CR, Knovich MA. Transplant-associated thrombotic microangiopathy: theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transplantation. (2021) 56(8):1805–17. doi: 10.1038/s41409-021-01283-0
9. Carreras E, Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplantation. (2011) 46:1495–502. doi: 10.1038/bmt.2011.65
10. Pagliuca S, Michonneau D, Sicre de Fontbrune F, Sutra Del Galy AL, Xhaard AN, Robin M, et al. Allogeneic reactivity-mediated endothelial cell complications after HSCT: a plea for consensual definitions. Blood Advances. (2019) 3(15):2424–35. doi: 10.1182/bloodadvances.2019000143
11. Ramgopal A, Sridar S, Dalal J, Kalpatthi R. Thrombotic microangiopathy: multi-institutional review of pediatric patients who underwent HSCT. J Personalized Med (2021) 11(6):467. doi: 10.3390/jpm11060467
12. Gloude NJ, Khandelwal P, Luebbering N, Lounder DT, Jodele S, Alder MN, et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood. (2017) 130(10):1259–66. doi: 10.1182/blood-2017-05-782870
13. Arai Y, Yamashita K, Mizugishi K, Watanabe T, Sakamoto S, Kitano T, et al. Serum neutrophil extracellular trap levels predict thrombotic microangiopathy after allogeneic stem cell transplantation. Biol Blood Marrow Transplantation. (2013) 19(12):1683–9. doi: 10.1016/j.bbmt.2013.09.005
14. Jodele S. Complement in pathophysiology and treatment of transplant-associated thrombotic microangiopathies. Semin Hematol (2018) 55(3):159–66. doi: 10.1053/j.seminhematol.2018.04.003
15. Gavriilaki E, Anagnostopoulos A, Mastellos DC. Complement in thrombotic microangiopathies: unraveling ariadne's thread into the labyrinth of complement therapeutics. Front Immunol (2019) 10:337(337). doi: 10.3389/fimmu.2019.00337
16. Chaturvedi S, Brodsky RA, McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol (2019) 10:449(449). doi: 10.3389/fimmu.2019.00449
17. Fletcher-Sandersjöö A, Bellander B-M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb Res (2020) 194:36–41. doi: 10.1016/j.thromres.2020.06.027
18. Zoja C, Buelli S, Morigi M. Shiga toxin triggers endothelial and podocyte injury: the role of complement activation. Pediatr Nephrology. (2019) 34(3):379–88. doi: 10.1007/s00467-017-3850-x
19. Khosla J, Yeh AC, Spitzer TR, Dey BR. Hematopoietic stem cell transplant-associated thrombotic microangiopathy: current paradigm and novel therapies. Bone Marrow Transplantation. (2018) 53(2):129–37. doi: 10.1038/bmt.2017.207
20. Dvorak CC, Higham C, Shimano KA. Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: A practical approach to diagnosis and management. Front Pediatrics. (2019) 7:133. doi: 10.3389/fped.2019.00133
21. Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC, et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood. (2016) 127(8):989–96. doi: 10.1182/blood-2015-08-663435
22. Douglas SPM, Lahtinen AK, Koski JR, Leimi L, Keränen MAI, Koskenvuo M, et al. Enrichment of cancer-predisposing germline variants in adult and pediatric patients with acute lymphoblastic leukemia. Sci Rep (2022) 12(1):10670. doi: 10.1038/s41598-022-14364-x
23. Katainen R, Donner I, Cajuso T, Kaasinen E, Palin K, Mäkinen V, et al. Discovery of potential causative mutations in human coding and noncoding genome with the interactive software BasePlayer. Nat Protoc (2018) 13(11):2580–600. doi: 10.1038/s41596-018-0052-3
24. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. (2015) 526(7571):68–74. doi: 10.1038/nature15393
25. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536(7616):285–91. doi: 10.1038/nature19057
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–23. doi: 10.1038/gim.2015.30
27. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet (2017) 100(2):267–80. doi: 10.1016/j.ajhg.2017.01.004
28. Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res (2019) 48(D1):D835–D44. doi: 10.1093/nar/gkz972
29. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res (2000) 28(1):235–42. doi: 10.1093/nar/28.1.235
30. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun (2016) 7(1):10588. doi: 10.1038/ncomms10588
31. Fernández FJ, Santos-López J, Martínez-Barricarte R, Querol-García J, Martín-Merinero H, Navas-Yuste S, et al. The crystal structure of iC3b-CR3 αI reveals a modular recognition of the main opsonin iC3b by the CR3 integrin receptor. Nat Commun (2022) 13(1):1955. doi: 10.1038/s41467-022-29580-2
32. Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet Epidemiol (2011) 35(7):606–19. doi: 10.1002/gepi.20609
33. Khan SQ, Khan I, Gupta V. CD11b activity modulates pathogenesis of lupus nephritis. Front Med (2018) 5:52. doi: 10.3389/fmed.2018.00052
34. Lokki AI, Teirilä L, Triebwasser M, Daly E, Bhattacharjee A, Uotila L, et al. Dysfunction of complement receptors CR3 (CD11b/18) and CR4 (CD11c/18) in pre-eclampsia: a genetic and functional study. BJOG: Int J Obstetrics Gynaecology. (2021) 128(8):1282–91. doi: 10.1111/1471-0528.16660
35. Zinter MS, Spicer A, Orwoll BO, Alkhouli M, Dvorak CC, Calfee CS, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiology-Lung Cell Mol Physiol (2016) 310(3):L224–L31. doi: 10.1152/ajplung.00336.2015
