 Jin Kyung Kim1†
Jin Kyung Kim1† Hui-Jung Jung1†
Hui-Jung Jung1† Miri Hyun2Ji Yeon Lee2Jong-Hwan Park3Seong-Il Suh1Won-Ki Baek1*
Miri Hyun2Ji Yeon Lee2Jong-Hwan Park3Seong-Il Suh1Won-Ki Baek1* Hyun ah Kim2*
Hyun ah Kim2*- 1Department of Microbiology, Keimyung University School of Medicine, Daegu, Republic of Korea
- 2Department of Infectious Diseases, Keimyung University Dongsan Hospital, Keimyung University School of Medicine, Daegu, Republic of Korea
- 3Laboratory Animal Medicine, College of Veterinary Medicine and Brain Korea 21 Plus Project Team, Chonnam National University, Gwangju, Republic of Korea
Introduction: Hypervirulent Klebsiella pneumoniae (hvKp) has emerged as a clinically significant global pathogen in the last decade. However, the host immune responses of the macrophages during hvKp infection are largely unknown. In the present study, we aimed to compare the cytotoxic effects of hvKp and classical K. pneumoniae (cKp) in murine macrophages.
Results: We found that the activation of caspase-1 -dependent pyroptosis was higher in cKp-infected macrophages compared with that in hvKp-infected macrophages. In Caspase-1 deficiency macrophages, pyroptosis diminished during infection. Both hvKp and cKp strains led to nucleotide-binding and oligomerization domain-like receptor protein 3 (NLRP3) inflammasome formation and lysosomal cathepsin B activation, thus resulting in pyroptosis. Compared with the cKp strain, the hvKp strain inhibited these phenomena in murine macrophages.
Conclusion: HvKp infection resulted in different levels of pyroptosis via the activation of cathepsin B-NLRP3-caspase-1 in murine macrophages. Therefore, the manipulation of pyroptotic cell death is a potential target for host response during hvKp infection in macrophages.
1 Introduction
Classical Klebsiella pneumoniae (cKp) usually presents as hospital-acquired pneumonia, urinary tract infection, and bacteremia in an immunocompromised host (1). It is a “critical concern” of the World Health Organization because of its carbapenem resistance (2). In contrast, hypervirulent K. pneumoniae (hvKp), which usually presents as community liver abscess, has emerged in East Asian countries in the last three decades. Usually, hvKp causes liver abscess, endophthalmitis, meningitis, and necrotizing fasciitis in healthy individuals with community origin (3). Recently, hvKp has spread globally and the prevalence of multidrug-resistant hvKp also increased (4, 5). Therefore, to respond to hvKp, we need to increase awareness of the pathogenesis of this strain. Metastatic infection due to hvKp is the hallmark clinical feature and it occurs through neutrophils (6). Additionally, hvKp is involved in cell death of neutrophils (6), but the mechanism has not been clearly identified.
Cell death is an important physiological process that plays a pivotal role in maintaining homeostasis in living organisms. Three types of cell death (pyroptosis, apoptosis, and necroptosis) have been well known and studied (7). Particularly, pyroptosis, called inflammatory cell death, is induced by inflammasome sensors such as the nucleotide-binding oligomerization domain-like receptor family, absent in myeloma 2 (AIM2), and pyrin receptor (8). In the canonical pyroptosis pathway, the inflammasome is induced by danger-associated molecular patterns and pathogen-associated molecular patterns. Consequently, pro-interleukin (IL)-1β, IL-18, and gasdermin D (GSDMD) are cleaved via activated caspase-1. The N-terminal from cleaved GSDMD forms pores in the plasma membrane, resulting in the secretion of IL-1β and IL-18 into the extracellular space (9). This pyroptosis is related to diverse human diseases, such as tumor (9), liver fibrosis (10), cardiovascular disease (11), and central nervous system disease (12).
Lysosomal cysteine cathepsins (B, C, F, H, K, L, O, S, V, Z, and W) are involved in nucleotide-binding and oligomerization domain-like receptor protein 3 (NLRP3) inflammasome activation (13). Among them, cathepsin B can interact with NLRP3 upon inflammasome activator treatment (14); thus, it activates the NLRP3 inflammasome under diverse stimuli (15, 16). Several studies have shown that pyroptosis is triggered by cathepsin B-mediated NLRP3 inflammasome activation (17, 18). Wang et al. suggested that coxsackievirus B3 infection activates cathepsin B and promotes pyroptosis (19); however, the crosstalk between cathepsin B and pyroptosis during pathogen infection is largely unknown.
Herein, we found that hvKp infection exhibited weakened pyroptosis compared to the cKp strain. Mechanistically, cathepsin B-mediated NLRP3 activation was significantly higher in cKp-infected macrophages than in hvKp-infected macrophages. In addition, Caspase-1-null macrophages showed reduced pyroptosis during cKp infection compared with wild-type cells.
2 Materials and methods
2.1 Ethics statements
This study was approved by the Institutional Research and Ethics Committee at Keimyung University (approval number: KM-2022-07R1). All animal experiments were performed in accordance with the guidelines of the Korean Food and Drug Administration.
2.2 Mice
Wild-type (WT) C57BL/6 female mice (age; 7–9 weeks, weight; 20–25 g) were purchased from Samtako BioKorea Co. (Osan, Korea). Caspase-1-/- female mice (age; 7–9 weeks, 20-25 g) were kindly provided by Prof. Gabriel Núñez (University of Michigan Medical School, Ann Arbor, MI, USA) (20). Mice were maintained under a 12-h light/dark cycle at an ambient temperature of 20–23°C and relative humidity of 40–60% and with ample food and water. All animals were maintained under barrier conditions in a biohazard animal room at the School of Medicine, Keimyung University, Daegu, Korea.
2.3 Cell culture
The murine macrophage cell lines RAW264.7 (American Type Culture Collection, TIB-71) were maintained in the Dulbecco’s Modified Eagle’s Medium (DMEM; Welgene, Gyeonsan, South Korea), supplemented with 10% fetal bovine serum (Welgene) and 1% penicillin/streptomycin (Gibco BRL, Grand Island, NY, USA). Cells were incubated in a humidified atmosphere at 37°C with 5% CO2. Primary bone marrow-derived macrophages (BMDMs) were isolated from C57BL/6 mice and cultured in DMEM for 3–5 days in the presence of macrophage colony-stimulating factor (M-CSF; R&D Systems, Minneapolis, MN, USA).
2.4 Bacterial strains and culture
Sixteen Kp isolates were obtained from the various clinical specimens of Keimyung University Dongsan Hospital. From these strains, we collected the hvKp strain from a blood specimen of a patient with a liver abscess, whereas cKp was collected from the sputum of a patient with hospital-acquired pneumonia. These isolates were identified using the automated microbial identification and susceptibility test system (VITEK 2 system, bioMerieux, Lyon, France). Extended-Spectrum ß-Lactamase (ESBL) production was confirmed by agar dilution test using cefotaxime and ceftazidime along with clavulanate, in accordance with the Clinical and Laboratory Standards Institute guidelines.
Capsular serotypes and genes of virulence factors were identified by polymerase chain reaction (PCR) (21, 22). Strains were serotyped as: K1, K2, K5, K20, K54, and K57, or as non-determining when no specific serotype could be identified. Hypervirulent characteristics were confirmed with a string test and gene amplification of rmpA, magA, and aerobactin. According to serotype analysis, the hvKp was a K1 serotype. HvKp had antibiotic susceptibility for most antibiotics except ampicillin. The cKp strain had a negative string test result, and, according to genetic analysis, it was negative for rmpA, magA, and aerobactin. The antibiotic susceptibility test result was positive for ESBL and showed resistance for most antibiotics except carbapenem and tigecycline.
