 Yoko Ueki
Yoko Ueki Riko Takimoto-Ito
Riko Takimoto-Ito Megumu K. Saito
Megumu K. Saito Hideaki Tanizaki
Hideaki Tanizaki Naotomo Kambe
Naotomo Kambe
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 21 June 2023
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1211240
This article is part of the Research Topic Basic, clinical, and translational studies of Yao syndrome and other NOD2 related diseases View all 8 articles
Introduction: Blau syndrome is a rare autosomal dominant autoinflammatory granulomatous disease caused by a mutation in the NOD2 gene. It is characterized by a clinical trial of granulomatous dermatitis, arthritis, and uveitis. Tofacitinib is a pan Janus kinase (JAK) inhibitor used for treatment of Blau syndrome and idiopathic sarcoidosis. Here, we evaluated its effect on inflammatory pathways associated with Blau syndrome. The effect of tofacitinib on downstream pathways regulated by mutant NOD2 was analyzed using luciferase assays with overexpression of NOD2 mutants.
Methods: The effect of tofacitinib on the upstream pathway for the induction of NOD2 expression and proinflammatory cytokine production was assessed using monocytic cell lines differentiated from Blau syndrome patient-derived induced pluripotent stem cells.
Results: Tofacitinib did not suppress the increased spontaneous transcriptional activity of NF-κB by mutant NOD2. In addition, mutant NOD2 was not involved in the transcription of ISRE and GAS, which are activated by type 1 and type 2 interferons (IFN), respectively. On the other hand, IFNγ induced the expression of NOD2, which led to the production of inflammatory cytokines by an autoinflammatory mechanism only in cells with mutant NOD2.
Discussion: Tofacitinib suppressed the induction of NOD2 by IFNγ, thereby inhibiting the production of pro-inflammatory cytokines. Thus, tofacitinib showed anti-inflammatory effects through suppression of NOD2 expression. The JAK inhibitor tofacitinib is a potential therapeutic agent for Blau syndrome because it suppresses the autoinflammation seen in Blau syndrome by inhibiting the expression of NOD2.
Sarcoidosis is a systemic inflammatory disease of unknown etiology that is characterized by granulomas in multiple organs. Less than 0.5% of all patients with sarcoidosis demonstrate distinct clinical manifestations, with onset before the age of 4 years, in the skin, joints, and eyes, without bilateral hilar lymphadenopathy (BHL). It was previously referred to as early-onset sarcoidosis (EOS) (1). Unlike sarcoidosis in older patients, EOS is progressive and causes severe complications such as joint destruction and blindness (2, 3). Heterozygous mutations in the nucleotide-binding oligomerization domain 2 gene (NOD2) were identified as the cause of this syndrome based on both familial and sporadic cases (1, 4, 5). This distinct pathology is referred to as Blau syndrome (6).
The NOD2 protein contains 3 domain structures: 2 sets of N-terminal caspase activation and recruitment domains (CARD) as signal transduction domains and C-terminal leucine-rich repeats (LRR) (7, 8). The domain located in the center is called the NACHT domain, named after the structure commonly found in NAIP, CIITA, HET-E, TEP1, and is evolutionarily conserved. This NACHT domain is also called the NOD domain in NLR family molecules. NOD2 works as an intra-cellular pattern recognition receptor where it recognizes muramyl dipeptide (MDP), a bacterial cell-wall component, leading to the activation of the nuclear factor κB (NF-κB) pathway. Activation causes the production and release of inflammatory cytokines and chemokines. Mutations in NOD2 are associated with two granulomatous disorders, Blau syndrome and Crohn disease. In patients with Crohn disease, a common bowel inflammatory disorder, loss-of-function mutations have been identified mainly in the LRR of NOD2 (9), whereas in patients with Blau syndrome mutations are in the centrally located NOD domain. In overexpression systems using plasmids expressing Blau syndrome-associated NOD2 mutants transfected into HEK293 cells, ligand-independent NF-κB activation was observed (1, 10, 11). Thus, the NOD2 mutations identified in Blau syndrome are likely gain-of-function mutations.
The third autoinflammatory disorder involving mutations in NOD2, Yao syndrome, has also been described, which is characterized by mutations in the intron of the NOD2 gene, IVS8 + 158, and may be associated with missense mutations in the NOD domain that differ from those in Blau syndrome (12–15). Yao syndrome is an independent disease from Blau syndrome because of the absence of uveitis, joint deformities, and granulomas on skin biopsy, although it presents with clinical symptoms such as periodic fever, dermatitis, polyarthritis, and serositis (12). Yao syndrome also differs from the clinical course of Blau syndrome in that it is an adult-onset, solitary disease. In this syndrome, a mutation in NOD2 is not responsible for the disease, but is presumed to be involved in the amplification and modification of inflammation (13).
We have been working on the analysis of Blau syndrome and have established induced pluripotent stem (iPS) cells from Blau syndrome patients. Functional analyses using these iPS cells revealed that interferon (IFN)γ is an important mediator for the initiation of the inflammatory development in Blau syndrome (16). This experimental finding is supported by the clinical observation that bacillus Calmette–Guesrin (BCG) vaccination is sometimes associated with disease onset (10, 17), since IFNγ is a major cytokine associated with BCG-mediated immune responses. However, the detailed molecular mechanisms of NOD2 mutations leading to granuloma formation remain unclear. Hence, no specific treatment for Blau syndrome has been established.
