
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 11 August 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1239251
This article is part of the Research Topic Inflammation in Skin-Related Diseases View all 16 articles
 Yutaka Hatano1*†
Yutaka Hatano1*† Peter M. Elias2†
Peter M. Elias2†Permeability barrier disruption has been shown to induce immunological alterations (i.e., an “outside-to-inside” pathogenic mechanism). Conversely, several inflammatory and immunological mechanisms reportedly interrupt permeability barrier homeostasis (i.e., an “inside-to-outside” pathogenic mechanism). It is now widely recognized that alterations of even a single molecule in keratinocytes can lead to not only permeability barrier dysfunction but also to immunological alterations. Such a simultaneous, bidirectional functional change by keratinocytes is herein named an “intrinsic” pathogenic mechanism. Molecules and/or pathways involved in this mechanism could be important not only as factors in disease pathogenesis but also as potential therapeutic targets for inflammatory cutaneous diseases, such as atopic dermatitis, psoriasis, and prurigo nodularis. Elevation of skin surface pH following permeability barrier abrogation comprises one of the key pathogenic phenomena of the “outside-to-inside” mechanism. Not only type 2 cytokines (e.g., IL-4, IL-13, IL-31) but also type 1 (e.g. IFN-γ), and type 3 (e.g., IL-17, IL-22) as well as several other inflammatory factors (e.g. histamine) can disrupt permeability barrier homeostasis and are all considered part of the “inside-to-outside” mechanism. Finally, examples of molecules relevant to the “intrinsic” pathogenic mechanism include keratin 1, filaggrin, and peroxisome proliferator-activated receptor-α (PPARα).
As previously described in numerous review articles, permeability barrier abrogation has been shown to induce immunological alterations (i.e., “outside-to-inside” pathogenic mechanism; Figure 1). Conversely, several inflammatory and immunological factors have been reported to disturb permeability barrier homeostasis (i.e., “inside-to-outside” pathogenic mechanism; Figure 1) (1). In this article, we will highlight additional associations between permeability barrier abrogation and inflammatory and/or immunological alterations.
