 Jaap G. Neels
Jaap G. Neels Claire Gollentz2
Claire Gollentz2 Giulia Chinetti
Giulia Chinetti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 04 July 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1215612
This article is part of the Research TopicDifferential Activation of Cell Death Pathways in Macrophages as a Result of Adaptation to Divergent MicroenvironmentView all 7 articles
Cell death is an important aspect of atherosclerotic plaque development. Insufficient efferocytosis of death cells by phagocytic macrophages leads to the buildup of a necrotic core that impacts stability of the plaque. Furthermore, in the presence of calcium and phosphate, apoptotic bodies resulting from death cells can act as nucleation sites for the formation of calcium phosphate crystals, mostly in the form of hydroxyapatite, which leads to calcification of the atherosclerotic plaque, further impacting plaque stability. Excessive uptake of cholesterol-loaded oxidized LDL particles by macrophages present in atherosclerotic plaques leads to foam cell formation, which not only reduces their efferocytosis capacity, but also can induce apoptosis in these cells. The resulting apoptotic bodies can contribute to calcification of the atherosclerotic plaque. Moreover, other forms of macrophage cell death, such as pyroptosis, necroptosis, parthanatos, and ferroptosis can also contribute by similar mechanisms to plaque calcification. This review focuses on macrophage death in atherosclerosis, and its potential role in calcification. Reducing macrophage cell death and/or increasing their efferocytosis capacity could be a novel therapeutic strategy to reduce the formation of a necrotic core and calcification and thereby improving atherosclerotic plaque stability.
Atherosclerosis and its clinical complications are still a leading cause for morbidity and mortality worldwide. Although significant improvements have been made in reducing atherosclerosis-associated mortality in western countries, this pathology remains a global health issue. Over decades, a plaque grows and narrows the arteries’ lumen, leading to clinical manifestations such as myocardial infarction, stroke, and peripheral artery disease. Atherosclerotic plaques build up inside the large and median arteries (aorta, carotids, femoral arteries) due to the deposition of fat, cholesterol, calcium, fibrotic tissue, cells, and cellular debris. The development of atherosclerosis involves the activation of various cell types (including endothelial cells, smooth muscle cells (SMC), lymphocytes, monocytes, and macrophages) in the intima of the arteries, which results in a local inflammatory response. An increase in circulating low density lipoprotein (LDL)-cholesterol levels, and the subsequent accumulation of oxidized LDL (Ox-LDL) in the subendothelial space, triggers the recruitment and retention of monocytes and lymphocytes in the arterial wall. In the intima, monocytes differentiate into macrophages, which then scavenge lipoproteins, particularly Ox-LDL, and accumulate lipids, mainly cholesterol (1). One of the most important functions of macrophages in the context of atherosclerosis is the handling of cholesterol. The maintenance of macrophage cholesterol homeostasis is of critical importance because an imbalance between cholesterol influx and efflux leads to an excessive accumulation of cholesterol in macrophages and their transformation into foam cells (2, 3).
Macrophages express different scavenger receptors, including SR-A1, CD36, and lectin-like oxLDL receptor-1 (LOX-1), allowing internalization of lipoproteins, particularly Ox-LDL. They also express cholesterol transporters, including ATP-binding cassette transporters (ABCA1, ABCG1), and SR-BI, involved in the reverse cholesterol transport allowing elimination of excess unesterified cholesterol. Esterified cholesterol contained in Ox-LDL is internalized by phagocytosis and transported to late endosomes/lysosomes, where lysosomal acid lipase (LAL) hydrolyzes it and releases unesterified cholesterol, that is subsequently processed by acetyl-CoA acetyltransferase (ACAT1), in the endoplasmic reticulum to generate cholesteryl esters that accumulate in the cytoplasm leading to the formation of foam cells. On the other hand, newly synthesized cholesteryl esters can be hydrolyzed by neutral cholesterol ester hydrolase (NCEH), to generate free cholesterol that can leave the cells through the cholesterol transporters. Furthermore, other cell types, particularly SMC, can also become foam cells (4). Moreover, large amounts of intracellular unesterified cholesterol accumulation in the endoplasmic reticulum (ER) alters the function of integral ER membrane proteins, induces the ER stress signal transduction pathway, leading to an apoptotic response in the macrophages. In vivo evidence suggests that this event may promote plaque destabilization in advanced atherosclerotic lesions (5). Additionally, these macrophage-derived foam cells secrete inflammatory molecules and factors that further promote lipoprotein retention, affect SMC phenotype, proliferation, and migration from the media to the intima and sustain inflammation (2). Infiltrated macrophages also secrete matrix metalloproteases (MMPs) and cysteine endoproteases leading to matrix degradation and plaque rupture (6).
Another important factor regulating plaque stability, in addition to the accumulation of foam cells, SMC differentiation/proliferation/migration, and extracellular matrix degradation, is vascular calcification (VC), corresponding to the deposition of calcium crystals in the vascular wall (7). VC is associated with increased overall mortality and cardiovascular events (8). An important initiator of VC is cell death and, in this review, that is part of the article collection “Differential Activation of Cell Death Pathways in Macrophages as a Result of Adaptation to Divergent Microenvironment”, we focus on the role of macrophage cell death in atherosclerosis, and its potential role in VC.
Apoptosis can occur in different cell types involved in atherosclerosis, such as SMC, endothelial cells, T lymphocytes and macrophages. Apoptotic macrophages account for more than 40% of dead cells (9). Apoptotic cell death progresses through several stages, initiating with nuclear chromatin condensation, followed by membrane blebbing, leading to disintegration of the cellular content into distinct membrane enclosed vesicles called apoptotic bodies (10). In addition, atherosclerotic plaque macrophages can undergo other types of cell death then apoptosis and necrosis. Indeed, while apoptosis remains the predominant form of macrophage death in atherosclerosis, less-well characterized types of regulated necrosis, such as pyroptosis, necroptosis, parthanatos and ferroptosis have been reported (11, 12) (Figure 1). Pyroptosis occurring in response to bacterial infection, is accompanied by NLRP3 inflammasome activation and by maturation of pro-inflammatory cytokines IL-1β and IL-18 (13, 14). Necroptosis is a form of cell death activated by death receptors, such as TNFR1, IFNR and TLR3/4 (15). The presence of lipopolysaccharide (LPS) from Escherichia Coli and Toll-like receptor 4 (TLR4) has been studied in specimens from carotids and controls matched patients. Immunochemistry analysis shown positive signals for LPS and TLR4 coincidentally with positivity for CD68 in the atherosclerotic plaque of carotids and the positivity for LPS and TLR4 was greater in the area with activated macrophages. These data provide the first evidence that LPS from Escherichia Coli localizes in human plaque and may contribute to atherosclerotic damage via TLR4-mediated oxidative stress (16). However, this link between infection and atherosclerosis is controversial and another mechanism was proposed where oxLDL can induce sterile inflammation through CD36-TLR4-TLR6 activation (17). Parthanatos is a mitochondrial-dependent cell death, triggered by DNA damaging stimuli, such as peroxynitrite or reactive oxygen species (ROS)-dependent activation of poly ADP-ribose polymerase (PARP)1 (18). Finally, ferropoptosis is a newly identified type of cell death caused by accumulation of iron-dependent lipid peroxides (19). The clearance of apoptotic cells is mostly mediated by macrophages, which recognize and internalize dead cells (endothelial cells, SMC, lymphocytes, senescent erythrocytes, neutrophils.) in a process termed efferocytosis (20–23).
