 Nick Giannoukakis
Nick Giannoukakis- 1Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, PA, United States
Tolerogenic dendritic cells (tDC) arrest the progression of autoimmune-driven dysglycemia into clinical, insulin-requiring type 1 diabetes (T1D) and preserve a critical mass of β cells able to restore some degree of normoglycemia in new-onset clinical disease. The safety of tDC, generated ex vivo from peripheral blood leukocytes, has been demonstrated in phase I clinical studies. Accumulating evidence shows that tDC act via multiple layers of immune regulation arresting the action of pancreatic β cell-targeting effector lymphocytes. tDC share a number of phenotypes and mechanisms of action, independent of the method by which they are generated ex vivo. In the context of safety, this yields confidence that the time has come to test the best characterized tDC in phase II clinical trials in T1D, especially given that tDC are already being tested for other autoimmune conditions. The time is also now to refine purity markers and to “universalize” the methods by which tDC are generated. This review summarizes the current state of tDC therapy for T1D, presents points of intersection of the mechanisms of action that the different embodiments use to induce tolerance, and offers insights into outstanding matters to address as phase II studies are imminent. Finally, we present a proposal for co-administration and serially-alternating administration of tDC and T-regulatory cells (Tregs) as a synergistic and complementary approach to prevent and treat T1D.
Introduction
With the recent news that Teplizumab (TZIELD™), a humanized CD3-targeting monoclonal antibody received FDA approval to delay the onset of Stage 3 type 1 diabetes mellitus (T1D) in adults and children =>8 years of age (1, 2), a media frenzy resulted in misrepresentation of the product’s actual efficacy and also misrepresented the careful comments and conclusions of scientists working with the antibody for more than 20 years. While the outcomes of important clinical studies over the past 10 years strongly support the efficacy of Teplizumab in a very selected population of pre-diabetic individuals (the formal characteristics of “responders” remain to be comprehensively defined (2–7);), T1D remains far from being cured and Teplizumab use has not resulted in an across-the-board delay or prevention of transition into Stage 3 or insulin-requiring diabetes (6, 8, 9). Thus, the search for a more comprehensive and more robust therapy remains to be discovered or developed. In this review, the case for tolerogenic dendritic cells (tDC) is once again [prior excellent reviews and expert opinion have already been published (10–12)] presented with more recent information that demonstrate that tDC act to re-enforce, re-make, and strengthen not one, but at least three interweaving immunosuppressive and immunoregulatory leukocyte networks. One can view tDC as the command center of a multi-level, multi-dimensional defense network (reviewed in (10–12) and illustrated in Figure 1 that no other T1D treatment has yet achieved.
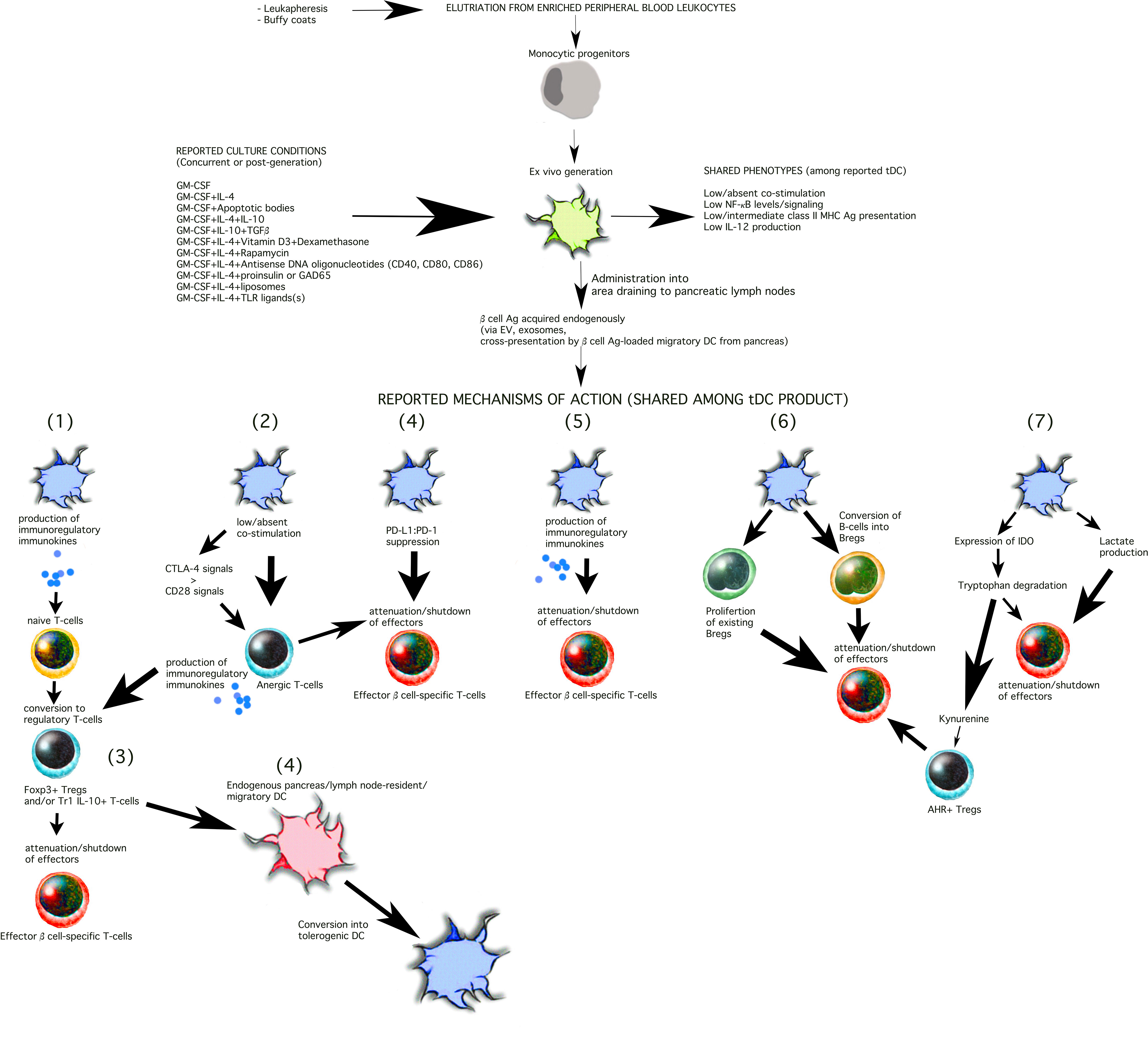
Figure 1 Shared phenotypes and mechanisms of action among T1D-targeting tDC. The current methods to generate tDC begin with peripheral blood leukocytes that are separated into monocytes either by traditional centrifugation methods or mechanical elutriation. The monocytes are then cultured ex vivo for 5-9 days towards differentiation into DC in the presence of either GM-CSF alone or with the additional of a number of other constituents illustrated in the Figure (top). These constituents are added into the differentiation cocktail at the start of the cell culture (e.g. IL-4, dexamethasone, vitamin D3, antisense DNA oligonucleotides targeting co-stimulation protein expression) and replenished during the generation period, or are added at the end of the differentiation period for an additional period of 1-5 days. While a universal purity marker for tDC has not yet been discovered, tDC clinical release criteria currently rely on low surface expression of CD86 and class II MHC (HLA) and low IL-12 production in vitro at the end of the cell generation process. The tDC have been administered i.v., s.c., or i.d. In some trials, the tDC have been administered into the abdomen at a site that drains, partially, to the pancreatic lymph nodes (13–15). Among the reported outcomes, verified in mouse models, tDC commonly re-establish and/or stimulate seven mechanisms of action as illustrated in the Figure 1) Conversion of naïve T-cells into regulatory T-cells that may belong to the Tr1 and/or Foxp3 T-cell populations. Tr1 and Foxp3+ induced Tregs are not mutually exclusive and may be β cell antigen-specific or polyclonal. tDC may achieve this by producing immunokines that convert T-cells into regulatory cells and/or via induction of anergic T-cells (mechanism 2, illustrated). Regulatory T-cells then suppress the activity or activation state of effector β cell autoreactive T-cells inside the lymph node space which they share with the tDC or inside the pancreas, close to the inflamed islets (mechanism 3, illustrated). While inside the pancreatic lymph nodes, tDC can also, on their own, or via regulatory T-cells, convert resident “immature/semi-mature” DC into tDC. β cell Ag can be acquired by the exogenously-supplied tDC in at least three possible ways: a) via extracellular vesicles draining from the inflamed pancreas; b) via cross-presentation/cross-dressing of Ag draining from the inflamed pancreas acquired by lymph node-resident “immature/semi-mature” DC; and/or c) migratory DC that arrive from inflamed pancreas carrying β cell-derived Ag and cross-presented/cross-dressed to tDC or endogenous tDC-”reprogrammed” lymph node-resident DC. Direct suppression of effector β cell autoreactive T-cells by the tDC (or tDC-directed, “reprogrammed” endogenous lymph node-resident DC) can additionally be achieved via PD-L1:PD-1 signaling (mechanism 4) and/or immunosuppressive immunokines produced by the tDC (mechanism 5, e.g. IL-10, TGFβ). A novel mechanism of action (mechanism 6) involves the differentiation of B-cells into Bregs and/or the proliferation of existing Bregs. Retinoic acid has been shown to be one of the tDC-produced molecules involved in the process. It is certain that other mechanisms inducing Bregs and/or that attenuate autoreactive B-cells remain to be discovered. Finally, in some instances, tDC express the enzyme IDO (mechanism 7, illustrated), which breaks down tryptophan, an amino acid necessary for T-cell survival and proliferation. Kynurenine, one of the breakdown products, can act as a ligand for the aryl hydrocarbon receptor (AHR), expressed in some regulatory T-cells to potentiate their suppressive capacity (mechanism 7).
tDC human use has now matured and entered the same space as tumor-fighting DC therapy. Table 1 presents the human clinical trials in the space of autoimmunity where safety outcomes have been reported. In all these studies, even those that are currently ongoing or recently completed, there were no reports of significant adverse events suggesting that the administration of the tDC could be well-tolerated, pending the anticipated reporting of the of the safety study outcomes.
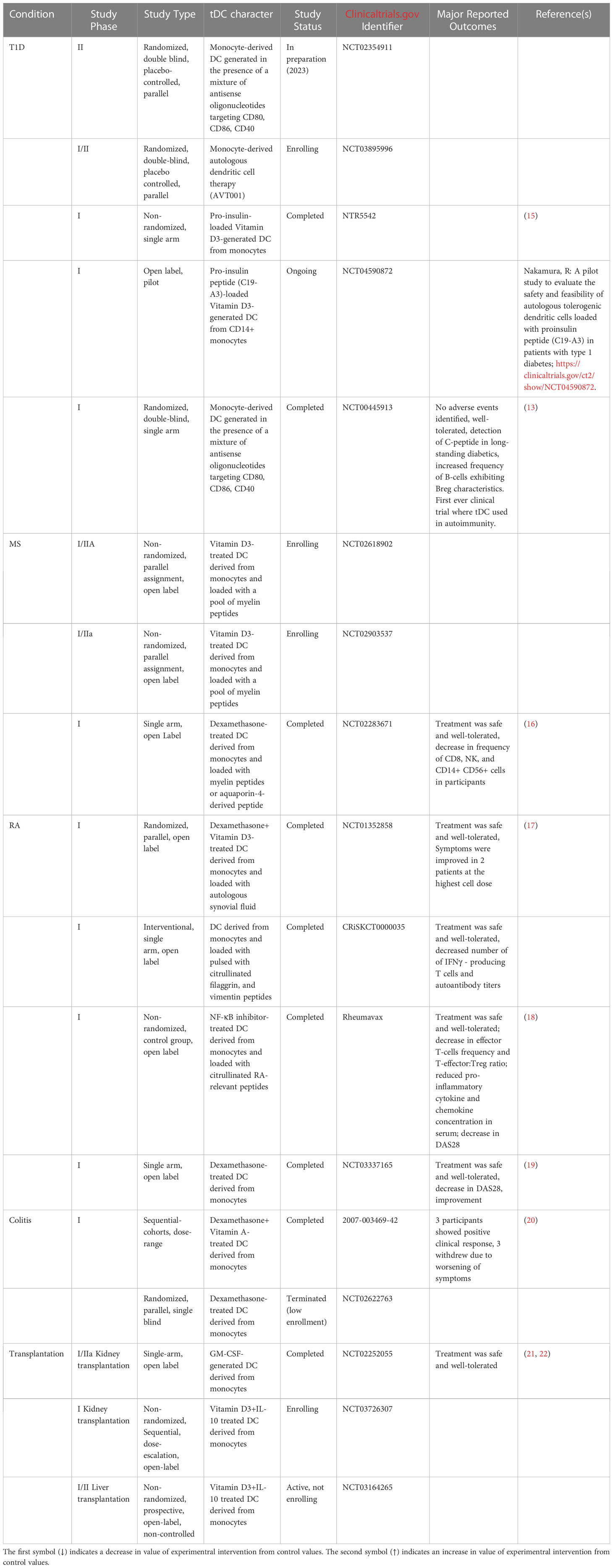
Table 1 Human tDC clinical trials in the space of autoimmunity.