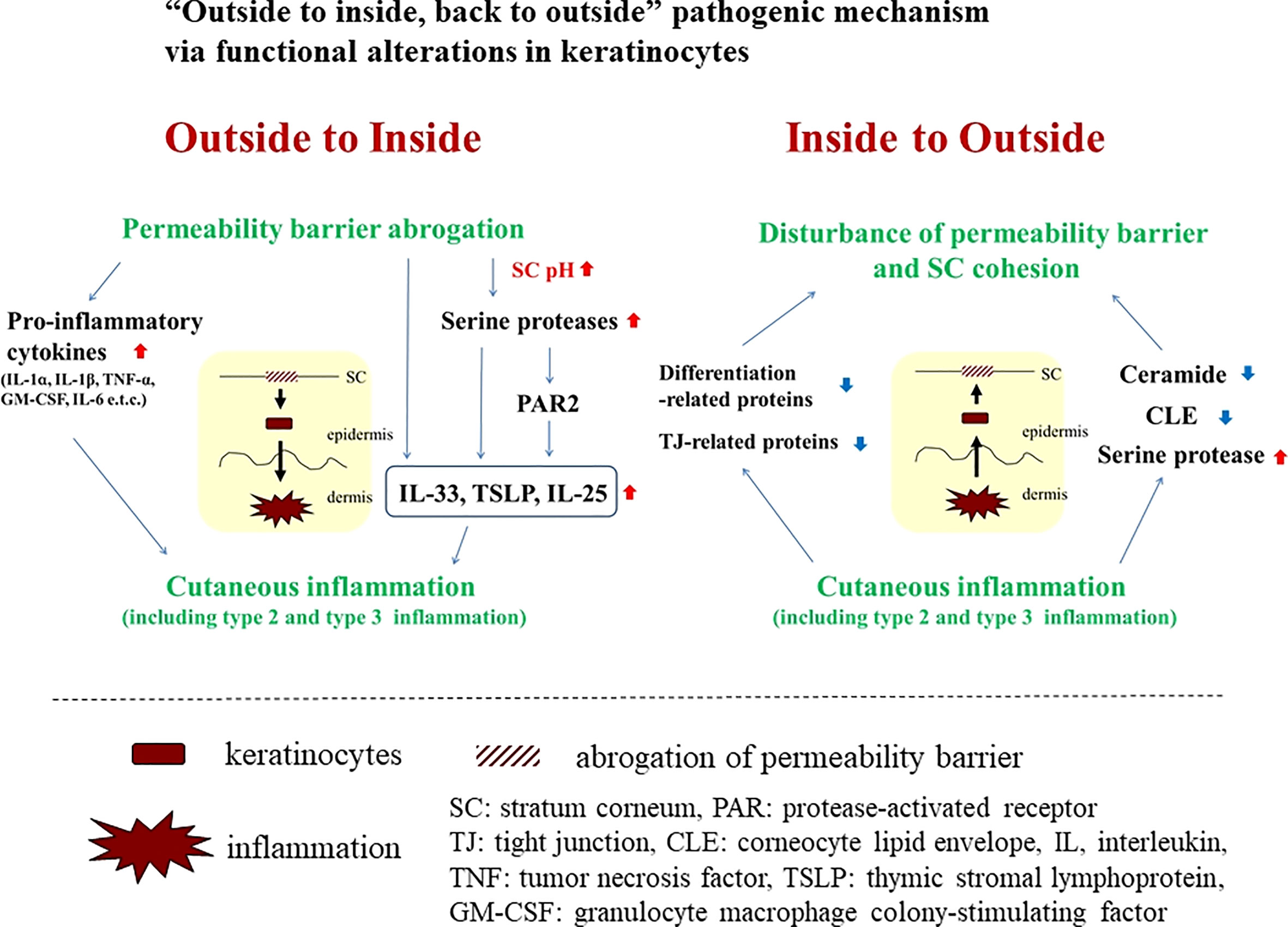
Figure 1 “Outside to inside, back to outside” pathogenic mechanism via functional alterations in keratinocytes.
Currently, multiple studies have revealed that modification in even a single molecule in keratinocytes can induce epidermal functional changes that not only disrupt permeability barrier homeostasis but also lead to immunological alterations. Here, we describe simultaneous functional changes in keratinocytes in two different directions as an “intrinsic” pathogenic mechanism (Figure 2).
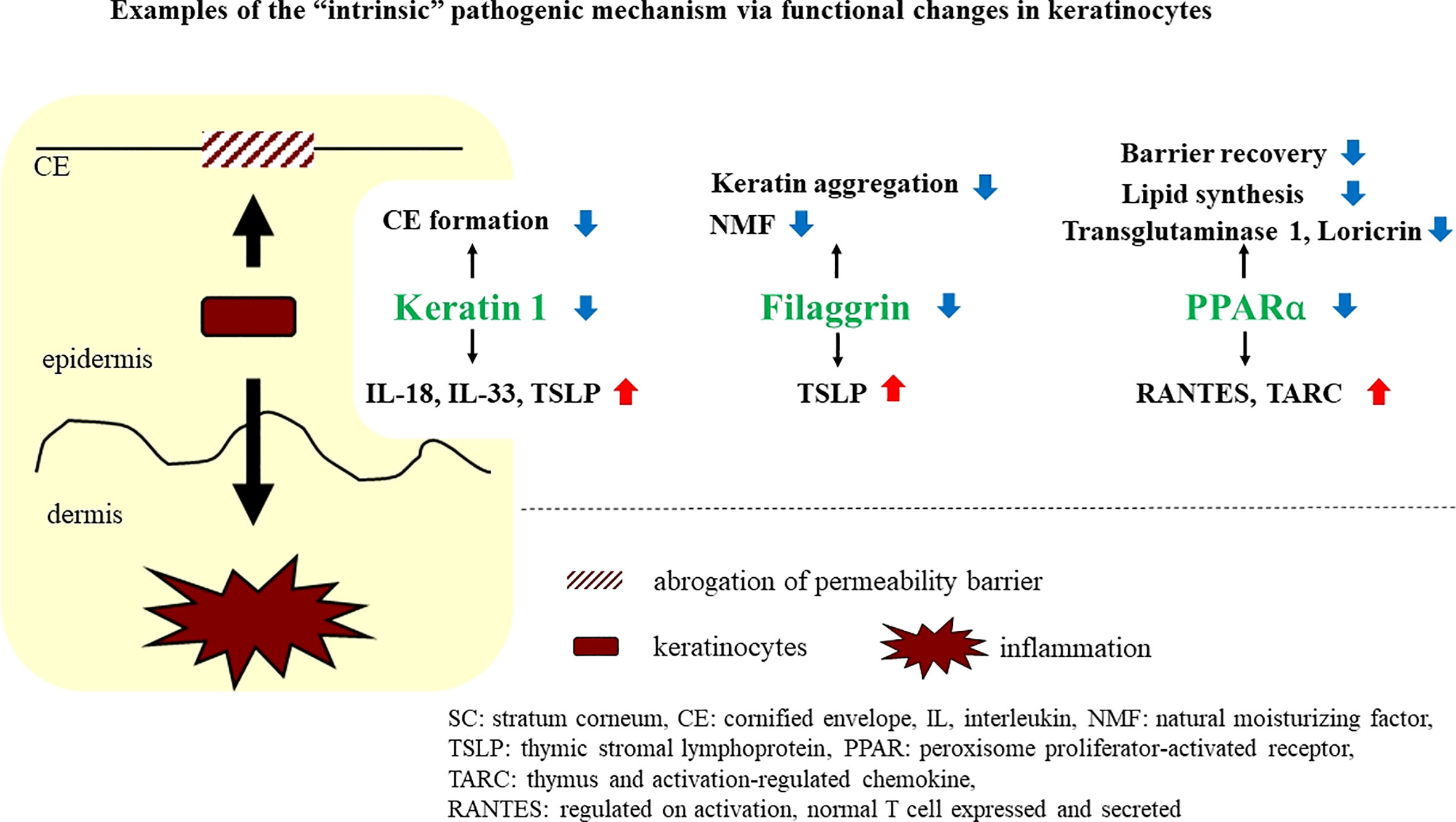
Figure 2 Examples of the “intrinsic” pathogenic mechanism via functional changes in keratinocytes.
Following epidermal permeability barrier abrogation, the “outside-to-inside” mechanism not only encompasses the induction and/or secretion of pro-inflammatory cytokines such as interleukin (IL)-1α, IL-1β, tumor necrosis factor (TNF)-α, and granulocyte macrophage colony-stimulating factor (GM-CSF) from epidermal keratinocytes (2), but also elevation of skin surface pH, accompanied by elevated kallikrein (KLK) activity (1), all key pathogenic phenomena for inducing inflammation, especially type 2. Initiation of production and/or secretion of so-called danger signals or alarmins, such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), lead to immunological alterations of type 2 inflammation with or without protease-activated receptor (PAR)2-dependent responses (3). In addition, permeability barrier abrogation reportedly induces type 2 and type 3 inflammation (e.g., IL-17 and IL-22) via activation of KLK5 and the PAR2 axis (4). In line with this theory, combined treatment with a PAR2 inhibitor and a lactobionic acid (LBA) (‘super acid’) exhibited therapeutic effects on hapten-induced atopic dermatitis (AD)-like dermatitis in