In 2018, Damsky et al. (18) reported a patient with cutaneous sarcoidosis who was successfully treated with tofacitinib, a pan Janus kinase (JAK) inhibitor, which acts mostly as a JAK1/3 inhibitor and has low/slight effect on JAK2. They suggested that JAK/signal transducer and activator of transcription (STAT) signaling plays a role in the pathogenesis of sarcoidosis. IFNγ activates the JAK/STAT signaling pathway, resulting in the up-regulation of STAT1 transcriptional targets. Several studies have shown that activation signatures of the JAK-STAT pathway, especially STAT1-dependent transcripts, are characteristic of the transcriptome in both peripheral blood mononuclear cells and other tissues in patients with sarcoidosis. They also reported that JAK inhibitors are effective in other granulomatous skin diseases (19). Another group in China recently reported the successful treatment of three Blau syndrome patients with tofacitinib (20). Therefore, in this study, we evaluated the effect of tofacitinib on inflammatory pathways associated with Blau syndrome as a potential therapeutic approach for the treatment of Blau syndrome.
The activities of NOD2 mutants were evaluated by dual luciferase reporter assays, as described previously (1, 10, 11). Briefly, mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and subcloned into the p3xFLAG-CMV vector (Sigma-Aldrich, St. Louis, MO). Two consensus elements in the IFN-stimulated gene promoters responsible for IFN induction have been detected: the ISRE (IFN-stimulated response element) for IFN-α/β activation and the GAS (IFN-γ-activated site) for IFN-γ activation (21). The plasmid of pLuc-MCS (Stratagene) containing 4 copies of the GAS element (5’-AGT GAT TTC TCG GAA AGA GAG-3’) or ISRE element (5′-CGA AGT ACT TTC AGT TTC ATA TTA GG-3′) (22) were kindly provided from Drs. Y. Takahashi and Y. Takakura (Kyoto University).
HEK293FT cells were transfected with a pGreenFire NF-kB reporter vector (System Biosciences, Mountain View, CA) or ISRE or GAS reporters, along with wild type (WT) or mutant NOD2 and as an internal control Renilla gene under phRL-Tk vector (Promega, Madison, WI). Transfected HEK293FT cells were cultured with or without 10 μg/mL of MDP (InvivoGen, San Diego, CA) and/or 1 μM tofacitinib (Sigma-Aldrich) for 24 h. As a positive control for the ISRE or GAS luciferase assay, cells were cultured with 1000 U/mL IFNα (PBL Assay Science, Piscataway, NJ) or 50 ng/mL IFNγ (PeproTech, Cranbury, NJ), respectively. The results were expressed as the ratio of firefly to renilla luciferase activity, and we set the activity of untreated cells transfected with mock vector as the standard.
Patient-derived iPS cells were established from peripheral blood cells from a Blau syndrome patient who had a p.R334W mutation in NOD2, as described previously (16). We also corrected the mutation in iPS cell clones by using CRISPR-Cas9–mediated targeting. Differentiation into the monocytic cell lineage was described previously (16). Immortalized proliferating monocytic cell lines (iPS-ML) were then established by introducing 3 transgenes (cMYC, BMI1, and MDM2) into the CSII-EF-RfA vector. They were kindly provided by Drs S. Senju (Kumamoto University) and H. Miyoshi (RIKEN Bio Resource Center), as also described previously (16). Then, iPS-ML cells were maintained in StemPro-34 serum-free medium (Thermo Fisher Scientific, Waltham, MA) containing 2 mmol/l L-glutamine with M-CSF (50 ng/mL, R&D Systems, Minneapolis, MN).
iPS-ML cells (1x106/mL) were cultured with 50 ng/mL IFNγ for 6 h. In some cases, 1 μM tofacitinib was added. RNA was prepared with the RNeasy Mini Kit (QIAGEN, Hilden, Germany) and subjected to reverse transcription with a PrimeScript RT reagent kit (Takara Bio, Shiga, Japan). qPCR was performed on the Light Cycler 480 System (Roche, Basel Switzerland). qPCR data were processed using the ΔΔcycle threshold method, and the quantities relative to the untreated sample are shown. The primers used in this study are listed in Table 1.

Table 1 List of primers.
Concentrations of cytokines in the culture supernatants were measured by using a human IL-8/CXCL8 DuoSet ELISA (R&D system) with DuoSet Ancillary Reagent Kit 2 (R&D system) in accordance with the manufacturer’s instructions. Quantification was done with a FlexStation3 (Molecular Devices, San Jose, CA).
The iPS-ML cells (1x106/mL) were cultured with 50 ng/ml of IFNγ for the time exhibited. In some cells, 1 µM tofacitinib were added. Cells were lysed with dithiothreitol and incubated at 95°C for 5 min. Proteins were then separated by using SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. After blocking with Blocking buffer (nacalai tesque, Kyoto, Japan), immunoblotting was performed with monoclonal antibodies to phospho-STAT1 (Cell Signaling Technology, Danvers, MA) and β-actin (MBL, Tokyo, Japan) and exposed with ECL Western Blotting Reagent (Cytiva, Marlborough, MA).
After confirming the superiority and normality distribution by two-way ANOVA, the Tukey method was used for multiple comparisons. All of the statistical analyses were performed with Prism software (GraphPad Software, La Jolla, CA). The values are presented as means ± the standard deviation, as indicated in the figure legends.