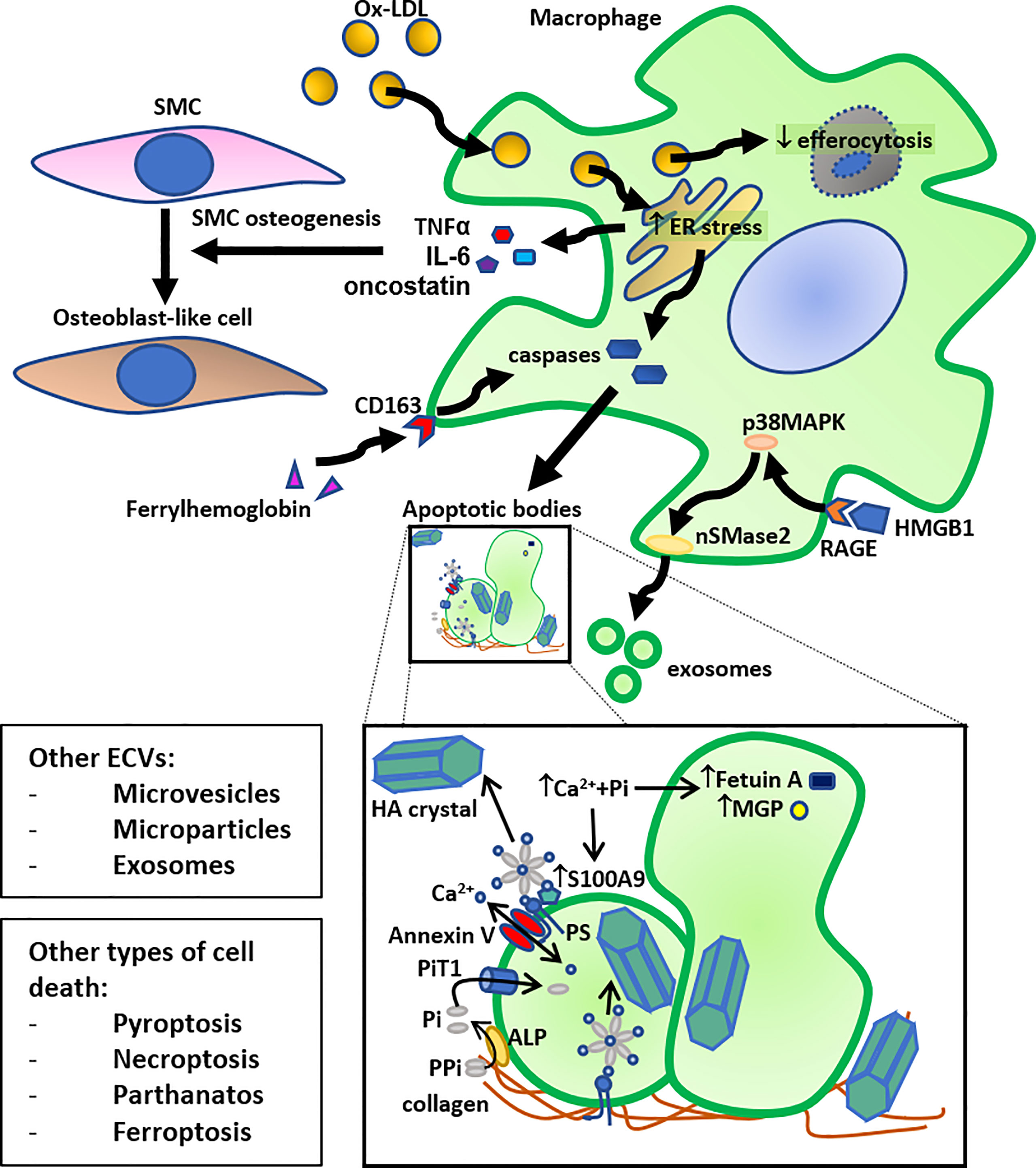
Figure 1 Schematic summary of the role of macrophage death in calcification. Macrophage cholesterol loading by oxidized LDL (Ox-LDL) (foam cell formation) leads to ER stress, followed by activation of caspases leading to apoptosis and release of apoptotic bodies. Another pathway that can induce macrophage apoptosis, followed by release of apoptotic bodies, is the ferrylhemoglobin internalization by CD163. Furthermore, high mobility group box 1 (HMGB1), released by dying cells, can induce exosome secretion by macrophages through activation of neutral sphingomyelinase-2 (nSMase2) by the receptor for advanced glycation end products (RAGE)/p38 mitogen-activated protein kinase (p38MAPK) pathway. The resulting apoptotic bodies and exosomes, or other secreted extra-cellular vesicles (ECVs), such as microvesicles and microparticles, bind to collagen in the extracellular matrix and act as calcium binding sites for hydroxyapatite (HA) crystals. More specifically, an annexin V - phosphatidylserine (PS) - S100A9/MRP14 membrane complex on the surface of ECVs acts as a nucleation site for HA crystal formation. More specifically, the anionic PS serves as a binding site for cationic Ca2+. HA crystals can also bind to collagen directly through electrostatic interactions; cationic moieties on collagen interact with negatively charged groups on HA crystal surface. Inorganic phosphate (Pi), derived from pyrophosphate (PPi) through alkaline phosphatase (ALP), can be transported into the ECV by the Pi transporter 1 (PiT1), and calcium can be transported by Annexin V. This accumulation of calcium and Pi also leads to HA crystal formation inside the ECV. An increase in the presence of calcium and inorganic phosphate (Pi) decreases the content of calcification inhibitors, such as Fetuin A and MGP, in these ECVs, and increases the presence of S100A9 on their surface, leading to further stimulation of HA crystal formation. Together, these mechanisms allow macrophages to contribute directly to VC. Macrophage foam cells also have reduced efferocytosis, and these lipid-loaded stressed macrophages secrete osteogenic factors, such as TNF-α, IL-6, and oncostatin, that stimulate trans-differentiation of smooth muscle cells (SMC) to osteoblast-like cells, thereby also indirectly contributing to VC. Lastly, other forms of cell death, such as pyroptosis, necroptosis, parthanatos, and ferroptosis can also lead to similar induction of calcification.
