
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 27 September 2023
Sec. Alloimmunity and Transplantation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1235661
This article is part of the Research TopicCommunity Series in Immune Tolerance Post Allogeneic Hematopoietic Cell Transplantation: Volume IIView all 3 articles
 Memnon Lysandrou1
Memnon Lysandrou1 Panagiota Stamou1
Panagiota Stamou1 Dionysia Kefala1
Dionysia Kefala1 Chryso Pierides2
Chryso Pierides2 Maria Kyriakou2Nikolaos Savvopoulos1Panayiota Christofi1
Maria Kyriakou2Nikolaos Savvopoulos1Panayiota Christofi1 Anastasia Papadopoulou3
Anastasia Papadopoulou3 Evangelia Yannaki3
Evangelia Yannaki3 Paul Costeas2†
Paul Costeas2† Alexandros Spyridonidis1*†
Alexandros Spyridonidis1*†Regulatory T cells (Tregs) are essential mediators of tolerance mitigating aberrant immune responses. While naturally occurring Treg (nTreg) development and function are directed by epigenetic events, induced Treg (iTreg) identity and mechanisms of action remain elusive. Mirroring the epigenetic circuits of nTregs, we and others have used hypomethylation agents (HAs) to ex vivo convert T cells into iTregs (HA-iTregs) and further showed that the suppressive properties of the HA-iTregs are predominantly confined in an emergent population, which de novo expresses the immunomodulatory molecule HLA-G, consequently providing a surface marker for isolation of the suppressive HA-iTreg compartment (G+ cells). We isolated the HA-induced G+ cells and their G− counterparts and employed high-throughput RNA-sequencing (RNA-seq) analyses to uncover the G+-specific transcriptomic changes guiding T cells toward a regulatory trajectory upon their exposure to HA. We found a distinct transcriptional upregulation of G+ cells accompanied by enrichment of immune-response–related pathways. Although single-cell RNA-seq profiling revealed regulatory G+ cells to have molecular features akin to nTregs, when assessed in conjunction with the comparative transcriptomic analysis and profiling of secreted cytokines against the non-suppressive G− cells, FOXP3 and other T-helper signatures appear to play a minor role in their suppressive phenotype. We found an ectopic expression of IDO-1 and CCL17/22 in G+ cells, denoting that in vitro exposure of T cells to HA may well unlock myeloid suppressor genes. This report provides transcriptional data shaping the molecular identity of a highly purified and potent HA-iTreg population and hints toward ectopic myeloid-specific molecular mechanisms mediating HA-iTreg function.
Regulatory T cells (Tregs) are essential mediators of immunological tolerance mitigating deviant or overt immune responses as illustrated by in vitro and in vivo studies and early phase clinical trials (1). Tregs can be encountered as naturally occurring cells in vivo (nTregs) or may be in vitro induced upon modulation of conventional T cells (iTregs) (2–5). While nTreg development and function are meticulously directed and orchestrated by epigenetic events ensuring specific and stable demethylation status of key genes, including the Forkhead Box P3 (FOXP3) transcriptional factor, iTregs identity and mechanisms of action remain largely ambiguous and probably highly dependent on the inherent variability of their generation protocol (3, 4, 6, 7). Seeking to mimic the epigenetic circuits of nTregs, several groups including ours have used hypomethylation agents (HAs) to ex vivo convert murine or human T cells into iTregs (HA-iTregs) (5, 8, 9). In an attempt to identify the exact molecular mechanism guiding the function of such murine HA-iTregs, Choi et al. elegantly demonstrated that their in vitro and in vivo suppressive abilities occur independently of FOXP3 (9). Human leukocyte antigen G (HLA-G) molecule is a well-known immunomodulatory molecule that shields the “semi-allogeneic” fetus from maternal immune rejection during pregnancy and is epigenetically repressed succeeding prenatal life (10). Naturally occurring HLA-G+ T cells are encountered in low percentages in human peripheral blood as part of the physiological repertoire of suppressor, distinct from nTregs, T cells and can putatively expand when needed to preserve immune tolerance (11–13). In our previous work, we hypothesized that HLA-G has an operative role in the suppressive action of HA-iTregs (5, 11). Indeed, by using the HA decitabine (Dec), we robustly and consistently produced human HA-iTregs with in vitro and in vivo suppressive activity that have now entered a phase I–II clinical trial against Graft-versus-Host disease (EudraCT 2021-006367-26), and showed that their suppressive function is mediated to a large extent but not exclusively by the HLA-G molecule (5, 14, 15). HA-induced de novo expression of HLA-G on T cells remains functionally stable upon removal of the HA as well as in pro-inflammatory environment (5). More importantly, we showed that the suppressive properties of the HA-iTregs are strictly confined in the HLA-G–expressing cellular compartment, henceforth, providing a surface marker for isolation of the suppressive HA-iTregs (termed G+ cells).
Aiming to unveil the immunosuppressive mechanisms of HA-iTregs and elucidate the epigenetic networks guiding T cells toward a regulatory trajectory upon their exposure to HA, we employed high-throughput analyses to uncover the G+-specific transcriptomic changes. Interestingly, we link de novo expression of established myeloid regulatory genes, such as indoleamine 2,3-dioxygenase 1 (IDO-1) with HA-iTregs emergence.
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers under informed consent and approval from the local ethics committee (Protocol Number: 5832). T cells were isolated by negative magnetic cell selection (MACS Pan-T Cell Isolation Kit II Human MiltenyiBiotec, Germany) except for the flow cytometry-based validation experiments where CD4+-selected T cells were used as starting material (RosetteSep Human CD4+ T-cell enrichment cocktail, StemCell Technologies, Canada), then activated for 3 days in the presence of anti-CD3/CD28 beads (Dynabeads Human T-Activator CD3/CD28, Gibco) at a 1:1 bead:cell ratio and subsequently incubated in the presence of 50U/mL human recombinant IL-2 (rhIL-2 PeproTech or Miltenyi Biotec) with 5 μM or 7.5 μM Dec (Sigma-Aldrich, Germany) for three additional days. Control T cells were treated with phosphate-buffered saline (PBS) solution and further denoted as PBS-treated controls.