T1D is the clinical outcome of a relapsing-remitting T-cell driven autoimmunity that, across three stages, gradually impairs and damages a mass of pancreatic β cells that requires exogenous insulin replacement to maintain normoglycemia (23, 24). While a combination of certain alleles at the HLA, INS, PTPN22, and IL2RA loci, together confer a substantial risk to develop the disease (25–27), there are many individuals who, in spite of inheriting high-risk alleles at one or more of these major loci, remain diabetes-free, even though evidence indicates that β cell-targeting autoantibodies and T-cells can be detected in the circulation many years prior to clinical onset (23). Mechanistically, the immunological basis of the relapsing-remitting T-cell-driven inflammation inside the pancreas is failure of central tolerance, where β cell-specific thymocytes are not deleted, as well as peripheral loss of tolerance to β-cell proteins such as insulin, glutamic acid decarboxylase 65 (GAD65), insulinoma-associated-2 (IA-2), and Zinc Transporter T8 (28, 29). T-cells reactive to these major β cell proteins are detected in the pancreata of newly-diagnosed humans as well as inside the pancreas of the NOD/LtJ mouse strain even weeks before the onset of clinical hyperglycemia (30–35). Pancreas-resident DC with markers associated with tolerogenic phenotype have been discovered, and their frequency disappears with the progression of T-cell infiltration around and into the pancreatic islets of Langerhans (33–40). This supports the hypothesis that, pancreas-resident tDC normally enforce immunoregulation (41–50) via maintenance of peripheral tolerogenic lymphocyte networks and their impairment/disappearance is permissive for the autoreactive T-cells to be unleashed around and inside the islets of Langerhans. This hypothesis, and its accumulating evidence in mouse, rat, and human tissues, motivated the consideration to use tDC to re-establish these immunoregulatory networks, or to create new networks of regulatory cells whose functional outcome would be some form of restored tolerance, inside newly-diagnosed T1D in order to “reverse” hyperglycemia and – more importantly – to erect these defenses inside the Stage 2-Stage 3 transition. Indeed, all the tDC embodiments studied and used to date to treat T1D, at least in the NOD/LtJ mouse model of the disease, have resulted in the complete prevention of hyperglycemia and varying success in “reversal” of hyperglycemia and maintenance of an insulin-free, stable, long-term, normoglycemic state (reviewed in (11, 12, 51–54) and listed in Table 2).
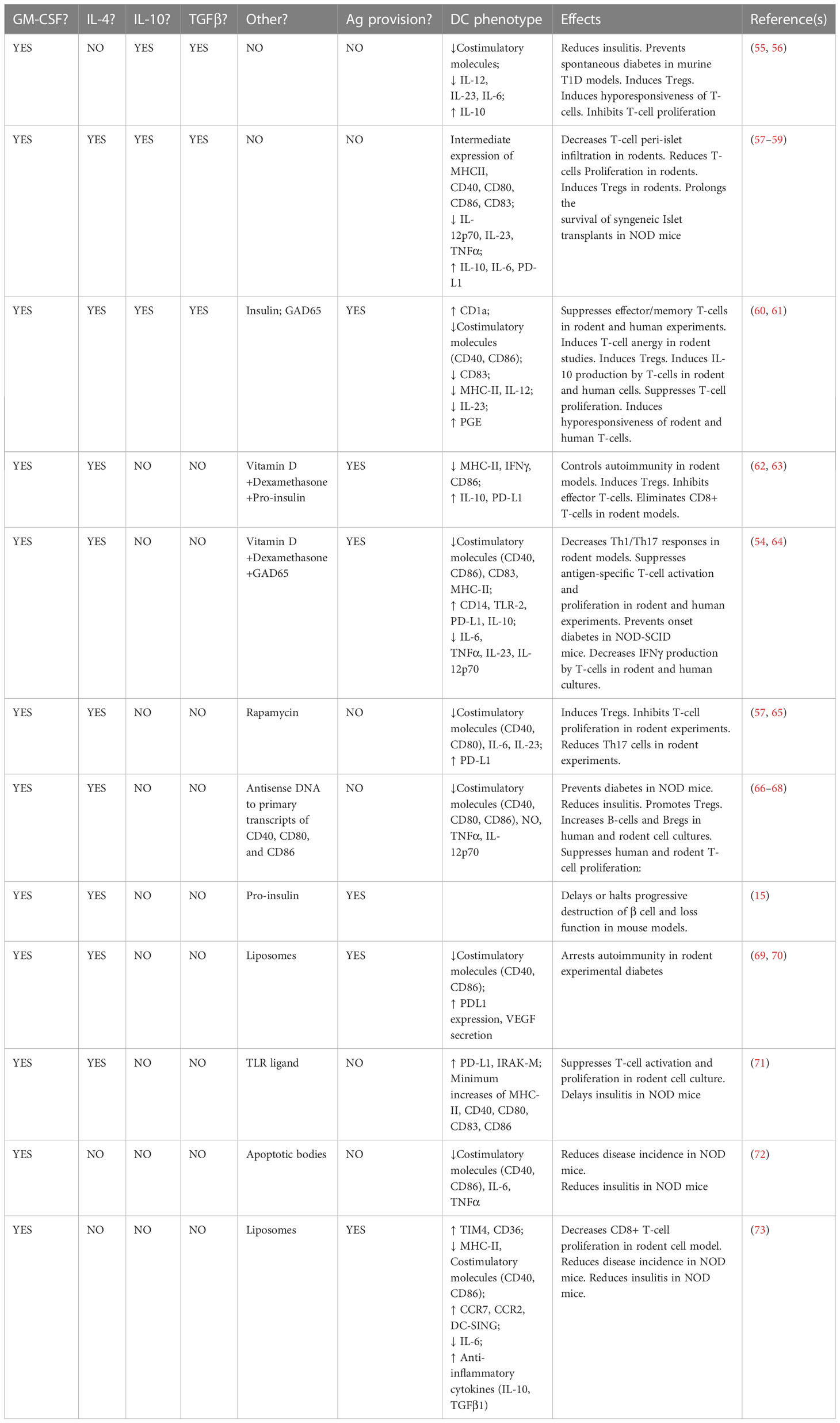
Table 2 Ex vivo approaches to generate tDC, phenotypes of the cells, and major effects.
What are tolerogenic DC? the state of the current knowledge
DC exist as a spectrum of phenotypes and immune actions between pro-inflammatory and anti-inflammatory cells at the population and single cell level (reviewed in (11, 12) and Table 3). At one end of this spectrum, they are widely referred to as “mature” as a consequence of their response to entering inside, or being subjected to a building micro-environment of tissue “damage/danger” (74–82). Under such micro-environmental conditions, tissue-resident and/or migratory DC increase the expression of major histocompatibility complex (MHC) proteins loaded with peptides acquired from cells and proteins from their microenvironment, and upregulated the cell surface levels of co-stimulatory proteins like CD40, CD86, and a series of cell-cell adhesion molecules – some with signaling capacity into and inside from T-cells (83–86). DC undergoing maturation will begin migrating towards the lymphoid organs that drain the particular anatomic site carrying with them, and presenting acquired proteins via the MHC. As they mature, they produce a spectrum of pro-inflammatory immunokines which will be required to prime the T-cells whose T-cell receptor(s) “match” the peptide:MHC complex inside the lymphoid organ(s) (10, 87).
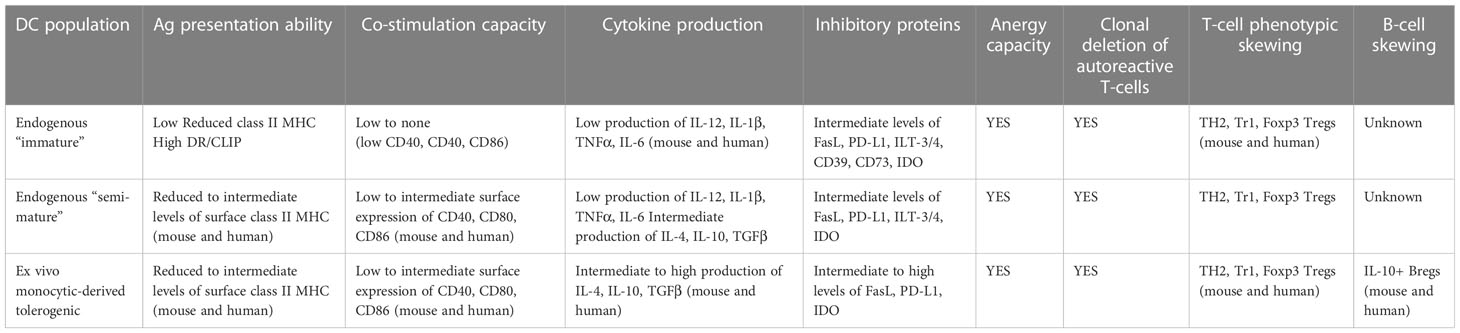
Table 3 Major phenotypes and actions of DC with inherent tolerogenic capacity.
At the other end of the phenotypic spectrum are DC exhibiting immunosuppressive, tolerogenic ability in vitro and in vivo (54, 88–93). The shared characteristics of these tDC include a low capability to stimulate T cells and low to absent IL-12 production in vitro, expression and production of immunoregulatory molecules such as anti-inflammatory cytokines [IL-10 and transforming growth factor (TGF) β], indolamine 2,3- dioxygenase (IDO), and the expression of surface inhibitors like programmed death-ligand 1 (PD-L1) (89, 94). tDCs act at different levels [reviewed in (10–12, 54, 88, 90)]: a) they can directly delete autoreactive T-cells; b) they can purge autoreactive CD8+ T cells through a mechanism referred to as peripheral cross-tolerance; c) they can enforce clonal deletion and clonal anergy; and d) they can convert naïve, and in some instances effector, T-cells in the periphery into regulatory T cells (Tregs).
Characteristics of tDC with tolerogenic potential
The ability of tDC to generate and to participate in establishing and maintaining tolerance inside the visceral and peripheral organs has been well-documented (38, 39, 43–49, 88, 89, 94). Migratory DC survey local and circulating antigens derived from the tissues into which they penetrate including the indigenous microbiota as well as foreign organisms such as viruses and bacteria (95–98). The antigens acquired from the cell constituents of these tissues as well as from the phagocytosis/endocytosis of the microbiota in the steady state are presented to T-cells inside the tissue-draining lymphoid organs under conditions where the presenting DC is in an immunologically “immature” or “semi-mature” state (92, 99–108) resulting in the induction and/or maintenance of T cell tolerance (10–12, 88, 89, 109, 110). Among the better understood mechanisms of how tDC achieve tissue- and organ-level immune tolerance to the tissue- and organ-restricted antigens is the induction and maintenance of Treg frequency and immunosuppressive stability. Peripheral tolerance as a function of tDC is partially-conditioned on the success of converting and maintaining T-cells into stable Tregs, many that express the Foxp3 transcription factor (54, 111–117). CCR7+ DC express a number of immunoregulatory molecules, including CD200, PD-L1 and PD-L2, facilitating tolerance in vivo (118, 119) even though it remains unclear if these DC facilitate the conversion of T-cells into Tregs or expand existing Tregs. In mice, migratory DC with tolerogenic potential carry acquired antigens from the parenchyma into the draining lymphoid organs, and in some cases even into the spleen (120, 121). While T-cells inside the lymphoid organs scan the arriving DC via TCR, another process is also active; where the antigens on the arriving DC are “cross-dressed” to tDC inside the lymphoid organs (10, 90, 122, 123). These antigens, irrespective of how they are delivered to the DC, can act as targets for T-cells that, in an environment of low pro-inflammatory signals, convert Tregs (90, 93, 124). This conversion is mainly driven by migratory and tissue-resident DC (that can migrate to the draining lymphoid organs) that have been positioned inside the family of Batf3+, XCR1+ type 1 subset (cDC1) (10, 90) as well as the more recently-described DC5 population which resemble plasmacytoid DC (125). Other distinguishing features of tDC include their expression of DEC-205, CLEC9A, BTLA (90, 124, 126–132). In addition to these characteristics, tDC induction of peripheral Tregs is dependent on their production of transforming growth factor β (TGFβ) which, alone or optimally together with DC-produced retinoic acid (RA) drives the expression of Foxp3 (127, 132). Others have reported that a subset of tDC expresses the enzyme indoleamine 2,3-dioxygenase (IDO), which prevents the expansion of effector T-cells by catabolism of tryptophan (133–143) and illustrated in Figure 1.
How do tDC relevant in modifying T1D autoimmunity work?