a murine model in which elevations of epidermal TSLP paralleled development of the dermatitis (5). Application of LBA during the induction phase was also reported to prevent the initial emergence of hapten-induced AD-like dermatitis (6). Notably, both type 2 and type 3 inflammation are reported to be involved in the pathogenesis of a hapten-induced AD-like dermatitis (7), supporting the concept that a combination of permeability barrier abrogation, elevation of stratum corneum pH, and PAR2 activation could lead to the induction of type 2 and type 3 inflammation.
Since we first described the “inside to outside” pathogenic mechanism (1), several inflammatory factors and pathways have been found to be involved in this mechanism.
Prototypic type 2 cytokines, IL-4 and IL-13, downregulate the expression of filaggrin (FLG), loricrin (LOR), and involucrin (IVL) (8, 9). IL-4 also reduces ceramide synthesis and compromises stratum corneum (SC) cohesion (10–12). Cutaneous permeability barrier recovery is suppressed by IL-4 (13). Recently, type 2 cytokines were reported to provoke shortening of ceramide carbon chains via down-regulation of elongase expression, such as elongation of very long chain fatty acids protein (ELOVL)3 (14). Type 2 cytokines also downregulate enzymes involved in cornified lipid envelope (CLE) formation, such as ALOX12B, ALOXE3, and ABHD9 (15). Likewise, IL-31 reportedly suppresses expression of FLG (16). Claudin-1, a tight junction-related protein, is downregulated by IL-4, IL-13, and IL-31 (17).
Considerable data demonstrating the harmful effects of a variety of inflammatory factors (besides type 2 cytokines) on the permeability barrier have been accumulated. Type 3 cytokines (e.g., IL-17 and IL-22) and histamines downregulate expression of FLG, LOR, and IVL (18, 19), R. Histamine reportedly downregulates the expression of claudin-1, claudin-4, occludin, and zonula occludens (ZO)-1 (19). Interferon (IFN)-γ, a prototypic type 1 cytokine, has been demonstrated to reduce expression of FLG, claudin-1, and ELOVL1 (20–22).
