 Jorge Santos-López
Jorge Santos-López Karla de la Paz1,2†
Karla de la Paz1,2† Francisco J. Fernández
Francisco J. Fernández M. Cristina Vega
M. Cristina Vega- 1Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
- 2Research & Development, Abvance Biotech SL, Madrid, Spain
The complement system plays crucial roles in a wide breadth of immune and inflammatory processes and is frequently cited as an etiological or aggravating factor in many human diseases, from asthma to cancer. Complement receptors encompass at least eight proteins from four structural classes, orchestrating complement-mediated humoral and cellular effector responses and coordinating the complex cross-talk between innate and adaptive immunity. The progressive increase in understanding of the structural features of the main complement factors, activated proteolytic fragments, and their assemblies have spurred a renewed interest in deciphering their receptor complexes. In this review, we describe what is currently known about the structural biology of the complement receptors and their complexes with natural agonists and pharmacological antagonists. We highlight the fundamental concepts and the gray areas where issues and problems have been identified, including current research gaps. We seek to offer guidance into the structural biology of the complement system as structural information underlies fundamental and therapeutic research endeavors. Finally, we also indicate what we believe are potential developments in the field.
1 Introduction to the complement receptors
The currently accepted list of complement receptors includes four broad structural classes of transmembrane proteins: complement control protein (CCP)/short consensus repeat (SCR) domain modular single-pass transmembrane receptors (CR1, CR2), β2 integrins (CR3, CR4), complement receptor of the immunoglobulin family (CRIg), and G-protein coupled receptors (GPCR) (C5aR1, C5aR2, C3aR) (Table 1). Various aspects of their sequence, function, structure, localization, regulation, activation, and implications for infection, pathogenesis, and therapy have been reviewed elsewhere (1–19). Complement receptors recognize either the C3 proteolytically activated fragments C3b, iC3b, and C3dg deposited on opsonized surfaces (CR1 to CR4 and CRIg) or the soluble anaphylatoxins released during alternative pathway activation (C3aR) or terminal pathway activation (C5aR1/C5aR2). The pattern of cellular expression and compartmentalization of the complement receptors underlies their various functions: leukocyte recruitment and migration, phagocytosis, and inflammation. A fascinating function of complement receptors is connecting the effector responses of the innate and adaptive branches of the immune system.
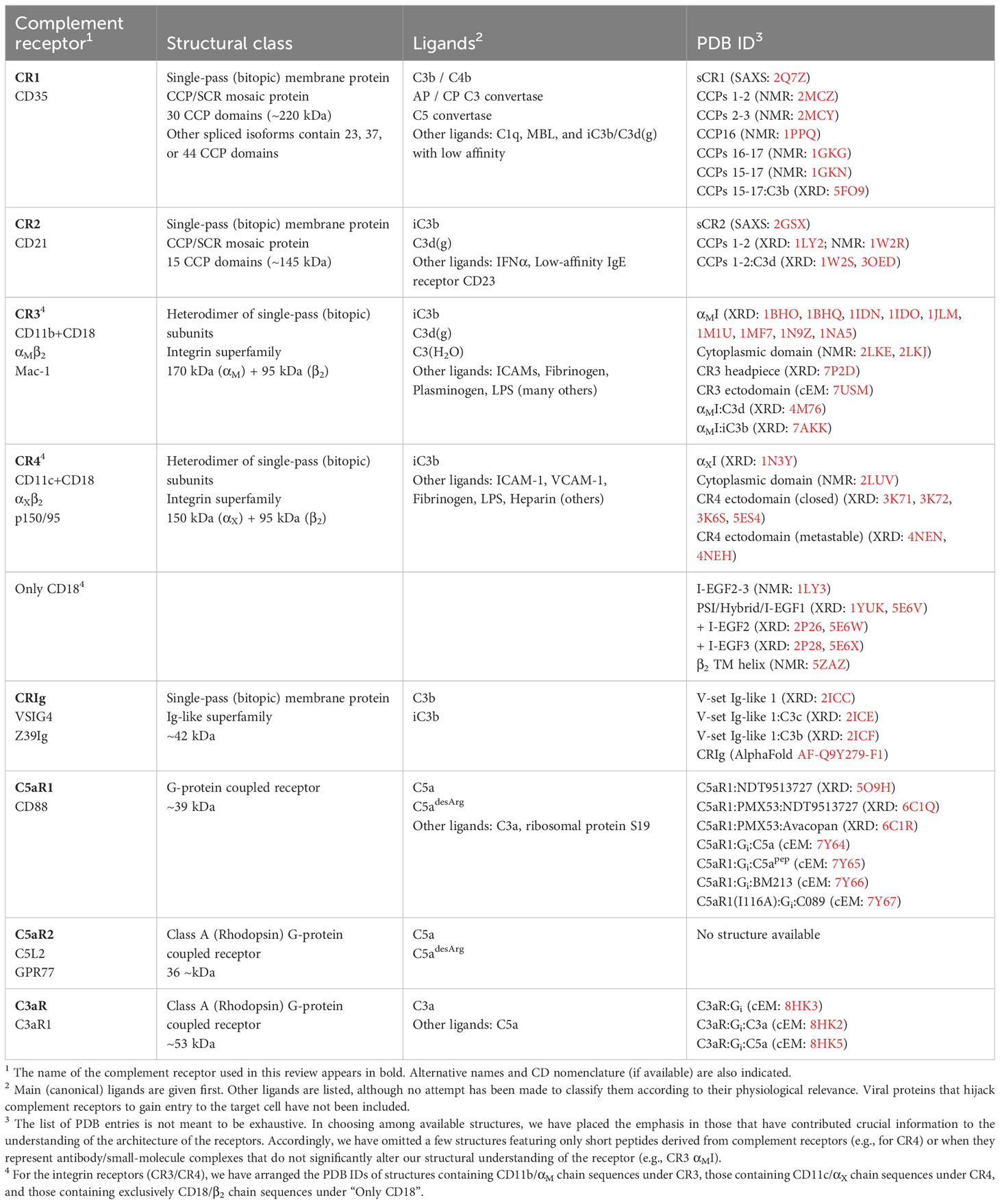
Table 1 Complement receptors and membrane-associated regulators.
The membrane-bound negative complement regulators membrane cofactor protein (MCP/CD46) and decay accelerating factor (DAF/CD55) are structurally related to CR1 and CR2 as they contain tandem repetitions of the CCP/SCR domain (20). However, they are not commonly considered complement receptors as their main function is to accelerate convertase decay or act as cofactor of complement factor I (FI) to prevent unregulated complement deposition on self-cell surfaces, and will not be discussed further (but see (21, 22) regarding MCP as a complement receptor in T-cell lymphocytes). Likewise, we will not cover three proteins that have been proposed as C1q receptors: C1qRp/CD93, cC1qR/calreticulin (CR), and gC1qbp. CD93, also known as C1q receptor protein (C1qRp), promotes cell adhesion, migration, and angiogenesis; however, its main ligand appears to be multimerin-2, not C1q (23, 24). cC1qR is a 62-kDa form of ecto-calreticulin that can bind the collagen domain in C1q with the assistance of other membrane proteins including β1 integrins, CD91, and the KDEL docking receptor (25). Finally, gC1qR is a 33-kDa binding protein for the globular head of C1q (gC1qbp) found in both the intracellular and cell surface compartments, where it can bind various proteins including vitronectin, thrombin, and fibrinogen; the location of gC1qR is mainly in the mitochondria, where its leader peptide is processed (24).
The renewed interest in the pharmacological modulation of the complement system (26–31) has contributed to a recent surge of structural information on the structure and function of complement receptors, especially CR3 (32–34) and the anaphylatoxin receptors C5aR1 and C3aR (35, 36). This review aims to inform and guide structurally aware basic and clinical research by providing an up-to-date synthesis of our current understanding of the structural biology of complement receptors.
An exciting topic that we will not cover in this review is the intracellular complement system, the complosome, despite the breadth of attributed effector functions and the important associations uncovered with prevalent diseases (reviewed in (37)). We have decided to leave the complosome outside the scope of this review as the available structural information is not specific to the intracellular location of complosome components (e.g., C3aR/C5aR1).
2 Highly modular complement receptors based on the CCP/SCR domain
2.1 The building block: CCP/SCR domain
The complement receptors CR1 and CR2 and the negative complement regulators FH, FH-related (FHR) proteins, C4b binding protein (C4BP), MCP, and DAF are all mosaic proteins comprised of several independently-folded modules known as the short consensus repeat (SCR), complement control protein (CCP), or Sushi domain (20, 38). Other complement factors that bind C3b or C4b, like FB or FI, also contain CCP domains and other structural and catalytic domains. Conversely, CCP domains are also found in complement factors lacking the ability to bind C3b or C3b, like the complement proteases C1r and C1s (39).
The dimensions of the CCP domain are approximately 1 nm in width and 3.6 nm in length (Figure 1A). Each CCP (61 amino acids) contains a hydrophobic core composed of up to eight-stranded antiparallel β-sheet stabilized by two conserved disulfide bridges (Cys1-Cys3 and Cys2-Cys4) and a buried conserved Trp residue (40) (Figure 1A). Individual CCP domains in mosaic sequences start with the first conserved Cys (Cys1) and end with the last conserved Cys (Cys4); the two-to-eight residues between Cys4 in the preceding CCP domain and Cys1 in the following CCP domain are denoted as inter-CCP linking regions. A structurally important feature of the inter-CCP linkers is that they allow a wide range of inter-domain orientations, thus adding to the structural variability of the mosaic proteins that contain them (41). CCP domains have been structurally classified into nine distinct groups (from A to I) according to sequence and structural features (42). This classification is intended to help the systematic structural analysis and the accurate homology modeling of CCP domain-containing proteins.
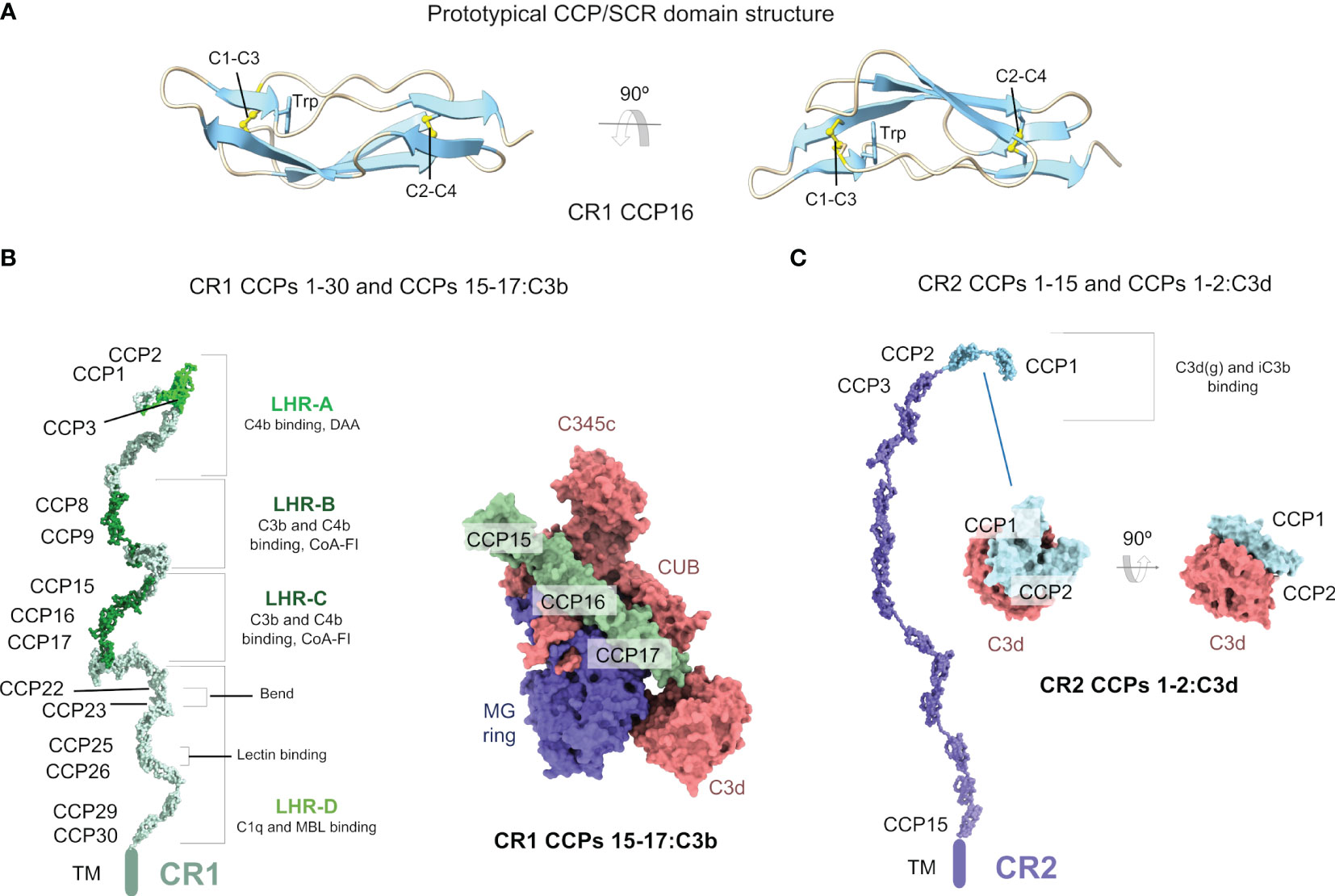
Figure 1 Complement receptors CR1 and CR2 are mosaic proteins built from CCP/SCR modules. (A) Structure of a prototypical CCP/SCR domain, the CR1 CCP16, taken from the structure of CR1 CCPs 15-17:C3b (PDB ID 5FO9). The domain is shown in cartoons in two orientations. The two most conserved features of CCP domains, the disulfide bonds between four conserved cysteine residues (C1-C3 and C2-C4) and a conserved tryptophan (W) residue, are shown in sticks and CPK atom colors. (B) Structure of the CR1 ectodomain (left) comprising CCPs 1-30 modeled from SAXS data (PDB ID 2Q7Z), with C3b/C4b interacting CCP domains colored in lime (C4b) and dark green (C3b/C4b). Structure of the CR1 CCPs 15-17:C3b (right) (PDB ID 5FOB) in molecular surface representation. C3b is colored according to chain (the α’ chain in red, the β chain in blue), and CR1 CCPs 15-17 is colored in dark green. (C) Structure of the CR2 ectodomain (left) comprising CCPs 1-15 modeled from SAXS data (PDB ID 2GSX), with C3b interacting CCP domains colored in cyan. Structure of the CR2 CCPs 1-2:C3d (right) (PDB ID 3OED) in molecular surface representation. C3d is colored in red, and CR2 CCPs 1-2 is colored in cyan.
2.2 Complement receptor 1
Complement receptor 1 (CR1, CD35, C3b/C4b receptor) is a type I transmembrane glycoprotein from the Regulators of Complement Activity (RCA) family. CR1 acts as a receptor for C3b/C4b, C1q (43), the mannan-binding lectin (MBL) (44), and as an immune adherence receptor (45). The biological functions of CR1 rely on its ability to bind to C3b and C4b reversibly, components of the C3 convertase of the alternative pathway (C3bBb) or the classical pathway (C4b2a), inactivating the C3 and C5 convertases, and promoting the dissociation of the catalytic subunits C3a or Bb (decay-accelerating activity) (46). CR1 also serves as a necessary cofactor for FI-mediated proteolytic cleavage of C3b and C4b to the breakdown products iC3b/C3dg and iC4b/C4dg, respectively (cofactor activity) (46). The cellular location of CR1 influences its biological functions. CR1 is mainly found on the surface of erythrocytes, where it is responsible for the Knops blood group (York and McCoy antigens), and on antigen-presenting (APC) cells (47). While in erythrocytes CR1 contributes to the clearance of complement-fixed immune complexes, in leukocytes its main role seems to be channeling the immune response to foreign antigens to other immune cell types bearing CR2, CR3, and CR4 receptors. In addition, CR1 acts as a B-cell receptor (BCR) inhibitor to prevent B cell activation (48). A soluble version of CR1 (sCR1) has also been identified with anti-inflammatory properties (43).
2.2.1 Structure of the largest complement receptor, CR1
CR1 is the largest member of the RCA family. In the most common allelic form, the extra-cellular component of CR1 (sCR1) contains 30 CCP domains and 14 occupied N-linked glycosylation sites; other allelic forms have 23, 37, or 44 CCP domains. The 30 CCP domains of CR1 are organized as four long homologous repeat regions spanning seven CCP domains (LHR-A to LHR-D) plus two C-terminal CCP domains. Three functionally relevant sites have been identified in CR1: Site 1 in CCPs 1-3 in LHR-A binds C4b and is the site harboring the decay-accelerating activity toward the classical and alternative pathway C3 convertases; site 2 in CCPs 8-10 in LHR-B and site 3 in CCPs 15-17 in LHR-C bind C3b and C4b and are the main sites for the FI cofactor activity.
Given the membrane location, size, glycosylation, and highly modular and flexible structure, obtaining high-resolution structural information about CR1 has been challenging. The first structural information about sCR1 was obtained by negative staining electron microscopy (NS-EM). Those early electron micrographs exposed the elongated structure of sCR1 in various CCP structural arrangements (49). However, the most comprehensive structural description of sCR1 has thus far been obtained by solution small-angle X-ray scattering (SAXS) coupled with biophysical techniques like analytical ultracentrifugation (AUC) and constrained computational modeling.
Even though at a moderate resolution, this approach has revealed critical structural features of CCP repeat proteins like FH (50), CR2 (51), and CR1 (52). Strikingly, sCR1, as FH and sCR2, does not adopt a fully extended structure, which would stick out of the cellular surfaces harboring it by a maximum theoretical length of 108 nm (compared with 17 nm for C3b/C4b). In contrast, SAXS data analysis revealed that sCR1 structure folds back onto itself to yield a more densely packed molecule with a maximum dimension Dmax = 55 nm and a radius of gyration RG = 13.4 nm (52) (PDB ID 2Q7Z) (Figure 1B). In comparison, FH has a Dmax = 40 nm instead of the maximum theoretical length of 73 nm, and sCR2 has a Dmax = 38 nm instead of the theoretically maximum length of 54 nm. Analysis of the SAXS cross-sectional radii of gyration for sCR1 (RXS-1 = 4.7 nm and RXS-2 = 1.2 nm) in combination with the RG and the frictional ratio RG/R0 = 3.76 provided further confirmation of the folded-back structure of sCR1, which resembles that of FH more closely than that of sCR2. For CCP-containing proteins, RXS-1 reports on the averaged medium-range folding back and RXS-2 on the averaged short-range inter-CCP orientation between adjacent CCP domains (50). Equilibrium sedimentation experiments with sCR1 resulted in a sedimentation coefficient s°20,w = 5.84, and a frictional coefficient anisotropy ratio f/f0 = 2.3 (compared to 1.25 for globular proteins), which agree with the extended but folded-back structural model derived from SAXS measurements.
SAXS-constrained modeling of the three-dimensional structure of sCR1 has shown that sCR1 can be best described as a family of partly folded-back CCP structural arrangements with a moderate degree of flexibility around the CCP inter-linking regions. SAXS and other biophysical techniques have comprehensively sampled the translational and rotational average structural ensemble. SAXS-constrained modeling has suggested that the four-residue linker between CCP22 and CCP23 might be responsible for the main bend in sCR1. Still, this inference awaits experimental corroboration as SAXS alone cannot prove where the kink occurs. A kink at CCP22-CCP23 would introduce an angle between the LHR-C and LHR-D regions, apparently without implications for ligand recognition, as the C3b/C4b binding sites are in the LHR-A, LHR-B, and LHR-C regions. This observation contrasts with the oft-cited statement that the folded-back structure of CR1 facilitates binding to multiple ligands on the surface of pathogens or immune complexes.
Higher resolution structures of unliganded CR1 domains are only available for a subset of CCP domains. The earliest structures were determined by multinuclear NMR for the three CCP domain tandem constructs CCPs 1-3, CCPs 8-10, and CCPs 22-24 (41), and the CCPs 15-16 (PDB ID 1GKN) and CCPs 16-17 (PDB ID 1GKG) domain pairs in site 3 of CR1 (53). The NMR structure of an individual CCP domain was also published for CCP16 (PDB ID 1PPQ) (54). The NMR structure of the N-terminal CCP domain pairs CCPs 1-2 (PDB ID 2MCZ) and CCPs 2-3 (PDB ID 2MCY) (55).
2.2.2 CR1 dampens C3 and C5 convertase activities, and transports opsonized cargo
CR1 uses CCP binding sites in LHR-A, LHR-B, and LHR-C to recognize and bind C3b and C4b. CR1 ligands include both monomeric ligands such as C3b, C4b, or the C3 convertase (C3bBb or C4b2a) and the bivalent C5 convertases, which contain a back-to-back arrangement of either C3b-C3b dimers (alternative pathway) or C3b-C4b dimers (classical pathway) (56, 57). This bivalent recognition sets CR1 apart from all other RCA proteins and it explains the 10-fold tighter binding affinity of CR1 for C5 convertases over C3 convertases (58).