There is no one specific mechanism of action through which tDC have demonstrated immune action and this is a significant advantage of tDC use for treatment of autoimmunity over single target, single mechanism approaches. Figure 1 illustrates those that are better characterized. First, is the general characteristic of immature DC that exhibit low co-stimulation capacity (i.e. antigen:MHC presentation to T-cells in the absence of, or very low binding of T-cell CD28 with CD80/CD86 on DC). This results in the impaired ability of the responding T-cells to produce IL-2 and proliferate (144). Second, is the increased provision of inhibitory signals by the DC to the T-cells via programmed death-ligand 1 (PD-L1) (145–149), triggering the activation of SHP-1 and SHP-2 which intercede to induce clonal anergy and abrogated Treg differentiation (150, 151). Further, CTLA-4 expressed by activated T-cells and Tregs, can bind CD80 and CD86 on DC and facilitate their degradation (152). This additional mechanism of action by immature (and inherently tolerogenic DC) acts at another level to impair the priming of naïve T cells (153). Within the scope of action of CTLA-4, tDC can directly promote antigen-specific suppressive capacity inside CD4+ and CD8+ T cells with high CTLA-4 expression (154, 155). Finally, some populations of tDC can directly kill T-cells directly via Fas-FasL or TRAIL-mediated mechanisms (156, 157).
tDC express an array of immunoregulatory immunokines and metabolites that transform the local microenvironment into a stroma that facilitates conversion and/or stabilization of leukocytes, including T-cells and B-cells into immunosuppressive cells. Many tDC embodiments produce IL-10 (57, 158) and elicit anergy in effector and memory CD4+ T cells, at least in vitro in both mice and humans (58, 155). tDC also produce TGF-β and IL-27, with or without retinoic acid (RA) which convert T-cells into, or cause the proliferation of existing Tregs, into IL-10 producing regulatory T-cells that express Foxp3 or are characterized as an additional Tr1 population (159–165). It is important, at this time, to note that many of the study outcomes referred to inside this section have been obtained mostly in experiments where the tDC and other leukocyte populations, including antigen-specific T-cells and T-cells or B-cells that acquire a tolerogenic capacity, are brought together in vitro. Demonstration of physical interactions in vivo between tDC and T-cells and/or B-cells in vivo, inside affected tissues or the lymph nodes into which they drain to remains to be confirmed and is of high experimental priority.
The tDC-B-regulatory cell system
In our phase I clinical trial to determine safety of tDC generated as monocyte progenitors in the presence of a mixture of antisense oligonucleotides targeting the primary transcripts of CD40, CD80, and CD86 (13), we discovered that these DC caused an increased frequency in circulating B-cells with characteristics shared with what were beginning to be referred to as “B-regulatory cells” (66, 67). This was the first in history report that tDC administration into humans could augment such cells and we further demonstrated that indeed B-cells are converted into Bregs and existing Bregs proliferate in the presence of tDC partly via retinoic acid (66). In a subsequent line of investigation, intravenous administration of vitamin D3-conditioned tDC reduced EAE in mice via Bregs (166).
Bregs were identified as a distinct population of immunosuppressive leukocytes in mice and humans and while they express IL-10, TGF-β, and IL-35 (167–182), these cytokines are not conditio sine qua non for their suppressive ability (183–186). In fact, IL-10-expressing B-cells can interact with DC in a contact-dependent manner in the NOD/LtJ mouse model of T1D conferring to them a regulatory capacity to suppress CD8+ diabetogenic T-cells (187). Bregs, remarkably, can inhibit the differentiation of T-cells into Th1, Th17, follicular helper T cells (Tfh), and the differentiation of B-cells into terminal B cells. They have been reported to also stimulate T cell anergy, expansion of Tregs, and to regulate the responses of invariant natural killer T-cells (iNKT) (181, 182, 188, 189). Vitamin D3-generated tDC have also demonstrated tolerogenic activity in the EAE mouse model of multiple sclerosis, where recipients exhibited clinical grade of the disease together with an increase in the frequency of Bregs (166).
These results reveal a second network of immunoregulatory leukocytes that can be upregulated and maintained by tDC to treat autoimmunity.
Interweaving networks of tDC-orchestrated immune regulation
“Infectious tolerance” and “linked suppression” refer to processes where a signal that triggers localized immune hyporesponsiveness, once established, causes a local loop of immunoregulatory action and “programs” other local leukocytes toward a tolerogenic phenotype, promoting localized peripheral tolerance. While Tregs are key to this phenomenon (190), significant and substantial evidence strongly supports tDCs as the “programmers” of local imprinting of T-cells into Tregs and B-cells into Bregs (as well as stimulators of the proliferation of existing Tregs and Bregs), resulting in cell-mediated suppression of effector autoreactive T-cells, deletion of naïve autoantigen-specific T-cells and tDC imprinting of the environment into an immunosuppressive state via tDC production of IL-10, IL-35, and TGF-β. Indeed, tissue-resident DC as well as migratory DC can be imprinted away from a potential “maturation” and into a tolerogenic state inside such an environment, thus adding a third network of leukocytes under the control of tDC (191). For example, VitD3 tDC induce autoantigen-specific Tregs that can not only repress autoreactive T-cells via linked suppression, but can also program semi-mature DC towards a state of immunosuppression, at least in vitro (192). Retinoic acid induces tDC in vitro, which – on their own - produce retinoic acid promoting IL-10-producing Tregs, amplifying a local environment of immune hyporesponsiveness (193). Given the multi-directional interaction among tDC-Trtegs-Bregs-and programming of other DC towards a state of tolerance induction potential, the process of “infectious tolerance” and “linked suppression” is established inside the lymphoid organs that drain tissues that are targets of autoimmunity and/or inside the autoimmunity target tissues themselves. This might explain why systemic administration of immunosuppressive cytokines, like IL-10, have not always resulted in efficacious treatment of autoimmunity; local IL-10 increase would maintain survival and stability of a network of tDC : Treg:Breg:imprinted DC. This would constitute a “tolerogenic feedback loop” which initiates and maintains a steady state of localized immune tolerance (194). Re-establishment of such a network is one of the bases of tDC therapy that we, and others, aim to achieve and stabilize in T1D.
Current methods to generate tDC relevant to modify T1D autoimmunity
By convention, DC characterized as “immature” or “semi-mature” cause immune hyporesponsiveness including antigen-specific immune hyporesponsiveness in vitro and in vivo (11, 12). Oftentimes, this translates into a tolerogenic capacity at the functional level. A specific population of DC with tolerogenic properties has been identified in humans (158) and these DC promote immune hyporesponsiveness in allogeneic CD4+ T cells, produce IL-10, and express the surface markers CD163, CD141, CD16, and CD14 (158). While their ontogeny is unknown, it is very possible that many more DC populations with tolerogenic phenotypes may exist and that this – as well as others waiting to be discovered - could in fact represent transient states captured as a consequence of time at sampling. Additionally, and more excitedly, these may be bona fide steady state tDC whose isolation and expansion would be a tremendous leap in tDC treatment of autoimmunity and transplantation tolerance induction.
While purity markers remain to be discovered for such DC populations, and given the phenotypic instability of “immature/semimature DC” in vivo (195), a number of methods have been developed to generate tDC in vitro and ex vivo, mostly based on monocytic progenitors (196). A variety of cell culture methods, shown in Table 2 and illustrated in Figure 1 have achieved cells with tolerogenic actions, underlied by mechanisms that involve Treg induction/preservation (196, 197).
The basis of generating tDC in vitro and/or ex vivo from monocytic progenitors rests on the discovery, more than 25 years ago, that GM-CSF and IL-4 caused the differentiation of monocytes into “immature” DC. These cells are inherently tolerogenic, eliciting T-cell immune hyporesponsiveness (92). These DC express low surface level class II MHC, CD80, and CD86 and by their low co-stimulation ability, they were shown to protect NOD/LtJ mice from T1D as they expressed IL-10 and dampened CD8+ effector function (116, 198–200). tDC were also generated from monocytic progenitors in the presence of IL-10 and TGFβ and these DC elicited antigen-specific immune hyporesponsiveness as well as efficient induction of anergy and generation of Tregs (58). tDC have been generated from monocytes only in the presence of IL-10 (57, 58) or TGFβ (59). These DC exhibit increased production of IL-10 and IL-6, reduced IL-12p70 production, and a “semi-mature” phenotype (expression of class II MHC, CD40, CD80, and CD86, at levels between “immature” and “mature” DC). Another GM-CSF-based method to generate tDC from monocytic progenitors involves the addition of IL-10 with the result that these DC can attenuate insulitis and spontaneous diabetes development in NOD/LtJ mice. These DC have also demonstrated efficacy in long-term survival of allogeneic islets of Langerhans transplants, but act in an antigen non-specific mechanism of action (55). In a similar manner, TGFβ-generated DC from monocytic progenitors also enhanced the survival of allogeneic islet transplants into autoimmune recipient diabetic mice (59).
As concerns the use of tDC to treat T1D, two DC embodiments have passed initial safety outcome measures and on the threshold for phase II trials; those generated with GM-CSF/IL-4 and a mixture of antisense oligonucleotides targeting the primary transcripts of CD40, CD80, and CD86, and those DC generated in the presence of 1,25-dihydroxyvitamin D3 and dexamethasone. While the former tDC are locked into a state of low/absent co-stimulation in vivo in a stable and long-term manner due to modification of transcription/translation (13, 52), the latter also exhibit low co-stimulation capacity (53). These cells exhibit high CD11c, DC-SIGN, PD-L1, and low class II MHC expression, supporting a state of “semi-mature” phenotype. Additionally, they also produce IL-10 (65, 201–203). A comparison of tDC generated with vitamin D3, IL-10, dexamethasone, TGF-β, or rapamycin demonstrated common features: low co-stimulation potential, and production of IL-10, conditions optimal for the induction of peripheral Tregs (57). Table 2 lists other approaches that resulted in shared phenotypes along the line of low co-stimulation potential (58, 196). Even though the maturation state of the tDC may not always distinguish functionally-tolerogenic DC (204), one additional feature that should be considered important towards tolerance capacity is the DC ability to migrate towards the lymphoid organs draining a tissue/organ targeted in an autoimmunity. This can explain the instances where therapeutic benefit was achieved in the absence of provision of disease-specific antigen(s) to the DC in vitro or ex vivo (57, 205).
Harmonizing tDC characteristics relevant for modifying T1D autoimmunity
A few years ago, an important publication offered some recommendations on what to consider as pre- and co-clinical measurements of tDC stability, potency, and stability (110). For example, the increased frequency of highly-suppressive Tregs generated in co-culture with IL-10-conditioned tDC (57, 205), restrained proliferation and activation of autoreactive CD4+ T-cells in the presence of 1,25-dihydroxyvitamin D3-conditioned tDC in vitro, and IL-10 expression in T cells are all hallmark capacities of tDC (206). Additional “readouts” of tDC can be Breg expansion (either expressing IL-10 or not) as well as their differentiation inside B-cell culture (67). Breg expansion/differentiation by tDCs is partly mediated by their production of retinoic acid, which incidentally, on its own, also causes the differentiation of Foxp3+ Tregs (66). Ex vivo generated tDCs with impaired costimulatory capability, delay and “reverse” new-onset hyperglycemia in association with increased frequency of Tregs in diabetes-free recipients (67, 68).
tDC with low co-stimulation potential have been mobilized effectively whether they are further modified by loading of specific Ag or not. Proinsulin, insulin, and GAD65 have been used alone or in combination as Ag added to tDC prior to cell administration in vivo. Vitamin D3/dexamethasone-generated and proinsulin-loaded tDC induce antigen-specific Tregs with various phenotypes in vitro (e.g. cell surface Lag-3, CD161, and inducible co-stimulator). These Ag-loaded tDC can suppress effector CD8+ and CD4+ T cells (62). In a humanized mouse model of T1D, the administration of tDC loaded with proinsulin controlled the progression of T1D, possibly via IL-10 production (63). In another study, vitamin D2/dexamethasone-generated GAD65-loaded tDCs were shown to prevent the adoptive transfer of diabetes by diabetogenic splenocytes to NOD-SCID recipients. However, in this study the Ag-loaded tDCs were not as effective as the parental non-Ag DC in protecting from T1D (64) supporting the model where Ag is acquired endogenously inside the lymph nodes draining the inflamed pancreas and where no ex vivo Ag loading is necessary.
While Ag loading of DC may or may not enhance the tolerogenic potential, treatment ex vivo with apoptotic target tissue may be an alternative method to provide a wide array of Ag to the tDC. In T1D, the increase in apoptotic pancreatic β-cells or defects in the process of capture and phagocytosis of apoptotic bodies (referred to as efferocytosis) contributes to the loss of tolerance (207). DC acquire a tolerogenic phenotype and functionality after ingestion of apoptotic β-cells and prevent T1D when transferred to NOD mice (72). The limitation of availability of freshly-collected human pancreas apoptotic bodies, however, necessitates different vehicles to promote DC efferocytosis. One such alternative are phosphatidyl-serine liposomes containing β-cell autoantigens and these were able to arrest autoimmunity and prevent T1D through the generation of tDCs. Liposome-exposed DC slowed the proliferation of autologous T cells, interfered with antigen presentation, and increased the expression of genes associated with a phenotype of tolerogenic state as well as anti-inflammatory pathways (69). Additionally, insulin-loaded liposome administration into NOD/LtJ mice reduced the severity of insulitis and expanded antigen-specific CD4+ T cells (73). Modulation of pattern recognition receptor signaling has been shown to be an innovative method to generate tDC [65]. DC exposure to a polyethylene glycol-conjugated TLR-7 ligand followed by the administration of these DC into NOD mice delayed the onset of T1D and insulitis (208). TLR-2 activation with its agonist, Pam3CSK4), resulted in DC-dependent suppression of T-cell activation (209) and combining this with a co-treatment with a dipeptidyl peptidase 4 inhibitor, which increases the mass of β-cells, “reversed” hyperglycemia in newly-diabetic NOD/LtJ mice (71).
Autoantigen- or auto-antigen-derived peptide-pulsing: is it required for Ag-specific tDC?
