 Zoya Kurago1†
Zoya Kurago1† Gang Guo2,3†
Gang Guo2,3† Huidong Shi2,3
Huidong Shi2,3 Roni J. Bollag3,4
Roni J. Bollag3,4 Michael W. Groves3,5
Michael W. Groves3,5 J. Kenneth Byrd3,5
J. Kenneth Byrd3,5 Yan Cui2,3*
Yan Cui2,3*- 1Department of Oral Biology and Diagnostic Sciences, Dental College of Georgia at Augusta University, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 2Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 3Georgia Cancer Center, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 4Department of Pathology, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 5Department of Otolaryngology, Medical College of Georgia, Augusta University, Augusta, GA, United States
The cell surface enzyme CD73 is increasingly appreciated as a pivotal non-redundant immune checkpoint (IC) in addition to PD-1/PD-L1 and CTLA-4. CD73 produces extracellular adenosine (eADO), which not only inhibits antitumor T cell activity via the adenosine receptor (AR) A2AR, but also enhances the immune inhibitory function of cancer-associated fibroblasts and myeloid cells via A2BR. Preclinical studies show that inhibition of the CD73-adenosinergic pathway in experimental models of many solid tumors either as a monotherapy or, more effectively, in combination with PD-1/PD-L1 or CTLA-4 IC blockades, improves antitumor immunity and tumor control. Consequently, approximately 50 ongoing phase I/II clinical trials targeting the CD73-adenosinergic IC are currently listed on https://clinicaltrials.gov. Most of the listed trials employ CD73 inhibitors or anti-CD73 antibodies alone, in combination with A2AR antagonists, and/or with PD-1/PD-L1 blockade. Recent evidence suggests that the distribution of CD73, A2AR and A2BR in tumor microenvironments (TME) is heterogeneous, and this distribution affects CD73-adenosinergic IC function. The new insights have implications for the optimally effective, carefully tailored approaches to therapeutic targeting of this essential IC. In the mini-review, we briefly discuss the cellular and molecular mechanisms of CD73/eADO-mediated immunosuppression during tumor progression and therapy in the spatial context of the TME. We include preclinical data regarding therapeutic CD73-eADO blockade in tumor models as well as available clinical data from completed trials that targeted CD73-adenosinergic IC with or without PD-1/PD-L1 inhibitors and discuss factors that are potentially important for optimal therapeutic outcomes in cancer patients.
1 Introduction
CD73 is a type I transmembrane glycoprotein widely expressed on cell surfaces of smooth muscle, epithelium, endothelium, fibroblasts, neurons, and the immune system (1–3). Functionally, CD73 is a rate-limiting ecto-5’-nucleotidase (NT5E), which together with other cell surface ectonucleoside triphosphate diphosphohydrolases, such as CD39 (ENTPDase 1), dephosphorylate ATP released from stressed/damaged cells and produce extracellular adenosine (eADO) (1–3). CD73 plays a critical role in tissue homeostasis under physiological and pathological conditions, including epithelial and endothelial barrier function, neuronal function, as well as immunity and inflammation (4–6). The roles of CD73 in modulating tumorigenesis, angiogenesis, and metastasis are increasingly appreciated (7–9) such that it is now recognized as a critical cancer immune checkpoint (IC) non-redundant to PD-1/PD-L1 and CTLA-4 (10–14). Preclinical studies and early clinical trials reveal important breakthroughs as well as challenges. Here, we briefly describe the cellular and molecular events associated with the CD73-adenosinergic pathway, discuss the current status of therapeutic interventions that target the CD73-ADO axis, and propose potential ways to enhance cancer treatment outcomes.
2 The CD73-adenosinergic pathway in the tumor microenvironment
Hypoxia is a hallmark of the TME (15–17). Hypoxia and therapy-induced cell death potentiate ATP release into the extracellular space, which is rapidly metabolized by the CD39/CD73 enzyme-pair to ADO (Figure 1A). ADO acts on specific adenosine receptors (AR), A1R, A2AR, A2BR, and A3R. Stimulatory A1R and A3R are coupled with Gi or Go proteins, whose activation suppresses cAMP with downstream immune-stimulatory effects. In contrast, A2AR and A2BR are coupled with Gs and/or Golf or Gq proteins, which promote cAMP signaling and thus inhibit anti-tumor immune responses (Figure 1B) (4, 18, 19).
