 Salma A. Rizo-Téllez
Salma A. Rizo-Téllez Meriem Sekheri
Meriem Sekheri János G. Filep
János G. Filep
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 26 July 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1237729
This article is part of the Research TopicBiology of C-reactive ProteinView all 17 articles
C-reactive protein (CRP) is well-recognized as a sensitive biomarker of inflammation. Association of elevations in plasma/serum CRP level with disease state has received considerable attention, even though CRP is not a specific indicator of a single disease state. Circulating CRP levels have been monitored with a varying degree of success to gauge disease severity or to predict disease progression and outcome. Elevations in CRP level have been implicated as a useful marker to identify patients at risk for cardiovascular disease and certain cancers, and to guide therapy in a context-dependent manner. Since even strong associations do not establish causality, the pathogenic role of CRP has often been over-interpreted. CRP functions as an important modulator of host defense against bacterial infection, tissue injury and autoimmunity. CRP exists in conformationally distinct forms, which exhibit distinct functional properties and help explaining the diverse, often contradictory effects attributed to CRP. In particular, dissociation of native pentameric CRP into its subunits, monomeric CRP, unmasks “hidden” pro-inflammatory activities in pentameric CRP. Here, we review recent advances in CRP targeting strategies, therapeutic lowering of circulating CRP level and development of CRP antagonists, and a conformation change inhibitor in particular. We will also discuss their therapeutic potential in mitigating the deleterious actions attributed to CRP under various pathologies, including cardiovascular, pulmonary and autoimmune diseases and cancer.
The prototypic acute-phase reactant C-reactive protein (CRP), discovered as a protein that precipitates C-polysaccharide of Streptococcus pneumoniae, has long been recognized as a sensitive biomarker of inflammation (1). Elevations in baseline serum CRP level have been detected in numerous pathologies, and have been suggested to being useful to monitor disease progression. CRP has received considerable attention as a diagnostic and prognostic marker in autoimmune diseases (2, 3), cardiovascular diseases (4–6), chronic kidney disease (7), cancer (8) and COVID-19 (9, 10) as well as for guiding therapy (8, 11). Although there is a continuing debate over whether CRP is primarily a passive indicator of inflammation or is a “culprit” mediating disease (12–18), CRP plays important roles in host defense against invading pathogens, autoimmunity and inflammation. CRP exhibits many, often conflicting pro- and anti-inflammatory activities (14, 19–21), which makes delineating its pathogenetic roles even more challenging. Results from structure-function studies challenge the long-held and rather simplistic view of CRP as a stable pentameric protein and identified conformationally distinct forms, including native pentameric CRP (pCRP) and modified/monomeric CRP (mCRP), which exhibit distinct functional properties and may explain many of the opposing biological activities attributed to CRP (21, 22). CRP synthesis, structure and biological activities have been reviewed in detail elsewhere (19, 21, 23–25). In this review, we aimed to critically assess the competing views on the role of CRP isomers in disease pathogenesis and therapy, focusing on recent advances that may provide a rationale basis for guiding therapy and/or therapeutic targeting of CRP isomers to limit inflammation underlying various diseases.
Native CRP is member of the pentraxin family, an evolutionarily highly conserved class of pattern recognition molecules. The human CRP gene, located on chromosome 1, q23-q24 encodes for a CRP subunit of a single polypeptide chain of 206 amino acids (23). CRP is composed of five identical non-covalently bound subunits forming a planar ring with a central pore (23). The two opposite faces of the pentamer, and thus each protomer, have distinct binding properties. The A-face binds and activates complement C1q, whereas the B-face contains the Ca2+-dependent binding pocket for phosphocholine, expressed by bacterial, fungal and eukaryotic cells (24, 26). CRP also binds to nuclear antigens, the oxidized LDL receptor, apoptotic cell membrane, glycan components of microorganisms, and many other ligands (24, 27, 28), though some of these interactions have been disputed.
Native CRP is predominantly synthesized in hepatocytes under transcriptional control by cytokines (IL-6 and to a lesser extent IL-1β and TNF-α), the transcription factors hepatic nuclear factor (HNF) 1α and HNF3 as part of the “reorchestration” of hepatic gene expression in response to infection or tissue injury (19), promoter methylation and a distal enhancer (29). Hepatic secretion of pCRP accounts for rapid increases in serum pCRP levels during the acute-phase reaction. The serum half-life of pCRP is about 19 h under both physiological and pathological conditions (30), thus directly reflecting the rate of its hepatic synthesis. Ethnicity, sex and polymorphism in the apoliprotein E and CRP genes are known to influence baseline serum pCRP levels in humans (31, 32). CRP gene polymorphism influences gene expression and may predispose to systemic lupus erythematosus (31), but do not appear to be associated with increased risk for cardiovascular diseases (33, 34). Additionally, the kidney has been reported as a second site of pCRP formation in humans (35). Expression of CRP mRNA and pCRP synthesis has also been detected in the diseased vessel wall, coronary artery bypass grafts and neurons (35–37). The contribution of these sites to circulating pCRP remains to be investigated.
Under physiological conditions, pCRP appears to exist in a NaCl concentration-dependent pentamer-decamer equilibrium (38). Another form of CRP, characterized by multiple-size lettuce-like structures of about 300-500kDa, were detected in the serum of obese patients (39). The origin and pathological significance of these CRP forms are not known.
Although not specific for a single disease process, CRP is commonly used as a static measurement and CRP levels have been correlated with disease activity and to some degree, severity and prognosis in several diseases. CRP has been promoted as an independent predictor of cardiovascular events and metabolic syndrome (40–42), though the association is considerably weaker than previously thought (43, 44). The data from Mendelian randomization studies (33, 34) coupled with animal studies with injection of human pCRP and transgenic mice over-expressing human CRP may support an association between CRP and cardiovascular disease, but provide no direct evidence for a causative role for pCRP. In the Dallas Heart study hs-CRP was not independently associated with atherosclerotic burden in the coronary artery and abdominal aorta (45), whereas the REVERSAL trial reported that lower hs-CRP levels were independently and significantly correlated with atherosclerosis progression (46). Other studies have concluded that hs-CRP likely serves as a biomarker of vascular inflammation underlying atherosclerosis (14, 44). Thus, whereas the potential of therapeutic targeting of pCRP in cardiovascular disease remains unresolved, hs-CRP has clinical usefulness in guiding therapy as discussed below.
Other clinical studies reported positive correlation between elevated plasma CRP levels and myocardial infarct size (47), reduced lung function in chronic obstructive pulmonary disease (48) or the severity of COVID-19-evoked respiratory distress (49, 50). Higher plasma CRP levels were found to predict flares in systemic lupus erythematosus (2) and to portend poor prognosis in melanoma (51). Patients with non-small cell lung cancer who received the immune checkpoint PD-1 inhibitor nivolumab, early increases in hs-CRP and IL-6 were predictive for the efficacy of treatment (52). Nivolumab evoked a CRP flare-response (defined as a rapid, more than twofold increase in CRP levels followed by decrease below baseline values within 3 months) in about 25% of patients with metastatic renal cell carcinoma and this was associated with significant tumor shrinkage and improved 1-year survival rate (53).
The association of CRP with prognosis should, however, be interpreted with caution as an indication of direct causal contribution of CRP to disease pathogenesis. A definitive way to test this is the use of compounds that specifically block binding of CRP to its ligands and/or receptors to assess its pro-inflammatory effects in vivo.