Flow cytometry was performed on BDFACS Canto II Flow Cytometer (BD Biosciences, USA) and data analysis was performed on FlowJo v10. Cell sorting was performed using BDFACSAria III (BD Biosciences) by gating on single viable HLA-G+CD4+ and HLA-G−CD4+ cells reaching a purity of sorted cell populations over 95%. The following monoclonal anti-human antibodies were used: HLA-G-PE (clone MEM- G9, Sigma-Aldrich), CD4 FITC/PerCP/APC-Cy7 (clone RPA-T4, BD Biosciences or BioLegend) and CD3-FITC (clone UCHT-1, BD Biosciences), IDO-1-Alexa Fluor 647 (clone V50-1886, BD Biosciences), CD45RA-PE-Cy7 (clone HI100, BioLegend), CD62L-V450 (clone DREG-56, BD Biosciences), CD25-PE (clone M-A251, BioLegend), CD127-PerCP-Cy5.5 (clone HIL-7R-M21, BD Biosciences), and GITR-BV421 (clone V27-580, BD Biosciences). Dead cell exclusion was performed with Zombie Aqua/NIR Fixable viability dye (BioLegend) or Far-Red Live/Dead Fixable dead cell stain kit (Invitrogen, USA). Fixation was performed using BD Phosflow Fix Buffer (BD Biosciences), and permeabilization using perm buffer (PBS, Triton 0.5%, and BSA 0.5%). Fluorescence minus one (FMO) control was used to set the threshold of positivity for HLA-G, CD45RA, CD62L, CD25, CD127, and GITR.
Total RNA was isolated from five paired samples (G+/G−) using the miRNeasy Micro kit (Qiagen, Germany), according to manufacturer’s instructions and was used for bulk RNA-seq. Illumina-compatible libraries were prepared according to manufacturer’s instructions and next generation sequencing (NGS) (paired-end and strand-specific) was performed on an Illumina HiSeq2000. For scRNA-seq, approximately 16.000 sorted G+ cells were loaded in a channel of a chromium controller (10× Genomics) for generation of gel-bead-in-emulsions (Greek Research Infrastructure for Personalized Medicine, pMedGR). The sequencing library was prepared using Single Cell 3′ Reagent Kits v3.1 (10× Genomics) and sequenced on an Illumina NextSeq2000.
Bioinformatic analysis for both RNA-seq and scRNA-seq data is described in detail in the Supplementary Data.
RNA was isolated from paired sorted G+ and G− cells using the RNeasy Micro kit (Qiagen, Germany), cDNA was synthesized (PrimeScript RT Reagent Kit, Takara) and TaqMan assays were performed for HLA-G, IDO-1, CCL22, CCL17, and ABL1. The reaction was performed on a BioRadCFX96, and each cDNA sample was assayed in duplicate. Quantification was performed using the ΔΔCT method and relative expression was calculated based on housekeeping gene (ABL1) expression.
Paired sorted G+, G−, and PBS-treated controls were plated at 4 × 105/ml in 48-well plates in the presence of IL-2 (100 U/ml) and were either left unstimulated or stimulated with anti-CD3/CD28 beads at a 1:1 bead:cell ratio for 48h. Cell-free supernatants were harvested and stored at −80°C. For cytokine/chemokine quantification, analysis was performed using the Bio-Plex Pro Human Cytokine Th1/Th2 Assay (Bio-Rad, USA) and a custom multiplex magnetic bead-based immunoassay (Protatonce, Ltd), according to manufacturer’s instructions on a Bio-Plex 200 system (Bio-Rad). Tryptophan (Trp) and kynurenine (Kyn) concentrations were assessed with the kynurenine/tryptophan ratio ELISA pack (ImmuSmol), according to manufacturer’s instructions, using the Infinite F50 system (Tecan, Switzerland). All samples were run in duplicates.
Statistical analyses were performed using paired two-tailed standard or ratio t-tests using Prism software (GraphPad Software Inc.). P values of less than 0.05 were considered significant.
We produced HA-iTregs by exposing human peripheral T cells to Dec in vitro (n = 5) and subsequently sorted the suppressive CD4+HLAG+ (G+) cells and their CD4+HLA-G− (G−) counterparts to perform comparative transcriptome analysis (Figure 1A). A total of 394 genes were identified to be differentially expressed in G+ cells as compared to G− cells [DEGs, |log2(fold change)| > 0.5 and q < 0.05]. Most of the DEGs (n = 358) in G+ cells were found to be upregulated, which may be either an intrinsic characteristic of these isolated cells or may indicate a more pronounced hypomethylation-induced gene activation in this population (Figure 1B). The principal components analysis (PCA) depicted that G+ cells are characterized by a distinct gene expression profile that is clearly separate from their G− counterparts (Figure 1C). We next conducted in silico analysis to identify pathways involved in the transcriptionally distinct HA-induced G+ population. Among the highest enrichments in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, the Gene Ontology (GO) molecular function annotations and the REACTOME data repository, several pathways involved in immune responses were uncovered (Figure 1D). In particular, chemokine/cytokine receptor interactions and chemokine signaling pathways appear to play a pivotal role in the transcriptional identity of G+ suppressive cells. We additionally found numerous pathways related to or containing HLA genes to be enriched in the G+ cells (Figure 1E). This is not unexpected since HLA-G expression was used to purify the G+ compartment. Surprisingly, the HLA-G gene was not identified as an upregulated DEG in the G+ cells RNA-seq analysis [HLA-G log2(fold change) = 0.3, p = 0.019, q = 0.087, Supplementary Figure S1A], which runs somewhat conflicting to our experimental design. This finding may be related to the shared homology of HLA-G with other DEGs of the HLA family resulting in a high overlap between HLA-G and the other HLA genes or may be due to the variability in HLA-G transcripts between experiments, which may have undermined the statistical power to detect a significant effect on the DEG analysis (Supplementary Figures 1A, B). On the contrary, RT-PCR confirmed that HLA-G is truly overexpressed on G+ cells (n = 5, median fold change-MdFC vs. G− cells 34.66, p = 0.0002) (Supplementary Figure S1C). Collectively, we show that HA-induced G+ cell emergence is closely accompanied by a unique and cohesive transcriptional upregulation of genes involved in immune response pathways.