Recent structural data have shed light on how CR1 recognizes its cognate ligand C3b (59). The crystallographic structure of C3b:CR1 CCPs 15-17 (PDB ID 5FO9) has shown how CR1 exploits a similar binding mode to FH CCPs 1-4 to exert cofactor activity (60) (Figure 1C). C3b domains engaged in this interaction include MG7, MG6, CUB, MG2, MG1, and TED, and the α’NT region over a region spanning ~100 Å with ~1910 Å2 buried surface area.
In contrast to C3b/C4b ligands, CR1 uses the LHR-D homologous repeat to recognize and bind C1q and MBL. Therefore, C1q and MBL must compete for binding to CR1. A structural characterization of the interaction between CR1 and C1q/MCP awaits further investigation.
2.2.3 Implications of CR1 structure for disease and immune evasion
sCR1 has been proposed for therapeutic use based on its anti-inflammatory properties and low immunogenicity. Possible applications include controlling inflammatory tissue damage in myocardial infarction (49), tissue damage suppression in complement-dependent autoimmune diseases (46), and the treatment of pemphigus foliaceus (61).
CR1 is also known as a receptor for various pathogens, including Plasmodium falciparum, the malaria agent, through direct interaction with erythrocyte membrane protein 1 (PfEMP1) and reticulocyte-binding homolog protein 4 (PfRh4) (55, 62–66). The interaction with PfEMP1 causes red blood cells to become “sticky” and rigid, displaying the parasite phenotype known as “rosetting” or adhesion of infected erythrocytes to uninfected erythrocytes, which maintains infected red blood cells in the microvasculature, and avoiding destruction in the spleen and liver. At the structural level, rosetting depends on C3b-binding sites on LHR-B and LHR-C homologous repeats, even though it does not involve C3b. In contrast, PfRh4 interacts with CR1 CCP1 inhibiting the decay-accelerating activity without affecting C3b/C4b binding (55).
2.3 Complement receptor 2
Complement receptor 2 (CR2, CD21) is a type I membrane glycoprotein found on the cell surface of mature B cells, follicular dendritic cells, epithelial cells, and some T cells. CR2 contains 15-16 CCP domains depending on alternative splicing (67), which makes it the third largest CCP repeat protein within the RCA proteins after CR1 and FH. CR2 is the only RCA protein lacking complement regulatory functions; instead, CR2 links the innate and adaptive immune response during the activation of B cells through binding to its primary ligand, C3d, in a complex with CD19, CD81, and mIgM, which is thought to reduce the threshold of immune activation. Besides C3d and the C3d-containing opsonin iC3b (but not C3b) (68, 69), CR2 has three known ligands: IFNα (70, 71), the low-affinity IgE receptor CD23 (72), the glycoprotein gp350 of the Epstein-Barr virus (73, 74).
2.3.1 CR2: a dynamic structure to bind opsonized surfaces
As sCR1 and FH, the structure of sCR2 has been studied by various structural and biophysical methods. The first images of sCR2’s elongated structure were obtained by electron microscopy (75). More detailed structural information has been obtained by a combination of SAXS and AUC (76). In solution, sCR2 has an RG = 11.5 nm (RG/R0 = 4.1), RXS-2 = 1.2 nm (and no RXS-1), and a Dmax = 38 nm (SAXS) and s020,w = 4.2 nm (f/f0 = 2) (AUC). In contrast to sCR1 and FH, the CCP overall structural arrangement of sCR2 is more extended (although not fully extended) and only folds back partially onto itself (PDB ID 2GSX) (Figure 1C). Accordingly, the Dmax derived from SAXS comes nearer to the theoretically maximum length of 54 nm. Another feature of CR2 concerns the length of the inter-CCP linkers, which is longer compared with CR1 and FH; in CR2, there are several inter-CCP linkers from four to eight amino-acid long. The greater length of the inter-CCP linkers gives CR2 a higher degree of flexibility, allowing it to reach a larger overall length than structurally similar proteins.
Isolated CR2 CCP domains have also been studied at the structural level. CCPs 1-2 have been shown to adopt a V-shaped structure by X-ray crystallography with implications for C3d/iC3b ligand binding (Figure 1C). Although the crystal structure of unbound CR2 CCPs 1-2 (PDB ID 1LY2) suggested a compact V-shaped arrangement with substantial flexibility at the junction between CCP1 and CCP2 domains (77), the solution NMR structures calculated for the same domains have shown a more open, but equally kinked, structure (PDB ID 1W2R) (78–80).
2.3.2 Ligand binding at the interface between innate and adaptive immunity
The CR2 structure binds three relatively small protein ligands: C3d, gp350, and IFNα at CCPs 1-2, while a fourth ligand, CD23, binds both CCPs 1-2 and CCPs 5-8. The maximum dimensions of these ligands are 6.0 nm (C3d), 10.2 nm (gp350), 5.3 nm (IFNα), and 6.8 nm (CD23). Although CD23 is not much larger than C3d, CR2 uses two sets of CCP binding sites to latch onto it, a process likely favored by the flexibility of CR2. Therefore, CR2 can bind to antigen-C3d complexes on the B-cell surface and CD23 to bring the N-terminal tip of CR2 closer to membrane-bound IgE molecules on the B-cell surface (76).
The first crystal structure of CR2 CCPs 1-2 in complex with C3d showed a compact V-shaped structure where only CCP2 interacts with C3d (81). This was later disputed by constrained molecular modeling in 50 mM NaCl and mutagenesis data in 137 mM NaCl, which provided compelling evidence that both the CCP1 and CCP2 domains bind to the surface of C3d in a kinked conformation (PDB ID 1W2S) (79, 82). Later, a new crystal structure was published for CR2 CCPs 1-2:C3d, revealing a V-shaped conformation for CR2 CCPs 1-2 with a more extensive interface comprising residues from both CCP domains (PDB ID 3OED) (Figure 1C) (83). Although the conformation of CR2 CCPs 1-2 is similar in the two C3d complexes (RMSD 1.2-1.5 Å), the region of C3d involved in the interaction with CR2 CCPs 1-2 found in the crystal structure is more consistent with available functional and mutagenesis data.
The first structural glimpse into the CR2:C3d complex, indeed, into any large CCP-containing protein and its ligand, was obtained by SAXS and AUC (51). While sCR2 structure and oligomeric state remained unchanged in 50-137 mM NaCl, unbound C3d was shown to exist as monomers only in 137 mM NaCl; in 50 mM NaCl, C3d exists in monomer-dimer and monomer-trimer equilibria. Interestingly, the sCR2:C3d complex could be analyzed by AUC only in 50 mM NaCl, where the sedimentation coefficient shifted from 4.0 S (sCR2 alone) to 4.5 S (sCR2:C3d). The models put forward to rationalize the CR2:C3d complex (PDB ID 1W2R, PDB ID 1W2S) provide a solid foundation for future work, even if the details of the C3d binding interface may be better captured by the CR2 CCPs 1-2:C3d crystallographic structure (PDB ID 3OED) (76, 83).
Several features of the interaction between CR2 and C3d are worth remembering. Firstly, the extended, flexible, and relatively fold-back structure of CR2 and the V-shaped arrangement of CCPs 1-2 that is ideally positioned to interact with C3d-antigen complexes. Secondly, the constellation of weak and electrostatically modulated interactions between CR2 and C3d becomes physiologically significant only by avidity effects driven by receptor clustering. This weak summation of specific interactions appears discriminatory for B cells to respond only to antigens presented as multimeric C3d molecules clustered through surface-bound CR2 molecules. This parallels the weak CR1 interaction with C3b/C4b molecules on neutrophils, which is enhanced by the polymerization or multimerization of the ligand and receptor clustering. Other examples of this structural principle will be seen later.
2.3.3 Implications of CR2 structure for diseases and immune evasion
CR2 was recognized as a receptor for the Epstein-Barr virus (EBV) very early on, mapping the CR2 binding region to the first two CCP domains and thereby in competition with C3d (68, 74). The gp350 protein on the surface of the viral membrane envelope interacts with CR2 during the first steps of EBV entry. Another virus that exploits CR2 as a receptor for viral entry is the human immunodeficiency virus 1 (HIV-1) (84). HIV-1 can infect B cell lymphocytes in a complement/C3-dependent and CD4-independent manner, thus facilitating viral dispersion and access to lymphoid organs (85). Besides viruses, the pathogenic yeast Cryptococcus neoformans has been shown to use an extracellular factor, the antiphagocytic protein 1 (App1), to bind to CR2 (and also CR3) and avoid complement-mediated phagocytosis by alveolar macrophages (86).
2.4 CR1 and CR2 are structurally selective receptors
CR1 and CR2 both recognize C3 activated fragments tethered to a biological surface by using a common structural framework, yet they accomplish a remarkable degree of selectivity through distinct binding modes and cell localization. As already seen, CR1’s main ligands are C3b, C4b, the AP/CP C3 convertases (monovalent binding sites), and the C5 convertases (bivalent binding sites), all markers of active complement opsonization. In contrast, CR2’s ligands are iC3b and C3d(g), both monovalent binding sites and markers of halted complement activation.
Although at face value CR1 and CR2 recognize distinct ligand sets, the fact that C3b, iC3b, and C3d(g) share the TED domain could potentially lead to overlapping binding sites. In fact, CR1 is known to bind iC3b and C3d with low affinity (87) in addition to the higher affinity binding to C3b/C4b. The solution to this apparent problem has been revealed by the X-ray crystal structures of CR1 CCPs 15-17:C3b (PDB ID 5FO9) (59) (Figure 1B) and CR2 CCPs 1-2:C3d (PDB ID 3OED) (83) (Figure 1C). By comparing these two complexes, which reflect the tightest receptor-ligand interactions (2 μM for CR1 CCPs 15-17:C3b (59) and 22 nM for CR2 CCPs 1-2:C3d (78)), it is straightforward to see that the molecular surfaces recognized by either receptor are rather distinct. A small overlap is, however, created by CR1 CCPs 15-17 interacting weakly with the CUB-TED domains in C3b (59), interactions that are likely lost in iC3b. Furthermore, interaction of CR2 with C3d occurs through surfaces that are partially occluded in C3b, but sterically unimpeded in iC3b/C3d(g).
3 Integrin receptors
Integrins link the extracellular matrix (ECM) to the cellular cytoskeleton and associated signal transduction pathways and mediate cell-cell, cell-ECM, and cell-pathogen adhesion (88). Integrin-mediated interactions participate in cytoskeletal remodeling, phagocytosis, and cell migration (89). Complement receptors 3 (CR3) and 4 (CR4) belong to the integrin superfamily of type I transmembrane heterodimers (90). CR3 and CR4 are co-expressed in myeloid cells like neutrophil granulocytes, monocytes, macrophages, activated T and B lymphocytes, and lymphoid natural killer cells (91). They mediate immune adhesion-dependent processes such as adhesion to endothelium, phagocytosis of opsonized foreign particles, and other activation events that promote the innate and adaptive branches of the immune system (92).
These leukocyte-specific receptors bind multiple ligands like iC3b (93–95), ICAMs (96), fibrinogen (97), or lipopolysaccharide (LPS) (98). Despite their sequence homology, structural similarities, and overlapping ligands, CR3 and CR4 are functionally specialized, presenting a “division of labor” that makes them nonredundant receptors (34, 99).
Integrin signaling involves bidirectional communication between the extracellular environment and the intracellular cytoskeleton and signal transduction pathways, and the most accepted mechanism invokes an “inside-out” signaling precedence (100, 101). In the resting state, integrins adopt a bent, “closed”, ligand-free inactive state; signals initiated in the actin cytoskeleton can trigger a dramatic conformational change in the extracellular region of integrins, which become extended, “open”, ready to engage the ligand if available. When appropriate ligands are nearby, they can engage the receptors, initiating signaling processes from the “outside-in”.
3.1 The overall structure of the β2 integrins
Integrins are comprised of two noncovalently-associated protein chains: the α subunit (150-172 kDa) and the smaller β subunit (95 kDa), which is glycosylated. The cytoplasmic regions of integrins are very small compared to the large ectodomains. CR3 and CR4 share the same β2 chain (CD18) and belong to the β2 integrin family of adhesion receptors (90, 102), differing in their α chain, which is αM in CR3 and αX in CR4. The full CR3 heterodimer is also known as Mac-1 (Macrophage-1 antigen), CD11b/CD18, or integrin αMβ2, and the CR4 heterodimer is also known as p150,95, CD11c/CD18, or integrin αXβ2.
CD18/β2 integrins are restricted to leukocytes, and except for mast cells, which lose CD18 expression during differentiation, all leukocytes express one or more CD18 integrins (91). CR3 and CR4 have found utility in biomedicine as their expression in NK cells enables complement-dependent cytotoxicity toward anti-CD20 (rituximab)-coated cancer B cells, contributing to the treatment’s efficacy (103). CR3 and CR4 belong to the class of inserted (I) domain-carrying receptors. Fittingly, the N-terminal end of the α chain contains the iC3b-binding von Willebrand type A (VWA) domain or α chain inserted domain (αI), a specialized region characterized by a modified Rossman-fold architecture and a metal ion (Mg2+)-dependent adhesion site (MIDAS) motif (7). This relatively small domain is inserted between β-sheets (blades) 2 and 3 of the next domain in the α chain, a seven-bladed β-propeller domain, which allows the relative orientation of the αI and β-propeller to adjust flexibly (104). The aptly named Thigh, Calf-1, and Calf-2 domains complete the α chains. The αM and αX chains are homologous, with an overall sequence identity of ~60% (~47% in the αI domain). The functional discrimination of ligands by CR3 and CR4 is even more intriguing because most of the ligand recognition seems to be mediated by the 320-amino-acid αI domain, with the crucial involvement of the Mg2+ in the MIDAS. A comparison of the nature of known ligands suggests that strongly negatively charged molecules tend to be recognized by CR3, whereas CR4 tends to bind positively charged species (7). CR3 and CR4 are also homologous to two additional β2 integrins, the lymphocyte function-associated antigen 1 (LFA1, CD11a/CD18, αLβ2) and αDβ2 (CD11d/CD18).
The sequence of domains of the β2 chain from the N to the C terminus includes I-like, Hybrid, plexin-semaphorin-integrin (PSI), integrin epidermal growth factor (I-EGF) 1-4, and β tail (BT) domains. The β2-chain I-like domain interacts with the α-chain β-propeller domain to form a broad platform that supports the ligand-binding αI domain. The overall structure of the β2 chain domains contributing to the headpiece will be discussed later with available structural data for CR3 and CR4. As for the stalk region of the β2 chain, there is structural data for a substantial part, even if piecemeal.
The crystal structure of the PSI/Hybrid domain/I-EGF1 segment from the human integrin β2 chain was solved by X-ray crystallography at 1.8-Å resolution (PDB ID 1YUK) (105). The structure of this first part of the stalk revealed an elongated molecule with a rigid architecture stabilized by nine disulfide bonds. The PSI domain is wedged between the Hybrid and I-EGF1 domains, with extensive interfaces stabilized by contacts between conserved arginine and tryptophan residues. Soon after, there appeared two additional structures containing the PSI/Hybrid domain/I-EGF1 and additional I-EGF modules, I-EGF2 (PDB ID 2P26) and I-EGF2 and I-EGF3 (PDB ID 2P28) (106). The I-EGF is a cysteine-rich repeat module with a nosecone shape; four copies are located in the stalk region, where they relay activation signals to the ligand-binding headpiece. In the first structure, there was a prominent kink between the I-EGF1 and I-EGF2 modules, whereas, in the second structure, the three I-EGF modules adopted an extended conformation. The NMR structure for I-EGF3 and the NMR analysis of the interface contacts between I-EGF2 and I-EGF3 had been previously studied (PDB ID 1LY3) (107). The interdomain contacts between I-EGF domains 2 and 3 could be measured by NMR and were interpreted in terms of an approximate two-fold screw axis. In the NMR structure, the I-EGF domains 2 and 3 adopt an extended conformation connected by the “genu”, a highly flexible linker that allows extreme bending. Based on these data, the authors posited that the release of contacts of the headpiece with I-EGF modules 2 and 3 could trigger a switchblade-like opening motion springing the integrin into its extended, active conformation. Reanalysis of these structures in the context of the structure of the αLβ2 headpiece in the closed conformation confirmed previous results while stressing the importance of proper disulfide pairing in the cysteine-rich I-EGF modules (PDB ID 5E6V, 5E6W, 5E6X) (108).
The structure of the single transmembrane helix of the β2 chain has been shown by NMR to use a membrane-snorkeling lysine residue (Lys702) to interact with acidic phospholipids in the membrane bilayer to stabilize the bent closed conformation (PDB ID 5ZAZ) (109). This interaction can be modulated by intracellular Ca2+, disrupting it and facilitating the acquisition of the extended open conformation. As this mechanism seems independent from the “inside-out” integrin signaling in T cell lymphocytes, it suggests a more prominent role for direct interactions of the β2 chain, membrane phospholipids, and Ca2+ in regulating integrin structure and conformational changes.
3.2 Complement receptor 3
The initial work on the structure of CR3 was carried out by negative-staining electron microscopy on the headpiece (110) and by X-ray crystallography on the ligand-binding αMI domain, which has been most thoroughly characterized (111–113). More recently, the crystal structure of the CR3 headpiece (33) and the cryoelectron microscopy structure of the CR3 ectodomain (114) have advanced the field significantly (Figure 2A, B).
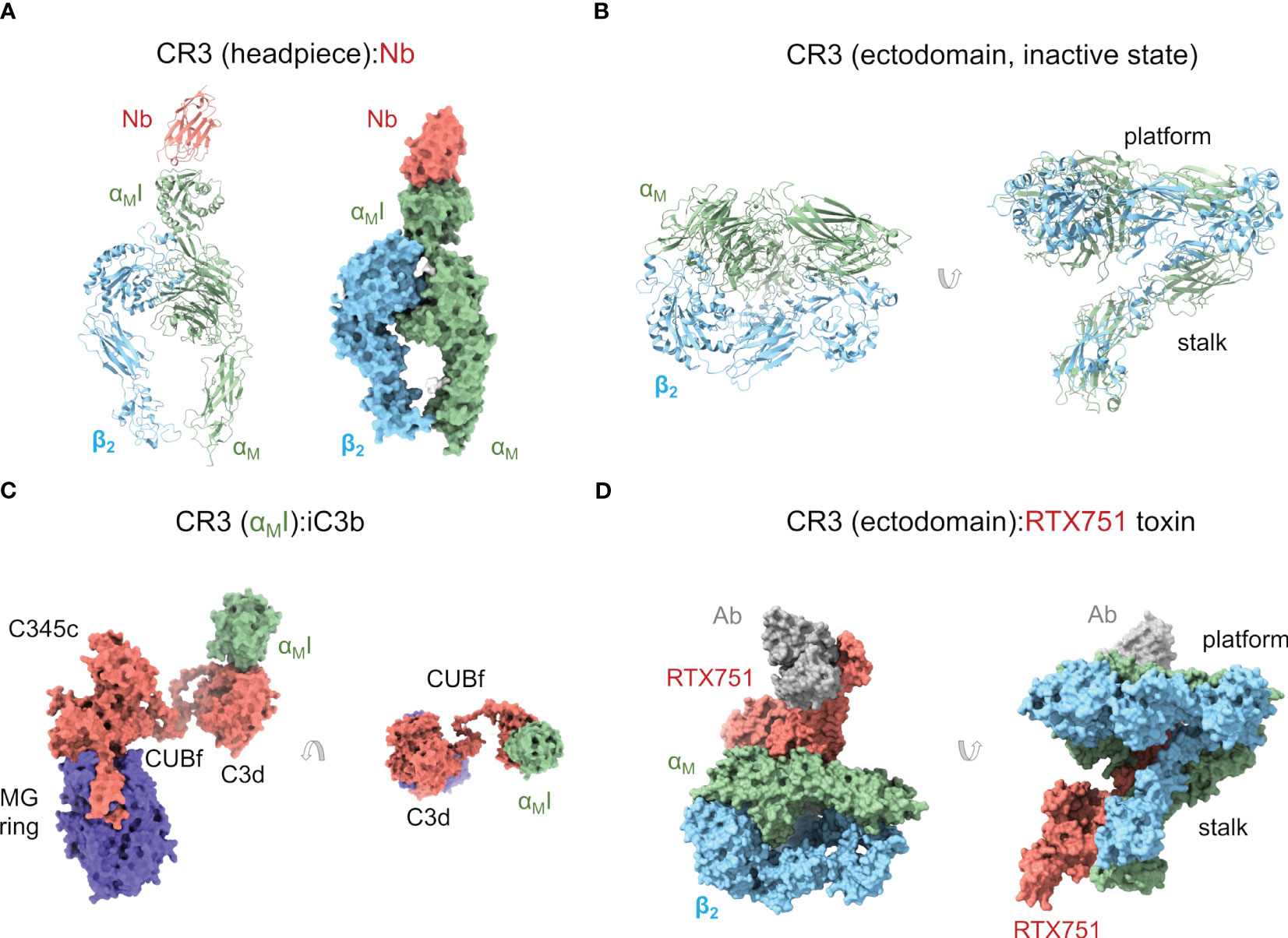
Figure 2 Complement receptor CR3. (A) Cartoon and molecular surface representations of the X-ray crystallographic structure of the CR3 headpiece in complex with a nanoantibody (Nb) (PDB ID 7P2D). The αMI domain is in the inactive, closed conformation. (B) Cartoon representation of the cryoelectron microscopy structure of the CR3 ectodomain (except for the αMI domain) in an inactive, closed conformation (PDB ID 7USM). (C) X-ray crystallographic structure of a complex between iC3b and CR3 αMI domain (PDB ID 7AKK) where iC3b adopts an extended conformation. The interaction between the TED/C3d domain of iC3b and CR3 αMI is shown in two orientations related by a 90° rotation. (D) Molecular surface representation of the cryoelectron microscopy structure of the CR3 ectodomain (except for the αMI domain) in complex with B. pertussis RTX751 toxin (PDB ID 7USL). In this structure, the CR3 ectodomain adopts a more extended conformation than in (B) through interactions with the toxin.