The underlying cause of tissue- and organ-specific autoimmunity is the failure of central tolerance, where AIRE+ medullary thymic epithelial cells together with medullary dendritic cells do not express autoantigens at levels adequate to present to thymocytes. As such, every tissue- and organ-specific autoimmunity has its specific spectrum of autoantigens together with a spectrum of thymocytes that react to those autoantigens – and that escaped the thymus. Some studies have shown that pulsing tDC with these autoantigens confer antigen specificity to immunosuppression by permitting those DC to be scanned by T-cells whose TCR are specific for that autoantigen:MHC complex (64, 105, 200). It stands to reason that this approach would be more effective in silencing autoreactive T-cells, minimizing the potential of restraining/anergizing T-cells that are not autoreactive. Where the autoantigens are well-characterized, they can be used in combinations to pulse tDC during or following ex vivo DC generation. GM-CSF+IL-4, dexamethasone (or Vitamin D3) tDC pulsed with myelin autoantigens have shown efficacy in treating multiple sclerosis in mouse models of disease (54). For rheumatoid arthritis, DC generated in the presence of GM-CSF+IL-4, and then supplemented with the NF-κB inhibitor Bay 11-7082, subsequently pulsed with citrullinated peptides of aggrecan, vimentin, collagen type II, and Aα and Bβ fibrinogen (putative RA autoantigens) conferred superior efficacy in vivo (210, 211). Given the heterogeneity of the autoimmunity progression, self-antigens are not necessarily universal. Different patients can exhibit T-cells reactive to different autoantigens. In T1D, different patients exhibit auto-antibodies different in antigen specificity and this is also seen in T-cell reactivty as well. The spectrum of autoantibodies and autoreactive T-cells also changes in the progression of the disease and different patients exhibit different changes in this progression (212–215). While insulin and GAD65 are the major T1D auto-antigens, rationally positioning them as the two key autoantigens to pulse tDC with in order to confer antigen-specific tolerance, as the autoimmunity progresses, T-cells reactive to other β cell proteins appear in the circulation, thus reflecting the process of antigen spreading. With disease progression, the autoimmunity would now be driven by the expanded set of autoantigens. Therefore, restricting the choice of autoantigens to reflect the common two (insulin and/or GAD65) could reduce the effectiveness of tDC treatment. This has already been demonstrated in T1D (200). A pre-emptive approach could be to serially-ascertain autoantigen profiles (e.g. serial assessment of T-cell reactivity to a panel of known autoantigens) in a patient-specific manner. This, although expensive, could improve antigen specificity in tDC-based tolerance, but would still not guarantee a complete coverage of all possible autoantigens (the list of all possible diabetes antigens remains under investigation as a growing number are post-translational modifications of previously-unexpected proteins (216–221). An alternative, autoantigen-agnostic, patient-specific approach could be informed by pivotal studies in the use of tDC for rheumatoid arthritis. Here, synovial fluid from inflamed joints of each patient was added to the cultured tDC prior to administration. The supposition was that all, possible autoantigens would be present inside that fluid (222, 223). Adaptation of this approach for T1D however is not feasible, as a patient-specific tailoring of tDC would require islet-containing pancreatic tissue resection from the patient. Another important consideration is based on the choice of the leading phase I clinical studies using tDC for T1D to administer the cells into an abdominal region that drains, partly, into the pancreatic lymph nodes (13, 14). An Ag-agnostic approach, i.e. to allow the Ag draining to those same lymph nodes from the pancreas, is a more natural process to allow the exogenously-supplied tDC to acquire those Ag. Recent studies have demonstrated that, even during the steady state, extracellular vesicles containing β cell Ag drain into the pancreatic lymph nodes (224). Under inflammatory conditions, such as autoimmunity, the rate of drainage is expected to increase resulting in an increase of uptake of these Ag by exogenously-supplied tDC which also drain inside these organs. As part of the process, these vesicles can also be aquired by lymph node-resident DC as well as by migratory DC that accumulate inside the pancreatic lymph nodes. By cross-presentation and/or cross-dressing, these Ag will be acquired by the tDC. This, we believe, is the basis for why our approach as well as those of others who have not “pulsed” tDC with specific Ag, have demonstrated outcomes supportive of a therapeutic benefit (13, 52, 198).
Human clinical trials: where are we now?
Our team in Pittsburgh was the first to historically demonstrate that tDC were safe and well-tolerated in adult insulin-requiring adult T1D (13). Remarkably, we detected C-peptide in 4/7 recipients of the tDC product and in one of the recipients of the parental GM-CSF/IL-4 DC. The observation of an increased frequency of B-cells inside which were elevated numbers of Bregs peri-treatment led us to demonstrate, in the NOD/LtJ mouse strain that DC directly promoted B-cell differentiation towards Bregs and existing Breg proliferation, partly via a retinoic acid-based mechanism (66, 67). In 2018, the NIDDK TrialNet Consortium approved a phase II study with these tDC in newly-diagnosed T1D participants, however at that time, NIDDK stopped supporting the production of study agents used in TrialNet studies. Thus, this phase II study, referred to as “TN-24”, even though it remains, will require some other support to generate the cell products. Work is underway to realize this over the next 12 months (Clinicaltrials.gov identifier: NCT02354911).
Where several pre-clinical studies have demonstrated T1D Ag-loaded tDC to induce Ag-specific tolerance, administration of pro-insulin peptide loaded tDC, while safe and well-tolerated, did not achieve any remarkable changes in C-peptide post-baseline (15). While this may appear disappointing, it is important to note that the trial was conducted in long-standing T1D patients, while Ag-loaded tDC are more effective in newly-diagnosed situations. Also, and of importance to all immunomodulation approaches in T1D, including the recently-approved Teplizumab, the extent of the progression of the inflammation as well as the impact of uncontrolled glycemia, uncompensated state and activation levels/numbers of β cell-targeting lymphocytes at the time of treatment, could be important determinants of effectiveness and efficacy (54, 60, 67). Additional points of consideration in tDC therapy include: a) Stability of the tDC once administered in vivo; b) progression of the disease state at the time of tDC administration; c) effect of T1D endotypes (225) on disease progression and response to tDC – this informing that tDC may be effective only in individuals exhibiting certain endotypes and at certain points in disease progression. In line with these determinant, a phase I study has been proposed to test pro-insulin peptide-loaded tDC in T1D participants who use insulin and do not exhibit any co-morbidity or other health condition (clinicaltrials. gov identifier: NCT04590872).
A side note: serial co- or alternating administration of autologous tDC and Tregs; lessons from the field of tumor immunotherapy
A number of tumor immunotherapy approaches involve ex vivo generation of tumor-specific T-cells generated in the presence of tumor antigen-pulsed DC ex vivo and administration of the expanded T-cells (226–229). This approach can be adapted for T1D leukocyte co-therapy as we describe herein. Ex vivo generation of Tregs is a technical reality and administration of autologous Tregs for T1D has shown some degree of efficacy (230–233). Emerging evidence indicates that these Tregs do not necessarily home to the pancreas and that their half life is not particularly well-characterized, especially inside the pancreas (230, 234). Another point of remaining interest is the capacity of these Tregs to directly suppress autoreactive T-effectors, or their participation in networks where other cells are indispensable to carry out the final acto of suppressing autoreactive lymphocytes. IL-2 supplementation in vivo may provide some degree of stability for the Treg half life (NCT02772679), however CD25 surface levels may limit the effect of the cytokine (235). In spite of these known or proposed limitations, Treg therapy remains under clinical investigation in T1D (CLBS03; NCT02691247) as well as in lupus (NCT02428309) and autoimmune hepatitis (NCT02704338)
Co-administration or serial/alternating administration of tDC with Tregs therefore becomes a question of clinical interest, especially at the time of new-onset T1D. The inter-relationship of tDC and Tregs facilitates inter-dependent stability of what we propose will be a very stable and robust network of peripheral tolerance. Serial/alternating administration, or co-administration of tDC and Tregs would stabilize Foxp3 expression and the stabilized Tregs would in turn reinforce the tDC tolerogenic state via cell-cell interactions and immunoregulatory cytokines acting in a paracrine manner. We envisage and propose an initial co-administration of tDC and Tregs followed by periodic “boosters” of serially-alternating tDC and Tregs. Or even serially co-administered cells. Logistically, we do not anticipate generation of tDC and Tregs in parallel from the same leukapheresis product technically challenging. From the common leukapheresis product, the same cell generation GMP facility could, in parallel, generate the tDC from the monocytic precursors as well as the Tregs from the PBMC population. At least pre-clinically, we believe the time is now to test this approach, certainly for T1D.
Clinical protocol design considerations in detecting efficacy in phase II studies
While the fundamental CMC among the currently clinically-considered tDC rely on GM-CSF and IL-4, there is a divergence in the remaining aspects of tDC generation and administration int humans. These differences could be the basis of efficacy/failure, different levels of efficacy – outcomes that are dependent on different mechanisms of action once the cells are administered. One important variable that might affect efficacy is cell dose (least number of cells and frequency of administration to achieve significant efficacy). Another variable is the administration site itself. We believe that this second variable is particularly important as the target site (inflamed anatomic region) of different autoimmunities is subserved by different anatomic points of entry for exogenously-introduced cells. There is a growing consensus that the ideal point of entry of exogenously-administered tDC is an anatomic site draining to lymphatics that also drain the autoimmunity target organ/tissue. Lymph nodes that drain the target tissue of autoimmunity often exhibit high concentrations of activated autoreactive T-cells (236). Examples include the cervical lymph nodes, targeted for tDC-based treatment of multiple sclerosis (clinicaltrials.gov identifier: NCT02618902) and the pancreatic lymph nodes for T1D, as we have demonstrated (13). Alternatively, if a target site of autoimmunity is well defined, a direct access to administer the tDC can be feasible as shown for Crohn’s disease (237).
As tDC are more widely-accepted for treating autoimmune diseases, some other variables for CMC harmonization should also be considered. For example, most, of not all the tDC preparations that are administered are not completely homogeneous; there are variable (albeit low) concentrations of “contaminating” undifferentiated or uncharacterized monocytic and granulocytic cells, carried over from the elutriation process. These carried-over cells could modify the tolerogenic capacity of the differentiated tDC once administered in vivo. Methods for further enrichment, removal of these carried-over cells should be pursued. A series of surface markers that identify only tolerogenic cells remains to be defined. It is also all but certain that a single dose of tDC will not result in stable remission of disease but a successful outcome will be a function of multiple administrations over time, at least in most individuals. Methods to expand an initial large batch of tDC for aliquoting into individual doses, stable to cryopreservation, without altering the phenotype and tolerogenic capacity of the individual aliquots, once thawed from cryopreservation, is an immediate CMC need. It remains unknown if thawed cryopreserved tDC are functionally-identical in vivo to the freshly-obtained cells. In this regard, international collaborations such as those that resulted in the first set of proposals for harmonization of tDC and Treg (Minimum Information about Tolerogenic Antigen-Presenting cells; MITAP) should be a priority and sharing of tDC generation protocols to verify and validate outcomes is critical (110).
Disease stage and patient-specific modifiers of tDC efficacy
Some of the mapped T1D-associated genetic polymorphisms have been shown to affect DC function (238). The state of activation of effector T-cells together with the stability of the network of tolerogenic leukocytes at the time of tDC administration could conceivably affect the efficacy outcomes, either in delaying the progression across Stage 3 and/or “reversal” of newly-diagnosed hyperglycemia. This information is currently not well-developed and can affect the in vivo stability of tDC, as well as their ability to strengthen weakened regulatory networks and/or delete/attenuate effector lymphocytes once administered into study subjects. For example, even as IL-10/TGFβ-generated tDC effectively induces insulin-specific tolerance in autologous effector/memory CD4+ T cells derived from T1D individuals, the degree of tolerance induction was dependent on the initial T-cell activation state of each study participant (60). These results were congruent with those of another study with IL-10/TGF-β-generated, insulin, or GAD65-loaded tDC from T1D patients (61). Furthermore, the variation of the differences in the evolving dysglycemic state among Stage 3 individuals as well as the newly-developed hyperglycemic state among T1D individuals, could affect the tolerogenic abilities of DC generated from their monocytic progenitors (239). For example, vitamin D2/dexamethasone-generated DC loaded with GAD65 from T1D patients exhibiting good or suboptimal glucose control exhibited significant variation in their ability to regulate TH1 and TH17 responses together with the suppression of antigen-specific T-cell activation, in vitro. When transferred into NOD-SCID mice, the tDC from these individuals, also exhibited a variability in diabetes delay dependent on the state of glucose control of the subject at the time of blood procurement (to generate the DC) (54). An understanding of genetic and metabolism-dependent differences in tDC activity and stability as well as the state of immune activation of the recipient at the time of tDC administration is therefore of some importance to dissect in the near future.
Irrespective of which tDC embodiment will be the finalist to demonstrate the most effective and resilient prevention of T1D and/or “reversal” of hyperglycemia, one critical point that must be taken into consideration is the resiliency and stability of the tolerogenic state of the DC once administered in vivo. The tolerogenic state and functions may not be guaranteed, especially should potently-inflammatory event arise at the site of tDC accumulation (e.g. viral infection). That is why, in our opinion, it is critical to ensure that any approach to generate tDC ex vivo considers ensuring a long-term stability of the tolerogenic state once the DC are administered. It is with this thought in mind that we began our investigations to generate tDC, targeting co-stimulation, more than 20 years ago (68).