In early lesions, phagocytes readily clear apoptotic cells, avoiding further progression of atherosclerosis, thus rendering plaques small and stable. During atherosclerotic plaque progression, ROS are abundantly produced by macrophages, leading to the formation of Ox-LDL that compete with apoptotic cells, interact with receptors of phagocytes thus leading to a reduced efferocytic capacity (24). This is one of the mechanisms by which Ox-LDL reduced efferocytosis. Moreover, it has been reported that the 7 keto-cholesterol (7KC) contained in Ox-LDL, and accumulating in macrophages and foam cells, impairs macrophage efferocytosis by a mechanism involving catabolism of phosphatidylinositol 4.5-biphosphate (PtdIns (4,5)P2) impairing actin depolymerization required for the completion of phagocytosis (25). Indeed,7KC inhibits both Fcγ receptor mediated and efferocytotic pathway by impairing phospholipase Cγ (PLCγ) thus preventing hydrolysis of PtdIns (4,5)P2.
Additionally, transformation of macrophages into foam cells also diminishes their capacity to clear apoptotic debris (26). Therefore, in chronic, advanced lesions, the combination of high levels of macrophage apoptosis and defective efferocytosis causes accumulation of dead pro-inflammatory macrophages that can undergo post-apoptotic necrosis, leading to the formation of a necrotic core which contributes to plaque inflammation, instability and rupture (21, 27). Moreover, in atherosclerotic lesions, macrophages are exposed to different environmental stimuli (such as modified lipids, cytokines, and senescent erythrocytes) that dictate their functional phenotype. Indeed, several sub-classes of macrophages have been described in atherosclerosis, defined by the expression of specific surface markers, production of specific factors and biological functions (28). In humans, pro-inflammatory macrophages have been detected in the necrotic core of the plaque, differentiated in the presence of Ox-LDL, cholesterol crystals and pro-inflammatory cytokines, and producing pro-inflammatory factors. Anti-inflammatory, reparative macrophages have also been identified in human plaques. Their differentiation is drawn by different stimuli (IL-4, IL-13, oxidized phospholipids, CXCL4). They have the property to produce anti-inflammatory factors and are generally resistant to lipid accumulation and foam cell formation (28). Foam cells have been described as being less inflammatory than non-foamy cells (29). Moreover, anti-inflammatory macrophages located in areas of neo-vascularization or outside the lipid core are very effective in efferocytosis and phagocytosis since they highly express opsonins and receptors (30, 31). Notably, anti-inflammatory macrophages express high levels of C1qa, C1qb, C1qc, GAS-6 and thrombospondin-1. Since C1q deficiency leads to defective clearance of apoptotic cells (32), the high levels of several opsonins expressed in anti-inflammatory macrophages provide the molecular basis for their high phagocytotic capacity and efferocytosis of apoptotic pro-inflammatory macrophages and senescent erythrocytes, thus contributing to inflammation resolution (30, 32). Interestingly, pro-inflammatory macrophages are abundant in unstable human atherosclerotic plaques, whereas more stable plaques are enriched in anti-inflammatory macrophages (33).
VC can be located in the media (34), especially in patients with type 2 diabetes (35) and chronic kidney disease (36), and originates from the osteochondrogenic differentiation of SMC which share a common origin with osteoblasts, cells responsible for bone mineralization (37). Medial calcification is characterized by diffuse mineral deposition throughout the vascular tree and occurs independently of atherosclerosis. When media calcification, also called Mönckeberg’s medial sclerosis (38), affects the aorta and large arteries (such as the iliac arteries and the arteries of the lower limbs), it decreases their elasticity and causes hemodynamic alterations (39), leading to hypertension, aortic stenosis and left ventricular hypertrophy (40). Intimal VC, associated with inflammation and oxidative stress, as well as the development of atherosclerotic plaques and occlusive lesions (41), is more frequent at the coronary and carotid level, and is largely influenced by traditional cardiovascular risk factors (41).
As mentioned above, diabetes plays a major role in VC in the media and in Mönckeberg’s disease (35). In addition, diabetes also contributes to the development of atherosclerosis since early stages, and is associated with atherosclerotic plaque calcification, which occurs in the advanced stages of atherosclerosis (42).
In the general population, VC has a high heritable component, suggesting that in certain cases there is a genetic factor contributing to the disease (43). However, there are few monogenic disorders presenting with early-onset cardiovascular disease that can be classified into disorders caused by an altered purine and phosphate/pyrophosphate metabolism (calcification of joints and arteries, generalized arterial calcification of infancy, Hutchinson-Gilford progeria syndrome, idiopathic basal ganglia calcification, pseudoxanthoma elasticum, interferonopathies (Singleton-Merten Syndrome), and Gaucher disease) (44).