Zhang et al. (20) studied the efficacy of tofacitinib in three patients with Blau syndrome. They proposed a model in which JAK, which acts downstream of the cytokine receptor, was activated by a mutated NOD2. To explore this possibility, we first examined whether the JAK-STAT signaling pathway was involved downstream from NOD2. Towards that end, we used HEK293FT cells transiently transfected with a plasmid carrying NOD2. Here, p.R334W is a representative mutation in Blau syndrome and p.R311W is a nonpathological single nucleotide variant (SNV) (10). First, we performed dual-luciferase assays of NF-κB. The result was the same as our previous report (1, 10, 11). That is, in mutant-p.R334W transfected cells, NF-κB was spontaneously activated in an MDP-independent manner (Figure 1A). In contrast, WT and non-pathogenic SNV, R311W, as well as pathogenic p.R334W, increased luciferase activity in response to MDP.
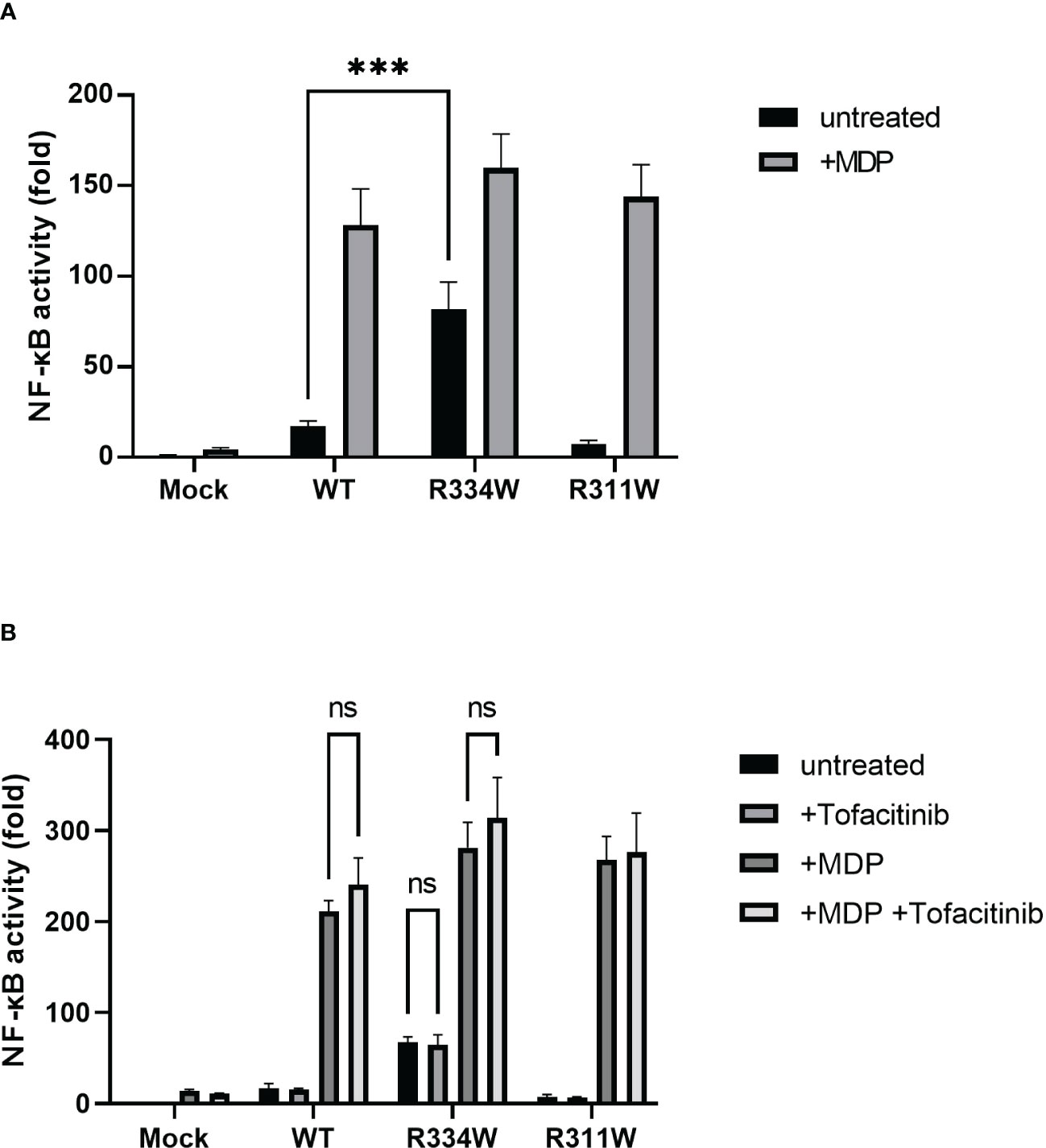
Figure 1 HEK293FT cells transfected with Blau syndrome-associated mutant NOD2 showed ligand-independent NF-κB activation, as evaluated with an NF-κB luciferase reporter system. There was no downregulation by tofacitinib. The relative fold-change from the untreated condition of Mock (set as 1) is shown. Bars indicate means ± SDs (n=3). This test was repeated at least three times, and the results of one representative test are shown, but the same results were obtained each time. (A) Cells were treated with MDP (10 µg/mL) for 24 (h) (B) Cells were treated with MDP (10 µg/mL) with or without tofacitinib (1 uM) for 24 (h) ***p <.001, ns. not significant.
Under the addition of tofacitinib, however, there was no attenuation of NF-κB activation in any type of NOD2-transfected cell, with or without the addition of MDP (Figure 1B). Those results indicated that the JAK-STAT pathway was not directly associated with the downstream pathway of NOD2, regardless of the presence or absence of NOD2 mutations associated with Blau syndrome.
