 Cécile Gouttefangeas
Cécile Gouttefangeas Reinhild Klein
Reinhild Klein Ana Maia1,2
Ana Maia1,2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 June 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1212546
This article is part of the Research Topic T cell specificity and Cross-reactivity – Implications in Physiology and Pathology View all 11 articles
T cells are main actors of the immune system with an essential role in protection against pathogens and cancer. The molecular key event involved in this absolutely central task is the interaction of membrane-bound specific T cell receptors with peptide-MHC complexes which initiates T cell priming, activation and recall, and thus controls a range of downstream functions. While textbooks teach us that the repertoire of mature T cells is highly diverse, it is clear that this diversity cannot possibly cover all potential foreign peptides that might be encountered during life. TCR cross-reactivity, i.e. the ability of a single TCR to recognise different peptides, offers the best solution to this biological challenge. Reports have shown that indeed, TCR cross-reactivity is surprisingly high. Hence, the T cell dilemma is the following: be as specific as possible to target foreign danger and spare self, while being able to react to a large spectrum of body-threatening situations. This has major consequences for both autoimmune diseases and cancer, and significant implications for the development of T cell-based therapies. In this review, we will present essential experimental evidence of T cell cross-reactivity, implications for two opposite immune conditions, i.e. autoimmunity vs cancer, and how this can be differently exploited for immunotherapy approaches. Finally, we will discuss the tools available for predicting cross-reactivity and how improvements in this field might boost translational approaches.
T cells are essential players of the adaptive immunity that are not only responsible for long-term immune memory, but also orchestrate innate and adaptive immune responses. Immature T cells undergo a strict selection in the thymus which leads to the release of mature, largely self-tolerant, T cells. Each of these cells bears several 10.000 copies of a unique kind of T cell receptor (TCR) that results from the assembly of two recombined TCR chains (in most cases α and β) (1, 2). The TCRαβ interacts with antigens presented as peptides by cell membrane-bound molecules of the major histocompatibility complex (pMHC) on the antigen presenting cell (APC) or on the target cell, for example, after pathogen infection (Figure 1A). Importantly, the binding of peptides to the various MHC allelic products is subject to specific rules (anchor or preferred residues) (4, 5). The diversity of the TCRαβ T cell repertoire is high, but not unlimited. Based on the V, D and J fragments´ recombination at the two chain loci, the theoretical number of single TCRs is estimated to reach at least 1015. In fact, the sum of all different TCRs present in the human blood has been estimated to be much less, in the range of 2.5 x 107 for naïve T cells and approximately 100-fold lower for memory T cells (6–8). The number of potential pathogen- (and tumour-) derived epitopes presented as pMHC throughout life might well exceed this number of T cell clones. It became therefore progressively clear that the clonal selection theory, which proposed that one lymphocyte/receptor is available for each single antigen, needed to be revised, and that cross-reactivity, i.e. the ability of single TCRs to recognise multiple peptide sequences, is a frequent event (9–11).
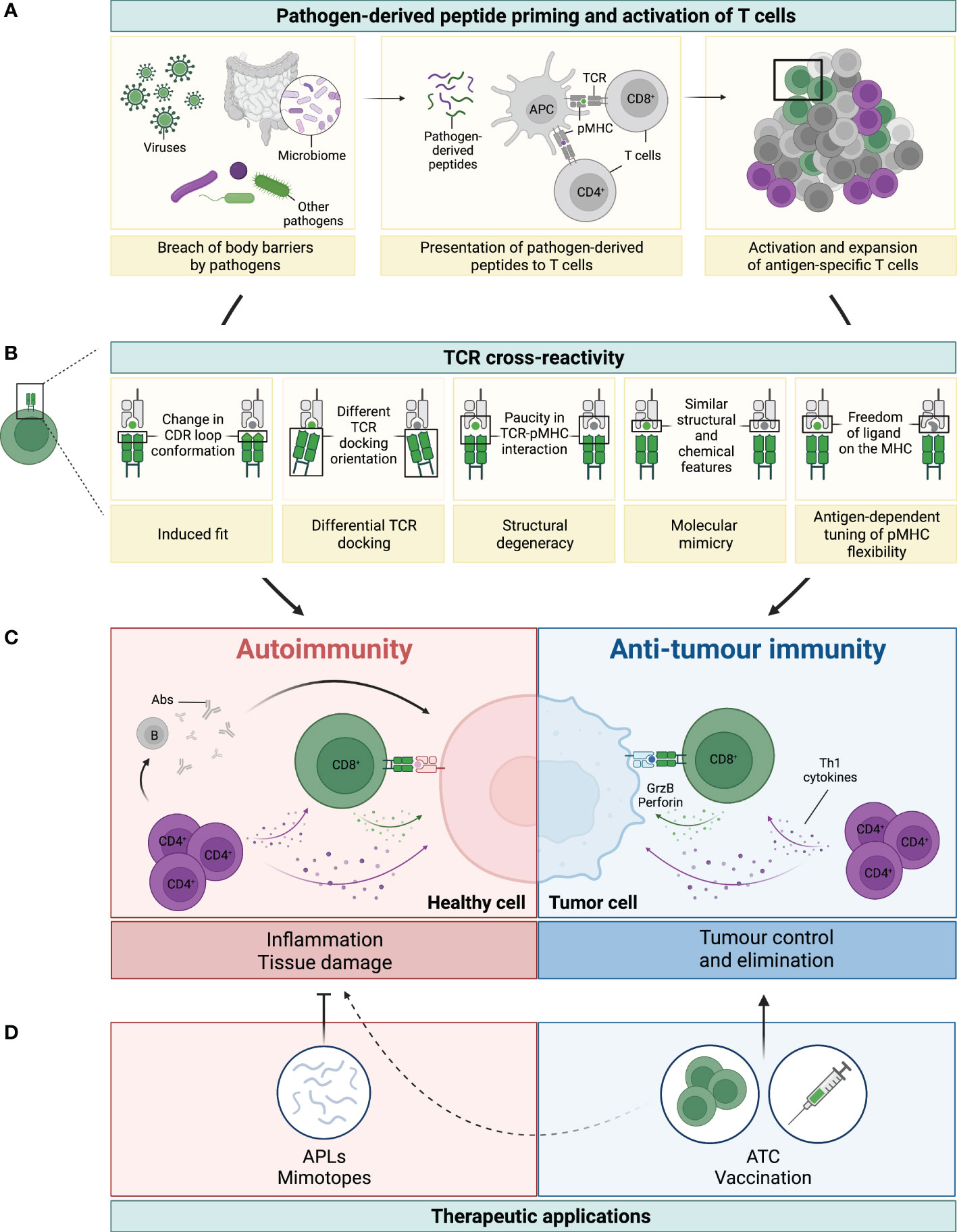
Figure 1 Legend: TCR cross-reactivity: a double-edged sword. (A) Microorganisms, such as viruses, microbiota or other pathogens, can penetrate body barriers and get into contact with our immune system (left panel). Processing and presentation of pathogen (foreign)-derived peptides by antigen-presenting cells (APCs) on MHC-class I (represented by the green dot) and MHC-class II (represented by the purple dot) primes and drives the activation of CD8+ and CD4+ T cells, respectively (middle panel). A polyclonal population of activated T cells then proliferates and expands to fight the invading microorganism (right panel). (B) Several mechanisms have been reported to be involved in TCR cross-reactivity. This figure has been adapted from reference (3). A representative CD8+ T cell and its TCR is shown in green interacting with the pathogen-derived peptide (in green) presented by an MHC-class I molecule (grey). Due to cross-reactivity, the same TCR can interact with another peptide (shown in grey). (C) If this peptide is presented by healthy cells (depicted in pink) or by cancer cells (depicted in blue) this can ultimately result in either autoimmunity or anti-tumour immunity, respectively. On the one hand, recognition of healthy tissues by cross-reactive TCRs (mainly from CD4+, but also from CD8+ T cells), leads to inflammation and tissue damage with deleterious consequences. Secretion of Th1 cytokines by CD4+ T cells (colored in purple) can directly affect healthy cells, but also support the activation of auto-reactive B (grey) and CD8+ T (green) cells, which then secrete Abs and cytotoxic molecules (i.e. granzyme B and perforin, illustrated with the green arrow and dots), respectively (left panel in C). On the other hand, recognition of tumour antigens by cross-reactive T cells can prompt cell killing and tumour elimination, highlighting the contrasting impact of TCR cross-reactivity in this setting (right panel in C). (D) TCR cross-reactivity can be exploited for therapeutic applications. Usage of mimotopes or APLs to drive a Th1 to Th2 or regulatory switch in autoimmunity is an attractive strategy to reduce tissue inflammation and its damage. In cancer, usage of cross-reactive TCRs in adoptive T cell therapy (ATC), or of pathogen-derived peptides for vaccination are promising nascent strategies. Potential side effects against healthy tissues of this novel anti-cancer therapies (represented by the dotted line) need to be carefully considered to prevent damage and severe toxicities.
Cross-reactivity is commonly observed when testing nearly identical peptides which differ only in 1 or 2 amino acids (aa) (for a total length of 8-10 aa for a CD8+ T cell epitope presented by MHC-class I). This is physiologically highly relevant for fighting rapidly mutating viruses like HIV, SARS-Cov-2 or dengue viruses (9, 12–14). An interesting example is that of HIV elite controllers who are often HLA-B*5701+, an allelic product which, according to in silico models, is recognised by T cells with high cross-reactive potential (15). Heterologous immunity, whereby T cells cross-react with different viruses, is also frequently reported and has been reviewed elsewhere (16). In addition, many examples of T cells reacting to very different aa sequences are known (17, 18). Kersh and colleagues claimed that a peptide is recognised as long as it contains a motif for binding to the MHC and one key residue for the TCR (18). This concept was refined by the observation that no single residue was strictly required for recognition, if the available residues allow for a sufficient affinity between the MHC and TCR molecules (19). Thus, peptides not sharing a single residue may productively interact with the same TCR. A similar flexibility was also observed for the length of the MHC-class II peptide, with some CD4+ T cells requiring as little as four aa for recognition as long as these optimally fit to the MHC and TCR (20). In contrast, in a more recent study based on a unique experimental approach, Birnbaum et al. tested a set of murine and human CD4+ T cell clones and observed that the diversity of the peptide sequences recognised by single TCRs could be smaller than previously thought (21). Still, pluriallelic restriction, as well as alloreactivity have also been experimentally observed (22–24), increasing the number of cross-reactivity scenarios. The structural features that rule cross-recognition have been described in detail (3, 14, 17, 23, 25). They include several mechanisms of conformational adaptation of the TCR and pMHC units (e.g. changes in the TCR docking, displacement of the CDR loop), as summarised in Figure 1B. Hence, TCR-pMHC interactions are not rigidly conserved, but rather allow for considerable flexibility within the confines of some general orientation and binding rules. It is also important to note that in vivo, T cell cross-reactivity is very likely fine-tuned by the set of co-receptors (inhibitory or activating) and adhesion molecules that the T cell expresses at a given time (26).
A very convincing hint that our T cell immunity is shaped by cross-reactivity was provided in the elegant study of Su et al. (27): a search for HLA-DRB1*0401 restricted CD4+ T cells specific for HIV-, CMV- and HSV-derived epitopes in the blood of virus-unexposed healthy donors revealed that although the frequency of such cells was very low (< 10 cells per million), a large fraction (variable between individuals but in average > 50%) were found in the CD45RO+ subset. Looking at HIV-specific cells more precisely, the authors confirmed that these CD45RO+ cells represent a memory cell pool by assessing IFN-γ production, sequencing the TCR, and analysing further memory markers by gene expression. In addition, such cells were not found in umbilical cord blood. Finally, cross-reactivity of HIV-specific T cell clones with a range of bacterial- or algae-derived peptides suggested that such cells had been primed by unrelated antigens. Also relevant for vaccination, the authors further showed that Influenza-specific clones derived after Flu vaccination were able to recognise related peptidic sequences derived from other microbes. Similar observations were done by the group of F. Sallusto that demonstrated that HIV-specific CD4+ T cells could be detected in both the naive and memory T cell subsets (defined with the two markers CD45RA and CCR7) of HIV-unexposed healthy donors (28). The large majority (>80%) of the HIV-epitopes activating memory T cells matched strongly with human microbiome aa sequences. A further notable observation in this report was that both the specificity and the frequency of these HIV-specific T cells were different across donors. This highlights the inter-individual variability of T cell responses, likely to be shaped by both MHC-polymorphism and the environment.