Crystal structures of the αMI domain in the open (PDB ID 1IDO) (112) and closed (PDB ID 1JLM) (113) conformations revealed the overall structure of the MIDAS motif and a ligand-binding regulatory mechanism whereby helix α7 plays a crucial role in the closed form by restricting access to negatively charged ligand residues. While in the open state a glutamic acid residue from a neighboring αMI domain completed the Mg2+ coordination sphere, in the closed form it was a key aspartic acid in helix α7 that played the role. Two other crystallographic structures of the αMI with the antagonist simvastatin (PDB ID 4XW2) (115) and with a ligand-mimicking antibody (PDB ID 3QA3) (116) have strengthened this idea by showing binding interfaces where the ligand contributes an acidic side chain to complete the metal coordination sphere.
CR3 and other β2 integrins show homotypic interactions or, at least, a tendency to form homotypic interactions. Tellingly, NS-EM studies on the CR3 headpiece showed large numbers of dimers (110). Even the αMI domain in the open conformation formed weak homodimeric interactions with crystallographic lattice neighboring molecules (112). Homotypic interactions may be desirable for β2 integrins as they must accommodate large concentrations during receptor clustering or result from the loose ligand specificity. The first structure of a β2 integrin with a complement factor ligand was that of CR3 αMI in complex with C3d (PDB ID 4M76), the main ligand for CR3 (111). This structure revealed the binding mode of C3d to αMI, characterized by an aspartate side chain of C3d chelating the MIDAS motif of αMI, which was occupied by a non-physiologic Ni2+ cation from the crystallization condition. Although the buried surface area is relatively small, the interaction is strong enough (affinity (KD) is in the micromolar range) to stabilize the complex and is comparable in area and affinity to other integrin complexes. A key insight from this structure showed that the C3d surface engaged by αMI is masked in C3b by a well-folded CUB domain, effectively ruling out the possibility of an αMI interaction with C3b. In iC3b, however, the cleavage by FI inside the CUB domain causes it to unfold, making the αMI binding motif accessible.
Another fascinating insight from the αMI:C3d structure was its compatibility with the binding of C3d by CR2 through its CCPs 1-2 domains (7). This hypothetical αMI:C3d:CR2 complex was interpreted as a hand-over or transfer of C3d-opsonized antigens from CR3-bearing macrophages to CR2-bearing B lymphocytes. This process might act like an MHC-independent antigen presentation mechanism bridging the innate and adaptive branches of immunity. It would be interesting to characterize the hand-over in more cellular structural detail as the involvement of three distinct surfaces (macrophages, B cells, and opsonized particles) renders the entire process challenging.
A recent structure by our group has shown a complex between the entire iC3b and the αMI domain (PDB ID 7AKK) (Figure 2C) (34). In this structure, the MIDAS motif of αMI was fully charged with Mg2+. The interaction between αMI and the TED domain of iC3b was identical to the interaction previously observed between αMI and C3d (111). More interestingly, the structure of CR3 αMI:iC3b revealed two potential interfaces for the αMI domain on the MG ring of the C3c fragment. This is relevant because the C3c moiety has been known to contribute to CR3 binding in interaction assays (111) and, perhaps more importantly, because iC3b-opsonized surfaces are phagocytosed by CR3-expressing macrophages at much lower opsonin concentration than C3d-opsonized surfaces (117), begging the question about what the role of C3c in CR3 binding might be. In one of these αMI:C3c interfaces involving C3c MG1-2 domains, the C3c moiety of iC3b would conserve an “original” orientation with respect to the surface-anchored TED domain (“upright”), while in the other one, involving the C3c MG3-4 domains, the C3c moiety would be required to turn around and lie “upside-down” with respect to the TED domain. Surface plasmon resonance (SPR) binding experiments and site-directed mutagenesis of interfacial residues on the αMI domain indicated that both interfaces may be relevant in vivo. Additional evidence will be necessary to clarify the physiological role of the C3c moiety of iC3b for CR3 recognition. Meanwhile, an enticing hypothesis is that iC3b behaves as a modular platform comprising a surface-anchored C3d and a more detached C3c moiety that collaborate to bind the CR3 ectodomain in the highly concentrated environment of the cell-particle interface. A modular iC3b would facilitate binding by increasing the number of low-affinity contacts (avidity effect), restricting the angular spread of CR3:C3d complexes to increase CR3:iC3b packing efficiency (alignment effect), and making it possible for C3d(g) and C3c moieties from the same or different iC3b molecules to collaborate in CR3 binding.
Although the role of CR3 as a complement receptor is well-attested, the capacity of CR3 (and CR4) to recognize and bind to a wide variety of other ligands remains puzzling. CR3, for example, interacts with many macromolecular components of the coagulation system (e.g., fibrinogen, fibrin, kininogen, plasminogen, heparin), denatured proteins, and oxidative and degrative products of lipids and glycans (7). Analysis of the electrostatic properties of the ligand-facing molecular surface has shown that it is a markedly negatively charged surface, which is in line with the preference of CR3 for cationic ligands like the intrinsically unstructured myelin basic protein (MBP) (118) and the antimicrobial peptide LL-37 (119), as long as they also have a carboxylate moiety. These properties have been used to justify including CR3 and CR4 as scavenging receptors, i.e., those receptors like CD36 and the receptor for advanced glycation end products (RAGE) that enable cellular removal of decayed macromolecules in extracellular space (7). With its preference for cationic ligands that can interact with cell membranes, CR3 could function as a receptor for clearing cellular debris associated with membrane damage. CR3’s multitude of ligands and the fact that its outside-in signaling dampens inflammatory responses are in line with this proposal; CR4, instead, lacks anti-inflammatory signaling, suggesting that it may be functionally distinct from CR3 in this context (7).
More recently, the crystal structure of the CR3 headpiece has been determined in complex with a nanoantibody at 3.2-Å resolution (PDB ID 7P2D) (33) (Figure 2A). This structure provides the first high-resolution structure of CR3 beyond the αMI domain. In this structure, the CR3 headpiece adopts the closed conformation, which resembles those of LFA1 and CR4 in the same state. The nanoantibody used for crystallization could sterically block C3d binding in an Mg2+-independent manner but, surprisingly, acted as an agonist for cell-bound αMβ2, thus apparently increasing affinity for the iC3b ligand. These seemingly contradictory observations could be reconciled by proposing that additional conformational flexibility on the integrin and iC3b might permit interactions in cell surface-bound integrin that are not observed in solution or binding experiments in vitro. Indeed, the current understanding of both αMβ2 and iC3b structures suggests that they can interact in complex, heterogeneous environments using multiple domains in the crowded cell surface environment (32, 34).
Within a few months of the publication of the previous structure, the cryoelectron microscopy structure of the CR3 ectodomain at 2.7-Å resolution was published (PDB ID 7USM), along with the structure of a complex with an adenylate cyclase toxin RTX751 from Bordetella pertussis and a stabilizing Fab (PDB ID 7USL) (114) (Figure 2D). While the unbound αMβ2 adopts the closed conformation seen for the CR3 headpiece and closed CR4 ectodomain (see below), RTX751-bound αMβ2 was able to achieve a more extended conformation through stabilizing interactions with RTX751, closely resembling the conformations of extended αXβ2 ectodomains (120, 121). The closed and extended conformations of CR3 were related by a hinge motion of the headpiece relative to the tailpiece, pivoting between the Thigh/β-propeller in the αM headpiece with the Calf-2 domain in the tailpiece. The observation of a partially extended conformation in the RTX751 complex suggests that CR3 must be able to spontaneously sample more extended conformations by built-in flexibility around the headpiece-tailpiece hinge angle, which is consistent with previous observations (107).
3.3 Complement receptor 4
Earlier structural work on the CR4 headpiece by NS-EM showed an overall structure like that of other integrins, particularly very similar to that of CR3 (120). Maintaining the inactive state of the αXI domain requires the correct pairing of the αX and β2 chains, as shown by several experimental approaches (e.g., 122). When CR4 adopts an extended conformation, an α-helix in the I-like domain exerts a pull on the αXI domain that opens it up for binding.
The αXI domain was the first αI domain to be elucidated by X-ray crystallography (121). The crystallographic structure of the αXI shows a similar overall fold to that of CR3. Studies of CR4 revealed the flexible connection of the αXI domain with the β-propeller domain, suggesting that rotational freedom was required for efficient ligand binding (108). This property might also be necessary for binding structurally diverse ligands since the chelation of acidic ligand groups by the core cation in the MIDAS motif is likely to restrict ligand movement to pivoting around the Mg2+ cation.
NS-EM micrographs of the CR4 headpiece bound to iC3b revealed up to two independent CR4 αXI binding sites on the iC3b MG ring (110). The dominant site, present in all complexes, was located close to the macroglobulin domains MG3-4, and a secondary site, less frequently occupied, was found near the C345c domain. Interestingly, the two αXI binding sites do not overlap with the MG binding sites identified for αMI (34), suggesting that the two receptors saturate all potential binding sites on the MG ring of iC3b without directly competing. This does not imply the potential for simultaneous binding of iC3b by CR4 and CR3, which would be sterically impeded, but it shows that the αI domains have some degree of selectivity in ligand binding.
A structural chemical feature of CR4 αXI is the presence of a “ridge” of positively charged residues on the ligand-facing molecular surface (7). The electrostatic properties of αXI are markedly distinct from those of the homologous αMI and αLI domains (7, 34). This property has been invoked to explain why CR4 can selectively bind polyanionic molecules more efficiently than CR3 (7). Indeed, highly negatively charged polymers/molecules like heparin, nucleic acids, LPS, and osteopontin are proficient ligands for CR4. In parallel to the potential role of CR3 as a scavenger receptor for polycationic species, a receptor like CR4 with a strong preference for polyanionic species could serve complementary functions as a scavenger receptor by clearing excessive amounts of negatively charged proteins, detecting the presence of the negatively charged bacterial cell wall and LPS-rich membranes, and potentially alerting the immune system about their presence.
The structure of the complete CR4 ectodomain has been solved by X-ray crystallography in a bent, resting state (PDB ID 3K71, 3K72, 3K6S, 5ES4) (121) and a metastable, transition state (PDB ID 4NEN, 4NEH) (120) (Figures 3A, B). In the resting structure, the αXI domain is in the inactive conformation, as expected for the ectodomain’s inactive state. Surprisingly, the αXI domain shows a high degree of flexibility around the loops connecting it to the β-propeller domain. This suggests a more dynamic coupling between the αI domain with implications for the allosteric transmission of information along the integrin’s body. The second set of structures of the αXβ2 ectodomain represents an intermediate or transition state from the bent, closed, to the open, extended conformations, with a crystal lattice contact stabilizing the αXI domain in an open conformation. A key feature of this structure consists in the unwinding of much of αX α7 helix and its insertion into the interface between the β-propeller and the βI domains. The elevation (lift-off) of the αXI domain above the headpiece’s platform facilitates large-scale extensional and rotational motions of sufficient amplitude to communicate allosteric changes across the length of CR4.
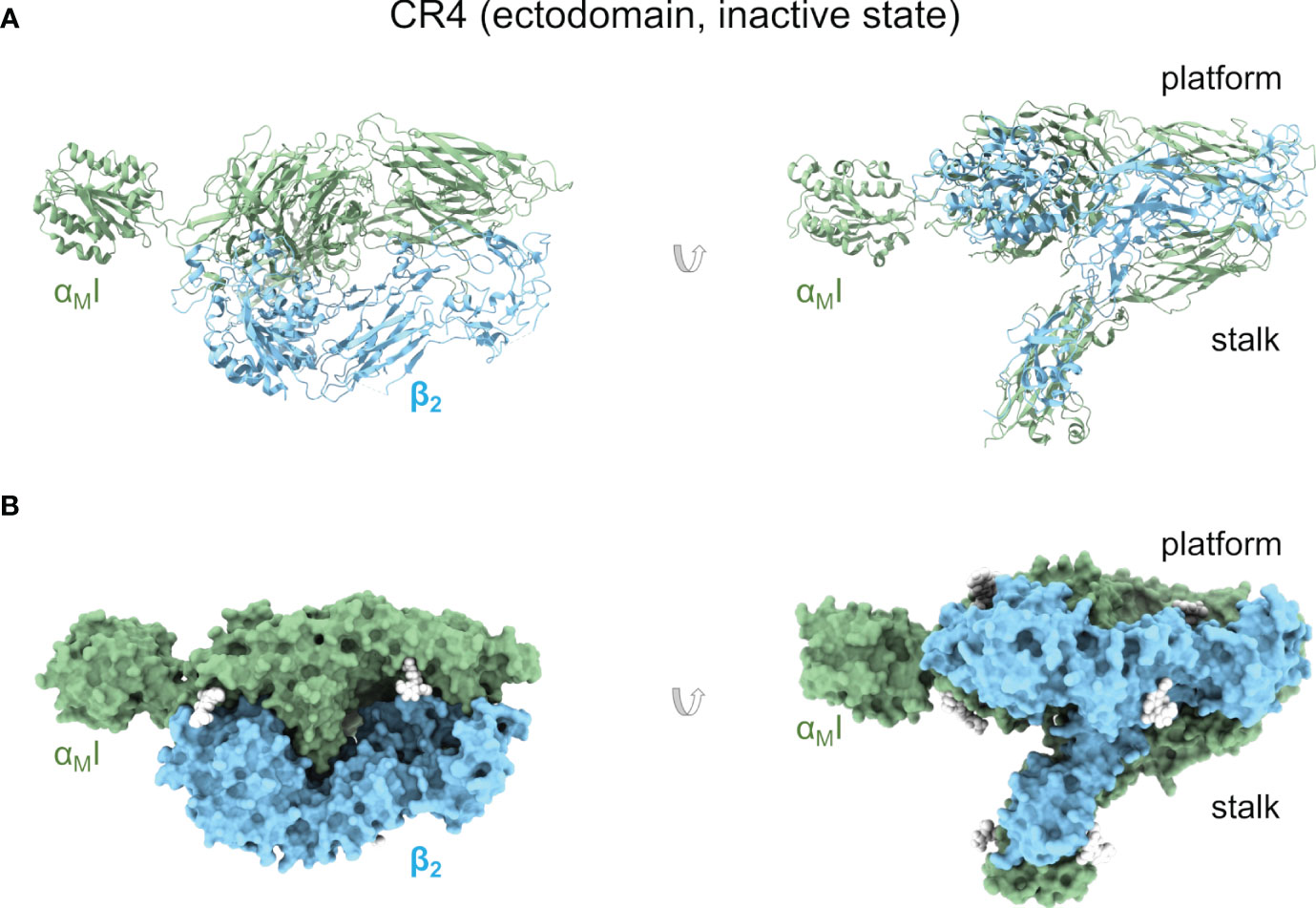
Figure 3 Complement receptor CR4. (A) Cartoon representation of an X-ray crystallographic structure of the CR4 ectodomain in an inactive but metastable structure that anticipates activation (PDB ID 4NEH). Two orientations related by a 90° rotation are shown. (B) As in (A) but as a molecular surface representation. Native glycan moieties are shown as white spheres.
4 Complement receptor of the immunoglobulin family
The complement receptor of the immunoglobulin (Ig) family (CRIg) is a type I transmembrane Ig superfamily member first cloned during a search for homologs of the junctional adhesion molecule (JAM) family (123). CRIg is also known as V-set and immunoglobulin domain-containing protein 4 (VSIG4), Z39Ig, UNQ317, and PRO362. In humans, there are three spliced isoforms of CRIg. The most common allelic form contains 399 amino acids and is known as huCRIg(L), whereas isoforms 2 and 3 are shorter, with 274 and 296 amino acids, respectively. Isoform 3 is also known as huCRIg(S). Full-length CRIg contains a signal peptide, a V-type Ig-like 1 domain, a C2-type Ig-like 2 domain, several potential O-glycosylation sites, and an intracellular domain with two potential phosphorylation sites, and is structurally related to the B7 family of immune regulatory proteins (124). Whereas huCRIg(L) comprises both Ig-like domains, huCRIg(S) contains only the V-type Ig-like 1 domain.
CRIg is expressed in tissue-resident and sinusoidal macrophages like the Kupffer cells, the resident macrophages in the liver, and it mediates phagocytosis of particles opsonized by any of its two ligands, C3b and iC3b (125). Besides stimulating pathogen opsonophagocytosis, CRIg is also known to be a potent inhibitor of the activation of the C3 convertase of the alternative pathway by binding to C3b, acting as a negative regulator of complement activation. In contrast to the other C3 fragment receptors (CR1 to CR4), CRIg is found on a constitutive recycling pool of membrane vesicles where it participates in the internalization of C3-opsonized particles from the bloodstream by Kupffer cells. Instead, CR1, CR3, and CR4 are located on secretory vesicles that fuse with the plasma membrane upon cytokine stimulation of the cells and internalize ligands through a macropinocytotic process only after receptor cross-linking (126). Besides its localization in the liver, CRIg is also abundantly expressed in resident macrophages from several fetal and adult tissues, with the highest expression attained in the lung and placenta. The main function of CRIg is to act as a potent negative regulator of T-cell proliferation and IL-2 production (127).
4.1 CRIg has two consecutive Ig-like domains
CRIg contains two Ig domains known as Ig-like 1 and Ig-like 2 domains. The crystallographic structure of Ig-like 1 (residues 19-137) was determined to 1.2-Å resolution, showing a V-set Ig-like fold that resembles the antibody variable domain, responsible for providing the binding specificity (PDB ID 2ICC) (128) (Figure 4A). Subsequent crystal structures of the same V-set Ig-like 1 domain from human and mouse have been determined in complex with a nanoantibody that blocks binding with C3c and C3b (PDB ID 5IMK, 5IML, 5IMM, 5IMO) (129).
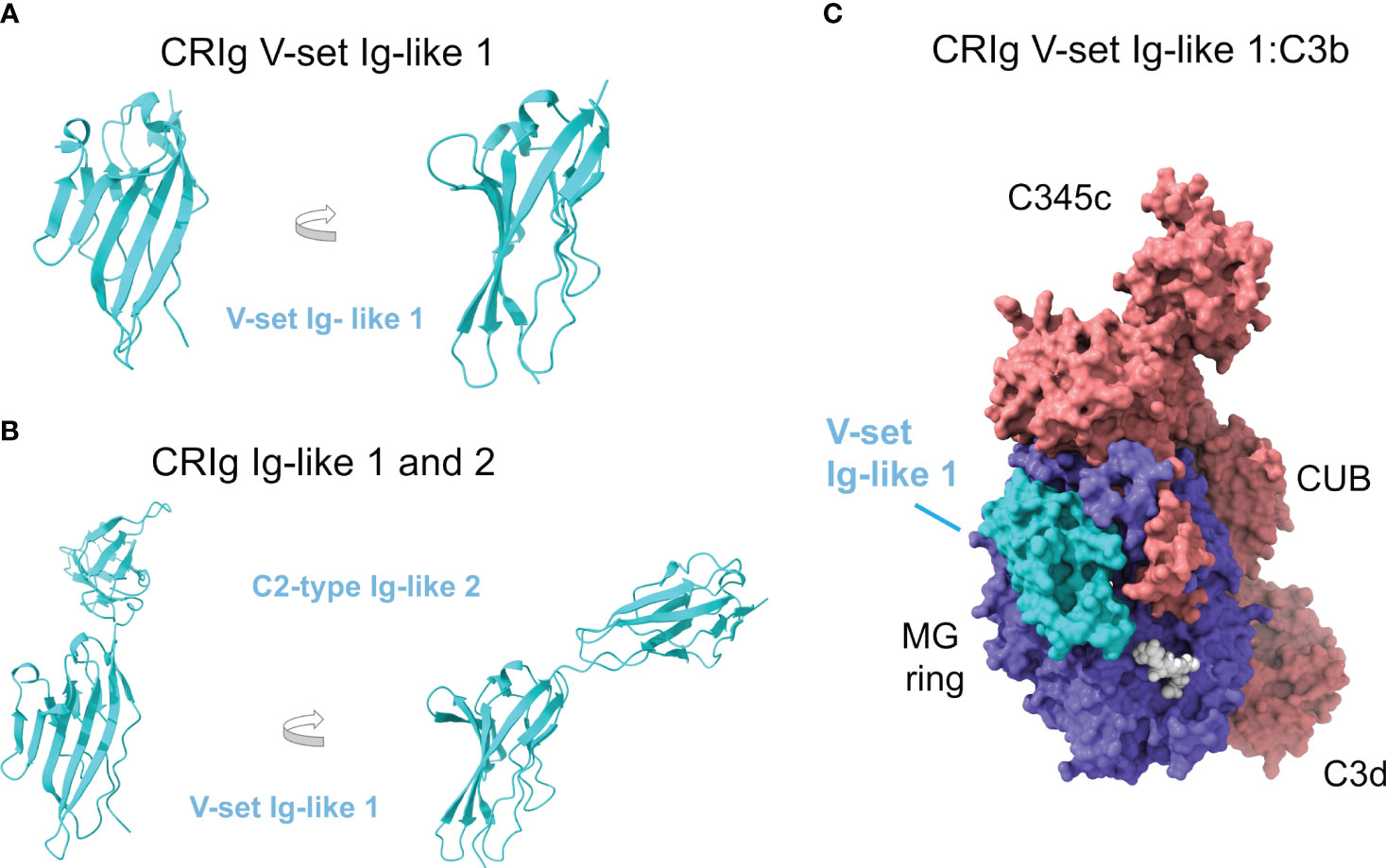
Figure 4 Complement receptor CRIg consists of two Ig-like domains and binds the β-chain of C3b. (A) Crystallographic structure of the V-set Ig-like 1 domain of CRIg, in cartoon representation and two orientations (PDB ID 2ICC). (B) AlphaFold predicts with high confidence the structural model of the C2-type Ig-like 2 domain of CRIg (AlphaFold AF-Q9Y279-F1). The consecutive Ig-like 1 and 2 domains are shown in cartoon representation and two orientations; the orientation of the V-set Ig-like 1 matches panel A for comparison. (C) Structure of CRIg V-set Ig-like 1:C3b (PDB ID 2ICF) in molecular surface representation. C3b is colored according to chain (the α chain in red, the β chain in blue), and CRIg V-set Ig-like 1 domain is in cyan. Native glycan chains in C3b are shown in white spheres.).