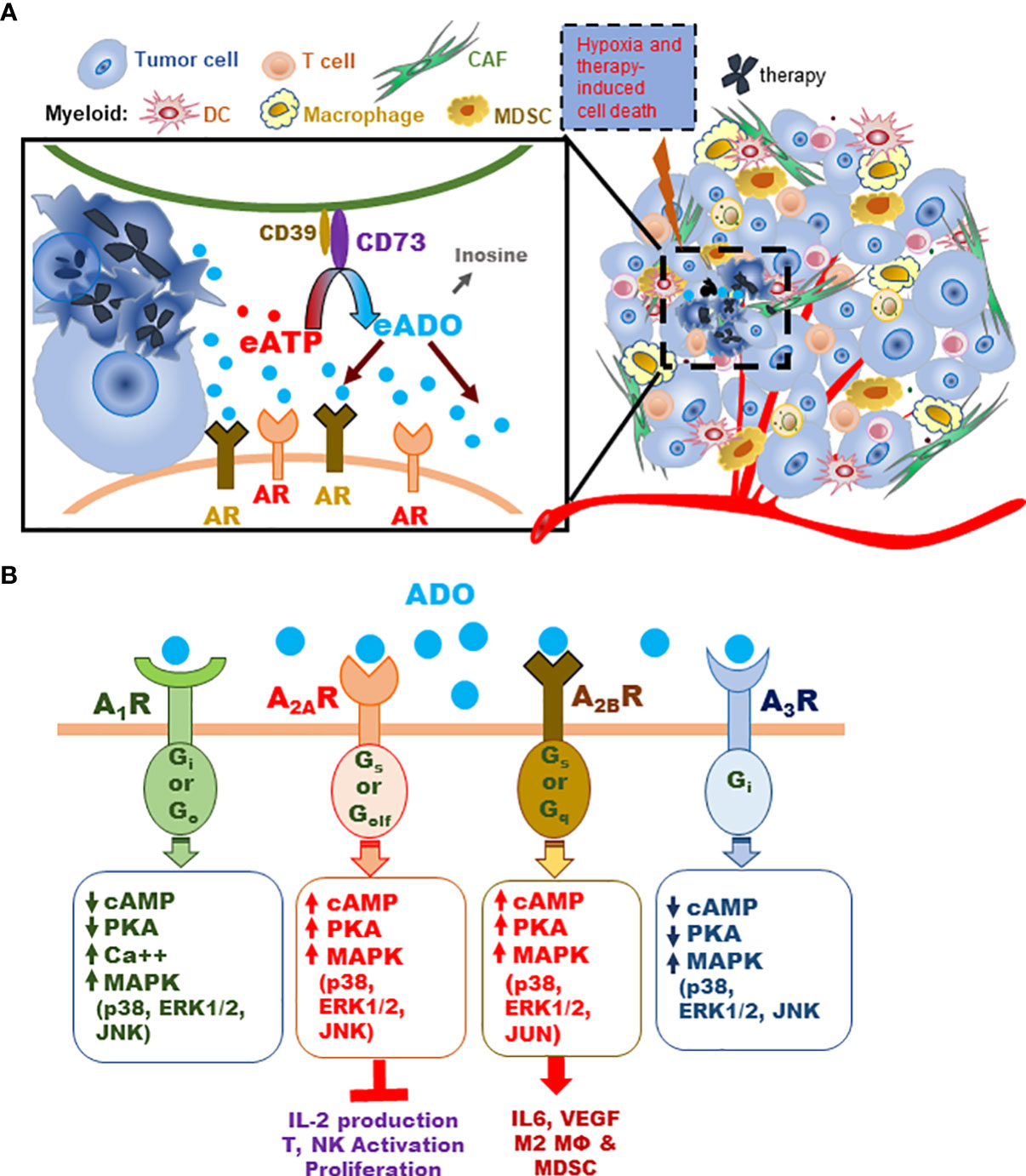
Figure 1 Schematic illustration of the CD73-adenosinergic pathway activity in the TME during tumor progression and treatment. (A) The CD39/CD73 enzyme pair converts ATP released by dying or stressed cells to immunosuppressive ADO, which inhibits antitumor immunity primarily by engaging A2AR and A2BR on various TME cells. (B) Schematic illustration of major AR signals activated by ADO. All four AR activate MAPK (p38, ERK1/2 and JNK or JUN) phosphorylation. Stimulatory A1R and A3R share several signaling events, including: A1R or A3R stimulation decreases adenylate cyclase activity and cAMP, inhibits protein kinase A (PKA), activates phospholipase C (PLCβ), and closes Ca++ channels. Stimulation of A2AR or A2BR produces opposite effects on adenylate cyclase, cAMP, and PKA (i.e. stimulates them) (4, 18, 19). Key functional impact of A2AR activity in TME effector T and NK cells are inhibition of activation, IL-2 production, and proliferation. The impact of ADO on A2BR-expressing myeloid and non-immune TME cells causes skewed pro-tumor phenotypes and activities that affect the TME and T/NK effector functions (4, 18, 19).
Preclinical and clinical studies show that in the TME, ADO mainly mediates immunosuppression via A2AR and A2BR due to hypoxia, inflammation and typically high ADO levels (4, 10, 13, 20). In particular, A2AR is highly expressed on T and NK cells and when activated, suppresses cell proliferation and effector function (13, 21–26). High A2BR levels on other TME cells potentiate immune suppressors including regulatory dendritic cells (DC), myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM) (13, 27–31), and cancer associated fibroblasts (CAF) (32). Moreover, A2BR augments CD73 expression on CAF via a CD73-A2BR-CD73 positive feedback loop, further exacerbating immunosuppression (13, 27–32).
3 CD73-adenosinergic pathway as a critical IC
3.1 Preclinical models
Preclinical studies targeting CD73 via genetic inactivation, neutralization, or small molecule inhibitors in numerous tumor models were reviewed extensively elsewhere (8–10, 13, 33). These studies reveal that the efficacy of anti-CD73 alone has limitations, in part because ADO can be generated by additional, though less prominent, pathways besides CD39/CD73. Similarly, clinical trials show that CD73 monotherapy is well-tolerated with moderate benefit in subsets of patients (9, 13, 34, 35). Treatment efficacy can be enhanced when CD73 targeting is combined with strategies to inhibit down-stream ADO signaling, among other methods. Here, we mainly focus on how these treatments impact specific AR-mediated cellular and molecular events that modulate the TME immune landscape.
3.1.1 A2AR activity on T and NK cells
The critical inhibitory role of A2AR in T cell activation and antitumor immunity was first revealed by Ohta et al. in 2006 (22). This seminal study demonstrated that genetic inactivation of A2AR enhanced CD8 T cell-dependent antitumor immunity leading to the rejection of immunogenic tumors in ~60% of hosts without affecting the progression of non-immunogenic tumors (22). Subsequent studies revealed that ADO-induced A2AR signaling suppressed TCR-induced T cell activation including decreased production of IL-2, IFN-γ and TNF-α, which subsequently disrupted T cell proliferation and CD4 differentiation to Th1 and Th17 effectors (24, 36). Instead, A2AR activation promoted the generation of FoxP3+ and Lag-3+ regulatory T cells (Treg) and persistent T cell unresponsiveness to subsequent stimuli (24). A2AR signaling in CD8 T cells also interfered with Notch-1 upregulation and granzyme B production following TCR stimuli (23).
Early studies suggested that NK cell-dependent cytotoxicity was inhibited by CD73+ tumors leading to enhanced tumor metastasis (37, 38). Subsequent research revealed that CD73-A2AR activity suppressed NK cell maturation (39) and inhibited IL-15-induced NK cytotoxicity (40). Moreover, CD73+ NK cells within large tumors possessed immune-regulatory function via STAT3-induced IL-10, which suppressed CD4 T cell proliferation and IFN-γ production (41).