Two types of calcifications have been reported in atherosclerotic plaques, based on their size. Initially, microcalcifications occur (45, 46) with a size lesser than 50 μm, indetectable by conventional imaging methods (47). They are identified in unstable plaques (48), favoring their rupture (49) and are associated with the presence of macrophages with pro-inflammatory activity (50). Secondarily, these microcalcifications coalesce to form macrocalcifications, with a size greater than 50 μm, present in stable plaques rather associated with anti-inflammatory macrophages (51). The calcium crystals present in atherosclerotic plaques, can be of different nature, including crystals of calcium phosphate (hydroxyapatite) which are predominant, calcium carbonate, calcium oxalate, dicalcium phosphate dihydrate and octacalcium phosphate (52). VC is a dynamic regulated process sharing many features with bone formation, implicating both stimulating and inhibitory factors, mediated by osteoblast-like and osteoclast-like cells, respectively, and involving particularly SMC and macrophages. Osteoclasts originate from hematopoietic lineage cells derived from bone marrow myeloid precursors or circulating monocytes (53). The presence of osteoclast-like cells in the vascular wall is rather limited, possibly because factors that inhibit/modulate the differentiation of monocyte/macrophages into osteoclast-like cells are present (54). In atherosclerotic lesions, macrophages that express both the MCSF and RANKL receptors (55) can be activated by their ligands produced by endothelial cells, SMC, and monocyte/macrophages themselves, and can thus potentially differentiate into osteoclast-like cells. Moreover, the presence of macrophages displaying an anti-inflammatory phenotype has been reported in areas surrounding calcium deposits in human atherosclerotic plaques (56). However, these anti-inflammatory macrophages express carbonic anhydrase type II (degrading the inorganic part of mineralized extra-cellular matrix), but relatively low levels of cathepsin K, involved in the degradation of the organic part of extracellular matrix, allowing them to be phenotypically defective to resorb calcification (56). Concerning the extra-cellular matrix mineralization, in the presence of Ox-LDL, macrophages produce TNFα, that enhances SMC osteoblastic-like trans-differentiation, alkaline phosphatase (ALP) expression and matrix mineralization (57). Moreover, macrophages express several bone-related proteins, such as matrix Gla protein (MGP), ALP, bone sialoprotein and osteopontin (58). In response to various pro-osteogenic signals such as high glucose, inflammation, and oxidative stress, SMC undergo an osteoblast-like trans-differentiation by acquiring an osteoblastic phenotype and become able to deposit calcium and other minerals within the plaque (59) a process that requires the expression of osteogenic transcription factors such as RUNX2 (60), as well as enzymes such as ALP.
Cell apoptosis from SMC and/or macrophages, is considered as one of the main drivers of calcification in humans. Indeed, post confluent human SMC spontaneously form nodules in cell culture and induce calcification, as detected by von Kossa’s method, Alizarin red S staining, and electron microscopy (61). Apoptosis induction of these cells with anti-Fas IgM and cycloheximide for 24 hours, demonstrated by Tunel labeling and phosphatidyl serine exposure, increased nodule calcification (61). Moreover, apoptosis inhibition with the cell permeable caspase inhibitor ZVAD.fmk decreases calcification, as assessed by alizarin red staining (61). Caspase inhibition can also lead to a reduced release of apoptotic bodies from cells (61). Interestingly, DNA damage characteristics were observed after 7 days of culture, while the calcification occurs after 28 days (61), thus suggesting that apoptosis precedes calcium crystal formation. Moreover, SMC-derived apoptotic bodies initiate calcification in a similar way as already reported for chondrocytes in cartilage (62). These apoptotic bodies accumulate calcium and need an integral membrane; phosphatidyl serine exposure by apoptotic bodies generates potential calcium binding sites suitable for hydroxyapatite deposition (61). Moreover SMC-derived matrix vesicles are also involved in the initiation of VC (63). These vesicles contain apoptotic proteins, such as BAX, suggesting that they may be remnants of apoptotic cells (64). Some studies have suggested that apoptotic bodies in atherosclerotic plaques are like matrix vesicles and may initiate calcification (65, 66). However, much controversy exists in the field concerning the classification and nomenclature used to classify different extra-cellular vesicles (ECVs), depending on the method of purification, the surface marker used, the morphology, selected size, etc … It’s actually admitted that the terms ECVs includes exosomes, microvesicles, microparticles, and apoptotic bodies (67). These ECVs, that can physiologically derive from macrophages, SMC, and endothelial cells, are microscopic phospholipid bilayer-enriched particles, of round or ovoid shape, covering size from approximatively 30 nm to 5 µm (68, 69). They are often associated with extracellular matrix components, particularly collagen, and displaying evidence of hydroxyapatite crystals on the inner membrane within the lumen, and on the outer membrane of the vesicle. Under physiological conditions, these ECVs are enriched in calcification inhibitors, such as fetuin A or MGP (70). When exposed to an excess of calcium and phosphate, the concentration of these inhibitors is reduced and ECVs become enriched in annexin V-and phosphatidylserine that allow calcification by inducing collagen binding (71) (Figure 1). Conditioned media collected from human coronary SMC cultured for 14 days in calcifying conditions (medium containing dexamethasone, L-ascorbic acid and β-glycerophosphate) was used to isolate ECVs by ultracentrifugation. The latter have been fluorescently labelled and added to collagen hydrogels, resulting in a progressive formation of ECV aggregates, between collagen fibers. In this way, calcium and phosphate nucleate to form hydroxyapatite leading to the appearance of dense calcifying structure like those observed in human calcified plaques (72). Thus, collagen serves as a scaffold directing ECVs aggregation and microcalcification formation. Calcifying ECVs have been reported in medial and intimal calcification (73), as well as in calcified aortic valves (74) and appear similar to the ECVs involved in physiological bone mineralization (75).
While the role of SMC apoptosis in initiation of calcium deposits is well documented and established in the literature (61), those of macrophages is still in its infancy, particularly because the direct role of macrophages in VC is quite novel. Indeed, while it is accepted that macrophages can modulate SMC mineralization by indirect paracrine mechanism, through the secretion of pro-osteogenic cytokines and factors (57), their direct role in VC, as cellular actors able to mineralize the extra-cellular matrix is more recent. Thus, further studies are necessary to understand the role of macrophage apoptosis in VC.