To ensure that the JAK-STAT pathway was not involved downstream from NOD2, we performed dual-luciferase assays of ISRE and GAS. When IFNα or IFNγ was added, activation of either ISRE or GAS was detected, respectively (Figures 2A, B). Those data confirmed that the assay system itself functioned properly. In both assays, we found no specific activation of ISRE or GAS in mutant-NOD2 transfected cells (Figures 2C, D). Even the stimulation of NOD2 with ligand MDP did not initiate ISRE or GAS activation. Taken together, we concluded that the JAK-STAT pathway is not directly involved downstream from NOD2.
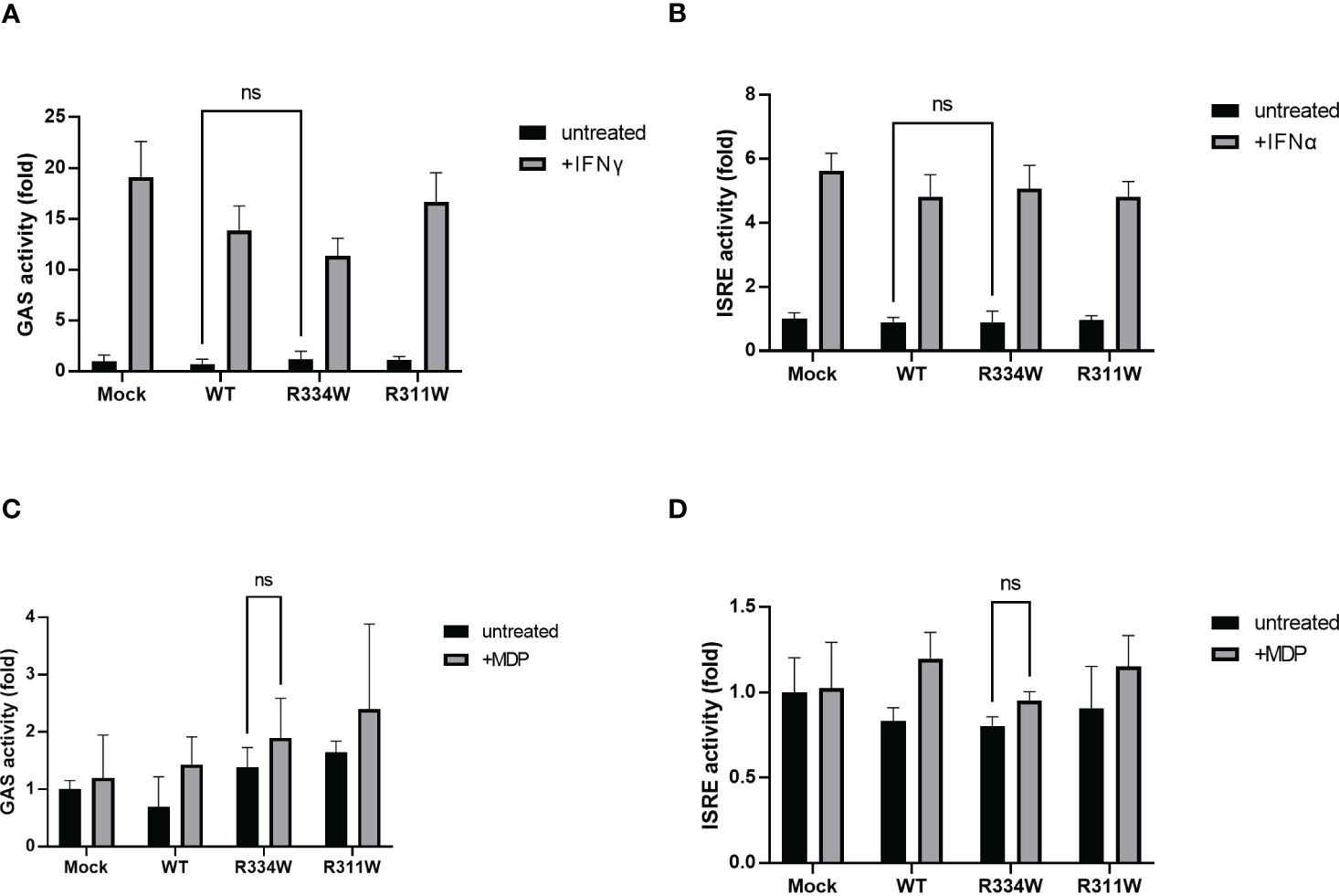
Figure 2 There was no significant difference in GAS or ISRE activation, as evaluated by a dual luciferase reporter system. The relative fold-change from the untreated condition of Mock (set as 1) is shown. Bars indicate means ± SDs (n=3). This test was repeated at least three times, and the results of one representative test are shown, but the same results were obtained each time. (A) Cells were treated with IFNγ (50 ng/mL) as a positive control for the activation of the GAS reporter. (B) Cells were treated with IFNα (1000 U/mL), as a positive control for the activation of the ISRE reporter. (C, D) Cells were treated with MDP (10 µg/mL) for 24 (h) Cells were treated with MDP (10 µg/mL) for 24 (h), ns. not significant.