The immunosuppressive role of A2AR in T and NK cells was validated in preclinical murine tumor models. Genetic inactivation as well as A2aR antagonists alleviated T and NK cell unresponsiveness and enhanced antitumor immunity (37, 39, 42–45). A recent study employed the CRISPR/Cas9-mediated A2AR knockout in engineered human chimeric antigen receptor (CAR)-T cells, which made them resistant to ADO (46) and enhanced effector function and antitumor immunity in vivo in a preclinical model (46). These exciting results warrant clinical application of targeted A2AR inhibition/inactivation in T and NK cells to improve antitumor immunity and treatment outcomes.
3.1.2 A2BR activity
Less is known about A2BR expression and function in various cell subsets and its relationship to CD73 in the TME. Unlike A2AR, A2BR has low affinity for ADO and is only activated by high ADO concentrations found under pathological conditions, which in the TME include hypoxia and therapy-induced cell stress or death (4, 5, 10, 47, 48). Notably, the expression of A2BR is markedly upregulated in response to hypoxia or inflammation (4, 20, 49). In the TME, A2BR is expressed in immune and non-immune cells, including myeloid cells, CAF, endothelium, and tumor cells (4, 18, 20, 32, 47). High levels of A2BR on some tumor cell types apparently promotes tumor proliferation, angiogenesis and metastasis (12, 47, 50, 51), which may be independent of immune regulation. In a glioblastoma model, a CD73-A2BR-CD73 positive feedback loop enhanced tumor chemoresistance (52).
So far, only myeloid cells, CAF and endothelium have been shown to exert ADO-A2BR-mediated immunosuppression. Moreover, a hypoxia-induced CD73-A2BR-CD73 positive feedback loop augmented CD73 and A2BR expression on endothelial cells (49). Much of this knowledge comes from in vitro studies and investigations of tissue damage in the absence of cancer (28, 48, 53), partly validated in the TME (27, 29, 47, 54). Activated A2BR in myeloid precursors promotes the immunosuppressive function of MDSC, differentiation of macrophages towards M2 phenotype, and induction of regulatory DC (27, 48, 54–57). These myeloid cells in turn inhibit antitumor T cell activity and promote angiogenesis and tumor metastasis potentially by reducing production of TNF-α and IL-12 while increasing IL-10, IL-6 and VEGF secretion (27, 58, 59).
Overall, these observations suggest an important immunosuppressive role for TME-associated A2BR and suggest that the CD73-A2BR-CD73 amplification loop may be a plausible therapeutic target.
3.1.3 Combinatory targeting of A2AR and/or A2BR together with other IC
Preclinical evidence showed that while targeting individual A2AR- or A2BR-axis each positively impacted antitumor immunity and generally delayed tumor progression (60–62), we found that combined anti-CD73, A2AR- and A2BR-axis blockade markedly improved anti-tumor immunity with tumor regression (32). The specific reasons for the observed additive/synergistic effects are incompletely understood. Evidence does suggest that the effectiveness of anti-IC strategies depends upon the tumor type and TME conditions (13, 32). Furthermore, combinatory regimens that inhibit various CD73-AR axes and PD-1/PD-L1 are more effective, substantiating the non-redundant roles of CD73-AR IC and the advantage of targeting multiple IC (32, 61, 63).
3.2 Clinical observations
A large body of clinical evidence supports the negative impact of TME CD73 on cancer patient outcomes. Markedly elevated CD73 levels found in numerous tumors, including colorectal cancer (CRC), triple negative breast cancer (TNBC), head and neck cancer (HNC) and ovarian cancer (OC), have been linked to poor patient survival (64–70). Moreover, cancer therapies, including PD-1 ICB (71, 72), upregulated CD73-expression and ADO-AR signaling in the TME, potentially amplifying the role of this IC in patient outcomes (70, 73–76).
Several clinical studies have suggested that high blood levels of soluble (s)CD73, potentially generated by shedding, MMP-mediated clipping, or exome secretion, may prognosticate poor clinical outcome (77–79). However, a positive correlation between sCD73 and CD73 levels in the TME has not been established (77). While sCD73 has enzymatic activity in circulation, its’ impact on T cell-mediated antitumor immunity in the TME might be limited due to the spatial impact (addressed in the discussion). A mechanistic insight concerning sCD73 production and distribution in the TME is needed in order to fully understand its impact on antitumor immunity.
The specific roles of A2AR or A2BR and their relationship to CD73 expression in patient outcomes are less clear and possibly depend upon specific-tumor type, immunogenicity, and the TME landscape. On one hand, recent reports suggest that in non-small cell lung cancer (NSCLC), high A2AR expression in the TME independently predicted better patient overall survival (OS), while high CD73 levels were associated with poor OS (69). Similarly, high A2AR expression on CD8 T cells within OC nests correlated with durable clinical benefit/response (CBR) during a clinical trial of PD-1 ICB and an epigenetic modifier (80). On the other hand, the negative impact of A2AR on anti-tumor immunity was shown in a combined trial of PD-L1 and A2AR inhibitors in renal cell carcinoma (RCC), demonstrating better clinical responses to A2AR inhibitors when tumors exhibited high adenosine signature profiles (81). Additional studies are necessary to dissect the relationships of various CD73-AR axes and application to treatments.
4 Clinical trials targeting the CD73-adenosinergic pathway
So far, approximately 50 active phase I/II cancer immunotherapy trials targeting the CD73-AR IC are listed on https://clinicaltrials.gov. Among these, more than 60% were designed to target CD73 by monoclonal antibodies or small molecule inhibitors, some of which were combined with PD-1/PD-L1 ICB regimens. The other 30-40% have employed small molecule inhibitors targeting A2AR alone or together with anti-CD73 and/or A2BR inhibitors (8, 61, 83–86). In addition to the safety (severity of adverse event, AE) and pharmacokinetic assessment of the therapeutic agents, a secondary objective was to collect data on clinical benefit rate (CBR), consisting of complete response (CR), partial response (PR) and stable disease (SD). Also, progression-free survival (PFS), objective response (OR), overall response rates (ORR) and overall survival (OS) were assessed based on standardized Response Evaluation Criteria in Solid Tumors version 1.1 (82) (for details, see legend to Table 1).