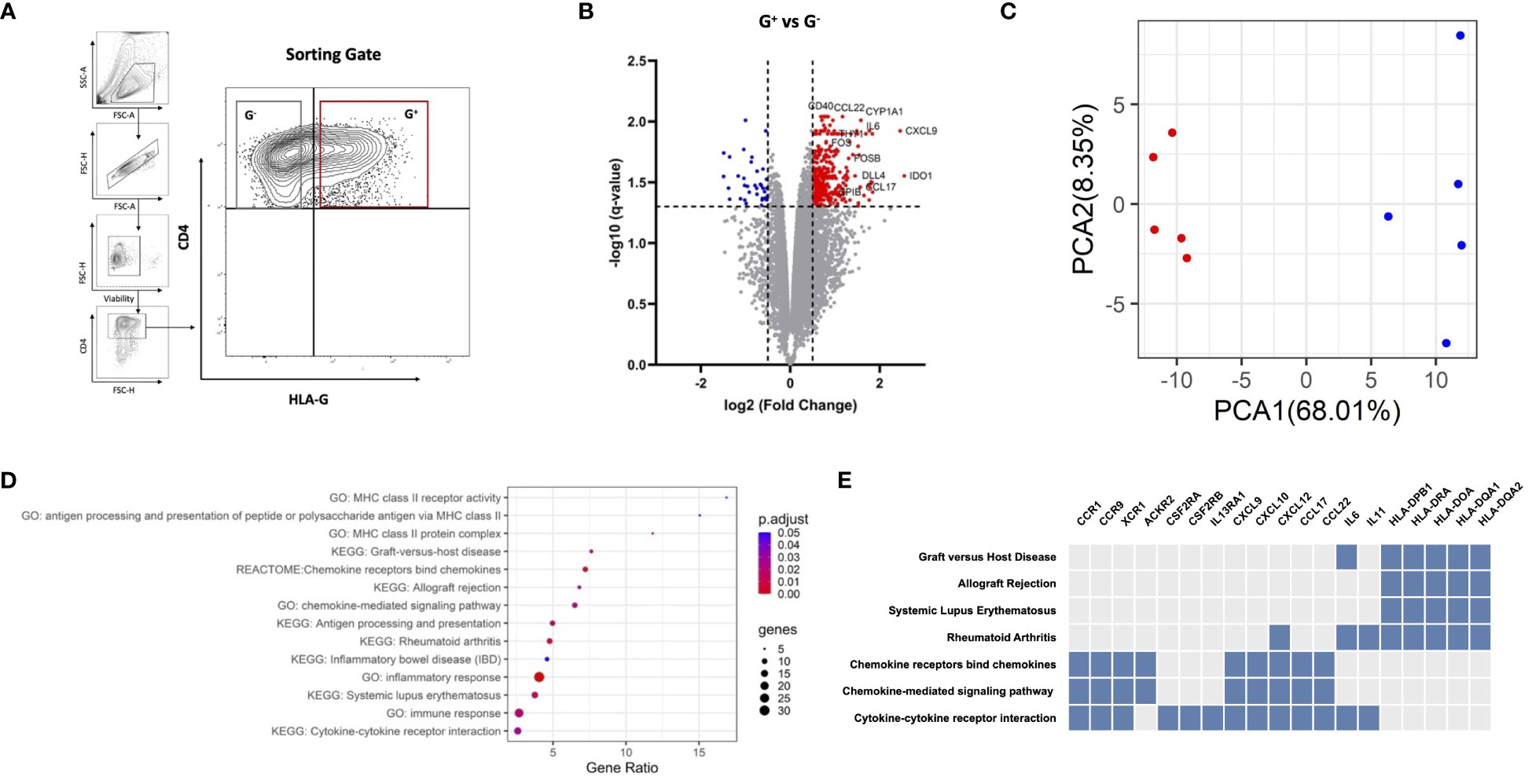
Figure 1 G+ cells represent a transcriptionally distinct population enriched in immune response pathways. (A) Representative plots of fluorescence-activated cell sorting (FACS) strategy for G+ and G− T-cell isolation. Cells were gated on alive singlets with CD4+HLA-G+ (red gate) or CD4+HLA-G+ (gray gate) phenotype, where the threshold of HLA-G positivity was set with a fluorescence minus one (FMO) control. (B) RNA sequencing of G+ T cells against their G− counterparts (n = 5 paired samples). Volcano plot of differentially expressed genes (DEGs) in G+ T cells, including IDO-1, CCL17, and CCL22. Vertical dotted lines represent |log2(fold change)= 0.5, and horizontal dotted lines represent q-value = 0.05. Red dots indicate upregulated DEGs, blue dots indicate downregulated DEGs, while gray dots represent genes that do not meet the criteria for differential expression [log2(fold change) > 0.5 and q < 0.05]. (C) Principal components analysis (PCA) plotted using the two main principal components (PCA1-2), depicting the transcriptional profile of the G+ population (red) entirely distinct from the G− counterpart (blue). (D) Dotplot of the pathway enrichment analysis showing pathways highly enriched in DEGs plotted based on gene ratio (number of DEGs/total genes in the corresponding pathway) using an adjusted p-value (p.adjust) threshold of 0.05. (E) Clustergram of pathway analysis depicting DEGs associated with enriched pathways of interest. Blue boxes indicate DEGs present in the corresponding pathway whereas gray boxes indicate the absence of the DEG.