Conclusion
With the announcement of the approval of Teplizumab as a treatment to delay the progression of Stage 3 dysglycemia to overt, insulin-requiring diabetes, the media is promoting “the end of type 1 diabetes”. A simple examination of the data using Teplizumab over two decades, indicates otherwise. Furthermore, all the other alternative immunomodulation approaches to delay the disease process and/or “reverse” hyperglycemia have either failed, or exhibit minimal efficacy – in specific subpopulations of patients, and/or are associated with significant toxicity limiting their use and broad consideration (240–246). tDC are different and substantially promising for the following reasons: a) they act to generate Tregs and Bregs concurrently; b) they can induce other DC inside the environment they accumulate into to acquire tolerogenic capacity; c) They imprint a tolerogenic environment inside which they accumulate; and d) they are safe and well-tolerated in many human phase I studies in the space of autoimmunity [reviewed in (12)] We believe that either alone or potentially in concert with Tregs, tDC will surmount what is more of a conceptual rather than a practical barrier that currently has stalled their consideration in phase II studies. While phase II studies in rheumatoid arthritis are closer to realization than in T1D, we are very confident that the time is nearing where the first phase II study in T1D will be announced.
Author contributions
NG wrote the original draft of the manuscript, edited all versions, and assumes responsibility of the final submitted draft.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Teplizumab (Tzield) to delay onset of type 1 diabetes. Med Lett Drugs Ther (2023) 65(1667):7–8. doi: 10.58347/tml.2023.1667c
2. Hirsch JS. FDA Approves teplizumab: a milestone in type 1 diabetes. Lancet Diabetes Endocrinol (2023) 11(1):18. doi: 10.1016/S2213-8587(22)00351-5
3. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med (2020) 382(6):586. doi: 10.1056/NEJMx190033
4. Abbasi J. Teplizumab improves beta cell function, delays type 1 diabetes. JAMA (2021) 325(14):1385. doi: 10.1001/jama.2021.4628
5. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. Type 1 diabetes TrialNet study: an anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
6. Nourelden AZ, Elshanbary AA, El-Sherif L, Benmelouka AY, Rohim HI, Helmy SK, et al. Safety and efficacy of teplizumab for treatment of type one diabetes mellitus: a systematic review and meta-analysis. Endocr Metab Immune Disord Drug Targets (2021) 21(10):1895–904. doi: 10.2174/1871530320999201209222921
7. Sims EK, Bundy BN, Stier K, Serti E, Lim N, Long SA, et al. Type 1 diabetes TrialNet study: teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci Transl Med (2021) 13(583). doi: 10.1126/scitranslmed.abc8980
8. Tooley JE, Vudattu N, Choi J, Cotsapas C, Devine L, Raddassi K, et al. Changes in T-cell subsets identify responders to FcR-nonbinding anti-CD3 mAb (teplizumab) in patients with type 1 diabetes. Eur J Immunol (2016) 46(1):230–41. doi: 10.1002/eji.201545708
9. Valle A, Barbagiovanni G, Jofra T, Stabilini A, Perol L, Baeyens A, et al. Heterogeneous CD3 expression levels in differing T cell subsets correlate with the in vivo anti-CD3-mediated T cell modulation. J Immunol (2015) 194(5):2117–27. doi: 10.4049/jimmunol.1401551
10. Iberg CA, Hawiger D. Natural and induced tolerogenic dendritic cells. J Immunol (2020) 204(4):733–44. doi: 10.4049/jimmunol.1901121
11. Morante-Palacios O, Fondelli F, Ballestar E, Martinez-Caceres EM. Tolerogenic dendritic cells in autoimmunity and inflammatory diseases. Trends Immunol (2021) 42(1):59–75. doi: 10.1016/j.it.2020.11.001
12. Rios-Rios WJ, Sosa-Luis SA, Torres-Aguilar H. Current advances in using tolerogenic dendritic cells as a therapeutic alternative in the treatment of type 1 diabetes. World J Diabetes (2021) 12(5):603–15. doi: 10.4239/wjd.v12.i5.603
13. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M, Phase I. (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi: 10.2337/dc11-0472
14. Nikolic T, Suwandi JS, Wesselius J, Laban S, Joosten AM, Sonneveld P, et al. Tolerogenic dendritic cells pulsed with islet antigen induce long-term reduction in T-cell autoreactivity in type 1 diabetes patients. Front Immunol (2022) 13:1054968. doi: 10.3389/fimmu.2022.1054968
15. Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol (2020) 8(6):470–2. doi: 10.1016/S2213-8587(20)30104-2
16. Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci U.S.A. (2019) 116(17):8463–70. doi: 10.1073/pnas.1820039116
17. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis (2017) 76(1):227–34. doi: 10.1136/annrheumdis-2015-208456
18. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med (2015) 7(290):290ra87. doi: 10.1126/scitranslmed.aaa9301
19. Leplina O, Kurochkina Y, Tikhonova M, Shevela E, Ostanin A, Chernykh E. Dendritic cells generated in the presence of interferon-alpha and modulated with dexamethasone as a novel tolerogenic vaccine platform. Inflammopharmacology (2020) 28(1):311–9. doi: 10.1007/s10787-019-00641-1
20. Willekens B, Presas-Rodriguez S, Mansilla MJ, Derdelinckx J, Lee WP, Nijs G, et al. R. consortium: tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open (2019) 9(9):e030309. doi: 10.1136/bmjopen-2019-030309
21. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
22. Moreau A, Kervella D, Bouchet-Delbos L, Braudeau C, Saiagh S, Guerif P, et al. D. consortium: a phase I/IIa study of autologous tolerogenic dendritic cells immunotherapy in kidney transplant recipients. Kidney Int (2023) 103(3):627–37. doi: 10.1016/j.kint.2022.08.037
23. Quattrin T, Mastrandrea LD, Walker LSK. Type 1 diabetes. Lancet (2023) S0140-6736(23):00223-4. doi: 10.1016/S0140-6736(23)00223-4
24. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet (2018) 391(10138):2449–62. doi: 10.1016/S0140-6736(18)31320-5
25. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Type 1 diabetes genetics: genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
26. Concannon P, Gogolin-Ewens KJ, Hinds DA, Wapelhorst B, Morrison VA, Stirling B, et al. A second-generation screen of the human genome for susceptibility to insulin-dependent diabetes mellitus. Nat Genet (1998) 19(3):292–6. doi: 10.1038/985
27. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Type 1 diabetes genetics: fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47(4):381–6. doi: 10.1038/ng.3245
28. Acharjee S, Ghosh B, Al-Dhubiab BE, Nair AB. Understanding type 1 diabetes: etiology and models. Can J Diabetes (2013) 37(4):269–76. doi: 10.1016/j.jcjd.2013.05.001
29. Pugliese A. Insulitis in the pathogenesis of type 1 diabetes. Pediatr Diabetes (2016) 17 Suppl 22(Suppl Suppl 22):31–6. doi: 10.1111/pedi.12388
30. Bonnet-Serrano F, Diedisheim M, Mallone R, Larger E. Decreased alpha-cell mass and early structural alterations of the exocrine pancreas in patients with type 1 diabetes: an analysis based on the nPOD repository. PloS One (2018) 13(1):e0191528. doi: 10.1371/journal.pone.0191528
31. Kaddis JS, Pugliese A, Atkinson MA. A run on the biobank: what have we learned about type 1 diabetes from the nPOD tissue repository? Curr Opin Endocrinol Diabetes Obes (2015) 22(4):290–5. doi: 10.1097/MED.0000000000000171
32. Pugliese A, Vendrame F, Reijonen H, Atkinson MA, Campbell-Thompson M, Burke GW. New insight on human type 1 diabetes biology: nPOD and nPOD-transplantation. Curr Diabetes Rep (2014) 14(10):530. doi: 10.1007/s11892-014-0530-0
33. in't Veld P. Rodent versus human insulitis: why the huge disconnect? Curr Opin Endocrinol Diabetes Obes (2015) 22(2):86–90. doi: 10.1097/MED.0000000000000135
34. Lundberg M, Seiron P, Ingvast S, Korsgren O, Skog O. Re-addressing the 2013 consensus guidelines for the diagnosis of insulitis in human type 1 diabetes: is change necessary? Diabetologia (2017) 60(4):756–7. doi: 10.1007/s00125-017-4212-8
35. Wedgwood KC, Richardson SJ, Morgan NG, Tsaneva-Atanasova K. Spatiotemporal dynamics of insulitis in human type 1 diabetes. Front Physiol (2016) 7:633. doi: 10.3389/fphys.2016.00633
36. Beumer W, Welzen-Coppens JM, van Helden-Meeuwsen CG, Gibney SM, Drexhage HA, Versnel MA. The gene expression profile of CD11c+ CD8alpha- dendritic cells in the pre-diabetic pancreas of the NOD mouse. PloS One (2014) 9(8):e103404. doi: 10.1371/journal.pone.0103404
37. Welzen-Coppens JM, van Helden-Meeuwsen CG, Drexhage HA, Versnel MA. Abnormalities of dendritic cell precursors in the pancreas of the NOD mouse model of diabetes. Eur J Immunol (2012) 42(1):186–94. doi: 10.1002/eji.201141770
38. Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. Reduced numbers of dendritic cells with a tolerogenic phenotype in the prediabetic pancreas of NOD mice. J Leukoc Biol (2012) 92(6):1207–13. doi: 10.1189/jlb.0312168
39. Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. The kinetics of plasmacytoid dendritic cell accumulation in the pancreas of the NOD mouse during the early phases of insulitis. PloS One (2013) 8(1):e55071. doi: 10.1371/journal.pone.0055071
40. Lee MH, Lee WH, Todorov I, Liu CP. CD4+ CD25+ regulatory T cells prevent type 1 diabetes preceded by dendritic cell-dominant invasive insulitis by affecting chemotaxis and local invasiveness of dendritic cells. J Immunol (2010) 185(4):2493–501. doi: 10.4049/jimmunol.1001036
41. Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of langerhans. Proc Natl Acad Sci U.S.A. (2011) 108(4):1561–6. doi: 10.1073/pnas.1018973108
42. Calderon B, Carrero JA, Miller MJ, Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci U.S.A. (2011) 108(4):1567–72. doi: 10.1073/pnas.1018975108
43. Calderon B, Carrero JA, Unanue ER. The central role of antigen presentation in islets of langerhans in autoimmune diabetes. Curr Opin Immunol (2014) 26:32–40. doi: 10.1016/j.coi.2013.10.011
44. Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci U.S.A. (2008) 105(16):6121–6. doi: 10.1073/pnas.0801973105
45. Calderon B, Unanue ER. Antigen presentation events in autoimmune diabetes. Curr Opin Immunol (2012) 24(1):119–28. doi: 10.1016/j.coi.2011.11.005
46. Carrero JA, Ferris ST, Unanue ER. Macrophages and dendritic cells in islets of langerhans in diabetic autoimmunity: a lesson on cell interactions in a mini-organ. Curr Opin Immunol (2016) 43:54–9. doi: 10.1016/j.coi.2016.09.004
47. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER. A minor subset of Batf3-dependent antigen-presenting cells in islets of langerhans is essential for the development of autoimmune diabetes. Immunity (2014) 41(4):657–69. doi: 10.1016/j.immuni.2014.09.012
48. Ferris ST, Carrero JA, Unanue ER. Antigen presentation events during the initiation of autoimmune diabetes in the NOD mouse. J Autoimmun (2016) 71:19–25. doi: 10.1016/j.jaut.2016.03.007
49. Unanue ER, Ferris ST, Carrero JA. The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse. Immunol Rev (2016) 272(1):183–201. doi: 10.1111/imr.12430
50. Vomund AN, Zinselmeyer BH, Hughes J, Calderon B, Valderrama C, Ferris ST, et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci U.S.A. (2015) 112(40):E5496–502. doi: 10.1073/pnas.1515954112
51. Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG. It's time to bring dendritic cell therapy to type 1 diabetes. Diabetes (2014) 63(1):20–30. doi: 10.2337/db13-0886
52. Giannoukakis N. Tolerogenic dendritic cells for type 1 diabetes. Immunotherapy (2013) 5(6):569–71. doi: 10.2217/imt.13.50
53. Phillips BE, Garciafigueroa Y, Engman C, Trucco M, Giannoukakis N. Tolerogenic dendritic cells and T-regulatory cells at the clinical trials crossroad for the treatment of autoimmune disease; emphasis on type 1 diabetes therapy. Front Immunol (2019) 10:148. doi: 10.3389/fimmu.2019.00148
54. Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N. Clinical tolerogenic dendritic cells: exploring therapeutic impact on human autoimmune disease. Front Immunol (2017) 8:1279. doi: 10.3389/fimmu.2017.01279