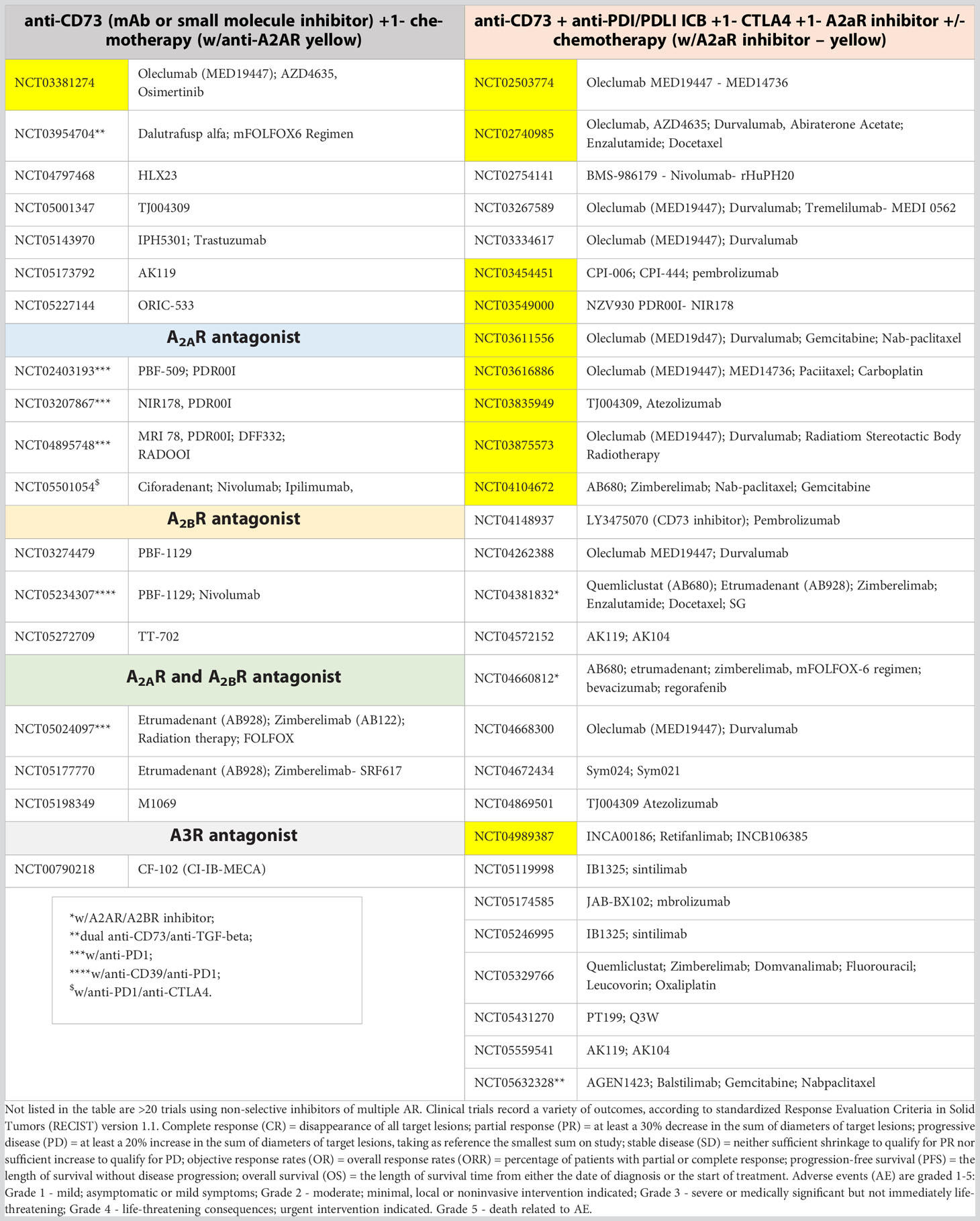
Table 1 List of registered clinical trials that target various aspects of CD73-adenosinergic axis.
4.1 Clinical trials targeting CD73
4.1.1 Anti-CD73 antibodies
In approximately 20 trials in a variety of solid tumors, anti-CD73 monoclonal antibodies have been employed alone or more often, in combination with anti-PD-L1 or anti-PD-1. These antibodies are listed as Oleclumab, MEDI9447, AK119, HLX23, IPH5301, Sym042, CPI-0006, IBI325, PT199, JAB-BX102, TJ004309, NZV930, INCA00186 and BMS-986179. Most of the trials were/are phase I/Ib for safety assessment with limited preliminary reports of clinical outcomes in publications, abstracts, or oral presentations at international conferences, briefly described below.
In general, anti-CD73 caused low-grade AE classified as manageable or acceptable tolerability (NCT02503774, NCT03381274, NCT03616886, NCT03611556 and NCT03334617). Early reported outcomes have been mixed, as some showed promising signs of disease control, while others lacked solid evidence of clinical benefits. For instance, the NCT02503774 phase I trial of anti-CD73 with or without anti-PD-L1 enrolled 77 patients with CRC, 73 with pancreatic adenocarcinoma (PDAC) and 42 with NSCLC positive for EGFR mutation (EGFRm). Among those with evaluable outcomes, one CRC, two PDAC and four EGFRm NSCLC patients had OR, while nine CRC, eight PDAC and nine EGFRm NSCLC patients had SD. Overall, the antitumor activity was promising in EGFRm NSCLC patients receiving anti-CD73/anti-PD-L1 therapy, whereas the effectiveness in CRC and PDAC is yet to be verified (34).
The NCT03381274 phase Ib/II study evaluated the effects of anti-CD73 combined with third-generation tyrosine kinase inhibitors (TKI) in advanced EGFRm NSCLC in previously treated patients and reported acceptable tolerability. Clinical observations up to July 2021 were published in 2023 for patients with T790M-negative EGFRm NSCLC and showed CBR of 75% and OR of 25% in five patients receiving 1500 mg anti-CD73 antibody; CBR of 82.4% and OR of 11.8% were noted in 21 patients administered 3000 mg anti-CD73 antibody (35). For patients on the higher dose of anti-CD73, the median PFS was 7.4 months as compared with PFS of 2.8 months without anti-CD73 (35).
Similar studies include NCT03616886 phase I/II trial testing anti-CD73, anti-PD-L1 and chemotherapy in subjects with advanced TNBC (87), NCT03611556 phase Ib/II trial testing anti-CD73 alone or combination with gemcitabine chemotherapy and anti-PD-L1 in 212 patients with metastatic PDAC, and NCT03334617 HUDSON Platform multi-arm phase II trial for NSCLC patients who previously failed anti-PD(L)1 immunotherapy. These trials have yet to report the results (NCT03611556), or else had limited patient numbers (87) (NCT03616886) or short treatment duration (NCT03334617) (88) insufficient to assess clinical benefits.