In contrast, the structure of Ig-like 2 (residues 143-226) has not been solved, although sequence homology has identified its fold as a C2-type Ig-like domain reminiscent of constant antibody domains that provide the effector functions. The AlphaFold model corresponding to full-length CRIg contains the predicted structure of the Ig-like 2 domain (AlphaFold AF-Q9Y279-F1) with a very high degree of confidence (Figure 4B).
The Ig-like domain consists of antiparallel β-strands arranged into two sheets linked by a disulfide bond. V-set domains can be distinguished from C2-type domains because they show the variability associated with antigen/ligand recognition, and the domain is longer with two extra strands tucked into the middle of the domain (C’ and C’’). The first β-sheet in Ig-like 1 comprises β-strands A’, G, F, C, C’, and C’’, and the second one of β-strands B, E, and D (Figures 3A, B).
4.2 CRIg binds to the β-chain of C3b and iC3b
C3b and iC3b are the main ligands of CRIg. Other C3 proteolytic fragments have been tested for CRIg binding, including C3, C3a, and C3d, and other homologous complement factors like C4 and C5, all with negative results (125).
The structures of CRIg Ig-like 1 domain bound to C3c (PDB ID 2ICE) and C3b (PDB ID 2ICF) (Figure 4C) have been determined by X-ray crystallography at 3.1-Å and 4.1-Å resolution, respectively (128). Since C3d is not a ligand for CRIg, the C3c moiety shared by C3b/iC3b becomes the most likely binding site for CRIg Ig-like 1 domain. Furthermore, C3c and C3b differ essentially in the lost TED domain (C3dg) while they share most of their structural core with minimal deviation: ~0.6 Å root mean square distance over 479 Cαs of the α-chain and 642 Cαs of the β-chain.
CRIg Ig-like 1 domain binds to C3c or C3b identically without restructuring or inducing conformational changes in the ligand. Residues from β-strands A’, F, G, C’, and C’’ from one of the β-sheets and β-strand B from the second β-sheet are engaged in the interaction, with β-strands C’ and C’’ contributing most of the interactions. The hairpin loop between β-strands C’ and C’’ sticks into the cavity in the center of the keyring-shaped β-chain of C3b.
The binding site is quite large (2,670 Å2 of solvent-accessible surface). It overlaps the cavity inside the C3c macroglobulin ring, crossing it diagonally and ending at the interfaces between consecutive MG domains MG3-MG4 and MG5-MG6 (Figure 3C). This binding site places CRIg on the opposite side of the surface-anchored C3dg/TED domain, a sterically accessible location facilitating binding. The most extensive contributions to binding are made by MG3 and MG6, supplemented by MG4, MG5, and LNK. Compared to C3, in C3c and C3b MG3 has rotated by 15° and there is a movement of the helical section in the LNK region, which appear to be necessary to form the CRIg binding site and thereby explain why CRIg cannot bind to C3.
The V-set Ig-like 1 domain of CRIg is sufficient for high-affinity binding to C3b/iC3b, even though this domain alone binds iC3b more strongly than to C3b. Although the presence of the C2-type Ig-like 2 domain is not required for binding, it restores high-affinity binding to C3b to the same level observed for iC3b (125). Interestingly, dimeric C3b (C3b2) (130) is bound more tightly to CRIg than monomeric C3b, a relevant result because multimeric C3b is supposed to represent the physiologic state of C3b on opsonized surfaces.
Significantly, CRIg binding to C3b inhibits the C3 and C5 convertases of the alternative pathway since CRIg blocks the generation of C3a and C3b by C3 convertase and C5b by C5 convertase. In a series of elegant experiments, site-directed mutations introduced in CRIg residues in contact with C3b MG3, MG5, or MG6 at the center, periphery, or outside the CRIg:C3b interface led to a strong, weak, or negligible effect on CRIg-mediated inhibition of the C3 convertase, evaluated in hemolytic assays with rabbit red blood cells (128). This proved that CRIg inhibition of the C3 convertase depended on its interaction with C3b.
In contrast to other complement negative regulators, CRIg does not inhibit the alternative pathway convertases by dissociating the catalytic subunit Bb (decay accelerating activity) or promoting C3b degradation by FI (cofactor activity). In contrast, it appears that CRIg, once bound to the β-chain of C3b, can sterically hamper association with the C3 and C5 alternative pathway convertases. Remarkably, CRIg fails to inhibit the C3 and C5 classical pathway convertases, indicating that the functional effect is confined to C3b and the alternative pathway.
4.3 Implications for diseases and immune evasion
CRIg is an essential receptor for the clearance of complement-opsonized particles, which are recognized and phagocytosed by Kupffer cells in the liver. Pathogens and immune complexes are shuttled in the circulation by CR1-bearing erythrocytes and handed over to CRIg-expressing Kupffer cells in the liver in a dynamic process relying on immune adherence that prevents systemic inflammation and immune complex diseases associated with aberrant vascular deposition (8). This extremely efficient mechanism of blood-borne pathogen clearance can catch Gram-positive and Gram-negative bacteria (8) and eukaryotic parasites (131).
5 Anaphylatoxin G-protein coupled receptors
The anaphylatoxins C5a and C3a, generated by the proteolytic activation of C5 and C3, respectively, are potent chemoattractants and pro-inflammatory mediators. The cognate receptors of C5a are C5aR1 (C5aR/CD88) and C5aR2 (C5L2/GPR77), and the cognate receptor of C3a is C3aR (C3aR). The analogous “anaphylatoxin” released by proteolytic activation of the C3-homologous zymogen C4, C4a, has thus far defied a functional description and, perhaps significantly, lacks a specific receptor (132, 133).
C5aR1/C5aR2 and C3aR belong to the G-protein coupled receptor (GPCR) family and, more precisely, to the rhodopsin family and class A, two equivalent groupings at the highest hierarchy level of the two main GPCR ordering systems (134–136). Based on sequence homology and functional similarity of GPCR, the three receptors are classified into the complement peptide group inside class A (135). In contrast, within the rhodopsin family defined by the phylogenetic classification of human GPCR, C5aR1/C5aR2 are members of the chemokine cluster of γ-group, while C3aR belongs to the purine receptor cluster of δ-group (134).
The three anaphylatoxin receptors share structural features common to all GPCR. They are comprised of an extracellular N-terminal region, seven transmembrane α-helices (TM1-7), connected sequentially by intracellular (ICL) or extracellular (ECL) loops, and an intracellular C-terminal region (137–139). To refer to TM residues, we will follow the Ballesteros-Weinstein numbering (140), and for the remaining residues, we will use the name of the containing motif (ECL, ICL, N-ter, or C-ter) as superscripts.
The anaphylatoxin receptors are widely expressed in immune cells from the myeloid and lymphoid lineages and nonimmune cells like epithelial cells and neurons (141). By binding their cognate ligands, the receptors are implicated in diverse cellular functions, physiological processes, and pathologies, mainly related to the immunological system (141–148).
The clinical relevance of these receptors in different acute and chronic disorders, mainly with an inflammatory etiology, has triggered a great interest in developing specific and effective modulators (149). In this context, an exhaustive investigation has aimed to reveal insights into the structure-function of these receptors to increase the understanding of how they carry out their functions and to support the finding of modulators with clinical potential.
5.1 Complement receptor 5a 1
5.1.1 Structure of C5aR1
C5aR1 was discovered in 1978 (150), and the coding sequence of its 350 amino acids was cloned and determined in 1991 (151, 152). While the unliganded receptor has defied structural determination, recently published high-resolution X-ray crystal (PDB ID 5O9H, 6C1Q, 6C1R) and cryoelectron microscopy (PDB ID 7Y64, 7Y65, 7Y66, 7Y67) structures (Figure 5A) have provided insights into the structural features of this receptor.
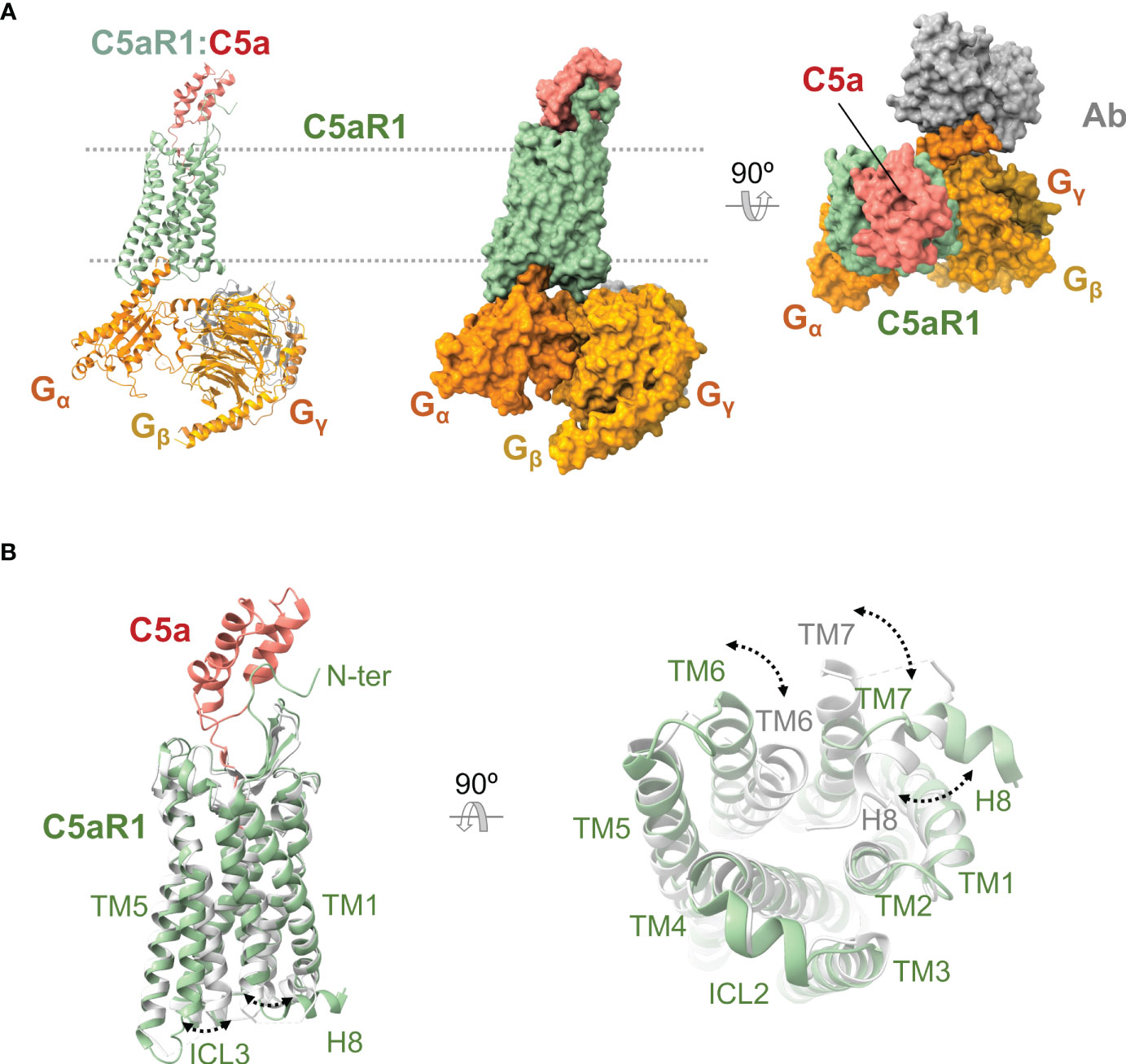
Figure 5 Anaphylatoxin receptor C5aR1 in complex with C5a and conformational changes underlying activation. (A) Cryoelectron microscopy structure of C5aR1:C5a bound to heterotrimeric G protein comprising the Gα, Gβ, and Gγ subunits (shown in orange, yellow, and gold), and a stabilizing antibody (shown in gray) (PDB ID 7I65). C5aR1 is shown in green, and C5a in red. The same view is shown of the cartoon and molecular surface representations of the complex. We show a 90° rotated view of the complex on the right in molecular surface representation. (B) Superposition of the active conformation of the C5aR1:C5a complex (PDB ID 7I65) (C5aR1 in green, C5a in red) and an inactive reference structure (PDB ID 6C1Q) (C5aR1 in grey; the antagonist PMX53 is not shown for clarity). Side (left) and bottom (right) views are shown. Conformational changes are indicated by dashed lines between and labeling of structural elements that occupy distinct positions in the active versus inactive conformations.
The structure of the core transmembrane region is conserved with other class A GPCR (138, 139, 153) including the overall helical arrangement and kinks, the placement of TM3 at the center of C5aR1 with a lower tilt-angle with respect to the plane of the lipid bilayer, W6.48 (W2556.48) near P6.50 (P2576.50), the PIF motif (P2145.50, I1243.40, F2516.44), an intrahelical sodium coordination site (D822.50, N2927.45, N2967.49) at the cytoplasmic side, the NPXXY motif (N2967.49, P2977.50, I2987.51, I2997.52, Y3007.53) and the DRY/F motif (D1333.49, R1343.50, and F1353.51). On the extracellular side, C5aR1 presents a conserved disulfide bond (C1093.25 and C188ECL2) and a β-hairpin conformation in ECL2 that resembles other peptide binding receptors (138, 154, 155).
At the intracellular region, the ICL2 exhibits a two-turn α-helical structure, and some C-terminal residues form the conserved eighth amphipathic α-helix (H8) of three turns long (35, 138, 154, 155). C5aR1 can oligomerize in vivo, forming homodimers and perhaps higher-order homo-oligomers (156, 157) and heterodimers with CCR5 (158) and C5aR2 (159).
Other post-translational modifications observed in C5aR1 are an N-glycosylation at N5 (160), tyrosine sulfation at Y11 and Y14 (161) at the N terminus, and serine phosphorylation at S314, S317, S327, S332, S334, and S338 in the C terminus (162); albeit to a lesser degree, threonine residues can also be phosphorylated (158).
5.1.2 C5a-binding orthosteric site
The cognate ligands of C5aR1 are the anaphylatoxin C5a and its dearginated product C5adesArg, which have been extensively researched (163, 164). Other ligands of the C5aR1 have been recently discovered (165).
The high-resolution structures of liganded C5aR1 have revealed an orthosteric and an allosteric binding site (35, 36, 154, 155). The orthosteric binding site consists of an interhelical solvent-exposed amphipathic cavity located at the extracellular side of the receptor (35, 36, 155) (Figure 5A). This site can be divided into a polar region and a hydrophobic cage. The polar region spans from the outer edge to one side of the binding site and comprises hydrophilic and charged residues recruited from TM3-7 and the membrane-proximal part of ECL2. The hydrophobic cage is mainly formed by hydrophobic residues from TM1-3, TM7, and ECL1, which extend from one side to the bottom of the binding site. The orthosteric site can bind peptide ligands of up to eight residues (amino acid positions numbered P1-P8) with various pharmacological effects, including C5a, BM213, C5apep, PMX53, and C089. Water molecules can be involved in ligand binding, forming a polar network connecting the ligand to residues from the polar surface (155). The allosteric binding site is an extra-helical hydrophobic cleft formed by residues from the middle regions of TM3, TM4, and TM5, which bind non-peptide ligands such as NDT9513727 or Avacopan through shape complementarity, hydrophobic interactions, and a key hydrogen bond with W2135.49 (154, 155). The N terminus and the turn region of the ECL2 can also participate in binding orthosteric ligands (35, 36).
The structures of C5aR1 in complex with C5a and the Gi protein show that C5a binds in a C-terminus-inside mode, and the receptor interacts with three distinct regions of the anaphylatoxin (35) (Figure 5A). Firstly, an amphipathic stretch of the receptor’s N terminus (L22-D27) interacts with a cavity between C5a α-helices H2 and H4 and the H3-H4 loop. Secondly, the orthosteric site accommodates the last eight residues of C5a. Inside the orthosteric site, the C5a C terminus adopts a hook-shaped conformation, establishing strong interactions with residues at the polar surface while accommodating inside the hydrophobic cage. In this orientation, the side chain of C5a LP6 interacts with a triad of receptor residues denominated IWI region (I912.59, W102ECL1, I1163.32) (35, 36). Finally, the amphipathic turn region of ECL2 (E180ECL2-P183ECL2) makes contact with C5a in a cleft assembled by α-helices H1, H2, and H4 and the H2-H3 loop.
Other C5aR1 ligand peptides occupy the orthosteric binding site in a similar fashion to the C5a C terminus (35). The main differences in binding mode between C5a and these peptides have been observed in the hydrophobic cage of the orthosteric site, particularly in the interactions established between the peptides’ P6 and P7 positions and the IWI region and surrounding residues (e.g., L922.60, S952.63, H100ECL1, P1133.29, and V2867.39) (35, 155). A unique feature of the cyclic peptide PMX53 is a hydrogen bond between WP5 and P1133.29 (155).
The recent flurry of C5aR1 structures has made important contributions to understanding the structure-function relationship of C5aR1 and how C5a binds and activates the receptor. However, a few questions remain unanswered. For example, receptor aspartic acid residues upstream of L22 (166, 167) and the sulfated Y11 and Y14 (161) had been shown to play important roles in C5a binding. Still, current structures have not revealed any direct interactions between these residues and the ligand. Therefore, how these residues contribute to C5a binding has not been explained. They may establish direct interactions with C5a (168) or be required for the structural conformation of the binding site (169).
The recent structural evidence agrees with a two binding sites model previously proposed (170). In this model, the first binding event with C5a engages a site 1 in C5aR1 composed of the N terminus and the ECL2, which directly contacts the helical core of C5a; this binding event favors a conformational change of C5a that promotes binding to the site 2, composed of the orthosteric pocket, which interacts with C5a C terminus. In support of this model, kinetic and thermodynamic analyses by atomic force microscopy of the binding interaction have confirmed that both sites 1 and 2 contribute to the high-affinity binding of C5aR1:C5a through a cooperative mechanism (171). The markedly different conformation adopted by bound C5a with respect to the free form lends further support to this model. In the bound conformation, C5a shows a rearrangement of the C terminus and the helical bundle, which changes from a 1.5-turn helix form placed near the helical bundle (as seen in free C5a) to an unfolded conformation in bound C5a (35). These changes have been rationalized as a bi-directional information flow between C5a and C5aR1 (166). According to this view, the first binding event would be characterized by C5aR1 site 1 promoting or favoring a conformational change in C5a, and the second binding event in a fully extended conformation of C5a inside site 2 would then activate the receptor.
Orthosteric ligands vary widely in their binding affinity for C5aR1. While C5a interacts with C5aR1 with a dissociation constant KD = 1-5 nM (151, 167, 172, 173), its dearginated version C5adesArg, which only differs from C5a in the last amino acid, R74 (174), has a 10 to 100-fold lower binding affinity. However, peptides derived from the C-terminal end of C5a show modest binding affinities, suggesting that the engagement of the receptor N terminus and the ligand coupling to the orthosteric site (35, 175) are important features for the strong binding of C5a. The lower affinity of C5aR1 for C5adesArg could be explained by the putative lack of interactions between R74 at P8 in the orthosteric site (35).