We then examined whether G+ cells follow a specific trajectory toward categorical T-helper (Th) effector (Th1, Th2, and Th17) or FOXP3+ nTreg subsets. Compared to G− cells, transcriptome analysis and cytokine secretion profiling at baseline and stimulated conditions did not reveal a polarization of G+ cells toward any trajectory (Supplementary Figures 2A, B). Intriguingly, FOXP3 was not found to be differentially expressed in the suppressive G+ cells[log2(fold change) = 0.26, p = 0.0067 and q = 0.056] suggesting a dispensable role in the G+ phenotype. Since our targeted comparison between the G+ and G− compartments does not allow the removal of confounding bias introduced by the HA effects on T-cell fate, we performed single-cell RNA sequencing (scRNA-seq) analysis on isolated G+ cells to decipher the underlying heterogeneity of the transcriptional programs of G+ cells in an unsupervised manner. Clustering analysis revealed six clusters (Figure 2A). Two clusters were clearly demarcated from the rest as a result of the differential expression of a plethora of cell cycle–related genes resulting in a high G2/M gene signature score (Supplementary Figure S2C). Cluster annotation using potential marker genes with known cell marker genes from a reference database (CellMatch) allocated four of the clusters to be strongly associated with nTreg cell signatures including FOXP3, IL2RA, and Glucocorticoid-Induced TNFR-related protein-GITR/TNFRSF18 (Regulatory T cell 1–4), while the other two clusters were marked as naïve T cells (Naïve T cell 1–2) (Figure 2A, Supplementary Figure S2C, and Supplementary Table S1). To validate this in silico scRNA-seq analysis, we performed flow cytometric analyses (n = 4) and showed that G+ cells are predominantly early differentiated naïve or central memory cells (Figure 2B) and bear a CD25highCD127low/-GITR+ immunophenotype, akin to that of thymic nTregs currently used in clinical practice (Figure 2C) (16). By performing in silico RNA velocity analysis, we could also uncover a connection between the naïve and regulatory clusters revealing that Regulatory T cell 2 and 3 clusters lie directly downstream of Naïve T cell 2 and 1 clusters, respectively, in the differentiation trajectory of G+ cells (Figure 2D). Finally, we interrogated our bulk RNA-seq data using the scRNA-seq–derived signature by applying the SCDC deconvolution algorithm and we found a differential composition of regulatory and naïve signatures between G+ and G− populations, with the G+ fraction to be enriched for regulatory clusters (Figures 2E, F). Taken together, although scRNA-seq profiling revealed that regulatory G+ cells have molecular features akin to nTregs, when this is assessed in conjunction with the comparative transcriptomic analysis and cytokine profiling against the non-suppressive G− cells, these nTregs signatures seem to play a minor role in the G+ identity, thus hinting toward alternate molecular mechanisms instigating the G+ suppressive phenotype.
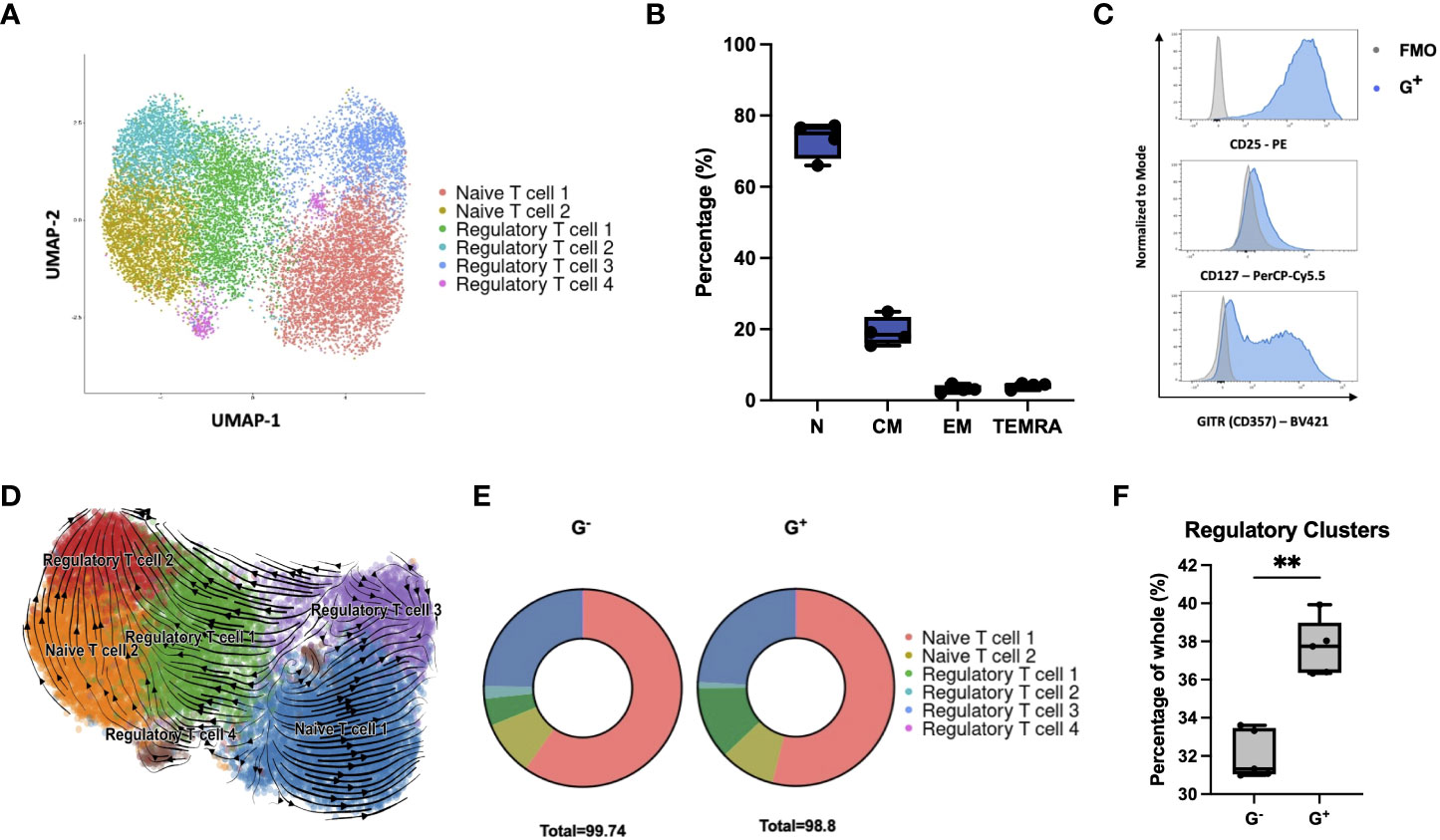
Figure 2 Single-cell RNA sequencing reveals G+ cells to be enriched in regulatory signatures. (A) Single-cell RNA-seq of G+ T cells (n = 15556 cells). Uniform manifold approximation and projection (UMAP) plot showing clustering into regulatory (Regulatory T cells 1–4) and naïve subpopulations (Naïve T cells 1–2) following cell annotation using the scCATCH algorithm. (B) Immunophenotypic validation of the early differentiated character of G+ cells (n = 4) [N: naïve (CD62L+CD45RA+) median 75%, range 66%–77.2%, CM: central memory (CD62L+CD45RA−) median 18.45%, range 15.4%–24.9%, EM: effector memory (CD62L−CD45RA−) median 3.04%, range 2.1%–4.72%, and TEMRA: terminal