However, deep histological analysis of human samples, revealed that apoptotic macrophages, more often apoptotic foam cells, are present in areas of calcium deposits as microcalcifications (76). These calcifications form into the lipid core in early fibroatheroma where macrophages infiltrate the plaque, undergo apoptosis and release matrix vesicles (77). Macrophage apoptosis may result in a different morphology of calcium deposits, compared to apoptotic SMC. Indeed, while SMC apoptosis led to fine microcalcifications, those derived from apoptotic macrophages are characterized by large, punctate and blocky appearance (76). These calcifications, generally observed in the deeper areas of the necrotic core, close to the internal elastic lamina (78), then coalesce to form larger calcification areas, called “fragmented” calcification, in a process involving both the necrotic core as well as the surrounding extra-cellular matrix (77). One of the more accepted hypothesis is that SMC apoptosis is the driving force for microcalcification (77), followed by macrophage infiltration into the lipid core where they also undergo cell death and calcification. Cell death provides phospholipid rich debris that serve as nucleation site of hydroxyapatite, a process that starts within lipid pools and progresses with inflammation and further cell death, leading to the development of necrotic core (78). Recently, an oxidative form of hemoglobin (ferrylhemoglobin) has been detected in complicated human atherosclerotic lesions. This ferrylhemoglobin, which is internalized by macrophages through the CD163 pathways, affected macrophage polarization, induced apoptosis as well as calcification (79) (Figure 1). Interestingly, while intraplaque hemorrhage has been associated with calcification and may facilitate its progression (80), a recent study demonstrated a mechanism by which CD163+ macrophages can inhibit VC through NF-κB–induced enhanced production of the anti-calcific glycosaminoglycan, hyaluronan (HA) (81).
Mechanistical data supporting the direct role of macrophage apoptosis/death in the process of VC are rare. Moreover, macrophages are an important source of ECVs that act as hydroxyapatite nucleation initiators. Indeed, microcalcifications in vulnerable plaques contain more macrophages than SMC in stable plaques (45, 73). When stimulated by phosphate and calcium, in vitro RAW 264.7 murine macrophages increase the number of released pro-calcifying EVCs, expressing exosomal markers (CD9 and TSG101) and being enriched in S100A9, also known as migration inhibitory factor-related protein 14 (MRP-14) and annexin V (Figure 1). Silencing S100A9 in vitro as well as deletion in S100A9-/- deficient mice reduced EVC calcification, whereas stimulation with S100A9 increased their calcification potential. The calcium/phosphate induction of phosphatidylserine-annexin V-S100A9 membrane complex in these EVCs facilitates the hydroxyapatite nucleation in the cell membrane (73). LPS-stimulation of RAW 264.7 murine macrophages decreased the number of ECVs, but these were able to induce inflammation and oxidative stress in SMC, resulting, under calcifying conditions, in an increased expression of osteogenic markers accompanied by an induced calcification and a decrease in contractile marker expression (82). These observations were specific to the LPS-induced ECVs, since LPS alone did not modulate calcification.
Moreover, the role of Transient Receptor Potential Canonical 3 (TRPC3), a non-selective calcium-permeable channel and obligatory signaling component of the ER stress-induced apoptosis (83) has been evaluated. Transfer of bone marrow with deficiency of the TRPC3into LDL-R KO recipient mice, led to a decrease in necrosis and number of apoptotic pro-inflammatory macrophages (84), compared to control mice, suggesting a more stable plaque phenotype. Interestingly, these mice were also characterized by a decreased calcification of the aortic roots, an effect accompanied by a significant reduction of the plaque expression of osteogenic markers, such as BMP-2, ALP and RUNX2 (85, 86). Since apoptotic bodies are important centers for nucleation, it can by hypothesized that the reduced calcification observed in this model is in part due to the reduced number of apoptotic macrophages present in the plaque. These data suggested that macrophage ER stress induced apoptosis can be targeted to control calcification. Disruption of ER homeostasis causes accumulation of unfolded and misfolded proteins in the ER lumen, leading to activation of ER stress signaling. ER stress signaling is composed of three signaling axes that are initiated by inositol-requiring protein-1 (IRE1), double-stranded RNA-dependent protein kinase-like ER kinase (PERK) and activating transcription factor 6 (ATF6). During ER homeostasis, PERK, ATF6, and IRE1 combine with the molecular chaperone GRP78T to form a stable complex. Under ER stress, these proteins disaggregate and stimulate three important signaling pathways, resulting in the expression of apoptosis protein CHOP and the activation of Jun-N-terminal kinase (JNK) to promote apoptosis (87).
PARP1 has been reported to contribute to foam cell death, particularly parthanatos, an important determinant of plaque composition (88). Interestingly, specific PARP1 macrophage deletion in diabetic apolipoprotein E deficient mice, attenuates the formation of calcium nodules in atherosclerotic lesion and significantly decreases the total calcium content of the aorta (89). In vitro experiments indicated that PARP1 inhibition reverses the high glucose-induced calcification as well as RUNX2 expression, by a mechanism involving STAT1 pathway. These results suggest that PARP1 inhibition decreases the osteogenic like potential of macrophages. Moreover, PARP1 inhibition leads to a shift of macrophage polarization toward an anti-inflammatory phenotype (89). Indeed, different PARP1 inhibitors have demonstrated atheroprotective effects by decreasing PARP activation, inflammatory markers, macrophage recruitment, endothelial dysfunction, foam cell death thus promoting plaque stability (88).
Finally, High mobility group box protein 1 (HMGB1) associated with macrophage pyroptosis (90), accumulated in areas of calcification (91) and has been identified as potential inducer of ECVs secretion by macrophages (92). HMGB1can induce exosome secretion by activating the neutral sphingomyelinase-2 (nSMase2) by the receptor for advanced glycation end products (RAGE)/p38 mitogen-activated protein kinase (p38MAPK) pathway. Indeed, failure in apoptotic bodies cleaning by macrophages allow calcium crystals to grow (93) and also result in the release of pro-inflammatory cytokines (such as TNFα, IL-6, and oncostatin M) able to modulate VC by inducing osteogenic gene expression in SMC (57, 94, 95). Interestingly, IL-1β released by macrophages during pyroptosis by a caspase-1 dependent mechanism, enhanced SMC osteogenic differentiation and subsequent calcification (96).
If we consider that in advanced atherosclerotic plaques the efferocytosis capacity of macrophages decreased, this will lead to a greater availability of apoptotic bodies and necrotic cells which could serve as a nucleation starter site for the formation of calcium crystals. Additionally, defective cleaning of apoptotic cell bodies by macrophages, derived from macrophages themselves or from SMC (97), will results in the release of inflammatory cytokines, also able to impact calcification (98). This may be a plausible explanation for the observation that calcification preferentially develops in advanced plaques.