Strains of Kp were grown in Luria-Bertani (LB) broth (Difco, Becton Dickinson, Sparks, MD, USA) for liquid culture and on MacConkey agar (Difco) as a solid medium at 37°C. All bacteria were stored at -80°C in LB broth supplemented with 15% v/v glycerol (Sigma-Aldrich, St. Louis, MO, USA). For infection stocks, bacteria were incubated overnight and then diluted in fresh LB broth and cultured to an optical density (OD 600 nm) of 0.1. Samples were frozen at -80°C with 15% glycerol, and one aliquot was counted prior to infection.
2.5 Infection
BMDMs were replated into 24- or 96-well plates in complete DMEM medium at a density of 2 × 105 cells/well and infected with the different strains at a multiplicity of infection (MOI) of 10 for 90 min. RAW264.7 cells were replated into 6-well or 96-well plates in complete DMEM medium at a density of 7 × 105 cells/well and infected with the different strains at MOI of 10 for 90 min. Cells were washed twice with phosphate-buffered saline (PBS) containing antibiotics (gentamicin 100 μg/ml) to remove non-phagocytosed bacteria.
2.6 Treatments
2.6.1 Inhibition of cathepsin B
CA-074 methyl ester (CA074-Me; S7420; Selleck Chemical, Houston, TX, USA) is a specific inhibitor of cathepsin B. To inhibit cathepsin B activity, CA074-Me was dissolved in dimethylsulfoxide (DMSO) at a concentration of 50 mM. Cells were treated in media containing 25 μM CA074-Me for 1 h prior to bacterial infection. BMDMs and RAW264.7 cells were infected with hvKp or cKp strains at MOI of 10 for 90 min, washed and then cultivated for 16 h.
2.6.2 Inhibition of caspase-1
Ac-YVAD-cmk was manufactured by the Sigma-Aldrich (SML0429; St. Louis, MO, USA) and solubilized in DMSO (stock 50 mM). Cells were grown in media containing 50 μM Ac-YVAD-cmk or an equivalent amount of vehicle (DMSO). Cells were treated in media containing 50 μM Ac-YVAD-cmk for 30 min prior to bacterial infection.
2.7 Immunoblotting
Cells were lysed in RIPA lysis buffer (20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM Na3VO4, 1 mM NaF, 2 mM ethylenediaminetetraacetic acid [EDTA], 200 nM aprotinin, 20 μM leupeptin, 50 mM phenanthroline, and 280 mM benzamidine-HCl) and supernatant fractions were collected. Protein concentrations were measured with bicinchoninic acid (Pierce, Rockford, IL, USA), and equal amounts of proteins (50 μg/lane) were separated by 13% sodium dodecyl-sulfate polyacrylamide gel electrophoresis and transferred to the nitrocellulose membranes (GE Healthcare Life Science, Pittsburgh, PA, USA) incubated with a specific antibody. The anti-caspase-1 antibody (3019-100; BioVision, Milpitas, CA, USA, and AG-20B-0042; AdipoGen, San Diego, CA, USA) and anti-GSDMD antibody (ab209845; Abcam PLC) were used as primary antibodies. These antibodies were used at a dilution of 1:1,000. The anti−ACTB antibody (A5441) was acquired from Sigma−Aldrich (Merck KGaA) and used at a dilution of 1:5000. Secondary antibodies (anti−rabbit IgG [1:2000 diluted; sc−2004] and anti−mouse IgG [1:2000 diluted; sc−2005]) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Bands were detected using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Darmstadt, Germany).
2.8 RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from infected cells using TRIzol (15596-026; Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Then, RNA was reverse transcribed to complementary DNA using the reverse transcriptase premix (Elpis Biotech, EBT-1515; Elpis Biotech, Lexington, MA, USA). Data were analyzed by the qTOWER3 PCR thermal cycler (Analytik Jena, Jena, Germany) using the TOPreal qPCR 2X SYBR Green PreMIX (Enzynomics, RT500M). The gene expression was calculated using the 2−ΔΔCt method and normalized to b-actin. The primer sequences (mouse) were as follows: Il1b forward: 5’- TACGGACCCCAAAAGATGA-3’, reverse: 5’-TGCTGCTGCGAGATTTGAAG-3’; Il18 forward: 5’-GACAGCCTGTGTTCGAGGATATG-3’, reverse: 5’-TGTTCTTACAGGAGAGGGTAGAC-3’; Nlrp3 forward: 5’-TCACAACTCGCCCAAGGAGGAA-3’, reverse: 5’-AAGAGACCACGGCAGAAGCTAG-3’; and b-actin forward: 5’- CCACCATGTACCCAGGCATT-3’, reverse: 5’- AGGGTGTAAAACGCAGCTCA-3’
2.9 Flow cytometric analysis
Cell cycle analysis was performed using flow cytometry. For DNA content analysis, approximately 106 cells were fixed in 80% ethanol for at least 1 h at 4°C. Ethanol-fixed cells were stained with propidium iodide (PI) staining solution (50 μg/ml PI, 0.1 mg/ml RNase A, 0.1% NP-40, 0.1% trisodium citrate) for 30 min, and analyzed using a FACS analyzer (BD Biosciences, San Diego, CA, USA).
2.10 Lactate dehydrogenase production assay
Lactate dehydrogenase (LDH) release into the culture supernatant was measured using the EZ-LDH assay kit (DoGenBio, Seoul, Korea), following the manufacturer’s instructions. Briefly, BMDMs or RAW264.7 cells were cultured and infected with hvKp or cKp. Then, the supernatant medium (10 µl) for each sample was transferred into a 96-well plate and was incubated with the LDH reaction mixture (100 µl) at 25°C for 30 min in a dark room. LDH levels in the culture medium were determined by measuring the absorbance (450 nm) using the Synergy/HTX (BioTek instrument, Inc. Winooski, VT, USA).
2.11 Enzyme-linked immunosorbent assay (ELISA)
BMDMs and RAW264.7 cells were cultured in 24-well culture plates at a density of 2 × 105/well. Media were collected after overnight culture and centrifuged at 800 rpm for 5 min at 4°C to remove cell debris. Culture supernatants were assayed for IL-1β using the mouse IL-1β/IL-1F2 Quantikine ELISA Kit from R&D Systems (MLB00C, Minneapolis, MN, USA) according to the manufacturer’s instructions.
2.12 Immunofluorescence and analysis
For NLRP3 and ASC staining, cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized with 0.25% Triton X-100 (Sigma-Aldrich) for 10 min. Next, cells were incubated with NLRP3 (1:400 diluted; 15101; Cell Signaling, Danvers, MA, USA), ASC (1:400 diluted; sc-514414; Santa Cruz Biotechnology, Inc.), and cleaved GSDMD (1:400 diluted; 34667; Cell Signaling, Danvers, MA, USA) primary antibodies overnight at 4°C. Then, cells were washed with PBS followed by an incubation with secondary Alexa Fluor 488 goat anti-rabbit IgG (H+L) (1:400 diluted; A-11034, Invitrogen) and Donkey anti-mouse IgG (H+L) Alexa Fluor Plus 594 (1:400 diluted; A32744, Invitrogen) at room temperature for 2 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole (D9542; Sigma-Aldrich) for 5 min at room temperature. The infected cells were stained with Lysotracker Yellow-HCK-123 (L12491; Thermo Fisher Scientific), according to the manufacturer’s instructions. Immunofluorescence images were observed and analyzed using the confocal laser microscope (TCS SP8; Leica Microsystems, Wetzlar, Germany) and Image J software (National Institutes of Health, Bethesda, MD, USA).