5.1.3 C5a-mediated activation of C5aR1
C5aR1 is activated by orthosteric agonists (170, 175–177), which cause rearrangements in the extracellular, transmembrane, and intracellular regions of the receptor (35, 36) (Figure 5B), leading to a conformation in which it can trigger intracellular signaling pathways by recruiting transducers (138, 139, 153). At the extracellular region, the N terminus appears not to be involved in the C5aR1 conformational changes since this stretch is not required for receptor activity (161, 166, 175, 177). The ECL1 and especially its WXFG motif perform a critical role in C5aR1 activation (178). The ECL2 accomplishes a striking function in ligand-mediated activation of C5aR1 beyond its role in ligand binding (35, 179). In contrast to ECL1 and ECL2, ECL3 seems not to be involved in C5aR1 activation (178). Other residues essential for C5aR1 activation were found at the core of the transmembrane helix bundle and inter-helical interfaces (178, 180, 181).
In line with the common mechanism of class A GPCR (153, 182), an activation mechanism has been proposed for C5aR1 based on the receptor conformational changes observed between an active state (bound to C5a and Gi) and an inactive state (bound to PMX53 and Avacopan), and the currently available receptor structure-function data (35, 36, 155). Initially, the coupling of C5a into the orthosteric site causes TM displacements that loosen a hydrophobic zipper formed by I1163.32, M1203.36, W2556.48, and Y2907.43 at the extracellular side of C5aR1. This zipper is proposed to act as a lock tethering TM3, TM6, and TM7 in an inactive configuration, and movements key for loosening it are an upward rotation of the extracellular region of TM3 and a TM6 downward displacement, which may be caused by the engagement of C5a LP6 by the IWI region and the RP8-Y2586.51 interaction, respectively. Next, TM shifts keep taking place disturbing the PIF motif at the hydrophobic core. Sequentially, the reorganization of the helical bundle core cause conformational changes at the cytoplasmic side of C5aR1 that include a pronounced outward displacement of the intracellular section of TM6, an H8 swing from its location between TM1 and TM7 oriented toward the receptor’s center to a classic conformation near to the lipid bilayer, alterations of NPXXY and DRY/F motifs, the sodium coordination site collapse, and the opening of an interhelical water-accessible cavity (Figure 5B). Eventually, the rearrangements promote receptor-transducer interactions and, ultimately, intracellular signaling (35, 36).
Other remarkable conformational changes associated with C5aR1 activation by C5a include an uncommon placement of Y3007.53 that may be important for the particular H8 reorientation, TM3 and TM7 distancing their extracellular sides and approaching their intracellular segments, and rearrangements of clusters of water molecules located into the interhelical cavities that are separated by W2556.48 and I1163.32 at the helical core of the receptor (35, 36, 155).
5.1.4 Modulation and selectivity of C5aR1 activity
C5aR1 exhibits functional selectivity as it triggers distinct downstream signaling and cellular responses in a ligand and cell-type-dependent fashion. The ability of C5aR1 to activate different transducers and the variety of mechanisms by which its activity can be modulated contribute to this functional selectivity. Agonist-activated C5aR1 can signal through heterotrimeric guanine nucleotide-binding proteins (G-proteins) with α-subunits of the Gi/o and Gα16 families (183–187). Coupling the Gαi1 subunit involves C5aR1 residues from ICL2, ICL3, TM3, TM5, and TM6, and Gαi1 residues from the β2-β3 loop, β6 strand, and α5 helix. Remarkably, the α5 helix of Gαi1 inserts into the receptor intracellular cavity taking up part of the space filled by H8 in the inactive C5aR1 (35, 36). The receptor segments involved in G-protein signaling include the DRY/F and NPXXY motifs, particularly the ICL3 (180, 188, 189).
The G-protein specificity of C5aR1 seems to lie in the three ICLs and particularly the receptor C terminus, which could work together to restrict the receptor interaction with G-proteins (188). Noteworthy, C5aR1 can bind Gi even in the absence of agonist stimulation, suggesting that, although G-protein pre-coupling to C5aR1 is not essential for ligand binding, the pre-coupling could increase the receptor affinity for ligands as C5a:C5aR1 binding fosters the G-protein coupling to the receptor (190–192).
The main phosphorylation sites identified in C5aR1 include serine (S314, S317, S327, S332, S334, and S338) and threonine residues at the C terminus (158, 162, 193). A likely PKC phosphorylation site has also been identified in the ICL3 (194). C5aR1 phosphorylation seems hierarchical as the initial modification of S332/S334 or S334/S338 is required for the efficient phosphorylation of the remaining sites (195). Several kinases, including GRK (GRK2, GRK3, GRK5, and GRK6) and PKC (PKCβ), can be implicated in C5aR1 phosphorylation (193, 196, 197). GRK2 can be translocated to the plasma membrane in response to C5a, and the receptor ICL3 is the intracellular stretch preferentially bound by GRK2 and PKCβII (198). Receptor phosphorylation is involved in β-arrestin recruitment (158, 193, 199), receptor internalization (158, 194, 199, 200), and desensitization (193, 195). Although the precise roles of these phosphorylation sites have not yet been elucidated, one or several of the C-terminal serine residues (S327, S332, S334, and S338) appear to be the most relevant phosphorylation sites for β-arrestin recruitment and C5aR1 internalization and desensitization (193). L318H8 plays an important role in the receptor conformation that can be efficiently phosphorylated and internalized (201).
Activated C5aR1 can recruit β-arrestins 1 and 2 (199). GRK 2, 3, 5, and 6 have all been shown to facilitate the recruitment of β-arrestins 1 and 2 to C5aR1 (187, 202). β-arrestins 1 and 2 recruitment by C5aR1 may be followed by receptor internalization and downstream signaling (199, 203). The C terminus of C5aR1 is required for efficient homologous receptor internalization (194). C5aR1 internalization has been observed as a clathrin-dependent process that is associated with the β-arrestin recruitment by the receptor. C5aR1 internalized in early endosomes can be targeted to lysosomes for degradation or recycled to the cell membrane. The trafficking of C5aR1 through these pathways appears to depend on the cell type (199–201). Recycling of C5aR1 likely requires its dephosphorylation by several phosphatases, including protein phosphatase 1 (PP1) (193, 200). Although a structure of C5aR1 binding to β-arrestins is lacking, impaired β-arrestin binding by different phosphorylation-deficient C5aR1 mutants indicates that a stable association between C5aR1 and β-arrestin likely requires a certain degree of receptor phosphorylation (158, 193, 199). C5aR1 can also couple with the Wiskott-Aldrich syndrome protein (WASP), which binds to the receptor’s C terminus (204).
Agonist-induced and unliganded C5aR1 receptor oligomerization have been observed (205). Oligomerization likely plays important functional or regulatory roles in vivo, but little is known about how C5aR1 assembles into oligomers and the implications for receptor activity. Structurally, TM1, 2, and 4 may be involved in C5aR1 dimerization (157, 181), while neither the N nor the C terminus is required for C5aR1 dimerization (205). Only in one of the solved structures with the extra-helical antagonist NDT9513727 (PDB ID 5O9H), C5aR1 has been observed in a non-crystallographic homodimeric organization, although this arrangement might not be physiologically relevant (154, 155); structural comparison with the homodimer of the smoothened (SMO) receptor (206) has lent support to the C5aR1 homodimer arrangement. Remarkably, the agonist activation of C5aR1 homodimers and phosphorylation of only a monomer can lead to the internalization of these dimers (205).
C5aR1 activity can also be modulated by antagonists and inverse agonists that bind to its orthosteric or allosteric site (154, 155). These allosteric modulators trap the receptor in the inactive state by stabilizing the network of hydrophobic interactions that maintain the receptor in the inactive state or by sterically hindering the helical rearrangements required for receptor activation. The structures of PMX53:C5aR1 (PDB ID 6C1Q, 6C1R) reveal how the cyclic peptide PMX53 sits in the orthosteric site, interacting with residues of the polar surface that interact with C5a and inserting the side chains of the d-cyclohexyl alanine (dChaP4) and WP5 in the vicinity to the IWI region (155). In the allosteric site, NDT9513727 (PDB ID 6C1Q) and Avacopan (PDB ID 6C1R) establish interactions with the hydrophobic core of the transmembrane region, including the PIF motif (155).
Several C5aR1 agonists display ligand bias, i.e., differences in receptor signaling via G-protein signaling and β-arrestin recruitment (35, 207, 208). Ligand bias may depend on interactions between the agonist and ECL2 and between the ligand P6 and the IWI region, which are important for C5a-mediated β-arrestin recruitment. Interestingly, C5aR1 inhibitors display ligand bias effects: while PMX53 preferentially inhibits G-protein coupling over β-arrestin recruitment, Avacopan shows the opposite trend (35).
5.2 Complement receptor 5a 2
5.2.1 Structural features of C5aR2
The gene encoding human C5aR2 (HsC5L2) was cloned and sequenced many years after HsC5aR1 (209), and what its specific functions may be is still an active research area. In particular, the structure of C5aR2 has not been elucidated experimentally. As C5aR1, C5aR2 belongs to the class A GPCR superfamily and therefore shares the overall structure of the model GPCR. Although the sequence identity and similarity with C5aR1 are relatively high (~36.4% and 57.1%, 128 identical and 73 similar residues), the difference is sufficiently extensive to make us cautious about extrapolating structural facts about C5aR1 onto C5aL2 without critical assessment.
The following putative and potential structural features have been recognized by sequence homology. Firstly, the seven transmembrane helical core that is a hallmark of all GPCR. Secondly, the N terminus contains an N-glycosylation site at N3 (143). Thirdly, the presence of a disulfide bond between C1073.25-C186ECL2. Fourthly, serine and threonine phosphorylation sites in the ICL3 and C terminus. And, finally, a remarkably shorter ICL3 compared with C5aR1 and other related GPCR.
As far as differences are concerned, C5aR2 lacks a canonical DRY/F motif at the end of TM3, which is substituted for by the sequence D1313.49, L1323.50, and C1333.51, thereby resulting in the obligate uncoupling of C5aR2 from heterotrimeric G proteins (210). It also lacks an S/T-X-R/K phosphorylation site in ICL3. Finally, the NPXXY motif in TM7 lacks the tyrosine residue, which has been replaced by F2917.53 (209, 211).
5.2.2 C5aR2 binds C5a and C5adesArg
C5aR2 has two high-affinity ligands: C5a and C5adesArg. Like C5aR1, C5aR2 exhibits a higher affinity for C5a over C5adesArg. While the affinity for C5a is similar for C5aR1 and C5aR2, the affinity of C5aR2 for C5adesArg is one order of magnitude tighter than C5aR1 (210, 211). This, together with the slower dissociation rates of C5a from C5aR2 (210), has motivated the proposal that the primary physiological role of C5aR2 may be modulating the activation through C5aR1.
C5aR2 does not seem to share the exact binding mechanism for orthosteric ligands proposed for C5aR1. Indeed, C5a and C5adesArg display distinct binding modes to C5aR2. The C5aR2 N terminus plays crucial roles in C5adesArg binding, especially through the acidic and tyrosine residues and the tyrosine sulfation posttranslational modification (212).
Underscoring this differential recognition of C5a by C5aR1 and C5aR2, two partial agonists (P32 and P59) that are selective for C5aR2 over C5aR1/C3aR have been discovered (213). Conversely, the well-characterized C5aR1 antagonist PMX53 is perfectly selective toward C5aR1 and biased toward Gi signaling and does not bind C5aR2 (159).
Besides the two cognate ligands, there has been some controversy over the possibility that C3a, C3adesArg (ASP), and C4a could also bind to C5aR2 (211). Although enticing, this proposal has not been supported by recent data. None of the three proteins activates the receptor (211, 214); it seems none binds to it (210, 213).
5.2.3 C5aR2: a C5a receptor that recruits β-arrestin
There are striking differences between C5aR2 and C5aR1 concerning localization, phosphorylation, internalization, and regulation.
To begin with, C5aR2 is mainly localized inside the cell rather than in the plasma membrane and does not show a rapid internalization in response to C5a binding (210, 215). The internalization mechanism of C5aR2 appears to be different from that of C5aR1 and is only clathrin-dependent. In fact, internalized C5aR2 can also be constitutively recycled to the plasma membrane by a clathrin-dependent process (215). These processes suggest that C5aR2-bearing cells can uptake C5a and C5adesArg and either keep them intracellularly, target them for degradation, or release them back into the extracellular environment. Since C5aR2 exhibits a greater efficiency in ligand uptake and processing than C5aR1, C5aR2 can be posited as the main receptor involved in internalizing, retaining, and degrading C5a in natively expressing C5aR2 cells (215).
Regarding phosphorylation status, C5aR2 is phosphorylated to a lesser degree than C5aR1 in response to C5a binding (210). C5aR2 can recruit β-arrestin 1 and 2 in an agonist-dependent manner (187, 213, 214), a process involving phosphorylation sites introduced by GRKs 5-6 (187). Besides C5a-mediated recruitment, C5aR2 can also pre-couple both β-arrestins in the absence of agonist (187, 216). The direct interaction of C5aR2 with β-arrestin 1 has been shown by NS-EM (187). Interestingly, both β-arrestins exhibit different conformations bound to C5aR2 than those bound to C5aR1 (187). In contrast with C5aR1, C5aR2 showed a greater β-arrestin 2 recruitment stimulated by C5a/C5adesArg that was equally strong for the two anaphylatoxins (214, 216).
An intriguing aspect of C5a receptors is that C5aR2 can form heterodimers with C5aR1 constitutively and also in an agonist-dependent manner (159). It has been hypothesized that through heterodimerization, C5aR2 might regulate C5aR1 activity and cooperate functionally (159, 216).
C5aR2 does not signal through the heterotrimeric G proteins (187, 210), and grafting the C5aR1 motifs that engage with G proteins such as the DRY/F motif, ICL3, and the NPXXY motif does not complement C5aR2 (215). The inability of C5aR2 to couple with G proteins explains that C5aR2 plays a much smaller role in intracellular signaling than C5aR1 (210, 214).
Based on the lack of G-protein signaling, clearing of extracellular C5a/C5adesArg, and β-arrestin recruitment and internalization properties, C5aR2 has been proposed to be a recycling decoy receptor (215). Although this is an attractive theory, C5aR2 still contributes to intracellular signaling processes in a variety of roles, including modulation of the phosphorylation state of several transducers (e.g., p90RSK and ERK1/2) and intracellular calcium mobilization (187, 217). Different functions attributed to C5aR2 consist in the modulation of other immunological receptors, like C5aR1, C3aR, chemokine-like receptor 1 (CMKLR1), or different pattern recognition receptors (PRRs) (217). Through these functions, C5aR2 is involved in regulating complex cellular responses, such as releasing cytokines from macrophages (213, 217).
5.3 Complement receptor 3a
5.3.1 Structure of C3aR
Like the other anaphylatoxin receptors, C3aR was classified as a GPCR when the encoding gene was cloned and its primary sequence determined (218, 219). Analysis of the primary sequence revealed a distinctive structural feature of C3aR: an unusually long ECL2 of about 172 amino acids, predicted to be highly flexible, with a disulfide bond formed between C953.25 and C172ECL2. In contrast, its N terminus is shorter than that of C5aR1.
C3aR has several posttranslational modifications, including N- and O-glycosylations and tyrosine sulfations (220). S266ECL2 is O-glycosylated (221), and N9N-ter and N194ECL2 have been predicted to be N-glycosylated. Y174, Y184, Y188, Y317, and Y318 in the ECL2 can carry sulfations. The ICL3 and C terminus of C3aR contain serine and threonine residues that can be phosphorylated (197).
The cryoelectron microscopy structures of C3a-free (PDB ID 8HK3) and C3a-bound C3aR coupled to a Gi heterotrimeric protein (PDB ID 8HK2) (Figure 6A) have only recently become available, representing a striking leap ahead in the field (36). As suspected, the long ECL2 loop is highly flexible, and only the first 16 residues (V159-K175) were resolved, adopting a β-hairpin conformation.
Figure 6 Anaphylatoxin receptor C3aR in complex with C3a and conformational changes underlying activation. (A) Cryoelectron microscopy structure of C3aR:C3a bound to heterotrimeric G protein comprising the Gα, Gβ, and Gγ subunits (shown in orange, yellow, and gold), and a stabilizing antibody (shown in gray) (PDB ID 8HZ2). C3aR is shown in green, and C3a in red. The same view is shown of the cartoon and molecular surface representations of the complex. We show a 90° rotated view of the complex on the right in molecular surface representation. (B) Superposition of the active conformation of the C3aR:C3a complex (PDB ID 8HZ2) (C3aR in green, C3a in red) and an inactive reference structure (PDB ID 6C1Q) (C5aR1 in grey; the antagonist PMX53 is not shown for clarity). Side (left) and bottom (right) views are shown. Conformational changes are indicated by dashed lines between and labeling of structural elements that occupy distinct positions in the active versus inactive conformations.
5.3.2 C3a last five amino acids are critical for C3aR interaction
The cognate ligand of C3aR is the C3a anaphylatoxin, which binds with high affinity (~1 nM) (218, 219, 222). Compared to C5aR1, C3aR does not bind C3adesArg (223). C3a recognition by C3aR has been proposed to follow a mechanism like that of C5aR1 with some differences. Like C5aR1, a two binding site model has been proposed (36, 222).
The primary or effector binding site (orthosteric site) consists of an interhelical amphipathic pocket found in the extracellular region of the receptor and formed by residues from TM3, TM5-7, and ECL2 (Figure 6A). The orthosteric site of C3aR is smaller than that of C5aR1, and it can only accommodate the last five residues of C3a (P1-P5). The C-terminal end of C3a inside the binding site adopts a hook-shaped conformation enabling multiple interactions with the amino acids in the cavity, including ionic and hydrophobic interactions and hydrogen bonds. Remarkably, R77 of C3a establishes some of the critical interactions, which include a salt bridge with D4177.35, a cation-π interaction with Y3936.51, and two hydrogen bonds with Y174ECL2 and R3405.42 (36).
The secondary binding site encompasses residues from the outermost segments of ECL2 and ECL3 that still make contact with the α4 helix of C3a. Even though this secondary binding site plays a less important role in engaging C3a, some functionally relevant contacts have been found in this region, e.g., the salt bridge between C3aR D404ECL3 and C3a R65 (36).
While the C3aR N terminus has shown little implication in C3a binding (36, 222, 224), its ECL2 plays an important role in the anaphylatoxin coupling. Site-directed mutagenesis studies have established that aspartic acid residues along the N-terminal and C-terminal ends of ECL2 (D183, D186, D325, D326, and D327) and sulfation of Y174 are required for high-affinity binding of C3a (36, 220, 222). Besides mediating C3a binding, the ECL2 appears vital for arranging the helical bundle functionally (224).
The selectivity toward their cognate anaphylatoxin ligands of C3aR and C5aR1 is in part due to the amino acid composition of the C terminal tails; in particular, C3a G74 seems to be essential for C3aR selectivity (36). Furthermore, the N terminus of these receptors also has shown a significant contribution to this selectivity, so the amino terminus of C3aR appears to hinder C5a binding (224). This is in accordance with the composition and implication of these N termini in the anaphylatoxin binding.
5.3.3 Activation of C3aR
Once liganded, C3aR can couple and signal by different heterotrimeric G proteins with α subunits from the Gi/o and Gq families (164). C3aR recognizes residues in the β2-β3 loop, β6 strand, and α5 helix of the Gαi subunit through residues from TM3-6, ICL2, and ICL3. The coupling to the heterotrimeric Gi to C3aR is similar to C5aR1, although the α5 helix of the Gαi subunit is inserted more deeply, TM6 is displaced by ~1.5 Å, and the ICL3 displays a distinct topology and broader interaction with Gαi (36) (Figure 6B).
C3a stimulation leads to C3aR phosphorylation on Ser and Thr residues in a dose-dependent manner by GRKs 2/3/5/6 and PKC (197). Removal of the C3a stimulus is followed by C3aR dephosphorylation. GRKs have shown different functions over C3aR signaling in mast cells. While GRK2/3 are involved in agonist-induced desensitization, GRK5/6 are implicated in cell degranulation (225). Phosphorylation at Ser459C-ter, Thr463C-ter, Ser465C-ter, Thr466C-ter, and Ser470C-ter is involved in β-arrestin 2 recruitment at C3aR and receptor desensitization in mast cells (226). Thr463C-ter, Ser465C-ter, Thr466C-ter, and Ser470C-ter have important roles in C3aR internalization, which occurs in an agonist-dependent manner (227). Agonist-induced phosphorylation of C3aR performs an important role in signal transduction from the receptor. In C3a-stimulated mast cells, phosphorylation is required for CCL2 production, but these modifications seem to attenuate a degranulation response (228).