The NCT03954704 phase I trial initiated in 2019 differs from others by testing a bi-functional antibody against CD73 and TGFβ (known as GS-1423 and AGEN1423) in patients with advanced solid tumors (89). Because TGF-β is an immunosuppressive cytokine that enhances ADO-mediated CD73 upregulation (76, 90–92), it is expected to improve treatment efficacy. Early assessment in 21 patients showed AE ranging from mild to severe, including death (89). In patients administered a high dose (20-45 mg/Kg), the circulating bi-functional antibody was durable and effectively bound to B cell CD73. Among the 17 patients who reached the first response assessment, 4.8% had a PR, 33.3% had SD, and 42.9% showed progressive disease (PD) (89).
4.1.2 CD73 small molecule inhibitors
Besides anti-CD73 antibody, small molecule inhibitors specific for CD73, AB680, ORIC-533 and LY3475070, have been employed. The NCT04104672 phase I/Ib trial was designed to evaluate safety and tolerability of AB680 with chemotherapy (paclitaxel and gemcitabine) and anti-PD-1 for treatment-naive patients with metastatic mPDAC (93). Preliminary observations in 13 patients receiving various doses of AB680 showed a manageable safety profile with AE up to grades 3-4. Early clinical responses among nine evaluable patients included three PR and five SD (93).
4.2 Trials targeting A2AR or A2BR
The A2AR antagonists AZD4635, NIR178 and Ciforadenant (CPI-444) were developed to block the A2AR-mediated inhibition of T and NK activity. The A2BR antagonists PBF-1129 and TT-702 as well as the dual A2AR/A2BR antagonists Etrumadenant (AB928) and M1069 were developed to target the myeloid, stromal and potential tumor cell-mediated immunosuppression for additive/synergistic effects of dual CD73-AR axes blockade.
Fong et al. reported the results of A2AR inhibitor CPI-444 phase I trial NCT03454451 in 68 patients with RCC (81), 33 of which received CPI-444 alone and 35 received both CPI-444 and anti-PD-L1. In both groups, the regimens were safe and improved overall survival with durable clinical benefit associated with increased CD8+ T cell recruitment into the tumors and broadened circulating T-cell repertoires (81). Remarkably, better clinical response was associated with enriched adenosine-related gene-expression profile in pre-treatment RCC specimens (81), supporting the hypothesis that elevated CD73-AR signaling is a targetable non-redundant IC, and its blockade may enhance antitumor immunity. Moreover, adenosine-regulated gene signature may be a useful marker to predict clinical prognosis (69, 80).
Early results of the NCT02740985 phase Ia/b trials using A2AR inhibitor AZD4635 alone or in combination with anti-PD-L1 antibody in 250 PD-1/PD-L1 inhibitor-naive patients with advanced solid tumors, including metastatic castration-resistant prostate cancer (mCRPC), CRC or NSCLC were reported recently (94). Both monotherapy and combination therapy were well tolerated with an overall <20% above grade 3 AE. ORR was observed in ~5% of the 39 mCRPC patients on AZD4635 monotherapy and ~16.2% of 37 patients on combination therapy (94). This trial also revealed a positive correlation between high adenosine signature in the blood and better clinical response, as 24-week PFS was noted in 48.9% of high adenosine-signature patients versus 20.8% of low adenosine-signature patients (94).
Clinical trials that target the A2BR are limited. The NCT04381832 phase Ib/II trial to evaluate the A2AR/A2BR dual antagonist AB928 with or without anti-PD-1 and chemotherapy in patients with mCRPC reported a manageable safety profile in 17 enrolled patients (95). Among 16 patients that continued with AB928 treatment, the composite ORR was 43% (95).
5 Discussion and future perspectives
5.1 Combinatory regimens of CD73-ICB with other therapies
The early clinical observations of CD73-AR ICB trials have demonstrated feasibility, manageable toxicity and promising potential for tumor control. The benefits of anti-CD73 monotherapy appear modest, but markedly improved when combined with PD-1/PD-L1 ICB and/or other cancer therapies. As CD73-IC is continuously activated and exacerbated by hypoxia and therapy-induced cell death (32, 80, 81, 94), targeting multiple CD73-AR axes in the context of conventional or advanced therapies will improve therapeutic benefits. In particular, CD73-ICB before and during cell death induced by therapy will promote eATP-mediated antitumor immunity (96, 97). While current CD73-AR ICB trials include chemotherapy-treated patients, future trials designed to target CD73-IC aspects specific to the patient and the TME may significantly improve outcomes.
5.2 Targeting strategies based on spatial context of CD73-AR axes
Productive antitumor immunity relies on direct interactions between effector and tumor cells. Recent studies have illustrated that close effector-target cell proximity in the TME directly affects clinical outcomes (98, 99). As eADO stability and diffusion are limited, the expression levels, distribution and proximity of CD73, A2AR and A2BR in the TME will determine the activity of specific CD73-AR axes and the mechanisms of ADO-mediated immunosuppression. For example, in the TME with few T and NK cells, CD73-A2AR axis might be insignificant despite high prevalence of CD73. Yet, in the absence of the spatial distribution mapping, it is unclear which aspects of ADO-mediated immunosuppression, and at what stage of treatment, would be most relevant. We propose that spatial distribution maps of these receptors, combined with the knowledge of relevant cellular compartments, could be important tools to identify key pathways of ADO-mediated immunosuppression operating in various TME over time and inform the design of CD73-IC targeting strategies.
In conclusion, tremendous advances have occurred in the area of CD73-AR ICB in the past decade. Combined ICB strategies targeting CD73-AR and PD-1/PD-L1 with conventional or advanced therapies remain a promising and exciting area of research. Further advances will be made possible through better understanding of the tumor-specific and treatment-specific TME, including the spatial distribution of CD73, A2AR and A2BR.