Despite clear evidences about the cross-reactive nature of the TCR, it remains unclear how many single peptides can a unique TCR recognise in “real life”. According to early estimates, it should be approx. 106 (29). Meanwhile, there is evidence from several studies that individual T cell clones can indeed sense over a million different peptides in the context of a single MHC molecule (30–32). Studying the cross-reactive repertoire of an autoimmune HLA-A*0201 CD8+ T cell clone which recognises a 10 aa-long preproinsulin-derived peptide, Sewell and co-workers showed that many of the “cross-reactive” peptides were better agonists than the original one, despite some sequences differing in up to 7 out of the 10 aa positions (31). Based on the assumption that only 1% of all peptides will end up being presented on MHC, they also estimated the “true” frequency of cross-reactivity to be approximately 1 in 104 peptides (11, 31). This is in the same range as the frequency of 1 in 3x104 found by Ishizuka et al. when using a peptide library derived from pathogen sequences (33).
The most obvious and detrimental consequence of T cell cross-reactivity to vast numbers of individual peptides is the risk of developing autoimmunity (Figure 1C, left panel). Although self-reactive T cells are deleted in the thymus, weakly cross-reactive T cells may survive and become activated in the periphery through the recognition of epitopes from infectious agents (microorganism antigens, MoAs), a phenomenon known as “molecular mimicry”. Memory T cells can be stimulated by peptide concentrations more than 50-fold lower than those required to stimulate naïve T cells (34, 35). It is, therefore, likely that a memory T cell could be stimulated by a cross-reactive self-peptide with an affinity for the TCR that is far lower than that of the original pathogen-derived peptide. This goes in line with the quite frequent observation that infections can precipitate autoimmune diseases (36), and is of particular interest for novel therapies (37, 38). In autoimmunity, preferentially TCR cross-reactivity of CD4+ T cells has been analysed as a consequence of their central role in the development of autoimmune disorders. This is in contrast to cancer where analysis of cytotoxic anti-tumour response, i.e. CD8+ T cells is more important.
Here, we mainly present three examples for the involvement of TCR cross-reactivity in the induction of autoimmune diseases: one resulting from a bacterial infection, i.e. rheumatic fever; another induced by a food component, i.e. celiac disease; and a third example representative for the many autoimmune disorders for which no clear connection to an environmental agent has been found, as for instance multiple sclerosis.
Acute rheumatic fever is a typical example of systemic autoimmunity which occurs subsequently to an infection, namely with group A β-haemolytic streptococci (39, 40). It can affect synovial joints, cardiac valves and the brain, resulting in clinical features as arthritis, carditis, chorea, erythema marginatum and subcutaneous nodules. Molecular mimicry between group A streptococci and heart tissue was first described by Kaplan in 1960 (41). In the early 1980s, the role of both humoral and cellular autoimmune responses was reported in several studies (42). The cross-reactive antibody (Ab) response against S. pyogenes has been well described (43, 44). Meanwhile, it is clear that also T cell-mediated immune reactions play an important role in RF (40, 44, 45). Three types of protein antigens present on the S. pyogenes surface are M, T, and R proteins. M protein is the most virulent one and shares structural similarities with various host proteins, including cardiac myosin, laminin, vimentin, and tropomyosin (43, 46, 47). During this cellular response, streptococcal antigens are presented via MHC-class II molecules and activate autoreactive T cells (40). Indeed, T cells from patients with RF recognise different alpha (α)-helical coiled-coil proteins such as streptococcal M protein, myosin, laminin, and tropomyosin, and identical epitopes on the N-terminal portions of both streptococcal M protein and cardiac myosin were identified (45). In addition, in the valvular tissue and myocardium of patients with RF, T cells with three patterns of cross-reactivity were found: 1) cardiac myosin and valve-derived proteins, 2) cardiac myosin and streptococcal M peptides, and 3) cardiac myosin, streptococcal M peptides and valve-derived proteins (48). Potential sites of mimicry were revealed in the S2- and light meromyosin (LMM)-region of human cardiac myosin peptides and distinct peptides in the B repeat region of streptococcal M protein (peptides B2 and B3A) (45). Other mechanisms which are involved in the pathogenesis of RF are epitope spreading and TCR degeneracy. Ellis et al. investigated the degeneracy of the cross-reactive T cell responses towards different α-helical proteins such as human cardiac myosin, laminin, tropomyosin, and streptococcal M protein, and observed a mosaic of different T cell clones reacting with at least six distinct α-helical proteins demonstrating different degrees of cross-reactivity (45). Moreover, T cells are activated in RF when auto-Abs interact with the endothelium cells, leading to upregulation of vascular cell adhesion molecule 1 (VCAM-1) and facilitating increased T cell infiltration into the heart valve (49). These activated auto-reactive T cells produce inflammatory cytokines and lead to valve damage but also promote activation of B cells which produce cross-reactive Abs. Due to the destruction of valvular tissue, epitope spreading may occur, thus enhancing the humoral and cellular autoimmune reaction.
Another crucial streptococcal antigen is N-acetyl ß D-glucosamine (GlcNac), a carbohydrate moiety of the bacteria cell wall (43). In a neurologic manifestation of RF, the Sydenham chorea, T cells as well as Abs that recognise this bacterial antigen have been shown to cross-react with the brain cell antigens lysogangliosides and tubulin (39, 50, 51). The humoral responses correlate with clinical symptoms and mediate neuronal cell signalling (52).
Celiac disease is highly interesting in view of the fact that autoimmune reactions are induced by a food component, i.e. dietary gluten (gliadin in wheat, hordein in barley, and secalin in rye are the most prominent examples). Antibodies against gliadin-peptides and the enzyme transglutaminase-2 (TG2) are highly-specific diagnostic markers of CeD, and a CD4+ T cell response towards post-translationally modified gluten peptides has been described. The disease shows a clear genetic association to the MHC-class II allelic products HLA-DQ2 (DQ2.5: DQA1*05:01-DQB1*02:01 or DQ2.2: DQA1*02:01-DQB1*02:02, approx. 95% of the patients) and HLA-DQ8 (DQA1*03:01-DQB1*03:02, approx. 5% of the patients) (53).
Interestingly, gliadin is a substrate for the TG2 enzyme which catalyses deamination at glutamine residues. The conversion of Q to E aa leads to increased binding affinity of peptides to the HLA-DQ2.5/2.2/8 molecules and enhanced recognition by gluten-specific CD4+ T cells (54–56). Hence, CeD-associated T cells preferably react with “self-produced mimotopes” that result from the deamidation of gliadin-derived peptides. Another level of cross-reactivity that has been documented in CeD is the recognition by a single DQ2.5-restricted TCR of peptides of similar, but not identical, aa sequences derived from various gliadins (i.e. α1a and ω1) (57). To which extend this cross-reactivity participates in the immune response against various gliadins and/or hordein or secalin is still not fully investigated, but is starting to be explored at large-scale (57–59). Altogether, the strong anti-gluten CD4+ T cell response present in CeD is providing help to B cells that bind TG2-gliadin complexes and deaminated gluten peptides to mature into plasma cells in the gut that in turn produce deaminated gluten-specific, as well as autoreactive, TG2-specific, Abs (60–62). In addition, gluten-specific CD4+ T cells are consistently found in the small intestine of celiac disease patients, where they activate intraepithelial CD8+ T cells (IELs) via the production of IFN-γ, IL-21 and IL-2 (62). Although these IELs are thought to largely contribute to disease pathogenesis, the link between the gliadin-specific CD4+ T cell response and the recruitment and activation of IELs in the gut remains obscure, especially because these IELs have not been shown to recognise gluten.
Even if there is ample evidence that HLA-DQ2.5, HLA-DQ2.2, or HLA-DQ8 molecules present gluten-derived peptides, expression of these allelic products alone is insufficient to cause disease. Other risk factors which may induce increased expression and activity of TG2 may also be involved. For instance, in vivo and in vitro studies support an association between gut microbiota alterations and celiac disease (63). First, the microbiota composition differs between individuals with active celiac disease, patients on a gluten-free diet, and normal controls in both oral, duodenal and faecal samples, with an increase in virulent strains noted in patients with active CeD (64). Bacteria can modify immunogenic food antigens resulting in an increase or decrease in antigenicity, and also utilise undigested particles as substrates, producing metabolites such as short-chain fatty acids that affect intestinal homeostasis. For instance, Pseudomonas aeruginosa, an opportunistic pathogen isolated from CeD patients, processes gluten to T cell reactive epitopes whereas bacterial species from healthy controls inactivate these reactive epitopes by further proteolytic breakdown (65). Second, and more relevant in the context of T cell cross-reactivity, peptides from common commensal and pathogenic bacteria, especially from several Pseudomonas and Bordetella species can mimic gliadin-derived peptides and activate gliadin-specific, HLA-DQ2.5-restricted T cells from CeD patients (66). It has been, therefore, hypothesised that celiac disease may be induced not only by gluten ingestion but also by infectious processes inducing pathogen-specific T cells that cross-react with gluten epitopes (66).
Multiple sclerosis is one of the most prevalent autoimmune disorders of the central nervous system (CNS), and is characterised by the loss of the protective myelin sheath that surrounds the axons of neurons (67, 68). Its pathophysiology has been extensively studied, especially in experimental allergic encephalomyelitis (EAE) which is a generally accepted animal model for the human disease. Nevertheless, the aetiology of MS is still unclear. Its association with an infection has been postulated already in the late 1800s, after it was first described (67). Nowadays, several factors such as genetic susceptibility, environment including infectious agents, obesity, lack of sun exposure and vitamin, have been suggested to be involved (69).
Autoantibodies specific for a variety of CNS proteins, as for instance myelin basic protein (MBP) or myelin oligodendrocyte glycoprotein (MOG), are present in the serum, cerebrospinal fluid (CSF), and brain of MS patients (70). Similarly, CD4+ T cells specific for myelin antigens are found in the blood. Studies on antigen recognition demonstrated that CD4+ autoreactive, MBP-specific, T cells from MS patients cross-react with peptides derived from bacterial or viral proteins (71, 72). As shown by structural analyses performed by Lang et al., the same TCR binds a MBP peptide presented by HLA-DRB1*1501 and an unrelated Epstein Barr virus (EBV)-derived peptide bound to HLA-DRB5*0101, a typical example of molecular mimicry (73, 74). A link between EBV infection and MS had already been suggested by the observation that the infection may precede MS pathology and the identification of cross-reactive Abs in MS patients (67). EBV is a well-investigated candidate for antigenic mimicry, from mimotope peptides recognised by T cells to cross-reactive Abs (75). Also, an altered anti-EBV T cell reaction was suggested in MS (76, 77).
These findings led to the concept that an immune response initially activated and expanded by an infectious agent may, in general, cross-react with autoantigens mediating CNS inflammation and induce destruction of the brain. To date, numerous infectious agents have been described to induce cross-reactive T cells against brain-specific epitopes. As an example, peptides from HSV and Pseudomonas aeruginosa bound to MHC molecules are recognised by cross reactive myelin-specific T cells (78). Furthermore, peptides from M. tuberculosis, S. typhimurium and E. coli lead to strong in vitro proliferation of MBP-specific T cells and induced EAE in mice with the same severity and incidence as the autoantigen peptide of MBP (79).
T cell clones isolated from the blood of patients with MS show high specificity for the immunodominant MBP epitope MBP85–99 (80). However, this specificity is not absolute. Indeed, changing the TCR contact residue lysine at position 93 to an arginine, or even just removing a hydroxyl group by changing a phenylalanine to a tyrosine at position 91, can totally abrogate T cell reactivity. This lysine-to-arginine substitution can also result in a more degenerate pattern of TCR recognition, in that a tyrosine or other aa residues can now be tolerated at positions 91 or even 90 (81). Hence, while a TCR appears to be highly specific in one situation, altering the peptide ligand can change the TCR conformation to yield a higher degree of T cell cross-reactivity. Analysis of a further series of MBP85–99 reactive T cell clones led to a similar conclusion, showing that a number of virus-derived epitopes can trigger autoreactive T cell clones in a manner that would not be predicted by simple algorithms (71). One of the studied MBP-reactive T cell clones recognised an epitope of MOG, an entirely different self-protein. Thus, a significant degree of functional degeneracy exists in the recognition of self-antigens by T cells.