C3aR activity can be modulated by β-arrestins 1 and 2. β-arrestins can inhibit C3a-induced ERK1/2 phosphorylation and perform other regulatory activities. For example, in mast cells, β-arrestin 2 is involved in C3aR desensitization, internalization, and inhibition of the C3a-induced NF-kB activation and CCL4 generation; β-arrestin 1 contributes mainly to cell degranulation (229).
6 Concluding remarks
The progressive recognition of the complement system as a driver of inflammatory and autoimmune diseases (11, 27, 28, 30, 31) has incentivized the functional and structural study of the complement system and, in particular, of the complement receptors, their ligands, and their complexes.
Work over many years has established the existence of at least four structurally distinct classes of complement receptors: the CCP/SCR mosaic receptors CR1 and CR2 (along with the negative regulators MCP and DAF), the Ig superfamily receptor CRIg, the β2 (CD18) integrin receptors CR3 and CR4, and the anaphylatoxin GPCR receptors C5aR1, C5aR2, and C3aR. This diversity likely reflects the enormous span of evolutionary time over which cellular immunity has coevolved with the complement system (230–232).
Although the diversity of functions carried out by the complement system and immune cells defies classification, specific unifying themes can be recognized.
Firstly, CCP/SCR mosaic receptors (CR1 and CR2) are long and flexible molecules that can survey considerable distances both in the plane of the membrane and above it to find their main ligands: C3b/C4b and C3 and C5 convertases (CR1) and C3d and iC3b (CR2). CR1 downregulates complement activation through the well-known decay-accelerating and cofactor activities, also exerted by the negative regulators MCP and DAF (membrane-bound) and FH and C4BP (fluid phase). In contrast, CR2 lacks complement regulatory activities but it has a more specialized role in bridging innate immunity with adaptive immunity by handing over complement-opsonized pathogens from macrophages to antigen-presenting cells in the spleen and other lymphoid organs.
Secondly, CRIg recognizes C3b (and iC3b) but does it with a structurally unrelated fold (Ig-like) and targets the β-chain of the proteolytically activated C3 fragments. Its unique binding mode is used by the tissue-resident macrophages that express it (mostly, Kupffer cells in the liver) to snatch complement-opsonized pathogens and cellular debris and facilitate their clearance by phagocytosis.
Thirdly, the β2 integrin receptors CR3 and CR4 have played an important role in establishing the structural basis for activation (e.g., the transition from a bent, closed, inactive conformation to an extended, open, active state) and ligand recognition by integrin receptors. Besides binding to iC3b (and to C3d in the case of CR3), these receptors have remarkable ligand promiscuity, employing a small but versatile domain (the αI domain) to recognize them. Their study has highlighted the degree to which surface concentration effects (the so-called 2D concentration, through avidity, compartmentalization, and clustering) and diversity in the structural presentation of ligands can influence the outcome of their interaction.
Fourthly, the anaphylatoxin GPCR receptors have aroused considerable interest as C5a and C3a have been associated with inflammatory diseases, and agonists/antagonists are available. Although the first structures of C5aR1 were obtained by X-ray crystallography, cryoelectron microscopy has allowed the determination of high-resolution structures of C5aR1:C5a and C3aR:C3a complexes within a short time. This acceleration promises a revolution in the structural understanding of anaphylatoxin receptors, how anaphylatoxins are recognized, and how binding triggers receptor activation and signal transduction. The detailed structural knowledge of receptor:anaphylatoxin complexes will also allow better agonists and antagonists to be engineered and validated as therapeutics.
The applications of the structural inquiry of the complement receptors and their ligands are important. Knowledge about the structural organization of the receptors and the ligand complexes advances a fundamental understanding of the immune system. It often results in unexpected and fascinating results, like the proposal that CR2 can hand over iC3b/C3d-opsonized surfaces from macrophages to antigen-presenting cells to stimulate (prime) adaptive immunity. More pragmatically, the structures of ligand complexes often make it possible to elucidate the mechanism of action of known (or suspected) receptor agonists and antagonists, with implications for developing more efficient treatments. It also paves the way for the rational or semi-rational discovery of new agonists and antagonists, increasing their utility, selectivity, and safety. For example, discovering new allosteric inhibitors/antagonists of the anaphylatoxin receptors could result in new treatments.
What are the challenges for the future regarding the structural biology of complement receptors?
The structural catalog of complement receptors is still incomplete. Although state-of-the-art protein structure prediction methods like AlphaFold (233) are filling in the gaps with enticing structural hypotheses (e.g., the C2-type domain of CRIg), it is important to elucidate the structures of receptors, ligands, ligand complexes, and complexes involving them and other immune system components and the host-pathogen machinery. In connection with this central endeavor, developing new methods for protein production and the characterization of protein-protein and protein-ligand complexes will continue to play a significant role as enabling technologies (234, 235).
Much remains to be elucidated regarding structural diversity and modularity, especially in 2D surfaces, highly concentrated clusters of receptors and ligands, and cell-cell interactions. The role of cryoelectron microscopy in advancing the field of structural biology of transmembrane receptors cannot be denied, and the trend is likely to strengthen further (236). A goal for the future should be to integrate cellular structural biology approaches (237) to gain insights into how complement receptors (and complement regulators, membrane-bound or otherwise) function in the physiological and pathological context.
As extracellular pathogens have evolved a seemingly endless repertoire of complement-evasive factors (238, 239), intracellular pathogens have evolved molecular mechanisms to hijack all the non-GPCR complement receptors, MCP, and DAF to gain entry to the cell (240, 241). Strategies aimed at restricting pathogen entry into their host cells targeting the virulence factors or their cognate complement receptors would greatly benefit from a complete understanding of the structural determinants of the interaction.
Finally, these advances should also result in a more subtle and precise appreciation of the differences between the various animal models in use in the field of complement. Applying structural methods and tools to the complement factors and complement receptors of mice, rats, and other animal models beside humans will allow a more informed understanding of the differences in the complement biology between these animals, with important implications for developing disease models and therapies.
Author contributions
FJF and MCV conceived the study. JS-L wrote the first draft of the sections on anaphylatoxin receptors. KdlP wrote the first draft of the sections on complement integrin receptors. FJF and MCV wrote the sections on CR1, CR2, and CRIg with support from KdlP. FJF and MCV wrote the final version of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was funded by Grants PDC2022-133713-I00 of the Spanish Ministerio de Ciencia e Innovación-Recovery, Transformation and Resilience Plan (PRTR), S2022/BMD-7278 of the Regional Government of Madrid, and the European Commission – NextGenerationEU through CSIC’s Global Health Platform (“PTI Salud Global”) (SGL2103020) (to MCV), and the CSIC Special Intramural Grant PIE-201620E064 (to MCV). It was additionally supported by the Network of Excellence Complement in Health and Disease (RED2022-134750-T). KdlP was supported by the Industrial Ph.D. Doctorate (DIN2018-010094) of the Spanish Ministry of Economy and Innovation.
Acknowledgments
JS-L acknowledges the support of the Ph.D. program in Molecular Biosciences of the Universidad Autónoma de Madrid (UAM) and the Ministry of Education, Culture and Sports of Spain (FPU Grant 17/06090). KdlP acknowledges the Ph.D. program in Biochemistry, Molecular Biology, and Biomedicine of the Universidad Complutense de Madrid (UCM).
Conflict of interest
Abvance Biotech SL provided salaries for KdlP and FJF.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brown EJ. Complement receptors and phagocytosis. Curr Opin Immunol (1991) 3:76–82. doi: 10.1016/0952-7915(91)90081-B
2. Erdei A, Kovács KG, Nagy-Baló Z, Lukácsi S, Mácsik-Valent B, Kurucz I, et al. New aspects in the regulation of human B cell functions by complement receptors CR1, CR2, CR3 and CR4. Immunol Lett (2021) 237:42–57. doi: 10.1016/j.imlet.2021.06.006
3. Lamers C, Plüss CJ, Ricklin D. The promiscuous profile of complement receptor 3 in ligand binding, immune modulation, and pathophysiology. Front Immunol (2021) 12:662164. doi: 10.3389/fimmu.2021.662164
4. Vandendriessche S, Cambier S, Proost P, Marques PE. Complement receptors and their role in leukocyte recruitment and phagocytosis. Front Cell Dev Biol (2021) 9:624025. doi: 10.3389/fcell.2021.624025
5. Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3-dependent effector functions of complement. Immunol Rev (2023) 313:120–38. doi: 10.1111/imr.13147
6. Ghebrehiwet B, Geisbrecht BV, Xu X, Savitt AG, Peerschke EIB. The C1q Receptors: Focus on gC1qR/p33 (C1qBP, p32, HABP-1). Semin Immunol (2019) 45:101338. doi: 10.1016/j.smim.2019.101338
7. Vorup-Jensen T, Jensen RK. Structural immunology of complement receptors 3 and 4. Front Immunol (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
8. Van Lookeren Campagne M, Verschoor A. Pathogen clearance and immune adherence “revisited”: Immuno-regulatory roles for CRIg. Semin Immunol (2018) 37:4–11. doi: 10.1016/j.smim.2018.02.007
9. Marin AV, Cárdenas PP, Jiménez-Reinoso A, Muñoz-Ruiz M, Regueiro JR. Lymphocyte integration of complement cues. Semin Cell Dev Biol (2019) 85:132–42. doi: 10.1016/j.semcdb.2018.02.005
10. Dustin ML. Complement receptors in myeloid cell adhesion and phagocytosis. Microbiol Spectr (2016) 4:4.6.04. doi: 10.1128/microbiolspec.MCHD-0034-2016
11. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol (2014) 32:433–59. doi: 10.1146/annurev-immunol-032713-120154
12. Kemper C, Köhl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol (2013) 56:181–90. doi: 10.1016/j.molimm.2013.05.223
13. Erdei A, Isaák A, Török K, Sándor N, Kremlitzka M, Prechl J, et al. Expression and role of CR1 and CR2 on B and T lymphocytes under physiological and autoimmune conditions. Mol Immunol (2009) 46:2767–73. doi: 10.1016/j.molimm.2009.05.181
14. Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunol Rev (2007) 219:157–66. doi: 10.1111/j.1600-065X.2007.00556.x
15. Van Lookeren Campagne M, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol (2007) 9:2095–102. doi: 10.1111/j.1462-5822.2007.00981.x
16. Villiers CL, Perrin-Cocon L, Marche PN, Villiers M-B. Complement receptors and B lymphocytes. Crit Rev Immunol (2004) 24:14. doi: 10.1615/CritRevImmunol.v24.i6.50
17. Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol (1998) 16:545–68. doi: 10.1146/annurev.immunol.16.1.545
18. Ember JA, Hugli TE. Complement factors and their receptors. Immunopharmacology (1997) 38:3–15. doi: 10.1016/S0162-3109(97)00088-X
19. Krych M, Atkinson JP, Michael Holers V. Complement receptors. Curr Opin Immunol (1992) 4:8–13. doi: 10.1016/0952-7915(92)90116-V
20. Soares DC, Barlow PN. Complement control protein modules in the regulators of complement activation. In: Structural biology of the complement system. Boca Raton, FL CRC Press (2005).
21. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science (2016) 352:aad1210. doi: 10.1126/science.aad1210
22. Arbore G, West EE, Rahman J, Le Friec G, Niyonzima N, Pirooznia M, et al. Complement receptor CD46 co-stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat Commun (2018) 9:4186. doi: 10.1038/s41467-018-06706-z
23. Norsworthy PJ, Fossati-Jimack L, Cortes-Hernandez J, Taylor PR, Bygrave AE, Thompson RD, et al. Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q-mediated enhancement of phagocytosis. J Immunol (2004) 172:3406–14. doi: 10.4049/jimmunol.172.6.3406
24. McGreal EP, Ikewaki N, Akatsu H, Morgan BP, Gasque P. Human C1qRp is identical with CD93 and the mNI-11 antigen but does not bind C1q. J Immunol (2002) 168:5222–32. doi: 10.4049/jimmunol.168.10.5222
25. Ghebrehiwet B. cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol (2004) 41:173–83. doi: 10.1016/j.molimm.2004.03.014
26. Kemper C, Ferreira VP, Paz JT, Holers VM, Lionakis MS, Alexander JJ. Complement: the road less traveled. J Immunol (2023) 210:119–25. doi: 10.4049/jimmunol.2200540
27. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol (2019) 114:341–52. doi: 10.1016/j.molimm.2019.07.030
28. Holers VM. Complement therapeutics are coming of age in rheumatology. Nat Rev Rheumatol (2023) 19:470–85. doi: 10.1038/s41584-023-00981-x
29. Lamers C, Ricklin D, Lambris JD. Complement-targeted therapeutics: An emerging field enabled by academic drug discovery. Am J Hematol (2023) 98, S29, S76–7. doi: 10.1002/ajh.26875
30. Ricklin D, Lambris JD. New milestones ahead in complement-targeted therapy. Semin Immunol (2016) 28:208–22. doi: 10.1016/j.smim.2016.06.001
31. Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol (2007) 25:1265–75. doi: 10.1038/nbt1342
32. Jensen RK, Bajic G, Sen M, Springer TA, Vorup-Jensen T, Andersen GR. Complement receptor 3 forms a compact high-affinity complex with iC3b. J Immunol (2021) 206:3032–42. doi: 10.4049/jimmunol.2001208
33. Jensen RK, Pedersen H, Lorentzen J, Laursen NS, Vorup-Jensen T, Andersen GR. Structural insights into the function-modulating effects of nanobody binding to the integrin receptor αMβ2. J Biol Chem (2022) 298:102168. doi: 10.1016/j.jbc.2022.102168
34. Fernández FJ, Santos-López J, Martínez-Barricarte R, Querol-García J, Martín-Merinero H, Navas-Yuste S, et al. The crystal structure of iC3b-CR3 alphaI reveals a modular recognition of the main opsonin iC3b by the CR3 integrin receptor. Nat Commun (2022) 13:1955. doi: 10.1038/s41467-022-29580-2
35. Feng Y, Zhao C, Deng Y, Wang H, Ma L, Liu S, et al. Mechanism of activation and biased signaling in complement receptor C5aR1. Cell Res (2023) 33:312–24. doi: 10.1038/s41422-023-00779-2
36. Wang Y, Liu W, Xu Y, He X, Yuan Q, Luo P, et al. Revealing the signaling of complement receptors C3aR and C5aR1 by anaphylatoxins. Nat Chem Biol (2023). doi: 10.1038/s41589-023-01339-w
37. West EE, Kemper C. Complosome — the intracellular complement system. Nat Rev Nephrol (2023) 19:426–39. doi: 10.1038/s41581-023-00704-1
38. Barlow PN, Baron M, Norman DG, Day AJ, Willis AC, Sim RB, et al. Secondary structure of the complement control protein module by two-dimensional proton NMR. Biochemistry (1991) 30:997–1004. doi: 10.1021/bi00218a016
39. Almitairi JOM, Venkatraman Girija U, Furze CM, Simpson-Gray X, Badakshi F, Marshall JE, et al. Structure of the C1r–C1s interaction of the C1 complex of complement activation. Proc Natl Acad Sci (2018) 115:768–73. doi: 10.1073/pnas.1718709115
40. Saunders RE, Abarrategui-Garrido C, Frémeaux-Bacchi V, Goicoechea de Jorge E, Goodship THJ, López Trascasa M, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat (2007) 28:222–34. doi: 10.1002/humu.20435
41. Perkins SJ, Gilbert HE, Aslam M, Hannan J, Holers VM, Goodship THJ. Solution structures of complement components by X-ray and neutron scattering and analytical ultracentrifugation. Biochem Soc Trans (2002) 30:996–1001. doi: 10.1042/bst0300996
42. Soares DC, Gerloff DL, Syme NR, Coulson AFW, Parkinson J, Barlow PN. Large-scale modelling as a route to multiple surface comparisons of the CCP module family. Protein Eng Des Sel (2005) 18:379–88. doi: 10.1093/protein/gzi039
43. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity (1997) 7:345–55. doi: 10.1016/s1074-7613(00)80356-8
44. GhIran I, Barbashov SF, Klickstein LB, Tas SW, Jensenius JC, Nicholson-Weller A. Complement receptor 1/cd35 is a receptor for mannan-binding lectin. J Exp Med (2000) 192:1797–808. doi: 10.1084/jem.192.12.1797
45. Nelson RA. The immune-adherence phenomenon: an immunologically specific reaction between microorganisms and erythrocytes leading to enhanced phagocytosis. Science (1953) 118:733–7. doi: 10.1126/science.118.3077.733
46. Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1: Active sites of CR1. Immunol Rev (2001) 180:112–22. doi: 10.1034/j.1600-065X.2001.1800110.x
47. Khera R, Das N. Complement Receptor 1: Disease associations and therapeutic implications. Mol Immunol (2009) 46:761–72. doi: 10.1016/j.molimm.2008.09.026
48. Kremlitzka M, Polgár A, Fülöp L, Kiss E, Poór G, Erdei A. Complement receptor type 1 (CR1, CD35) is a potent inhibitor of B-cell functions in rheumatoid arthritis patients. Int Immunol (2013) 25:25–33. doi: 10.1093/intimm/dxs090
49. Weisman HF, Bartow T, Leppo MK, Marsh HC, Carson GR, Concino MF, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science (1990) 249:146–51. doi: 10.1126/science.2371562
50. Aslam M, Perkins SJ. Folded-back solution structure of monomeric factor H of human complement by synchrotron X-ray and neutron scattering, analytical ultracentrifugation and constrained molecular modelling. J Mol Biol (2001) 309:1117–38. doi: 10.1006/jmbi.2001.4720
51. Li K, Okemefuna AI, Gor J, Hannan JP, Asokan R, Holers VM, et al. Solution structure of the complex formed between human complement C3d and full-length complement receptor type 2. J Mol Biol (2008) 384:137–50. doi: 10.1016/j.jmb.2008.08.084
52. Furtado PB, Huang CY, Ihyembe D, Hammond RA, Marsh HC, Perkins SJ. The partly folded back solution structure arrangement of the 30 SCR domains in human complement receptor type 1 (CR1) permits access to its C3b and C4b ligands. J Mol Biol (2008) 375:102–18. doi: 10.1016/j.jmb.2007.09.085
53. Smith BO, Mallin RL, Krych-Goldberg M, Wang X, Hauhart RE, Bromek K, et al. Structure of the C3b binding site of CR1 (CD35), the immune adherence receptor. Cell (2002) 108:769–80. doi: 10.1016/s0092-8674(02)00672-4
54. O’Leary JM, Bromek K, Black GM, Uhrinova S, Schmitz C, Wang X, et al. Backbone dynamics of complement control protein (CCP) modules reveals mobility in binding surfaces. Protein Sci (2004) 13:1238–50. doi: 10.1110/ps.03582704
55. Park HJ, Guariento M, Maciejewski M, Hauhart R, Tham W-H, Cowman AF, et al. Using mutagenesis and structural biology to map the binding site for the plasmodium falciparum merozoite protein pfRh4 on the human immune adherence receptor. J Biol Chem (2014) 289:450–63. doi: 10.1074/jbc.M113.520346