While pCRP has been postulated to be stable under physiological conditions (24), compelling evidence indicates that CRP exists in conformationally distinct forms and conformational changes in pCRP results in expression of potent pro-inflammatory activities (Figure 1) (21). The mild acidic environment within inflamed tissues confers pCRP binding specificities for factor H (55) and conformationally altered proteins, such as oxidized LDL and complement C3b (56) that do not bind to pCRP at physiological pH. Binding of circulating pCRP to phosphocholine or phosphoethanolamine head groups of membrane lipids expressed on the surface of activated platelets or apoptotic cells induces the formation of a partially dissociated pentamer (pCRP*), which then dissociates into the monomeric subunits, mCRP (57–59). pCRP* and mCRP exhibit potent pro-inflammatory activities, including stimulation of IL-8 secretion from neutrophils and human coronary artery endothelial cells (60, 61), promote neutrophil adhesion to platelets and endothelial cells (62, 63), delay neutrophil apoptosis (64) and trigger extrusion of neutrophil extracellular traps (65), characteristic features of the inflammatory response. pCRP* binds and activate complement C1q (66), which, in turn, can amplify pre-existing inflammation and tissue damage (21, 57, 67). Accumulation of pro-inflammatory CRP isoforms with in inflamed/injured but not in healthy tissues, and local expression of mCRP, for example within arteriosclerotic plaques (68) and in circulating microparticles (69, 70) would ensure localization of inflammation (57) and precipitate tissue injury (21).
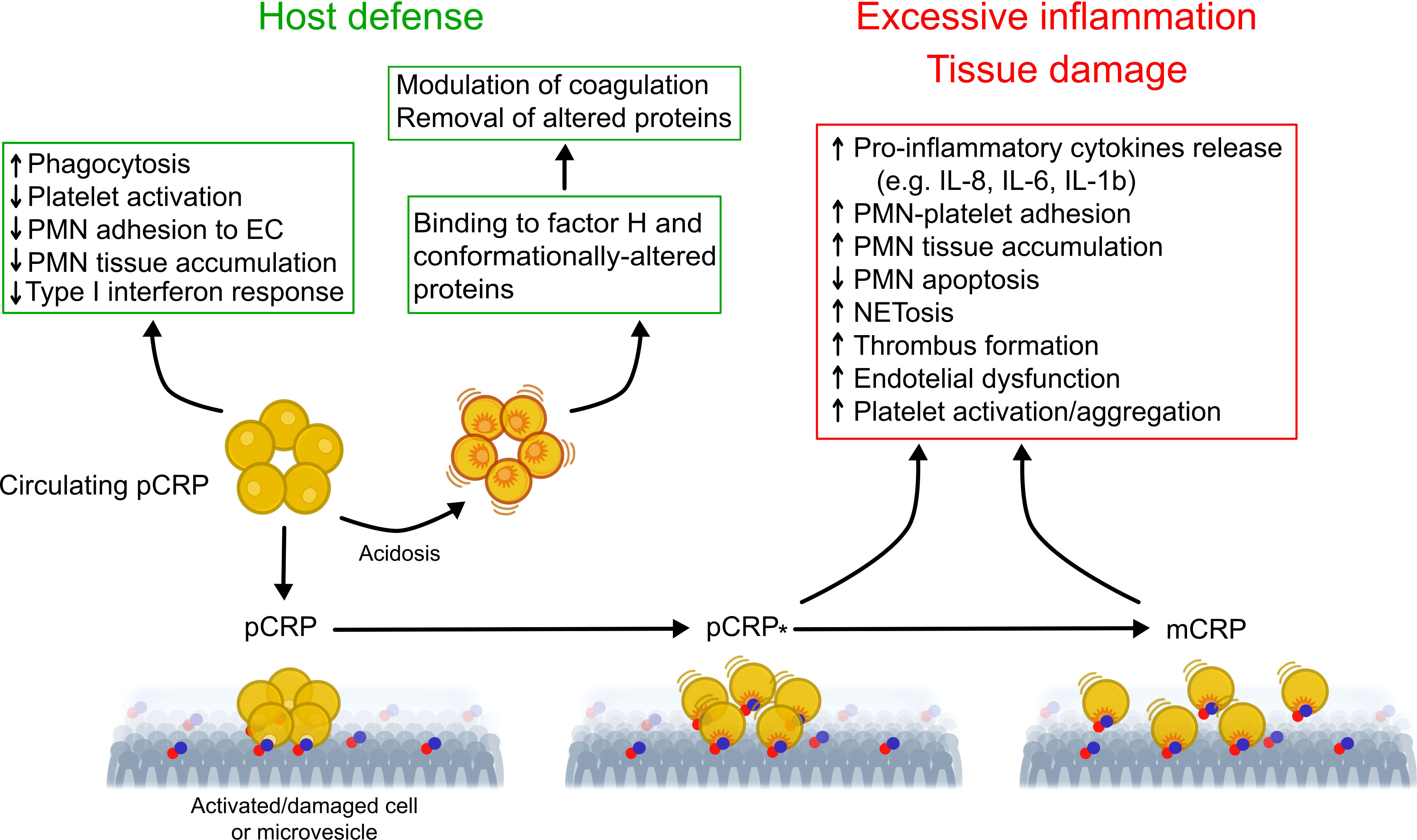
Figure 1 Proposed model for regulation of the inflammatory response by conformationally distinct CRP isomers. In the circulation, native CRP exists in a disc-shaped pentameric form (pCRP) and exhibits predominantly anti-inflammatory activities (e.g. opsonization) that are critical for clearance of invading bacteria and damaged cells by phagocytes. Thus, pCRP may limit further damage and prevent autoimmunity. Mild acidosis within the inflammatory locus unmasks additional binding sites in pCRP for complement factor H, which modulate coagulation and for conformationally altered proteins, thereby promoting their removal. Binding of pCRP to phosphocholine or phosphoethanolamine head groups exposed on the surface of activated or damaged cells, or microvesicles, leads to formation of a partially dissociated pentamer (pCRP*) followed by dissociation into its monomeric subunits (mCRP). Unlike pCRP, pCRP*/mCRP exhibit potent pro-inflammatory actions, including stimulation of thrombus formation, activation of endothelial cells, monocytes, platelets and neutrophils, neutrophil and monocyte adhesion to endothelial cells, enhanced formation of neutrophil-platelet and platelet-monocyte aggregates, production of pro-inflammatory cytokines IL-1β and IL-6, and extrusion of neutrophil extracellular traps (NET). These may contribute to excessive, non-resolving inflammation and to aggravation of tissue injury. This multistep mechanism would uncouple the local effects of pCRP*/mCRP from those of circulating pCRP, therefore contribute to localization of inflammation. Of note, microvesicle-associtaed pCRP*/mCRP may contribute spreading the inflammatory reaction to distant sites. EC, endothelial cells; mCRP, monomeric CRP; NETosis, extrusion of neutrophil extracellular traps; pCRP, pentameric CRP; pCRP*, partially dissociated pentameric CRP; PMN, polymorphonuclear neutrophil granulocytes. Modified from Wu et al. (21) and Filep (54).