The link between infection and autoimmunity via molecular mimicry has also been investigated in other inflammatory CNS diseases, particularly in chronic Lyme disease. Following acute infection with Borrelia burgdorferi (Bb), a chronic inflammatory disease can emerge which targets joints or the CNS in the absence of residual bacterial infection. In this condition, an autoimmune response to self-antigens (similarly as described above for RF) may arise from bacterial-specific T cells (82). Indeed, in Lyme arthritis, CD4+ T cells isolated from the synovial fluid of patients were shown to recognise a 9mer peptide from an outer surface antigen from Bb (OspA165–173) and an analogous, but not identical, sequence from the human LFA-1 molecule (CD11a332–340) (83). Similarly, Bb-specific T cells from the CSF of a patient with CNS manifestation of borreliosis cross-reacted with several self-antigens, one of them being a myelin antigen (84).
In uveitis, it has also been shown that peptides with similar structure rather than similar aa sequences can induce cross-reactive T cell responses. For instance, similarities of 6 to 7 aa with the 14mer autoantigen peptide from retinal S-antigen (PDSAg) with peptides of 11 or 12 aa in length from different environmental proteins is sufficient to induce autoreactive CD4+ T cell recognition and experimental anterior uveitis in rats (85). Although the pathogenic cells in uveitis are MHC-class II restricted CD4+ T lymphocytes, statistical associations with HLA-class I molecules (B*27, B*51) are well known. Interestingly, the HLA-class I molecule seems to serve as an autoantigen itself, being presented as a peptide (B27125–138, termed B27PD) on HLA-class II and mimicking the retinal PDSAg peptide (86). Oral administration of B27BP peptide to patients was also shown to improve uveitis symptoms, suggesting that cross-reactivity could be even exploited for inducing oral tolerance to autoimmune antigens (87). In a recent study including patients with acute anterior uveitis and ankylosing spondylitis, an HLA-B27-linked rheumatic disease frequently associated with uveitis, TCRs responding to HLA-B*27-bound peptides derived from microbial antigens or from self-antigens were identified. These peptides shared common TCR binding motifs, supporting the idea that HLA-B*27-presented microbial peptides could act as trigger for autoimmunity by activating anti-self CD8+ T cells (88). Interestingly, the ankylosing spondylitis-associated TCRs showed weaker affinity for the human peptide ligands than for a peptide from a conserved bacterial inner membrane protein. Evaluating the structures of seven of the HLA-B*27:05 peptide-TCR complexes, the authors showed that in all of these structures, the TCRs used a similar solution to interact with the conserved motifs in the self and bacterial peptides (89).
In type I diabetes, a T cell-mediated, HLA-DQ2 (DQA1*05:01-DQB1*02:01) and -DQ8 (DQA1*03:01-DQB1*03:02)-associated autoimmune disease directed at pancreatic β cells, insulin B-chain9-23 (B:9-23) is a key epitope presented by MHC-class II to CD4+ T cells targeting pancreatic β-cells. Lack of an acidic aa residue (i.e. aspartic acid and glutamic acid) at position 57 of the DQ8 β chain of the MHC molecule favours binding of the insulin-B peptide and is associated with increased risk of developing the disease (90). Without this acidic residue, the presented peptide repertoire is typically negatively charged (91, 92). Mimotopes with acidic aa substitutions at P9 have been shown to detect self-reactive, IFNγ-producing T cells much stronger than the wild-type peptide (93, 94). Interestingly, an immune response to this mimotope was also observed in control subjects without diabetes, but in these individuals, rather IL-10 producing, hence, anti-inflammatory CD4+ T cells were activated (93).
Distinct cytokine patterns of T cell subsets make them unique and define their role in host defence or their contribution in disease pathogenesis. In autoimmune diseases, the role of Th1 and Th2 cells along with their cytokine profiles is well documented. In particular, the priming signal (specificity, affinity and avidity of the pMHC/TCR, APC/T cell interaction) controls the maturation, differentiation and function (i.e. cytokine profile) of the T cell (37). Alteration of peptides and of their binding to MHC may, therefore, influence the strength of the immune response. For the development of therapeutic agents in autoimmune diseases, silencing the armful anti-self T cell activity by either shifting the inflammatory Th1 response towards a Th2 profile, inducing regulatory T cells (Tregs), or even completely inhibiting T cells using strong antagonists are all strategies of interest (Figure 1D, left panel). Many of such “mimotopes” have been meanwhile designed based on in vitro testing of the responsiveness of T cells isolated from patients or in vivo using animal models (38). Especially those reducing pathogenic responses have been tested for therapeutic purposes in clinical trials.
The ability of altered peptide ligands (modified peptide sequences derived from an original antigenic peptide, i.e APLs) to shift an unfavourable Th1- in a more favourable Th2-response in the murine EAE model of MS has first been shown by Nicholson and colleagues (95). The authors used an analogue of the encephalitogenic myelin proteolipid PLP139-151 (the common T cell antigen in EAE) with substitutions at the two main TCR contact residues (L144/R147) which had been shown to be a powerful TCR antagonist for the encephalitogenic PLP-specific T cell clones in vitro (96). Injection of this analogue protected the animals from developing EAE. Kuchroo et al. showed that this APL can activate IL-4 secretion by both encephalitogenic T cells and naive T cell clones that cross-react with self-antigens and inhibit autoimmunity by the induction of Tregs leading to bystander suppression of EAE (96). In further animal models of EAE, APLs have been proven to have a significant therapeutic value (97). Meanwhile, autoreactive human T cell clones have been shown to secrete the anti-inflammatory cytokines IL-4 and TGF-β after TCR engagement by APLs (98). However, application of APLs in MS may be a double-edged sword. On the one hand, it was shown that an altered MBP85–99 peptide induces Th2 cytokine secretion by MBP-reactive T cells isolated from the peripheral blood of MS patients while on the other hand, it can induce disease in some patients by activating these MBP-reactive T cells against the patient’s own tissues (99). Moreover, in a phase II clinical trial with this peptide, two out of seven MS patients developed high frequencies of MBP-reactive T cells, and these responses were associated with significant increases in MRI-detectable lesions (100). In contrast, patients treated with lower doses of the same APL experienced some degree of immune deviation towards increases in IL-4 secretion by MBP-reactive T cells (101, 102).
A mimotope was also developed for patients with diabetes mellitus type 1 in order to preserve pancreatic β cell function. It was modified from the human insulin peptide B:9-23 which binds to HLA-DQ8 and is recognised by CD4+ T cells present in the islets of organ donors with type 1 diabetes ((103) and section 2.4). The substitutions in this modified peptide are known to be important in the diabetes-prone NOD mouse model (104, 105). However, a four-arm phase II clinical study conducted by Walter et al. could not show any clinical improvement (as measured by C-peptide concentrations, a measure of pancreatic β cell function), after subcutaneous administration of the mimotopes over two years compared to the placebo (103).
For celiac disease, and as mentioned in section 2.2, disease-associated T cells preferably react with “naturally produced mimotopes” that result from deamidation of gliadin-derived peptides. Epitope-specific immunotherapies are, therefore, a logical translational step. In HLA-DQ2.5-positive celiac disease patients, clinical trials using a combination of three gluten-derived peptides, which contain at least five gliadin-specific T cell epitopes presented by HLA-DQ2.5 (Nexvax2) were conducted. While the phase I studies showed preferable outcomes in terms of safety and tolerability, the recent Nexvax2 phase II trial had to be discontinued due to lack of protection to gluten challenge.
An alternative approach to the use of a single APL is the administration of peptide mixtures that contain many different antigen specificities. Random copolymers that contain aa commonly used as MHC anchors and TCR contact residues have been proposed as possible “universal APLs.” The synthetic immuno-active copolymer glatiramer acetate (GA) is comprised of four aa in random order with an average length of 40-100 residues which resemble MBP (106). It was first synthesised in 1967 to induce EAE in murine models, but was then unexpectedly found to reduce signs and progression of the disease (107). Rather than inducing an autoimmune disease, GA was found to induce regulatory and protective neuroimmune responses. In most patients, daily injection with GA causes a striking loss of responsiveness to this polymer antigen, accompanied by greater secretion of IL-5 and IL-13 by CD4+ T cells, indicating a shift towards a Th2 response (108, 109). In addition, the GA-reactive T cells exhibit a high degree of degeneracy, as measured by their ability to cross-react with a large variety of peptides represented in a combinatorial library (108). GA-induced migration of those highly cross-reactive Th2 (and perhaps regulatory FoxP3- Th3) cells to the sites of inflammation may allow their highly degenerate TCRs to contact self-antigens, which they recognise as weak agonists. These T cells then apparently secrete suppressive, Th2/Th3 cytokines, thus restricting local inflammation (108). Due to these beneficial effects, GA was approved for therapeutic use in 1996 and is since then a first-line treatment of relapsing remitting MS (110, 111).
With the notable exception of rare antigenic aberrant sequences, e.g. mutated antigens, tumours generally present self-antigens on their MHC molecules and are poorly immunogenic (112). This can be globally seen as the result of the thymic negative selection where highly self-reactive T cells are eliminated to prevent the development of autoimmune diseases, leaving us with a TCR repertoire with only low to moderate affinity to self-antigens (113). Although this is beneficial in a healthy state, it makes tumour targeting by T cells a hard task, as it impairs the mounting of an effective and strong immune response. Hence, in contrast to the situation in autoimmune diseases, cross-reactivity of potential pathogen-specific T cells against self-antigens specifically presented by tumour cells is not only desirable, but would likely result in favourable anti-tumour immunity (114).
An early and staggering example of TCR cross-reactivity was described by the group of P. Romero for the tumour-associated antigen (TAA) Melan-A. While the frequency of any antigen-reactive T cell in the peripheral immune naïve repertoire is generally extremely low (< 1 in 100.000 T cells), up to 1 out of 1000 CD8+ T cells bind the immunodominant peptide from Melan-A26-35 (the modified A27L ligand), when presented by HLA-A*0201, both in healthy donors as in melanoma patients (115). Although numerous T cells were able to bind the pMHC, as assessed by MHC-tetramer staining, a subgroup failed to be significantly activated by the Melan-A peptide in a cytotoxicity assay. In contrast, several other tested peptides, which included proteins of self- or pathogen- origin, generated a strong response in the same assay, hinting at the highly cross-reactive nature of this repertoire of T cells (116). Further supporting this, a following study of the same group showed that a tumour-reactive CD8+ T cell clone, also specific to the same immunodominant peptide mentioned above, was able to cross-recognise numerous peptides and that stimulation of this clone with these peptides drove the expansion of a heterogeneous CD8+ T cell population, with only a fraction actually reacting to the Melan-A peptide (117). Importantly, immunisation with Melan-A peptide through vaccination leads to a reduction on the population of cross-reactive T cells and an enrichment of antigen-restricted T cells that can react with the tumour (118). These early works on TCR cross-reactivity demonstrated its relevance not only in tumour biology but also in the design of effective anti-cancer immunotherapies.
Studies have described viral-specific T cells within the microenvironment of several tumour entities with no prior known viral aetiology (119). Although there is experimental evidence for the presence of intracellular bacteria or viruses in tumour cells (120–122), this local pathogen load might not be the only reason for the presence of pathogen-specific T cells within tumours. After sequencing the TCRs of tumour-infiltrating lymphocytes (TILs) in non-small cell lung carcinoma (NSCLC), Chiou et al. identified a novel TAA derived from the epithelial protein TMEM161A. A TCR recognising this peptide was shown to readily cross-react with epitopes from EBV (and E. coli). Specific T cells were not only found in NSCLC patients, but also in healthy donors, an observation which the authors offer as an explanation to the presence of virus-specific T cells within NSCLCs, but possibly also in other tumours (123). In another in silico-based approach, Ragone et al. examined the cancer peptide database and identified numerous TAAs with shared homology with viral sequences. The viruses whose sequences were most commonly shared with the tumour antigens were HIV type 1 (HIV-1), HSV, and human papillomaviruses (HPV). In addition to sequence homology, the authors also report that these peptides share structural similarities with comparable patterns of contact between the HLA molecule and the TCR (114). A recent case report has also described tumour reduction in three metastatic colorectal cancer patients upon SARS-CoV-2 infection (124). Altogether, these studies point out to the fact that pathogen- and tumour antigen- cross-reactive T cell responses might play an important role in anti-cancer immunity, and that the immune repertoire of each patient, shaped by previous infections, might be a crucial factor in disease control.