56. Takata Y, Kinoshita T, Kozono H, Takeda J, Tanaka E, Hong K, et al. Covalent association of C3b with C4b within C5 convertase of the classical complement pathway. J Exp Med (1987) 165:1494–507. doi: 10.1084/jem.165.6.1494
57. Kinoshita T, Takata Y, Kozono H, Takeda J, Hong KS, Inoue K. C5 convertase of the alternative complement pathway: covalent linkage between two C3b molecules within the trimolecular complex enzyme. J Immunol Baltim Md 1950 (1988) 141:3895–901. doi: 10.4049/jimmunol.141.11.3895
58. Krych-Goldberg M, Hauhart RE, Subramanian VB, Yurcisin BM, Crimmins DL, Hourcade DE, et al. Decay accelerating activity of complement receptor type 1 (CD35). J Biol Chem (1999) 274:31160–8. doi: 10.1074/jbc.274.44.31160
59. Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J (2016) 35:1133–49. doi: 10.15252/embj.201593673
60. Wu J, Wu Y-Q, Ricklin D, Janssen BJC, Lambris JD, Gros P. Structure of complement fragment C3b–factor H and implications for host protection by complement regulators. Nat Immunol (2009) 10:728–33. doi: 10.1038/ni.1755
61. Oliveira LC, Kretzschmar GC, Dos Santos ACM, Camargo CM, Nisihara RM, Farias TDJ, et al. Complement receptor 1 (CR1, CD35) polymorphisms and soluble CR1: A proposed anti-inflammatory role to quench the fire of “Fogo selvagem” Pemphigus foliaceus. Front Immunol (2019) 10:2585. doi: 10.3389/fimmu.2019.02585
62. Krych-Goldberg M, Moulds JM, Atkinson JP. Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol Med (2002) 8:531–7. doi: 10.1016/S1471-4914(02)02419-X
63. Tham W-H, Wilson DW, Lopaticki S, Schmidt CQ, Tetteh-Quarcoo PB, Barlow PN, et al. Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc Natl Acad Sci (2010) 107:17327–32. doi: 10.1073/pnas.1008151107
64. Reiling L, Richards JS, Fowkes FJI, Wilson DW, Chokejindachai W, Barry AE, et al. The Plasmodium falciparum Erythrocyte Invasion Ligand Pfrh4 as a Target of Functional and Protective Human Antibodies against Malaria. PloS One (2012) 7:e45253. doi: 10.1371/journal.pone.0045253
65. Prajapati SK, Borlon C, Rovira-Vallbona E, Gruszczyk J, Menant S, Tham W-H, et al. Complement Receptor 1 availability on red blood cell surface modulates Plasmodium vivax invasion of human reticulocytes. Sci Rep (2019) 9:8943. doi: 10.1038/s41598-019-45228-6
66. Jensen AR, Adams Y, Hviid L. Cerebral Plasmodium falciparum malaria: The role of PfEMP1 in its pathogenesis and immunity, and PfEMP1-based vaccines to prevent it. Immunol Rev (2020) 293:230–52. doi: 10.1111/imr.12807
67. Prodinger WM. Complement receptor type two (CR2,CR21): A target for influencing the humoral immune response and antigen-trapping. Immunol Res (1999) 20:187–94. doi: 10.1007/BF02790402
68. Lowell CA, Klickstein LB, Carter RH, Mitchell JA, Fearon DT, Ahearn JM. Mapping of the Epstein-Barr virus and C3dg binding sites to a common domain on complement receptor type 2. J Exp Med (1989) 170:1931–46. doi: 10.1084/jem.170.6.1931
69. Carel JC, Myones BL, Frazier B, Holers VM. Structural requirements for C3d,g/Epstein-Barr virus receptor (CR2/CD21) ligand binding, internalization, and viral infection. J Biol Chem (1990) 265:12293–9. doi: 10.1016/S0021-9258(19)38344-9
70. Delcayre AX, Salas F, Mathur S, Kovats K, Lotz M, Lernhardt W. Epstein Barr virus/complement C3d receptor is an interferon alpha receptor. EMBO J (1991) 10:919–26. doi: 10.1002/j.1460-2075.1991.tb08025.x
71. Asokan R, Hua J, Young KA, Gould HJ, Hannan JP, Kraus DM, et al. Characterization of human complement receptor type 2 (CR2/CD21) as a receptor for IFN-α: A potential role in systemic lupus erythematosus. J Immunol (2006) 177:383–94. doi: 10.4049/jimmunol.177.1.383
72. Aubry J-P, Pochon S, Graber P, Jansen KU, Bonnefoy J-Y. CD21 is a ligand for CD23 and regulates IgE production. Nature (1992) 358:505–7. doi: 10.1038/358505a0
73. Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci (1984) 81:4510–4. doi: 10.1073/pnas.81.14.4510
74. Nemerow GR, Wolfert R, McNaughton ME, Cooper NR. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor (CR2). J Virol (1985) 55:347–51. doi: 10.1128/jvi.55.2.347-351.1985
75. Moore MD, DiScipio RG, Cooper NR, Nemerow GR. Hydrodynamic, electron microscopic, and ligand-binding analysis of the epstein-barr virus/C3dg receptor (CR2). J Biol Chem (1989) 264:20576–82. doi: 10.1016/S0021-9258(19)47101-9
76. Gilbert HE, Asokan R, Holers VM, Perkins SJ. The 15 SCR flexible extracellular domains of human complement receptor type 2 can mediate multiple ligand and antigen interactions. J Mol Biol (2006) 362:1132–47. doi: 10.1016/j.jmb.2006.08.012
77. Prota AE, Sage DR, Stehle T, Fingeroth JD. The crystal structure of human CD21: Implications for Epstein–Barr virus and C3d binding. Proc Natl Acad Sci (2002) 99:10641–6. doi: 10.1073/pnas.162360499
78. Guthridge JM, Rakstang JK, Young KA, Hinshelwood J, Aslam M, Robertson A, et al. Structural studies in solution of the recombinant N-terminal pair of short consensus/complement repeat domains of complement receptor type 2 (CR2/CD21) and interactions with its ligand C3dg. Biochemistry (2001) 40:5931–41. doi: 10.1021/bi0101749
79. Gilbert HE, Eaton JT, Hannan JP, Holers VM, Perkins SJ. Solution structure of the complex between CR2 SCR 1-2 and C3d of human complement: an X-ray scattering and sedimentation modelling study. J Mol Biol (2005) 346:859–73. doi: 10.1016/j.jmb.2004.12.006
80. Gilbert HE, Aslam M, Guthridge JM, Holers VM, Perkins SJ. Extended flexible linker structures in the complement chimaeric conjugate CR2-ig by scattering, analytical ultracentrifugation and constrained modelling: implications for function and therapy. J Mol Biol (2006) 356:397–412. doi: 10.1016/j.jmb.2005.11.050
81. Szakonyi G, Guthridge JM, Li D, Young K, Holers VM, Chen XS. Structure of complement receptor 2 in complex with its C3d ligand. Science (2001) 292:1725–8. doi: 10.1126/science.1059118
82. Hannan JP, Young KA, Guthridge JM, Asokan R, Szakonyi G, Chen XS, et al. Mutational analysis of the complement receptor type 2 (CR2/CD21)–C3d interaction reveals a putative charged SCR1 binding site for C3d. J Mol Biol (2005) 346:845–58. doi: 10.1016/j.jmb.2004.12.007
83. Van Den Elsen JMH, Isenman DE. A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science (2011) 332:608–11. doi: 10.1126/science.1201954
84. Montefiori DC, Stewart K, Ahearn JM, Zhou J, Zhou J. Complement-mediated binding of naturally glycosylated and glycosylation-modified human immunodeficiency virus type 1 to human CR2 (CD21). J Virol (1993) 67:2699–706. doi: 10.1128/jvi.67.5.2699-2706.1993
85. Montefiori DC, Zhou J, Shaff DI. CD4-independent binding of HIV-1 to the B lymphocyte receptor CR2 (CD21) in the presence of complement and antibody. Clin Exp Immunol (2008) 90:383–9. doi: 10.1111/j.1365-2249.1992.tb05855.x
86. Stano P, Williams V, Villani M, Cymbalyuk ES, Qureshi A, Huang Y, et al. App1: an antiphagocytic protein that binds to complement receptors 3 and 2. J Immunol (2009) 182:84–91. doi: 10.4049/jimmunol.182.1.84
87. Medof ME, Iida K, Mold C, Nussenzweig V. Unique role of the complement receptor CR1 in the degradation of C3b associated with immune complexes. J Exp Med (1982) 156:1739–54. doi: 10.1084/jem.156.6.1739
88. Bajtay Z. Biologia Futura: stories about the functions of β2-integrins in human phagocytes. Biol Futura (2021) 72:7–13. doi: 10.1007/s42977-020-00063-z
90. Springer TA. Adhesion receptors of the immune system. Nature (1990) 346:425–34. doi: 10.1038/346425a0
91. Kishimoto TK, Larson RS, Corbi AL, Dustin ML, Staunton DE, Springer TA. The leukocyte Integrins. In: Advances in Immunology:Academic Press, New York. Elsevier (1989). p. 149–82. doi: 10.1016/S0065-2776(08)60653-7
92. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
93. Rosen H, Law SKA. The Leukocyte Cell Surface Receptor(s) for the iC3b Product of Complement. In: Lambris JD, editor. The Third Component of Complement. Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer Berlin Heidelberg (1990). p. 99–122. doi: 10.1007/978-3-642-74977-3_6
94. Ross GD, Větvička V. CR3 (CD11b, CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol (2008) 92:181–4. doi: 10.1111/j.1365-2249.1993.tb03377.x
95. Myones BL, Dalzell JG, Hogg N, Ross GD. Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. J Clin Invest (1988) 82:640–51. doi: 10.1172/JCI113643
96. Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol (1993) 120:1031–43. doi: 10.1083/jcb.120.4.1031
97. Lishko VK, Yakubenko VP, Hertzberg KM, Grieninger G, Ugarova TP. The alternatively spliced αEC domain of human fibrinogen-420 is a novel ligand for leukocyte integrins αMβ2 and αXβ2. Blood (2001) 98:2448–55. doi: 10.1182/blood.V98.8.2448
98. Wright SD, Jong MT. Adhesion-promoting receptors on human macrophages recognize Escherichia coli by binding to lipopolysaccharide. J Exp Med (1986) 164:1876–88. doi: 10.1084/jem.164.6.1876
99. Erdei A, Lukácsi S, Mácsik-Valent B, Nagy-Baló Z, Kurucz I, Bajtay Z. Non-identical twins: Different faces of CR3 and CR4 in myeloid and lymphoid cells of mice and men. Semin Cell Dev Biol (2019) 85:110–21. doi: 10.1016/j.semcdb.2017.11.025
100. Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol (2012) 24:107–15. doi: 10.1016/j.ceb.2011.10.004
101. Wen L, Lyu Q, Ley K, Goult BT. Structural basis of β2 integrin inside—Out activation. Cells (2022) 11:3039. doi: 10.3390/cells11193039
102. Whittaker CA, Hynes RO. Distribution and evolution of von willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell (2002) 13:3369–87. doi: 10.1091/mbc.e02-05-0259
103. Lee C-H, Romain G, Yan W, Watanabe M, Charab W, Todorova B, et al. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat Immunol (2017) 18:889–98. doi: 10.1038/ni.3770
104. Springer TA. Folding of the N-terminal, ligand-binding region of integrin α-subunits into a β-propeller domain. Proc Natl Acad Sci (1997) 94:65–72. doi: 10.1073/pnas.94.1.65
105. Shi M, Sundramurthy K, Liu B, Tan S-M, Law SKA, Lescar J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/I-EGF1 segment from the human integrin β2 subunit at 1.8-Å Resolution. J Biol Chem (2005) 280:30586–93. doi: 10.1074/jbc.M502525200
106. Shi M, Foo SY, Tan S-M, Mitchell EP, Law SKA, Lescar J. A structural hypothesis for the transition between bent and extended conformations of the leukocyte β2 integrins. J Biol Chem (2007) 282:30198–206. doi: 10.1074/jbc.M701670200
107. Beglova N, Blacklow SC, Takagi J, Springer TA. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat Struct Biol (2002) 9:282–7. doi: 10.1038/nsb779
108. Sen M, Springer TA. Leukocyte integrin αLβ2 headpiece structures: The αI domain, the pocket for the internal ligand, and concerted movements of its loops. Proc Natl Acad Sci (2016) 113:2940–5. doi: 10.1073/pnas.1601379113
109. Guo J, Zhang Y, Li H, Chu H, Wang Q, Jiang S, et al. Intramembrane ionic protein–lipid interaction regulates integrin structure and function. PloS Biol (2018) 16:e2006525. doi: 10.1371/journal.pbio.2006525
110. Xu S, Wang J, Wang J-H, Springer TA. Distinct recognition of complement iC3b by integrins αXβ2 and αMβ2. Proc Natl Acad Sci (2017) 114:3403–8. doi: 10.1073/pnas.1620881114
111. Bajic G, Yatime L, Sim RB, Vorup-Jensen T, Andersen GR. Structural insight on the recognition of surface-bound opsonins by the integrin I domain of complement receptor 3. Proc Natl Acad Sci U.S.A. (2013) 110:16426–31. doi: 10.1073/pnas.1311261110
112. Lee J-O, Rieu P, Arnaout MA, Liddington R. Crystal structure of the A domain from the a subunit of integrin CR3 (CD11 b/CD18). Cell (1995) 80:631–8. doi: 10.1016/0092-8674(95)90517-0
113. Baldwin ET, Sarver RW, Bryant GL, Curry KA, Fairbanks MB, Finzel BC, et al. Cation binding to the integrin CD11b I domain and activation model assessment. Structure (1998) 6:923–35. doi: 10.1016/S0969-2126(98)00093-8
114. Goldsmith JA, DiVenere AM, Maynard JA, McLellan JS. Structural basis for non-canonical integrin engagement by Bordetella adenylate cyclase toxin. Cell Rep (2022) 40:111196. doi: 10.1016/j.celrep.2022.111196
115. Jensen MR, Bajic G, Zhang X, Laustsen AK, Koldsø H, Skeby KK, et al. Structural basis for simvastatin competitive antagonism of complement receptor 3. J Biol Chem (2016) 291:16963–76. doi: 10.1074/jbc.M116.732222
116. Mahalingam B, Ajroud K, Alonso JL, Anand S, Adair BD, Horenstein AL, et al. Stable coordination of the inhibitory ca2+ Ion at the metal ion-dependent adhesion site in integrin CD11b/CD18 by an antibody-derived ligand aspartate: implications for integrin regulation and structure-based drug design. J Immunol (2011) 187:6393–401. doi: 10.4049/jimmunol.1102394
117. Gaither TA, Vargas I, Inada S, Frank MM. The complement fragment C3d facilitates phagocytosis by monocytes. Immunology (1987) 62:405–11.
118. Stapulionis R, Pinto Oliveira CL, Gjelstrup MC, Pedersen JS, Hokland ME, Hoffmann SV, et al. Structural insight into the function of myelin basic protein as a ligand for integrin αMβ2. J Immunol (2008) 180:3946–56. doi: 10.4049/jimmunol.180.6.3946
119. Zhang X, Bajic G, Andersen GR, Christiansen SH, Vorup-Jensen T. The cationic peptide LL-37 binds Mac-1 (CD11b/CD18) with a low dissociation rate and promotes phagocytosis. Biochim Biophys Acta BBA - Proteins Proteomics (2016) 1864:471–8. doi: 10.1016/j.bbapap.2016.02.013
120. Sen M, Yuki K, Springer TA. An internal ligand-bound, metastable state of a leukocyte integrin, αXβ2. J Cell Biol (2013) 203:629–42. doi: 10.1083/jcb.201308083
121. Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an αI domain, complement receptor type 4. EMBO J (2010) 29:666–79. doi: 10.1038/emboj.2009.367
122. Bilsland CA, Diamond MS, Springer TA. The leukocyte integrin p150,95 (CD11c/CD18) as a receptor for iC3b. Activation by a heterologous beta subunit and localization of a ligand recognition site to the I domain. J Immunol Baltim Md 1950 (1994) 152:4582–9. doi: 10.1101/gr.1293003
123. Clark HF, Gurney AL, Abaya E, Baker K, Baldwin D, Brush J, et al. The secreted protein discovery initiative (SPDI), a large-scale effort to identify novel human secreted and transmembrane proteins: A bioinformatics assessment. Genome Res (2003) 13:2265–70. doi: 10.1101/gr.1293003
124. Smith DK, Xue H. Sequence profiles of immunoglobulin and immunoglobulin-like domains. J Mol Biol (1997) 274:530–45. doi: 10.1006/jmbi.1997.1432
125. Helmy KY, Katschke KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
126. Carpentier JL, Lew DP, Paccaud JP, Gil R, Iacopetta B, Kazatchkine M, et al. Internalization pathway of C3b receptors in human neutrophils and its transmodulation by chemoattractant receptors stimulation. Cell Regul (1991) 2:41–55. doi: 10.1172/JCI25673
127. Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, Behnke S, et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest (2006) 116:2817–26. doi: 10.1172/JCI25673
128. Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, Fairbrother WJ, et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature (2006) 444:217–20. doi: 10.1038/nature05263
129. Wen Y, Ouyang Z, Schoonooghe S, Luo S, De Baetselier P, Lu W, et al. Structural evaluation of a nanobody targeting complement receptor Vsig4 and its cross reactivity. Immunobiology (2017) 222:807–13. doi: 10.1016/j.imbio.2016.11.008
130. Arnaout MA, Melamed J, Tack BF, Colten HR. Characterization of the human complement (c3b) receptor with a fluid phase C3b dimer. J Immunol Baltim Md 1950 (1981) 127:1348–54. doi: 10.1073/pnas.1913443116
131. Liu G, Fu Y, Yosri M, Chen Y, Sun P, Xu J, et al. CRIg plays an essential role in intravascular clearance of bloodborne parasites by interacting with complement. Proc Natl Acad Sci (2019) 116:24214–20. doi: 10.1073/pnas.1913443116
132. Barnum SR. C4a: an anaphylatoxin in name only. J Innate Immun (2015) 7:333–9. doi: 10.1159/000371423
133. Han X, de la Fuente M, Nieman MT. Complement factor C4a does not activate protease-activated receptor 1 (PAR1) or PAR4 on human platelets. Res Pract Thromb Haemost (2021) 5:104–10. doi: 10.1002/rth2.12459
134. Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol (2003) 63:1256–72. doi: 10.1124/mol.63.6.1256
135. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Mathie A, Peters JA, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein-coupled receptors. Br J Pharmacol (2021) 178, S27–156. doi: 10.1111/bph.15538
136. Kolakowski LF. GCRDb: a G-protein-coupled receptor database. Receptors Channels (1994) 2:1–7. doi: 10.1038/nrm908
137. Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol (2002) 3:639–50. doi: 10.1038/nrm908
138. Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature (2013) 494:185–94. doi: 10.1038/nature11896
139. Weis WI, Kobilka BK. The molecular basis of G protein–coupled receptor activation. Annu Rev Biochem (2018) 87:897–919. doi: 10.1146/annurev-biochem-060614-033910
140. Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Methods in Neurosciences:Academic Press, New York. Elsevier (1995). p. 366–428. doi: 10.1016/S1043-9471(05)80049-7
141. Laumonnier Y, Karsten CM, Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol Immunol (2017) 89:44–58. doi: 10.1016/j.molimm.2017.05.019
142. Klos A, Tenner AJ, Johswich K-O, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
143. Zhang T, Garstka MA, Li K. The controversial C5a receptor C5aR2: its role in health and disease. J Immunol Res (2017) 2017:1–16. doi: 10.1155/2017/8193932
144. Wang Y, Zhang H, He Y-W. The complement receptors C3aR and C5aR are a new class of immune checkpoint receptor in cancer immunotherapy. Front Immunol (2019) 10:1574. doi: 10.3389/fimmu.2019.01574
145. Gao S, Cui Z, Zhao M. The complement C3a and C3a receptor pathway in kidney diseases. Front Immunol (2020) 11:1875. doi: 10.3389/fimmu.2020.01875
146. Giorgio C, Zippoli M, Cocchiaro P, Castelli V, Varrassi G, Aramini A, et al. Emerging role of C5 complement pathway in peripheral neuropathies: current treatments and future perspectives. Biomedicines (2021) 9:399. doi: 10.3390/biomedicines9040399
147. Ruocco A, Sirico A, Novelli R, Iannelli S, Van Breda SV, Kyburz D, et al. The role of C5a-C5aR1 axis in bone pathophysiology: A mini-review. Front Cell Dev Biol (2022) 10:957800. doi: 10.3389/fcell.2022.957800
148. Schanzenbacher J, Köhl J, Karsten CM. Anaphylatoxins spark the flame in early autoimmunity. Front Immunol (2022) 13:958392. doi: 10.3389/fimmu.2022.958392
149. Hawksworth OA, Li XX, Coulthard LG, Wolvetang EJ, Woodruff TM. New concepts on the therapeutic control of complement anaphylatoxin receptors. Mol Immunol (2017) 89:36–43. doi: 10.1016/j.molimm.2017.05.015
150. Chenoweth DE, Hugli TE. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc Natl Acad Sci (1978) 75:3943–7. doi: 10.1073/pnas.75.8.3943
151. Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature (1991) 349:614–7. doi: 10.1038/349614a0
152. Boulay F, Mery L, Tardif M, Brouchon L, Vignais P. Expression cloning of a receptor for C5a anaphylatoxin on differentiated HL-60 cells. Biochemistry (1991) 30:2993–9. doi: 10.1021/bi00226a002
153. Hilger D. The role of structural dynamics in GPCR-mediated signaling. FEBS J (2021) 288:2461–89. doi: 10.1111/febs.15841
154. Robertson N, Rappas M, Doré AS, Brown J, Bottegoni G, Koglin M, et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature (2018) 553:111–4. doi: 10.1038/nature25025