While purified human pCRP itself does not evoke inflammation when injected into healthy individuals (71), it can amplify tissue injury in animal models, induce the expression of adhesion molecules and production of IL-6 and IL-8 (24, 72–75). Caution should be exercised in interpreting these observations because many of these effects can be attributed to contaminants (endotoxin or the preservative sodium azide) in commercial CRP preparations not to pCRP itself (13, 21). By contrast, other studies documented pCRP protection against the assembly of the terminal complement attack complex (27) and bacterial sepsis (76), reversal of proteinuria in autoimmune mice (77), and prevention of autoimmunity (78). Recently, pCRP was found to reduce immune complex-triggered type I interferon response, consistent with a protective action in systemic lupus erythematosus (79). This action was lost in mCRP, further highlighting the complexity of regulation of immune response by CRP isomers in autoimmune diseases.
CRP isomers bind to distinct receptors. Thus, pCRP binds primarily to the low affinity Ig receptor FcγRIIa (CD32) and to some extent to the high affinity IgG receptor FCγRI (CD64) on phagocytes and endothelial cells (21, 80), and αIIbβ3integrin on platelets (81), whereas mCRP binds to FcγRIIIb (CD64) on phagocytes and lipid rafts on human endothelial cells (82).
The concept of activation-induced conformational changes could explain why pCRP itself is not pro-inflammatory in the absence of infection or tissue injury. Conformational changes in pCRP, and generation of pCRP* and mCRP, would unmask “hidden” pro-inflammatory activities that may collectively amplify the initial inflammatory signal evoked by infection or tissue injury, leading to exacerbation of tissue damage and more severe disease (21, 73). However, further studies are needed to explore the clinical importance of mCRP or other CRP conformers.
The association of persisting modest elevations in plasma CRP level (detected by high sensitivity assays) with chronic diseases has attracted considerable clinical interest and often contradictory interpretations. CRP is generally recognized as a biomarker of ongoing inflammation and to a varying degree as a predictor of clinical outcome. Although conclusive evidence for a causal role for pCRP (and/or CRP isomers) is still lacking, lowering pCRP levels is widely anticipated to reduce the adverse effects attributed to CRP. Indeed, several approaches have been developed and tested for lowering plasma pCRP level or countering CRP`s actions (Table 1).
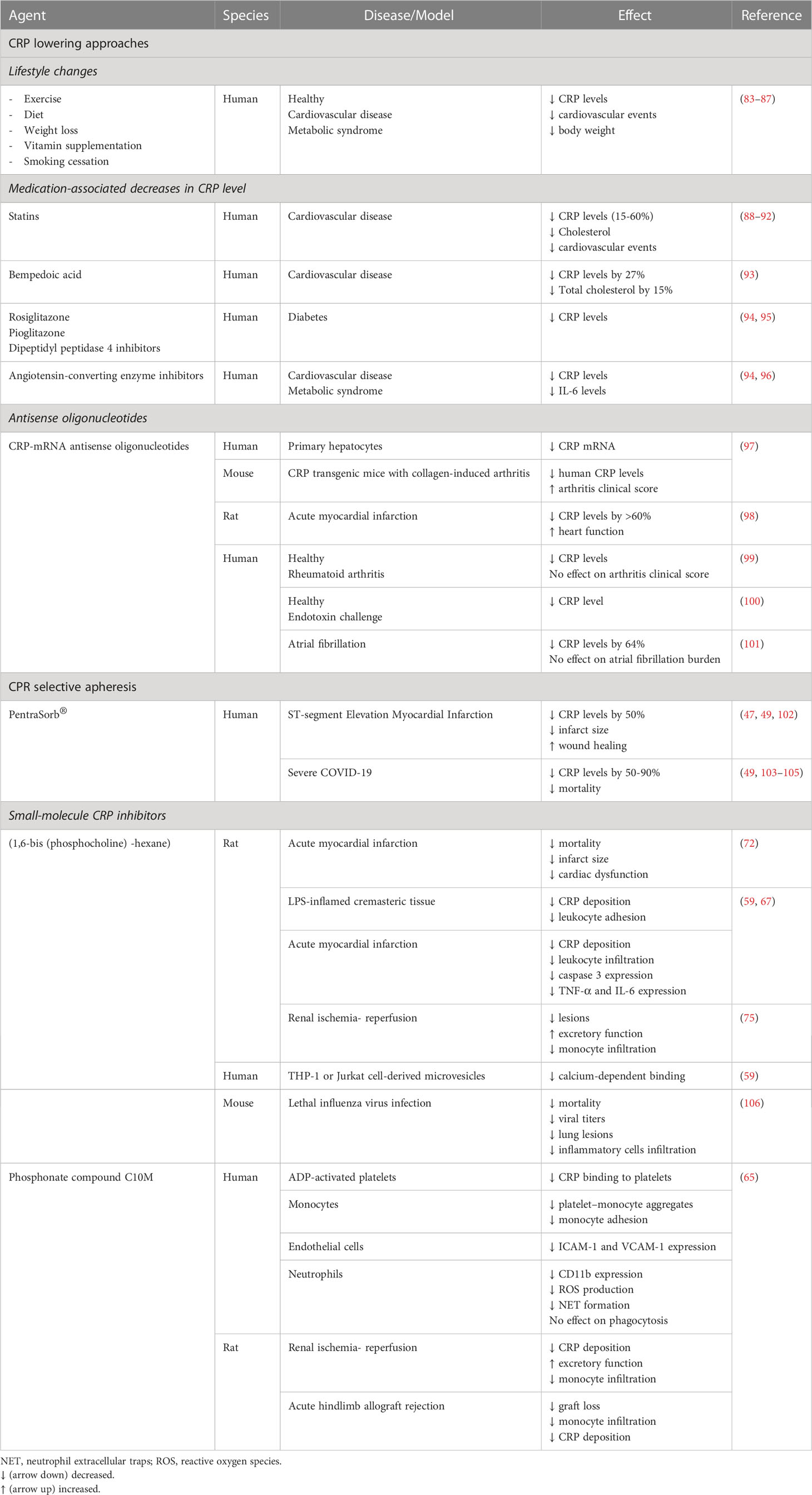
Table 1 CRP targeting strategies.
Observational studies reported a relationship between life style changes, encouraging cessation of smoking, weight loss, more physical activity and Mediterranean diet, with concurrent reduction in hs-CRP levels and the risk for future cardiovascular events (83–86). Studies with angiotensin converting enzyme inhibitors, angiotensin II (type I) receptor blockers, vitamins E and C, and the anti-platelet agents clopidogrel and aspirin, yielded conflicting results in regard with their efficiency to lowering hs-CRP levels (94, 107). Unlike insulin, the anti-diabetic drugs rosiglitazone and pioglitazone have been found to significantly decrease serum CRP, though the molecular mechanisms and the potential clinical benefits remain largely unknown (94).
Evidence derived mainly from trials with statins support the potential value of hs-CRP for primary and secondary prevention of cardiovascular disease, though this notion still remains controversial. The landmark JUPITER trial demonstrated the benefits of rosuvastatin therapy in primary prevention as well as the utility of hs-CRP for identifying a population at risk for cardiovascular disease (88–90). A new meta-analysis from the PROMINENT, REDUCE-IT and STRENGTH trials (which were originally designed to test triglyceride-lowering) showed that inflammation, and thus hs-CRP is more tightly linked than LDL cholesterol to future adverse effects in patients already on statins (91). Limitations of this analysis include the effects of confounding bias (e.g. high intensity statin use and diabetes) and lack of attention to primary versus secondary prevention (108). These concerns notwithstanding, the meta-analysis would argue for routine hs-CRP testing to assess residual inflammatory risk (109) and a combined approach to aggressive lipid-lowering and inflammation-inhibiting therapy with colchicine, IL-1 or IL-6 inhibitors or bempedoic acid (91, 93).