2.13 Caspase-1 activity assay
Caspase-1 activity was measured using the FAM-FLICA Caspase-1 (YVAD) Assay Kit (#98; ImmunoChemistry Technologies, Davis, CA, USA) according to the manufacturer’s instructions. Murine macrophages on coverslips were infected and stained with a staining solution at 37°C for 1 h. After washing with 1× apoptosis wash buffer, cells were stained with Hoechst 33342 at 37°C for 10 min and fixed with a fixative. The samples were acquired and analyzed with a confocal laser microscope (TCS SP8; Leica Microsystems, Wetzlar, Germany) and Image J software.
2.14 Detection of cathepsin B activation
The infected cells were stained using the Magic Red Cathepsin B assay kit (#938; ImmunoChemistry Technologies) at 37°C for 1 h and nuclei were stained with Hoechst 33342 at 37°C for 10 min. Cells were fixed with 4% paraformaldehyde and images were visualized by a confocal laser microscope (TCS SP8; Leica Microsystems, Wetzlar, Germany).
2.15 Statistical analysis
Data are presented as mean ± standard deviation and were obtained from three independent experiments. The Student t-test or analysis of variance was used to analyze the data. Statistical analyses were performed, and graphs were made using GraphPad Prism 7.0 software for Windows (GraphPad Software Inc., La Jolla, CA, USA). P-values <0.05, 0.01, and 0.001 were considered statistically significant.
3 Results
3.1 The hvKp infection suppresses the GSDMD-derived pyroptosis in BMDMs compared with cKp infection
To compare the host responses to hvKp and cKp infection within the murine macrophages, clinical hvKp and cKp strains were isolated from blood and sputum samples, respectively (Supplementary Table 1). Since a previous study reported that two clinical strains of Kp showed different cell death patterns (23), we investigated whether hvKp and cKp affected the different cell fates in murine macrophages. To examine this, we infected BMDMs with hvKp and cKp strains and measured the sub-G1 population, which represents the apoptotic cells. As shown in Figure 1A, cKp infection induced excessive cell death, while hvKp strains exhibited weaker responses in the BMDMs. As expected, hvKp infection induced lower levels of LDH release in an MOI-dependent manner compared with cKp infection (Figure 1B). These data indicate that cKp infection triggers severe cell death in BMDMs compared with hvKp infection.
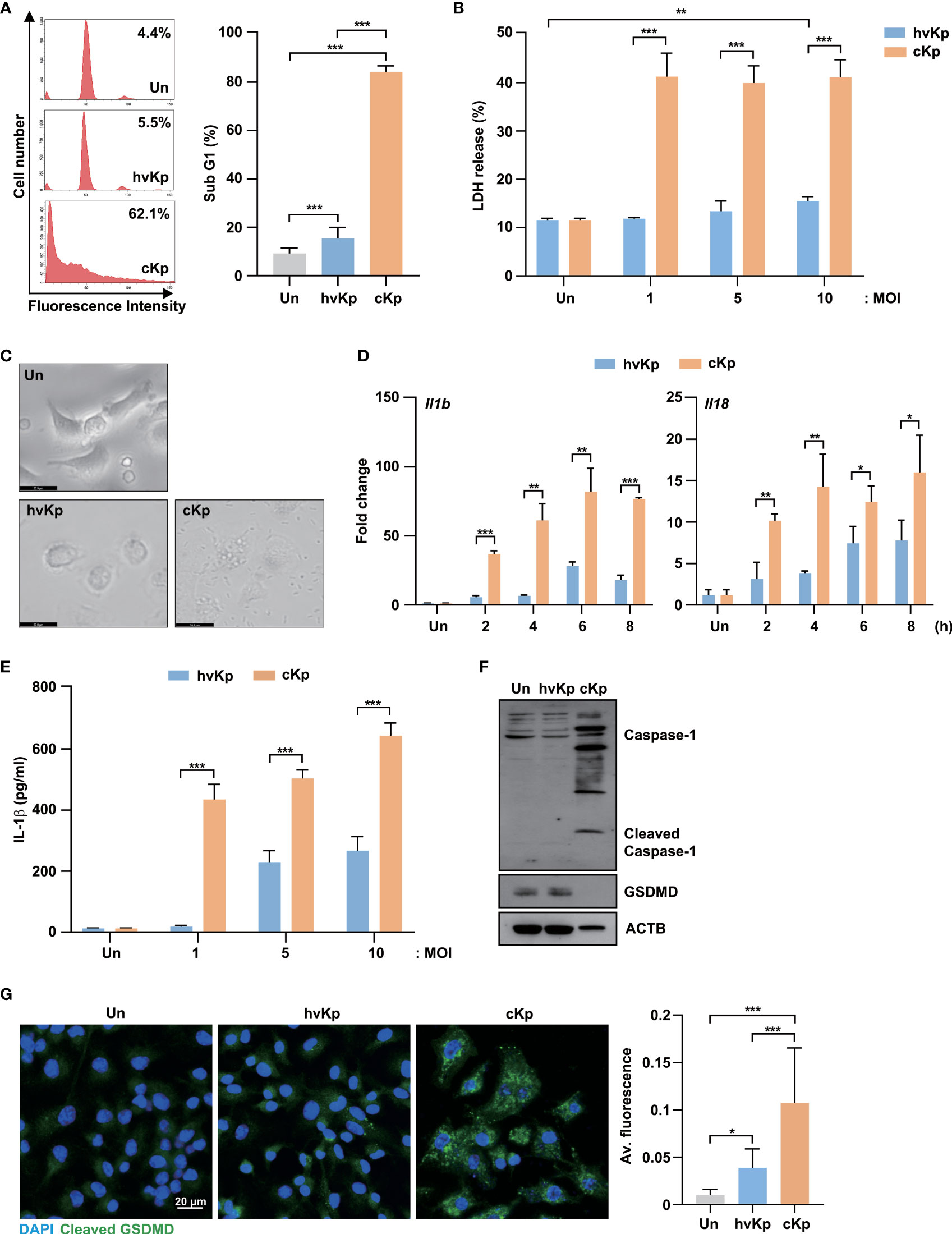
Figure 1 HvKp and cKp strains induce pyroptotic cell death in BMDMs. (A–C) BMDMs were infected with hvKp or cKp (MOI of 10) for 16 h. Cell death was analyzed using (A) flow cytometry with propidium iodide (PI) staining and (B) LDH release in the supernatant. The number of PI+ macrophages was determined using flow cytometry and is shown by a representative value from the three independent experiments. (C) The cell morphologies were determined by bright-field microscopy. Images were magnified ×400. Scale bar, 20 μm. (D) BMDMs were infected with hvKp or cKp (MOI of 10) for the indicated times, and the mRNA levels of Il1b and Il18 were measured by qRT-PCR. (E) BMDMs were infected with hvKp or cKp at different MOI for 16 h. The IL-1β levels were determined using ELISA in cell culture medium. (F) BMDMs were infected with hvKp or cKp (MOI of 10) for 16 h. Cells were lysed and subjected to immunoblotting using antibodies against caspase-1, GSDMD and ACTB. (G) BMDMs were infected with hvKp or cKp (MOI of 10) for 16 h. Cells were stained with cleaved GSDMD (green) and DAPI (for nuclei). Representative images were analyzed using confocal microscopy. Scale bar, 20 μm (left). The quantification of cleaved GSDMD fluorescence (right). Data are expressed as the mean ± standard deviation of the three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 according to analysis of variance. Un, uninfected; Av, average.