Abundant evidence leads us to recognition of the association between permeability barrier dysfunction and inflammatory reactions. Alteration of even one molecule in epidermal keratinocytes can induce functional changes, leading to simultaneous permeability barrier dysfunction and inflammatory and/or immunological dysregulation. Here, such a molecule is called an “intrinsic” participant, and the pathogenic mechanism induced by the alteration of these molecules is also called “intrinsic.” Examples of “intrinsic” molecules are described below (Figure 2).
Keratins constitute the intermediate filament cytoskeleton in keratinocytes and play an important role in the mechanical integrity of corneocytes, during linkage to cornified envelopes, which is a critical process for competent permeability barrier formation (23, 24). In fact, permeability barrier abnormalities have been demonstrated in keratin (KRT)-deficient conditions (25, 26). Meanwhile, KRT1 abnormalities reportedly lead to cutaneous inflammation, accompanied by elevations of IL-18, IL-33, and TSLP (27), which are well-known danger signals in type 2 inflammation induction, a hallmark in AD pathogenesis (3, 28–30). Interestingly, IL-18 secretion is induced in KRT1 knockout-cultured keratinocytes in a caspase-1-dependent manner, suggesting that the secretion of IL-18 in KRT1-deficient mice could be a primary effect of KRT1 depletion, rather than a secondary effect, following permeability barrier abrogation (27). Together, downregulation of KRT1 could cause functional, dual directional alterations in keratinocytes, leading to both permeability barrier abrogation and allergic inflammation in AD. Accordingly, expression of KRT1 reportedly is downregulated in atopic lesions due to elevated levels of inflammatory cytokines, such as IL-33, IL-4, and IL-13 (31, 32). Thus, the downregulation of KRT1 could augment not only permeability barrier abrogation but also allergic inflammation.
FLG is an epidermal differentiation-related molecule, which plays important roles in both permeability barrier homeostasis and SC hydration (33, 34). In fact, epidermis in which the filaggrin gene is knocked down exhibits substantial alterations in permeability barrier function (35). Interestingly, it has been recently shown that keratinocytes transfected with siRNA against the profilaggrin gene are able to produce greater quantities (versus control) of TSLP, which is one of the essential cytokines in the induction of type 2 immunological reactions (36). Knockdown of FLG reportedly increases the production of interleukin (IL)-1α, IL-8, IL-18 and GM-CSF in stratified human keratinocytes (37). It has also been reported that keratinocytes of flaky tail (versus wild-type mice), in which FLG is deficient due to a loss-of-function mutation in profilaggrin, produce more of the proinflammatory cytokine, IL-1β (38). In ichthyosis vulgaris, which is caused by loss of function mutations in FLG, expression of pro-inflammatory cytokines increases. Together, these results show that an abnormality in FLG, which has been mainly regarded as a barrier-related molecule, could simultaneously modulate processes leading to allergic inflammation in skin (39). Whether such a functional change in keratinocytes towards a proinflammatory phenotype is attributable to dysfunction of FLG in KRT aggregation remains undetermined, although in cultured epidermal keratinocytes knocked down of FLG, KRT1 expression was reported to be unaffected (40).
Peroxisome proliferators-activated receptors (PPARs) belong to the nuclear hormone receptor class II and have three subtypes, PPARα, PPARβ/δ and PPARγ (41). They are called liposensors because their ligands are lipids or lipid derivatives. Generally, PPAR signaling has positive effects on barrier homeostasis, but it can also have anti-inflammatory effects, although there are some differences in the impact of their subtypes (41). The activation of PPARs stimulates lipid synthesis and epidermal differentiation, while also accelerating recovery after permeability barrier disruption (41). Moreover, epidermal barrier development is delayed in PPARα-deficient mice (42).
Activators of PPARα suppress both allergic and irritant cutaneous inflammation in vivo (43). Interestingly, it has been reported that PPARα expression in the skin is reduced in patients with AD and that PPARα-deficient mice develop more severe hapten-induced AD-like dermatitis than wild-type mice (44). RNA sequence analysis also revealed that PPARα expression is down-regulated in AD-like lesions compared with those in non-lesional flaky tail mice skin (45). In addition, PPARα expression in epidermis is reduced in similar hapten-induced murine AD models, and topical activation of PPARα exhibits a substantial therapeutic effect on murine AD, by restoring permeability barrier function and by dampening allergic inflammation (46, 47).