Another approach to reduce CRP production is the use of antisense oligonucleotides (ASO) to specifically inhibit mRNA translation in particular in the liver, the predominant site of pCRP synthesis (23) where ASOs have a propensity to accumulate (110). Lowering plasma CRP level with rat-specific ASO ISIS 197178 was associated with reduction of infarct size and improved cardiac function in a rat model of myocardial infarction (111). Human-specific ASO ISIS 353512 reduced neointima formation in human CRP transgenic mice subjected to carotid artery ligation (98). In healthy subjects, ASO ISIS 329993 (ISIS-CRPRx) reduced by about 70% of the peak plasma CRP response to endotoxin challenge without affecting cytokine production and coagulation (100). Treatment with ISIS-CRPRx of patients with rheumatoid arthritis also decreased plasma CRP level, but did not reduce disease activity (99). Unexpectedly, another ASO, ISIS 353512 increased IL-6 and CRP levels in healthy volunteers (112), illustrating the challenges with CRP-ASO therapy.
Another strategy to investigate pathogenetic roles for pCRP (and arguably other CRP isomers) is reducing plasma pCRP level by selective apheresis, which appears to be a relatively simple, efficient and clinically safe approach (49, 113). In this protocol, patients are subjected to a 4-6 h extracorporeal circuit and blood plasma is applied to phosphocholine-linked resin. A case report and studies on small cohorts of patients with ST elevation myocardial infarction (STEMI) demonstrated efficient lowering of plasma CRP levels (47, 102, 114, 115). However, results from the multi-center pilot CAMI-1 (CRP Apheresis in Acute Myocardial Infarction-1) study were inconclusive in regard with correlation of reduced CRP levels with myocardial infarct size (47) because the study was not randomized. The ongoing trials on the effects of CRP apheresis on the course of STEMI (NCT04939805) and ischemic stroke (CASTRO-B, NCT03884153) are anticipated to address this issue. There are several case reports with mixed results on reducing lung injury with CRP apheresis in patients with COVID-19-evoked respiratory distress syndrome (49, 103–105). The subsequently planned trial on pulmonary, myocardial and/or renal injury in COVID-19 (NCT04898062) has been withdrawn [https://www.clinical.trials.gov, as of June 4, 2023].
Similar to other CRP lowering strategies, the fundamental question whether the beneficial effects can be attributed to lowering pCRP level directly or to reduction of formation of conformationally altered CRP secondary to reduced availability of the parent molecule pCRP remains unanswered. While short-term reductions of plasma CRP levels may be beneficial under certain circumstances, markedly lower CRP levels over prolonged periods of time may impair antimicrobial defense, and thus augmenting the risk of bacterial or viral infection. Whether this would limit the clinical utility of CRP apheresis and what plasma CRP levels after CRP apheresis will be still sufficient to support innate defense functions remain to be investigated.
Two distinct CRP targeting strategies have been developed. In 2006, the Pepys group has designed and synthesized the first small-molecule inhibitor of CRP (72). The bivalent compound, 1,6-bis(phosphocholine)-hexane (bis-PC) bridges the phosphocholine-binding pockets on the B-face of two separate CRP pentamers, bringing the phosphocholine-binding surfaces together in a parallel fashion (72). The resulting decamer structure stabilizes conformation of pCRP and prevents binding of other ligands to the B-face. Bis-PC has also been reported to inhibit dissociation of pCRP to mCRP on the surface of circulating microparticles isolated from the blood of patients with myocardial infarction (69). Pretreatment with bis-PC abolished the increase in infarct size and cardiac dysfunction produced by injection of human pCRP in a rat model of myocardial infarction (72). A controversy exists whether rat CRP can activate rat complement and whether rat factor H, the native complement-control protein, could interact with human CRP (116, 117). Hence, the translational implication of these observations remains to be clarified. Binding of CRP decamers to Fcγ receptors or possible deposition of large CRP complexes might trigger immune reactions, thereby limiting the therapeutic use of bis-PC. This compound is apparently no longer being considered for clinical development [http://pentraxin.word-press.com/rd-programs/].
Considering the role of native CRP in host defense, therapies aimed at reducing serum pCRP level would likely impair defense mechanisms and predispose to infection. Thus, an attractive alternative approach is to selectively block expression of pro-inflammatory properties “hidden” in pCRP without interfering with its protective functions. As a proof of this concept, Zeller et al. (65) developed a low molecular weight monovalent compound C10M [3-(dibutyl amino)propyl) phosphonic acid]. C10M binds to the phosphocholine binding pocket of pCRP and prevents pCRP binding to phosphocholine residues exposed on the surface of activated or damaged cells or microvesicles, and subsequently the formation of pCRP*/mCRP (65). Apart from the occupied phosphocholine binding pocket, the B-face remains accessible to other ligands, including misfolded or aggregated proteins or proteins whose secondary structure is predominantly β-sheet (56) as well as neutrophils. Consistently, C10M inhibited pCRP*/mCRP-stimulated activation of monocytes and neutrophils, extrusion of neutrophil extracellular traps (NETs), monocyte adhesion to activated endothelial cells, and formation of platelet-monocyte aggregates (65). C10M reduction of pCRP*/mCRP-triggered, presumably ROS-dependent NET release (i.e. via the suicidal pathway) could contribute to preventing NET-mediated tissue damage under pathological conditions (54, 118). Importantly, C10M did not impair antibacterial host defense as evidenced by unaltered pCRP opsonization-mediated phagocytosis of bacteria by monocytes and neutrophils (65). Furthermore, C10M efficiently suppressed tissue deposition of human pCRP*/mCRP and monocyte accumulation within the affected organs in murine models of renal ischemia-reperfusion and allograft rejection of hindlimb transplants (65). Consistently, C10M significantly improved renal function following ischemia-reperfusion, and prevented premature loss of allograft transplants driven by human pCRP. While these findings would indicate the functional importance of mCRP, further studies are needed to distinguish the effects of CRP antagonists on endogenous CRP and injected human CRP in these and other experimental models.
CRP is a well-established biomarker of inflammation and much written about its association with disease state. Circulating hs-CRP may identify patients at risk, predict disease progression and outcome, and guide therapy in a context-dependent manner. Nevertheless, since even strong associations do not establish causality, further studies are clearly warranted to elucidate its potential pathogenic roles. Activation-induced conformational changes in pCRP would unmask “hidden” pro-inflammatory properties as opposed to the largely protective role of pCRP. CRP lowering strategies yielded promising, but often inconclusive data on altering disease progression. Development of CRP antagonists, and in particular recent development of a phosphocholine mimetic that binds to pCRP and inhibits conformation change-mediated expression of pro-inflammatory activities without impairing pCRP’s defense function, should facilitate future investigations into the full spectrum of the roles of CRP isomers in inflammatory pathologies. This approach has the potential of opening a novel therapeutic avenue for preventing or limiting the deleterious actions attributed to CRP.
All authors contributed to the article and approved the submitted work.