155. Liu H, Kim HR, Deepak RNVK, Wang L, Chung KY, Fan H, et al. Orthosteric and allosteric action of the C5a receptor antagonists. Nat Struct Mol Biol (2018) 25:472–81. doi: 10.1038/s41594-018-0067-z
156. Floyd DH, Geva A, Bruinsma SP, Overton MC, Blumer KJ, Baranski TJ. C5a receptor oligomerization. J Biol Chem (2003) 278:35354–61. doi: 10.1074/jbc.M305607200
157. Klco JM, Lassere TB, Baranski TJ. C5a receptor oligomerization. J Biol Chem (2003) 278:35345–53. doi: 10.1074/jbc.M305606200
158. Hüttenrauch F, Pollok-Kopp B, Oppermann M. G protein-coupled receptor kinases promote phosphorylation and β-arrestin-mediated internalization of CCR5 homo- and hetero-oligomers. J Biol Chem (2005) 280:37503–15. doi: 10.1074/jbc.M500535200
159. Croker DE, Halai R, Fairlie DP, Cooper MA. C5a, but not C5a-des Arg, induces upregulation of heteromer formation between complement C5a receptors C5aR and C5L2. Immunol Cell Biol (2013) 91:625–33. doi: 10.1038/icb.2013.48
160. Pease JE, Barker MD. N-linked glycosylation of the C5a receptor. Biochem Mol Biol Int (1993) 31:719–26. doi: 10.1084/jem.193.9.1059
161. Farzan M, Schnitzler CE, Vasilieva N, Leung D, Kuhn J, Gerard C, et al. Sulfated tyrosines contribute to the formation of the C5a docking site of the human C5a anaphylatoxin receptor. J Exp Med (2001) 193:1059–66. doi: 10.1084/jem.193.9.1059
162. Giannini E, Brouchon L, Boulay F. Identification of the major phosphorylation sites in human C5a anaphylatoxin receptor in vivo. J Biol Chem (1995) 270:19166–72. doi: 10.1074/jbc.270.32.19166
163. Monk PN, Scola A-M, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol (2007) 152:429–48. doi: 10.1038/sj.bjp.0707332
164. Klos A, Wende E, Wareham KJ, Monk PN. International union of basic and clinical pharmacology. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev (2013) 65:500–43. doi: 10.1124/pr.111.005223
165. Pandey S, Maharana J, Li XX, Woodruff TM, Shukla AK. Emerging insights into the structure and function of complement C5a receptors. Trends Biochem Sci (2020) 45:693–705. doi: 10.1016/j.tibs.2020.04.004
166. DeMartino JA, Van Riper G, Siciliano SJ, Molineaux CJ, Konteatis ZD, Rosen H, et al. The amino terminus of the human C5a receptor is required for high affinity C5a binding and for receptor activation by C5a but not C5a analogs. J Biol Chem (1994) 269:14446–50. doi: 10.1016/S0021-9258(17)36643-7
167. Mery L, Boulay F. The NH2-terminal region of C5aR but not that of FPR is critical for both protein transport and ligand binding. J Biol Chem (1994) 269:3457–63. doi: 10.1016/S0021-9258(17)41884-9
168. Hagemann IS, Narzinski KD, Floyd DH, Baranski TJ. Random mutagenesis of the complement factor 5a (C5a) receptor N terminus provides a structural constraint for C5a docking. J Biol Chem (2006) 281:36783–92. doi: 10.1074/jbc.M607686200
169. Chen Z, Zhang X, Gonnella NC, Pellas TC, Boyar WC, Ni F. Residues 21–30 within the extracellular N-terminal region of the C5a receptor represent a binding domain for the C5a anaphylatoxin. J Biol Chem (1998) 273:10411–9. doi: 10.1074/jbc.273.17.10411
170. Siciliano SJ, Rollins TE, DeMartino J, Konteatis Z, Malkowitz L, Van Riper G, et al. Two-site binding of C5a by its receptor: analternative binding paradigm for G protein-coupled receptors. Proc Natl Acad Sci (1994) 91:1214–8. doi: 10.1073/pnas.91.4.1214
171. Dumitru AC, Deepak RNVK, Liu H, Koehler M, Zhang C, Fan H, et al. Submolecular probing of the complement C5a receptor–ligand binding reveals a cooperative two-site binding mechanism. Commun Biol (2020) 3:786. doi: 10.1038/s42003-020-01518-8
172. Pease JE, Burton DR, Barker MD. Generation of chimeric C5a/formyl peptide receptors: towards the identification of the human C5a receptor binding site. Eur J Immunol (1994) 24:211–5. doi: 10.1002/eji.1830240133
173. Gerard C, Gerard NP. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol (1994) 12:775–808. doi: 10.1146/annurev.iy.12.040194.004015
174. Bokisch VA, Müller-Eberhard HJ. Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J Clin Invest (1970) 49:2427–36. doi: 10.1172/JCI106462
175. Kawai M, Quincy DA, Lane B, Mollison KW, Or YS, Luly JR, et al. Structure-function studies in a series of carboxyl-terminal octapeptide analogs of anaphylatoxin C5a. J Med Chem (1992) 35:220–3. doi: 10.1021/jm00080a004
176. Kawai M, Quincy DA, Lane B, Mollison KW, Luly JR, Carter GW. Identification and synthesis of a receptor binding site of human anaphylatoxin C5a. J Med Chem (1991) 34:2068–71. doi: 10.1021/jm00111a022
177. Higginbottom A, Cain SA, Woodruff TM, Proctor LM, Madala PK, Tyndall JDA, et al. Comparative agonist/antagonist responses in mutant human C5a receptors define the ligand binding site *. J Biol Chem (2005) 280:17831–40. doi: 10.1074/jbc.M410797200
178. Klco JM, Nikiforovich GV, Baranski TJ. Genetic analysis of the first and third extracellular loops of the C5a receptor reveals an essential WXFG motif in the first loop. J Biol Chem (2006) 281:12010–9. doi: 10.1074/jbc.M600548200
179. Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nat Struct Mol Biol (2005) 12:320–6. doi: 10.1038/nsmb913
180. Baranski TJ, Herzmark P, Lichtarge O, Gerber BO, Trueheart J, Meng EC, et al. C5a receptor activation. J Biol Chem (1999) 274:15757–65. doi: 10.1074/jbc.274.22.15757
181. Geva A, Lassere TB, Lichtarge O, Pollitt SK, Baranski TJ. Genetic mapping of the human C5a receptor. J Biol Chem (2000) 275:35393–401. doi: 10.1074/jbc.M005602200
182. Zhou Q, Yang D, Wu M, Guo Y, Guo W, Zhong L, et al. Common activation mechanism of class A GPCRs. eLife (2019) 8:e50279. doi: 10.7554/eLife.50279
183. Amatruda TT, Gerard NP, Gerard C, Simon MI. Specific interactions of chemoattractant factor receptors with G-proteins. J Biol Chem (1993) 268:10139–44. doi: 10.1016/S0021-9258(18)82183-4
184. Buhl AM, Eisfelder BJ, Worthen GS, Johnson GL, Russell M. Selective coupling of the human anaphylatoxin C5a receptor and α16 in human kidney 293 cells. FEBS Lett (1993) 323:132–4. doi: 10.1016/0014-5793(93)81464-B
185. Vanek M, Hawkins LD, Gusovsky F. Coupling of the C5a receptor to Gi in U-937 cells and in cells transfected with C5a receptor cDNA. Mol Pharmacol (1994) 46:832–9. doi: 10.1006/bbrc.1995.1327
186. Shum JK, Allen RA, Wong YH. The human chemoattractant complement C5a receptor inhibits cyclic AMP accumulation through gi and gz proteins. Biochem Biophys Res Commun (1995) 208:223–9. doi: 10.1006/bbrc.1995.1327
187. Pandey S, Kumari P, Baidya M, Kise R, Cao Y, Dwivedi-Agnihotri H, et al. Intrinsic bias at non-canonical, β-arrestin-coupled seven transmembrane receptors. Mol Cell (2021) 81:4605–4621.e11. doi: 10.1016/j.molcel.2021.09.007
188. Matsumoto ML, Narzinski K, Kiser PD, Nikiforovich GV, Baranski TJ. A comprehensive structure-function map of the intracellular surface of the human C5a receptor. J Biol Chem (2007) 282:3105–21. doi: 10.1074/jbc.M607679200
189. Auger GA, Smith BM, Pease JE, Barker MD. The use of membrane translocating peptides to identify sites of interaction between the C5a receptor and downstream effector proteins. Immunology (2004) 112:590–6. doi: 10.1111/j.1365-2567.2004.01919.x
190. Siciliano SJ, Rollins TE, Springer MS. Interaction between the C5a receptor and Gi in both the membrane-bound and detergent-solubilized states. J Biol Chem (1990) 265:19568–74. doi: 10.1016/S0021-9258(17)45409-3
191. Wennogle LP, Conder L, Winter C, Braunwalder A, Vlattas S, Kramer R, et al. Stabilization of C5a receptor-G-protein interactions through ligand binding. J Cell Biochem (1994) 55:380–8. doi: 10.1002/jcb.240550316
192. Raffetseder U, Roper D, Mery L, Gietz C, Klos A, Grotzinger J, et al. Site-directed mutagenesis of conserved charged residues in the helical region of the human C5a receptor. Arg206 determines high-affinity binding sites of C5a receptor. Eur J Biochem (1996) 235:82–90. doi: 10.1111/j.1432-1033.1996.00082.x
193. Pollok-Kopp B, Hüttenrauch F, Rethorn S, Oppermann M. Dynamics of protein kinase C-mediated phosphorylation of the complement C5a receptor on serine 334. J Biol Chem (2007) 282:4345–53. doi: 10.1074/jbc.M601317200
194. Bock D, Martin U, Gärtner S, Rheinheimer C, Raffetseder U, Arseniev L, et al. The C terminus of the human C5a receptor (CD88) is required for normal ligand-dependent receptor internalization. Eur J Immunol (1997) 27:1522–9. doi: 10.1002/eji.1830270631
195. Christophe T, Rabiet M-J, Tardif M, Milcent M-D, Boulay F. Human complement 5a (C5a) anaphylatoxin receptor (CD88) phosphorylation sites and their specific role in receptor phosphorylation and attenuation of G protein-mediated responses. J Biol Chem (2000) 275:1656–64. doi: 10.1074/jbc.275.3.1656
196. Tardif M, Mery L, Brouchon L, Boulay F. Agonist-dependent phosphorylation of N-formylpeptide and activation peptide from the fifth component of C (C5a) chemoattractant receptors in differentiated HL60 cells. J Immunol Baltim Md 1950 (1993) 150:3534–45. doi: 10.1002/(SICI)1521-4141(199909)29:09<3035::AID-IMMU3035>3.0.CO;2-Z
197. Langkabel P, Zwirner J, Oppermann M. Ligand-induced phosphorylation of anaphylatoxin receptors C3aR and C5aR is mediated by G protein-coupled receptor kinases. Eur J Immunol (1999) 29:3035–46. doi: 10.1002/(SICI)1521-4141(199909)29:09<3035::AID-IMMU3035>3.0.CO;2-Z
198. Suvorova ES, Gripentrog JM, Oppermann M, Miettinen HM. Role of the carboxyl terminal di-leucine in phosphorylation and internalization of C5a receptor. Biochim Biophys Acta BBA - Mol Cell Res (2008) 1783:1261–70. doi: 10.1016/j.bbamcr.2008.02.004
199. Braun L, Christophe T, Boulay F. Phosphorylation of key serine residues is required for internalization of the complement 5a (C5a) anaphylatoxin receptor via a β-arrestin, dynamin, and clathrin-dependent pathway. J Biol Chem (2003) 278:4277–85. doi: 10.1074/jbc.M210120200
200. Naik N, Giannini E, Brouchon L, Boulay F. Internalization and recycling of the C5a anaphylatoxin receptor: evidence that the agonist-mediated internalization is modulated by phosphorylation of the C-terminal domain. J Cell Sci (1997) 110:2381–90. doi: 10.1242/jcs.110.19.2381
201. Suvorova ES, Gripentrog JM, Miettinen HM. Different endocytosis pathways of the C5a receptor and the N-formyl peptide receptor: different targeting of activated C5aR vs. FPR Traffic (2005) 6:100–15. doi: 10.1111/j.1600-0854.2004.00256.x
202. Drube J, Haider RS, Matthees ESF, Reichel M, Zeiner J, Fritzwanker S, et al. GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat Commun (2022) 13:540. doi: 10.1038/s41467-022-28152-8
203. Wu K-C, Condon ND, Hill TA, Reid RC, Fairlie D, Lim J. Ras related protein Rab5a regulates complement C5a receptor trafficking, chemotaxis and chemokine secretion in human macrophages. J Innate Immun (2023) 15, (1): 468–84. doi: 10.1159/000530012
204. Tardif M, Brouchon L, Rabiet M-J, Boulay F. Direct binding of a fragment of the Wiskott-Aldrich syndrome protein to the C-terminal end of the anaphylatoxin C5a receptor. Biochem J (2003) 372:453–63. doi: 10.1042/bj20021803
205. Rabiet M-J, Huet E, Boulay F. Complement component 5a receptor oligomerization and homologous receptor down-regulation. J Biol Chem (2008) 283:31038–46. doi: 10.1074/jbc.M805260200
206. Wang C, Wu H, Katritch V, Han GW, Huang X-P, Liu W, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature (2013) 497:338–43. doi: 10.1038/nature12167
207. Pandey S, Li XX, Srivastava A, Baidya M, Kumari P, Dwivedi H, et al. Shukla AK. Partial ligand-receptor engagement yields functional bias at the human complement receptor, C5aR1. J Biol Chem (2019) 294:9416–29. doi: 10.1074/jbc.RA119.007485
208. Gorman DM, Li XX, Lee JD, Fung JN, Cui CS, Lee HS, et al. Development of potent and selective agonists for complement C5a receptor 1 with in vivo activity. J Med Chem (2021) 64:16598–608. doi: 10.1021/acs.jmedchem.1c01174
209. Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol Immunol (2000) 37:407–12. doi: 10.1016/S0161-5890(00)00067-5
210. Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry (2003) 42:9406–15. doi: 10.1021/bi034489v
211. Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-arg74. J Biol Chem (2002) 277:7165–9. doi: 10.1074/jbc.C100714200
212. Scola A-M, Higginbottom A, Partridge LJ, Reid RC, Woodruff T, Taylor SM, et al. The role of the N-terminal domain of the complement fragment receptor C5L2 in ligand binding. J Biol Chem (2007) 282:3664–71. doi: 10.1074/jbc.M609178200
213. Croker DE, Monk PN, Halai R, Kaeslin G, Schofield Z, Wu MC, et al. Discovery of functionally selective C5aR2 ligands: novel modulators of C5a signalling. Immunol Cell Biol (2016) 94:787–95. doi: 10.1038/icb.2016.43
214. Van Lith LHC, Oosterom J, Van Elsas A, Zaman GJR. C5a-stimulated recruitment of β-arrestin2 to the nonsignaling 7-transmembrane decoy receptor C5L2. SLAS Discovery (2009) 14:1067–75. doi: 10.1177/1087057109341407
215. Scola A-M, Johswich K-O, Morgan BP, Klos A, Monk PN. The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol (2009) 46:1149–62. doi: 10.1016/j.molimm.2008.11.001
216. Croker DE, Halai R, Kaeslin G, Wende E, Fehlhaber B, Klos A, et al. C5a2 can modulate ERK1/2 signaling in macrophages via heteromer formation with C5a1 and β-arrestin recruitment. Immunol Cell Biol (2014) 92:631–9. doi: 10.1038/icb.2014.32
217. Li XX, Clark RJ, Woodruff TM. C5aR2 activation broadly modulates the signaling and function of primary human macrophages. J Immunol (2020) 205:1102–12. doi: 10.4049/jimmunol.2000407
218. Ames RS, Li Y, Sarau HM, Nuthulaganti P, Foley JJ, Ellis C, et al. Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem (1996) 271:20231–4. doi: 10.1074/jbc.271.34.20231
219. Crass T, Raffetseder U, Martin U, Grove M, Klos A, Köhl J, et al. Expression cloning of the human C3a anaphylatoxin receptor (C3aR) from differentiated U-937 cells. Eur J Immunol (1996) 26:1944–50. doi: 10.1002/eji.1830260840
220. Gao J, Choe H, Bota D, Wright PL, Gerard C, Gerard NP. Sulfation of tyrosine 174 in the human C3a receptor is essential for binding of C3a anaphylatoxin. J Biol Chem (2003) 278:37902–8. doi: 10.1074/jbc.M306061200
221. Halim A, Rüetschi U, Larson G, Nilsson J. LC–MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res (2013) 12:573–84. doi: 10.1021/pr300963h
222. Chao T-H, Ember JA, Wang M, Bayon Y, Hugli TE, Ye RD. Role of the second extracellular loop of human C3a receptor in agonist binding and receptor function. J Biol Chem (1999) 274:9721–8. doi: 10.1074/jbc.274.14.9721
223. Wilken H-C, Götze O, Werfel T, Zwirner J. C3a(desArg) does not bind to and signal through the human C3a receptor. Immunol Lett (1999) 67:141–5. doi: 10.1016/S0165-2478(99)00002-4
224. Crass T, Ames RS, Sarau HM, Tornetta MA, Foley JJ, Köhl J, et al. Chimeric receptors of the human C3a receptor and C5a receptor (CD88). J Biol Chem (1999) 274:8367–70. doi: 10.1074/jbc.274.13.8367
225. Guo Q, Subramanian H, Gupta K, Ali H. Regulation of C3a receptor signaling in human mast cells by G protein coupled receptor kinases. PloS One (2011) 6:e22559. doi: 10.1371/journal.pone.0022559
226. Gupta K, Subramanian H, Klos A, Ali H. Phosphorylation of C3a receptor at multiple sites mediates desensitization, β-arrestin-2 recruitment and inhibition of NF-κB activity in mast cells. PloS One (2012) 7:e46369. doi: 10.1371/journal.pone.0046369
227. Settmacher B, Rheinheimer C, Hamacher H, Ames RS, Wise A, Jenkinson L, et al. Structure-function studies of the C3a-receptor: C-terminal serine and threonine residues which influence receptor internalization and signaling. Eur J Immunol (2003) 33:920–7. doi: 10.1002/eji.200323293
228. Ahamed J, Haribabu B, Ali H. Cutting edge: differential regulation of chemoattractant receptor-induced degranulation and chemokine production by receptor phosphorylation. J Immunol (2001) 167:3559–63. doi: 10.4049/jimmunol.167.7.3559
229. Vibhuti A, Gupta K, Subramanian H, Guo Q, Ali H. Distinct and shared roles of β-arrestin-1 and β-arrestin-2 on the regulation of C3a receptor signaling in human mast cells. PloS One (2011) 6:e19585. doi: 10.1371/journal.pone.0019585
230. Ramirez MD, Pairett AN, Pankey MS, Serb JM, Speiser DI, Swafford AJ, et al. The last common ancestor of most bilaterian animals possessed at least nine opsins. Genome Biol Evol (2016) 8:3640–52. doi: 10.1093/gbe/evw248
231. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev (2016) 274:9–15. doi: 10.1111/imr.12474
232. Nonaka M. Evolution of the complement system. Subcell Biochem (2014) 80:31–43. doi: 10.1007/978-94-017-8881-6_3
233. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
234. Fernández FJ, Vega MC. Technologies to keep an eye on: alternative hosts for protein production in structural biology. Curr Opin Struct Biol (2013) 23:365–73. doi: 10.1016/j.sbi.2013.02.002
235. Fernández FJ, Vega MC. Choose a suitable expression host: A survey of available protein production platforms. In: Vega MC, editor. Advanced Technologies for Protein Complex Production and Characterization. Advances in Experimental Medicine and Biology. Cham: Springer International Publishing (2016). p. 15–24. doi: 10.1007/978-3-319-27216-0_2
236. García-Nafría J, Tate CG. Cryo-electron microscopy: moving beyond X-ray crystal structures for drug receptors and drug development. Annu Rev Pharmacol Toxicol (2020) 60:51–71. doi: 10.1146/annurev-pharmtox-010919-023545
237. Ito Y, Selenko P. Cellular structural biology. Curr Opin Struct Biol (2010) 20:640–8. doi: 10.1016/j.sbi.2010.07.006
238. Querol-García J, Fernández FJ, Marin AV, Gómez S, Fullà D, Melchor-Tafur C, et al. Crystal structure of glyceraldehyde-3-phosphate dehydrogenase from the gram-positive bacterial pathogen A. vaginae, an immunoevasive factor that interacts with the human C5a anaphylatoxin. Front Microbiol (2017) 8:541. doi: 10.3389/fmicb.2017.00541
239. Gómez S, Querol-García J, Sánchez-Barrón G, Subias M, González-Alsina À, Franco-Hidalgo V, et al. The antimicrobials anacardic acid and curcumin are not-competitive inhibitors of gram-positive bacterial pathogenic glyceraldehyde-3-phosphate dehydrogenase by a mechanism unrelated to human C5a anaphylatoxin binding. Front Microbiol (2019) 10:326. doi: 10.3389/fmicb.2019.00326
240. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6:132–42. doi: 10.1038/nrmicro1824
Keywords: complement, complement receptors, structural biology, CR1/CR2, CR3/CR4, CRIg, C5aR1/C5L2/C3aR, host-pathogen interactions
Citation: Santos-López J, de la Paz K, Fernández FJ and Vega MC (2023) Structural biology of complement receptors. Front. Immunol. 14:1239146. doi: 10.3389/fimmu.2023.1239146
Received: 12 June 2023; Accepted: 16 August 2023;
Published: 11 September 2023.
Edited by:
Hyun Ho Park, Chung-Ang University, Republic of KoreaReviewed by:
Laure Yatime, UMR5235 Laboratoire des Interactions Pathogène Hôte (LPHI), FranceBrandon L. Garcia, East Carolina University, United States
Copyright © 2023 Santos-López, de la Paz, Fernández and Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco J. Fernández, ZmpmZXJuYW5kZXpAYWJ2YW5jZS5jb20=; M. Cristina Vega, Y3ZlZ2FAY2liLmNzaWMuZXM=
†These authors have contributed equally to this work