In murine melanoma models, Chiaro et al. showed that similarities between tumour- and viral- derived antigens can influence the clearance of tumours upon peptide cancer vaccination as a consequence of cross-reactive T cell activity. Upon immunisation with viral peptide pools previously selected based on their homology to tyrosinase related protein (TRP2180–188) or glycoprotein 100 (gp10025–33), a strong reduction in tumour growth was seen. Interestingly, the authors further argue that viral molecular mimicry is an important factor that dictates immune response also in metastatic human melanoma by showing a direct correlation between pre-existing Abs against CMV, and response to the immune checkpoint inhibitor (ICI) anti-PD-1 (125). TCRβ sequencing experiments further suggested that the same T cell clone recognised similar peptide sequences of MAGE-A10 and CMV. Further studies using pre-clinical murine models suggest the relevance of activating virus-specific T cells for tumour growth control (119, 126). The authors describe the formation of an immune-permissive microenvironment upon in vivo virus-peptide vaccination, whereby cross-reactivity of these viral-specific T cells with tumour antigens, although not tested, could be responsible for the effect observed. Interestingly, another study simulating immunisation of mice with the TAA and homologous viral peptides predicted a similar clearance of tumour cells in both scenarios, suggesting equivalent anti-tumour efficacy of the effector T cell response (114).
Cross-reactivity of tumour-specific T cells with bacterial epitopes has also been described. In melanoma, a MAGE-A6-derived peptide (MAGE-A6172-187) was shown to be cross-reactive with its highly immunogenic homolog HF-2216-229. This mycoplasma-derived peptide and MAGE-A6 can drive the formation of memory CD8+ T cells. Interestingly, in vitro priming with dendritic cells loaded with the bacterial-derived peptide resulted in CD8+ T cells with 100-fold higher avidity to the MAGE-A6 peptide compared to that of cells primed with the MAGE-A6 peptide itself (127).
The main in vivo source of bacteria-derived antigens is the microbiota. The human gut is colonised by approximately 1014 microbes (128). The sheer number of colonising microorganisms means that exposure of immune cells to these bacteria throughout life is unavoidable, which results in the generation of an immune response against commensal-derived peptides. In a similar analysis to the one performed earlier, Ragone et al. compared all TAAs from the cancer peptide database against the microbiota species Firmicutes (taxid:1239) and Bacteroidetes (taxid:976) sequences. The authors demonstrated a high level of homology of tumour antigens and peptides derived from these species, which account for 90% of all gut microbiota (129). Flückiger et al. showed that T cell clones that recognise the cancer antigen protein glycerol-3-phosphate dehydrogenase 1-like (GPD1-L) and cross-react with epitopes derived from the tail tape measure protein (TMP) of an Enterococcus hirae (E. hirae) bacteriophage, could be detected in melanoma patients. Importantly, the authors further observed an association between the presence of this prophage in the stools of patients with renal and lung cancer, expression of GPD1-L by tumour cells, and a long-term benefit to PD-1 checkpoint blockade (130). Interestingly, in the same study, cyclophosphamide treatment of tumour-bearing mice, which induces the translocation of E. hirae from the gut lumen to the mesenteric and splenic immune tissues, resulted in improved anti-cancer CD8+ T cell responses. This anti-tumour effect was abrogated once the mice were given antibiotics and rescued by administration of E. hirae isolates. Moreover, lack of expression of the TAA by the tumour cells also abolished any anti-tumour immunity previously observed.
Other studies have also shown a favourable clinical outcome in cancer patients presenting CD4+ and CD8+ T cells specific for E. hirae, Bacteroides fragilis, Ruminococcaceae (131), and Akkermansia muciniphila (131–134). The immune repertoire, namely the frequency of precursor T cells prior to antigen exposure, is a critical factor in determining the magnitude of an immune response. Based on the aforementioned observations of cross-reactivity between numerous pathogen-derived epitopes and tumour antigens, it is plausible that the gut microbiome is an important modulator and dictator of how individuals will mount an immune response to tumours but also how they will respond to immunotherapies.
In contrast to the demonstrated potential of T cells to be cross-reactive (11), neoantigens generally activate specific T cells that react only very weakly against the wild-type (wt) peptide which often differs only in 1 aa (135–138). This apparent contradiction may be explained when considering the position of the mutated aa in the peptide sequence (e.g. if a novel anchor residue for binding to the MHC molecule is created by the new aa) or its structural properties (e.g. changes in peptide charge which renders the peptide “visible” to the TCR). Still, the large majority of predicted neoantigens probably activate similar TCRs to that specific for the self-peptide and are, therefore, not of interest. If this is the case, these neoantigens do not trigger a strong anti-tumour response as a result of central tolerance. On the other hand, neoantigens can share homology to pathogen-derived antigens. In this case, these neoantigens could elicit an efficient response against tumours by activating cross-reactive pre-existing memory T cells that have been previously generated against such pathogens, as discussed above for wt tumour antigens (139). Bessel et al. identified an epitope (SVYRYYGL (SVY)) derived from the genome of the commensal Bifidobacterium breve (B. breve), homologous to the neoepitope expressed by the murine model B16-SIY (SIYRYYGL (SIY)) (140). They further demonstrate that B. breve promotes the expansion of SVY-specific CD8+ T cells and that these are able of effective tumour control in SIY-expressing tumours, although comparison with SIY-specific T cells was not performed. In pancreatic cancer patients, Balachandran et al. demonstrate that the quality of the tumour neoantigens, namely the similarity to pathogen-derived epitopes, rather than the quantity, greatly associates with long-term survival (141).
Importantly, cross-reactive neoantigens seem to be a critical predictive factor for checkpoint inhibitor therapy efficacy. In the seminal study by Snyder et al. which first identified mutated antigens as T cell targets during checkpoint blockade, the authors observed that patients with long-term benefit to anti-CTLA-4 therapy share neoepitopes homologous to more viral and bacterial antigens, in contrast to patients with minimal or no benefit (142). These intriguing findings strongly suggest that cross-reactive T cells specific for pathogens can get activated upon checkpoint inhibition and participate in a clinically significant anti-tumour response. This is in line with the different studies presented above where the importance of the gut microbiome in checkpoint therapy responsiveness has been highlighted (143).
In addition to being able to dictate the outcome of immunotherapies such as checkpoint inhibition and therapeutic cancer vaccination with tumour-derived antigens, TCR cross-reactivity is currently being exploited for the development of novel and more potent cancer therapies, which we will discuss in more detail below.
If numerous T cell clones recognise the same epitope, affinity and avidity for this epitope will be inevitable highly variable. Using checkpoint inhibitors will unleash the inhibition in all lymphocytes present in the tumour microenvironment (TME), high or low functional ones. Differently, the goal of therapeutic vaccination is to selectively drive the recruitment of high-avidity T cells and promote strong and long-lasting anti-tumour responses. As mentioned above, high-affinity T cells against TAAs are usually lacking as a consequence of negative selection in the thymus, which leaves us only with a low-affinity repertoire. This tolerance is observed when A2xneu mice (Her2/neu mice crossed with A2.1/Kb mice) are injected with the immunodominant Her2773-782 peptide, which results in little to no tumour control (144). A similar tolerance was observed when mice were injected with p53-derived peptides. In this case, the authors demonstrated that using the p53261-269 self-epitope led to the expansion of cytotoxic T lymphocytes (CTLs) in p53 wt mice with an avidity more than 10-fold lower than the ones obtained from p53 null mice (145). This nicely shows the importance of circumventing tolerance to achieve an effective cancer vaccination.
One way to improve the immunogenicity of TAAs would be to exploit the cross-reactive nature of TCRs. Identification of peptides that are not naturally processed and presented but that can be used to elicit strong cross-reactive T cell responses against the original TAAs is already an old idea. The design of such heteroclitic peptides, where the stability of interaction between the peptide and MHC molecule is improved by replacement of certain aa was shown to be a powerful strategy for both improving CTL reactivity in vitro and controlling tumor growth in mice (144, 146–150). Importantly, these heteroclitic peptides need to be recognised by T cells that cross-react with the native sequence and can, therefore, drive the killing of tumour cells naturally presenting the original peptide. Despite the encouraging results seen in pre-clinical models, this concept has failed yet to lead to the development of an effective cancer therapeutic vaccine (151, 152). A famous example was the observation by Speiser et al. that immunisation of melanoma patients with the wt Melan-A26-35 (together with CpG as adjuvant) was superior in generating high avidity, tumour-reactive T cells, compared to the Melan-A26-35 modified peptide (152). Since the only difference between the two peptides is one aa substitution at an anchoring position (A27L), it suggests that increasing pMHC binding properties is not the ultimate key for improving T cell reactivity to TAAs.
Recently, a novel concept exploiting TCR cross-reactivity for therapeutic purposes has emerged. It is based on the identification of natural analogue peptides capable of inducing strong T cell responses against the tumour antigen. The shared homology between pathogen-derived peptides and tumour antigens and the aforementioned correlations between cross-reactive T cells and clinical outcome makes this an attractive and promising strategy that is currently being further investigated.
We have introduced in sections 3.1 and 3.2 that tumour antigens share homology with numerous pathogen-derived epitopes which, as a consequence, can drive the activation of T cells that share the same TCR. In other words, T cells that have been activated upon exposure to a certain pathogen can cross-react with tumour antigens (Figures 1A–C). The reasons for exploiting this cross-reactivity in the context of therapeutic cancer vaccination are manifold: first, it allows to overcome the low immunogenicity and affinity of natural TAAs, since TCRs that recognise MoAs have not been depleted from the T cell repertoire. Second, memory T cells can be activated by much lower peptide concentrations as compared to their naïve counterparts (see section 2). Third, recalling T cell responses upon immunisation is obviously easier to achieve than priming new effectors, especially when considering the current lack of gold-standard strong adjuvants. Fourth, exploiting “natural” T cells that were already expanded in the body after infection should present less risk of autoimmunity, although, as exemplified in section 2, autoimmunity cannot be fully excluded.
In summary, activation of viral- or commensal- specific T cells that cross-react to the tumour cells have shown promising results in a couple of pre-clinical models. Furthermore, correlations between the presence of these T cells and clinical outcome in patients have also been drawn. All this is opening a new field of research, to identify tumour antigens and MoAs that share high homology for the developing of novel T cell-based immunotherapies for cancer (Figure 1D). Since the presence of MoAs-specific memory T cells depends on prior infections, the composition of the microbiota, and the MHC-allotype, one could speculate that the development of such strategies should be done in an individualised manner to guarantee a high success rate and decrease the risk of side effects. Combination of such therapeutic vaccinations with ICIs could unleash the expansion of potent effector memory cells that readily target the tumour antigen and are able to control tumour growth.
TCR cross-reactivity undoubtedly opens large avenues for developing more potent cancer therapies. However, there are important bottlenecks to consider. The possible side effects in immunotherapy, especially in adoptive T cell therapies, where optimised TCRs with high affinity against a certain peptide are administered to patients is a serious issue. Side effects with these engineered TCRs are not rare, due to the strong interaction between the TCR and its target. Very low expression levels of the antigen in healthy tissue, which was initially dismissed as potentially dangerous led to severe consequences (153, 154). This on-target toxicity is not that unexpected (Figure 1D). However, overlooking off-target effects due to TCR cross-reactivity can have similarly severe and fatal adverse effects as it was observed in the case of anti-MAGE-A3 TCR engineered T cells. Due to its restrictive expression to immune privileged sites such as placenta and testis which lack the expression of HLA molecules, MAGE-A3 was considered a genuinely tumour-specific target, since it is found to be overexpressed in multiple tumours. This bona fide target attracted the attention and promptly immunotherapies that target this molecule were developed. Contrary to the expectations, severe cases of toxicity were observed, despite the lack of antigen expression in any of the tissues affected. In the first of two well-known incidents, engineered anti-MAGE-A3112–120 (KVAELVHFL) T cells were adoptively transferred to cancer patients after nonmyeloablative lymphodepletion, who then received high doses of IL-2. This led to severe neurological damages, and even to a patient death. This fatal toxicity was attributed to a cross-reactivity of the effector TCRs with a MAGE-A12 sequence (KMAELVHFL) which has a superior binding affinity for HLA-A*0201 than MAGE-A3112–120. MAGE-A12 was found a posteriori to be expressed in the brain (153). In the second, even less predictable case, engineered lymphocytes with affinity-enhanced TCRs against the HLA-A*01-restricted MAGE-A3168-176 peptide (EVDPIGHLY) drove cardiotoxicity and patient death due to recognition of an unrelated peptide derived from the muscle protein titin (ESDPIVAQY) which is presented by cardiomyocytes (155, 156) (Figure 1D). Experimental and computational tools for prediction of potential toxicities have been improved since then and will be presented in section 4.