The authors apologize to those experts whose articles have not been cited due to space limitations. This work was supported by a grant from the Canadian Institutes of Health Research (PJT-169075 to JF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340(6):448–54. doi: 10.1056/NEJM199902113400607.1999
2. Gaitonde S, Samols D, Kushner I. C-reactive protein and systemic lupus erythematosus. Arth Rheum (2008) 59(12):1814–20. doi: 10.1002/art.24316
3. Pope JE, Choy EH. C-reactive protein and implications in rheumatoid arthritis and associated comorbidities. . Semin Arthritis Rheum (2021) 51(1):219–29. doi: 10.1016/j.semarthrit.2020.11.005
4. Tsimikas S, Willerson JT. Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol (2006) 47(8 Suppl):C19–31. doi: 10.1016/j.jacc.2005.10.066
5. Casas JP, Shah T, Hingorani AD, Danesh J, Pepys MB. C-reactive protein and coronary heart disease: a critical review. J Intern Med (2008) 264(4):295–314. doi: 10.1111/j.1365-2796.2008.02015.x
6. Kushner I, Elyan M. Why does C-reactive protein predict coronary events? Am J Med (2008) 121(7):e11. doi: 10.1016/j.amjmed.2008.03.004
7. Menon V, Greene T, Wang X, Pereira AA, Marcovina SM, Beck GJ, et al. C-reactive protein and albumin as predictors of all-cause and cardiovascular mortality in chronic kidney disease. Kidney Int (2005) 68(2):766–72. doi: 10.1111/j.1523-1755.2005.00455.x
8. Allin KH, Nordestgaard BG. Elevated C-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit Rev Clin Lab Sci (2011) 48(4):155–70. doi: 10.3109/10408363.2011.599831
9. Luo X, Zhou W, Yan X, Guo T, Wang B, Xia H, et al. Prognostic value of C-reactive protein in patients with COVID-19. Clin Infect Dis (2020) 71(16):2174–9. doi: 10.1093/cid/ciaa641
10. Smilowitz NR, Kunichoff D, Garshick M, Shah B, Pillinger M, Hochman JS, et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur J Heart (2021) 42:2270–9. doi: 10.1093/eurheartj/ehaa1103
11. Koenig W. High-sensitivity C-reactive protein and atherosclerotic disease: from improved risk prediction to risk-guided therapy. Int J Cardiol (2013) 168(5):5126–134. doi: 10.1016/j.ijcard.2013.07.113
12. Bisoendial RJ, Kastelein JJ, Levels JH, Zwaginga JJ, van den Bogaard B, Reitsma PH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res (2005) 96(7):714–6. doi: 10.1161/01.RES.0000163015.67711.AB
13. Lowe GDO, Pepys MB. C-reactive protein and cardiovascular disease: weighing the evidence. Curr Atheroscler Rep (2006) 8(5):421–8. doi: 10.1007/s11883-006-0040-x
14. Scirica BM, Morrow DA. Is C-reactive protein an innocent bystander of proatherogenic culprit? Circulation (2006) 113(17):2128–34. doi: 10.1161/CIRCULATIONAHA.105.611350
15. Verma S, Devaraj S, Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation (2006) 113(17):2135–50. doi: 10.1161/CIRCULATIONAHA.105.611350
16. McGrath EE, Memon SS, Anderson PB. C-reactive protein – marker of inflammation or future therapeutic target? Am J Respir Crit Care Med (2007) 176(6):624. doi: 10.1164/ajrccm.176.6.624
17. Yousuf O, Mohanty BD, Martin SS, Joshi PH, Blaha MJ, Nasir K, et al. High-sensitivity C-reactive protein and cardiovascular disease. A resolute belief or an elusive link? J Am Coll Cardiol (2013) 62(5):397–408. doi: 10.1016/j.jacc.2013.05.016
18. Zimmerman MA, Selzman CH, Cothre C, Sorensen AC, Raeburn CD, Harken AH. Diagnostic implications of C-reactive protein. Arch Surg (2003) 138:220–4. doi: 10.1001/archsurg.138.2.220
19. Black S, Kushner I, Samols D. C-reactive protein. J Biol Chem (2004) 279(47):48487–90. doi: 10.1074/jbc.R400025200
20. Kushner I, Agrawal A. CRP can play both pro-inflammatory and anti-inflammatory roles. Mol Immunol (2007) 44(4):670–1. doi: 10.1016/j.molimm.2006.02.001
21. Wu Y, Potempa LA, El Kebir D, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem (2015) 396(11):1181–97. doi: 10.1515/hsz-2015-0149
22. Ullah N, Wu Y. Regulation of conformational changes in C-reactive protein alters its bioactivity. Cell Biochem Biophys (2022) 80(4):595–608. doi: 10.1007/s12013-022-01089-x
23. Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol (2001) 38(2-3):189–97. doi: 10.1016/S0161-5890(01)00042-6
24. Pepys MB, Hirschfield GM. C-reactive protein: A critical update. J Clin Invest (2003) 111(12):1805–12. doi: 10.1172/JCI18921
25. Kushner I. C-reactive protein - My perspective on its first half century, 1930-1982. Front Immunol (2023) 14:1150103. doi: 10.3389/fimmu.2023.1150103
26. Agrawal A, Shrive AK, Greenhough TJ, Volanakis JE. Topology and structure of the C1q-binding site on C-reactive protein. J Immunol (2001) 166(6):3998–4004. doi: 10.4049/jimmunol.166.6.3998
27. Gershov D, Kim SJ, Brot N, Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from the assembly of the terminal complement components, and sustains an antiinflamamtory innate immune response. J Exp Med (2000) 192(9):1353–64. doi: 10.1084/jem.192.9.1353
28. Singh SK, Suresh MV, Prayhter DC, Moorman JP, Rusinol AE, Agrawal A. Exposing of hidden functional site of C-reactive protein by site-directed mutagenesis. J Biol Chem (2012) 287(5):3550–8. doi: 10.1074/jbc.M111.310011
29. Wielscher M, Mandaviya PR, Kuehnel B, Joehanes R, Mustafa R, Robinson O, et al. DNA methylation signature of chronic low-grade inflammation and its role in cardio-respiratory diseases. Nat Commun (2022) 13(1):2408. doi: 10.1038/s41467-022-29792-6
30. Aziz N, Fahey JL, Detels R, Butch AW. Analytical performance of a highly sensitive C-reactive protein-based immunoassay and the effects of laboratory variables on levels of protein in blood. Clin Diagn Lab Immunol (2003) 10(4):652–7. doi: 10.1128/cdli.10.4.652-657.2003
31. Russell AI, Cunninghame Graham DS, Shepherd C, Roberton CA, Whittaker J, Meeks J, et al. Polymorphism in the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet (2004) 13(1):137–47. doi: 10.1093/hmg/ddh021
32. März W, Scharnagl H, Hoffmann MM, Boehm BO, Winkelmann BR. The apolipoprotein E polymorphism is associated with circulating C-reactive protein (the Ludwigshafen risk and cardiovascular health study). Eur Heart J (2004) 25(23):2109–19. doi: 10.1016/j.ehj.2004.08.024
33. Crawford DC, Sanders CL, Qin X, Smith JD, Shephard C, Wong M, et al. Genetic variation is associated with C-reactive protein levels in the third national health and nutrition examination survey. Circulation (2006) 114(23):2458–65. doi: 10.1161/CIRCULATIONAHA.106.615740