To distinguish the types of cell death, we observed the morphological changes in BMDMs during hvKp and cKp infection using bright-field microscopy. The cKp infection caused severe cell membrane rupture and cell swelling (Figure 1C). Therefore, we measured the expression mRNA levels of interleukin (Il)1b and Il18, which are induced by pyroptosis activation, in BMDMs during infection. Similarly, the cKp-infected cells showed higher mRNA expression levels of Il1b and Il18 compared with hvKp-infected cells (Figure 1D). Moreover, ELISA showed the low level of IL-1β production in hvKp-infected BMDMs (Figure 1E). During pyroptosis, full-length GSDMD, a pyroptosis-associated protein, was cleaved by inflammatory caspases, and its N-terminus assembled to form pores in the plasma membrane (24). To confirm the occurrence of GSDMD-mediated pyroptosis during hvKp and cKp infection, we measured the GSDMD protein levels. Full-length GSDMD expression level was significantly decreased in cKp-infected cells compared with that in hvKp-infected cells, while the levels of activated caspase-1 increased in the cKp-infected BMDMs (Figure 1F). Confocal analysis showed that the expression level of cleaved GSDMD was significantly increased in hvKp- and cKp-infected BMDMs (Figure 1G). The cKp infection exhibited higher the fluorescence of cleaved GSDMD when compared to hvKp infection (Figure 1G). These results demonstrate that hvKp infection in BMDMs reduces the levels of GSDMD-mediated pyroptosis compared with that during cKp infection.
3.2 The cKp infection promotes pyroptosis in RAW264.7 cells compared with hvKp infection
To evaluate the effects of hvKp and cKp infection in the murine macrophage cell line, we infected RAW264.7 cells with hvKp and cKp strains and measured the sub-G1 population. As shown in Figure 2A, both strains exhibited an increase in the sub-G1 phase cells in a MOI-dependent manner. Consistent with the BMDMs results, cKp infection increased the percentage of the sub-G1 population in RAW264.7 cells. Similarly, the level of LDH release from cKp-infected RAW264.7 cells was significantly higher than that from hvKp-infected cells at different MOIs (Figure 2B). In addition, qRT-PCR analysis showed a higher expression level of Il1b and Il18 in cKp-infected RAW264.7 cells than in hvKp-infected RAW264.7 cells, in a MOI-dependent manner (Figure 2C). By contrast, hvKp infection induced relatively low levels of Il1b and Il18 expression in these cells (Figure 2C). Like in infected BMDMs, the full-length GSDMD was cleaved, and the level of activated caspase-1 was higher in cKp-infected RAW264.7 cells compared to that observed in hvKp-infected RAW264.7 cells (Figure 2D). These data indicate that the level of cKp-induced pyroptosis is higher than that of hvKp-induced pyroptosis in RAW264.7 murine macrophage cell line.
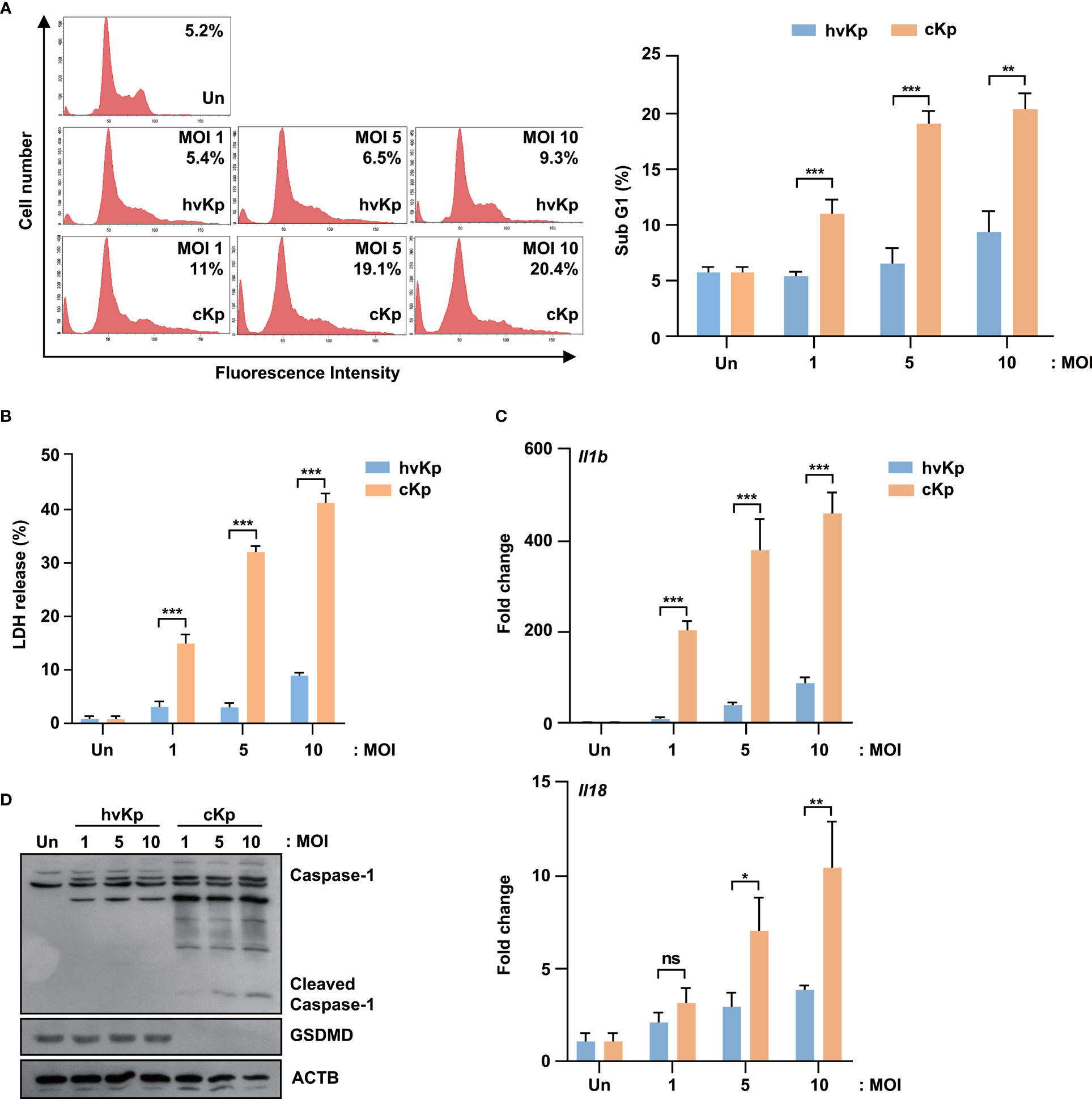
Figure 2 HvKp and cKp strains induce pyroptotic cell death in the RAW264.7 murine macrophage cell line. (A, B) RAW264.7 cells were infected with hvKp or cKp (MOI of 1, 5, and 10) for 16 h. Cell death was analyzed using (A) flow cytometry with propidium iodide (PI) staining and (B) LDH release in the supernatant. The number of PI+ macrophages was determined by flow cytometry and is shown by a representative value from the three independent experiments. (C) RAW264.7 cells were infected with hvKp and cKp for 2 h, and the mRNA levels of Il1b and Il18 were measured by qRT-PCR. (D) RAW264.7 cells were infected with hvKp or cKp (MOI of 1, 5, and 10). After incubation for 16 h, the cells were lysed and subjected to immunoblotting using antibodies against caspase-1, GSDMD, and ACTB. Data are expressed as the mean ± standard deviation of the three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 according to analysis of variance. Un, uninfected; ns, not significant.