Author contributions
ZK, GG and YC reviewed the literature and wrote the manuscript. HS, RB, MG, and KB participated in the discussion and revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by 1R21DE028716 from the NIH/NIDCR and funds from AU-Georgia Cancer Center, Augusta University to YC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alcedo KP, Bowser JL, Snider NT. The elegant complexity of mammalian ecto-5’-nucleotidase (CD73). Trends Cell Biol (2021) 31(10):829–42. doi: 10.1016/j.tcb.2021.05.008
2. Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a multi-signalling guardian angel in human diseases: when, where and how does it exert its protective effects? Trends Pharmacol Sci (2016) 37(6):419–34. doi: 10.1016/j.tips.2016.02.006
3. Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic Signal (2006) 2(2):351–60. doi: 10.1007/s11302-005-5302-5
4. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi: 10.1038/nri.2016.4
5. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18(10):601–18. doi: 10.1038/s41568-018-0037-0
6. Yuan X, Ferrari D, Mills T, Wang Y, Czopik A, Doursout MF, et al. Editorial: purinergic signaling and inflammation. Front Immunol (2021) 12:699069. doi: 10.3389/fimmu.2021.699069
7. Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell (2019) 36(6):582–96. doi: 10.1016/j.ccell.2019.10.007
8. Antonioli L, Novitskiy SV, Sachsenmeier KF, Fornai M, Blandizzi C, Hasko G. Switching off CD73: a way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discovery Today (2017) 22(11):1686–96. doi: 10.1016/j.drudis.2017.06.005
9. Chen S, Wainwright DA, Wu JD, Wan Y, Matei DE, Zhang Y, et al. CD73: an emerging checkpoint for cancer immunotherapy. Immunotherapy (2019) 11(11):983–97. doi: 10.2217/imt-2018-0200
10. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol (2020) 17(10):611–29. doi: 10.1038/s41571-020-0382-2
11. Chiarella AM, Ryu YK, Manji GA, Rustgi AK. Extracellular ATP and adenosine in cancer pathogenesis and treatment. Trends Cancer (2021) 7(8):731–50. doi: 10.1016/j.trecan.2021.04.008
12. Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discovery (2006) 5(3):247–64. doi: 10.1038/nrd1983
13. Thompson EA, Powell JD. Inhibition of the adenosine pathway to potentiate cancer immunotherapy: potential for combinatorial approaches. Annu Rev Med (2021) 72(1):331–48. doi: 10.1146/annurev-med-060619-023155
14. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17(12):765–. doi: 10.1038/nrc.2017.110
15. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
16. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
17. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol (2015) 15(11):669–82. doi: 10.1038/nri3902
18. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13(12):842–57. doi: 10.1038/nrc3613
19. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of adenosine receptors: the state of the art. Physiol Rev (2018) 98(3):1591–625. doi: 10.1152/physrev.00049.2017
20. Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu Rev Immunol (2019) 37:325–47. doi: 10.1146/annurev-immunol-051116-052406
21. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res (2014) 74(24):7239–49. doi: 10.1158/0008-5472.CAN-13-3581
22. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
23. Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Osborne BA, et al. Adenosine A2A receptor stimulation inhibits TCR-induced Notch1 activation in CD8+T-cells. Front Immunol (2019) 10:162. doi: 10.3389/fimmu.2019.00162
24. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111(1):251–9. doi: 10.1182/blood-2007-03-081646
25. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res (2014) 74(24):7250–9. doi: 10.1158/0008-5472.CAN-13-3583
26. Chambers AM, Matosevic S. Immunometabolic dysfunction of natural killer cells mediated by the hypoxia-CD73 axis in solid tumors. Front Mol Biosci (2019) 6:60. doi: 10.3389/fmolb.2019.00060
27. Chen S, Akdemir I, Fan J, Linden J, Zhang B, Cekic C. The expression of adenosine A2B receptor on antigen-presenting cells suppresses CD8+ T-cell responses and promotes tumor growth. Cancer Immunol Res (2020) 8(8):1064–74. doi: 10.1158/2326-6066.CIR-19-0833
28. Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26(1):376–86. doi: 10.1096/fj.11-190934
29. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Activation of the A2B adenosine receptor in B16 melanomas induces CXCL12 expression in FAP-positive tumor stromal cells, enhancing tumor progression. Oncotarget (2016) 7(39):64274–88. doi: 10.18632/oncotarget.11729
30. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol (2011) 187(11):6120–9. doi: 10.4049/jimmunol.1101225
31. Sorrentino C, Morello S. Role of adenosine in tumor progression: focus on A2B receptor as potential therapeutic target. J Cancer Metastasis Treat (2017) 3:127–38. doi: 10.20517/2394-4722.2017.29
32. Yu M, Guo G, Huang L, Deng L, Chang CS, Achyut BR, et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Commun (2020) 11(1):515. doi: 10.1038/s41467-019-14060-x
33. Azambuja JH, Ludwig N, Braganhol E, Whiteside TL. Inhibition of the adenosinergic pathway in cancer rejuvenates innate and adaptive immunity. Int J Mol Sci (2019) 20(22):5698. doi: 10.3390/ijms20225698
34. Bendell JC, LoRusso P, Overman MJ, Noonan AM, Kim D-W, Strickler J, et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab ± durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J Clin Oncol (2021) 39(15_suppl):9047. doi: 10.1200/JCO.2021.39.15_suppl.9047
35. Kim D-W, Kim S-W, Camidge DR, Shu CA, Marrone KA, Le X, et al. CD73 inhibitor oleclumab plus osimertinib in previously treated patients with advanced T790M-negative EGFR-mutated NSCLC: a brief report. J Thorac Oncol (2023) 18(5):650–56. doi: 10.1016/j.jtho.2022.12.021
36. Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol (2005) 174(2):1073–80. doi: 10.4049/jimmunol.174.2.1073
37. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A (2013) 110(36):14711–6. doi: 10.1073/pnas.1308209110
