 Hongfan Zhao1‡
Hongfan Zhao1‡ Dashan Wu1‡
Dashan Wu1‡ Michael Adu Gyamfi1
Michael Adu Gyamfi1 Pinchao Wang1
Pinchao Wang1 Christian Luecht1
Christian Luecht1 Anna Maria Pfefferkorn2
Anna Maria Pfefferkorn2 Muhammad Imtiaz Ashraf2
Muhammad Imtiaz Ashraf2 Julian Kamhieh-Milz3
Julian Kamhieh-Milz3 Janusz Witowski4
Janusz Witowski4 Duska Dragun1†
Duska Dragun1† Klemens Budde1
Klemens Budde1 Ralf Schindler1
Ralf Schindler1 Daniel Zickler1
Daniel Zickler1 Guido Moll1,5*§‖
Guido Moll1,5*§‖ Rusan Catar1*§‖
Rusan Catar1*§‖- 1Department of Nephrology and Internal Intensive Care Medicine, at Charité Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Berlin, Germany
- 2Department of Surgery, at Charité Universitätsmedizin Berlin, Berlin, Germany
- 3Institute of Transfusion Medicine, at Charité Universitätsmedizin Berlin, Berlin, Germany
- 4Department of Pathophysiology, Poznan University of Medical Sciences, Poznan, Poland
- 5BIH Center for Regenerative Therapies (BCRT) and Berlin-Brandenburg School for Regenerative Therapies (BSRT), at Charité Universitätsmedizin Berlin, Berlin, Germany
Aims: Expanded hemodialysis (HDx) therapy with improved molecular cut-off dialyzers exerts beneficial effects on lowering uremia-associated chronic systemic microinflammation, a driver of endothelial dysfunction and cardiovascular disease (CVD) in hemodialysis (HD) patients with end-stage renal disease (ESRD). However, studies on the underlying molecular mechanisms are still at an early stage. Here, we identify the (endothelial) transcription factor Krüppel-like factor 2 (KLF2) and its associated molecular signalling pathways as key targets and regulators of uremia-induced endothelial micro-inflammation in the HD/ESRD setting, which is crucial for vascular homeostasis and controlling detrimental vascular inflammation.
Methods and results: First, we found that human microvascular endothelial cells (HMECs) and other typical endothelial and kidney model cell lines (e.g. HUVECs, HREC, and HEK) exposed to uremic serum from patients treated with two different hemodialysis regimens in the Permeability Enhancement to Reduce Chronic Inflammation II (PERCI-II) crossover clinical trial - comparing High-Flux (HF) and Medium Cut-Off (MCO) membranes - exhibited strongly reduced expression of vasculoprotective KLF2 with HF dialyzers, while dialysis with MCO dialyzers led to the maintenance and restoration of physiological KLF2 levels in HMECs. Mechanistic follow-up revealed that the strong downmodulation of KLF2 in HMECs exposed to uremic serum was mediated by a dominant engagement of detrimental ERK instead of beneficial AKT signalling, with subsequent AP1-/c-FOS binding in the KLF2 promoter region, followed by the detrimental triggering of pleiotropic inflammatory mediators, while the introduction of a KLF2 overexpression plasmid could restore physiological KLF2 levels and downmodulate the detrimental vascular inflammation in a mechanistic rescue approach.
Conclusion: Uremia downmodulates vasculoprotective KLF2 in endothelium, leading to detrimental vascular inflammation, while MCO dialysis with the novel improved HDx therapy approach can maintain physiological levels of vasculoprotective KLF2.
1 Introduction
Chronic kidney disease (CKD) and end-stage renal disease (ESRD) are a major public health burden despite recently improved global awareness and better access to kidney care (1–4). In addition to kidney transplantation (KTx) (5), several renal replacement therapies (RRTs), such as peritoneal dialysis (PD) (6) and hemodialysis (HD) (7), offer well-established life-sustaining therapies for ESRD patients (4), which are nonetheless still associated with a considerable need for improvements considering their long-term morbidity and mortality outcomes (8–12).
Even under optimal care, both, CKD and in particular more advanced ESRD patients, suffer from multiple strong cardiovascular disease (CVD) risk factors, which are a leading cause for the underlying high morbidity and mortality (13). These complications can project into the manifestation of chronic microvascular injury, which in turn promotes the cardiovascular and post-inflammatory risks and detrimentally affects patient morbidity and mortality in the long run (14, 15). Thus, in order to optimize existing RRT treatments, intense research is currently ongoing to stepwise overcome any evident RRT-related chronic complications (8–12).
In the case of HD, where uremic toxins are removed via membrane-filter systems, uremic interaction with the endothelium and incomplete removal of uremic macromolecules promotes endothelial dysfunction and systemic microinflammation (15, 16). However, the novel expanded hemodialysis therapy (HDx) approach (9–11), which aims for a more efficient removal of middle-sized uremic toxins through use of novel improved middle cut-off (MCO) dialyzers (17), may offer better endothelial protection than the traditional high-flux (HF) dialyzers (7, 17, 18). Thus, more detailed mechanistic studies in this scientific context are of great interest.
As shown in the PERCI-II-MCO study (NCT02084381) (17), compared to HF-dialyzed patients the uremic serum from MCO-dialyzed patients exhibits reduces levels of typical pro-inflammatory mediators and less maladaptive angiogenesis in functional readouts (7, 17). In the current study, we have investigated in detail the role of Krüppel-like factor 2 (KLF2), an important zinc-finger transcription factor expressed in endothelial cells (ECs) (19, 20), considering its putative role in regulating and maintaining vascular homeostasis in the HDx setting.
KLF2 maintains vascular homeostasis and protection against endothelial dysfunction and atherosclerosis by multiple mechanisms (20), e.g. by blocking leukocyte adhesion to endothelium, by inhibiting endothelial inflammation, and by modulating thrombotic pathways and detrimental vascular calcification (20–24). Importantly, KLF2 exerts inhibitory effects on various pro-inflammatory stimuli, such as interleukin-1β (IL-1β), tumor-necrosis-factor-α (TNF-α), transforming-growth-factor-β (TGF-β), and thrombin, thus suggesting a pleiotropic anti-inflammatory and vasculo-protective effect (20, 23, 25). In turn, KLF2-suppression can promote endothelial inflammation and damage of glomerular endothelium (20, 26, 27).
On the transcriptional level, KLF2 has been shown earlier to suppress pulmonary fibrosis and inflammation, and to strongly impede TGF-β signals in the endothelium, through the inhibition of the activator protein 1 (AP-1) (28, 29). However, the potential role of KLF2 in uremia-induced endothelial inflammation, maladaptation, and dysfunction has not been studied in detail yet. Indeed, the modulation of vasculo-protective KLF2 may provide both, an interesting prognostic to either monitor the impact of inflammation and vascular health in advanced CKD/ESRD patients, but also an interesting therapeutic target to restore vascular health and integrity in uremic patients within vascular crisis.
In this study, we have investigated in detail the effect of uremia-induced regulation of KLF2 on the impact of pro-inflammatory mediators and endothelial function. First of all, we found that endothelial KLF2 expression is strongly downmodulated by uremia compared to healthy serum. Mechanistically, we have explored the underlying signaling mechanisms, and indicating that KLF2 signaling is mediated via regulation of AP-1. Eventually, we found that KLF2 expression differs in ECs exposed to uremic serum from patients dialyzed with conventional HF membranes compared to novel MCO dialyzers, thus associating with a reduced release of proinflammatory mediators with the novel improved HDx therapy.
2 Methods
2.1 Study patient description and serum samples
The uremic serum samples were obtained during the Permeability Enhancement to Reduce Chronic Inflammation-II clinical trial (PERCI-II-MCO; ClinicalTrials.gov NCT02084381; https://clinicaltrials.gov/ct2/show/NCT02084381) (17). The clinical study was conducted in accordance with the ethical principles of the Declaration of Helsinki and approved by the Ethics Committees of the Martin-Luther-University Halle-Wittenberg and the Charité Berlin, and written informed consent was given prior to inclusion of subjects into the study, as outlined previously (7, 17). The mean age of the patients in the PERCI-II study was as follows: MCO-first (58.1 +/- 16.5 years, n=23 patients) and HF-first (59.8 +/- 16.5 years, n=25). More detailed study patient parameters, serum sampling procedures, and its experimental use are described in greater detail in our prior publications (7, 17). Briefly, for mechanistic experiments, samples of 20 patients were pooled at equal volumes to obtain a uremic serum pool (USP) and non-uremic serum from 14 healthy donors (Age 36 ± 9.2 years; nine males, five females) was collected at our department, to generate a healthy serum pool (HSP) for comparison (30).
2.2 Endothelial cell culture and experimental readout
The disposable cell culture plastic materials were purchased from Becton Dickinson (Falcon; Franklin Lakes, USA) and basic chemicals from Sigma (St Luis, USA). The specific blocking reagents employed in the cell culture experiments were as follows: AP1-blocker (AP1B; SR-11302), AKT-blocker (AKTB; MK-2206), ERK-Blocker (ERKB; PD-184352), all obtained from Tocris Bioscience (Wiesbaden Germany) (6, 7, 25). The basic media and buffers (e.g. phosphate buffered saline; PBS) and penicillin/streptomycin were purchased from Biochrom-AG (Berlin, Germany) and the fetal calf serum (FCS) from Invitrogen (Darmstadt, Germany). The following specific cell types and respective specialized culture media were used for cell culture expansion and different experimental readouts.
1) Human microvascular endothelial cells (HMECs; catalogue no. CRL-3243; purchased from ATCC®, Manassas, VA, USA) were cultured in MCDB131 basic media for HMECs (Invitrogen and Gibco, Waltham, MA, USA) supplemented with 5% FCS, 10 mM L-glutamine, 10 ng/ml recombinant human EGF, 10nM hydrocortisone, and 1% penicillin/streptomycin (100 U/ml and 100 ug/ml, respectively) (31).
2) Human umbilical vein endothelial cells (HUVECs; Cat. No.: P10961, purchased from Innoprot; Bizkaia, Spain; https://innoprot.com) were cultured in Endothelial Growth Medium for HUVECs (Ca No: CCM027 from Biotechne/R&D Systems, www.rndsystems.com) supplemented with 5% Endothelial Growth Supplement (ECGS; containing human FGF-basic, LR3-IGF-1, VEGF165, EGF, heparin, hydrocortisone, l-ascorbic acid, and FCS, as described in detail by the supplier) and 1% penicillin/streptomycin (100 U/ml and 100 ug/ml, respectively).
3) Human reticular endothelial cells (HRECs; Cat. No.: P10880, from Innoprot) were cultured in Endothelial Cell Growth Medium HRECs (Ca No: P60104 from Innoprot), containing endothelial basic medium, supplemented with 5% FCS, 1% of ECGS (as provided by manufacturer), and 1% penicillin/streptomycin (100 U/ml and 100 ug/ml, respectively).
4) Human embryonic kidney cells (HEK293; Cat. No.: CRL-1573, from ATCC®) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Cat. No.: 30-2003 from ATCC®) supplemented with 10% FCS, 2mM GlutaMax (Gibco), 1mM sodium pyruvate, 10mM HEPES, and 100 U/ml penicillin/100 ug/ml streptomycin (31).
For experimental readout, the different ECs were seeded at standardized densities (20.000 cells per well) and cultured in basic medium with 0.5% FCS, supplemented with or without 5% (vol/vol) HSP, USP, or different types of individual uremic patient serum fractions from the MCO study, as described in detail in figure legends and reported earlier (7). The cell viability was assessed by quantifying mitochondrial activity with the water-soluble tetrazolium (WST-8) salt assay according to the manufacturer’s instructions (PromoCell) (30).
2.3 Gene expression analysis with qRT-PCR and Profiler PCR arrays
Gene expression was assessed with reverse transcription and quantitative real-time polymerase chain reaction (qRT-PCR), as outlined earlier (30, 32–37). Total RNA was extracted by using the PerfectPure RNA Cultured Cell Kit (5 Prime, Hamburg, Germany), its concentration and purity was estimated with a spectrophotometer (Nanodrop; Thermo Fisher Scientific), and the RNA reverse transcribed into cDNA with random hexamer primers, and qRT-PCRs performed on a 7500 Fast Block Real-Time PCR system (Applied Biosystems). Human primer sequences can be found in Table 1. The specificity of the qRT-PCR reaction was verified with melting curve analysis and the relative amount of transcript calculated with the cycle threshold method, using the Applied Biosystems 7500 System v.1.2.3 software and gene expression normalized relative to the endogenous reference gene β2-microglobulin (30). The Human Inflammatory Response and Autoimmunity RT2 Profiler PCR Array kit (Qiagen, Cat. No: PAHS-077ZA-6) was used to evaluate the expression of an exploratory panel of n=84 selected genes involved in the inflammatory response, according to the manufacturer’s instructions, as reported earlier (33–35).