34. Kardys I, de Maat MP, Uitterlinden AG, Hofman A, Witteman JC. C-reactive protein gene haplotypes and risk of coronary heart disease: the Rotterdam Study. Eur Heart J (2006) 27(11):1331–7. doi: 10.1093/eurheartj/ehl018
35. Jabs WJ, Logering BA, Gerke P, Kreft B, Wolber EM, Klinger MH, et al. The kidney as a second site of human C-reactive protein formation in vivo. Eur J Immunol (2003) 33(1):152–61. doi: 10.1002/immu.200390018
36. Sun H, Koike T, Ichikawa T, Hatakeyama K, Shiomi M, Zhang B, et al. C-reactive protein in atherosclerotic lesions: its origin and pathophysiological significance. Am J Pathol (2005) 167:1139–48. doi: 10.1016/S0002-9440(10)61202-3
37. Slevin M, Matou-Nasri S, Turu M, Luque A, Rovira N, Badimon L, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol (2010) 20(1):151–65. doi: 10.1111/j.1750-3639.2008.00256.x
38. Okemefuna AI, Stach L, Rana S, Buetas AJ, Gor J, Perkins SJ. C-reactive protein exists in an NaCl concentration-dependent pentamer-decamer equilibrium in physiological buffer. J Biol Chem (2010) 285(2):1041–52. doi: 10.1074/jbc.M109.044495
39. Asztalos BF, Horan MS, Horvath KV, Mcdermott AY, Chalasani NP, Schaefer EJ. Obesity associated molecular forms of C-reactive protein in human. PloS One (2014) 9(10):e109238. doi: 10.1371/journal.pone.0109238
40. Oh J, Teoh H, Leiter LA. Should C-reactive protein be a target of therapy? Diabetes Care (2011) 34(Suppl.2):S155–60. doi: 10.2337/dc11-s211
41. Burger PM, Pradhan AD, Dorresteijn JAN, Koudstaal S, Teraa M, de Borst GJ, et al. Utrecht Cardiovascular Cohort-Second Manifestations of ARTerial disease study group. C-reactive protein and risk of cardiovascular events and mortality in patients with various cardiovascular disease locations. . Am J Cardiol (2023) 197:13–23. doi: 10.1016/j.amjcard.2023.03.025
42. Dietrich M, Jialal I. The effect of weight loss on a stable biomarker of inflammation, C-reactive protein. Nutr Rev (2005) 63:22–8. doi: 10.1111/j.1753-4887.2005.tb00107.x
43. Levinson SS, Miller JJ, Elin RJ. Poor predictive value of high-sensitivity C-reactive protein indicates need for reassessment. Clin Chem (2004) 50:1733–5. doi: 10.1373/clinchem.2004.037895
44. Genest J. C-reactive protein: Risk factor, biomarker and/or therapeutic target? Can J Cardiol (2010) 26(Suppl A):41A–4A. doi: 10.1016/s0828-282x(10)71061-8
45. Khera A, De Lemos JA, Peshock RM, Lo HS, Stanek HG, Murphy SA, et al. Relationship between C-reactive protein and subclinical atherosclerosis: the Dallas Heart Study. Circulation (2006) 113(1):38–43. doi: 10.1161/CIRCULATIONAHA.105.575241
46. Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA (2004) 291(9):1071–80. doi: 10.1001/jama.291.9.1071
47. Ries W, Torzewski J, Heigl F, Pfluecke C, Kelle S, Darius H, et al. C-reactive protein apheresis as anti-inflammatory therapy in acute myocardial infarction: Results of the CAMI-1 Study. Front Cardiovasc Med (2021) 8:591714. doi: 10.3389/fcvm.2021.591714
48. Donaldson GC. C-reactive protein: does it prevent mortality? Am J Respir Crit Care Med (2007) 175(3):209–10. doi: 10.1164/rccm.200610-1565ED
49. Torzewski J, Brunner P, Ries W, Garlichs CD, Kayser S, Heigl F, et al. Targeting C-reactive protein by selective apheresis in humans: Pros and Cons. J Clin Med (2022) 11:1771. doi: 10.3390/jcm11071771
50. Pepys MB. C-reactive protein predicts outcome in COVID-19: is it also a therapeutic target? Eur Heart J (2021) 42:2280–3. doi: 10.1093/eurheartj/ehab169
51. Fang S, Wang Y, Sui D, Liu H, Ross MI, Gershenwald JE, et al. C-reactive protein as a marker of melanoma progression. J Clin Oncol (2015) 33(12):1389–96. doi: 10.1200/JCO.2014.58.0209
52. Ozawa Y, Amano Y, Kanata K, Hasegwa H, Matsui T, Kakutani T, et al. Impact of early inflammatory cytokine elevation after commencement of PD-1 inhibitors to predict efficacy in patients with non-small cell lung cancer. Med Oncol (2019) 36(4):33. doi: 10.1007/s12032-019-1255-3
53. Fukuda S, Saito K, Yasuda Y, Kijima T, Yoshida S, Yokoyama M, et al. Impact of C-reactive protein flare-response on oncological outcomes in patients with metastatic renal cell carcinoma treated with nivolumab. J Immuno Ther Cancer (2021) 9:e001564. doi: 10.1136/jitc-2020-001564
54. Filep JG. Targeting conformational changes in C-reactive protein to inhibit pro-inflammatory actions. EMBO Mol Med (2023) 15(1):e17003. doi: 10.15252/emmm.202217003
55. Perkins SJ, Okemefuna AI, Nan R. Unravelling protein-protein interactions between complement factor H and C-reactive protein using a multidisciplinary strategy. Biochem Soc Trans (2010) 38(4):894–900. doi: 10.1042/BST0380894
56. Hammond DJ Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, Pangburn MK, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem (2010) 285(46):36235–44. doi: 10.1074/jbc.M110.142026
57. Eisenhardt SU, Habersberger J, Murphy A, Chen Y-C, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105(2):128–37. doi: 10.1161/CIRCRESAHA.108.190611
58. Molins B, Pena E, de la Torre R, Badimon L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc Res (2011) 92(2):328–37. doi: 10.1093/cvr/cvr226
59. Braig D, Nero TL, Koch HG, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun (2017) 8:14188. doi: 10.1038/ncomms14188
60. Khreiss T, József L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation (2004) 109(16):2016–22. doi: 10.1161/01.CIR.0000125527.41598.68
61. Khreiss T, József L, Potempa LA, Filep JG. Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ Res (2005) 97(7):690–7. doi: 10.1161/01.RES.0000183881.11739.CB
62. Khreiss T, József L, Potempa LA, Filep JG. Opposing effects of C-reactive protein isoforms on shear-induced neutrophil-platelet adhesion and neutrophil aggregation in whole blood. Circulation (2004) 110(17):2713–20. doi: 10.1161/01.CIR.0000146846.00816.DD
63. Zouki C, Haas B, Chan JSD, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J Immunol (2001) 167(9):5355–61. doi: 10.4049/jimmunol.167.9.5355
64. Khreiss T, József L, Hossain S, Chan JS, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J Biol Chem (2002) 277(43):40775–81. doi: 10.1074/jbc.M205378200
65. Zeller J, Cheung Tung Shing KS, Nero TL, McFadyen JD, Krippner G, Bogner B, et al. A novel phosphocholine-mimetic inhibits a pro-inflammatory conformational change of C-reactive protein. EMBO Mol Med (2023) 15(1):e16236. doi: 10.15252/emmm.202216236