38. Qin L, Thompson LF, Kuzel TM, Zhang B. Requirement of NK cells for selective A2A receptor blockade to suppress CD73+ tumor metastasis. Immunotherapy (2014) 6(1):19–21. doi: 10.2217/imt.13.154
39. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
40. Kang G, Zhao X, Sun J, Cheng C, Wang C, Tao L, et al. A2AR limits IL-15-induced generation of CD39(+) NK cells with high cytotoxicity. Int Immunopharmacol (2023) 114:109567. doi: 10.1016/j.intimp.2022.109567
41. Neo SY, Yang Y, Record J, Ma R, Chen X, Chen Z, et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest (2020) 130(3):1185–98. doi: 10.1172/JCI128895
42. Hatfield SM, Sitkovsky M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr Opin Pharmacol (2016) 29:90–6. doi: 10.1016/j.coph.2016.06.009
43. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother (2018) 67(8):1271–84. doi: 10.1007/s00262-018-2186-0
44. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother (2012) 61(6):917–26. doi: 10.1007/s00262-011-1155-7
45. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia (2017) 19(7):530–6. doi: 10.1016/j.neo.2017.02.004
46. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun (2021) 12(1):3236. doi: 10.1038/s41467-021-23331-5
47. Gao Z-G, Jacobson KA. A2B adenosine receptor and cancer. Int J Mol Sci (2019) 20(20):5139. doi: 10.3390/ijms20205139
48. Haskó G, Csóka B, Németh ZH, Vizi ES, Pacher P. A(2B) adenosine receptors in immunity and inflammation. Trends Immunol (2009) 30(6):263–70. doi: 10.1016/j.it.2009.04.001
49. Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med (2003) 198(5):783–96. doi: 10.1084/jem.20030891
50. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res (2016) 76(15):4372–82. doi: 10.1158/0008-5472.CAN-16-0544
51. Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, et al. Identification of a pharmacologically tractable fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci (2013) 110(13):5139–44. doi: 10.1073/pnas.1222085110
52. Yan A, Joachims ML, Thompson LF, Miller AD, Canoll PD, Bynoe MS. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A(2B) adenosine receptor signaling. J Neurosci (2019) 39(22):4387–402. doi: 10.1523/JNEUROSCI.1118-18.2019
53. El-Naccache DW, Chen F, Palma MJ, Lemenze A, Fischer MA, Wu W, et al. Adenosine metabolized from extracellular ATP promotes type 2 immunity through triggering A2BAR signaling in intestinal epithelial cells. Cell Rep (2022) 40(5):111150. doi: 10.1016/j.celrep.2022.111150
54. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia (2013) 15(12):1400–9. doi: 10.1593/neo.131748
55. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112(5):1822–31. doi: 10.1182/blood-2008-02-136325
56. Xaus J, Mirabet M, Lloberas J, Soler C, Lluis C, Franco R, et al. IFN-gamma up-regulates the A2B adenosine receptor expression in macrophages: a mechanism of macrophage deactivation. J Immunol (1999) 162(6):3607–14. doi: 10.4049/jimmunol.162.6.3607
57. Belikoff BG, Hatfield S, Georgiev P, Ohta A, Lukashev D, Buras JA, et al. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J Immunol (2011) 186(4):2444–53. doi: 10.4049/jimmunol.1001567
58. Németh ZH, Lutz CS, Csóka B, Deitch EA, Leibovich SJ, Gause WC, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol (2005) 175(12):8260–70. doi: 10.4049/jimmunol.175.12.8260
59. Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, et al. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol (2009) 182(8):4616–23. doi: 10.4049/jimmunol.0801279
60. Sequeira I, Neves JF, Carrero D, Peng Q, Palasz N, Liakath-Ali K, et al. Immunomodulatory role of keratin 76 in oral and gastric cancer. Nat Commun (2018) 9(1):3437. doi: 10.1038/s41467-018-05872-4
61. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
62. Dziedzic K, Węgrzyn P, Gałęzowski M, Bońkowska M, Grycuk K, Satała G, et al. Release of adenosine-induced immunosuppression: comprehensive characterization of dual A(2A)/A(2B) receptor antagonist. Int Immunopharmacol (2021) 96:107645. doi: 10.1016/j.intimp.2021.107645
63. Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and anti-CTLA-4 in preclinical models. Cancer Immunol Res (2018) 6(10):1136–49. doi: 10.1158/2326-6066.CIR-18-0056
64. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res (2016) 22(1):158–66. doi: 10.1158/1078-0432.CCR-15-1181
65. Liu N, Fang XD, Vadis Q. CD73 as a novel prognostic biomarker for human colorectal cancer. J Surg Oncol (2012) 106(7):918–9; author reply 20. doi: 10.1002/jso.23159
66. Ren ZH, Lin CZ, Cao W, Yang R, Lu W, Liu ZQ, et al. CD73 is associated with poor prognosis in HNSCC. Oncotarget (2016) 7(38):61690–702. doi: 10.18632/oncotarget.11435
67. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res (2015) 75(21):4494–503. doi: 10.1158/0008-5472.CAN-14-3569
68. Sidders B, Zhang P, Goodwin K, O’Connor G, Russell DL, Borodovsky A, et al. Adenosine signalling is prognostic for cancer outcome and has predictive utility for immunotherapeutic response. Clin Cancer Res (2020) 26(9):2176–87. doi: 10.1158/1078-0432.Ccr-19-2183
69. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget (2017) 8(5):8738–51. doi: 10.18632/oncotarget.14434
70. Buisseret L, Pommey S, Allard B, Garaud S, Bergeron M, Cousineau I, et al. Clinical significance of CD73 in triple-negative breast cancer: multiplex analysis of a phase III clinical trial. Ann Oncol (2018) 29(4):1056–62. doi: 10.1093/annonc/mdx730
71. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19(20):5626–35. doi: 10.1158/1078-0432.CCR-13-0545
72. Beavis PA, Slaney CY, Milenkovski N, Henderson MA, Loi S, Stagg J, et al. CD73: a potential biomarker for anti-PD-1 therapy. OncoImmunology (2015) 4(11):e1046675. doi: 10.1080/2162402X.2015.1046675
73. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med (2020) 26(1):39–+. doi: 10.1038/s41591-019-0694-x