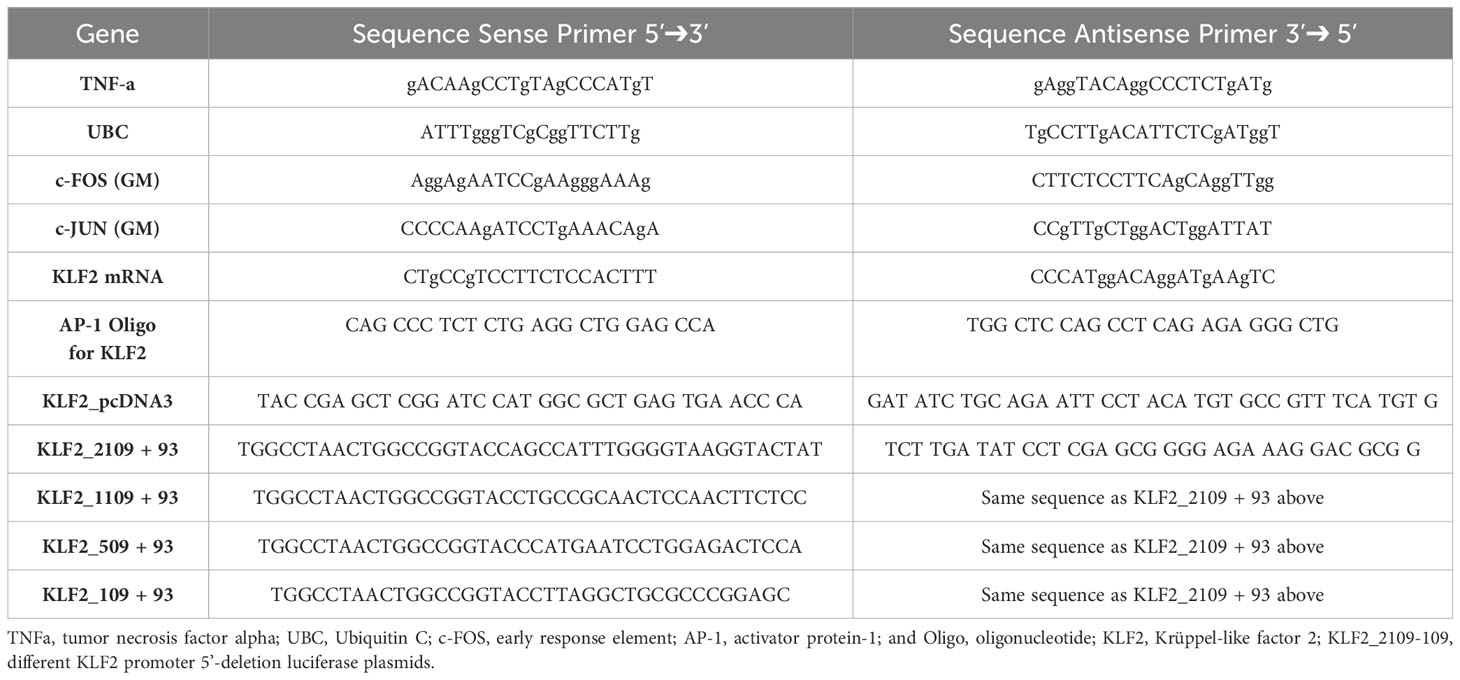
Table 1 Sequences of primers used in quantitative-real-time-PCR analysis.
2.4 DNA constructs, transient transfection and luciferase assay
The DNA constructs of progressive KLF2 5’-putative promoter region deletion fragments (KLF2_2109, KLF2_1109, KLF2_509, and KLF2_109) were cloned into luciferase plasmid vector (pGL4.10, Promega, Madison, WI, USA), and the isolated plasmids were sent for sequencing (LC Genomics, Houston, TX, USA) to ensure the correct length of promoter segments. The HMECs were cultured in 12-well plates at a density that allowed them to reach 70-80% confluence after 24 hours. Transfections were performed using the TurboFect transfection reagent (Fermentas, Darmstadt, Germany) according to the manufacturer’s instructions. The cells were transfected with the constructed reporter plasmid (400ng/well) and co-transfected with the reference pRL-TK renilla plasmid (20ng/well), or the KLF2 overexpression gene, as described earlier (7). Luciferase activity was assessed with the dual-luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Luciferase activity was measured using a microplate luminometer (Fluostar Optima, BMG Labtech, Ortenberg, Germany) and normalized to background levels of Renilla luciferase activity from co-transfected control vectors. The primers sequences are provided in Table 2. The human KLF2 gene promoter region -509 to -110 (Figure S1; GenBank AY738222.1) was analysed with the PROMO virtual laboratory program for the presence and location of potential transcription factor binding sites (http://gene-regulation.com/pub/programs/alibaba2/), accessed on 23 of March 2023 (25).
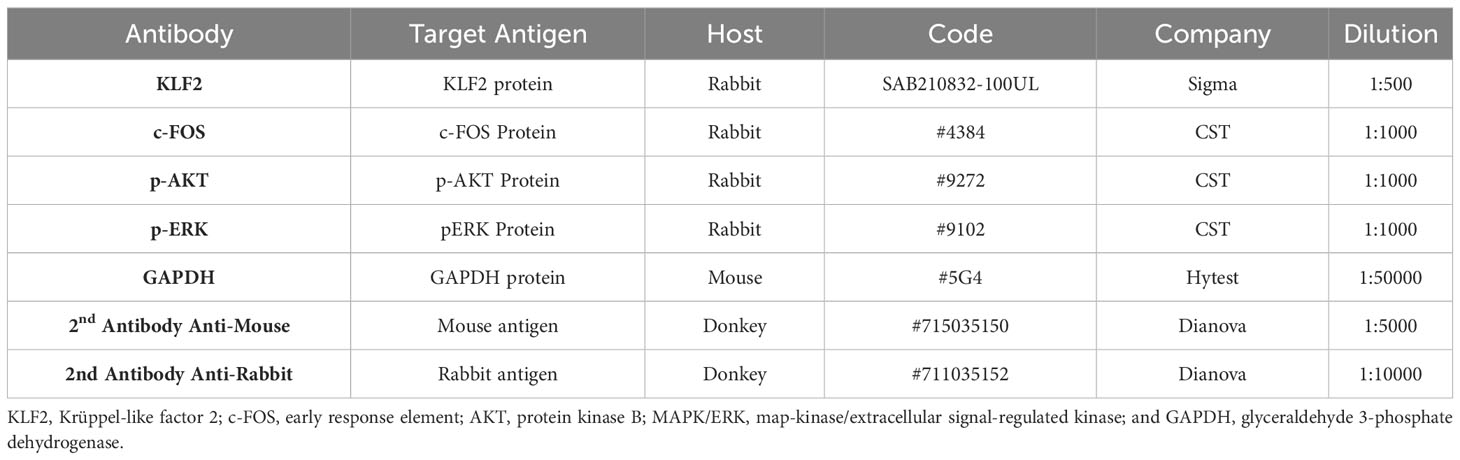
Table 2 Antibodies and reagents used for EMSA and western blot.
2.5 Nuclear extracts, electrophoretic mobility shift assay, and immunoassays
Nuclear extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit and oligonucleotide probes labelled with Biotin 3’ End DNA Labeling Kit (ThermoFisher). For the EMSA (37), the following probe was used (promoter region provided in parentheses): AP-1 - 5’-CAGGCTTCACTGAGCGTCCGCAG-3’ (-441 to -464) (Table 2). Each EMSA binding mixture (20 µl) contained 5 µg of nuclear extract, 20 fmol of labeled double-stranded probe, 1 µg of poly (deoxyinosinicdeoxycytidylic) acid, and 2 µl of 10 x reaction buffer, and was incubated at room temperature for 30 min. Protein–DNA complexes were analyzed by electrophoresis in 6% non-denaturing polyacrylamide gels and visualized using a LightShift Chemiluminescent EMSA Kit (ThermoFisher). The Pierce BCA Protein Assay Reagent (Perbio Science, Bonn, Germany) was used to determine the protein concentration for NE-PER and Western Blot analysis.
Cell extracts for KLF2, c-FOS, pAKT and PERK1/2 protein detection in Western blot analysis were prepared as described earlier (38). The cell lysates were electrophoresed on sodium dodecyl sulfate (SDS)-polyacrylamide gels and analyzed by Western blotting using primary antibodies directed against the target proteins KLF2, c-FOS, pAKT and pERK1/2 (Merck-Sigma-Aldrich, Taufenkirchen, Germany, and Cell Signaling Technology, Frankfurt, Germany, respectively) and GAPDH (Hytest, Turku, Finland), and respective secondary peroxidase-conjugated IgG (Dianova, Hamburg, Germany) (Table 2). The bands were visualized with an Enhanced Chemiluminescence Detection System (Thermo Scientific) and analzed Image J 1.43 software (National Institues of Health, Bethesda, MD, USA). The levels of IL-6, IL-8, and TNF-α in the supernatant of HMEC-conditioned media were measure by enzyme-linked immune assay (ELISA) using respective antibody pair buffer kits (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
2.6 Statistical analysis
Data are expressed as mean ± SEM. Non-parametric data are presented as medians. Statistical analysis and visualization was performed using GraphPad Prism (GraphPad®, San Diego, US) and R (version 3.5.1). Analyses of multiple variables were performed by one-way analysis of variance with Student–Newman–Keuls post- or Kruskal–Wallis with Müller–Dunn post-test. A P-value < 0.05 was considered statistically significant.
3 Results
3.1 Uremic serum reduces the transcription and expression of KLF2 in HMECs
The expression of KLF2 in ECs confers endothelial integrity by regulating anti-inflammatory and antithrombotic effects (21), while reduced transcriptional activity of KLF2 exacerbates chronic inflammation and endothelial damage in renal diseases (39). Thus, it is of great interest to study in detail the signaling events and functional effects related to KLF2.
For this purpose, we established a model system (Figure 1), to study the KLF2 response of microvascular HMECs to uremic serum pool (USP) from ESRD patients, as compared to healthy serum pool (HSP), at both the transcriptional mRNA and the translational protein level, employing USP from a well-defined clinical study cohort that has tested novel medium cut-off (MCO) dialyzers with increased molecular cut-off (Figures 1A, B) (7, 17, 18).