To evaluate the anti-inflammatory effect of tofacitinib in Blau syndrome, we focused our attention on the upstream pathway of NOD2. For this evaluation, we used iPS-ML cell lines established from a Blau syndrome patient; one line had the p.R334W mutation in NOD2 and the other mutation was corrected back to WT by CRISPR-Cas9–mediated targeting. qPCR experiments in both iPS-ML cell lines showed that stimulation with IFNγ for 2 h induced NOD2, regardless of the presence or absence of the Blau syndrome-associated mutation (Figure 3A). Similar results were obtained in our previous experiments with macrophages generated by culturing iPS-ML cells in RPMI-1640 medium supplemented with 10% FCS containing 100 ng/mL M-CSF for 7 days (16, 23). The same results were observed when IFNγ was used to induce NOD2 in iPS-ML cells established from the control iPS line 201B7E1 (data not shown). In the iPS-ML cells from 201B7E1, we previously confirmed that NOD2 expression was induced by IFNγ using macrophages induced by further differentiation, similar to observations obtained with iPS-ML cells generated from Blau syndrome patient-derived iPS cells (16). These results suggested that it was possible to evaluate the effect of tofacitinib on the NOD2-mediated inflammatory pathway associated with Blau syndrome without differentiating iPS-ML cells further into macrophages.
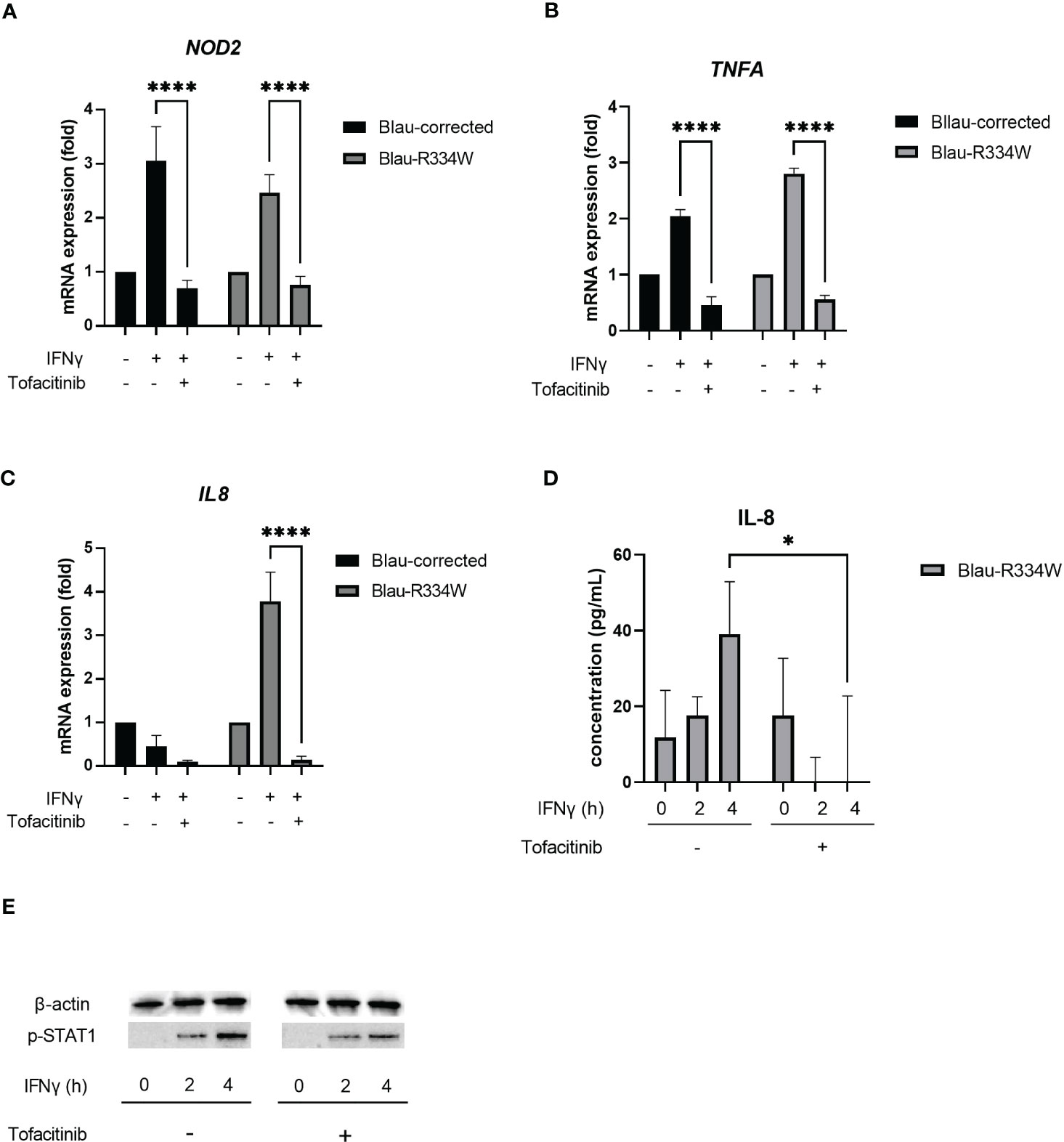
Figure 3 Tofacitinib downregulated NOD2 induction, inflammatory cytokine production and phosphorylation of STAT1 by IFNγ. This test was repeated at least three times, and the results of one representative test are shown, but the same results were obtained each time. Bars indicate means ± SDs (n=3). *p <.05, ****p <.0001. Quantitative RT-PCR validation of NOD2 (A), TNFA (B) and IL8 expression (C) in the patient-derived iPS-MLs with corrected wild-type NOD2 (Blau-corrected) or disease-associated mutant NOD2 (Blau-R334W). Cells were treated with IFNγ (50 ng/mL) for 6 h with or without tofacitinib (1 µM). In Quantitative RT-PCR, the relative fold-change from each untreated condition (set as 1) is shown. (D) Secretion of cytokine IL-8 from Blau-R334W evaluated by ELISA. (E) Phosphorylation of STAT1 (pSTAT1) by Western Blotting.