effector memory T cells expressing CD45RA (CD62L−CD45RA+) median 4.43%, range 2.87%–4.82%]. (C) Immunophenotypic validation of the nTreg characteristics arising from the scRNA-seq data in G+ cells. Representative histograms are shown from one out of four independent experiments. FMO controls (gray) are overlaid to G+ cells (blue) as negative controls (n = 4). (D) In silico RNA velocity analysis, projected on UMAP plots, revealing that Regulatory T cell clusters lie downstream of Naïve T-cell clusters in the differentiation trajectory of G+ cells. Arrows point toward the predicted course of cell differentiation dynamics while arrow sizes indicate the strength of calculated directionality. (E) Deconvolution analysis depicting deconvoluted bulk RNA-seq data of G+ and G- T cells using the scRNA-seq–derived dataset. (F) Enrichment of G+ T cells in Regulatory T-cell cluster signatures compared to G- cells upon deconvolution of bulk RNA-seq data (n = 5, Mann–Whitney test, p = 0.0079). **p ≤ 0.01 and ns, not significant.
Strikingly, the RNA-seq comparison of G+ versus G− cells showed that the top upregulated DEG in G+ cells were IDO-1 [log2(fold change) = 2.55, q = 0.028, Figure 1B], a finding which was further validated through RT-PCR (n = 4, MdFC = 10.82) (Supplementary Figure S1C). IDO-1 was also equally detected in all clusters from the scRNA-seq dataset. IDO-1 is a potent, epigenetically controlled, immunosuppressive molecule mostly associated with myeloid cells and scarcely reported on lymphoid cells (17, 18). This prompted us to further investigate this rather ectopic IDO-1 gene upregulation in T cells by validating its protein expression over both G− cells and paired cells not exposed to Dec (PBS-treated controls). Interestingly, we found a significantly high expression of IDO-1 in G+ cells over both G− cells (n = 8, p = 0.001) and PBS-treated controls (n = 5, p = 0.0145) (Figure 3A). IDO-1 mediates suppression by catabolizing the essential amino acid Tryptophan (Trp) to the immunomodulatory Kynurenine (Kyn) leading to T-cell anergy due to tryptophan starvation and to nTreg induction by Kyn (19, 20). To find a possible functional significance of transcriptional and protein IDO-1 expression in G+ cells we measured these metabolites in culture supernatants and calculated the Kyn/Trp ratio as a surrogate marker of IDO-1 enzymatic activity. Indeed, Kyn was found to be highly and significantly produced in G+ cells as compared to PBS-treated counterparts (n = 3, p = 0.03), resulting in a trend toward an increased Kyn/Trp ratio in the G+ cell cultures (p = 0.06) (Figure 3B). Intriguingly, beyond IDO-1 we observed additional mediators of myeloid-driven immune regulation, namely, C–C motif chemokine ligand (CCL) 17 and CCL22, to be among the top 15 upregulated DEGs on G+ cells over G− cells [log2(fold change) > 1.5]. The transcriptional overexpression of CCL17 and CCL22 chemokines in G+ cells was validated by RT-PCR (n = 4, MdFC = 4.82, p = 0.014 and MdFC = 9.52, p = 0.003, respectively) (Supplementary Figure S1C). Moreover, the scRNA-seq analysis revealed CCL17 transcripts to be present within all the cluster, with Regulatory T cell 4 cluster displaying the higher level of expression. When compared to G− cells and PBS-treated controls, we found an increased baseline secretion of CCL17 (n = 4, p < 0.05) from G+ cells and excessive amounts of CCL17 (n = 4, p < 0.001) and of CCL22 (n = 4, p < 0.001) upon stimulation in both G− and G+ supernatants over PBS-treated cells (Figure 3C). Taken together, we report that in vitro pharmacological hypomethylation of T cells unleashes epigenetically repressed myeloid-specific suppressor genes, such as IDO-1 and CCL17/22.