3.3 Association between the level of hvKp- and cKp-induced pyroptosis and caspase-1 expression in murine macrophages
Similar to western blot analysis, confocal microscopy showed that caspase-1 expression level was significantly higher in cKp-infected macrophages compared with that in hvKp-infected macrophages (Figure 3A for BMDMs and Supplementary Figure 1A for RAW264.7 cells). Therefore, we examined the effects of a caspase-1 inhibitor (Ac-YVAD-cmk) on the production of LDH upon infection. The caspase-1 inhibitor reduced the levels of LDH release from the supernatant of infected macrophages (Supplementary Figure 1B for BMDMs and Supplementary Figure 1C for RAW264.7 cells), which indicated the involvement and level of caspase-1 activity in pyroptosis.
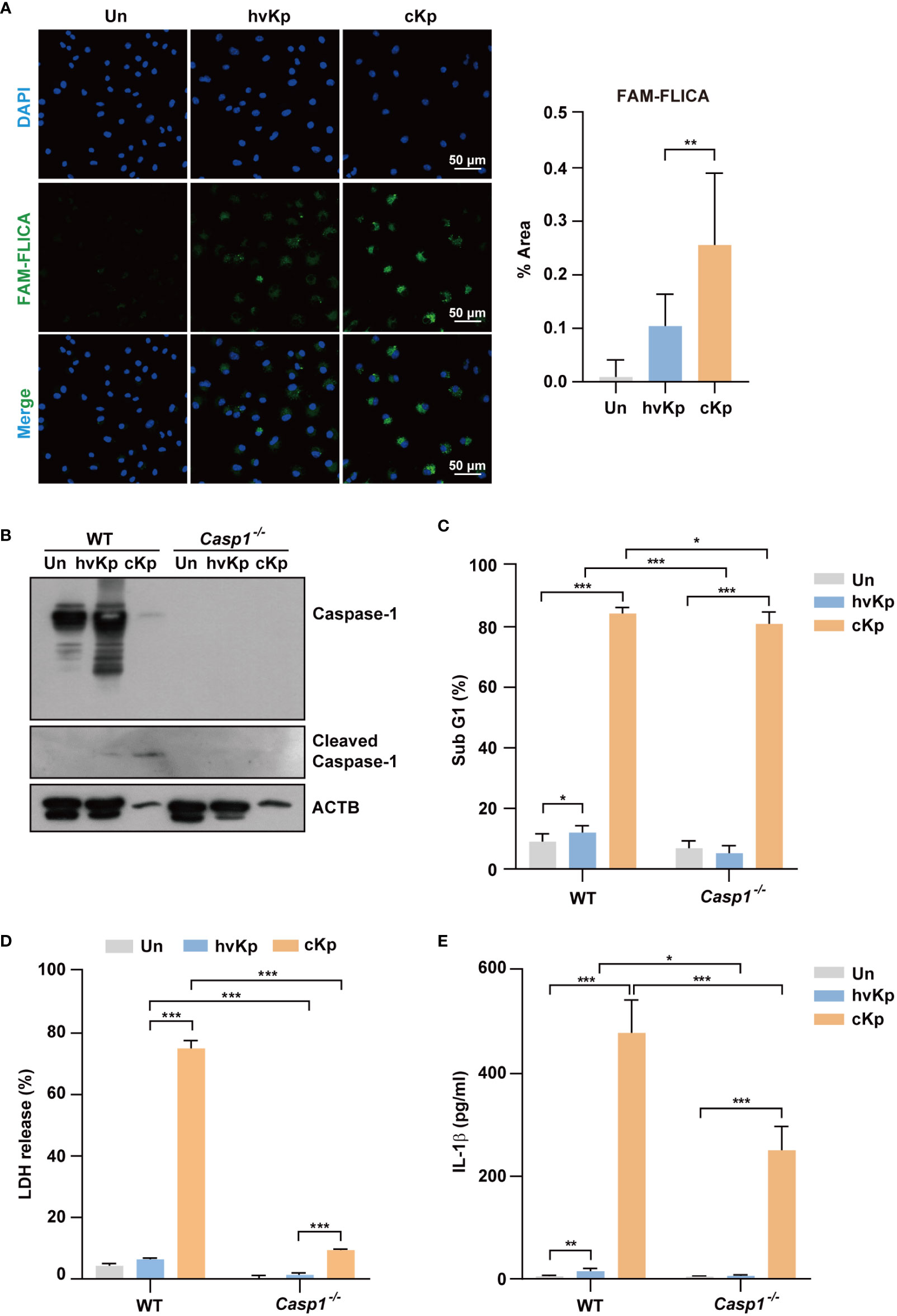
Figure 3 Infection of hvKp and cKp induces caspase-1-dependent pyroptosis in BMDMs. (A) BMDMs were infected with hvKp or cKp (MOI of 10) for 8 h, and cells were stained with FAM-FLICA for 1 h. Nuclei were stained with Hoechst 33342. The images were visualized using confocal microscopy. Scale bar, 50 μm (left). Quantification of the FAM-FLICA area (right). (B–E) BMDMs were differentiated from WT and Caspase-1-/- mice and infected with the hvKp or cKp strains (MOI of 10) for 16 h. (B) Proteins were detected using immunoblotting. Cell death induced by the different strains was determined by (C) PI incorporation and (D) LDH release in the supernatant. (E) The secretion of IL-1β was measured by ELISA. Data are expressed as the mean ± standard deviation of the three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 according to analysis of variance. Un, uninfected; Casp1, caspase-1.
We further investigated the effects of caspase-1 in the cell death in macrophages from WT and Caspase-1-/- mice. The cleavage of caspase-1 following cKp infection in WT BMDMs was diminished in Caspase-1-/- BMDMs (Figure 3B). Furthermore, the cytotoxic effect of cKp infection in Caspase-1-/- BMDMs remarkably reduced compared with that in WT BMDMs (Figures 3C, D). In addition, the increased percentage of the sub-G1 population and level of LDH release following hvKp infection in WT BMDMs were inhibited in Caspase-1-/- BMDMs (Figures 3C, D). However, the involvement of caspase-1 in the cytotoxicity assays was higher in cKp-infected BMDMs compared with that in hvKp-infected BMDMs. ELISA result showed that the IL-1β production was reduced in Caspase-1-/- BMDMs compared with that in WT BMDMs during infection (Figure 3E). Collectively, these data demonstrate that caspase-1 is required for the activation of pyroptosis in macrophages during infection.
3.4 The cKp infection promotes the activation of NLRP3 inflammasome compared with hvKp infection
The NLRP3 inflammasome is involved in the induction of pyroptosis during pathogen infection (25–27). Therefore, we explored whether the NLRP3 inflammasome was activated during hvKp and cKp infection. The qRT-PCR analysis showed that hvKp and cKp infection upregulated the mRNA level of Nlrp3 in BMDMs (Figure 4A), which indicates that hvKp and cKp infections enhance the occurrence of pyroptosis via the formation of NLRP3 inflammasome in BMDMs. However, the expression level of Nlrp3 inflammasome gene was different. As expected, the expression level of Nlrp3 was significantly lower in hvKp-infected BMDMs (Figure 4A). Using confocal microscopy, we confirmed that the number of ASC, an adaptor protein in the NLRP3 inflammasome (28). The percentage of speck positive cells and the colocalization of NLRP3 with ASC were lower after hvKp infection compared with that after cKp infection (Figures 4B–D). Therefore, hvKp and cKp infections enhance the occurrence of pyroptosis via the formation of NLRP3 inflammasome in BMDMs.