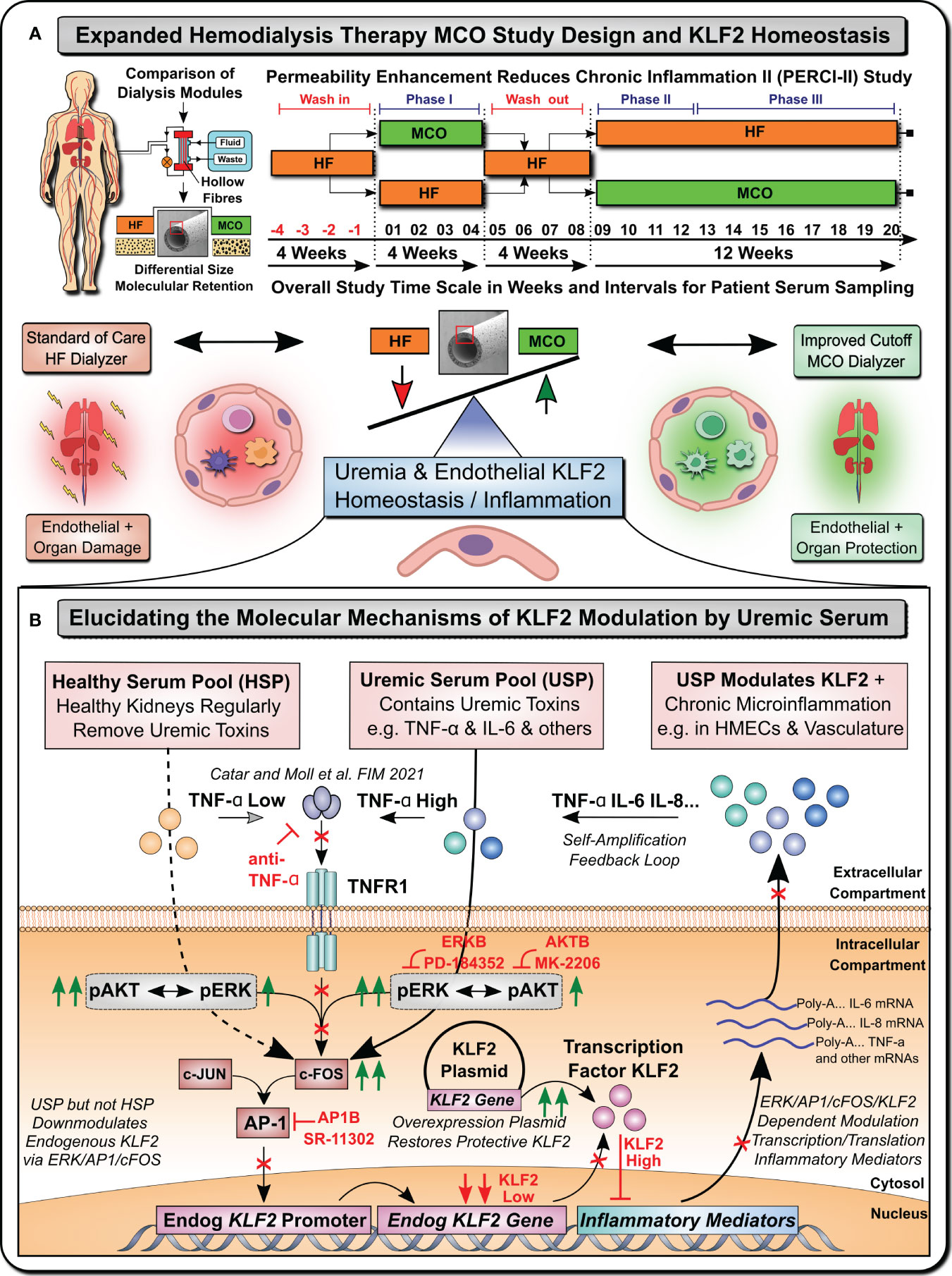
Figure 1 Uremia Modulates Vasculoprotective KLF2 Expression (Graphical Abstract). (A) Expanded Hemodialysis Therapy (HDx) MCO Study Design and KLF2 Homeostasis: Recently, particular attention has been placed into lowering chronic treatment-associated adverse cardiovascular diseases (CVD) and new optimized treatment concepts, such as “Expanded Hemodialysis Therapy” with improved molecular cut-off hemodialyzers (7, 9, 10). Within the PERCI-II study n=48 hemodialysis patients underwent crossover randomized multi-center comparison employing novel medium-cut-off (MCO; MCOI-Ci400, Gambro) dialyzers in comparison to standard of care high-flux (HF) hemodialyzers (PERCI-II-MCO; ClinicalTrials.gov: NCT02084381) (7, 17). These novel MCO dialyzers have an improved molecular size cut-off, which positively modulates systemic microinflammation and may thus be protective to lower uremia associated endothelial dysfunction and organ damage (7, 17). A key factor in regulating endothelial homeostasis and inflammation is Krüppel-like factor 2 (KLF2), which is the main focus of this mechanistic follow-up study to the PERCI-II clinical trial (7). (B) Elucidating molecular mechanisms of vasculoprotective KLF2 modulation by uremic serum: During the biomarker follow-up of the initial PERCI-II clinical trial KLF2 was identified as a potential target for mechanistic follow-up of the molecular signalling mechanisms underlying KLF2 impairment by uremia associated with the conventional HF dialysis regimen, but its improved KLF2 maintenance or restoration with the novel improved MCO dialyzer approach. For the mechanistic studies, to have sufficient amounts of standardized uremic serum available, we compared in detailed signalling pathway analysis and concomitant blocking experiments the response of microvascular endothelial cells (HMECs) to either healthy serum pools (HSP) or uremic serum pools (USP), as described in more detail in an earlier publication that identified TNF-signalling/triggering as a crucial mediator of USP activation in HMECs (7). In the current study, we first-of-all documented a strong impairment of KLF2 expression and concomitant pleiotropic upregulation of a panel of proinflammatory mediators in response to USP but not HSP, thereby triggering a vicious self-amplification loop to trigger the release of more inflammatory uremic toxins from HMECs (e.g. TNF-a, IL-6 etc). Signalling pathway analysis found that this was mediated by preferential engagement of map-kinase/extracellular signal-regulated kinase (MAPK/ERK) instead of protein kinase B (AKT) signalling by employing the respective molecular inhibitors (ERKB: PD-184352 and AKTB: MK-2206). Next, we found that this is mediated by modulation of activator protein 1 (AP-1) and early response element (c-FOS) transcription factor complex (AP-1/c-FOS) signalling (Employing AP1B: SR-11302) and its concomitant binding to the respective promoter region in endothelial KLF2 gene. Importantly, the introduction of a KLF2 overexpression plasmid could restore physiological KLF2 levels and downmodulate the detrimental vascular inflammation in a mechanistic rescue approach. These findings provide new avenues for molecular targets and treatment modalities to reduce chronic microinflammation in the context of hemodialysis.
First of all, we found a continuous time-dependent downmodulation of KLF2 mRNA in HMECs after 3-24 hour exposure to 5% USP, but not 5% HSP, with a 50% reduction as early as 6 hours post initiation (and 64% decrease by 24 hours), which was thus chosen as the default optimal exposure time for the assessment of changes in KLF2 mRNA transcription in the subsequent experiments (P<0.05 and P<0.1, respectively, Figure 2A).
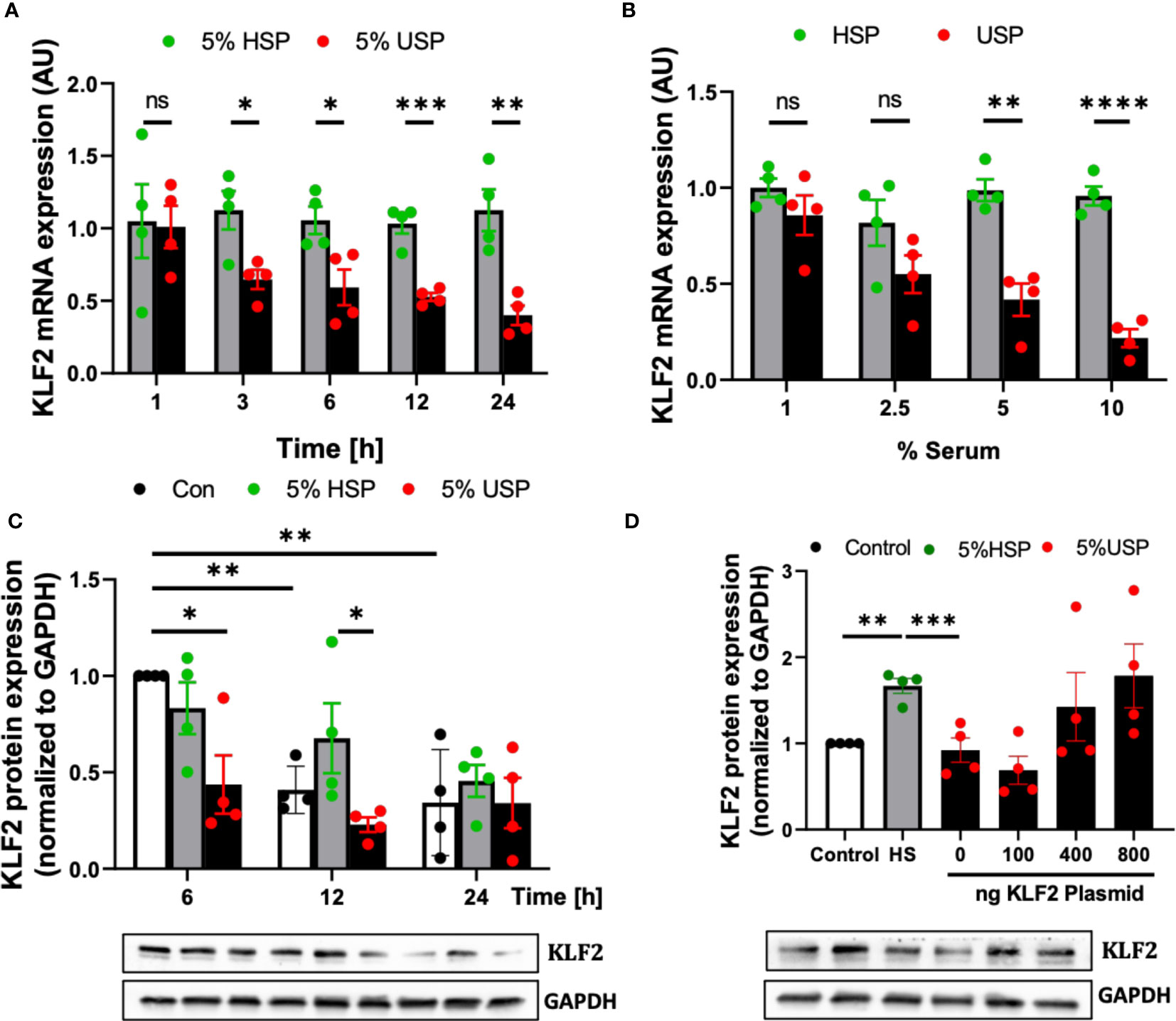
Figure 2 Uremic serum inhibits transcription and translation of KLF2 in HMECs. (A) Time-dependent downregulation of KLF2 upon USP exposure: KLF2 mRNA expression levels detected in HMECs (AU, arbitrary units, n=4) upon stimulation with either 5% healthy serum pool (HSP) or 5% uremic serum pool (USP), with strong downmodulation of KLF2 mRNA at 3-24 hours (50% reduction at 6 hours); (B) Dose-dependent downregulation of KLF2 upon HMEC exposure to USP: KLF2 mRNA expression levels (AU, n=4) after a 6-hours exposure of HMECs to USP or HSP at different serum doses (1%, 2.5%, 5% and 10%), respectively, verifying a strong downmodulation of KLF2 upon exposure to 2.5-10% USP; (C) Western blot quantification of time-dependent expression changes of KLF2 protein: KLF2 protein is shown relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) house-keeping gene control (AU, n=4) upon cell exposure to either 5% HSP or 5% USP for either 6, 12, and 24 hours (optimal differential readout at 6 hours); and (D) Antagonizing KLF2 Downmodulation by USP: Western blot quantification of KLF2 protein expression in HMECs (AU, n=4) after transfection with different concentrations of KLF2 plasmids (100, 400 and 800 ng of KLF2 plasmid) to counteract the KLF2 downmodulation observed upon stimulation with 5% USP. Statistical comparison with ANOVA, Mean ± SEM, with *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, ns, not significant.
Next, we tested HMEC exposure to different doses of serum and found that a 6-hour incubation with 5-10% USP, but not similar doses of HSP, led to a strong downmodulation of KLF2 mRNA transcript (around 50% reduction with 5% USP, P<0.01, and more than 80% reduction with 10% USP, P<0.001, Figure 2B). Due to the scarcity of these samples, we chose 5% USP as our standard working concentrations for the subsequent experiments.
Similarly, the exposure of HMECs to 5% USP resulted in a 50% and 66% reduction in KLF2 protein levels at 6 and 12 hours compared to HSP (Both P<0.5, Figure 2C). These results indicate that USP-exposure represses both, the transcriptional and post-transcriptional activity of KLF2 in endothelial cells, which may ultimately aggravate renal injury.
To better understand the mechanism of USP-induced inhibition of KLF2 activity, an exogenous pcDNA3.0-KLF2 (KLF2-OE) plasmid was constructed for this study and applied in our model to restore the inhibition of KLF2 induced by USP (Figure 2D). An amount of 400 ng of plasmid transfected into 50.000 HMECs was found to be the optimal amount to restore KLF2 protein levels in the USP compared to the HSP group.
3.2 Uremic serum induces KLF2 promoter inhibition via AP-1/c-FOS signaling
KLF2 transcriptional activity can be influenced by many regulatory elements and co-dependent genes, including the ubiquitous activator protein-1 (AP-1/c-FOS) family (40–42). Typical transcription factors and signaling cascades that may influence KLF2 transcription and consequent down-stream-signaling in HMECs may include cellular signaling pathways involving the transcription factor AP-1/c-FOS, but also the protein kinase B (AKT) and map-kinase/extracellular signal-regulated kinase 1/2 (MAPK/ERK1/2) signaling (7, 32).
Employing our well-established luciferase-reporter-system (7), we found that treatment of HMECs containing full-length KLF2 promoter constructs with 5% USP, but not 5% HSP, resulted in a strong decrease of luciferase activity in the USP-group (P<0.001, Figure 3A), while stepwise 5’-deletions of the KLF2 promoter region demonstrate that the deletion of positions from -509 to -109 could fully restore the USP-stimulated inhibitory effect on KLF2 promoter activity (Line 1-3, P<0.001, vs. line 4, ns, Figure 3A). These results suggested that key regulatory elements for KLF2 transcription are located in the -509 to -109 region.