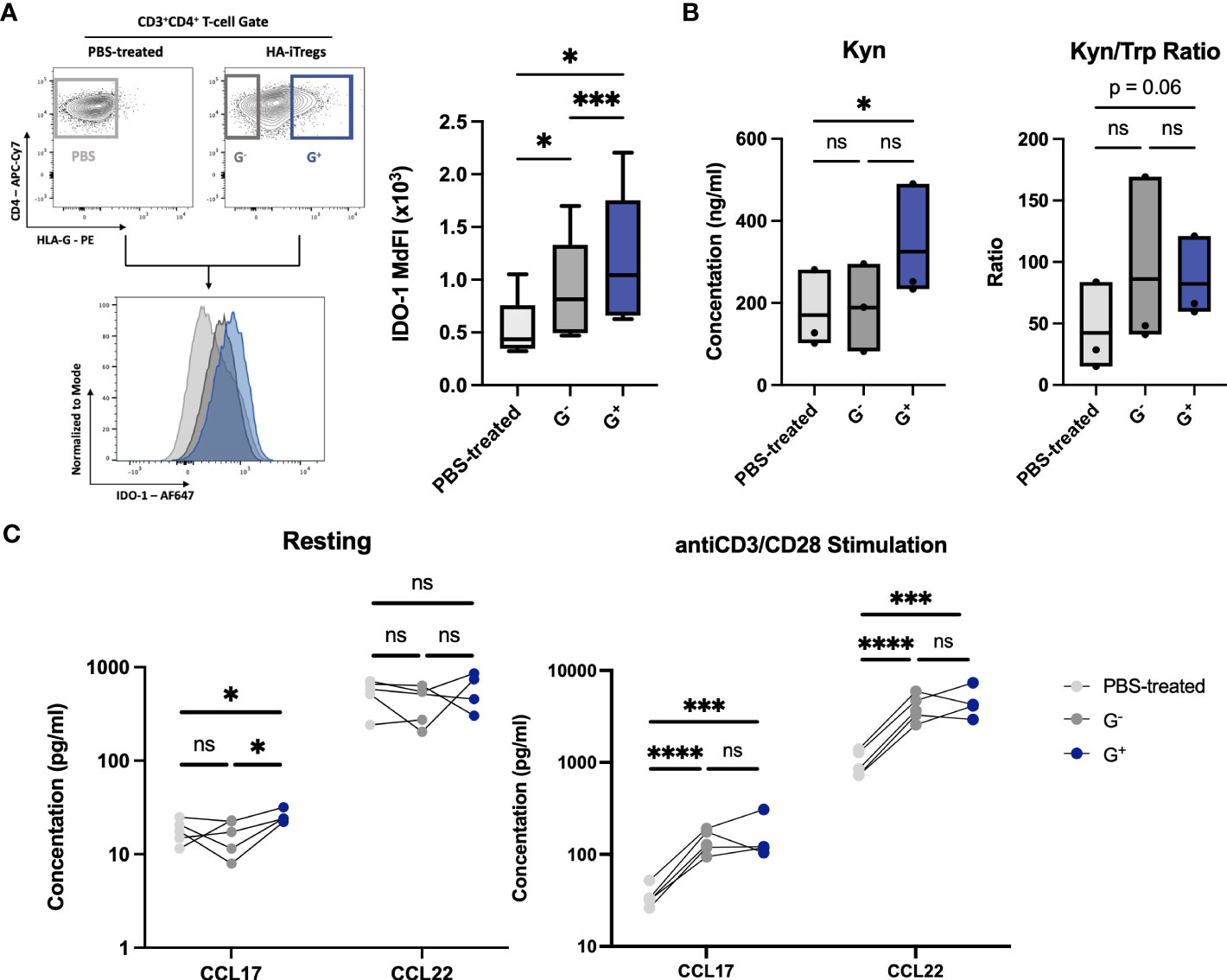
Figure 3 Validation of ectopically expressed myeloid-specific suppressive genes uncovered by G+-specific transcriptome. (Α) IDO-1 protein expression in G+ cells. Representative flow cytometry plots for intracellular IDO-1 detection (left) and collective data of IDO-1 Median Fluorescence Intensity (MdFI) in G+ T-cells (blue) over G- T-cells (dark grey) (n=8, p=0.001) and PBS-treated controls (light grey) (n=5, p=0.014) (right). (B) Enzyme-Linked Immunosorbent Assay (ELISA) for quantitative analysis of the Tryptophan (Trp) catabolite Kynurenine (Kyn) concentrations (left) and Kyn to Trp ratio (Kyn/Trp) (right) depicting the enzymatic activity of IDO-1 by the means of increased Kyn and Kyn/Trp ratio in G+ cell culture supernatants. (C) Luminex assay for quantitative assessment of chemokines in cell culture supernatants of G+, G- and PBS-treated control cells (n=4-5) at rest (left) or after anti-CD3/CD28 stimulation (right) revealing the increased secretion of CCL17 and CCL22. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, and ns: not significant.
In this study, we employed G+ cells as a model to unravel the molecular underpinnings of the epigenetic reprogramming driving suppressive function in HA-iTregs. Through transcriptome analysis of the suppressive G+ cell against the non-suppressive G− cells we show that even though pharmacological hypomethylation takes place in a stochastic fashion, the G+ compartment is characterized by a unique gene expression profile that is noticeably distinct from their negative counterparts. These findings are in line with previous work highlighting that isolated suppressive populations of murine and human nTregs and in vitro-induced regulatory type 1 (Tr1)- iTregs display a distinct gene expression profile that is clearly separated from their biological counterparts and/or parental cells (3, 4, 6, 7).
Moreover, we identified several pathways implicated in immune responses to be enriched, with chemokines/cytokine receptor interactions and chemokine signaling pathways to be highly represented, which is in agreement with transcriptomic analyses of both isolated suppressive nTregs and iTregs populations (3, 21). We also highlight inherent restrictions of in silico tools for accurate HLA inference derived from NGS data that may have hampered previous studies in identifying HLA-G as a key mediator and subsequently marker for iTreg function and identity, respectively (3, 4). Such tools focused solely on HLA genes routinely assessed for clinical HLA typing (HLA-A, -B, -C, -DPA1, -DPB1, -DQA1, -DQB1, and -DRB1-5), thus excluding nonclassical HLA genes, which share homology with genes in the HLA family such as HLA-G, and more specific algorithms to tackle this issue are warranted (22–24).