In summary, tofacitinib suppressed the expression of NOD2 induced by IFNγ in all types of cells, regardless of the presence or absence of the p.R334W mutation in NOD2 (Figure 3A).
Next, we examined the proinflammatory cytokines produced by iPS-ML cells. Along with the induction of NOD2 expression by IFNγ treatment, TNFA expression was induced not only in cells with the p.R334W mutation associated with Blau syndrome, but also in WT cells (Figure 3B). This IFNγ-induced expression of TNFA was suppressed by tofacitinib.
In contrast, expression of IL8, also known as CXCL8, was observed only in mutant iPS-ML cells with the p.R334W mutation, suggesting that IL8 is a suitable indicator of mutant NOD2 activation (Figure 3C). The increased expression of IL8 was attenuated by the addition of 1 μM tofacitinib.
Upon induction of IL8 expression, sufficient amounts of IL-8 were released into the cell supernatant in mutant iPS-ML cells 4 h after IFNγ treatment, and this production was suppressed by tofacitinib treatment (Figure 3D). When STAT1 phosphorylation (pSTAT1) was confirmed by western blotting, pSTAT1 was not observed before IFNγ stimulation in the mutant iPS-ML cells, but only after IFNγ treatment. This induction was observed to be suppressed by tofacitinib treatment, although not completely (Figure 3E). These results confirm our findings that tofacitinib reduces inflammatory cytokines in Blau syndrome by suppressing NOD2 expression.
JAK inhibitors are commonly used for the treatment of inflammatory diseases such as rheumatoid arthritis. Recently, those inhibitors have attracted interest as therapies for inflammatory skin disorders, such as atopic dermatitis and psoriasis. Furthermore, there are reports suggesting their efficacy against granulomatous diseases (18, 19, 24–27) and, very recently, Blau syndrome (20).
The molecular mechanism of granulomatosis is unknown. One report showed that a patient with cutaneous sarcoidosis was successfully treated with tofacitinib. Moreover, staining for pSTAT1 and pSTAT3 was significantly higher in samples obtained from patients with sarcoidosis than in normal control skin samples. In addition, staining for pSTAT1 was strongest in the center of the granuloma and more prominent in macrophages than in T cells, whereas staining for pSTAT3 was strongest in the nuclei of lymphocytes within and between granulomas (14). However, precisely how a JAK inhibitor suppresses cutaneous granuloma formation is unclear.
As a NOD2-dependent autoinflammatory granulomatosis, Blau syndrome is a good model to investigate the mechanism of granuloma formation. We evaluated the effect of tofacitinib on Blau syndrome associated with pro-inflammatory responses. Tofacitinib did not suppress the increased transcriptional activity of NF-κB induced by mutant NOD2. In contrast, IFNγ induced NOD2 expression both in the presence and absence of mutations. Instead, expression of NOD2 results in the production of pro-inflammatory cytokines by an autoinflammatory mechanism in cells with mutant NOD2. Tofacitinib showed anti-inflammatory effects by suppressing the expression of NOD2. This suggests that tofacitinib will not affect granulomas once they have formed. However, it is unclear how granulomas are maintained. If persistent inflammation is required, this drug may be effective in some situations by suppressing NOD2 expression and preventing the development of inflammation. In Blau syndrome, the skin rash goes through periodic exacerbations and remissions over a period of several months, with some cases spontaneously regressing with age. Thus, even if the therapeutic strategy is to suppress NOD2 expression, if it is effective in preventing inflammatory flare-ups, it may be effective in preventing the maintenance of granulomas as a result. Based on our experimental results and these discussions, we present the mechanism of action of tofacitinib in Blau syndrome as Figure 4.
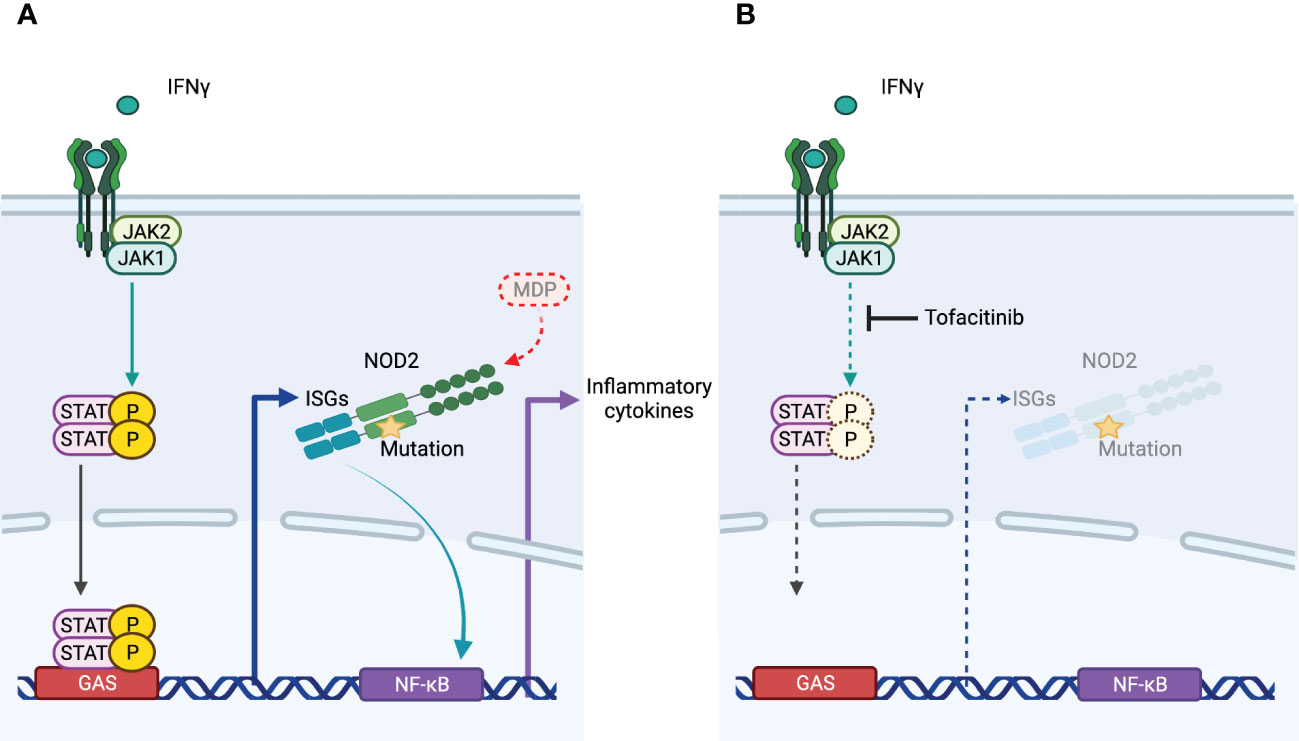