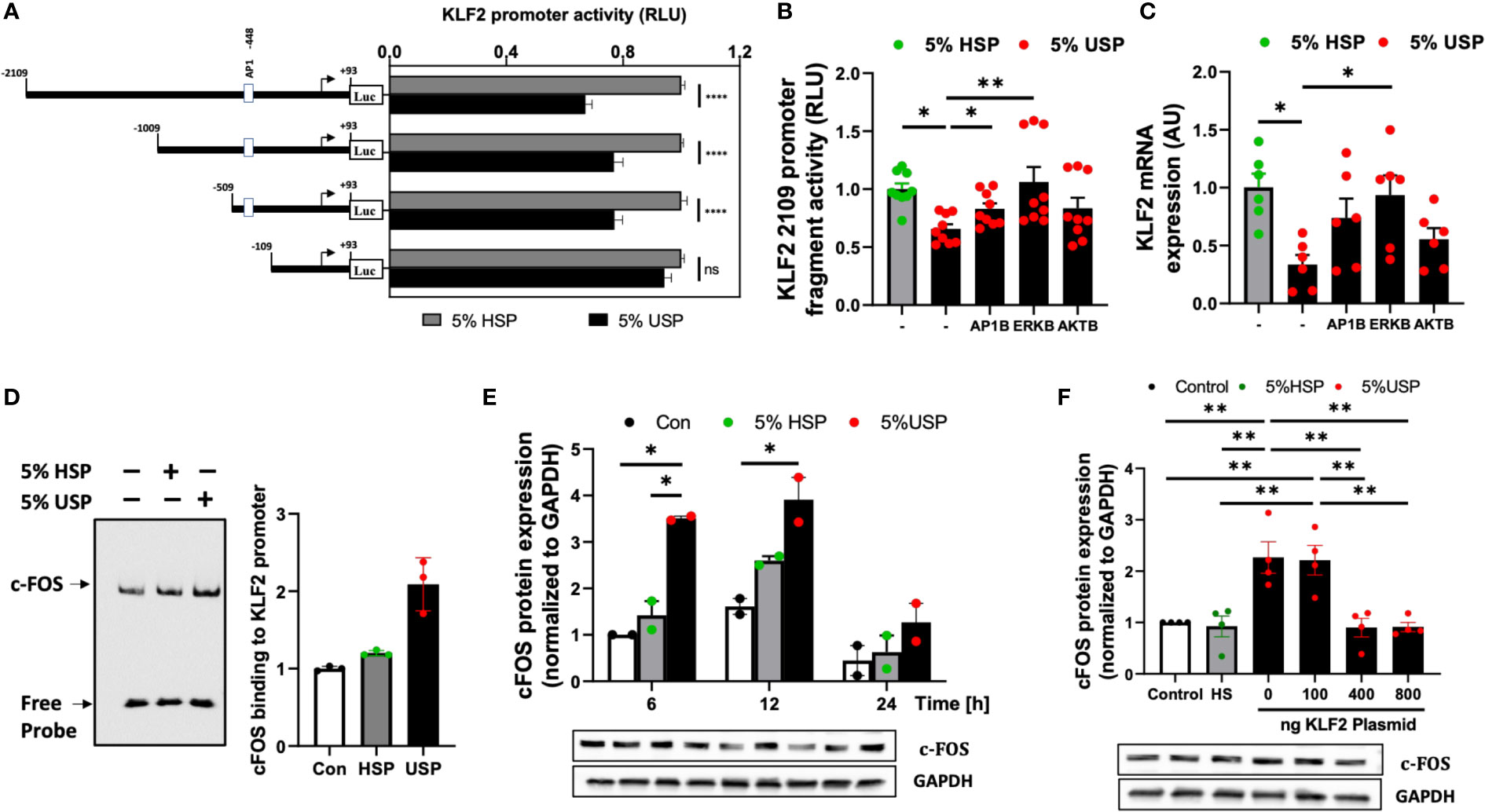
Figure 3 USP-inhibition of KLF2 in HMECs occurs in AP-1/c-FOS dependent manner. (A) KLF2 Promoter Analysis: Luciferase reporter assays indicate a functional loss of KLF2 activity upon deletion of the -509 to -109 promoter region containing the AP1-/c-FOS transcription factor binding site. HMECs were first transfected with different KLF2 reporter plasmid constructs containing progressive promoter region 5’-deletions, employing either the full-length -2109 construct, or different deletion constructs -1009, -509, -109, and +93, and then incubated with either 5% uremic serum pool (USP) or 5% healthy serum pool (HSP) for 6 hours, to quantify KLF2 promoter activity via readout of luciferase activity (RLU, relative luciferase activity, n=12 independent experiments); (B, C) Signaling Pathways: Inhibition of KLF2 expression upon a 6-hour exposure to 5% USP can be partially restored by blocking different signaling pathways: e.g. activator protein 1 (AP1B: SR-11302, 1000 nM), protein kinase B (AKTB: MK-2206, 100 nM), or map-kinase/extracellular signal-regulated kinase (MAPK/ERKB: PD-184352, 100 nM) with respective blocking agents (1-hour preincubation) (7, 25, 30), (B) Luciferase activity of HMECs containing full-length KLF2 reporter constructs (RLU, n=6) and (C) Expression of KLF2 mRNA (AU, arbitrary units, n=6); (D) AP-1/c-FOS transcription factor-binding site in the KLF2 promoter region: Biotin-labelled double-stranded oligonucleotides targeting the calculated AP-1/c-FOS positions -464 to -441 of the corresponding KLF2 promoter region were validated using the gel electrophoresis mobility shift assay (EMSA, one representative experiments is shown); (E) USP exposure induces a time-dependent upregulation of c-FOS expression in HMECs: Western blot analysis to quantify the expression of c-FOS protein in HMECs (AU, relative to GAPDH, n=2) which were left either untreated (baseline), or incubated for different time intervals (6, 12, and 24 hours) with either 5% USP or 5% HSP, demonstrating the strongest differential response at 6-12 hours; and (F) KLF2 overexpression downmodulates c-FOS expression upon USP exposure of HMECs: Different concentrations of KLF2 containing plasmid (100, 400 and 800 ng) were transfected into HMECs, followed by a 6-hour stimulation with 5% USP or 5% HSP, and western blot detection of KLF2 protein expression (AU, relative to GAPDH, n=4). Statistical comparison with ANOVA, Mean ± SEM, with *P<0.05, and **P<0.01, ns, not significant.
Next, we employed AP-1-, AKT-, and ERK-targeted pharmacological blockade to identify the signaling pathways in HMECs transfected with full-length KLF2-2109 promoter construct that are responsive to stimulation with 5% USP vs. 5% HSP baseline control. The blockade of AP-1 and ERK signaling most effectively restored the USP-induced downmodulation of KLF2 promoter activity (P<0.05 and P<0.01, Figure 3B) and KLF2 mRNA expression (P<0.05, Figure 3C), while blockade of AKT was less effective.
This demonstrates that the USP-induced decrease in KLF2 expression and activity that is depended on the promoter regions -509 to -109 bp engages both AP-1 and ERK signaling. Since AP-1 is a combinatorial transcription factor, the observed promoter region of KLF2 (Entailing the -509 to -109 bp promoter region) was analyzed using the PROMO virtual laboratory (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) to determine the DNA-binding switch between AP-1 transcription factor and KLF2 gene (40, 43), and we found that AP-1/c-FOS is a likely candidate could mediate AP-1 action on KLF2.
The specificity of the AP-1/c-FOS binding sites in the KLF2 promoter was confirmed by EMSA (Figure 3D). Semiquantitative EMSA analysis revealed that nuclear extracts from HMECs stimulated with 5% USP contained a DNA-protein complex with more efficient c-FOS probe binding compared to that in the HSP or unstimulated group (Figure 3D). We observed an approximately 2-fold increased intensity of c-FOS-probe in 5% USP-stimulated cells compared to 5% HSP stimulated cells and an approximately 3-fold increased intensity compared to unstimulated HMECs relative to the amount of free probe (P<0.05, Figure 3D). Computational analysis of probe sequences in the EMSA revealed that an AP-1/c-FOS DNA-protein binding site is located at the promoter region -464 to -441 bp (Figure S1).
Time-dependent stimulation of HMECs with 5% USP demonstrated a strong 2-3-fold up-regulation of c-FOS protein compared to stimulation with 5% HSP or untreated baseline, at 6 and 12 hours, but not at 24 hours (First two P<0.05, Figure 3E). Furthermore, we found that transfection and overexpression of 400-800 ng of KLF2 plasmid was effective to reverse the AP1-/c-FOS activity seen with 5% USP stimulation down to the level seen with 5% HSP or baseline control (P<0.01 for both 400 and 800 ng of KLF2 plasmid, Figure 3F).
Altogether, these data indicate that USP-induced inhibition of KLF2 includes binding of AP-1/c-FOS within the KLF2 promoter region -469 to -446 bp (Figures 3A–D), that the AP-1/c-FOS engagement is strongest at 6-12 hours post USP-stimulation (Figure 3E), and that it could be antagonized by overexpression of KLF2 plasmid in HMECs (Figure 3F).
3.3 Uremic serum modulates KLF2 via c-FOS, AKT and MAPK/ERK signaling
Another important component of AP-1 signaling is c-JUN, that can form the alternative transcription factor complex AP1-/c-JUN, as contrasted to the AP-1/c-FOS complex (25, 32, 44). In addition, it is known that ERK activation contributes to EC homeostasis and contributes to the release of inflammatory factors (7, 25, 45), while the role of AKT-signaling in HMECs, is not yet clearly elucidated in the uremic state and conflicting results were reported in the past (46–48).
First, we studied the mRNA expression of c-FOS and c-JUN in response to USP in combination with pharmacological blockers of AKT and MAPK/ERK (Figures 4A, B). We found that c-FOS mRNA was strongly upregulated in response to stimulation with 5% USP (P<0.001, Figure 4A), while c-JUN mRNA was not affected (not significant, Figure 4B). Furthermore, the upregulation of c-FOS mRNA could be inhibited by blocking both AKT and MAPK/ERK signaling pathways (P<0.001, Figure 4A). This indicates that AP-1/c-FOS, but not AP-1/c-JUN, is the key transcription factor complex involved in uremic induction.
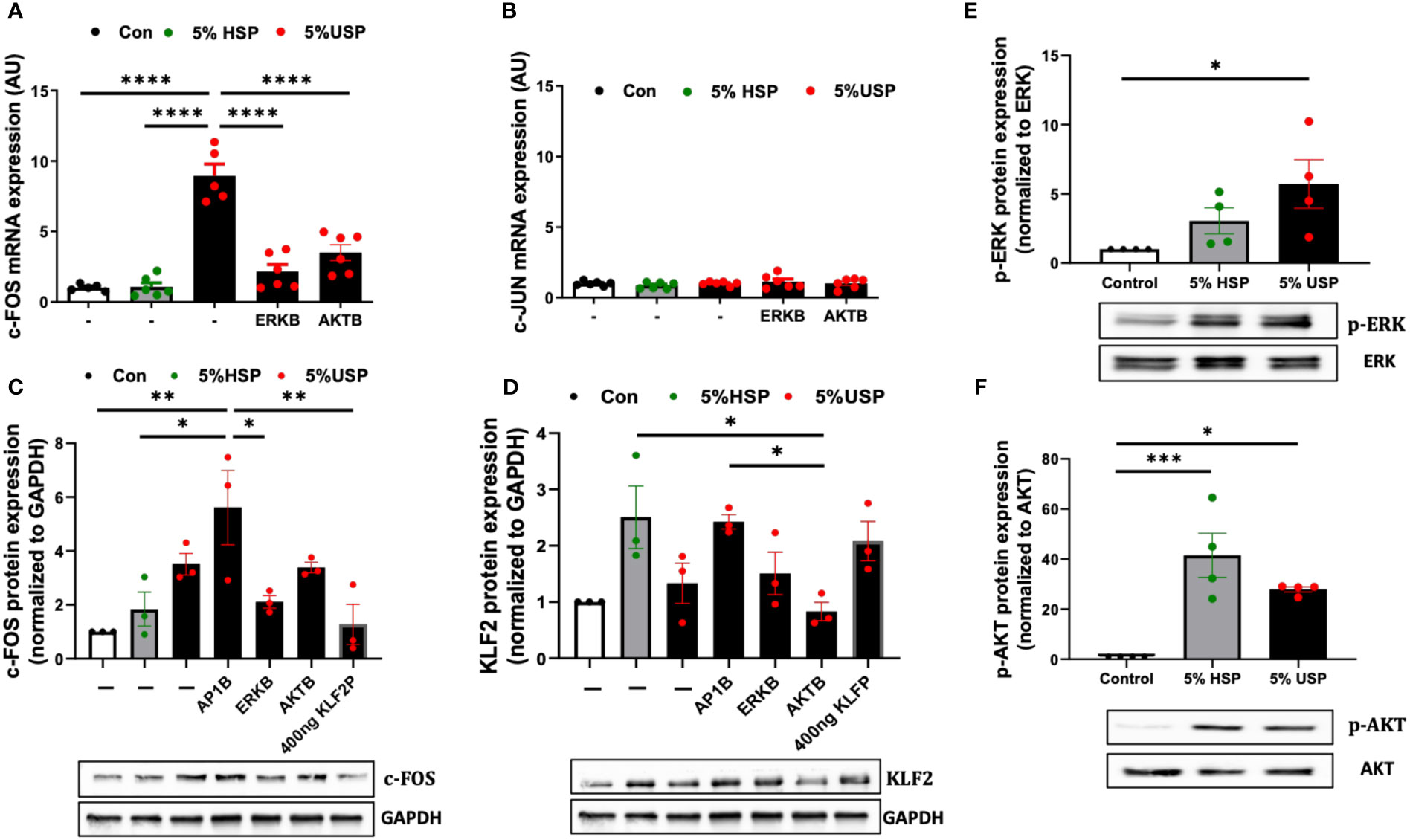
Figure 4 USP-induced KLF2 inhibition engages ERK and AKT signaling via c-FOS. (A, B) Engagement of c-FOS vs. c-JUN: qRT-PCR quantification of mRNA expression levels (AU, n=6) of the key transcription factors c-FOS and c-JUN upon a 6-hour stimulation with either 5% USP or 5% HSP, with or without 1-hour preincubation with blockers of AKT or ERK signaling pathways: protein kinase B (AKTB: MK-2206, 100 nM), or map-kinase/extracellular signal-regulated kinase (MAPK/ERKB: PD-184352, 100 nM), respectively, and (C, D) c-FOS and KLF2: Western blot quantification of protein expression levels for KLF2 and c-FOS (AU, relative to GAPDH, n=3 each) after a 12-hour stimulation with either 5% USP or 5% HSP, following a 1-hour preincubation of HMECs with blocking agents against either activator protein 1 (AP1B: SR-11302, 1000 nM), (AKTB: MK-2206, 100 nM), or (MAPK/ERKB: PD-184352, 100 nM), respectively. (E, F) Engagement of p-AKT and p-ERK: Western blot quantification for detection of phosphorylated p-AKT and p-ERK, normalized relative to their non-phosphorylated forms (Ratio of pERK/ERK and pAKT/AKT, n=4), upon a 12-hour stimulation with either 5% USP or 5% HSP. Statistical comparison with ANOVA, Mean ± SEM, with *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, ns, not significant.