55. Tai N, Yasuda H, Xiang Y, Zhang L, Rodriguez-Pinto D, Yokono K, et al. IL-10-conditioned dendritic cells prevent autoimmune diabetes in NOD and humanized HLA-DQ8/RIP-B7. 1 mice. Clin Immunol (2011) 139(3):336–49. doi: 10.1016/j.clim.2011.03.003
56. Haase C, Ejrnaes M, Juedes AE, Wolfe T, Markholst H, von Herrath MG. Immunomodulatory dendritic cells require autologous serum to circumvent nonspecific immunosuppressive activity in vivo. Blood (2005) 106(13):4225–33. doi: 10.1182/blood-2005-03-0975
57. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin Immunol (2012) 142(3):332–42. doi: 10.1016/j.clim.2011.11.011
58. Torres-Aguilar H, Aguilar-Ruiz SR, Gonzalez-Perez G, Munguia R, Bajana S, Meraz-Rios MA, et al. Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4+ T cells. J Immunol (2010) 184(4):1765–75. doi: 10.4049/jimmunol.0902133
59. Thomas DC, Wong FS, Zaccone P, Green EA, Wallberg M. Protection of islet grafts through transforming growth factor-beta-induced tolerogenic dendritic cells. Diabetes (2013) 62(9):3132–42. doi: 10.2337/db12-1740
60. Torres-Aguilar H, Sanchez-Torres C, Jara LJ, Blank M, Shoenfeld Y. IL-10/TGF-beta-treated dendritic cells, pulsed with insulin, specifically reduce the response to insulin of CD4+ effector/memory T cells from type 1 diabetic individuals. J Clin Immunol (2010) 30(5):659–68. doi: 10.1007/s10875-010-9430-5
61. Segovia-Gamboa N, Rodriguez-Arellano ME, Rangel-Cruz R, Sanchez-Diaz M, Ramirez-Reyes JC, Faradji R, et al. Tolerogenic dendritic cells induce antigen-specific hyporesponsiveness in insulin- and glutamic acid decarboxylase 65-autoreactive T lymphocytes from type 1 diabetic patients. Clin Immunol (2014) 154(1):72–83. doi: 10.1016/j.clim.2014.06.009
62. Suwandi JS, Laban S, Vass K, Joosten A, van Unen V, Lelieveldt BPF, et al. Multidimensional analyses of proinsulin peptide-specific regulatory T cells induced by tolerogenic dendritic cells. J Autoimmun (2020) 107:102361. doi: 10.1016/j.jaut.2019.102361
63. Gibson VB, Nikolic T, Pearce VQ, Demengeot J, Roep BO, Peakman M. Proinsulin multi-peptide immunotherapy induces antigen-specific regulatory T cells and limits autoimmunity in a humanized model. Clin Exp Immunol (2015) 182(3):251–60. doi: 10.1111/cei.12687
64. Funda DP, Golias J, Hudcovic T, Kozakova H, Spisek R, Palova-Jelinkova L. Antigen loading (e.g., glutamic acid decarboxylase 65) of tolerogenic DCs (tolDCs) reduces their capacity to prevent diabetes in the non-obese diabetes (NOD)-severe combined immunodeficiency model of adoptive cotransfer of diabetes as well as in NOD mice. Front Immunol (2018) 9:290. doi: 10.3389/fimmu.2018.00290
65. Navarro-Barriuso J, Mansilla MJ, Naranjo-Gomez M, Sanchez-Pla A, Quirant-Sanchez B, Teniente-Serra A, et al. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci Rep (2018) 8(1):14985. doi: 10.1038/s41598-018-33248-7
66. Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive b lymphocytes. Clin Exp Immunol (2013) 174(2):302–17. doi: 10.1111/cei.12177
67. Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Involvement of suppressive b-lymphocytes in the mechanism of tolerogenic dendritic cell reversal of type 1 diabetes in NOD mice. PloS One (2014) 9(1):e83575. doi: 10.1371/journal.pone.0083575
68. Machen J, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol (2004) 173(7):4331–41. doi: 10.4049/jimmunol.173.7.4331
69. Rodriguez-Fernandez S, Pujol-Autonell I, Brianso F, Perna-Barrull D, Cano-Sarabia M, Garcia-Jimeno S, et al. Phosphatidylserine-liposomes promote tolerogenic features on dendritic cells in human type 1 diabetes by apoptotic mimicry. Front Immunol (2018) 9:253. doi: 10.3389/fimmu.2018.00253
70. Rodriguez-Fernandez S, Murillo M, Villalba A, Perna-Barrull D, Cano-Sarabia M, Gomez-Munoz L, et al. Impaired phagocytosis in dendritic cells from pediatric patients with type 1 diabetes does not hamper their tolerogenic potential. Front Immunol (2019) 10:2811. doi: 10.3389/fimmu.2019.02811
71. Kim DH, Lee JC, Lee MK, Kim KW, Lee MS. Treatment of autoimmune diabetes in NOD mice by toll-like receptor 2 tolerance in conjunction with dipeptidyl peptidase 4 inhibition. Diabetologia (2012) 55(12):3308–17. doi: 10.1007/s00125-012-2723-x
72. Marin-Gallen S, Clemente-Casares X, Planas R, Pujol-Autonell I, Carrascal J, Carrillo J, et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clin Exp Immunol (2010) 160(2):207–14. doi: 10.1111/j.1365-2249.2009.04082.x
73. Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, et al. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PloS One (2015) 10(6):e0127057. doi: 10.1371/journal.pone.0127057
74. Fuchs EJ, Matzinger P. Does the danger model shed any light on central tolerance?: a response to Al-yassin. Scand J Immunol (2018) 88(3):e12660. doi: 10.1111/sji.12660
75. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
76. Matzinger P. Essay 1: the danger model in its historical context. Scand J Immunol (2001) 54(1–2):4–9. doi: 10.1046/j.1365-3083.2001.00974.x
77. Matzinger P. Introduction to the series. Danger Model immunity. Scand J Immunol (2001) 54(1-2):2–3. doi: 10.1046/j.1365-3083.2001.00973.x
78. Matzinger P. An innate sense of danger. Ann N Y Acad Sci (2002) 961:341–2. doi: 10.1111/j.1749-6632.2002.tb03118.x
79. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296(5566):301–5. doi: 10.1126/science.1071059
80. Matzinger P. The evolution of the danger theory. Interview by Lauren Constable Commissioning Editor. Expert Rev Clin Immunol (2012) 8(4):311–7. doi: 10.1586/eci.12.21
81. Matzinger P. Autoimmunity: are we asking the right question? Front Immunol (2022) 13:864633. doi: 10.3389/fimmu.2022.864633
82. Shaw ER, Matzinger P. Transient autoantibodies to danger signals. Front Immunol (2023) 14:1046300. doi: 10.3389/fimmu.2023.1046300
83. Beyersdorf N, Kerkau T, Hunig T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther (2015) 4:111–22. doi: 10.2147/ITT.S61647
84. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
85. Imanishi T, Saito T. T Cell Co-stimulation and functional modulation by innate signals. Trends Immunol (2020) 41(3):200–12. doi: 10.1016/j.it.2020.01.003
86. Scheinecker C. Blockade of co-stimulation in chronic inflammatory diseases. Wien Med Wochenschr (2015) 165(1-2):23–7. doi: 10.1007/s10354-014-0313-x
87. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
88. Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol (2017) 39(2):113–20. doi: 10.1007/s00281-016-0587-8
89. Jia L, Lu J, Zhou Y, Tao Y, Xu H, Zheng W, et al. Tolerogenic dendritic cells induced the enrichment of CD4(+)Foxp3(+) regulatory T cells via TGF-beta in mesenteric lymph nodes of murine LPS-induced tolerance model. Clin Immunol (2018) 197:118–29. doi: 10.1016/j.clim.2018.09.010
90. Iberg CA, Jones A, Hawiger D. Dendritic cells as inducers of peripheral tolerance. Trends Immunol (2017) 38(11):793–804. doi: 10.1016/j.it.2017.07.007
91. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol (2001) 19:47–64. doi: 10.1146/annurev.immunol.19.1.47
92. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol (2002) 23(9):445–9. doi: 10.1016/s1471-4906(02)02281-0
93. Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest (2013) 123(2):844–54. doi: 10.1172/JCI65260
94. George MM, Subramanian Vignesh K, Landero Figueroa JA, Caruso JA, Deepe GS Jr. Zinc induces dendritic cell tolerogenic phenotype and skews regulatory T cell-Th17 balance. J Immunol (2016) 197(5):1864–76. doi: 10.4049/jimmunol.1600410
95. Lecuyer E, Le Roy T, Gestin A, Lacombe A, Philippe C, Ponnaiah M, et al. Tolerogenic dendritic cells shape a transmissible gut microbiota that protects from metabolic diseases. Diabetes (2021) 70(9):2067–80. doi: 10.2337/db20-1177
96. Magrone T, Jirillo E. The interplay between the gut immune system and microbiota in health and disease: nutraceutical intervention for restoring intestinal homeostasis. Curr Pharm Des (2013) 19(7):1329–42. doi: 10.2174/138161213804805793
97. Swiatczak B, Rescigno M. How the interplay between antigen presenting cells and microbiota tunes host immune responses in the gut. Semin Immunol (2012) 24(1):43–9. doi: 10.1016/j.smim.2011.11.004
98. Wang J, Zhu N, Su X, Gao Y, Yang R. Gut-Microbiota-Derived metabolites maintain gut and systemic immune homeostasis. Cells (2023) 12(5). doi: 10.3390/cells12050793
99. Dudek AM, Martin S, Garg AD, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol (2013) 4:438. doi: 10.3389/fimmu.2013.00438
100. Frick JS, Grunebach F, Autenrieth IB. Immunomodulation by semi-mature dendritic cells: a novel role of toll-like receptors and interleukin-6. Int J Med Microbiol (2010) 300(1):19–24. doi: 10.1016/j.ijmm.2009.08.010
101. Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c+CD8a–dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol (2009) 21(3):269–82. doi: 10.1093/intimm/dxn147
102. Gordon JR, Ma Y, Churchman L, Gordon SA, Dawicki W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front Immunol (2014) 5:7. doi: 10.3389/fimmu.2014.00007
103. Haase C, Yu L, Eisenbarth G, Markholst H. Antigen-dependent immunotherapy of non-obese diabetic mice with immature dendritic cells. Clin Exp Immunol (2010) 160(3):331–9. doi: 10.1111/j.1365-2249.2010.04104.x
104. Lutz MB. Therapeutic potential of semi-mature dendritic cells for tolerance induction. Front Immunol (2012) 3:123. doi: 10.3389/fimmu.2012.00123
105. Morel PA, Turner MS. Dendritic cells and the maintenance of self-tolerance. Immunol Res (2011) 50(2-3):124–9. doi: 10.1007/s12026-011-8217-y
106. Nikolic T, Roep BO. Regulatory multitasking of tolerogenic dendritic cells - lessons taken from vitamin d3-treated tolerogenic dendritic cells. Front Immunol (2013) 4:113. doi: 10.3389/fimmu.2013.00113
107. Nouri-Shirazi M, Thomson AW. Dendritic cells as promoters of transplant tolerance. Expert Opin Biol Ther (2006) 6(4):325–39. doi: 10.1517/14712598.6.4.325
108. Sato K, Yamashita N, Matsuyama T. Human peripheral blood monocyte-derived interleukin-10-induced semi-mature dendritic cells induce anergic CD4(+) and CD8(+) T cells via presentation of the internalized soluble antigen and cross-presentation of the phagocytosed necrotic cellular fragments. Cell Immunol (2002) 215(2):186–94. doi: 10.1016/s0008-8749(02)00021-7
109. Marin E, Bouchet-Delbos L, Renoult O, Louvet C, Nerriere-Daguin V, Managh AJ, et al. Human tolerogenic dendritic cells regulate immune responses through lactate synthesis. Cell Metab (2019) 30(6):1075–1090 e8. doi: 10.1016/j.cmet.2019.11.011
110. Lord P, Spiering R, Aguillon JC, Anderson AE, Appel S, Benitez-Ribas D, et al. Minimum information about tolerogenic antigen-presenting cells (MITAP): a first step towards reproducibility and standardisation of cellular therapies. PeerJ (2016) 4:e2300. doi: 10.7717/peerj.2300
111. Schuster C, Jonas F, Zhao F, Kissler S. Peripherally induced regulatory T cells contribute to the control of autoimmune diabetes in the NOD mouse model. Eur J Immunol (2018) 48(7):1211–6. doi: 10.1002/eji.201847498
112. Zhu Y, Winer D, Goh C, Shrestha A. Injectable thermosensitive hydrogel to modulate tolerogenic dendritic cells under hyperglycemic condition. Biomater Sci (2023) 11(6):2091–102. doi: 10.1039/d2bm01881k
113. Sgouroudis E, Kornete M, Piccirillo CA. IL-2 production by dendritic cells promotes Foxp3(+) regulatory T-cell expansion in autoimmune-resistant NOD congenic mice. Autoimmunity (2011) 44(5):406–14. doi: 10.3109/08916934.2010.536795
114. Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med (2007) 204(1):191–201. doi: 10.1084/jem.20061631
115. Luo X, Tarbell KV, Yang H, Pothoven K, Bailey SL, Ding R, et al. Dendritic cells with TGF-beta1 differentiate naive CD4+CD25- T cells into islet-protective Foxp3+ regulatory T cells. Proc Natl Acad Sci U.S.A. (2007) 104(8):2821–6. doi: 10.1073/pnas.0611646104
116. Gaudreau S, Guindi C, Menard M, Besin G, Dupuis G, Amrani A. Granulocyte-macrophage colony-stimulating factor prevents diabetes development in NOD mice by inducing tolerogenic dendritic cells that sustain the suppressive function of CD4+CD25+ regulatory T cells. J Immunol (2007) 179(6):3638–47. doi: 10.4049/jimmunol.179.6.3638
117. DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol (2007) 179(7):4685–93. doi: 10.4049/jimmunol.179.7.4685
118. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature (2020) 580(7802):257–62. doi: 10.1038/s41586-020-2134-y
119. Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, et al. Dendritic cells coordinate the development and homeostasis of organ-specific regulatory T cells. Immunity (2016) 44(4):847–59. doi: 10.1016/j.immuni.2016.01.025
120. Sichien D, Lambrecht BN, Guilliams M, Scott CL. Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol (2017) 10(4):831–44. doi: 10.1038/mi.2017.8
121. Jenkins MM, Bachus H, Botta D, Schultz MD, Rosenberg AF, Leon B, et al. Lung dendritic cells migrate to the spleen to prime long-lived TCF1(hi) memory CD8(+) T cell precursors after influenza infection. Sci Immunol (2021) 6(63):eabg6895. doi: 10.1126/sciimmunol.abg6895
122. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol (2019) 19(2):89–103. doi: 10.1038/s41577-018-0088-1
123. Iberg CA, Hawiger D. Advancing immunomodulation by in vivo antigen delivery to DEC-205 and other cell surface molecules using recombinant chimeric antibodies. Int Immunopharmacol (2019) 73:575–80. doi: 10.1016/j.intimp.2019.05.037
124. Jones A, Bourque J, Kuehm L, Opejin A, Teague RM, Gross C, et al. Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity (2016) 45(5):1066–77. doi: 10.1016/j.immuni.2016.10.008
125. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (2017) 356(6335). doi: 10.1126/science.aah4573
126. Iberg CA, Bourque J, Fallahee I, Son S, Hawiger D. TNF-alpha sculpts a maturation process in vivo by pruning tolerogenic dendritic cells. Cell Rep (2022) 39(2):110657. doi: 10.1016/j.celrep.2022.110657
127. Bourque J, Hawiger D. Immunomodulatory bonds of the partnership between dendritic cells and T cells. Crit Rev Immunol (2018) 38(5):379–401. doi: 10.1615/CritRevImmunol.2018026790
128. Henderson JG, Opejin A, Jones A, Gross C, Hawiger D. CD5 instructs extrathymic regulatory T cell development in response to self and tolerizing antigens. Immunity (2015) 42(3):471–83. doi: 10.1016/j.immuni.2015.02.010
129. Gargaro M, Scalisi G, Manni G, Briseno CG, Bagadia P, Durai V, et al. Indoleamine 2,3-dioxygenase 1 activation in mature cDC1 promotes tolerogenic education of inflammatory cDC2 via metabolic communication. Immunity (2022) 55(6):1032–1050 e14. doi: 10.1016/j.immuni.2022.05.013
130. Tabansky I, Keskin DB, Watts D, Petzold C, Funaro M, Sands W, et al. Targeting DEC-205(-)DCIR2(+) dendritic cells promotes immunological tolerance in proteolipid protein-induced experimental autoimmune encephalomyelitis. Mol Med (2018) 24(1):17. doi: 10.1186/s10020-018-0017-6
131. Price JD, Hotta-Iwamura C, Zhao Y, Beauchamp NM, Tarbell KV. DCIR2+ cDC2 DCs and Zbtb32 restore CD4+ T-cell tolerance and inhibit diabetes. Diabetes (2015) 64(10):3521–31. doi: 10.2337/db14-1880
132. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
133. Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol Appl Pharmacol (2010) 246(1-2):18–28. doi: 10.1016/j.taap.2010.03.013
134. Fallarino F, Vacca C, Orabona C, Belladonna ML, Bianchi R, Marshall B, et al. Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol (2002) 14(1):65–8. doi: 10.1093/intimm/14.1.65
135. Huang L, Baban B, Johnson 3BA, Mellor AL. Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. Int Rev Immunol (2010) 29(2):133–55. doi: 10.3109/08830180903349669
136. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol (2000) 164(7):3596–9. doi: 10.4049/jimmunol.164.7.3596
137. Liu K, Yang Y, Chen Y, Li S, Gong Y, Liang Y. The therapeutic effect of dendritic cells expressing indoleamine 2,3-dioxygenase (IDO) on an IgA nephropathy mouse model. Int Urol Nephrol (2020) 52(2):399–407. doi: 10.1007/s11255-019-02365-1
138. Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, et al. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut (2010) 59(5):595–604. doi: 10.1136/gut.2009.185108
139. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
140. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science (2002) 297(5588):1867–70. doi: 10.1126/science.1073514
141. Sittig SP, van Beek JJP, Florez-Grau G, Weiden J, Buschow SI, van der Net MC, et al. Human type 1 and type 2 conventional dendritic cells express indoleamine 2,3-dioxygenase 1 with functional effects on T cell priming. Eur J Immunol (2021) 51(6):1494–504. doi: 10.1002/eji.202048580
142. Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med (2002) 196(4):447–57. doi: 10.1084/jem.20020052
143. Wolf B, Posnick D, Fisher JL, Lewis LD, Ernstoff MS. Indoleamine-2,3-dioxygenase enzyme expression and activity in polarized dendritic cells. Cytotherapy (2009) 11(8):1084–9. doi: 10.3109/14653240903271230
144. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med (1987) 165(2):302–19. doi: 10.1084/jem.165.2.302
145. Vander Lugt B, Riddell J, Khan AA, Hackney JA, Lesch J, DeVoss J, et al. Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J Cell Biol (2017) 216(3):779–92. doi: 10.1083/jcb.201512012
146. Hoffmann C, Noel F, Grandclaudon M, Massenet-Regad L, Michea P, Sirven P, et al. PD-L1 and ICOSL discriminate human secretory and helper dendritic cells in cancer, allergy and autoimmunity. Nat Commun (2022) 13(1):1983. doi: 10.1038/s41467-022-29516-w
147. Mazerolles F, Rieux-Laucat F. : PD-L1 is expressed on human activated naive effector CD4+ T cells. regulation by dendritic cells and regulatory CD4+ T cells. PloS One (2021) 16(11):e0260206. doi: 10.1371/journal.pone.0260206
148. Moreira TG, Mangani D, Cox LM, Leibowitz J, Lobo ELC, Oliveira MA, et al. PD-L1(+) and XCR1(+) dendritic cells are region-specific regulators of gut homeostasis. Nat Commun (2021) 12(1):4907. doi: 10.1038/s41467-021-25115-3
149. Tamburini BAJ. Contributions of PD-L1 reverse signaling to dendritic cell trafficking. FEBS J (2022) 289(20):6256–66. doi: 10.1111/febs.16084
150. Song S, Yuan P, Wu H, Chen J, Fu J, Li P, et al. Dendritic cells with an increased PD-L1 by TGF-beta induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int Immunopharmacol (2014) 20(1):117–23. doi: 10.1016/j.intimp.2014.02.027
151. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic cell PD-L1 limits autoimmunity and follicular T cell differentiation and function. J Immunol (2018) 200(8):2592–602. doi: 10.4049/jimmunol.1701231
152. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
153. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U.S.A. (2010) 107(4):1524–8. doi: 10.1073/pnas.0910341107
154. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood (2002) 99(7):2468–76. doi: 10.1182/blood.v99.7.2468
155. Pletinckx K, Vaeth M, Schneider T, Beyersdorf N, Hunig T, Berberich-Siebelt F, et al. Immature dendritic cells convert anergic nonregulatory T cells into Foxp3- IL-10+ regulatory T cells by engaging CD28 and CTLA-4. Eur J Immunol (2015) 45(2):480–91. doi: 10.1002/eji.201444991
156. Fanger NA, Maliszewski CR, Schooley K, Griffith TS. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Exp Med (1999) 190(8):1155–64. doi: 10.1084/jem.190.8.1155
157. Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med (1996) 183(4):1789–96. doi: 10.1084/jem.183.4.1789
158. Comi M, Avancini D, Santoni de Sio F, Villa M, Uyeda MJ, Floris M, et al. Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10). Cell Mol Immunol (2020) 17(1):95–107. doi: 10.1038/s41423-019-0218-0
159. Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol (2007) 8(12):1380–9. doi: 10.1038/ni1541
160. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol (2013) 14(10):1054–63. doi: 10.1038/ni.2695
161. Karakhanova S, Bedke T, Enk AH, Mahnke K. IL-27 renders DC immunosuppressive by induction of B7-H1. J Leukoc Biol (2011) 89(6):837–45. doi: 10.1189/jlb.1209788
162. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol (2008) 181(4):2277–84. doi: 10.4049/jimmunol.181.4.2277
163. Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma-Garcia I, et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity (2015) 42(3):499–511. doi: 10.1016/j.immuni.2015.02.003
164. Mucida D, Cheroutre H. TGFbeta and retinoic acid intersect in immune-regulation. Cell Adh Migr (2007) 1(3):142–4. doi: 10.4161/cam.1.3.5062
165. Mucida D, Pino-Lagos K, Kim G, Nowak E, Benson MJ, Kronenberg M, et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) treg cell conversion of naive T cells. Immunity (2009) 30(4):471–2. doi: 10.1016/j.immuni.2009.03.008
166. Mansilla MJ, Contreras-Cardone R, Navarro-Barriuso J, Cools N, Berneman Z, Ramo-Tello C, et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J Neuroinflamm (2016) 13(1):113. doi: 10.1186/s12974-016-0584-9
167. Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent b-cell subset in humans that parallels mouse regulatory B10 cells. Blood (2011) 117(2):530–41. doi: 10.1182/blood-2010-07-294249
168. Kalampokis I, Venturi GM, Poe JC, Dvergsten JA, Sleasman JW, Tedder TF. The regulatory b cell compartment expands transiently during childhood and is contracted in children with autoimmunity. Arthritis Rheumatol (2017) 69(1):225–38. doi: 10.1002/art.39820
169. Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory b cells (B10 cells) in autoimmune disease. Arthritis Res Ther (2013) 15 Suppl 1(Suppl 1):S1. doi: 10.1186/ar3907
170. Lykken JM, Candando KM, Tedder TF. Regulatory B10 cell development and function. Int Immunol (2015) 27(10):471–7. doi: 10.1093/intimm/dxv046
171. Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory b cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol (2010) 185(4):2240–52. doi: 10.4049/jimmunol.1001307
172. Matsushita T, Tedder TF. Identifying regulatory b cells (B10 cells) that produce IL-10 in mice. Methods Mol Biol (2011) 677:99–111. doi: 10.1007/978-1-60761-869-0_7
173. Tedder TF. Introduction: regulatory b cell special issue-making all the pieces fit. Int Immunol (2015) 27(10):467–70. doi: 10.1093/intimm/dxv047
174. Tedder TF. B10 cells: a functionally defined regulatory b cell subset. J Immunol (2015) 194(4):1395–401. doi: 10.4049/jimmunol.1401329
175. Tedder TF, Leonard WJ. Autoimmunity: regulatory b cells–IL-35 and IL-21 regulate the regulators. Nat Rev Rheumatol (2014) 10(8):452–3. doi: 10.1038/nrrheum.2014.95
176. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory b cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi: 10.1038/nature11501
177. Mauri C. Novel frontiers in regulatory b cells. Immunol Rev (2021) 299(1):5–9. doi: 10.1111/imr.12964
178. Mauri C, Menon M. Human regulatory b cells in health and disease: therapeutic potential. J Clin Invest (2017) 127(3):772–9. doi: 10.1172/JCI85113
179. Menon M, Rosser EC, Mauri C. Identification and isolation of regulatory b cells in mouse and human. Methods Mol Biol (2019) 1899:55–66. doi: 10.1007/978-1-4939-8938-6_5
180. Rosser EC, Mauri C. The emerging field of regulatory b cell immunometabolism. Cell Metab (2021) 33(6):1088–97. doi: 10.1016/j.cmet.2021.05.008
181. Achour A, Simon Q, Mohr A, Seite JF, Youinou P, Bendaoud B, et al. Human regulatory b cells control the T(FH) cell response. J Allergy Clin Immunol (2017) 140(1):215–22. doi: 10.1016/j.jaci.2016.09.042
182. Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory b cells that suppress autoimmune disease. Nat Med (2014) 20(6):633–41. doi: 10.1038/nm.3554
183. Figueiro F, Muller L, Funk S, Jackson EK, Battastini AM, Whiteside TL. Phenotypic and functional characteristics of CD39(high) human regulatory b cells (Breg). Oncoimmunology (2016) 5(2):e1082703. doi: 10.1080/2162402X.2015.1082703
184. Luo Y, Luo F, Zhang K, Wang S, Zhang H, Yang X, et al. Elevated circulating IL-10 producing breg, but not regulatory b cell levels, restrain antibody-mediated rejection after kidney transplantation. Front Immunol (2020) 11:627496. doi: 10.3389/fimmu.2020.627496
185. Mohd Jaya FN, Garcia SG, Borras FE, Guerrero D, Chan GCF, Franquesa M. In vitro characterization of human CD24(hi)CD38(hi) regulatory b cells shows CD9 is not a stable breg cell marker. Int J Mol Sci (2021) 22(9). doi: 10.3390/ijms22094583