Figure 4 The mechanism of action of tofacitinib in Blau syndrome. (A) NOD2 expression is induced by IFNγ. NOD2 with mutations associated with Blau syndrome is activated in the absence of its ligand MDP, leading to increased transcription of NF-κB and production of proinflammatory cytokines. (B) Tofacitinib did not inhibit downstream signaling of NOD2. As an inhibitor of JAK, which acts downstream of IFNγ, resulting in the suppression of IFNγ-induced NOD2 expression, the inflammation of Blau syndrome is regulated.
Precisely how NOD2 activation induces granuloma formation in Blau syndrome remains to be elucidated. In this study, we focused on ex vivo studies using a luciferase assay system overexpressing the NOD2 mutant and iPS-ML cells differentiated from iPS cells derived from a Blau syndrome patient. In this respect, it would be of interest to isolate and culture monocytes from the blood of patients and perform similar experiments. However, most patients with Blau syndrome are children, and frequent invasive testing is not practical. The fact that we were able to elucidate the effects of tofacitinib without performing invasive procedures is an advantage of using patient-derived iPS cells.
The second limitation of this study is that a variety of immune cells, including cells resident in the tissue, may be involved in the formation of granulomas. Therefore, it will be necessary to examine whether tofacitinib affects immune cells within the granuloma, especially those that may produce IFNγ in the tissue. In addition, as an indicator of inflammatory cytokines in this study we used IL-8, a chemokine that induces neutrophil migration. However, the extent to which IL-8 and neutrophil migration are actually involved in the formation of granulomas that characterize Blau syndrome has not yet been addressed.
In this study, we demonstrated that tofacitinib suppresses inflammatory cytokine production by inhibiting the expression of NOD2. We hope that tofacitinib will become a treatment option for Blau syndrome in the future and that the mechanism of granuloma formation will be elucidated.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by CiRA18-06 in Kyoto University and 2017104 in Kansai Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
YU, RT-I, and NK designed the experiments. MKS and HT supported the planning and validated the strategy. YU and RT-I performed experiments. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
This work was supported in part by the Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine (JP21bm0104001) from AMED (MS), the Acceleration Program for Intractable Diseases Research Utilizing Disease-Specific iPS Cells (22bm0804004h0006) from AMED (MS), JSPS KAKENHI Grant Number 19K08784 and 22K08380 (NK), a research grant from MHLW (NK), and a Lexi’s Legacy Research Grant from Cure Blau Syndrome Foundation (NK).
We would like to thank Drs. Y. Takahashi and Y. Takakura (Kyoto University Graduate School of Pharmaceutical Sciences) for kindly providing us GAS and ISRE reporter plasmid for luciferase assay, Drs S. Senju (Kumamoto University) and H. Miyoshi (RIKEN Bio Resource Center) for cMYC, BMI1, and MDM2 in the CSII-EF-RfA vector, and Dr. S. Yamanaka (Kyoto University) for the control iPS line 201B7E1. We also thank all the patients, their parents, and volunteers for participating in the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with blau syndrome. Blood (2005) 105(3):1195–7. doi: 10.1182/blood-2004-07-2972
2. Sarens IL, Casteels I, Anton J, Bader-Meunier B, Brissaud P, Chédeville G, et al. Blau syndrome-associated uveitis: preliminary results from an international prospective interventional case series. Am J Ophthalmol (2018) 187:158–66. doi: 10.1016/j.ajo.2017.08.017
3. Rose CD, Pans S, Casteels I, Anton J, Bader-Meunier B, Brissaud P, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatol (Oxford) (2015) 54(6):1008–16. doi: 10.1093/rheumatology/keu437
4. Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in blau syndrome. Nat Genet (2001) 29(1):19–20. doi: 10.1038/ng720
5. Rose CD, Doyle TM, McIlvain-Simpson G, Coffman JE, Rosenbaum JT, Davey MP, et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J Rheumatol (2005) 32(2):373–5.
6. Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr (1985) 107(5):689–93. doi: 10.1016/s0022-3476(85)80394-2
7. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem (2001) 276(7):4812–8. doi: 10.1074/jbc.M008072200
8. Kambe N, Nishikomori R, Kanazawa N. The cytosolic pattern-recognition receptor Nod2 and inflammatory granulomatous disorders. J Dermatol Sci (2005) 39(2):71–80. doi: 10.1016/j.jdermsci.2005.04.001
9. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to crohn's disease. Nature (2001) 411(6837):603–6. doi: 10.1038/35079114
10. Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical characteristics and treatment of 50 cases of blau syndrome in Japan confirmed by genetic analysis of the NOD2 mutation. Ann Rheum Dis (2020) 79(11):1492–9. doi: 10.1136/annrheumdis-2020-217320
11. Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S, et al. Role of the NOD2 genotype in the clinical phenotype of blau syndrome and early-onset sarcoidosis. Arthritis Rheum (2009) 60(1):242–50. doi: 10.1002/art.24134
12. Yao Q, Zhou L, Cusumano P, Bose N, Piliang M, Jayakar B, et al. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther (2011) 13(5):R148. doi: 10.1186/ar3462
13. McDonald C, Shen M, Johnson EE, Kabi A, Yao Q. Alterations in nucleotide-binding oligomerization domain-2 expression, pathway activation, and cytokine production in yao syndrome. Autoimmunity (2018) 51(2):53–61. doi: 10.1080/08916934.2018.1442442
14. Yao Q, Su LC, Tomecki KJ, Zhou L, Jayakar B, Shen B. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol (2013) 68(4):624–31. doi: 10.1016/j.jaad.2012.09.025
15. Yao Q, Shen M, McDonald C, Lacbawan F, Moran R, Shen B. NOD2-associated autoinflammatory disease: a large cohort study. Rheumatol (Oxford) (2015) 54(10):1904–12. doi: 10.1093/rheumatology/kev207
16. Takada S, Kambe N, Kawasaki Y, Niwa A, Honda-Ozaki F, Kobayashi K, et al. Pluripotent stem cell models of blau syndrome reveal an IFN-gamma-dependent inflammatory response in macrophages. J Allergy Clin Immunol (2018) 141(1):339–49.e11. doi: 10.1016/j.jaci.2017.04.013
17. Arakawa A, Kambe N, Nishikomori R, Tanabe A, Ueda M, Nishigori C, et al. 2 mutation-associated case with blau syndrome triggered by BCG vaccination. Children (Basel) (2021) 8(2). doi: 10.3390/children8020117
18. Damsky W, Thakral D, Emeagwali N, Galan A, King B. Tofacitinib treatment and molecular analysis of cutaneous sarcoidosis. N Engl J Med (2018) 379(26):2540–6. doi: 10.1056/NEJMoa1805958
19. Damsky W, Thakral D, McGeary MK, Leventhal J, Galan A, King B. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol (2020) 82(3):612–21. doi: 10.1016/j.jaad.2019.05.098
20. Zhang S, Cai Z, Mo X, Zeng H. Tofacitinib effectiveness in blau syndrome: a case series of Chinese paediatric patients. Pediatr Rheumatol (2021) 19(1). doi: 10.1186/s12969-021-00634-x
21. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi: 10.1126/science.8197455
22. Miyakawa N, Nishikawa M, Takahashi Y, Ando M, Misaka M, Watanabe Y, et al. Prolonged circulation half-life of interferon gamma activity by gene delivery of interferon gamma-serum albumin fusion protein in mice. J Pharm Sci (2011) 100(6):2350–7. doi: 10.1002/jps.22473
23. Kitagawa Y, Kawasaki Y, Yamasaki Y, Kambe N, Takei S, Saito MK. Anti-TNF treatment corrects IFN-gamma-dependent proinflammatory signatures in blau syndrome patient-derived macrophages. J Allergy Clin Immunol (2021) 149(1):176–88.e7. doi: 10.1016/j.jaci.2021.05.030
24. Friedman MA, Le B, Stevens J, Desmarais J, Seifer D, Ogle K, et al. Tofacitinib as a steroid-sparing therapy in pulmonary sarcoidosis, an open-label prospective proof-of-Concept study. Lung (2021) 199(2):147–53. doi: 10.1007/s00408-021-00436-8
25. Kerkemeyer KL, Meah N, Sinclair RD. Tofacitinib for cutaneous and pulmonary sarcoidosis: a case series. J Am Acad Dermatol (2021) 84(2):581–3. doi: 10.1016/j.jaad.2020.10.016
26. Singh K, Wang A, Heald P, McNiff JM, Suozzi K, King B, et al. Treatment of angiolupoid sarcoidosis with tofacitinib ointment 2% and pulsed dye laser therapy. JAAD Case Rep (2021) 7:122–4. doi: 10.1016/j.jdcr.2020.11.016
Keywords: autoinflammatory disease, Blau syndrome, NOD2, JAK-STAT signaling pathway, tofacitinib, IFNγ
Citation: Ueki Y, Takimoto-Ito R, Saito MK, Tanizaki H and Kambe N (2023) Tofacitinib, a suppressor of NOD2 expression, is a potential treatment for Blau syndrome. Front. Immunol. 14:1211240. doi: 10.3389/fimmu.2023.1211240
Received: 24 April 2023; Accepted: 06 June 2023;
Published: 21 June 2023.
Edited by:
Christine McDonald, Cleveland Clinic, United StatesReviewed by:
Thomas El Jammal, Hospices Civils de Lyon, FranceCopyright © 2023 Ueki, Takimoto-Ito, Saito, Tanizaki and Kambe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naotomo Kambe, bmthbWJlQGt1aHAua3lvdG8tdS5hYy5qcA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.