Moreover, it has been reported that anti-inflammatory macrophages located around the calcium deposits in human plaques, are highly efferocytotic (30). However, the capacity of these macrophages to directly participate to VC is under investigated, even though they display a defective osteoclast-like phenotype but appear to be able to participate to extracellular matrix mineralization (56) and we can hypothesize that their efferocytosis effectiveness could be also modified by the presence of calcium deposits. It would thus be necessary to better study the impact of osteoclast defective phenotype and enhanced efferocytosis of these macrophages in terms of calcification. We can thus hypothesize that macrophages can be crucial in the control of calcium deposition in atherosclerosis, since they are cells highly specialized in efferocytosis, thus reducing the number of environmental apoptotic cells. Considering that inhibition of apoptosis results in inhibition of calcification (61), we can speculate that targeting macrophage efferocytosis may be a promise way to reduce VC as well as general plaque progression.
Bibliography search, writing and editing: JN, CG and GC. All authors have read and agreed to the published version of the manuscript.
This work was supported by the “Agence Nationale de la Recherche” (ANR-16-CE14-0001) and the “Fédération française de cardiologie” to GC and by the European Foundation for the Study of Diabetes (EFSD, to JN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Libby P, Aikawa M, Schonbeck U. Cholesterol and atherosclerosis. Biochim Biophys Acta (2000) 1529:299–309. doi: 10.1016/S1388-1981(00)00161-X
2. Libby P. Inflammation in atherosclerosis. Nat (London) (2002) 420:868–74. doi: 10.1038/nature01323
3. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med (2015) 21:628–37. doi: 10.1038/nm.3866
4. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl) (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
5. Tabas I. Apoptosis and plaque destabilization in atherosclerosis: the role of macrophage apoptosis induced by cholesterol. Cell Death Differ (2004) 11 Suppl 1:S12–6. doi: 10.1038/sj.cdd.4401444
6. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nat (London) (2011) 473:317–25. doi: 10.1038/nature10146
7. Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation (2008) 117:2938–48. doi: 10.1161/CIRCULATIONAHA.107.743161
8. Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag (2009) 5:185–97. doi: 10.2147/VHRM.S4822
9. Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol (2000) 157:1259–68. doi: 10.1016/S0002-9440(10)64641-X
10. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26:239–57. doi: 10.1038/bjc.1972.33
11. Yan G, Dawood M, Bockers M, Klauck SM, Fottner C, Weber MM, et al. Multiple modes of cell death in neuroendocrine tumors induced by artesunate. Phytomedicine (2020) 79:153332. doi: 10.1016/j.phymed.2020.153332
12. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol (2019) 10:306. doi: 10.3389/fphar.2019.00306
13. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
14. Xu XD, Chen JX, Zhu L, Xu ST, Jiang J, Ren K. The emerging role of pyroptosis-related inflammasome pathway in atherosclerosis. Mol Med (2022) 28:160. doi: 10.1186/s10020-022-00594-2
15. Zhang X, Ren Z, Xu W, Jiang Z. Necroptosis in atherosclerosis. Clin Chim Acta (2022) 534:22–8. doi: 10.1016/j.cca.2022.07.004
16. Carnevale R, Nocella C, Petrozza V, Cammisotto V, Pacini L, Sorrentino V, et al. Localization of lipopolysaccharide from escherichia coli into human atherosclerotic plaque. Sci Rep (2018) 8:3598. doi: 10.1038/s41598-018-22076-4
17. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol (2010) 11:155–61. doi: 10.1038/ni.1836
18. Robinson N, Ganesan R, Hegedus C, Kovacs K, Kufer TA, Virag L. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
19. Ma J, Zhang H, Chen Y, Liu X, Tian J, Shen W. The role of macrophage iron overload and ferroptosis in atherosclerosis. Biomolecules (2022) 12:1702. doi: 10.3390/biom12111702
20. Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol (2005) 25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f
21. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol (2010) 10:36–46. doi: 10.1038/nri2675
22. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
23. Wang L, Li H, Tang Y, Yao P. Potential mechanisms and effects of efferocytosis in atherosclerosis. Front Endocrinol (Lausanne) (2020) 11:585285. doi: 10.3389/fendo.2020.585285
24. Song G, Wu X, Zhang P, Yu Y, Yang M, Jiao P, et al. High-density lipoprotein inhibits ox-LDL-induced adipokine secretion by upregulating SR-BI expression and suppressing ER stress pathway. Sci Rep (2016) 6:30889. doi: 10.1038/srep30889
25. Lu SM, Fairn GD. 7-ketocholesterol impairs phagocytosis and efferocytosis via dysregulation of phosphatidylinositol 4,5-bisphosphate. Traffic (2018) 19:591–604. doi: 10.1111/tra.12576
26. Su YR, Dove DE, Major AS, Hasty AH, Boone B, Linton MF, et al. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein e-deficient mice lacking macrophage-derived ACAT1. Circulation (2005) 111:2373–81. doi: 10.1161/01.CIR.0000164236.19860.13
27. Gonzalez L, Trigatti BL. Macrophage apoptosis and necrotic core development in atherosclerosis: a rapidly advancing field with clinical relevance to imaging and therapy. Can J Cardiol (2017) 33:303–12. doi: 10.1016/j.cjca.2016.12.010
28. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol (2015) 12:10–7. doi: 10.1038/nrcardio.2014.173
29. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
30. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPAR{gamma} and LXRα pathways. Circ Res (2011) 108:985–95. doi: 10.1161/CIRCRESAHA.110.233775
31. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (2012) 189:3508–20. doi: 10.4049/jimmunol.1200662
32. Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med (2000) 192:359–66. doi: 10.1084/jem.192.3.359
33. Li H, Cao Z, Wang L, Liu C, Lin H, Tang Y, et al. Macrophage subsets and death are responsible for atherosclerotic plaque formation. Front Immunol (2022) 13:843712. doi: 10.3389/fimmu.2022.843712
34. Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J (2014) 35:1515–25. doi: 10.1093/eurheartj/ehu163
35. Yahagi K, Kolodgie FD, Lutter C, Mori H, Romero ME, Finn AV, et al. Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol (2017) 37:191–204. doi: 10.1161/ATVBAHA.116.306256
36. Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation (2007) 116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342
37. Kassem M, Abdallah BM, Saeed H. Osteoblastic cells: differentiation and trans-differentiation. Arch Biochem Biophys (2008) 473:183–7. doi: 10.1016/j.abb.2008.03.028
38. Chauhan A, Sandal R, Jandial A, Mishra K. Diabetes mellitus, monckeberg's sclerosis and cardiovascular disease. BMJ Case Rep (2022) 15(2):e245778. doi: 10.1136/bcr-2021-245778
39. Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol (2004) 15:2959–64. doi: 10.1097/01.ASN.0000145894.57533.C4
40. Niederhoffer N, Marque V, Lartaud-Idjouadiene I, Duvivier C, Peslin R, Atkinson J. Vasodilators, aortic elasticity, and ventricular end-systolic stress in nonanesthetized unrestrained rats. Hypertension (1997) 30:1169–74. doi: 10.1161/01.HYP.30.5.1169
41. Pugliese G, Iacobini C, Blasetti Fantauzzi C, Menini S. The dark and bright side of atherosclerotic calcification. Atherosclerosis (2015) 238:220–30. doi: 10.1016/j.atherosclerosis.2014.12.011
42. Kronmal RA, McClelland RL, Detrano R, Shea S, Lima JA, Cushman M, et al. Risk factors for the progression of coronary artery calcification in asymptomatic subjects: results from the multi-ethnic study of atherosclerosis (MESA). Circulation (2007) 115:2722–30. doi: 10.1161/CIRCULATIONAHA.106.674143
43. Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res (2011) 109:578–92. doi: 10.1161/CIRCRESAHA.111.247965
44. Rutsch F, Buers I, Nitschke Y. Hereditary disorders of cardiovascular calcification. Arterioscler Thromb Vasc Biol (2021) 41:35–47. doi: 10.1161/ATVBAHA.120.315577
45. Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation (2007) 116:2841–50. doi: 10.1161/CIRCULATIONAHA.107.732867
46. Kawtharany L, Bessueille L, Issa H, Hamade E, Zibara K, Magne D. Inflammation and microcalcification: a never-ending vicious cycle in atherosclerosis? J Vasc Res (2022) 59(3):137–50. doi: 10.1159/000521161
47. Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, Fletcher AM, et al. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol (2012) 59:1539–48. doi: 10.1016/j.jacc.2011.12.037
48. Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation (2004) 110:3424–9. doi: 10.1161/01.CIR.0000148131.41425.E9
49. Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, et al. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol (2012) 303:H619–28. doi: 10.1152/ajpheart.00036.2012
50. Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. J Atheroscler Thromb (2018) 25:294–303. doi: 10.5551/jat.RV17020
51. Cho KY, Miyoshi H, Kuroda S, Yasuda H, Kamiyama K, Nakagawara J, et al. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J Stroke Cerebrovasc Dis (2013) 22:910–8. doi: 10.1016/j.jstrokecerebrovasdis.2012.11.020
52. Perrotta I, Perri E. Ultrastructural, elemental and mineralogical analysis of vascular calcification in atherosclerosis. Microsc Microanal (2017) 23:1030–9. doi: 10.1017/S1431927617012533
53. Massy ZA, Mentaverri R, Mozar A, Brazier M, Kamel S. The pathophysiology of vascular calcification: are osteoclast-like cells the missing link? Diabetes Metab (2008) 34 Suppl 1:S16–20. doi: 10.1016/S1262-3636(08)70098-3
54. Mozar A, Haren N, Chasseraud M, Louvet L, Maziere C, Wattel A, et al. High extracellular inorganic phosphate concentration inhibits RANK-RANKL signaling in osteoclast-like cells. J Cell Physiol (2008) 215:47–54. doi: 10.1002/jcp.21283
55. Sherr CJ, Rettenmier CW, Sacca R, Roussel MF, Look AT, Stanley ER. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell (1985) 41:665–76. doi: 10.1016/S0092-8674(85)80047-7
56. Chinetti-Gbaguidi G, Daoudi M, Rosa M, Vinod M, Louvet L, Copin C, et al. Human alternative macrophages populate calcified areas of atherosclerotic lesions and display impaired RANKL-induced osteoclastic bone resorption activity. Circ Res (2017) 121:19–30. doi: 10.1161/CIRCRESAHA.116.310262
57. Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation (2000) 102:2636–42. doi: 10.1161/01.CIR.102.21.2636
58. Shanahan CM, Cary NR, Metcalfe JC, Weissberg PL. High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J Clin Invest (1994) 93:2393–402. doi: 10.1172/JCI117246
59. Jaminon A, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci (2019) 20(22):5694. doi: 10.3390/ijms20225694
60. Abbasian N. Vascular calcification mechanisms: updates and renewed insight into signaling pathways involved in high phosphate-mediated vascular smooth muscle cell calcification. Biomedicines (2021) 9(7):804. doi: 10.3390/biomedicines9070804
61. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res (2000) 87:1055–62. doi: 10.1161/01.RES.87.11.1055
62. Hashimoto S, Ochs RL, Rosen F, Quach J, McCabe G, Solan J, et al. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad Sci U.S.A. (1998) 95:3094–9. doi: 10.1073/pnas.95.6.3094
63. Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix gla protein and low levels of osteopontin expression. Arterioscler Thromb Vasc Biol (1998) 18:379–88. doi: 10.1161/01.ATV.18.3.379
64. Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation (1998) 97:2307–15. doi: 10.1161/01.CIR.97.23.2307
65. Kockx MM. Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol (1998) 18:1519–22. doi: 10.1161/01.ATV.18.10.1519
66. Bauriedel G, Hutter R, Welsch U, Bach R, Sievert H, Luderitz B. Role of smooth muscle cell death in advanced coronary primary lesions: implications for plaque instability. Cardiovasc Res (1999) 41:480–8. doi: 10.1016/S0008-6363(98)00318-6
67. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles (2015) 4:27066. doi: 10.3402/jev.v4.27066
68. Buffolo F, Monticone S, Camussi G, Aikawa E. Role of extracellular vesicles in the pathogenesis of vascular damage. Hypertension (2022) 79:863–73. doi: 10.1161/HYPERTENSIONAHA.121.17957
69. Hutcheson JD, Aikawa E. Extracellular vesicles in cardiovascular homeostasis and disease. Curr Opin Cardiol (2018) 33:290–7. doi: 10.1097/HCO.0000000000000510
70. Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol (2005) 16:2920–30. doi: 10.1681/ASN.2004100895
71. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res (2011) 109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808
72. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater (2016) 15:335–43. doi: 10.1038/nmat4519
73. New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res (2013) 113:72–7. doi: 10.1161/CIRCRESAHA.113.301036
74. Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc (1976) 35:156–62.
75. Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest (2010) 90:1549–57. doi: 10.1038/labinvest.2010.152
76. Jinnouchi H, Sato Y, Sakamoto A, Cornelissen A, Mori M, Kawakami R, et al. Calcium deposition within coronary atherosclerotic lesion: implications for plaque stability. Atherosclerosis (2020) 306:85–95. doi: 10.1016/j.atherosclerosis.2020.05.017
77. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol (2014) 34:724–36. doi: 10.1161/ATVBAHA.113.302642
78. Mori H, Torii S, Kutyna M, Sakamoto A, Finn AV, Virmani R. Coronary artery calcification and its progression: what does it really mean? JACC Cardiovasc Imaging (2018) 11:127–42. doi: 10.1016/j.jcmg.2017.10.012
79. Potor L, Hendrik Z, Patsalos A, Katona E, Mehes G, Poliska S, et al. Oxidation of hemoglobin drives a proatherogenic polarization of macrophages in human atherosclerosis. Antioxid Redox Signal (2021) 35:917–50. doi: 10.1089/ars.2020.8234
80. Lin R, Chen S, Liu G, Xue Y, Zhao X. Association between carotid atherosclerotic plaque calcification and intraplaque hemorrhage: a magnetic resonance imaging study. Arterioscler Thromb Vasc Biol (2017) 37:1228–33. doi: 10.1161/ATVBAHA.116.308360
81. Sakamoto A, Kawakami R, Mori M, Guo L, Paek KH, Mosquera JV, et al. CD163+ macrophages restrain vascular calcification, promoting the development of high-risk plaque. JCI Insight (2023) 8(5):e154922. doi: 10.1172/jci.insight.154922
82. Yaker L, Tebani A, Lesueur C, Dias C, Jung V, Bekri S, et al. Extracellular vesicles from LPS-treated macrophages aggravate smooth muscle cell calcification by propagating inflammation and oxidative stress. Front Cell Dev Biol (2022) 10:823450. doi: 10.3389/fcell.2022.823450
83. Solanki S, Dube PR, Tano JY, Birnbaumer L, Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am J Physiol Cell Physiol (2014) 307:C521–31. doi: 10.1152/ajpcell.00369.2013
84. Solanki S, Dube PR, Birnbaumer L, Vazquez G. Reduced necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice with macrophage-specific loss of Trpc3. Sci Rep (2017) 7:42526. doi: 10.1038/srep42526
85. Dube PR, Chikkamenahalli LL, Birnbaumer L, Vazquez G. Reduced calcification and osteogenic features in advanced atherosclerotic plaques of mice with macrophage-specific loss of TRPC3. Atherosclerosis (2018) 270:199–204. doi: 10.1016/j.atherosclerosis.2017.12.025
86. Dube PR, Birnbaumer L, Vazquez G. Evidence for constitutive bone morphogenetic protein-2 secretion by M1 macrophages: constitutive auto/paracrine osteogenic signaling by BMP-2 in M1 macrophages. Biochem Biophys Res Commun (2017) 491:154–8. doi: 10.1016/j.bbrc.2017.07.065
87. Duang S, Zhang M, Liu C, Dong Q. Parathyroid hormone-induced vascular smooth muscle cells calcification by endoplasmic reticulum stress. J Physiol Pharmacol (2022) 73(5). doi: 10.26402/jpp.2022.5.03
88. Xu S, Bai P, Little PJ, Liu P. Poly(ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev (2014) 34:644–75. doi: 10.1002/med.21300
89. Li P, Wang Y, Liu X, Liu B, Wang ZY, Xie F, et al. Loss of PARP-1 attenuates diabetic arteriosclerotic calcification via Stat1/Runx2 axis. Cell Death Dis (2020) 11:22. doi: 10.1038/s41419-019-2215-8
90. Shang J, Zhao F, Cao Y, Ping F, Wang W, Li Y. HMGB1 mediates lipopolysaccharide-induced macrophage autophagy and pyroptosis. BMC Mol Cell Biol (2023) 24:2. doi: 10.1186/s12860-023-00464-7
91. Passmore M, Nataatmadja M, Fung YL, Pearse B, Gabriel S, Tesar P, et al. Osteopontin alters endothelial and valvular interstitial cell behaviour in calcific aortic valve stenosis through HMGB1 regulation. Eur J Cardiothorac Surg (2015) 48:e20–9. doi: 10.1093/ejcts/ezv244
92. Chen Q, Bei JJ, Liu C, Feng SB, Zhao WB, Zhou Z, et al. HMGB1 induces secretion of matrix vesicles by macrophages to enhance ectopic mineralization. PloS One (2016) 11:e0156686. doi: 10.1371/journal.pone.0156686
93. Waring OJ, Skenteris NT, Biessen EAL, Donners M. Two-faced janus: the dual role of macrophages in atherosclerotic calcification. Cardiovasc Res (2021) 118(13):2768–2777 doi: 10.1093/cvr/cvab301
94. Li G, Qiao W, Zhang W, Li F, Shi J, Dong N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J Thorac Cardiovasc Surg (2017) 153:1318–1327 e1. doi: 10.1016/j.jtcvs.2017.01.052
95. Deuell KA, Callegari A, Giachelli CM, Rosenfeld ME, Scatena M. RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: dependence on IL-6 and TNF-alpha. J Vasc Res (2012) 49:510–21. doi: 10.1159/000341216
96. Awan Z, Denis M, Roubtsova A, Essalmani R, Marcinkiewicz J, Awan A, et al. Reducing vascular calcification by anti-IL-1beta monoclonal antibody in a mouse model of familial hypercholesterolemia. Angiology (2016) 67:157–67. doi: 10.1177/0003319715583205
97. Kockx MM, Knaapen MW. The role of apoptosis in vascular disease. J Pathol (2000) 190:267–80. doi: 10.1002/(SICI)1096-9896(200002)190:3<267::AID-PATH523>3.0.CO;2-A
Keywords: macrophages, cell death, calcification, atherosclerosis, plaque stability, cardiovascular disease
Citation: Neels JG, Gollentz C and Chinetti G (2023) Macrophage death in atherosclerosis: potential role in calcification. Front. Immunol. 14:1215612. doi: 10.3389/fimmu.2023.1215612
Received: 02 May 2023; Accepted: 20 June 2023;
Published: 04 July 2023.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Aloke Finn, CVPath Institute, United StatesCopyright © 2023 Neels, Gollentz and Chinetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Chinetti, Z2l1bGlhLmNoaW5ldHRpQHVuaXYtY290ZWRhenVyLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.