Awareness of physiological properties of PPARα in the skin and the association of decreased PPARα expression with AD suggests that PPARα might be one of the macromolecules that participates in “intrinsic” cross talk. In fact, reduction of PPARα expression by transfection with siRNA against PPARα not only up-regulates expression of the Regulated-upon-activation, normal-t cell-expressed-and-presumably-secreted cytokine (RANTES) and the Thymus- and activation-regulated chemokine (TARC) in cultured keratinocytes, but also down-regulates expression of transglutaminase 1 and LOR (48), further suggesting that PPARα modulates functions associated both with inflammation and with permeability barrier homeostasis in skin.
A vicious cycle involving permeability barrier dysfunction and allergic inflammation is one of the basic mechanisms leading to the pathogenesis of AD. In this article, in addition to discussing the concept of the so-called “outside-to-inside, and back-to-outside” paradigm, we offer an idea that links permeability dysfunction and allergic inflammation in the pathogenesis of AD. In the “outside-to-inside and back-to-outside” model, keratinocyte functions are modified secondarily by external stimuli, such as SC pH, or certain inflammatory factors. On the other hand, in our “intrinsic” model, primary functional changes relevant to both permeability barrier and inflammation can occur in keratinocytes by alteration of even a single molecule. Secondary alteration of such a molecule may also contribute to augment the vicious cycle between permeability barrier dysfunction and allergic inflammation. This concept demonstrates the importance of keratinocytes as a key player in the pathogenesis of AD, although it is unclear whether this concept applies equally to extrinsic and intrinsic AD.
Keratinocytes are well known to have functions related to both permeability barrier dysfunction and inflammation, meaning that keratinocytes likely are involved in both of these processes in the pathogenesis of AD. The concept “intrinsic” highlights molecules in keratinocytes which are simultaneously involved in both permeability barrier function and inflammation, and such molecules could be candidates as therapeutic targets in AD, as in the case of PPARα activators in the murine AD model. Interestingly, the Janus kinase inhibitor, JTE-052, now the main ingredient in an ointment called Delgocitinib, was originally reported to induce filaggrin expression, and this ointment has been deployed as topical therapy for AD (49, 50), suggesting that targeting keratinocyte functions is one potential strategy for treating AD.
Knowledge that changes in one molecule in keratinocytes can lead to both permeability barrier abrogation and inflammation has been already reported, as in the case of KRT1 mutations (27). Moreover, Akiyama et al. have described an autoinflammatory keratinization disease paradigm (51–53), and this disease concept seems almost identical to inflammatory aspects in our “intrinsic” mechanism described here. Therefore, seeking the pathomechanisms of autoinflammatory keratinization diseases could help to clarify the mechanism underlining our “intrinsic” paradigm, and AD might be recognized as an autoinflammatory keratinization disease in the future.
YH wrote the first draft of the manuscript. PE revised the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Grant-in-Aid for Scientific Research (C).
We sincerely appreciate Ms. Joan S. Wakefield’s (San Francisco Veterans Affairs Health Care System) excellent editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
IL, interleukin; TNF, tumor necrosis factor; GM-CSF, granulocyte macrophage colony-stimulating factor; KLK, kallikrein; TSLP, thymic stromal lymphopoietin; PAR, protease-activated receptor; LBA, lactobionic acid; AD, atopic dermatitis; FLG, filaggrin; LOR, loricrin; IVL, involucrin; ELOVL, elongation of very long chain fatty acids protein; CLE, cornified lipid envelope; ALOX12B, arachidonate 12-lipoxygenase, 12R type; ALOXE3, epidermis-type lipoxygenase 3; ABHD9, epoxide hydrolase 3; ZO, zonula occludens; IFN, Interferon; KRT, keratin; SC, stratum corneum; PPARs, Peroxisome proliferators-activated receptors; RANTES, Regulated upon activation, normal t cell expressed and presumably secreted cytokine; TARC, Thymus- and activation-regulated chemokine.