186. Wang M, Gu Z, Yang J, Zhao H, Cao Z. Changes among TGF-beta1(+) breg cells and helper T cell subsets in a murine model of allergic rhinitis with prolonged OVA challenge. Int Immunopharmacol (2019) 69:347–57. doi: 10.1016/j.intimp.2019.01.009
187. Boldison J, Da Rosa LC, Davies J, Wen L, Wong FS. Dendritic cells license regulatory b cells to produce IL-10 and mediate suppression of antigen-specific CD8 T cells. Cell Mol Immunol (2020) 17(8):843–55. doi: 10.1038/s41423-019-0324-z
188. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi b cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med (2013) 5(173):173ra23. doi: 10.1126/scitranslmed.3005407
189. Oleinika K, Rosser EC, Matei DE, Nistala K, Bosma A, Drozdov I, et al. CD1d-dependent immune suppression mediated by regulatory b cells through modulations of iNKT cells. Nat Commun (2018) 9(1):684. doi: 10.1038/s41467-018-02911-y
190. Kendal AR, Chen Y, Regateiro FS, Ma J, Adams E, Cobbold SP, et al. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med (2011) 208(10):2043–53. doi: 10.1084/jem.20110767
191. Ochando J, Ordikhani F, Jordan S, Boros P, Thomson AW. Tolerogenic dendritic cells in organ transplantation. Transpl Int (2020) 33(2):113–27. doi: 10.1111/tri.13504
192. Kleijwegt FS, Laban S, Duinkerken G, Joosten AM, Koeleman BP, Nikolic T, et al. Transfer of regulatory properties from tolerogenic to proinflammatory dendritic cells via induced autoreactive regulatory T cells. J Immunol (2011) 187(12):6357–64. doi: 10.4049/jimmunol.1101638
193. Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol (2015) 8(2):265–78. doi: 10.1038/mi.2014.64
194. Min WP, Zhou D, Ichim TE, Strejan GH, Xia X, Yang J, et al. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J Immunol (2003) 170(3):1304–12. doi: 10.4049/jimmunol.170.3.1304
195. Tureci O, Bian H, Nestle FO, Raddrizzani L, Rosinski JA, Tassis A, et al. Cascades of transcriptional induction during dendritic cell maturation revealed by genome-wide expression analysis. FASEB J (2003) 17(8):836–47. doi: 10.1096/fj.02-0724com
196. Svajger U, Rozman PJ. Recent discoveries in dendritic cell tolerance-inducing pharmacological molecules. Int Immunopharmacol (2020) 81:106275. doi: 10.1016/j.intimp.2020.106275
197. Cauwels A, Tavernier J. Tolerizing strategies for the treatment of autoimmune diseases: from ex vivo to in vivo strategies. Front Immunol (2020) 11:674. doi: 10.3389/fimmu.2020.00674
198. Funda DP, Palova-Jelinkova L, Golias J, Kroulikova Z, Fajstova A, Hudcovic T, et al. Optimal tolerogenic dendritic cells in type 1 diabetes (T1D) therapy: what can we learn from non-obese diabetic (NOD) mouse models? Front Immunol (2019) 10:967. doi: 10.3389/fimmu.2019.00967
199. Creusot RJ, Chang P, Healey DG, Tcherepanova IY, Nicolette CA, Fathman CG. A short pulse of IL-4 delivered by DCs electroporated with modified mRNA can both prevent and treat autoimmune diabetes in NOD mice. Mol Ther (2010) 18(12):2112–20. doi: 10.1038/mt.2010.146
200. Lo J, Xia CQ, Peng R, Clare-Salzler MJ. Immature dendritic cell therapy confers durable immune modulation in an antigen-dependent and antigen-independent manner in nonobese diabetic mice. J Immunol Res (2018) 2018:5463879. doi: 10.1155/2018/5463879
201. Kleijwegt FS, Jansen DT, Teeler J, Joosten AM, Laban S, Nikolic T, et al. Tolerogenic dendritic cells impede priming of naive CD8(+) T cells and deplete memory CD8(+) T cells. Eur J Immunol (2013) 43(1):85–92. doi: 10.1002/eji.201242879
202. Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res (2012) 11(2):941–71. doi: 10.1021/pr200724e
203. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol (2009) 39(11):3147–59. doi: 10.1002/eji.200839103
204. Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood (2006) 108(5):1435–40. doi: 10.1182/blood-2006-03-006403
205. Kryczanowsky F, Raker V, Graulich E, Domogalla MP, Steinbrink K. IL-10-Modulated human dendritic cells for clinical use: identification of a stable and migratory subset with improved tolerogenic activity. J Immunol (2016) 197(9):3607–17. doi: 10.4049/jimmunol.1501769
206. Ferreira GB, Gysemans CA, Demengeot J, da Cunha JP, Vanherwegen AS, Overbergh L, et al. 1,25-dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J Immunol (2014) 192(9):4210–20. doi: 10.4049/jimmunol.1302350
207. Vives-Pi M, Rodriguez-Fernandez S, Pujol-Autonell I. How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis (2015) 20(3):263–72. doi: 10.1007/s10495-015-1090-8
208. Hayashi T, Yao S, Crain B, Promessi VJ, Shyu L, Sheng C, et al. Induction of tolerogenic dendritic cells by a PEGylated TLR7 ligand for treatment of type 1 diabetes. PloS One (2015) 10(6):e0129867. doi: 10.1371/journal.pone.0129867
209. Kim DH, Lee JC, Kim S, Oh SH, Lee MK, Kim KW, et al. Inhibition of autoimmune diabetes by TLR2 tolerance. J Immunol (2011) 187(10):5211–20. doi: 10.4049/jimmunol.1001388
210. Raptopoulou A, Sidiropoulos P, Katsouraki M, Boumpas DT. Anti-citrulline antibodies in the diagnosis and prognosis of rheumatoid arthritis: evolving concepts. Crit Rev Clin Lab Sci (2007) 44(4):339–63. doi: 10.1080/10408360701295623
211. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/S0140-6736(16)30173-8
212. Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol (2001) 166(8):5265–70. doi: 10.4049/jimmunol.166.8.5265
213. Phillips BE, Giannoukakis N, Trucco M. Dendritic cell mediated therapy for immunoregulation of type 1 diabetes mellitus. Pediatr Endocrinol Rev PER (2008) 5(4):873–9.
214. Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes (2005) 54 Suppl 2:S52–61. doi: 10.2337/diabetes.54.suppl_2.S52
215. Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med (2012) 2(4):a007781. doi: 10.1101/cshperspect.a007781
216. Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Chromogranin a is an autoantigen in type 1 diabetes. Nat Immunol (2010) 11(3):225–31. doi: 10.1038/ni.1844
217. Garg R, Agarwal A, Katekar R, Dadge S, Yadav S, Gayen JR. Chromogranin a-derived peptides pancreastatin and catestatin: emerging therapeutic target for diabetes. Amino Acids (2023). doi: 10.1007/s00726-023-03252-x
218. Crawford SA, Wiles TA, Wenzlau JM, Powell RL, Barbour G, Dang M, et al. Cathepsin d drives the formation of hybrid insulin peptides relevant to the pathogenesis of type 1 diabetes. Diabetes (2022) 71(12):2793–803. doi: 10.2337/db22-0303
219. Podojil JR, Genardi S, Chiang MY, Kakade S, Neef T, Murthy T, et al. Miller: tolerogenic immune-modifying nanoparticles encapsulating multiple recombinant pancreatic beta cell proteins prevent onset and progression of type 1 diabetes in nonobese diabetic mice. J Immunol (2022) 209(3):465–75. doi: 10.4049/jimmunol.2200208
220. Parras D, Sole P, Delong T, Santamaria P, Serra P. Recognition of multiple hybrid insulin peptides by a single highly diabetogenic T-cell receptor. Front Immunol (2021) 12:737428. doi: 10.3389/fimmu.2021.737428
221. Piganelli JD, Mamula MJ, James EA. The role of beta cell stress and neo-epitopes in the immunopathology of type 1 diabetes. Front Endocrinol (Lausanne) (2020) 11:624590. doi: 10.3389/fendo.2020.624590
222. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheumatic Dis (2016) 76(1):227–34. doi: 10.1136/annrheumdis-2015-208456
223. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis (2010) 69(11):2042–50. doi: 10.1136/ard.2009.126383
224. Aguirre RS, Kulkarni A, Becker MW, Lei X, Sarkar S, Ramanadham S, et al. Extracellular vesicles in beta cell biology: role of lipids in vesicle biogenesis, cargo, and intercellular signaling. Mol Metab (2022) 63:101545. doi: 10.1016/j.molmet.2022.101545
225. Petrelli A, Giovenzana A, Insalaco V, Phillips BE, Pietropaolo M, Giannoukakis N. Autoimmune inflammation and insulin resistance: hallmarks so far and yet so close to explain diabetes endotypes. Curr Diabetes Rep (2021) 21(12):54. doi: 10.1007/s11892-021-01430-3
226. Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med (1990) 172(2):631–40. doi: 10.1084/jem.172.2.631
227. Simon T, Fonteneau JF, Gregoire M. Requirement of tumor-associated antigen-specific CD4+ T cells for an efficient dendritic cell vaccine in antitumor immunotherapy. Immunotherapy (2013) 5(6):565–7. doi: 10.2217/imt.13.45
228. Marten A, Greten T, Ziske C, Renoth S, Schottker B, Buttgereit P, et al. Generation of activated and antigen-specific T cells with cytotoxic activity after co-culture with dendritic cells. Cancer Immunol Immunother (2002) 51(1):25–32. doi: 10.1007/s00262-001-0251-5
229. Tsai V, Kawashima I, Keogh E, Daly K, Sette A, Celis E. In vitro immunization and expansion of antigen-specific cytotoxic T lymphocytes for adoptive immunotherapy using peptide-pulsed dendritic cells. Crit Rev Immunol (1998) 18(1-2):65–75. doi: 10.1615/critrevimmunol.v18.i1-2.80
230. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
231. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35(9):1817–20. doi: 10.2337/dc12-0038
232. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med (2016) 14(1):332. doi: 10.1186/s12967-016-1090-7
233. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
234. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PloS Med (2016) 13(10):e1002139. doi: 10.1371/journal.pmed.1002139
235. Hotta-Iwamura C, Benck C, Coley WD, Liu Y, Zhao Y, Quiel JA, et al. Low CD25 on autoreactive tregs impairs tolerance via low dose IL-2 and antigen delivery. J Autoimmun (2018) 90:39–48. doi: 10.1016/j.jaut.2018.01.005
236. Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J Exp Med (2006) 203(4):1045–54. doi: 10.1084/jem.20051954
237. Jauregui-Amezaga A, Cabezon R, Ramirez-Morros A, Espana C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory crohn's disease: a phase I study. J Crohn's colitis (2015) 9(12):1071–8. doi: 10.1093/ecco-jcc/jjv144
238. Hotta-Iwamura C, Tarbell KV. Type 1 diabetes genetic susceptibility and dendritic cell function: potential targets for treatment. J Leukoc Biol (2016) 100(1):65–80. doi: 10.1189/jlb.3MR1115-500R
239. Danova K, Grohova A, Strnadova P, Funda DP, Sumnik Z, Lebl J, et al. Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen-specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J Immunol (2017) 198(2):729–40. doi: 10.4049/jimmunol.1600676
240. Ludvigsson J, Linkoping Diabetes Immune Intervention Study G. The role of immunomodulation therapy in autoimmune diabetes. J Diabetes Sci Technol (2009) 3(2):320–30. doi: 10.1177/193229680900300213
241. Robert S, Korf H, Gysemans C, Mathieu C. Antigen-based vs. systemic immunomodulation in type 1 diabetes: the pros and cons. Islets (2013) 5(2):53–66. doi: 10.4161/isl.24785
242. Skyler JS. Immunomodulation for type 1 diabetes mellitus. Int J Clin Pract (2010) Suppl(166):59–63. doi: 10.1111/j.1742-1241.2009.02280.x
243. Waldron-Lynch F, Herold KC. Advances in type 1 diabetes therapeutics: immunomodulation and beta-cell salvage. Endocrinol Metab Clin North Am (2009) 38(2):303–17, viii. doi: 10.1016/j.ecl.2009.01.005
244. Insel R, Dutta S, Hedrick J. Type 1 diabetes: disease stratification. BioMed Hub (2017) 2(Suppl 1):111–26. doi: 10.1159/000481131
245. Skyler JS, Bakris GL, Bonifacio E, Darsow T, Eckel RH, Groop L, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes (2017) 66(2):241–55. doi: 10.2337/db16-0806
Keywords: type 1 diabetes, dendritic cells, tolerance, immunomodulation, autoimmunity
Citation: Giannoukakis N (2023) Tolerogenic dendritic cells in type 1 diabetes: no longer a concept. Front. Immunol. 14:1212641. doi: 10.3389/fimmu.2023.1212641
Received: 26 April 2023; Accepted: 31 May 2023;
Published: 14 June 2023.
Edited by:
Silvia Gregori, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), ItalyReviewed by:
James A. Hutchinson, University Medical Center Regensburg, GermanyAurelie Moreau, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2023 Giannoukakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick Giannoukakis, bmduMUBhbmRyZXcuY211LmVkdQ==