Next, we studied the protein levels of c-FOS and KLF2 in response to USP-stimulation in combination with using specific AP-1, AKT, and MAPK/ERK pathway blocking agents (Figures 4C, D). Interestingly, the preincubation of HMECs with AP-1-blocker resulted in a 2-fold amplification of USP-induced c-FOS protein expression compared to HSP or baseline control (P<0.05 and P<0.01, Figure 4C), and the AP-1-blocker could also completely reverse the USP-induced downmodulation of KLF2 protein expression (Figure 4D).
Similar to preventing upregulation of c-FOS mRNA expression (P<0.001, Figure 4A), the blockade of MAPK/ERK-signaling was more effective in preventing upregulation of c-FOS protein upon USP-induction (P<0.05, Figure 4C), while the AKT-blocker was less effective on c-FOS protein than mRNA level, thus indicating that USP-induced c-FOS signaling is more dependent on ERK than AKT signaling. The upregulation of c-FOS protein by USP could be lowered to HSP baseline by transfection of 400 ng KLF2 plasmid (P<0.01, Figure 4C), which could also fully restore KLF2 protein expression (Figure 4D).
Interestingly, stimulation with 5% USP induced a 5-fold increase in phosphorylation of ERK1/2 compared to baseline (P<0.05, Figure 4E), which was approximately 2-fold higher than for stimulation with 5% HSP. In contrast, stimulation with 5% USP induced a 2-fold lower expression of phosphorylated AKT compared to stimulation with 5% HSP (P<0.05 and P<0.001, Figure 4F). Thus, USP and HSP are acting directly opposed on ERK and AKT phosphorylation in HMECs, which may explain the results obtained in Figures 4A–D.
In summary, the diverse components contained in USP compared to HSP (e.g. higher levels of uremic mediators and toxins with on overall more dysbalanced profile vs. healthy blood environment containing less inflammatory or much less uremic mediators) may result in triggering of multiple signaling cascades (e.g. HMEC response to uremic vs. healthy serum). Thereby, USP and HSP trigger differential AP-1/c-FOS signaling through engaging the KLF2 promoter region, and consequently AKT and MAPK/ERK1/2 canonic signaling pathways, in controlling the expression and regulation of KLF2 expression.
3.4 Uremic serum induces a proinflammatory response in HMECs
Endothelial microinflammation and its beneficial modulation during RRT is critical to the pathophysiology and management of uremia in CKD and ESRD (7). Thus, we studied in representative fashion, the expression changes of a panel of typical inflammatory mediators (e.g. pro/anti-inflammatory cytokines and chemokines) in HMECs in response to stimulation with USP vs HSP, by using the well-defined PCR array technology (33–35).
Out of a panel of 84 target genes (Figure 5A, left panel) a number of proinflammatory activation markers were upregulated in the USP-treated compared to the HSP-treated group (Figure 5A, right panel, cluster analysis: group 2 upregulated transcripts shown in red vs. group 1 unchanged or downmodulated transcripts shown in green, vs. non-treated baseline control group), including targets such as IL-6, CCL2, CCL5, CCL7, CCR2, CXCL1,CXCL8, NFKB1, TLR4 et, while ITGB2 was downregulated compared to the HSP group.
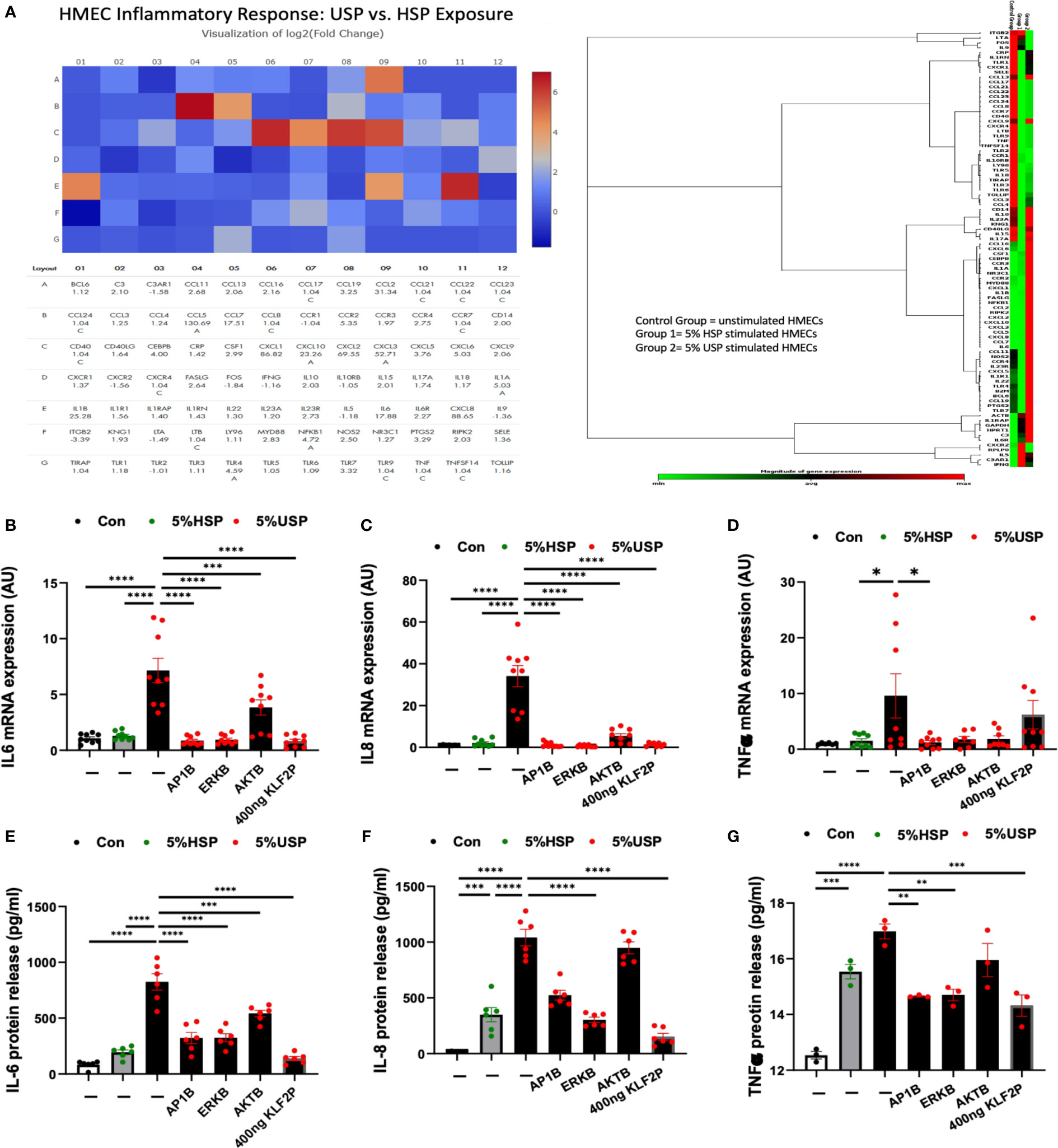
Figure 5 KLF2 modulates USP-induced inflammatory response and cytokine release. (A) QPCR-Array Readout: differential proinflammatory response of HMECs upon a 6-hour exposure to either 5% USP or 5% HSP, depicting on the left a Heatmap visualization of log2-fold change for n=84 selected genes typically involved in the inflammatory response and to the right the corresponding gene expression cluster analysis to verify relevant inflammatory gene families associated with USP vs HSP stimulation in HMECs; and (B–G) Analysis of Key Inflammatory Markers: (B, D, F) qRT-PCR analysis of mRNA expression and (C, E, G) ELISA quantification of cytokine release for three selected key cytokines (IL-6, IL-8, and TNF-α, respectively) measured after a 6-hour or 24-hour stimulation, respectively, with either 5% USP or 5% HSP, with or without 1-hour preincubation of HMECs with AP1B, ERKB and AKTB blockers. Statistical comparison with ANOVA, Mean ± SEM, with *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. ns, not significant.
To quantify the changes in specific targets, we measured representative cytokines on mRNA and protein level (e.g. IL6, IL8 and TNF-a, Figures 5B–G) in the presence or absence of the blocking agents or KLF2 plasmid. Indeed, we could confirm that IL-6, IL8, and TNF-a are strongly upregulated in HMECs upon incubation with 5% USP compared to 5% HSP or untreated baseline, on mRNA level (P<0.05 to P0.0001, Figures 5B, D, F) and also on the protein level (P<0.01 to P<0.0001, Figures 5C, E, G).
Incubation with blocking agents directed against AP-1, AKT, or MAPK/ERK1/2 was effective in downmodulating the proinflammatory cytokine mRNA production (P<0.05 to P<0.0001, Figures 5B, D, F), and in particular AP-1- and ERK-blockers were effective in also downmodulating the respective protein production (P<0.01 and P<0.0001, Figures 5C, E, G). The AKT-blocker was effective in blocking IL-6 protein release (P<0.001, Figure 5C), but less effective in blocking IL-8 and TNF-a production (both not significant).
The introduction of the KLF2 overexpression plasmid strongly reduced HMECs’ mRNA production of IL-6 and IL-8 in presence of 5% USP (P<0.0001, Figures 5B, D), while TNF-a mRNA expression was unchanged (Figure 5F). The protein production of all three mediators, IL-6, IL-8, and TNF-a was strongly downmodulated upon introduction of KLF2 plasmid (Figures 5C, E, G).
3.5 Compared to HF dialyzers, expanded hemodialysis therapy with novel improved MCO dialyzers normalizes KLF2 transcription in endothelial cells
Our experimental results demonstrate, that USP exposure promotes the inhibition of KLF2 transcription in HMECs by engaging the AP-1/c-FOS transcription factor complex, with activation of ERK and AKT signaling, which is followed by the induction of typical proinflammatory markers such as IL-6. In turn, the blockade of AP1/ERK/AKT-signaling or supplementation of KLF2 overexpression plasmid could reverse the USP-induced activation of inflammatory markers. We next wondered how this would translate in the clinical setting.
We have previously already demonstrated in the PERCI-II clinical study that novel improved medium cut-off (MCO) dialyzers reduce uremic serum (USP)-induced systemic micro-inflammation and endothelial dysfunction (7, 17). Intriguingly, we can here report for the first time that this also translates into beneficial modulation of KLF2 expression (Figure 6).