In general, therapeutic cancer vaccines are safe and no severe side effects have been observed to date. This may arise from the relatively low affinity of the induced T cells. The potential of MoAs to be used in immunisation approaches against tumour antigens, renders caution to what kind of side effects can arise. In a recent study, Gil-Cruz et al. showed that microbiota-derived peptide mimicry can induce lethal cardiomyopathy through the activation of heart-specific (MYH6-specific TCR) Th17 CD4+ T cells (157). In their mouse model, cross-reactive CD4+ T cells are primed in the intestine and later circulate and infiltrate the myocardium where they can damage myosin-expressing cells. In the context of checkpoint inhibition, it is tempting to speculate that not only self-, but also cross-reactive pathogen-specific T cells could be responsible for driving lethal cases of myocarditis that were observed in some patients (158, 159). The large number of auto-immune diseases that are associated with pathogen infection itself (section 2) demonstrate the delicate balance in the selection of these MoAs for therapeutic intervention.
A number of the examples of TCR cross-reactivity discussed so far have been brought to light using in vitro systems based on the testing of T cell activity against synthetic peptides. In early works, epitopic peptides of interest were modified by introducing aa substitutions at various positions. Later advances, supported by increased automatization of peptide synthesis, led to the development of synthetic peptide libraries. One common approach is to generate combinatorial (sub)libraries of peptides with each of the 20 aa fixed at one position while all other positions can be occupied by all other aa (160). Such approach can theoretically generate all possible aa sequences for a given peptide length and allows screening of up to 1012 peptides. In vitro testing of agonists or antagonists´ effects on T cell activity can be performed either by measuring cytokine secretion, killing of loaded target cells (for CD8+ T cells), or proliferation (for CD4+ and CD8+ T cells) (32, 161). Once a library has been shown to activate the T cell of interest, sub-libraries can be consecutively tested until the sequence(s) responsible for cross-reactivity is (are) identified. Subsequent database search can finally reveal whether the random peptide is indeed part of a known protein.
Together with the development of TCR engineering and adoptive transfer therapies, currently most advanced in the oncology clinical setting, high through-put and comprehensive approaches for testing TCR cross-reactivity have become mandatory for pre-clinical development. The main interest here is to assess TCR-mediated toxicity, i.e. the potential of transferred T cells to exert deleterious effects in vivo via recognition of non-related pMHC expressed on healthy tissues (Figure 1D). This is particularly relevant when the TCR has been manipulated for increasing its affinity to the cognate pMHC or has been obtained from HLA-unmatched donors (allorestricted). Challenges for the safe use of engineered TCRs in solid tumours have been very recently reviewed (162), and we have presented examples of fatal toxicities in section 3.3. In the context of clinical development, in vitro testing of T cell reactivity against random peptide sequences, as mentioned above, is the most straight-forward approach to assess cross-reactivity. DNA-tagged pMHC multimers, which allow to address TCR-pMHC affinity more easily is an elegant alternative method (163, 164). From the point of view of experimental feasibility, all these assays require high amount of material (e.g. T cell clones), which might be circumvented by modern methods. TCR cloning and subsequent transfer in reporter cells or MHC-matched PBMCs, and possibly the use of soluble TCRs and yeast pMHC libraries can overcome the aforementioned limitations (21). By titrating the peptide concentrations, TCR affinities can be more precisely assessed.
Which threshold of reactivity will lead to in vivo toxicity is likely impossible to predict with high accuracy and might even vary between individuals. One weakness of synthetic peptide testing is that recognition of a particular sequence by a certain TCR as measured in vitro cannot ultimately predict in vivo reactivity, since it is unknown whether this aa sequence is indeed processed and to which extent it is presented on body tissues. More sophisticated platforms try to overcome these limitations. First, testing primary normal cells from a range of organs representing essential human tissues (e.g. cardiovascular, gastrointestinal, brain, liver, among other systems) and/or a panel of tumour cell lines will assess potential off-target recognition (22, 165). Second, alloreactivity against MHC-mismatched cell lines can also be assessed (22, 166). As an example, reactivity of a TCR specific for a MAGEA4-derived epitope presented by HLA-A*0201 was found to recognise HLA-A*0205 (in the absence of MAGEA4), indicating alloreactivity; hence, patients bearing the HLA-A*0205 allelic product should be excluded from the clinical study using this TCR (22). Third, recognition of similar, but not identical synthetic peptides (containing aa substitutions), can also be tested in vitro, and the occurrence of potentially recognised sequences in the human proteome predicted. This combined approach could advantageously replace combinatorial peptide libraries (167).
Lastly, a comprehensive view of all peptides presented by MHC molecules in normal cells is needed and of utmost importance. The typical experimental setting for assessing the MHC ligand “landscape”, is to perform peptide immunoprecipitation followed by mass spectrometry analysis. First milestones steps have been engaged, with the Human Immunopeptidome Project (HIPP) and the HLA ligand atlas which both aim at deciphering the entire MHC-ligandome landscape of human healthy tissues (168, 169). In addition, quantitative analysis of peptide presentation by mass spectrometry has become possible. Using this method, it was recently shown that a peptide derived from collagen type VI A3 is present in 41% of the tumour samples analysed at an average of 228 (max 1928) copies per cell, but only in 6% of the normal tissues with an average of 28 copies (max of 49) per cell (165).
All these approaches are so far imperfect, since it cannot be excluded that an organ subpart, or specialised cells at a certain stage of differentiation or activation, may be targeted by cross-reactive T cells. As discussed earlier, the recognition by MAGE-A3 specific T cells of a titin-derived peptide expressed only in beating cardiomyocytes showed to be fatal for treated patients (155, 156). However, combining and refining them will decrease the chance of unexpected in vivo TCR cross-reactivity and toxicity. Possibly, tissue engineering and the development of 3D in vitro culture systems which better recapitulate the complexity of human organs and can be used in T cell assays might become a relevant addition to the testing pipelines.
In complement to experimental approaches, many efforts are ongoing for developing reliable in silico pipelines for predicting T cell cross-reactivity. It should be noted that many of such tools are not developed specifically for addressing cross-reactivity, but more generally to predict peptide immunogenicity (170). In principle, two aspects can be investigated: on the one hand, the probability for a peptidic sequence to be presented by various MHC allelic products, and on the other hand, the interaction of a specific TCR with a pMHC complex.
Regarding peptide MHC binding, the simplest strategy would be to start from the original peptide and deduce which altered sequence could or not bind to the presenting MHC allelic product. NetMHC and syfpeithi, which are essential publicly available tools, can deliver robust MHC-binding predictions, but they cannot directly interrogate TCR cross-reactivity. In addition, immunogenicity prediction tools based on aa properties (size, charge, aromaticity, gravy score) are also being developed (171, 172). In the tool available at the Immune Epitope Database (IEDB) (173), TCR preferences were deduced from the study of 600 immunogenic and 181 non-immunogenic 9mer peptides: the authors found out that peptides containing aromatic and large side chains aa (in particular phenylalanine) were preferentially recognised by T cells, and that positions 4-6 were the most critical, confirming previous findings. The task is much more complex when addressing the direct binding of a specific TCR to pMHC (see also section 1). Current approaches aiming at modelling such interactions in 3D are based on x-ray crystallography data (174–176). In addition, pMTnet, NetTCR and ERGO are neural networks that predict pMHC-TCR (CDR3 regions of the TCR β, and more recently, α chains) and are in continuous refinement (177–179).
In silico tools rely on the exploitation of experimental data. Hence, in vitro testing of e.g. peptide library scanning is not only useful for current assessment of T cell cross-reactivity, it is also needed for training and improving prediction tools. In this respect, repository of TCR-pMHC interactions and affinities, as well as 3D information, such as those available at the IEDB, Altered TCR Ligand Affinities and Structures (Atlas) (180), Structural T-cell Receptor Database (STCRDab) (181) or TCR associated with pathology conditions (McPAS-TCR) (182) and PMID databases are essential. Implementation of more information, in particular for rare MHC allelic products, is still necessary and will help improving the robustness of these approaches in the next years.
Cross-reactivity is a very smart property of our adaptive immune system to cope with the large pathogen universe. It also plays a significant role in pathological conditions as different as autoimmunity and cancer. While it has been longer discussed that virus- or bacteria-specific T cells are associated with some autoimmune diseases, more recent research is uncovering their role in cancer. This knowledge can be exploited for therapy in both diseases. Application of mimotopes in the treatment of autoimmune diseases will depend to a large extent upon their ability to suppress immunoreactivity, for instance by stimulating regulatory anti-inflammatory CD4+ T cells, or by directly inhibiting pathogenic cytotoxic CD8+ T cells. This is obviously a complex task, and identification and prediction of self-epitopes and mimotopes recognised by particular TCRs is, therefore, important to make such antigen-specific approaches successful in autoimmune diseases. In cancer immunotherapy, TCR cross-reactivity is becoming an essential consideration, not only for designing more efficient T cell-based treatments, but also for preventing severe side effects. Considering the ongoing personalisation of therapeutic approaches, the upstream TCR selection process needs to be speed up. The development of novel and refined prediction methods is of utmost importance, but is a challenging process due to the numerous aspects that can impact cross-reactivity.
CG, AM, and RK conceived the review and jointly wrote the manuscript. AM prepared the figure which was reviewed by all authors. All authors contributed to the article and approved the submitted version.
CG and AM are supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy EXC 2180, 390900677.