By performing bulk and single-cell transcriptomic analysis along with cytokine profiling, we show that regulatory G+ cells do not follow a specific trajectory toward polarization while displaying selected molecular and phenotypic features of nTregs. These features, nonetheless, appear to play a secondary role in the G+ identity, since by comparing the G+ suppressive compartment against their equally HA-treated G− counterparts, we show that FOXP3 is not upregulated in G+ cells. However, this result does not rule out that FOXP3 is upregulated upon HA treatment as shown by others, but points toward the dispensable role of FOXP3 in the suppressive function of Tregs (8, 9). Similarly, Choi et al. reported that HA-iTregs derived from FOXP3 knockout mice maintained their suppressive abilities in vitro and in vivo (9), and Lam et al. demonstrated that ablation of FOXP3 in mature nTregs was associated with concurrent demethylated DNA patterns, albeit retention of their suppressive function (21).
Our data hint toward previously unrecognized molecular mechanisms prompting the suppressive HA-iTregs phenotype in G+ cells. We report the transcriptional and protein expression of enzymatically active IDO-1 and CCL17/22 by G+ cells, which are both atypically expressed in T cells. IDO-1 is a potent regulatory mediator mostly associated with myeloid and mesenchymal stromal cells with implications in feto-maternal tolerance, tumor immune escape, autoimmunity and alloimmune responses such as graft-versus-host disease (18, 25–28). Interestingly, we could identify limited literature on IDO-1 transcriptional expression in T-cells, found to be induced either by IFNa2b or CTLA-4-Fc on CD4+ cells (29, 30). Furthermore, a CCL17/22-rich milieu orchestrated by myeloid cells leads to local nTreg chemoattraction, which has been shown to enable tumor immune evasion, prevent autoimmune diabetes and promote donor-specific tolerance (31–34). Collectively, we show that in vitro exposure of T-cells to HA may unlock myeloid-specific suppressor genes, but their level compared to naturally expressing myeloid populations remains to be answered. Since these molecules were found predominantly in the suppressive G+ compartment, we postulate that they contribute to the suppressive properties of HA-iTregs, something which must be confirmed in future functional studies.
In summary, our study provides compelling evidence that the G+ suppressive compartment of HA-iTregs possesses a unique transcriptional profile with prominent regulatory signatures. Our results add to the hitherto literature detaching iTreg function from FOXP3 expression and hint toward novel molecular mechanisms mediating HA-iTregs function, namely the ectopically expressed myeloid-specific molecules IDO-1 and CCL17/22. In conclusion, this report extends our understanding of the molecular features that shape the transcriptional identity of HA-iTregs and provides valuable transcriptional data of a highly purified and potent iTreg population that can be compared with other Tregs (35).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE229057. https://www.ncbi.nlm.nih.gov/geo/, GSE229170.
The studies involving humans were approved by University of Patras Ethics Committee and University General Hospital of Patras Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
ML performed the study and wrote the manuscript which was designed and supervised by AS; PS performed sorting experiments for bulk RNA sequencing. CP, DK, MK, PCh, and NS performed additional experiments; PCo supervised sequencing experiments; AP and EY provided critical reviews and edits. All authors contributed to the article and approved the submitted version.
The research work was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (HFRI-FM17-2971, Project Number: 81233).
We would like to express our gratitude to the Choose Life Foundation for its generous and continuous support. We would also like to thank D. Karamitros, M. Sakkou, A. Patmanidi and R. Calogero for their valuable advice and M. Tsima for the administrative support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1235661/full#supplementary-material
1. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
2. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med (2003) 9(9):1144–50. doi: 10.1038/nm915
3. Liu JMH, Chen P, Uyeda MJ, Cieniewicz B, Sayitoglu EC, Thomas BC, et al. Pre-clinical development and molecular characterization of an engineered type 1 regulatory T-cell product suitable for immunotherapy. Cytotherapy (2021) 23(11):1017–28. doi: 10.1016/j.jcyt.2021.05.010
4. Chen PP, Cepika AM, Agarwal-Hashmi R, Saini G, Uyeda MJ, Louis DM, et al. Alloantigen-specific type 1 regulatory T cells suppress through CTLA-4 and PD-1 pathways and persist long-term in patients. Sci Transl Med (2021) 13(617):eabf5264. doi: 10.1126/scitranslmed.abf5264
5. Stamou P, Marioli D, Patmanidi AL, Sgourou A, Vittoraki A, Theofani E, et al. Simple in vitro generation of human leukocyte antigen-G–expressing T-regulatory cells through pharmacological hypomethylation for adoptive cellular immunotherapy against graft-versus-host disease. Cytotherapy (2017) 19(4):521–30. doi: 10.1016/j.jcyt.2017.01.004
6. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37(5):785–99. doi: 10.1016/j.immuni.2012.09.010
7. Ohkura N, Yasumizu Y, Kitagawa Y, Tanaka A, Nakamura Y, Motooka D, et al. Regulatory T cell-specific epigenomic region variants are a key determinant of susceptibility to common autoimmune diseases. Immunity (2020) 52(6):1119–1132.e4. doi: 10.1016/j.immuni.2020.04.006
8. Stübig T, Badbaran A, Luetkens T, Hildebrandt Y, Atanackovic D, Binder TMC, et al. 5-azacytidine promotes an inhibitory T-cell phenotype and impairs immune mediated antileukemic activity. Mediators Inflamm (2014) 2014:e418292. doi: 10.1155/2014/418292