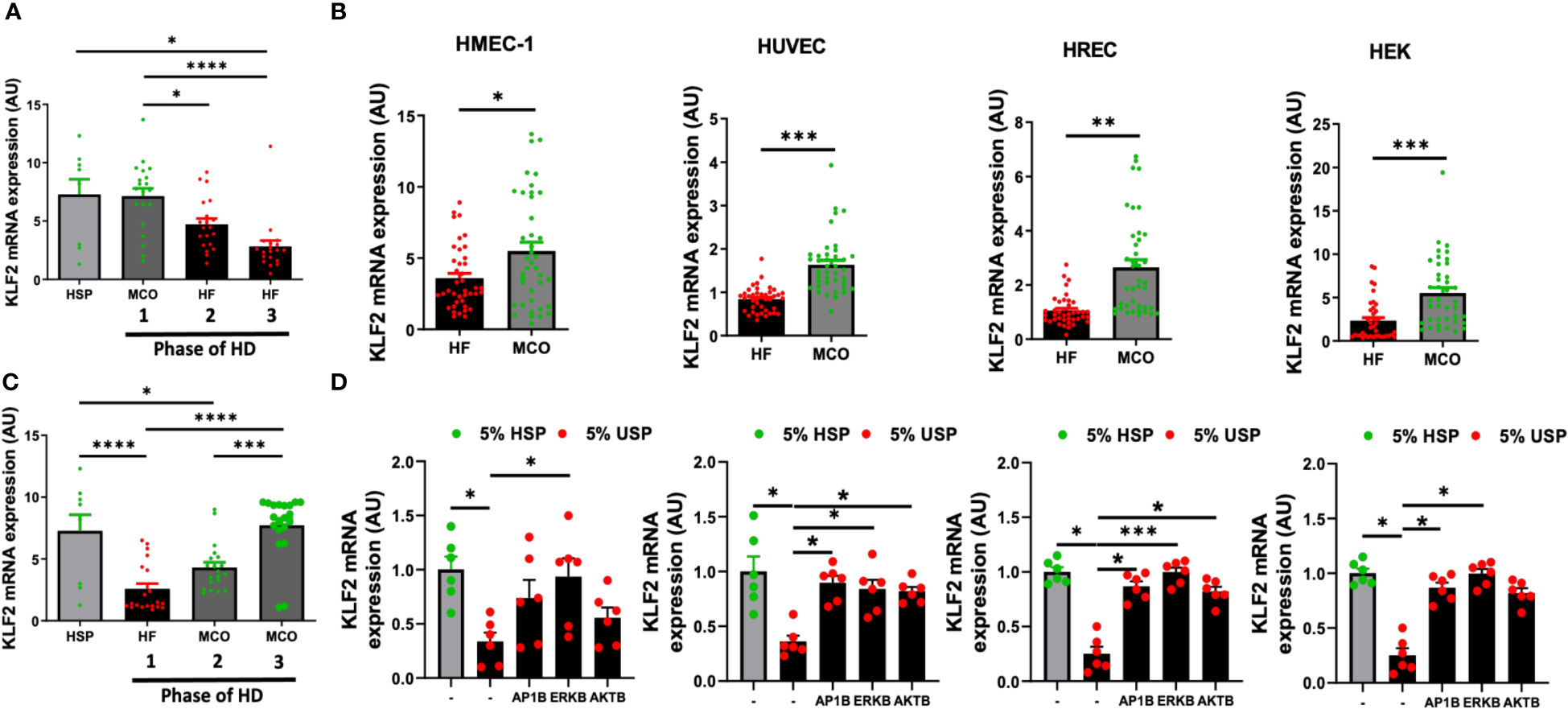
Figure 6 Expanded hemodialysis (HDx) therapy with novel improved MCO dialyzers normalizes KLF2 transcription in HMEC, HUVEC, HREC, and HEK cells, by modulating AP-1/c-FOS and AKT/ERK-dependent intracellular signaling. The effect of uremic serum fractions collected from patients undergoing hemodialysis with two different dialyzers/dialysis regimens in the PERCI-II clinical trial on KLF2 expression in different endothelial cell types. The serum fractions were collected from patients first dialyzed with high flux (HF) dialysis during the run-in phase of the trial and then dialyzed either with the conventional HF or the novel improved medium cut-off (MCO) dialyzer for comparison of the two different dialysis regimens in a cross-over clinical study design (n=40 patients included in analysis, comparison of 20 patients in each phase, the inclusion target was 50 patients) (7, 17). (A, B) KLF2 mRNA transcript levels at different trial stages were measured after stimulation of HMECs with healthy serum pool HSP reference or individual uremic serum samples from different stages of the PERCI-II trial (n=20 per group), as we previously described in PERCI-II study (A: MCO/HF/HF vs. B: HF/MCO/MCO) (7, 17), (C, D) To study if this can be generalized to other cell types, we also compared the effect of HF and MCO on HMECs, HUVECs, HRECs, and HEKs in presence or absence of different inhibitors (AP1B, ERKB, AKTB). KLF2 mRNA transcript levels were measured after a 6-hour incubation of HMECs with 5% uremic serum (n=40 patients) collected at the end of the first HF or MCO phase (each cycle lasted 4 weeks), using ANOVA, Mean ± SEM, with *P<0.05, **P<0.01, ***P<0.001, and****P<0.0001.
We first of all found that exposure of HMECs to uremic serum from patients treated either with conventional high-flux (HF) or novel improved MCO dialyzers resulted in normalization of KLF2 mRNA expression with MCO dialyzers (P<0.5 to P<0.001, Figures 6A, B).
When analyzing the individual trial phases, we first of all found that HMECs incubated with uremic serum from patients in the MCO/HF/HF-regimen, starting with MCO dialyzer, had similar KLF2 expression upon incubation MCO serum (end of the first 4-weeks cycle) than upon incubation with HSP, which indicates a beneficial effect of the MCO dialyzer (Figure 6A). In turn, the incubation of HMECs with uremic serum from the following two conventional HF cycles (4 and 8 weeks, respectively, in total 12 weeks long) lead to a strong downmodulation of KLF2 expression in the HMECs (P<0.05 to P<0.0001, Figure 6A).
Vice versa, HMECs incubated with uremic serum from patients in the cross-over HF/MCO/MCO-regimen, starting with HF dialyzer, had a strong downmodulation of KLF2 mRNA expression upon incubation of HMECs with the HF uremic serum collected at the end of the first 4-weeks cycle (P<0.0001, Figure 6B). However, incubation of HMECs with uremic serum from the following two MCO cycles (again 4 and 8 weeks, respectively) was effective in normalizing KLF2 expression to the prior baseline levels observed with HSP (P<0.001 and P<0.0001, Figure 6B).
We next wondered if similar protective effects of MCO-dialysis could be observed with other commonly used endothelial and kidney model related cell types, such as HUVECs, HRECs, and HEK cells (Figures 6C, D). Indeed, we found that in pair-wise analysis improved KLF2 expression could be observed in HMECs, HUVECs, HRECs, and HEKs (P<0.05, P<0.001, P<0.001, and P<0.01, respectively, Figure 6C), comparable to that of HMECs.
In addition, we found that similarly to the signaling pathways employed in HMECs, any detrimental endothelial down-modulation of KLF2 upon exposure to conventional uremic serum from HF dialysis could be mostly reversed by employing selective AP-1-, ERK-, and AKT-blockade in HUVECs, HRECs, and HEKs, respectively (P<0.05 to 0.001, Figure 6D). The ERK-blockade was slightly more effective than the AKT-blockade, thus demonstrating a more dominant signaling through the AP-1/ERK pathway (Figure 6D). Intriguingly, the dominance of ERK signaling was strongest in the HMECs, while this was less evident in the other three cell lines tested, although in general similar trends considering the dominance of AP-1/ERK signaling could be observed (Figure 6D).
In conclusion, this indicates that MCO dialyzers may offer a better treatment outcome compared to the HF dialyzer, through normalizing KLF2 transcription, which is important for mediating anti-inflammatory and angiogenic effects.
4 Discussion
High cardiovascular morbidity and mortality, caused in part by chronic inflammation associated with endothelial dysfunction, remains a major challenge in the management of CKD and ESRD patients with underlying uremia and systemic microinflammation (8–12). Endothelial cells (ECs) - as a direct barrier to the uremic substances in the blood - are essential for maintaining vascular homeostasis (49). There is an urgent unmet medical need to reverse endothelial dysfunction and reduce chronic inflammation in the clinical setting of renal replacement therapies (RRTs) (7–13). Thus, we here conducted a detailed study to decipher the major role of KLF2, a key vascular mediator and regulator of inflammatory process (20).
Krüppel-like factor 2 (KLF2), a key transcription factor involved in anti-inflammatory processes in the vasculature, has been proposed to be a critical target in the pathobiology of both CKD and ESRD (39, 50). Intriguingly, the effect of uremic serum (in particular uremic serum from well controlled clinical trials, such as PERCI-I/II) on the regulation and signaling of KLF2 has not been explicitly studied to date. Therefore, we focused in the present study on investigating uremic serum induced transcriptional regulation of KLF2 expression and the engagement of concomitant signaling pathways in microvascular endothelial cells (HMECs), since these cells are typically also a major target of uremic microinflammation (7).
First of all, we found that exposure of HMECs to uremic serum inhibits KLF2 mRNA transcription via engaging activating protein-1 (AP-1)/c-FOS signaling, thus resulting in the activation of several canonical signaling pathways, such as MAPK/ERK and AKT signaling, which in turn leads to a strong release of proinflammatory mediators by HMECs, including interleukin-6 (IL-6) and IL-8 and a panel of chemokines, among others. This is in line with our prior results on the response of ECs to uremic microinflammation in the context of endothelial maladaptation (7). Linking the modulation of KLF2, a key vascular transcription factor, to this study provides further insight into the mechanisms underlying the beneficial effect of expanded hemodialysis with improved medium cut-off (MCO) dialyzers (8–12).
Indeed, KLF2 expression is essential for renal vascular integrity and function (20, 39), and upregulation of KLF2 restores glomerular endothelial damage in mice undergoing unilateral nephrectomy (51). In turn, reduced expression of KLF2 potentiates podocyte injury in diabetic nephropathy and increases renal proteinuria (27, 52). Loss of KLF2 expression is also associated with leading to excessive accumulation of uremic solutes in patients with ESRD (53, 54). In support of these results, our current study found a substantial dose- and time-dependent repression of KLF2 transcriptional activity and protein expression in USP-treated HMECs. Thus, chronic accumulation of uremic toxins and inflammatory mediators in USP can lead to a loss of protective anti-inflammatory KLF2 expression in ECs exposed to uremia.
The regulation of KLF2 is complex and may be orchestrated by a variety of genes (39). Thus, the observed downmodulation of KLF2 in response to USP may be coregulated by a multiplicity of genes and signaling pathways. Indeed, biologically, it is not uncommon for genes to share or co-ordinate with similar regulatory binding elements to counter-influence transcriptional regulation. Interestingly, a comparative de-novo analysis of podocyte slit diaphragm molecules revealed that many genes share KLF promoter binding sites (55).
Similarly, downregulation of KLF2 transcription is known to up-regulate several genes and transcription factors such as Binding Endothelial Cell Precursor-Derived Regulator (BMPER) and Suppressor of Mothers against Decapentaplegic (SMAD1) (56, 57), thus also suppressing vascular calcification (57). However, the KLF2 promoter sequence to which regulatory factors bind is still not studied in great detail. Interestingly, an anticalcifying effect on vascular smooth muscle cells (VSMCs) was also reported for the use of uremic serum obtained from patients dialyzed with high- and medium-cut-off HCO/MCO dialyzers (58, 59).
Indeed, early reports suggested that KLF2 promoter activity is influenced by histone methylation or acetylated histones H3 and H4 associated with bare stress or other signals (60, 61). In addition, nuclear factors, including heterogeneous nuclear ribonucleoprotein (hnRNP)-U, hnRNP-D, and P300/CBP-associated factor (PCAF), can also bind to the human KLF2 promoter region (in position -138 and -111 bp) and regulate its transcription (62), while sequestration of the transcriptional coactivator p300/CBP by KLF2 suppresses inflammation through preventing NF-kB/p300 interaction and subsequent activation of the vascular cell adhesion molecule-1 (VCAM-1) (20, 41).
KLF2-p300 interaction also permits KLF2 binding to the endothelial nitric oxide synthase (eNOS) promoter to induce the transcription of this vasculoprotective enzyme (41). These early studies demonstrated KLF2’s ability to influence transcription through both, direct DNA-binding and/or indirect cofactor sequestration mechanisms (20). Furthermore, the KLF2 promoter region from -157 to -95 bp is also essential for the induction of fluid shear stress in the endothelium (63). In addition, p53 can also target the KLF2 promoter and repress its transcription in microglial cells (64).
In our study, USP-stimulated HMECs were found to downregulate KLF2 expression through the engagement of the KLF2 promoter sequence region -509 to -109 as a crucial regulatory region. Interestingly, we identified an AP-1 binding site in this region and studied, whether AP-1 is involved in the regulation of KLF2 transcription. Previously, KLF2 was shown to be involved in endothelial inflammation by negatively regulating AP-1 (28), but the specific promoter binding region was unknown. Indeed, targeted deletion analysis in our study indicates that AP-1 and the constitutive element c-FOS rather than c-JUN bind to the KLF2 promoter sequence -464 to -441 to regulate the inhibition of KLF2 in uremia.
Transcription of KLF2 is regulated by proliferative and inflammatory pathways (21, 41). Independent studies have demonstrated the regulation of KLF2 by mitogenic markers, e.g. MAPK/ERK-signaling or through its specific inhibitor PD184352 in mast cells (65). Interestingly, glucose-loading of human umbilical vein endothelial cells (HUVECs) also represses KLF2 transcription via attenuated ERK 1/2 activation (66). Shimizu et al. reported that the uremic toxin indoxyl-sulfate can increase monocyte chemoattractant protein-1 (MCP-1) production by activating reactive oxygen species (ROS) and the MAPK/ERK1/2 signaling (67).
MAPK/ERK-signaling is involved in a ricin-induced severe inflammatory response in mouse hemolytic-uremic syndrome in mice (45), thereby suggesting that ERK-signaling may act as a deleterious mediator in the response to uremic toxins. Similarly, enhanced phosphorylation of p-AKT has been observed in different stages of both, hemodialysis- and peritoneal-dialysis-treated uremia (68). Interestingly, the uremic toxin indoxyl-sulfate was found to suppress the expression of p-AKT, but not ERK in HUVECs (69), while P-cresol, another uremic toxin, was found to activate p-AKT and p-ERK in VSMCs (70), indicating a certain cell type dependency.
The molecular analysis in our current study indicates, that there exits some crosstalk between USP-stimulation-induced regulation of KLF2 transcription and both ERK and AKT pathway activation. This crosstalk is at least in part mediated via AP-1/c-FOS-signaling. Indeed, the suppression of mRNA expression of c-FOS, but not c-JUN, by pharmacological inhibition of ERK-signaling clearly indicated that c-FOS is crucially involved in USP-induced inhibition of KLF2 transcription. Binding of the AP-1/c-FOS complex to the KLF2 promoter region appears to mediate both, the activation of ERK and AKT pathways. Nevertheless, global transcriptomic analysis of KLF2 signaling may be necessary to further uncover additional potential DNA binding elements of the KLF2 promoter sequence.