1. Elias PM, Hatano Y, Williams ML. Basis for the barrier abnorMality in atopic dermatitis. outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol (2008) 121:1337–43. doi: 10.1016/j.jaci.2008.01.022
2. Denda M, Wood LC, Emami S, Calhoun C, Brown BE, Elias PM, et al. The epidermal hyperplasia associated with repeated barrier disruption by acetone treatment or tape stripping cannot be attributed to increased water loss. Arch Dermatol Res (1996) 288:230–8. doi: 10.1007/BF02530090
3. Nakahara T, Kido-Nakahara M, Tsuji G, Furue M. Basics and recent advances in the pathophysiology of atopic dermatitis. J Dermatol (2021) 48:130–9. doi: 10.1111/1346-8138.15664
4. Kishibe M. Physiological and pathological roles of kallikrein-related peptidases in the epidermis. J Dermatol Sci (2019) 95:50–5. doi: 10.1016/j.jdermsci.2019.06.007
5. Sakai T, Hatano Y, Matsuda-Hirose H, Zhang W, Takahashi D, Jeong SK, et al. Combined benefits of a PAR2 inhibitor and stratum corneum acidification for murine atopic dermatitis. J Invest Dermatol (2016) 136:538–41. doi: 10.1016/j.jid.2015.11.011
6. Hatano Y, Man MQ, Uchida Y, Crumrine D, Scharschmidt TC, Kim EG, et al. Maintenance of an acidic stratum corneum prevents emergence of murine atopic dermatitis. J Invest Dermatol (2009) 129:1824–35. doi: 10.1038/jid.2008.444
7. Heo WI, Lee KE, Hong JY, Kim MN, Oh MS, Kim YS, et al. The role of interleukin-17 in mouse models of atopic dermatitis and contact dermatitis. Clin Exp Dermatol (2015) 40:665–71. doi: 10.1111/ced.12567
8. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol (2007) 120:150–5. doi: 10.1016/j.jaci.2007.04.031
9. Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol (2008) 126:332–7. doi: 10.1016/j.clim.2007.11.006
10. Hatano Y, Terashi H, Arakawa S, Katagiri K. Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-alpha and interferon-gamma in human epidermis. J Invest Dermatol (2005) 124:786–92. doi: 10.1111/j.0022-202X.2005.23651.x
11. Hatano Y, Katagiri K, Arakawa S, Fujiwara S. Interleukin-4 depresses levels of transcripts for acid-sphingomyelinase and glucocerebrosidase and the amount of ceramide in acetone-wounded epidermis, as demonstrated in a living skin equivalent. J Dermatol Sci (2007) 47:45–7. doi: 10.1016/j.jdermsci.2007.02.010
12. Hatano Y, Adachi Y, Elias PM, Crumrine D, Sakai T, Kurahashi R, et al. The Th2 cytokine, interleukin-4, abrogates the cohesion of normal stratum corneum in mice. implications for pathogenesis of atopic dermatitis. Exp Dermatol (2013) 22:30–5. doi: 10.1111/exd.12047
13. Kurahashi R, Hatano Y, Katagiri K. IL-4 suppresses the recovery of cutaneous permeability barrier functions in vivo. J Invest Dermatol (2008) 128:1329–31. doi: 10.1038/sj.jid.5701138
14. Berdyshev E, Goleva E, Bronova I, Dyjack N, Rios C, Jung J, et al. Lipid abnorMalities in atopic skin are driven by type 2 cytokines. JCI Insight (2018) 22:e98006. doi: 10.1172/jci.insight.98006
15. Chiba T, Nakahara T, Kohda F, Ichiki T, Manabe M, Furue M. Measurement of trihydroxy-linoleic acids in stratum corneum by tape-stripping. Possible biomarker of barrier function in atopic dermatitis. PloS One (2019) 14:e0210013. doi: 10.1371/journal.pone.0210013
16. Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Lüscher-Firzlaff J, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol (2012) 129:426–33. doi: 10.1016/j.jaci.2011.10.042
17. Gruber R, Börnchen C, Rose K, Daubmann A, Volksdorf T, Wladykowski E, et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am J Pathol (2015) 185:2777–89. doi: 10.1016/j.ajpath.2015.06.021
18. Furue M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2. Pathogenic implications in atopic dermatitis. Int J Mol Sci (2020) 21:5382. doi: 10.3390/ijms21155382
19. Gschwandtner M, Mildner M, Mlitz V, Gruber F, Eckhart L, Werfel T, et al. Histamine suppresses epidermal keratinocyte differentiation and impairs skin barrier function in a human skin model. Allergy (2013) 68:37–47. doi: 10.1111/all.12051