36. Yehya N, Thomas NJ, Meyer NJ, Christie JD, Berg RA, Margulies SS. Circulating markers of endothelial and alveolar epithelial dysfunction are associated with mortality in pediatric acute respiratory distress syndrome. Intensive Care Med (2016) 42(7):1137–45. doi: 10.1007/s00134-016-4352-1
37. Zinter MS, Delucchi KL, Kong MY, Orwoll BE, Spicer AS, Lim MJ, et al. Early plasma matrix metalloproteinase profiles. A novel pathway in pediatric acute respiratory distress syndrome. Am J Respir Crit Care Med (2019) 199(2):181–9. doi: 10.1164/rccm.201804-0678OC
38. Fitch T, Myers KC, Dewan M, Towe C, Dandoy C. Pulmonary complications after pediatric stem cell transplant. Front Oncol (2021) 11:755878. doi: 10.3389/fonc.2021.755878
39. Elbahlawan L, McArthur J, Morin CE, Abdelhafeez H, McCarville MB, Ruiz RE, et al. Pulmonary complications in children following hematopoietic cell transplantation: A case report and review of the diagnostic approach. Front Oncol (2021) 11:772411. doi: 10.3389/fonc.2021.772411
40. Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their rOmance. Blood. (2016) 127(13):1719–27. doi: 10.1182/blood-2015-10-676114
41. Frame D, Scappaticci GB, Braun TM, Maliarik M, Sisson TH, Pipe SW, et al. Defibrotide therapy for SARS-coV-2 ARDS. Chest. (2022) 162(2):346–55. doi: 10.1016/j.chest.2022.03.046
42. Hess K, Ajjan R, Phoenix F, Dobó J, Gál P, Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PloS One (2012) 7(4):e35690. doi: 10.1371/journal.pone.0035690
43. Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemostasis. (2015) 13:S121–S32. doi: 10.1111/jth.12950
44. Amara U, Flierl Ma, Rittirsch D, Klos A, Klos A Fau - Chen H, Chen H Fau - Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol (2010) 185(9):5628–36. doi: 10.4049/jimmunol.0903678. FauRittirsch D Fau.
45. Ali YM, Ferrari M, Lynch NJ, Yaseen S, Dudler T, Gragerov S, et al. Lectin pathway mediates complement activation by SARS-CoV-2 proteins. Front Immunol (2021) 12:714511. doi: 10.3389/fimmu.2021.714511
46. Khaled SK, Claes K, Goh YT, Kwong YL, Leung N, Mendrek W, et al. Narsoplimab, a mannan-binding lectin-associated serine protease-2 inhibitor, for the treatment of adult hematopoietic stem-cell transplantation–associated thrombotic microangiopathy. J Clin Oncol (2022) 40(22):2447–57. doi: 10.1200/JCO.21.02389
47. Guzzo G, Kissling S, Pantaleo G, Pascual M, Sadallah S, Teta D. Complement activation and blockade in massive post-partum haemorrhage, thrombotic microangiopathy and acute kidney injury: a case report. BMC Nephrology. (2021) 22(1):252. doi: 10.1186/s12882-021-02456-1
48. Anker-Møller T, Hvas A-M, Sunde N, Thiel S, Troldborg A. Proteins of the Lectin Pathway of complement activation at the site of injury in subarachnoid hemorrhage compared with peripheral blood. Brain Behavior. (2020) 10(8):e01728. doi: 10.1002/brb3.1728
49. Holste K, Xia F, Garton HJL, Wan S, Hua Y, Keep RF, et al. The role of complement in brain injury following intracerebral hemorrhage: A review. Exp Neurology. (2021) 340:113654. doi: 10.1016/j.expneurol.2021.113654
50. Alshareef M, Mallah K, Vasas T, Alawieh A, Borucki D, Couch C, et al. A role of complement in the pathogenic sequelae of mouse neonatal germinal matrix hemorrhage. Int J Mol Sci (2022) 23(6):2943. doi: 10.3390/ijms23062943
51. Massri M, Foco L, Würzner R. Comprehensive update and revision of nomenclature on complement C6 and C7 variants. J Immunol (2022) 208(12):2597–612. doi: 10.4049/jimmunol.2200045
52. Tseng M-H, Tsai J-D, Tsai IJ, Huang S-M, Huang J-L, Fan W-L, et al. Whole-exome sequencing detects mutations in pediatric patients with atypical hemolytic uremic syndrome in Taiwan. Clinica Chimica Acta (2019) 494:143–50. doi: 10.1016/j.cca.2019.03.1623
53. Gange WS, Haghighi A, Toy BC. Purtscher-like retinopathy associated with atypical hemolytic uremic syndrome: case report and review of outcomes. Retinal cases Brief Rep (2023) 17(2):154–9. doi: 10.1097/ICB.0000000000001126
Keywords: hematopoietic stem cell transplantation, hematopoietic stem cell transplantation adverse effects, acute toxicity, vascular complications, complement system, endothelial damage
Citation: Leimi L, Koski JR, Kilpivaara O, Vettenranta K, Lokki AI and Meri S (2023) Rare variants in complement system genes associate with endothelial damage after pediatric allogeneic hematopoietic stem cell transplantation. Front. Immunol. 14:1249958. doi: 10.3389/fimmu.2023.1249958
Received: 29 June 2023; Accepted: 29 August 2023;
Published: 13 September 2023.
Edited by:
Tomomi Toubai, Yamagata University, JapanReviewed by:
Thomas Luft, Heidelberg University Hospital, GermanyKunihiko Moriya, Tohoku University, Japan
Copyright © 2023 Leimi, Koski, Kilpivaara, Vettenranta, Lokki and Meri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lilli Leimi, bGlsbGkubGVpbWlAaGVsc2lua2kuZmk=
†These authors have contributed equally to this work and share senior authorship