We found that optimized amounts of KLF2 plasmid transfected into HMECs could attenuate the negative effects of USP-induced proinflammatory cytokine release, e.g. including IL-6, IL-8, and TNF-α. Remarkably, exogenous KLF2 could also inhibit c-FOS expression, indicating a concurrent effect on other regulatory signaling mechanisms, such as ERK and AKT activation. Thus, targeting the modulation of endogenous KLF2 expression (or its activity) could motivate novel therapies to treat the vascular microinflammation induced by uremia. Indeed, several pharmacological approaches, e.g. the use of statins, suberanilohydroxamic acid, tannic acid, and resveratrol, have all been shown to exert atheroprotective and anti-inflammatory effects on the endothelium through induction/targeting of KLF2 (19, 71–75).
The effect of USP on the inhibition of KLF2 mRNA transcription suggested, that dialysis membranes with improved molecular cutoff could offer a better therapeutic outcome. To answer this question, we retrospectively studied USP of patients treated with either high-flux (HF) or medium-cut-off (MCO) dialyzers (7, 41), observing significant improvements in the expression of KLF2 in the MCO group, which is in line with improvements in other inflammatory pathways and reduced endothelial maladaptation (7). This suggests an improved effectiveness of MCO to not only remove small toxic molecules (<0.5kD), but also middle-sized molecules and pro-inflammatory mediators, such as TNF-α, interleukins, β-2-microglobulin, and others (76), compared to the conventional HF hemodialyzers.
In agreement with our prior study (7), MCO may offer a better vasculoprotective role through improved maintenance/upregulation of KLF2 transcription in uremic patients. Indeed, prolonged HD with MCO (for up to 21 weeks) restored KLF2 expression in USP-stimulated HMECs to that found in HMECs stimulated with HSP, which may indicate a beneficial effect for the respective MCO-treated patients. Conversely, extended application of HF gradually reduced KLF2 to pre-treatment levels. Thus, our study demonstrates that uremic solutes down-regulates KLF2 transcription, leading to upregulation of proinflammatory mediators such as IL-6 via triggering AP-1/c-FOS, ERK and AKT signaling. However, maintaining/restoring KLF2 expression levels by use of beneficial MCO dialyzers (or exogenous KLF2 induction) could be a novel treatment approach to restore renal integrity and function in CKD/ESRD.
5 Conclusions and study limitations
Here, we identified that uremia downmodulates vasculoprotective KLF2 expression in common endothelial/kidney model cell lines, such as HMECs, HUVECs, HRECs, and HEKs. We then examined in detail the regulatory signaling mechanisms underlying modulation of KLF2 expression in response to uremic serum (USP). First, we identified the signaling events triggered upon USP-stimulation, including AP-1/c-FOS, MAPK/ERK1/2 and AKT signaling, as respective upstream molecules to regulate the expression of KLF2 in HMECs, and found that this was also applicable to other typical cell lines, such as HUVECs, HRECs, and HEKs.
Next, we demonstrated that any adverse effects of USP-exposure can be effectively attenuated by modulating endogenous KLF2 expression, which was achieved by specific inhibitors or transfection with an exogenous KLF2 plasmid. Further, we compared the inflammatory response to uremic and healthy serum, identifying a panel of mediators involved in uremia-induced chronic vascular inflammation.
Importantly, clinical verification of our results in a well-controlled clinical trial cohort, dialyzed with novel improved medium-cutoff (MCO)-dialysis membranes, was found to better maintain, or even restore KLF2 expression (cross-over design). Indeed, prolonged MCO dialysis eliminated any prior downmodulation of endothelial KLF2 expression observed with conventional HF dialysis membranes or uremic serum before HD treatment.
Thus, novel clinical trials with focus on comparing the therapeutic effects of MCO and HF membranes, but also well-designed pharmacological studies to targeting the respective signaling pathways, may provide new insights for future research and future improvements for patients depending on RRTs, to further reduce the detrimental long-term effects of uremia, chronic vascular inflammation and concomitant organ damage.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committees of the Martin-Luther-University Halle-Wittenberg and the Charité Berlin. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
Experiments and investigation: HZ, DW, MG, CL, PW, AP. Writing, review, and editing: HZ, DW, MA, JK-M, JW, GM, RC. Conception and Supervision: JW, DD, KB, RS, DZ, GM, RC. All authors contributed to the article and approved the submitted version.
Funding
The results in this paper have not been published previously, but they are in part complementary to a previously published mechanistic follow-up study to the concomitant clinical trial (7). This manuscript is a follow-up of the published clinical trial: PERCI-II-Medium-Cut-Off (MCO), which is registered at ClinicalTrials.gov NCT02084381 https://clinicaltrials.gov/ct2/show/NCT02084381) (7, 17). The current manuscript focuses on the mechanistic side studies that aim to verify promising biological leads identified in the prior clinical trial. The “lab research” was funded by academic grants through BMBF, DFG, BCRT/BSRT and EU-Project Grants and CGS/CSC-Scholarships, as outlined in detail below. The contributions by GM and RC were made possible by funding from the German Federal Ministry for Education and Research (BMBF) and German Research Foundation (DFG project Nephroproteciton #934046635, subproject A03, as part of CRC 1365, and EXPAND-PD; CA2816/1-1) through the Berlin Institute of Health (BIH)-Center for Regenerative Therapies (BCRT) and the Berlin-Brandenburg School for Regenerative Therapies (BSRT, GSC203), respectively, and in part by the European Union’s Horizon 2020 Research and Innovation Program under grant agreements No 733006 (PACE) and 779293 (HIPGEN) and 754995 (EUTRAIN). We also acknowledge financial support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin and the German Research Foundation (DFG). HZ, DW, and PW were supported by Chinese Scholarship Council (CSC) Stipends (HZ: 202008080251, DW: 202108080082, and PW 202108440113, respectively): https://www.chinesescholarshipcouncil.com/. The 2.1 Million Euro academic financing for the prior PERCI-II clinical study came from the German Ministry for Education and Research (BMBF, Project Codes 1313N11786, 13N11787, 13N11788, and 13N11789) headed on the academic side by RS and DZ at Charité and Prof. Dr. Girndt in Halle, while the clinical study was coordinated by Gambro Dialysatoren GmbH, Hechingen (Gambro AB, including all direct and indirect subsidiaries is now part of Baxter International Inc.) who sponsored parts of the PERCI-II clinical trial (e.g. disposable dialysis materials and standard of care versus newly developed types of MCO dialysis filters from Gambro/Baxter). The subsequent academic research was done independently from Gambro/Baxter, as an academic follow-up study: https://www.gesundheitsindustrie-bw.de/fachbeitrag/pm/dialyseforschung-bmbf-foerdert-von-gambro-koordiniertes-verbundprojekt.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1209464/full#supplementary-material
Supplementary Figure 1 | Analysis of the KLF2 promoter region and AP-1 binding site.
References
1. Eckardt KU, Coresh J, Devuyst O, Johnson RJ, Kottgen A, Levey AS, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet (2013) 382(9887):158–69. doi: 10.1016/S0140-6736(13)60439-0
2. Remuzzi G, Benigni A, Finkelstein FO, Grunfeld JP, Joly D, Katz I, et al. Kidney failure: aims for the next 10 years and barriers to success. Lancet (2013) 382(9889):353–62. doi: 10.1016/S0140-6736(13)60438-9
3. Bikbov B, Purcell CA, Levey AS, Smith M, Abdoli A, Abebe M, et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (2020) 395(10225):709–33. doi: 10.1016/S0140-6736(20)30045-3
4. Thurlow JS, Joshi M, Yan G, Norris KC, Agodoa LY, Yuan CM, et al. Global epidemiology of end-stage kidney disease and disparities in kidney replacement therapy. Am J Nephrol (2021) 52(2):98–107. doi: 10.1159/000514550
5. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. Bmj (2020) 371:m3734. doi: 10.1136/bmj.m3734
6. Catar RA, Bartosova M, Kawka E, Chen L, Marinovic I, Zhang C, et al. Angiogenic role of mesothelium-derived chemokine CXCL1 during unfavorable peritoneal tissue remodeling in patients receiving peritoneal dialysis as renal replacement therapy. Front Immunol (2022) 13:821681. doi: 10.3389/fimmu.2022.821681
7. Catar R, Moll G, Kamhieh-Milz J, Luecht C, Chen L, Zhao H, et al. Expanded hemodialysis therapy ameliorates uremia-induced systemic microinflammation and endothelial dysfunction by modulating VEGF, TNF-α and AP-1 signaling. Front Immunol (2021) 12:774052. doi: 10.3389/fimmu.2021.774052
8. García-Prieto A, de la Flor JC, Coll E, Iglesias E, Reque J, Valga F. Expanded hemodialysis, what´s up, Doc? Clin Kidney J (2023). doi: 10.1093/ckj/sfad033
9. Ronco C, Marchionna N, Brendolan A, Neri M, Lorenzin A, Martínez Rueda AJ. Expanded haemodialysis: from operational mechanism to clinical results. Nephrol Dialysis Transplant (2018) 33(suppl_3):iii41–7. doi: 10.1093/ndt/gfy202
10. Ronco C, Clark WR. Haemodialysis membranes. Nat Rev Nephrol (2018) 14(6):394–410. doi: 10.1038/s41581-018-0002-x
11. Ronco C, La Manna G. Expanded hemodialysis: A new therapy for a new class of membranes. Contributions to Nephrol (2017) 190:124–33. doi: 10.1159/000468959
12. Ronco C. The rise of expanded hemodialysis. Blood Purif (2017) 44(2):I–VIII. doi: 10.1159/000476012
13. Jankowski J, Floege J, Fliser D, Bohm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation (2021) 143(11):1157–72. doi: 10.1161/CIRCULATIONAHA.120.050686
14. de Mattos AM, Prather J, Olyaei AJ, Shibagaki Y, Keith DS, Mori M, et al. Cardiovascular events following renal transplantation: role of traditional and transplant-specific risk factors. Kidney Int (2006) 70(4):757–64. doi: 10.1038/sj.ki.5001628
15. Floege J, Gillespie IA, Kronenberg F, Anker SD, Gioni I, Richards S, et al. Development and validation of a predictive mortality risk score from a European hemodialysis cohort. Kidney Int (2015) 87(5):996–1008. doi: 10.1038/ki.2014.419
16. Zimmermann J, Herrlinger S, Pruy A, Metzger T, Wanner C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int (1999) 55(2):648–58. doi: 10.1046/j.1523-1755.1999.00273.x
17. Zickler D, Schindler R, Willy K, Martus P, Pawlak M, Storr M, et al. Medium cut-off (MCO) membranes reduce inflammation in chronic dialysis patients-A randomized controlled clinical trial. PloS One (2017) 12(1):e0169024. doi: 10.1371/journal.pone.0169024
18. Kirsch AH, Lyko R, Nilsson LG, Beck W, Amdahl M, Lechner P, et al. Performance of hemodialysis with novel medium cut-off dialyzers. Nephrol Dial Transplant (2017) 32(1):165–72. doi: 10.1093/ndt/gfw310
19. Xu S, Liu Y, Ding Y, Luo S, Zheng X, Wu X, et al. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct Target Ther (2021) 6(1):266. doi: 10.1038/s41392-021-00690-5
20. Sweet DR, Fan L, Hsieh PN, Jain MK. Krüppel-like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med (2018) 5:6. doi: 10.3389/fcvm.2018.00006
21. Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res (2005) 96(5):e48–57. doi: 10.1161/01.RES.0000159707.05637.a1
22. Lin Z, Natesan V, Shi H, Dong F, Kawanami D, Mahabeleshwar GH, et al. Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol (2010) 30(10):1952–9. doi: 10.1161/ATVBAHA.110.211474
23. Nayak L, Goduni L, Takami Y, Sharma N, Kapil P, Jain MK, et al. Kruppel-like factor 2 is a transcriptional regulator of chronic and acute inflammation. Am J Pathol (2013) 182(5):1696–704. doi: 10.1016/j.ajpath.2013.01.029
24. Fan Y, Lu H, Liang W, Hu W, Zhang J, Chen YE. Kruppel-like factors and vascular wall homeostasis. J Mol Cell Biol (2017) 9(5):352–63. doi: 10.1093/jmcb/mjx037
25. Catar R, Moll G, Hosp I, Simon M, Luecht C, Zhao H, et al. Transcriptional regulation of thrombin-induced endothelial VEGF induction and proangiogenic response. Cells (2021) 10(4). doi: 10.3390/cells10040910