We acknowledge support from the Open Access Publishing Fund of the University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schodin BA, Tsomides TJ, Kranz DM. Correlation between the number of T cell receptors required for T cell activation and TCR-ligand affinity. Immunity (1996) 5(2):137–46. doi: 10.1016/s1074-7613(00)80490-2
2. Labrecque N, Whitfield LS, Obst R, Waltzinger C, Benoist C, Mathis D. How much TCR does a T cell need? Immunity (2001) 15(1):71–82. doi: 10.1016/s1074-7613(01)00170-4
3. Yin Y, Mariuzza RA. The multiple mechanisms of T cell receptor cross-reactivity. Immunity (2009) 31(6):849–51. doi: 10.1016/j.immuni.2009.12.002
4. Falk K, Rötzschke O, Stevanović S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature (1991) 351(6324):290–6. doi: 10.1038/351290a0
5. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics (1999) 50(3-4):213–9. doi: 10.1007/s002510050595
6. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science (1999) 286(5441):958–61. doi: 10.1126/science.286.5441.958
7. Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol (2004) 4(2):123–32. doi: 10.1038/nri1292
8. Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol (2013) 4:485. doi: 10.3389/fimmu.2013.00485
9. Hu C, Shen M, Han X, Chen Q, Li L, Chen S, et al. Identification of cross-reactive CD8(+) T cell receptors with high functional avidity to a SARS-CoV-2 immunodominant epitope and its natural mutant variants. Genes Dis (2022) 9(1):216–29. doi: 10.1016/j.gendis.2021.05.006
10. Jerne NK. The natural-selection theory of antibody formation. Proc Natl Acad Sci USA (1955) 41(11):849–57. doi: 10.1073/pnas.41.11.849
11. Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol (2012) 12(9):669–77. doi: 10.1038/nri3279
12. Lineburg KE, Grant EJ, Swaminathan S, Chatzileontiadou DSM, Szeto C, Sloane H, et al. CD8(+) T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope cross-react with selective seasonal coronaviruses. Immunity (2021) 54(5):1055–65.e5. doi: 10.1016/j.immuni.2021.04.006
13. Imrie A, Meeks J, Gurary A, Sukhbataar M, Kitsutani P, Effler P, et al. Differential functional avidity of dengue virus-specific T-cell clones for variant peptides representing heterologous and previously encountered serotypes. J Virol (2007) 81(18):10081–91. doi: 10.1128/JVI.00330-07
14. Su LF, Davis MM. Antiviral memory phenotype T cells in unexposed adults. Immunol Rev (2013) 255(1):95–109. doi: 10.1111/imr.12095
15. Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature (2010) 465(7296):350–4. doi: 10.1038/nature08997
16. Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev (2010) 235(1):244–66. doi: 10.1111/j.0105-2896.2010.00897.x
17. Borbulevych OY, Piepenbrink KH, Gloor BE, Scott DR, Sommese RF, Cole DK, et al. T Cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity (2009) 31(6):885–96. doi: 10.1016/j.immuni.2009.11.003
18. Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med (1996) 184(4):1259–68. doi: 10.1084/jem.184.4.1259
19. Hemmer B, Vergelli M, Gran B, Ling N, Conlon P, Pinilla C, et al. Predictable TCR antigen recognition based on peptide scans leads to the identification of agonist ligands with no sequence homology. J Immunol (1998) 160(8):3631–6. doi: 10.4049/jimmunol.160.8.3631
20. Hemmer B, Kondo T, Gran B, Pinilla C, Cortese I, Pascal J, et al. Minimal peptide length requirements for CD4(+) T cell clones–implications for molecular mimicry and T cell survival. Int Immunol (2000) 12(3):375–83. doi: 10.1093/intimm/12.3.375
21. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell (2014) 157(5):1073–87. doi: 10.1016/j.cell.2014.03.047
22. Sanderson JP, Crowley DJ, Wiedermann GE, Quinn LL, Crossland KL, Tunbridge HM, et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology (2020) 9(1):1682381. doi: 10.1080/2162402X.2019.1682381
23. Macdonald WA, Chen Z, Gras S, Archbold JK, Tynan FE, Clements CS, et al. T Cell allorecognition via molecular mimicry. Immunity (2009) 31(6):897–908. doi: 10.1016/j.immuni.2009.09.025
24. Doherty DG, Penzotti JE, Koelle DM, Kwok WW, Lybrand TP, Masewicz S, et al. Structural basis of specificity and degeneracy of T cell recognition: pluriallelic restriction of T cell responses to a peptide antigen involves both specific and promiscuous interactions between the T cell receptor, peptide, and HLA-DR. J Immunol (1998) 161(7):3527–35. doi: 10.4049/jimmunol.161.7.3527
25. Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, et al. Crossreactive T cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity (2008) 28(3):324–34. doi: 10.1016/j.immuni.2008.01.008
26. Attaf M, Legut M, Cole DK, Sewell AK. The T cell antigen receptor: the Swiss army knife of the immune system. Clin Exp Immunol (2015) 181(1):1–18. doi: 10.1111/cei.12622
27. Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity (2013) 38(2):373–83. doi: 10.1016/j.immuni.2012.10.021
28. Campion SL, Brodie TM, Fischer W, Korber BT, Rossetti A, Goonetilleke N, et al. Proteome-wide analysis of HIV-specific naive and memory CD4(+) T cells in unexposed blood donors. J Exp Med (2014) 211(7):1273–80. doi: 10.1084/jem.20130555
29. Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today (1998) 19(9):395–404. doi: 10.1016/s0167-5699(98)01299-7
30. Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity (2005) 22(1):81–92. doi: 10.1016/j.immuni.2004.11.015
31. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287(2):1168–77. doi: 10.1074/jbc.M111.289488
32. Hiemstra HS, van Veelen PA, Willemen SJ, Benckhuijsen WE, Geluk A, de Vries RR, et al. Quantitative determination of TCR cross-reactivity using peptide libraries and protein databases. Eur J Immunol (1999) 29(8):2385–91. doi: 10.1002/(SICI)1521-4141(199908)29:08<2385::AID-IMMU2385>3.0.CO;2-B
33. Ishizuka J, Grebe K, Shenderov E, Peters B, Chen Q, Peng Y, et al. Quantitating T cell cross-reactivity for unrelated peptide antigens. J Immunol (2009) 183(7):4337–45. doi: 10.4049/jimmunol.0901607
34. Curtsinger JM, Lins DC, Mescher MF. CD8+ memory T cells (CD44high, ly-6C+) are more sensitive than naive cells to (CD44low, ly-6C-) to TCR/CD8 signaling in response to antigen. J Immunol (1998) 160(7):3236–43. doi: 10.4049/jimmunol.160.7.3236
35. Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation. in vivo. Nat Immunol (2000) 1(1):47–53. doi: 10.1038/76907
36. Rose NR. Infection, mimics, and autoimmune disease. J Clin Invest (2001) 107(8):943–4. doi: 10.1172/JCI12673
37. Hafler DA. Degeneracy, as opposed to specificity, in immunotherapy. J Clin Invest (2002) 109(5):581–4. doi: 10.1172/JCI15198
38. Slansky JE, Nakayama M. Peptide mimotopes alter T cell function in cancer and autoimmunity. Semin Immunol (2020) 47:101395. doi: 10.1016/j.smim.2020.101395
39. Cunningham MW. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol (2014) 33(4):314–29. doi: 10.3109/08830185.2014.917411
40. Toor D, Sharma N. T Cell subsets: an integral component in pathogenesis of rheumatic heart disease. Immunol Res (2018) 66(1):18–30. doi: 10.1007/s12026-017-8978-z
41. Kaplan MH. The concept of autoantibodies in rheumatic fever and in the postcommissurotomy state. Ann N Y Acad Sci (1960) 86:974–91. doi: 10.1111/j.1749-6632.1960.tb42854.x
42. Kaplan MH. Rheumatic fever, rheumatic heart disease, and the streptococcal connection: the role of streptococcal antigens cross-reactive with heart tissue. Rev Infect Dis (1979) 1(6):988–86. doi: 10.1093/clinids/1.6.988
43. Cunningham MW. Pathogenesis of group a streptococcal infections. Clin Microbiol Rev (2000) 13(3):470–511. doi: 10.1128/CMR.13.3.470
44. Guilherme L, Kalil J. Rheumatic fever and rheumatic heart disease: cellular mechanisms leading autoimmune reactivity and disease. J Clin Immunol (2010) 30(1):17–23. doi: 10.1007/s10875-009-9332-6
45. Ellis NM, Li Y, Hildebrand W, Fischetti VA, Cunningham MW. T Cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J Immunol (2005) 175(8):5448–56. doi: 10.4049/jimmunol.175.8.5448
46. Guilherme L, Ramasawmy R, Kalil J. Rheumatic fever and rheumatic heart disease: genetics and pathogenesis. Scand J Immunol (2007) 66(2-3):199–207. doi: 10.1111/j.1365-3083.2007.01974.x
47. Krisher K, Cunningham MW. Myosin: a link between streptococci and heart. Science (1985) 227(4685):413–5. doi: 10.1126/science.2578225
48. Fae KC, da Silva DD, Oshiro SE, Tanaka AC, Pomerantzeff PM, Douay C, et al. Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol (2006) 176(9):5662–70. doi: 10.4049/jimmunol.176.9.5662
49. Roberts S, Kosanke S, Terrence Dunn S, Jankelow D, Duran CM, Cunningham MW. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis (2001) 183(3):507–11. doi: 10.1086/318076
50. Taranta A, Stollerman GH. The relationship of sydenham's chorea to infection with group a streptococci. Am J Med (1956) 20(2):170–5. doi: 10.1016/0002-9343(56)90186-3
51. Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in sydenham chorea. Nat Med (2003) 9(7):914–20. doi: 10.1038/nm892
52. Ben-Pazi H, Stoner JA, Cunningham MW. Dopamine receptor autoantibodies correlate with symptoms in sydenham's chorea. PloS One (2013) 8(9):e73516. doi: 10.1371/journal.pone.0073516
53. Ciacchi L, Reid HH, Rossjohn J. Structural bases of T cell antigen receptor recognition in celiac disease. Curr Opin Struct Biol (2022) 74:102349. doi: 10.1016/j.sbi.2022.102349
54. Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol (2009) 9(12):858–70. doi: 10.1038/nri2670
55. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med (1998) 4(6):713–7. doi: 10.1038/nm0698-713
56. van de Wal Y, Kooy Y, van Veelen P, Peña S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol (1998) 161(4):1585–8. doi: 10.4049/jimmunol.161.4.1585
57. Ciacchi L, Farenc C, Dahal-Koirala S, Petersen J, Sollid LM, Reid HH, et al. Structural basis of T cell receptor specificity and cross-reactivity of two HLA-DQ2.5-restricted gluten epitopes in celiac disease. J Biol Chem (2022) 298(3):101619. doi: 10.1016/j.jbc.2022.101619
58. Dahal-Koirala S, Ciacchi L, Petersen J, Risnes LF, Neumann RS, Christophersen A, et al. Discriminative T-cell receptor recognition of highly homologous HLA-DQ2-bound gluten epitopes. J Biol Chem (2019) 294(3):941–52. doi: 10.1074/jbc.RA118.005736
59. Chlubnová M, Christophersen AO, Sandve GKF, Lundin KEA, Jahnsen J, Dahal-Koirala S, et al. Identification of gluten T cell epitopes driving celiac disease. Sci Adv (2023) 9(4):eade5800. doi: 10.1126/sciadv.ade5800
60. Hoydahl LS, Richter L, Frick R, Snir O, Gunnarsen KS, Landsverk OJB, et al. Plasma cells are the most abundant gluten peptide MHC-expressing cells in inflamed intestinal tissues from patients with celiac disease. Gastroenterology (2019) 156(5):1428–39 e10. doi: 10.1053/j.gastro.2018.12.013
61. Ting YT, Dahal-Koirala S, Kim HSK, Qiao SW, Neumann RS, Lundin KEA, et al. A molecular basis for the T cell response in HLA-DQ2.2 mediated celiac disease. Proc Natl Acad Sci USA (2020) 117(6):3063–73. doi: 10.1073/pnas.1914308117
62. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol (2011) 29:493–525. doi: 10.1146/annurev-immunol-040210-092915
63. Caminero A, Meisel M, Jabri B, Verdu EF. Mechanisms by which gut microorganisms influence food sensitivities. Nat Rev Gastroenterol Hepatol (2019) 16(1):7–18. doi: 10.1038/s41575-018-0064-z
64. Sacchetti L, Nardelli C. Gut microbiome investigation in celiac disease: from methods to its pathogenetic role. Clin Chem Lab Med (2020) 58(3):340–9. doi: 10.1515/cclm-2019-0657
65. Caminero A, Galipeau HJ, McCarville JL, Johnston CW, Bernier SP, Russell AK, et al. Duodenal bacteria from patients with celiac disease and healthy subjects distinctly affect gluten breakdown and immunogenicity. Gastroenterology (2016) 151(4):670–83. doi: 10.1053/j.gastro.2016.06.041
66. Petersen J, Ciacchi L, Tran MT, Loh KL, Kooy-Winkelaar Y, Croft NP, et al. T Cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat Struct Mol Biol (2020) 27(1):49–61. doi: 10.1038/s41594-019-0353-4
67. Soldan SS, Lieberman PM. Epstein-Barr Virus and multiple sclerosis. Nat Rev Microbiol (2023) 21(1):51–64. doi: 10.1038/s41579-022-00770-5
68. Steinman L. Antigen-specific therapy of multiple sclerosis: the long-sought magic bullet. Neurotherapeutics (2007) 4(4):661–5. doi: 10.1016/j.nurt.2007.07.007
69. Zarghami A, Li Y, Claflin SB, van der Mei I, Taylor BV. Role of environmental factors in multiple sclerosis. Expert Rev Neurother (2021) 21(12):1389–408. doi: 10.1080/14737175.2021.1978843
70. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med (1999) 5(2):170–5. doi: 10.1038/5532
71. Hemmer B, Fleckenstein BT, Vergelli M, Jung G, McFarland H, Martin R, et al. Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J Exp Med (1997) 185(9):1651–9. doi: 10.1084/jem.185.9.1651
72. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell (1995) 80(5):695–705. doi: 10.1016/0092-8674(95)90348-8
73. Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol (2002) 3(10):940–3. doi: 10.1038/ni835
74. Vergelli M, Hemmer B, Kalbus M, Vogt AB, Ling N, Conlon P, et al. Modifications of peptide ligands enhancing T cell responsiveness imply large numbers of stimulatory ligands for autoreactive T cells. J Immunol (1997) 158(8):3746–52. doi: 10.4049/jimmunol.158.8.3746
75. Robinson WH, Steinman L. Epstein-Barr Virus and multiple sclerosis. Science (2022) 375(6578):264–5. doi: 10.1126/science.abm7930
76. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (2022) 375(6578):296–301. doi: 10.1126/science.abj8222
77. Schneider-Hohendorf T, Gerdes LA, Pignolet B, Gittelman R, Ostkamp P, Rubelt F, et al. Broader Epstein-Barr virus-specific T cell receptor repertoire in patients with multiple sclerosis. J Exp Med (2022) 219(11). doi: 10.1084/jem.20220650