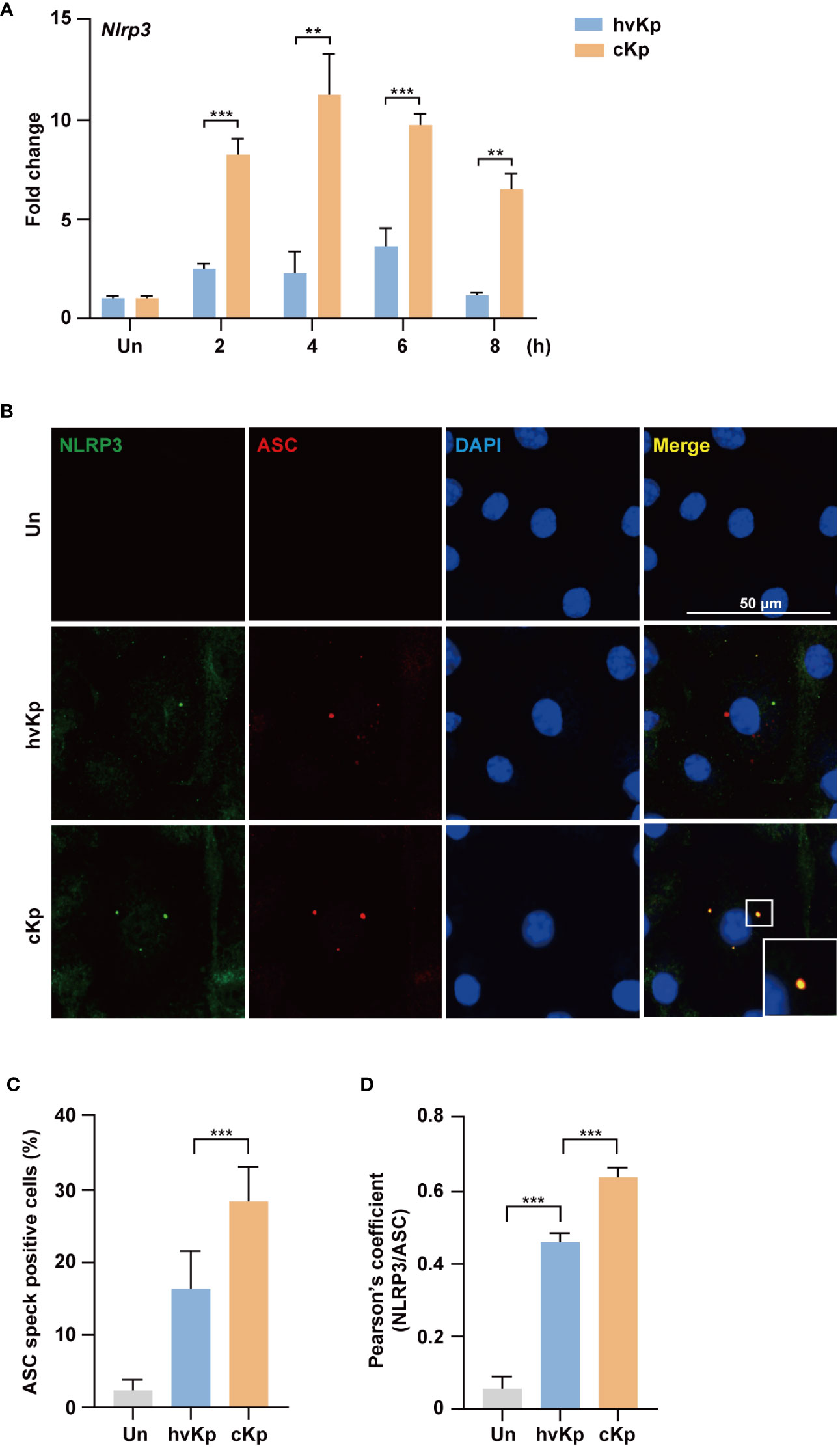
Figure 4 HvKp and cKp infections induce NLRP3 inflammasome formation. (A) BMDMs were infected with hvKp or cKp (MOI of 10) for the indicated times and the mRNA level of Nlrp3 was measured by qRT-PCR. (B–D) BMDMs were infected with hvKp or cKp (MOI of 10) for 8 h and cells were stained with NLRP3 (green), ASC (red), and DAPI (for nuclei). (B) Representative images were analyzed using confocal microscopy. Scale bar, 50 μm. (C) The quantification of ASC speck positive cells. (D) NLRP3 and ASC colocalization were analyzed by calculating the Pearson’s correlation coefficient. Data are expressed as the mean ± standard deviation of the three independent experiments. **P < 0.01 and ***P < 0.001 according to analysis of variance. Un, uninfected.
3.5 Involvement of cathepsin B in NLRP3-mediated pyroptosis during infection
As lysosomal cathepsin B is involved in NLRP3 inflammasome activation (14, 29), we examined whether cathepsin B activity contributes to the elevated NLRP3 inflammasome formation and pyroptosis during infection. During hvKp and cKp infection, Magic Red, which measures the cathepsin B activity, increased in BMDMs (Figure 5A). The cells were stained with LysoTracker, and their fluorescence intensity showed a remarkable reduction in cKp-infected macrophages compared with that in hvKp-infected cells (Supplementary Figure 2A for BMDMs and Supplementary Figure 2B for RAW264.7 cells); this finding suggests that hvKp and cKp infections induce cathepsin B leakage from lysosomes in macrophages.