74. Rocha P, Salazar R, Zhang J, Ledesma D, Solorzano JL, Mino B, et al. CD73 expression defines immune, molecular, and clinicopathological subgroups of lung adenocarcinoma. Cancer Immunol Immunother (2021) 70(7):1965–76. doi: 10.1007/s00262-020-02820-4
75. Ott M, Tomaszowski K-H, Marisetty A, Kong L-Y, Wei J, Duna M, et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight (2020) 5(17). doi: 10.1172/jci.insight.134386
76. Sidders B, Zhang P, Goodwin K, O’Connor G, Russell DL, Borodovsky A, et al. Adenosine signaling is prognostic for cancer outcome and has predictive utility for immunotherapeutic response. Clin Cancer Res (2020) 26(9):2176–87. doi: 10.1158/1078-0432.CCR-19-2183
77. Messaoudi N, Cousineau I, Arslanian E, Henault D, Stephen D, Vandenbroucke-Menu F, et al. Prognostic value of CD73 expression in resected colorectal cancer liver metastasis. Oncoimmunology (2020) 9(1):1746138. doi: 10.1080/2162402X.2020.1746138
78. Morello S, Capone M, Sorrentino C, Giannarelli D, Madonna G, Mallardo D, et al. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J Trans Med (2017) 15(1):244. doi: 10.1186/s12967-017-1348-8
79. Turiello R, Capone M, Giannarelli D, Morretta E, Monti MC, Madonna G, et al. Serum CD73 is a prognostic factor in patients with metastatic melanoma and is associated with response to anti-PD-1 therapy. J Immunother Cancer (2020) 8(2):e001689. doi: 10.1136/jitc-2020-001689
80. Chen S, Xie P, Cowan M, Huang H, Cardenas H, Keathley R, et al. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J Clin Invest (2022) 132(14). doi: 10.1172/JCI158800
81. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discovery (2020) 10(1):40–53. doi: 10.1158/2159-8290.CD-19-0980
82. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (2009) 45(2):228–47. doi: 10.1016/j.ejca.2008.10.026
83. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
84. Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Hasko G. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer (2016) 2(2):95–109. doi: 10.1016/j.trecan.2016.01.003
85. Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti–PD-(L)1 and anti–CTLA-4 in preclinical models. Cancer Immunol Res (2018) 6(10):1136–49. doi: 10.1158/2326-6066.CIR-18-0056
86. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17(12):709–24. doi: 10.1038/nrc.2017.86
87. Eiger D, Maurer C, Brandao M, Aftimos PG, Punie K, Taylor D, et al. 348P first findings from SYNERGY, a phase I/II trial testing the addition of the anti-CD73 oleclumab (O) to the anti-PD-L1 durvalumab (D) and chemotherapy (ChT) as first line therapy for patients (pts) with metastatic triple-negative breast cancer (mTNBC). Ann Oncol (2020) 31:S386–S7. doi: 10.1016/j.annonc.2020.08.450
88. Besse B, Awad M, Forde P, Thomas M, Park K, Goss G, et al. OA07.08 HUDSON: an open-label, multi-drug, biomarker-directed, phase II platform study in patients with NSCLC, who progressed on anti-PD(L)1 therapy. J Thorac Oncol (2021) 16(3, Supplement):S118–S9. doi: 10.1016/j.jtho.2021.01.299
89. Tolcher AW, Gordon M, Mahoney KM, Seto A, Zavodovskaya M, Hsueh C-H, et al. Phase 1 first-in-human study of dalutrafusp alfa, an anti–CD73-TGF-β-trap bifunctional antibody, in patients with advanced solid tumors. J Immunother Cancer (2023) 11(2):e005267. doi: 10.1136/jitc-2022-005267
90. Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, et al. CD73 expression on effector T cells sustained by TGF-beta facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat Commun (2019) 10(1):150. doi: 10.1038/s41467-018-08123-8
91. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol (2014) 193(6):3155–64. doi: 10.4049/jimmunol.1400578
92. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
93. Manji GA, Wainberg ZA, Krishnan K, Giafis N, Udyavar A, Quah CS, et al. ARC-8: phase I/Ib study to evaluate safety and tolerability of AB680 + chemotherapy + zimberelimab (AB122) in patients with treatment-naive metastatic pancreatic adenocarcinoma (mPDAC). J Clin Oncol (2021) 39(3_suppl):404. doi: 10.1200/JCO.2021.39.3_suppl.404
94. Lim EA, Bendell JC, Falchook GS, Bauer TM, Drake CG, Choe JH, et al. Phase ia/b, open-label, multicenter study of AZD4635 (an adenosine A2A receptor antagonist) as monotherapy or combined with durvalumab, in patients with solid tumors. Clin Cancer Res (2022) 28(22):4871–84. doi: 10.1158/1078-0432.CCR-22-0612
95. Subudhi SK, Bendell JC, Carducci MA, Kopp LM, Scott J, Grady MM, et al. ARC-6: a phase 1b/2, open-label, randomized platform study to evaluate efficacy and safety of etrumadenant (AB928)-based treatment combinations in patients with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol (2021) 39(15_suppl):5039. doi: 10.1200/JCO.2021.39.15_suppl.5039
96. Wennerberg E, Spada S, Rudqvist NP, Lhuillier C, Gruber S, Chen Q, et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol Res (2020) 8(4):465–78. doi: 10.1158/2326-6066.CIR-19-0449
97. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer (2020) 20(4):203–17. doi: 10.1038/s41568-020-0246-1
98. Phillips D, Matusiak M, Gutierrez BR, Bhate SS, Barlow GL, Jiang S, et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nat Commun (2021) 12(1):6726. doi: 10.1038/s41467-021-26974-6
Keywords: CD73, NT5E, CD39, adenosine, A2AR, A2BR, immune checkpoint inhibitor, combination therapy
Citation: Kurago Z, Guo G, Shi H, Bollag RJ, Groves MW, Byrd JK and Cui Y (2023) Inhibitors of the CD73-adenosinergic checkpoint as promising combinatory agents for conventional and advanced cancer immunotherapy. Front. Immunol. 14:1212209. doi: 10.3389/fimmu.2023.1212209
Received: 25 April 2023; Accepted: 31 May 2023;
Published: 26 June 2023.
Edited by:
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United StatesReviewed by:
Santosh Bhosale, Cedars Sinai Medical Center, United StatesRanjan Nanda, International Centre for Genetic Engineering and Biotechnology, India
Copyright © 2023 Kurago, Guo, Shi, Bollag, Groves, Byrd and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Cui, eWN1aUBhdWd1c3RhLmVkdQ==
†These authors have contributed equally to this work