20. Hvid M, Johansen C, Deleuran B, Kemp K, Deleuran M, Vestergaard C. Regulation of caspase 14 expression in keratinocytes by inflammatory cytokines–a possible link between reduced skin barrier function and inflammation? Exp Dermatol (2011) 20:633–6. doi: 10.1111/j.1600-0625.2011.01280.x
21. Mizutani Y, Takagi N, Nagata H, Inoue S. Interferon-γ downregulates tight junction function, which is rescued by interleukin-17A. Exp Dermatol (2021) 30:1754–63. doi: 10.1111/exd.14425
22. Tawada C, Kanoh H, Nakamura M, Mizutani Y, Fujisawa T, Banno Y, et al. Interferon-γ decreases ceramides with long-chain fatty acids. possible involvement in atopic dermatitis and psoriasis. J Invest Dermatol (2014) 134:712–8. doi: 10.1038/jid.2013.364
23. Schweizer J, Bowden PE, Coulombe PA, Langbein L, Lane EB, Magin TM, et al. New consensus nomenclature for mamMalian keratins. J Cell Biol (2006) 174:169–74. doi: 10.1083/jcb.200603161
24. Candi E, Schmidt R, Melino G. The cornified envelope. a model of cell death in the skin. Nat Rev Mol Cell Biol (2005) 6:328–40. doi: 10.1038/nrm1619
25. Lane EB, McLean WH. Keratins and skin disorders. J Pathol (2004) 204:355–66. doi: 10.1002/path.1643
26. Schmuth M, Yosipovitch G, Williams ML, Weber F, Hintner H, Ortiz-Urda S, et al. Pathogenesis of the permeability barrier abnorMality in epidermolytic hyperkeratosis. J Invest Dermatol (2001) 117:837–47. doi: 10.1046/j.0022-202x.2001.01471.x
27. Roth W, Kumar V, Beer HD, Richter M, Wohlenberg C, Reuter U. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J Cell Sci (2012) 125:5269–79. doi: 10.1242/jcs.116574
28. Ihim SA, Abubakar SD, Zian Z, Sasaki T, Saffarioun M, Maleknia S, et al. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity. Biological role in induction, regulation, and treatment. Front Immunol (2022) 13:919973. doi: 10.3389/fimmu.2022.919973
29. Konishi H, Tsutsui H, Murakami T, Yumikura-Futatsugi S, Yamanaka K, Tanaka M, et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc Natl Acad Sci U S A (2002) 99:11340–5. doi: 10.1073/pnas.152337799
30. Trzeciak M, Gleń J, Bandurski T, Sokołowska-Wojdyło M, Wilkowska A, Roszkiewicz J. Relationship between serum levels of interleukin-18, IgE and disease severity in patients with atopic dermatitis. Clin Exp Dermatol (2011) 36:728–32. doi: 10.1111/j.1365-2230.2011.04113.x
31. Dai X, Utsunomiya R, Shiraishi K, Mori H, Muto J, Murakami M, et al. Nuclear IL-33 plays an important role in the suppression of FLG, LOR, keratin 1, and keratin 10 by IL-4 and IL-13 in human keratinocytes. J Invest Dermatol (2021) 141:2646–2655.e6. doi: 10.1016/j.jid.2021.04.002
32. Totsuka A, Omori-Miyake M, Kawashima M, Yagi J, Tsunemi Y. Expression of keratin 1, keratin 10, desmoglein 1 and desmocollin 1 in the epidermis. possible downregulation by interleukin-4 and interleukin-13 in atopic dermatitis. Eur J Dermatol (2017) 27:247–53. doi: 10.1684/ejd.2017.2985
33. Brown SJ, McLean WH. One remarkable molecule. filaggrin. J Invest Dermatol (2012) 132:751–62. doi: 10.1038/jid.2011.393
34. Thyssen JP, Kezic S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol (2014) 134:792–9. doi: 10.1016/j.jaci.2014.06.014
35. Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol (2012) 129:1538–46.e6. doi: 10.1016/j.jaci.2012.01.068
36. Hönzke S, Wallmeyer L, Ostrowski A, Radbruch M, Mundhenk L, Schäfer-Korting M, et al. Influence of Th2 cytokines on the cornified envelope, tight junction proteins, and ß-defensins in filaggrin-deficient skin equivalents. J Invest Dermatol (2016) 136:631–9. doi: 10.1016/j.jid.2015.11.007
37. Sakai T, Hatano Y, Zhang W, Fujiwara S, Nishiyori R. Knockdown of either filaggrin or loricrin increases the productions of interleukin (IL)-1α, IL-8, IL-18 and granulocyte macrophage colony-stimulating factor in stratified human keratinocytes. J Dermatol Sci (2015) 80:158–60. doi: 10.1016/j.jdermsci.2015.09.002
38. Kezic S, O'Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol (2012) 129:1031–9. doi: 10.1016/j.jaci.2011.12.989
39. Sakabe J, Kamiya K, Yamaguchi H, Ikeya S, Suzuki T, Aoshima M, et al. Proteome analysis of stratum corneum from atopic dermatitis patients by hybrid quadrupole-orbitrap mass spectrometer. Allergy Clin Immunol (2014) 134:957–60.e8. doi: 10.1016/j.jaci.2014.07.054