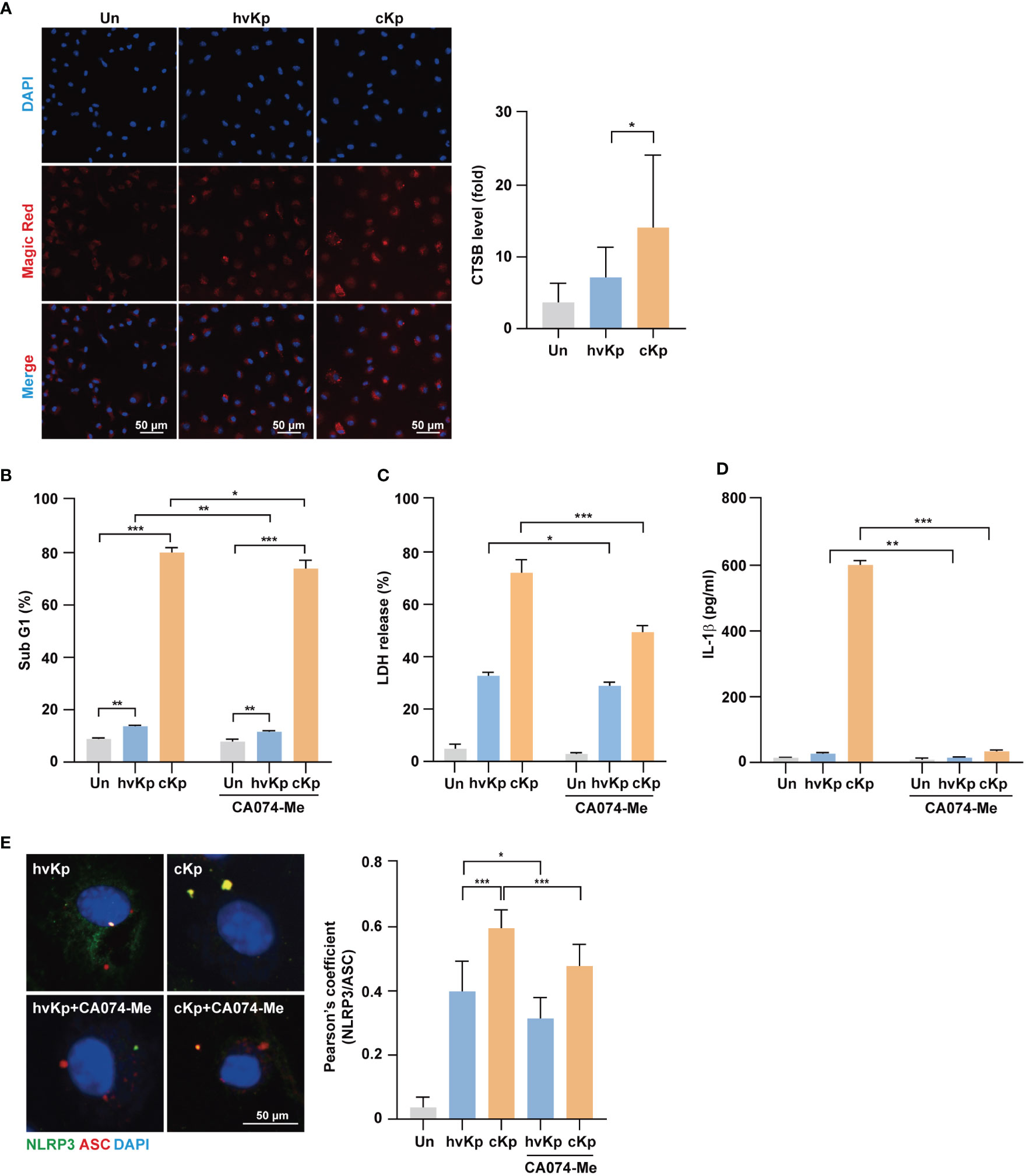
Figure 5 Activation of cathepsin B is involved in pyroptosis induced by hvKp and cKp infections. (A) BMDMs were infected with hvKp or cKp (MOI of 10) for 8 h, and cells were stained with Magic Red for 1 h. Nuclei were stained with Hoechst 33342. The images were visualized using confocal microscopy. Scale bar, 50 μm (left). Quantification of cathepsin B activity level (right). (B) BMDMs were pretreated with CA074-Me (25 μM) for 1 h, followed by infection with hvKp or cKp (MOI of 10). After incubation for 24 h (hvKp) or 16 h (cKp), cells were stained with PI and subjected to flow cytometry. (C, D) BMDMs were incubated in the presence or absence of CA074-Me for 1 h and then infected with the hvKp or cKp (MOI of 10), and after 24 h (hvKp) or 16 h (cKp), the supernatant was collected. (C) LDH release was measured in the supernatant and (D) IL-1β levels were determined using ELISA. (E) BMDMs were incubated in the presence or absence of CA074-Me for 1 h and then infected with hvKp or cKp (MOI of 10) for 8 h. Cells were stained with NLRP3 (green), ASC (red), and DAPI (for nuclei). Scale bar, 50 μm (left). NLRP3 and ASC colocalization were analyzed by calculating Pearson’s correlation coefficient (right). Data are expressed as the mean ± standard deviation of the three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 according to analysis of variance. Un, uninfected; CTSB, cathepsin B.
Next, we investigated whether increased cathepsin B activity due to infection enhances cell death during infection. BMDMs were infected with hvKp and cKp strains in the presence or absence of CA074-Me, an irreversible cathepsin B inhibitor, and cell death was assessed by fluorescence-activated cell sorting and LDH release assays. During infection, the cytotoxic effect was diminished by cathepsin B inhibition in BMDMs (Figures 5B, C) and RAW264.7 cells (Supplementary Figure 2C). In cKp-infected macrophages, the cytotoxic effect decreased robustly. Moreover, CA074-Me treatment reduced the production of IL-1β (Figure 5D) and caspase-1 activity in macrophages (Supplementary Figures 2D, E for BMDMs and Supplementary Figure 2F for RAW264.7 cells) during infection. In addition, we examined whether cathepsin B inhibition blocked the formation of NLRP3 inflammasome. As shown in Figure 5E, the colocalization between NLRP3 and ASC decreased following CA074-Me treatment. Taken together, these data demonstrate that cathepsin B release from lysosomes contributes to the NLRP3 inflammasome induction, caspase-1 activity, and cell death during hvKp and cKp infections.
4 Discussion
Since the first community-onset invasive liver abscess reported in East Asia caused by Kp exhibited a hypervirulent characteristic (30, 31), studies reporting hvKp have dramatically increased worldwide (32). The cKp strain has also been considered as an important pathogen over the last two decades (33) as it has shown carbapenem resistance. Clinically, hvKp infection causes severe inflammatory reactions, induces abscess formation, and is frequently accompanied by metastatic lesions such as endophthalmitis and myositis compared with cKp infection (34). Over time, hvKp strains have become increasingly resistant to antibiotics. The hvKp strains acquire antimicrobial resistance genes by inserting resistance plasmids or resistance elements into the hyKp virulence plasmid (35, 36). These bacterial strains can cause severe infections. However, it remains unknown why hvKp exhibits pathogenicity with severe inflammatory reactions. Therefore, a mechanistic understanding of the immunological responses is warranted to understand the pathogenesis of hvKp infection.
Cell death has a crucial role in aspects of immune responses against pathogen infection. Pyroptosis, a type of regulated cell death, controls microbial survival and contributes to the regulation of the host immune response (37–39). Recent studies of the role of pyroptosis in Kp infection have suggested that Kp-induced pyroptosis is important for the development of host immunity. When HEp-2 cells were treated with OmpA, the transmembrane porin protein of Kp (ATCC 13883), OmpA induced cell cycle arrest, apoptosis, and pyroptosis. These events may act as host defense mechanisms against Kp infection (40). In addition, caspase-11-mediated IL-1α and IL-1β secretion, and pyroptosis were involved in the early stages of Kp infection (41). However, Codo et al. showed that different clinical strains of Kp exhibited opposite behaviors within the macrophages (23). A28006, a Kp carbapenemase-2-producing clinical strain, triggered high levels of pyroptosis and IL-1β production, leading to bacterial clearance via efferocytosis (23). Meanwhile, the A54970 clinical strain inhibited pyroptosis and inflammasome activation through the production of IL-10 (23). We also observed differential levels of pyroptosis between the hvKp and cKp strains in the macrophages. Similar to the A28006 Kp strain, cKp induced high levels of pyroptosis and IL-1β production in murine macrophages. The induction of pyroptosis can trigger pore-induced intracellular traps (PITs), which clear pathogens by efferocytosis and increase the susceptibility to antibiotics and oxidative stress (42). Based on this finding, cKp-infected macrophages may form PITs that clear pathogens and block their dissemination through the induction of pyroptosis.