26. Wang XQ, Nigro P, World C, Fujiwara K, Yan C, Berk BC. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing Kruppel-like factor 2. Circ Res (2012) 110(4):560–8. doi: 10.1161/CIRCRESAHA.111.256362
27. Zhong F, Chen H, Wei C, Zhang W, Li Z, Jain MK, et al. Reduced Kruppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int (2015) 87(2):382–95. doi: 10.1038/ki.2014.286
28. Shi J, Zhou LR, Wang XS, Du JF, Jiang MM, Song Z, et al. KLF2 attenuates bleomycin-induced pulmonary fibrosis and inflammation with regulation of AP-1. Biochem Biophys Res Commun (2018) 495(1):20–6. doi: 10.1016/j.bbrc.2017.10.114
29. Boon RA, Fledderus JO, Volger OL, van Wanrooij EJ, Pardali E, Weesie F, et al. KLF2 suppresses TGF-beta signaling in endothelium through induction of Smad7 and inhibition of AP-1. Arterioscler Thromb Vasc Biol (2007) 27(3):532–9. doi: 10.1161/01.ATV.0000256466.65450.ce
30. Zickler D, Luecht C, Willy K, Chen L, Witowski J, Girndt M, et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transplant (2018) 33(4):574–85. doi: 10.1093/ndt/gfx316
31. Philippe A, Kleinau G, Gruner JJ, Wu S, Postpieszala D, Speck D, et al. Molecular effects of auto-antibodies on angiotensin II type 1 receptor signaling and cell proliferation. Int J Mol Sci (2022) 23(7). doi: 10.3390/ijms23073984
32. Catar R, Witowski J, Wagner P, Annett Schramm I, Kawka E, Philippe A, et al. The proto-oncogene c-Fos transcriptionally regulates VEGF production during peritoneal inflammation. Kidney Int (2013) 84(6):1119–28. doi: 10.1038/ki.2013.217
33. Connolly-Andersen AM, Moll G, Andersson C, Akerstrom S, Karlberg H, Douagi I, et al. Crimean-Congo hemorrhagic fever virus activates endothelial cells. J Virol (2011) 85(15):7766–74. doi: 10.1128/JVI.02469-10
34. Moll G, Rasmusson-Duprez I, von Bahr L, Connolly-Andersen AM, Elgue G, Funke L, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells (2012) 30(7):1565–74. doi: 10.1002/stem.1111
35. Moll G, Ignatowicz L, Catar R, Luecht C, Sadeghi B, Hamad O, et al. Different procoagulant activity of therapeutic mesenchymal stromal cells derived from bone marrow and placental decidua. Stem Cells Dev (2015) 24(19):2269–79. doi: 10.1089/scd.2015.0120
36. Moll G, Alm JJ, Davies LC, von Bahr L, Heldring N, Stenbeck-Funke L, et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells (2014) 32(9):2430–42. doi: 10.1002/stem.1729
37. Catar R, Witowski J, Zhu N, Lucht C, Derrac Soria A, Uceda Fernandez J, et al. IL-6 trans-signaling links inflammation with angiogenesis in the peritoneal membrane. J Am Soc Nephrol JASN (2017) 28(4):1188–99. doi: 10.1681/ASN.2015101169
38. Hegner B, Weber M, Dragun D, Schulze-Lohoff E. Differential regulation of smooth muscle markers in human bone marrow-derived mesenchymal stem cells. J Hypertens (2005) 23(6):1191–202. doi: 10.1097/01.hjh.0000170382.31085.5d
39. Mallipattu SK, Estrada CC, He JC. The critical role of Krüppel-like factors in kidney disease. Am J Physiol Renal Physiol (2017) 312(2):F259–f265. doi: 10.1152/ajprenal.00550.2016
40. Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol (1997) 9(2):240–6. doi: 10.1016/s0955-0674(97)80068-3
41. SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med (2004) 199(10):1305–15. doi: 10.1084/jem.20031132
42. Turpaev KT. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochem (Mosc) (2020) 85(1):54–67. doi: 10.1134/S0006297920010058
43. Wisdom R. AP-1: one switch for many signals. Exp Cell Res (1999) 253(1):180–5. doi: 10.1006/excr.1999.4685
44. Meng Q, Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell (2011) 2(11):889–98. doi: 10.1007/s13238-011-1113-3
45. Korcheva V, Wong J, Corless C, Iordanov M, Magun B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am J Pathol (2005) 166(1):323–39. doi: 10.1016/S0002-9440(10)62256-0
46. Sindhu RK, Ehdaie A, Vaziri ND, Roberts CK. Effects of chronic renal failure on caveolin-1, guanylate cyclase and AKT protein expression. Biochim Biophys Acta (2004) 1690(3):231–7. doi: 10.1016/j.bbadis.2004.06.013
47. Li Y, Takemura G, Okada H, Miyata S, Maruyama R, Esaki M, et al. Molecular signaling mediated by angiotensin II type 1A receptor blockade leading to attenuation of renal dysfunction-associated heart failure. J Card Fail (2007) 13(2):155–62. doi: 10.1016/j.cardfail.2006.11.005
48. Semple D, Smith K, Bhandari S, Seymour AM. Uremic cardiomyopathy and insulin resistance: a critical role for akt? J Am Soc Nephrol JASN (2011) 22(2):207–15. doi: 10.1681/ASN.2009090900
49. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation (2007) 115(10):1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859
50. Dabravolski SA, Sukhorukov VN, Kalmykov VA, Grechko AV, Shakhpazyan NK, Orekhov AN. The role of KLF2 in the regulation of atherosclerosis development and potential use of KLF2-targeted therapy. Biomedicines (2022) 10(2). doi: 10.3390/biomedicines10020254
51. Zhong F, Mallipattu SK, Estrada C, Menon M, Salem F, Jain MK, et al. Reduced kruppel-like factor 2 aggravates glomerular endothelial cell injury and kidney disease in mice with unilateral nephrectomy. Am J Pathol (2016) 186(8):2021–31. doi: 10.1016/j.ajpath.2016.03.018
52. Guo Y, Pace J, Li Z, Ma'ayan A, Wang Z, Revelo MP, et al. Podocyte-specific induction of kruppel-like factor 15 restores differentiation markers and attenuates kidney injury in proteinuric kidney disease. J Am Soc Nephrol JASN (2018) 29(10):2529–45. doi: 10.1681/ASN.2018030324
53. Brunet P, Gondouin B, Duval-Sabatier A, Dou L, Cerini C, Dignat-George F, et al. Does uremia cause vascular dysfunction? Kidney Blood Press Res (2011) 34(4):284–90. doi: 10.1159/000327131
54. Saum K, Campos B, Celdran-Bonafonte D, Nayak L, Sangwung P, Thakar C, et al. Uremic advanced glycation end products and protein-bound solutes induce endothelial dysfunction through suppression of krüppel-like factor 2. J Am Heart Assoc (2018) 7(1). doi: 10.1161/jaha.117.007566
55. Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, et al. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc Natl Acad Sci USA (2006) 103(15):5682–7. doi: 10.1073/pnas.0511257103
56. Jha P, Das H. KLF2 in regulation of NF-kappaB-mediated immune cell function and inflammation. Int J Mol Sci (2017) 18(11). doi: 10.3390/ijms18112383
57. Huang J, Pu Y, Zhang H, Xie L, He L, Zhang CL, et al. KLF2 mediates the suppressive effect of laminar flow on vascular calcification by inhibiting endothelial BMP/SMAD1/5 signaling. Circ Res (2021) 129(4):e87–e100. doi: 10.1161/CIRCRESAHA.120.318690
58. Zickler D, Willy K, Girndt M, Fiedler R, Martus P, Storr M, et al. High cut-off dialysis in chronic haemodialysis patients reduces serum procalcific activity. Nephrol Dial Transplant (2016) 31(10):1706–12. doi: 10.1093/ndt/gfw293
59. Willy K, Hulko M, Storr M, Speidel R, Gauss J, Schindler R, et al. In vitro dialysis of cytokine-rich plasma with high and medium cut-off membranes reduces its procalcific activity. Artif organs (2017) 41(9):803–9. doi: 10.1111/aor.12884
60. Xu M, Chen X, Lin K, Zeng K, Liu X, Pan B, et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer (2018) 17(1):141. doi: 10.1186/s12943-018-0894-x
61. Huddleson JP, Ahmad N, Srinivasan S, Lingrel JB. Induction of KLF2 by fluid shear stress requires a novel promoter element activated by a phosphatidylinositol 3-kinase-dependent chromatin-remodeling pathway. J Biol Chem (2005) 280(24):23371–9. doi: 10.1074/jbc.M413839200
62. Ahmad N, Lingrel JB. Kruppel-like factor 2 transcriptional regulation involves heterogeneous nuclear ribonucleoproteins and acetyltransferases. Biochemistry (2005) 44(16):6276–85. doi: 10.1021/bi050018s
63. Huddleson JP, Srinivasan S, Ahmad N, Lingrel JB. Fluid shear stress induces endothelial KLF2 gene expression through a defined promoter region. Biol Chem (2004) 385(8):723–9. doi: 10.1515/bc.2004.088
64. Zhang X, Liu T, Xu S, Gao P, Dong W, Liu W, et al. A pro-inflammatory mediator USP11 enhances the stability of p53 and inhibits KLF2 in intracerebral hemorrhage. Mol Ther Methods Clin Dev (2021) 21:681–92. doi: 10.1016/j.omtm.2021.01.015
65. Marschall JS, Wilhelm T, Schuh W, Huber M. MEK/Erk-based negative feedback mechanism involved in control of Steel Factor-triggered production of Krüppel-like factor 2 in mast cells. Cell Signal (2012) 24(4):879–88. doi: 10.1016/j.cellsig.2011.12.007
66. Liu YS, Xu DL, Huang ZW, Hao L, Wang X, Lu QH. Atorvastatin counteracts high glucose-induced Krüppel-like factor 2 suppression in human umbilical vein endothelial cells. Postgrad Med (2015) 127(5):446–54. doi: 10.1080/00325481.2015.1039451
67. Shimizu H, Bolati D, Higashiyama Y, Nishijima F, Shimizu K, Niwa T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-κB, p53, ERK, and JNK in proximal tubular cells. Life Sci (2012) 90(13-14):525–30. doi: 10.1016/j.lfs.2012.01.013
68. Martin-Rodriguez S, Caballo C, Gutierrez G, Vera M, Cruzado JM, Cases A, et al. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur J Clin Invest (2015) 45(2):160–9. doi: 10.1111/eci.12392
69. Adelibieke Y, Shimizu H, Saito S, Mironova R, Niwa T. Indoxyl sulfate counteracts endothelial effects of erythropoietin through suppression of Akt phosphorylation. Circ J (2013) 77(5):1326–36. doi: 10.1253/circj.cj-12-0884
70. Lee CH, Hung YJ, Shieh YS, Chien CY, Hsu YJ, Lin CY, et al. Cilostazol inhibits uremic toxin-induced vascular smooth muscle cell dysfunction: role of Axl signaling. Am J Physiol Renal Physiol (2017) 312(3):F398–f406. doi: 10.1152/ajprenal.00258.2016
71. Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, et al. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation (2005) 112(5):720–6. doi: 10.1161/circulationaha.104.525774
72. Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA Jr., García-Cardeña G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem (2005) 280(29):26714–9. doi: 10.1074/jbc.C500144200
73. Tuomisto TT, Lumivuori H, Kansanen E, Häkkinen SK, Turunen MP, van Thienen JV, et al. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, Kruppel-like factor 2. Cardiovasc Res (2008) 78(1):175–84. doi: 10.1093/cvr/cvn007
74. Xu Y, Xu S, Liu P, Koroleva M, Zhang S, Si S, et al. Suberanilohydroxamic acid as a pharmacological kruppel-like factor 2 activator that represses vascular inflammation and atherosclerosis. J Am Heart Assoc (2017) 6(12). doi: 10.1161/jaha.117.007134
75. Gracia-Sancho J, Villarreal G Jr., Zhang Y, Yu JX, Liu Y, Tullius SG, et al. Flow cessation triggers endothelial dysfunction during organ cold storage conditions: strategies for pharmacologic intervention. Transplantation (2010) 90(2):142–9. doi: 10.1097/TP.0b013e3181e228db
Keywords: chronic kidney disease (CKD), end-stage renal disease (ESRD), uremic toxins, cytokine signaling, systemic inflammation, cardiovascular disease (CVD), Krüppel-like factor 2 (KLF2), and expanded hemodialysis therapy (HDx)
Citation: Zhao H, Wu D, Gyamfi MA, Wang P, Luecht C, Pfefferkorn AM, Ashraf MI, Kamhieh-Milz J, Witowski J, Dragun D, Budde K, Schindler R, Zickler D, Moll G and Catar R (2023) Expanded Hemodialysis ameliorates uremia-induced impairment of vasculoprotective KLF2 and concomitant proinflammatory priming of endothelial cells through an ERK/AP1/cFOS-dependent mechanism. Front. Immunol. 14:1209464. doi: 10.3389/fimmu.2023.1209464
Received: 20 April 2023; Accepted: 31 August 2023;
Published: 19 September 2023.
Edited by:
Sina Naserian, Hôpital Paul Brousse, FranceReviewed by:
Mohamed Essameldin Abdelgawad, Wake Forest University, United StatesSara Shamdani, Hôpital Paul Brousse, France
Copyright © 2023 Zhao, Wu, Gyamfi, Wang, Luecht, Pfefferkorn, Ashraf, Kamhieh-Milz, Witowski, Dragun, Budde, Schindler, Zickler, Moll and Catar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido Moll, Z3VpZG8ubW9sbEBjaGFyaXRlLmRl; Rusan Catar, cnVzYW4uY2F0YXJAY2hhcml0ZS5kZQ==
†In memory of Prof. Duska Dragun
‡These authors have contributed equally to this work and share first authorship
§These authors have contributed equally to this work and share senior authorship
‖ORCID: Guido Moll, orcid.org/0000-0001-6173-5957
Rusan Catar, orcid.org/0000-0002-7000-7860