40. Mildner M, Jin J, Eckhart L, Kezic S, Gruber F, Barresi C, et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J Invest Dermatol (2010) 130:2286–94. doi: 10.1038/jid.2010.115
41. Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series. skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res (2008) 9:499–509. doi: 10.1194/jlr.R800001-JLR200
42. Schmuth M, Schoonjans K, Yu QC, Fluhr JW, Crumrine D, Hachem JP, et al. Role of peroxisome proliferator-activated receptor alpha in epidermal development in utero. J Invest Dermatol (2002) 119:1298–303. doi: 10.1046/j.1523-1747.2002.19605.x
43. Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J, et al. Topical peroxisome proliferators-activated receptor-alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol (2002) 118:94–101. doi: 10.1046/j.0022-202x.2001.01626.x
44. Staumont-Salle D, Abboud G, Brenuchon C, Kanda A, Roumier T, Lavogiez C, et al. Peroxisome proliferator-activated receptor alpha regulates skin inflammation and humoral response in atopic dermatitis. J Allergy Clin Immunol (2008) 121:962–8. doi: 10.1016/j.jaci.2007.12.1165
45. Sakai T, Aoki C, Mori Y, Yamate T, Matsuda-Hirose H, Hatano Y. Site-specific microarray evaluation of spontaneous dermatitis in flaky tail mice. J Invest Dermatol (2019) 139:2554–2557.e5. doi: 10.1016/j.jid.2019.04.024
46. Hatano Y, Man MQ, Uchida Y, Crumrine D, Mauro TM, Feingold KR, et al. Murine atopic dermatitis responds to peroxisome proliferator-activated receptors alpha and beta/delta (but not gamma) and liver X receptor activators. J Allergy Clin Immunol (2010) 125:160–9. doi: 10.1016/j.jaci.2009.06.049
47. Chiba T, Takeuchi S, Esaki H, Yamamura K, Kurihara Y, Moroi Y, et al. Topical application of PPARα (but not β/δ or γ) suppresses atopic dermatitis in NC/Nga mice. Allergy (2012) 67:936–42. doi: 10.1111/j.1398-9995.2012.02844.x
48. Adachi Y, Hatano Y, Sakai T, Fujiwara S. Expressions of peroxisome proliferator-activated receptors (PPARs) are directly influenced by permeability barrier abrogation and inflammatory cytokines and depressed PPARα modulates expressions of chemokines and epidermal differentiation-related molecules in keratinocytes. Exp Dermatol (2013) 22:606–8. doi: 10.1111/exd.12208
49. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol (2015) 136:667–677.e7. doi: 10.1016/j.jaci.2015.03.051
50. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kabashima K, Oda M, et al. Delgocitinib ointment in pediatric patients with atopic dermatitis. A phase 3, randomized, double-blind, vehicle-controlled study and a subsequent open-label, long-term study. J Am Acad Dermatol (2021) 85:854–62. doi: 10.1016/j.jaad.2021.06.014
51. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. J Allergy Clin Immunol (2017) 140:1545–7. doi: 10.1016/j.jaci.2017.05.019
52. Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. An emerging concept encompassing various inflammatory keratinization disorders of the skin. J Dermatol Sci (2018) 90:105–11. doi: 10.1016/j.jdermsci.2018.01.012
Keywords: atopic dermatitis, keratinocyte, permeability barrier dysfunction, allergic inflammation, filaggrin, keratin 1, PPAR alpha
Citation: Hatano Y and Elias PM (2023) “Outside-to-inside,” “inside-to-outside,” and “intrinsic” endogenous pathogenic mechanisms in atopic dermatitis: keratinocytes as the key functional cells involved in both permeability barrier dysfunction and immunological alterations. Front. Immunol. 14:1239251. doi: 10.3389/fimmu.2023.1239251
Received: 13 June 2023; Accepted: 31 July 2023;
Published: 11 August 2023.
Edited by:
Chunmeng Shi, First Affiliated Hospital of Army Medical University, ChinaReviewed by:
Xia Dou, Peking University, ChinaCopyright © 2023 Hatano and Elias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yutaka Hatano, aGF0YW5vQG9pdGEtdS5hYy5qcA==
†ORCID: Yutaka Hatano, orcid.org/0000-0002-1349-755X
Peter M. Elias, orcid.org/0000-0001-7989-4032
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.