66. Braig D, Kaiser B, Thiele JR, Bannasch H, Peter K, Stark GB, et al. A conformational change of C-reactive protein in burn wounds unmasks its proinflammatory properties. Int Immunol (2014) 26(8):467–78. doi: 10.1093/intimm/dxu056
67. Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and new anti-inflammatory strategy. Circulation (2014) 130(1):35–50. doi: 10.1161/CIRCULATIONAHA.113.007124
68. Wang MY, Ji S-R, Bai C-J, El Kebir D, Li H-Y, Shi J-M, et al. A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J (2011) 25(9):3186–96. doi: 10.1096/fj.11-182741
69. Habersberger J, Strang F, Scheichl A, Htun N, Bassler N, Merivirta RM, et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res (2012) 96(1):64–72. doi: 10.1093/cvr/cvs237
70. Crawford JR, Trial J, Nambi V, Hoogeveen RC, Taffet GE, Entman ML. Plasma levels of endothelial microparticles bearing monomeric C-reactive protein are increased in peripheral artery disease. J Cardiovasc Transl Res (2016) 9(3):184–93. doi: 10.1007/s12265-016-9678-0
71. Lane T, Wassef N, Poole S, Mistry Y, Lachmann HJ, Gillmore JD, et al. Infusion of pharmaceutical-grade natural human C-reactive protein is not proinflammatory in healthy adult human volunteers. Circ Res (2014) 114(4):672–6. doi: 10.1161/CIRCRESAHA.114.302770
72. Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature (2006) 440(7088):1217–21. doi: 10.1038/nature04672
73. McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, et al. Dissociation of C-reactive protein localizes and amplifies inflammation: Evidence for a direct biological role of C-reactive protein and its conformational changes. Front Immunol (2018) 9:1351. doi: 10.3389/fimmu.2018.01351
74. Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab (2004) 24(11):1214–8. doi: 10.1097/01.WCB.0000136517.61642.99
75. Thiele JR, Zeller J, Kiefer J, Braig D, Kreuzaler S, Lenz Y, et al. A conformational change in C-Reactive protein enhances leukocyte recruitment and reactive oxygen species generation in Ischemia/Reperfusion injury. Front Immunol (2018) 9:675. doi: 10.3389/fimmu.2018.00675
76. Xia D, Samols D. Transgenic mice expressing rabbit C-reactive protein are resistant to endotoxemia. Proc Natl Acad Sci USA (1997) 94(6):2575–80. doi: 10.1073/pnas.94.6.2575
77. Rodriguez W, Mold C, Kataranovski M, Hutt JA, Marnell LL, Verbeek JS, et al. C-reactive protein mediated suppression of nephrotoxic nephritis: role of macrophages, complement, and Fcγ receptors. J Immunol (2007) 178(1):530–8. doi: 10.4049/jimmunol.178.1.530
78. Du Clos TW, Mold C. C-reactive protein: an activator of innate immunity and a modulator of adaptive immunity. Immunol Res (2004) 30(3):261–77. doi: 10.1385/IR:30:3:261
79. Svanberg C, Enocsson H, Govender M, Martinsson K, Potempa LA, Rajab IM, et al. Conformational state of C-reactive protein is critical for reducing immune complex-triggered type I interferon response: Implications for pathogenic mechanisms in autoimmune diseases imprinted by type I interferon gene dysregulation. J Autoimmun (2023) 135:102998. doi: 10.1016/j.jaut.2023.102998
80. Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature (2008) 456(7224):989–92. doi: 10.1038/nature07468
81. Brennan MP, Moriarty RD, Grennan S, Chubb AJ, Cox D. C-reactive protein binds to αIIbβ3. J Thromb Haemost (2008) 6(7):1239–41. doi: 10.1111/j.1538-7836.2008.02993.x
82. Ji SR, Ma L, Bai CJ, Shi JM, Li HY, Potempa LA, et al. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J (2009) 23(6):1806–16. doi: 10.1096/fj.08-116962
83. Fedewa MV, Hathaway ED, Ward-Ritacco C, Ward-Ritacco CL. Effect of exercise training on C reactive protein: a systematic review and meta-analysis of randomised and non-randomised controlled trials. Br J Sports Med (2017) 1(8):670–76. doi: 10.1136/bjsports-2016-095999
84. Selvin E, Paynter NP, Erlinger TP. The effect of weight loss on C-Reactive protein: A systematic review. Arch Intern Med (2007) 167:31–9. doi: 10.1001/ARCHINTE.167.1.31
85. Scheurig AC, Thorand B, Fischer B, Heier M, Koenig W. Association between the intake of vitamins and trace elements from supplements and C-reactive protein: results of the MONICA/KORA Augsburg study. Eur J Clin Nutr (2008) 62:127–37. doi: 10.1038/sj.ejcn.1602687
86. Smidowicz A, Regula J. Effect of nutritional status and dietary patterns on human serum C-reactive protein and interleukin-6 concentrations. Adv Nutr (2015) 6:738–47. doi: 10.3945/AN.115.009415
87. Gallus S, Lugo A, Suatoni P, Taverna F, Bertocchi E, Boffi R, et al. Effect of tobacco smoking cessation on C-Reactive protein levels in A cohort of low-dose computed tomography screening participants. Sci Rep (2018) 8:12908. doi: 10.1038/s41598-018-29867-9
88. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJP, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med (2008) 359(21):2195–207. doi: 10.1056/NEJMoa0807646
89. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJP, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet (2009) 373(9670):1175–82. doi: 10.1016/S0140-6736(09)60447-5
90. Zhang J, Wang X, Tian W, Wang T, Jia J, Lai R, et al. The effect of various types and doses of statins on C-reactive protein levels in patients with dyslipidemia or coronary heart disease: A systematic review and network meta-analysis. Front Cardiovasc Med (2022) 9:936817. doi: 10.3389/fcvm.2022.936817
91. Ridker PM, Bhatt DL, Pradhan AD, Glynn RJ, McFadyen JG, Nissen SE, et al. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomised trials. . Lancet (2023) 401(10384):1293–1301. doi: 10.1016/S0140-6736(23)00215-5
92. Kandelouei T, Abbasifard M, Imani D, Aslani S, Razi B, Fasihi M, et al. Effect of Statins on Serum level of hs-CRP and CRP in Patients with Cardiovascular Diseases: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Mediators Inflammation (2022) 2022:8732360. doi: 10.1155/2022/8732360
93. Cicero AFG, Fogacci F, Hernandez AV, Banach M, Alnouri F, Amar F, et al. Efficacy and safety of bempedoic acid for the treatment of hypercholesterolemia: A systematic review and meta-analysis. PloS Med (2020) 17:e1003121. doi: 10.1371/JOURNAL.PMED.1003121
94. Prasad K. C-reactive protein (CRP)-lowering agents. Cardiovasc Drug Rev (2006) 24(1):33–50. doi: 10.1111/j.1527-3466.2006.00033.x