9. Choi J, Ritchey J, Prior JL, Holt M, Shannon WD, Deych E, et al. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood (2010) 116(1):129–39. doi: 10.1182/blood-2009-12-257253
10. Carosella ED, Favier B, Rouas-Freiss N, Moreau P, LeMaoult J. Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood (2008) 111(10):4862–70. doi: 10.1182/blood-2007-12-127662
11. Lazana I, Zoudiari A, Kokkinou D, Themeli M, Liga M, Papadaki H, et al. Identification of a novel HLA-G+ regulatory population in blood: expansion after allogeneic transplantation and de novo HLA-G expression at graft-versus-host disease sites. Haematologica (2012) 97(9):1338–47. doi: 10.3324/haematol.2011.055871
12. Feger U, Tolosa E, Huang YH, Waschbisch A, Biedermann T, Melms A, et al. HLA-G expression defines a novel regulatory T-cell subset present in human peripheral blood and sites of inflammation. Blood (2007) 110(2):568–77. doi: 10.1182/blood-2006-11-057125
13. Pankratz S, Bittner S, Herrmann AM, Schuhmann MK, Ruck T, Meuth SG, et al. Human CD4+HLA-G+ regulatory T cells are potent suppressors of graft-versus-host disease in vivo. FASEB J (2014) 28(8):3435–45. doi: 10.1096/fj.14-251074
14. Kefala D, Lysandrou M, Christofi P, Pieridou C, Papayanni PG, Savvopoulos N, et al. Immunotherapy: pre-clinical development of a decitabine-induced regulatory Hlag+Cd4+-t cell-enriched cell product (Ig-Treg) against graft-vs-host-disease. Cytotherapy (2022) 24(5):S118–9. doi: 10.1016/S1465-3249(22)00322-X
15. Lysandrou M, Kefala D, Christofi P, Savvopoulos N, Papayanni PG, Theodorellou R, et al. Study protocol: Phase I/II trial of induced HLA-G+ regulatory T cells in patients undergoing allogeneic hematopoietic cell transplantation from an HLA-matched sibling donor. Front Med (2023) 10:1166871. doi: 10.3389/fmed.2023.1166871
16. Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, Sahaf B, et al. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight (2019) 4(10). doi: 10.1172/jci.insight.127244
17. NooNepalle SK, Gu F, Lee EJ, Choi J-H, Han Q, Kim J, et al. Promoter methylation modulates indoleamine 2,3-dioxygenase 1 induction by activated T cells in human breast cancers. Cancer Immunol Res (2017) 5(4):330–44. doi: 10.1158/2326-6066.CIR-16-0182
18. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124(5):750–60. doi: 10.1182/blood-2013-12-546416
19. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
20. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117(5):1147–54. doi: 10.1172/JCI31178
21. Lam AJ, Lin DTS, Gillies JK, Uday P, Pesenacker AM, Kobor MS, et al. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human Treg identity. Cell Rep (2021) 36(5). doi: 10.1016/j.celrep.2021.109494
22. Boegel S, Löwer M, Schäfer M, Bukur T, de Graaf J, Boisguérin V, et al. HLA typing from RNA-Seq sequence reads. Genome Med (2012) 4(12):102. doi: 10.1186/gm403
23. Orenbuch R, Filip I, Comito D, Shaman J, Pe’er I, Rabadan R. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics (2020) 36(1):33–40. doi: 10.1093/bioinformatics/btz474
24. Chelysheva I, Pollard AJ, O’Connor D. RNA2HLA: HLA-based quality control of RNA-seq datasets. Briefings Bioinf (2021) 22(5):bbab055. doi: 10.1093/bib/bbab055
25. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281(5380):1191–3. doi: 10.1126/science.281.5380.1191
26. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med (2005) 11(3):312–9. doi: 10.1038/nm1196
27. Szántó S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res Ther (2007) 9(3):R50. doi: 10.1186/ar2205
28. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood (2009) 114(24):5062–70. doi: 10.1182/blood-2009-06-227587
29. Curreli S, Romerio F, MIrandola P, Barion P, Bemis K, Zella D. Human primary CD4 + T cells activated in the presence of IFN-alpha 2b express functional indoleamine 2,3-dioxygenase. J Interferon Cytokine Res (2001) 21(6):431–7. doi: 10.1089/107999001750277916
30. Boasso A, Herbeuval JP, Hardy AW, Winkler C, Shearer GM. Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4+ T cells. Blood (2005) 105(4):1574–81. doi: 10.1182/blood-2004-06-2089
31. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res (2009) 69(5):2000–9. doi: 10.1158/0008-5472.CAN-08-2360
32. Montane J, Bischoff L, Soukhatcheva G, Dai DL, Hardenberg G, Levings MK, et al. Prevention of murine autoimmune diabetes by CCL22-mediated Treg recruitment to the pancreatic islets. J Clin Invest (2011) 121(8):3024–8. doi: 10.1172/JCI43048
33. Montane J, Obach M, Alvarez S, Bischoff L, Dai DL, Soukhatcheva G, et al. CCL22 prevents rejection of mouse islet allografts and induces donor-specific tolerance. Cell Transpl (2015) 24(10):2143–54. doi: 10.3727/096368914X685249
34. Fisher JD, Zhang W, Balmert SC, Aral AM, Acharya AP, Kulahci Y, et al. In situ recruitment of regulatory T cells promotes donor-specific tolerance in vascularized composite allotransplantation. Sci Adv (2020) 6(11):eaax8429. doi: 10.1126/sciadv.aax8429
Keywords: regulatory T-cells, hypomethylating agents, HLA-G, RNA-Seq, ScRNA-seq, IDO-1
Citation: Lysandrou M, Stamou P, Kefala D, Pierides C, Kyriakou M, Savvopoulos N, Christofi P, Papadopoulou A, Yannaki E, Costeas P and Spyridonidis A (2023) Hypomethylation-induced regulatory programs in T cells unveiled by transcriptomic analyses. Front. Immunol. 14:1235661. doi: 10.3389/fimmu.2023.1235661
Received: 06 June 2023; Accepted: 21 August 2023;
Published: 27 September 2023.
Edited by:
Everett Meyer, Stanford University, United StatesReviewed by:
Tatiana Akimova, University of Pennsylvania, United StatesCopyright © 2023 Lysandrou, Stamou, Kefala, Pierides, Kyriakou, Savvopoulos, Christofi, Papadopoulou, Yannaki, Costeas and Spyridonidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandros Spyridonidis, c3B5cmlkb25pZGlzQHVwYXRyYXMuZ3I=
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.