78. Sethi DK, Gordo S, Schubert DA, Wucherpfennig KW. Crossreactivity of a human autoimmune TCR is dominated by a single TCR loop. Nat Commun (2013) 4:2623. doi: 10.1038/ncomms3623
79. Grogan JL, Kramer A, Nogai A, Dong L, Ohde M, Schneider-Mergener J, et al. Cross-reactivity of myelin basic protein-specific T cells with multiple microbial peptides: experimental autoimmune encephalomyelitis induction in TCR transgenic mice. J Immunol (1999) 163(7):3764–70. doi: 10.4049/jimmunol.163.7.3764
80. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-Cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature (1990) 346(6280):183–7. doi: 10.1038/346183a0
81. Ausubel LJ, Kwan CK, Sette A, Kuchroo V, Hafler DA. Complementary mutations in an antigenic peptide allow for crossreactivity of autoreactive T-cell clones. Proc Natl Acad Sci USA (1996) 93(26):15317–22. doi: 10.1073/pnas.93.26.15317
82. Martin R, Gran B, Zhao Y, Markovic-Plese S, Bielekova B, Marques A, et al. Molecular mimicry and antigen-specific T cell responses in multiple sclerosis and chronic CNS Lyme disease. J Autoimmun (2001) 16(3):187–92. doi: 10.1006/jaut.2000.0501
83. Gross DM, Forsthuber T, Tary-Lehmann M, Etling C, Ito K, Nagy ZA, et al. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science (1998) 281(5377):703–6. doi: 10.1126/science.281.5377.703
84. Hemmer B, Gran B, Zhao Y, Marques A, Pascal J, Tzou A, et al. Identification of candidate T-cell epitopes and molecular mimics in chronic Lyme disease. Nat Med (1999) 5(12):1375–82. doi: 10.1038/70946
85. Wildner G, Diedrichs-Mohring M. Autoimmune uveitis induced by molecular mimicry of peptides from rotavirus, bovine casein and retinal s-antigen. Eur J Immunol (2003) 33(9):2577–87. doi: 10.1002/eji.200324058
86. Wildner G. Antigenic mimicry - the key to autoimmunity in immune privileged organs. J Autoimmun (2022), 102942. doi: 10.1016/j.jaut.2022.102942
87. Thurau SR, Diedrichs-Mohring M, Fricke H, Burchardi C, Wildner G. Oral tolerance with an HLA-peptide mimicking retinal autoantigen as a treatment of autoimmune uveitis. Immunol Lett (1999) 68(2-3):205–12. doi: 10.1016/s0165-2478(99)00071-1
88. Yang X, Garner LI, Zvyagin IV, Paley MA, Komech EA, Jude KM, et al. Autoimmunity-associated T cell receptors recognize HLA-B*27-bound peptides. Nature (2022) 612(7941):771–7. doi: 10.1038/s41586-022-05501-7
89. Bordon Y. Autoimmune TCRs cross-react with microbial and self-antigens. Nat Rev Immunol (2023) 23(2):72. doi: 10.1038/s41577-022-00831-z
90. Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen WM, et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet (2015) 47(8):898–905. doi: 10.1038/ng.3353
91. Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest (2005) 115(8):2268–76. doi: 10.1172/JCI25350
92. van Lummel M, van Veelen PA, Zaldumbide A, de Ru A, Janssen GM, Moustakas AK, et al. Type 1 diabetes-associated HLA-DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem (2012) 287(12):9514–24. doi: 10.1074/jbc.M111.313940
93. Nakayama M, McDaniel K, Fitzgerald-Miller L, Kiekhaefer C, Snell-Bergeon JK, Davidson HW, et al. Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects. Proc Natl Acad Sci USA (2015) 112(14):4429–34. doi: 10.1073/pnas.1502967112
94. Spanier JA, Sahli NL, Wilson JC, Martinov T, Dileepan T, Burrack AL, et al. Increased effector memory insulin-specific CD4(+) T cells correlate with insulin autoantibodies in patients with recent-onset type 1 diabetes. Diabetes (2017) 66(12):3051–60. doi: 10.2337/db17-0666
95. Nicholson LB, Murtaza A, Hafler BP, Sette A, Kuchroo VK. A T cell receptor antagonist peptide induces T cells that mediate bystander suppression and prevent autoimmune encephalomyelitis induced with multiple myelin antigens. Proc Natl Acad Sci USA (1997) 94(17):9279–84. doi: 10.1073/pnas.94.17.9279
96. Kuchroo VK, Greer JM, Kaul D, Ishioka G, Franco A, Sette A, et al. A single TCR antagonist peptide inhibits experimental allergic encephalomyelitis mediated by a diverse T cell repertoire. J Immunol (1994) 153(7):3326–36. doi: 10.4049/jimmunol.153.7.3326
97. Brocke S, Gijbels K, Allegretta M, Ferber I, Piercy C, Blankenstein T, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature (1996) 379(6563):343–6. doi: 10.1038/379343a0
98. Ausubel LJ, Krieger JI, Hafler DA. Changes in cytokine secretion induced by altered peptide ligands of myelin basic protein peptide 85-99. J Immunol (1997) 159(5):2502–12. doi: 10.4049/jimmunol.159.5.2502
99. Ausubel LJ, Bieganowska KD, Hafler DA. Cross-reactivity of T-cell clones specific for altered peptide ligands of myelin basic protein. Cell Immunol (1999) 193(1):99–107. doi: 10.1006/cimm.1998.1447
100. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med (2000) 6(10):1167–75. doi: 10.1038/80516
101. Crowe PD, Qin Y, Conlon PJ, Antel JP. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann Neurol (2000) 48(5):758–65. doi: 10.1002/1531-8249(200011)48:5<758::AID-ANA9>3.0.CO;2-2
102. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. the altered peptide ligand in relapsing MS study group. Nat Med (2000) 6(10):1176–82. doi: 10.1038/80525
103. Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R, Group NBIS. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care (2009) 32(11):2036–40. doi: 10.2337/dc09-0449
104. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA (2011) 108(40):16729–34. doi: 10.1073/pnas.1113954108
105. Nakayama M, Beilke JN, Jasinski JM, Kobayashi M, Miao D, Li M, et al. Priming and effector dependence on insulin B:9-23 peptide in NOD islet autoimmunity. J Clin Invest (2007) 117(7):1835–43. doi: 10.1172/JCI31368
106. Kasindi A, Fuchs DT, Koronyo Y, Rentsendorj A, Black KL, Koronyo-Hamaoui M. Glatiramer acetate immunomodulation: evidence of neuroprotection and cognitive preservation. Cells (2022) 11(9). doi: 10.3390/cells11091578
107. Keith AB, Arnon R, Teitelbaum D, Caspary EA, Wisniewski HM. The effect of cop 1, a synthetic polypeptide, on chronic relapsing experimental allergic encephalomyelitis in guinea pigs. J Neurol Sci (1979) 42(2):267–74. doi: 10.1016/0022-510x(79)90058-3
108. Duda PW, Krieger JI, Schmied MC, Balentine C, Hafler DA. Human and murine CD4 T cell reactivity to a complex antigen: recognition of the synthetic random polypeptide glatiramer acetate. J Immunol (2000) 165(12):7300–7. doi: 10.4049/jimmunol.165.12.7300
109. Neuhaus O, Farina C, Yassouridis A, Wiendl H, Then Bergh F, Dose T, et al. Multiple sclerosis: comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA (2000) 97(13):7452–7. doi: 10.1073/pnas.97.13.7452
110. Bell C, Anderson J, Ganguly T, Prescott J, Capila I, Lansing JC, et al. Development of Glatopa(R) (Glatiramer acetate): the first FDA-approved generic disease-modifying therapy for relapsing forms of multiple sclerosis. J Pharm Pract (2018) 31(5):481–8. doi: 10.1177/0897190017725984
111. Rommer PS, Milo R, Han MH, Satyanarayan S, Sellner J, Hauer L, et al. Immunological aspects of approved MS therapeutics. Front Immunol (2019) 10:1564. doi: 10.3389/fimmu.2019.01564
112. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
113. Rose NR. Negative selection, epitope mimicry and autoimmunity. Curr Opin Immunol (2017) 49:51–5. doi: 10.1016/j.coi.2017.08.014
114. Ragone C, Manolio C, Cavalluzzo B, Mauriello A, Tornesello ML, Buonaguro FM, et al. Identification and validation of viral antigens sharing sequence and structural homology with tumor-associated antigens (TAAs). J Immunother Cancer (2021) 9(5). doi: 10.1136/jitc-2021-002694
115. Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Liénard D, Lejeune F, et al. High frequencies of naive melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med (1999) 190(5):705–15. doi: 10.1084/jem.190.5.705
116. Dutoit V, Rubio-Godoy V, Pittet MJ, Zippelius A, Dietrich PY, Legal FA, et al. Degeneracy of antigen recognition as the molecular basis for the high frequency of naive A2/Melan-a peptide multimer(+) CD8(+) T cells in humans. J Exp Med (2002) 196(2):207–16. doi: 10.1084/jem.20020242
117. Rubio-Godoy V, Dutoit V, Zhao Y, Simon R, Guillaume P, Houghten R, et al. Positional scanning-synthetic peptide library-based analysis of self- and pathogen-derived peptide cross-reactivity with tumor-reactive melan-a-specific CTL. J Immunol (2002) 169(10):5696–707. doi: 10.4049/jimmunol.169.10.5696
118. Appay V, Speiser DE, Rufer N, Reynard S, Barbey C, Cerottini JC, et al. Decreased specific CD8+ T cell cross-reactivity of antigen recognition following vaccination with melan-a peptide. Eur J Immunol (2006) 36(7):1805–14. doi: 10.1002/eji.200535805
119. Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun (2019) 10(1):567. doi: 10.1038/s41467-019-08534-1
120. Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature (2021) 592(7852):138–43. doi: 10.1038/s41586-021-03368-8
121. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science (2017) 358(6369):1443–8. doi: 10.1126/science.aal5240
122. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
123. Chiou SH, Tseng D, Reuben A, Mallajosyula V, Molina IS, Conley S, et al. Global analysis of shared T cell specificities in human non-small cell lung cancer enables HLA inference and antigen discovery. Immunity (2021) 54(3):586–602.e8. doi: 10.1016/j.immuni.2021.02.014
124. Ottaiano A, Scala S, D'Alterio C, Trotta A, Bello A, Rea G, et al. Unexpected tumor reduction in metastatic colorectal cancer patients during SARS-Cov-2 infection. Ther Adv Med Oncol (2021) 13:17588359211011455. doi: 10.1177/17588359211011455
125. Chiaro J, Kasanen HHE, Whalley T, Capasso C, Grönholm M, Feola S, et al. Viral molecular mimicry influences the antitumor immune response in murine and human melanoma. Cancer Immunol Res (2021) 9(8):981–93. doi: 10.1158/2326-6066.Cir-20-0814
126. Ning J, Gavil NV, Wu S, Wijeyesinghe S, Weyu E, Ma J, et al. Functional virus-specific memory T cells survey glioblastoma. Cancer Immunol Immunother (2022) 71(8):1863–75. doi: 10.1007/s00262-021-03125-w
127. Vujanovic L, Shi J, Kirkwood JM, Storkus WJ, Butterfield LH. Molecular mimicry of MAGE-A6 and mycoplasma penetrans HF-2 epitopes in the induction of antitumor CD8(+) T-cell responses. Oncoimmunology (2014) 3(8):e954501. doi: 10.4161/21624011.2014.954501
128. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J (2017) 474(11):1823–36. doi: 10.1042/bcj20160510
129. Ragone C, Manolio C, Mauriello A, Cavalluzzo B, Buonaguro FM, Tornesello ML, et al. Molecular mimicry between tumor associated antigens and microbiota-derived epitopes. J Transl Med (2022) 20(1):316. doi: 10.1186/s12967-022-03512-6
130. Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science (2020) 369(6506):936–42. doi: 10.1126/science.aax0701
131. Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med (2021) 27(8):1432–41. doi: 10.1038/s41591-021-01406-6
132. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84. doi: 10.1126/science.aad1329
133. Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity (2016) 45(4):931–43. doi: 10.1016/j.immuni.2016.09.009
134. Rong Y, Dong Z, Hong Z, Jin Y, Zhang W, Zhang B, et al. Reactivity toward bifidobacterium longum and enterococcus hirae demonstrate robust CD8(+) T cell response and better prognosis in HBV-related hepatocellular carcinoma. Exp Cell Res (2017) 358(2):352–9. doi: 10.1016/j.yexcr.2017.07.009
135. Bräunlein E, Lupoli G, Füchsl F, Abualrous ET, de Andrade Krätzig N, Gosmann D, et al. Functional analysis of peripheral and intratumoral neoantigen-specific TCRs identified in a patient with melanoma. J Immunother Cancer (2021) 9(9). doi: 10.1136/jitc-2021-002754
136. Li F, Deng L, Jackson KR, Talukder AH, Katailiha AS, Bradley SD, et al. Neoantigen vaccination induces clinical and immunologic responses in non-small cell lung cancer patients harboring EGFR mutations. J Immunother Cancer (2021) 9(7). doi: 10.1136/jitc-2021-002531
137. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med (2015) 21(1):81–5. doi: 10.1038/nm.3773
138. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase ib glioblastoma trial. Nature (2019) 565(7738):234–9. doi: 10.1038/s41586-018-0792-9
139. Petrizzo A, Tagliamonte M, Mauriello A, Costa V, Aprile M, Esposito R, et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J Transl Med (2018) 16(1):286. doi: 10.1186/s12967-018-1662-9
140. Bessell CA, Isser A, Havel JJ, Lee S, Bell DR, Hickey JW, et al. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight (2020) 5(8). doi: 10.1172/jci.insight.135597
141. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551(7681):512–6. doi: 10.1038/nature24462
142. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
143. Boesch M, Baty F, Rothschild SI, Tamm M, Joerger M, Früh M, et al. Tumour neoantigen mimicry by microbial species in cancer immunotherapy. Br J Cancer (2021) 125(3):313–23. doi: 10.1038/s41416-021-01365-2
144. Lustgarten J, Dominguez AL, Pinilla C. Identification of cross-reactive peptides using combinatorial libraries circumvents tolerance against her-2/neu-immunodominant epitope. J Immunol (2006) 176(3):1796–805. doi: 10.4049/jimmunol.176.3.1796
145. Theobald M, Biggs J, Hernández J, Lustgarten J, Labadie C, Sherman LA. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med (1997) 185(5):833–41. doi: 10.1084/jem.185.5.833
146. Carrabba MG, Castelli C, Maeurer MJ, Squarcina P, Cova A, Pilla L, et al. Suboptimal activation of CD8(+) T cells by melanoma-derived altered peptide ligands: role of melan-A/MART-1 optimized analogues. Cancer Res (2003) 63(7):1560–7.
147. Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol (1996) 157(6):2539–48. doi: 10.4049/jimmunol.157.6.2539
148. Chen JL, Dunbar PR, Gileadi U, Jäger E, Gnjatic S, Nagata Y, et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol (2000) 165(2):948–55. doi: 10.4049/jimmunol.165.2.948
149. Bae J, Samur M, Munshi A, Hideshima T, Keskin D, Kimmelman A, et al. Heteroclitic XBP1 peptides evoke tumor-specific memory cytotoxic T lymphocytes against breast cancer, colon cancer, and pancreatic cancer cells. Oncoimmunology (2014) 3(12):e970914. doi: 10.4161/21624011.2014.970914
150. Dao T, Korontsvit T, Zakhaleva V, Jarvis C, Mondello P, Oh C, et al. An immunogenic WT1-derived peptide that induces T cell response in the context of HLA-A*02:01 and HLA-A*24:02 molecules. Oncoimmunology (2017) 6(2):e1252895. doi: 10.1080/2162402x.2016.1252895
151. Filipazzi P, Pilla L, Mariani L, Patuzzo R, Castelli C, Camisaschi C, et al. Limited induction of tumor cross-reactive T cells without a measurable clinical benefit in early melanoma patients vaccinated with human leukocyte antigen class I-modified peptides. Clin Cancer Res (2012) 18(23):6485–96. doi: 10.1158/1078-0432.Ccr-12-1516
152. Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, et al. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc Natl Acad Sci USA (2008) 105(10):3849–54. doi: 10.1073/pnas.0800080105
153. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36(2):133–51. doi: 10.1097/CJI.0b013e3182829903
154. Jones HF, Molvi Z, Klatt MG, Dao T, Scheinberg DA. Empirical and rational design of T cell receptor-based immunotherapies. Front Immunol (2020) 11:585385. doi: 10.3389/fimmu.2020.585385
155. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122(6):863–71. doi: 10.1182/blood-2013-03-490565
156. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5(197):197ra03. doi: 10.1126/scitranslmed.3006034
157. Gil-Cruz C, Perez-Shibayama C, De Martin A, Ronchi F, van der Borght K, Niederer R, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science (2019) 366(6467):881–6. doi: 10.1126/science.aav3487
158. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med (2016) 375(18):1749–55. doi: 10.1056/NEJMoa1609214
159. Zhang L, Jones-O'Connor M, Awadalla M, Zlotoff DA, Thavendiranathan P, Groarke JD, et al. Cardiotoxicity of immune checkpoint inhibitors. Curr Treat Options Cardiovasc Med (2019) 21(7):32. doi: 10.1007/s11936-019-0731-6
160. Nino-Vasquez JJ, Allicotti G, Borras E, Wilson DB, Valmori D, Simon R, et al. A powerful combination: the use of positional scanning libraries and biometrical analysis to identify cross-reactive T cell epitopes. Mol Immunol (2004) 40(14-15):1063–74. doi: 10.1016/j.molimm.2003.11.005
161. Szomolay B, Liu J, Brown PE, Miles JJ, Clement M, Llewellyn-Lacey S, et al. Identification of human viral protein-derived ligands recognized by individual MHCI-restricted T-cell receptors. Immunol Cell Biol (2016) 94(6):573–82. doi: 10.1038/icb.2016.12
162. Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: state of the art and perspectives. Sci Adv (2023) 9(7):eadf3700. doi: 10.1126/sciadv.adf3700
163. Bentzen AK, Hadrup SR. T-Cell-receptor cross-recognition and strategies to select safe T-cell receptors for clinical translation. Immunooncol Technol (2019) 2:1–10. doi: 10.1016/j.iotech.2019.06.003
164. Bentzen AK, Such L, Jensen KK, Marquard AM, Jessen LE, Miller NJ, et al. T Cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat Biotechnol (2018). doi: 10.1038/nbt.4303
165. Kim GB, Fritsche J, Bunk S, Mahr A, Unverdorben F, Tosh K, et al. Quantitative immunopeptidomics reveals a tumor stroma-specific target for T cell therapy. Sci Transl Med (2022) 14(660):eabo6135. doi: 10.1126/scitranslmed.abo6135
166. de Rooij MAJ, Remst DFG, van der Steen DM, Wouters AK, Hagedoorn RS, Kester MGD, et al. A library of cancer testis specific T cell receptors for T cell receptor gene therapy. Mol Ther Oncolytics (2023) 28:1–14. doi: 10.1016/j.omto.2022.11.007
167. Bijen HM, van der Steen DM, Hagedoorn RS, Wouters AK, Wooldridge L, Falkenburg JHF, et al. Preclinical strategies to identify off-target toxicity of high-affinity TCRs. Mol Ther (2018) 26(5):1206–14. doi: 10.1016/j.ymthe.2018.02.017
168. Vizcaíno JA, Kubiniok P, Kovalchik KA, Ma Q, Duquette JD, Mongrain I, et al. The human immunopeptidome project: a roadmap to predict and treat immune diseases. Mol Cell Proteomics (2020) 19(1):31–49. doi: 10.1074/mcp.R119.001743
169. Marcu A, Bichmann L, Kuchenbecker L, Kowalewski DJ, Freudenmann LK, Backert L, et al. HLA ligand atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J Immunother Cancer (2021) 9(4). doi: 10.1136/jitc-2020-002071
170. Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell (2020) 183(3):818–34.e13. doi: 10.1016/j.cell.2020.09.015
171. Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PloS Comput Biol (2013) 9(10):e1003266. doi: 10.1371/journal.pcbi.1003266
172. Klatt MG, Mack KN, Bai Y, Aretz ZEH, Nathan LI, Mun SS, et al. Solving an MHC allele-specific bias in the reported immunopeptidome. JCI Insight (2020) 5(19). doi: 10.1172/jci.insight.141264
173. Mendes M, Mahita J, Blazeska N, Greenbaum J, Ha B, Wheeler K, et al. IEDB-3D 2.0: structural data analysis within the immune epitope database. Protein Sci (2023) 32(4):e4605. doi: 10.1002/pro.4605
174. Hellman LM, Foley KC, Singh NK, Alonso JA, Riley TP, Devlin JR, et al. Improving T cell receptor on-target specificity via structure-guided design. Mol Ther (2019) 27(2):300–13. doi: 10.1016/j.ymthe.2018.12.010
175. Antunes DA, Rigo MM, Freitas MV, Mendes MFA, Sinigaglia M, Lizée G, et al. Interpreting T-cell cross-reactivity through structure: implications for TCR-based cancer immunotherapy. Front Immunol (2017) 8:1210. doi: 10.3389/fimmu.2017.01210
176. Riley TP, Hellman LM, Gee MH, Mendoza JL, Alonso JA, Foley KC, et al. T Cell receptor cross-reactivity expanded by dramatic peptide-MHC adaptability. Nat Chem Biol (2018) 14(10):934–42. doi: 10.1038/s41589-018-0130-4
177. Springer I, Tickotsky N, Louzoun Y. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction. Front Immunol (2021) 12:664514. doi: 10.3389/fimmu.2021.664514
178. Montemurro A, Schuster V, Povlsen HR, Bentzen AK, Jurtz V, Chronister WD, et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun Biol (2021) 4(1):1060. doi: 10.1038/s42003-021-02610-3
179. Lu T, Zhang Z, Zhu J, Wang Y, Jiang P, Xiao X, et al. Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat Mach Intell (2021) 3(10):864–75. doi: 10.1038/s42256-021-00383-2
180. Borrman T, Cimons J, Cosiano M, Purcaro M, Pierce BG, Baker BM, et al. ATLAS: a database linking binding affinities with structures for wild-type and mutant TCR-pMHC complexes. Proteins (2017) 85(5):908–16. doi: 10.1002/prot.25260
181. Leem J, de Oliveira SHP, Krawczyk K, Deane CM. STCRDab: the structural T-cell receptor database. Nucleic Acids Res (2018) 46(D1):D406–d12. doi: 10.1093/nar/gkx971
Keywords: T cell receptor, cross-reactivity, autoimmunity, cancer, immunotherapy
Citation: Gouttefangeas C, Klein R and Maia A (2023) The good and the bad of T cell cross-reactivity: challenges and opportunities for novel therapeutics in autoimmunity and cancer. Front. Immunol. 14:1212546. doi: 10.3389/fimmu.2023.1212546
Received: 26 April 2023; Accepted: 24 May 2023;
Published: 19 June 2023.
Edited by:
Tomasz Piotr Wypych, Polish Academy of Sciences, PolandReviewed by:
Kenichi Hanada, National Institutes of Health (NIH), United StatesCopyright © 2023 Gouttefangeas, Klein and Maia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cécile Gouttefangeas, Y2VjaWxlLmdvdXR0ZWZhbmdlYXNAdW5pLXR1ZWJpbmdlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.