95. Liu D, Jin B, Chen W, Yun P. Dipeptidyl peptidase 4 (DPP-4) inhibitors and cardiovascular outcomes in patients with type 2 diabetes mellitus (T2DM): a systematic review and meta-analysis. BMC Pharmacol Toxicol (2019) 20(1):18. doi: 10.1186/S40360-019-0293-Y
96. Awad K, Zaki MM, Mohammed M, Lewek J, Lavie CJ, Banach M. Effect of the renin-angiotensin system inhibitors on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc (2022) 97:1808–23. doi: 10.1016/J.MAYOCP.2022.06.036
97. Jones NR, Pegues MA, McCrory MA, Singleton W, Bethune C, Baker BF, et al. A selective inhibitor of human C-reactive protein translation is efficacious in vitro and in C-reactive protein transgenic mice and humans. Mol Ther Nucleic Acids (2012) 1:e52. doi: 10.1038/MTNA.2012.44
98. Szalai AJ, McCrory MA, Xing D, Hage FG, Miller A, Oparil S, et al. Inhibiting C-reactive protein for the treatment of cardiovascular disease: promising evidence from rodent models. Med Inflammation (2014) 2014:353614. doi: 10.1155/2014/353614
99. Warren MS, Hughes SG, Singleton W, Yamashita M, Genovese MC. Results of a proof of concept, double-blind, randomized trial of a second-generation antisense oligonucleotide targeting high-sensitivity C-reactive protein (hs-CRP) in rheumatoid arthritis. Arthritis Res Ther (2015) 17(1):80. doi: 10.1186/s13075-015-0578-5
100. Noveck R, Stroes ESG, Flaim JD, Baker BF, Hughes S, Graham MJ, et al. Effects of an antisense oligonucleotide inhibitor of C-reactive protein synthesis on the endotoxin challenge response in healthy human male volunteers. J Am Heart Assoc (2014) 3(4):e001084. doi: 10.1161/JAHA.114.001084
101. Sugihara C, Freemantle N, Hughes SG, Furniss S, Sulke N. The effect of C-reactive protein reduction with a highly specific antisense oligonucleotide on atrial fibrillation assessed using beat-to-beat pacemaker Holter follow-up. J Interv Card Electrophysiol (2015) 43:91–8. doi: 10.1007/S10840-015-9986-3/METRICS
102. Skarabis H, Torzewski J, Ries W, Heigl F, Garlichs CD, Kunze R, et al. Sustainability of C-Reactive protein apheresis in acute myocardial infarction—Results from a supplementary data analysis of the exploratory C-Reactive protein in acute myocardial infarction-1 study. J Clin Med (2022) 11:6446. doi: 10.3390/JCM11216446
103. Ringel J, Ramlow A, Bock C, Sheriff A. Case report: C-Reactive protein apheresis in a patient with COVID-19 and fulminant CRP increase. Front Immunol (2021) 12:708101. doi: 10.3389/fimmu.2021.708101
104. Schumann C, Heigl F, Rohrbach IJ, Sheriff A, Wagner L, Wagner F, et al. A report on the first 7 sequential patients treated within the C-Reactive protein apheresis in COVID (CACOV) registry. Am J Case Rep (2021) 23:e935263. doi: 10.12659/AJCR.935263
105. Esposito F, Matthes H, Schad F. Seven COVID-19 patients treated with C-Reactive protein (CRP) apheresis. J Clin Med (2022) 11:1956. doi: 10.3390/JCM11071956/S1
106. Gao R, Wang L, Bai T, Zhang Y, Bo H, Shu Y. C-Reactive protein mediating immunopathological lesions: A potential treatment option for severe influenza A diseases. EBioMedicine (2017) 22:133. doi: 10.1016/J.EBIOM.2017.07.010
107. Block G, Jansen CD, Dalvi TB, Norkus EP, Hudes M, Crawford PB, et al. Vitamin C treatment reduces elevated C-reactive protein. Free Radic Biol Med (2009) 46(1):70–7. doi: 10.1016/j.freeradbiomed.2008.09.030
108. Tardif J-C, Samuel M. Inflammation contributes to cardiovascular risk in patients receiving statin therapy. Lancet (2023) 401(10384):1245–7. doi: 10.1016/S0140-6736(23)00454-3
109. Aday AW, Ridker PM. Targeting residual inflammatory risk: A shifting paradigm for atherosclerotic disease. Front Cardiovasc Med (2019) 6:16. doi: 10.3389/fcvm.2019.00016
110. Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, et al. Antisense oligonucleotides: An emerging area in drug discovery and development. J Clin Med (2020) 9(6):2004. doi: 10.3390/jcm9062004
111. Xing D, Hage FG, Chen Y-F, McCrory MA, Feng W, Skibinski GA, et al. Exaggerated neointima formation in human C-reactive protein transgenic mice is IgG Fc receptor type I (FcgRI)-dependent. Am J Pathol (2008) 172:22–30. doi: 10.2353/ajpath.2008.070154
112. Burel SA, Machemer T, Baker BF, Kwoh TJ, Paz S, Younis H, et al. Early-stage identification and avoidance of antisense oligonucleotides causing species-specific inflammatory responses in human volunteer peripheral blood mononuclear cells. Nucleic Acid Ther (2022) 32(6):457–72. doi: 10.1089/NAT.2022.0033
113. Sheriff A, Schindler R, Vogt B, Abdel-Aty H, Unger JK, Bock C, et al. Selective apheresis of C-reactive protein: A new therapeutic option in myocardial infarction? J Clin Apher (2014) 30:15–21. doi: 10.1002/jca.21344
114. Ries W, Sheriff A, Heigl F, Zimmermann O, Garlichs CD, Torzewski J. “First in man”: case report of selective C-Reactive protein apheresis in a patient with acute ST segment elevation myocardial infarction. Case Rep Cardiol (2018) 2018:4767105. doi: 10.1155/2018/4767105
115. Ries W, Heigl F, Garlichs C, Sheriff A, Torzewski J. Selective C-reactive protein-apheresis in patients. Ther Apher Dial (2019) 23(6):570–4. doi: 10.1111/1744-9987.12804
116. Kraus VB, Jordan JM. Serum C-reactive protein (CRP), Target for therapy or trouble? biomark Insights (2006) I:77–80. doi: 10.1177/117727190600100020
117. Diaz Padilla N, Bleeker WK, Lubbers Y, Rigter GMM, Van Mierlo GJ, Daha MR, et al. Rat C-reactive protein activates the autologous complement system. Immunology (2003) 109(4):564–71. doi: 10.1046/J.1365-2567.2003.01681.X
Keywords: C-reactive protein, monomeric CRP, CRP antagonists, CRP lowering therapies, inflammation, cardiovascular disease, autoimmunity, cancer
Citation: Rizo-Téllez SA, Sekheri M and Filep JG (2023) C-reactive protein: a target for therapy to reduce inflammation. Front. Immunol. 14:1237729. doi: 10.3389/fimmu.2023.1237729
Received: 09 June 2023; Accepted: 07 July 2023;
Published: 26 July 2023.
Edited by:
Yi Wu, Xi’an Jiaotong University, ChinaReviewed by:
Shang-Rong Ji, Lanzhou University, ChinaCopyright © 2023 Rizo-Téllez, Sekheri and Filep. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: János G. Filep, amFub3MuZy5maWxlcEB1bW9udHJlYWwuY2E=
†ORCID: Salma A. Rizo-Téllez, orcid.org/0000-0003-0587-5167
Meriem Sekheri, orcid.org/0000-0001-9394-0875
János G. Filep, orcid.org/0000-0001-5022-3534
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.