However, pathogens have evolved counter-strategies to avoid or exploit pyroptosis to maintain survival (43). Mycobacterium tuberculosis induces type VII secretion system-dependent plasma membrane damage and pyroptosis, thus allowing M. tuberculosis to spread to neighboring cells (26). Li et al. showed that Shigella flexneri can escape caspase-11/4-mediated pyroptosis using OspC3, a type III secretion system effector (44). Because Kp also regulates cell death including pyroptosis, it is important to understand cell death caused by Kp infection. A54970 Kp strain inhibited pyroptosis to survive in the host, leading to bacterial dissemination (23). In addition, Kp infection triggered apoptosis and changed cellular ATP levels in HepG2 cells (45). In neutrophil, Kp delayed the induction of apoptosis, which could restrict intracellular bacteria replication (46). Moreover, Kp induced necroptosis of neutrophil, but inhibited apoptosis, resulting in the reduction of efferocytosis (47). Importantly, our study found that the levels of pyroptosis and NLRP3 inflammasome formation were lower in hvKp-infected macrophages compared with those in cKp-infected macrophages. These data suggest that hvKp can control cell death to escape from pyroptosis-induced efferocytosis and PIT formation. Although pyroptosis can be induced by a non-canonical pathway that is activated by the direct interaction between caspase-4/5/11 and cytosolic lipopolysaccharides (9), we focused on investigating the canonical pathway during infection. Notably, we found that the cytotoxic effects were decreased in Caspase-1-/- BMDMs and after caspase-1 inhibitor treatment. These findings demonstrate that hvKp and cKp induce the occurrence of pyroptosis via the activation of caspase-1. We also showed that cytotoxic effects were significantly reduced in hvKp-infected Caspase-1-/- BMDMs, compared with cKp-infected cells.
To further understand the mechanisms underlying the activation of pyroptosis, we investigated whether cathepsin B influences the occurrence of pyroptosis during infection. Pathogens such as M. tuberculosis and Legionella pneumophila upregulate the expression and enzymatic activity of cathepsin B (18, 48). During hvKp infection, the transcription of cysteine cathepsin inhibitor was upregulated (49). Additionally, thrombospondin-1-deficient mice showed increased bacterial clearance and survival compared with WT during Kp infection (50). These mice showed enhanced cathepsin G enzymatic activity in neutrophil (50). Although these results suggest that cathepsins can participate in immune responses upon Kp infection, there are no reports that cathepsins are involved in the ability of Kp to survive in macrophages. We found that hvKp and cKp infection increased the enzymatic activity of cathepsin B in murine macrophages. Previous studies have indicated that lysosomal cathepsin B activated pyroptosis through the formation of NLRP3 inflammasome in various cell types (13) and the Kp 43816 strain could trigger pyronecrosis requiring activity of cathepsin B (51). We also found that cathepsin B activation is involved in the induction of pyroptosis and NLRP3 inflammasome formation during infection. The collective findings of this study show that hvKp significantly inhibits induction of cathepsin B-NLRP3-dependent pyroptosis in murine macrophages compared with those in cKp-infected macrophages (Figure 6).
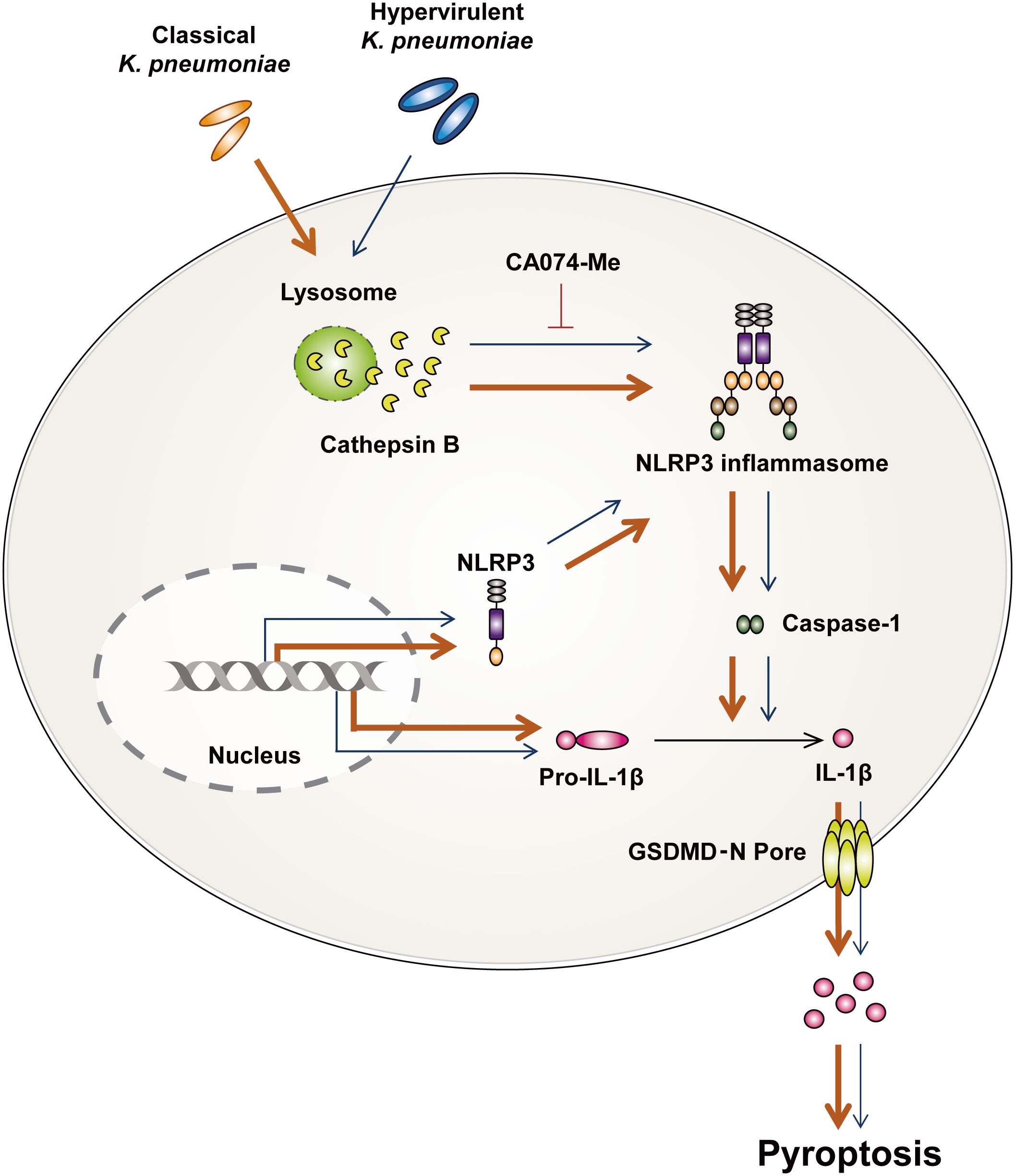
Figure 6 Summarized illustration of hvKp- and cKp-induced pyroptosis in murine macrophages. HvKp and cKp infections induce cathepsin B activation, NLRP3 inflammasome formation, and caspase-1-dependent pyroptosis. All phenomena are inhibited in hvKp-infected macrophages.
The important virulence factors of hvKp were identified. The most frequent contributors to the hypervirulent characteristics are rmpA and aerobactin; these virulence genes are associated with a virulence plasmid, regardless of the capsular serotype (52). Since earlier study reported that strains with K1-type capsules delayed apoptosis of neutrophil compared with acapsular mutants (53), we can speculate that the induction of pyroptosis may be delayed in hvKp-infected macrophages. However, further studies are needed to identify the factors that cause these differences in host cells. This study was the first to report the occurrence of pyroptosis during hvKp infection in macrophages. Based on these findings, cathepsin B-mediated pyroptosis is a potential new target for regulating host immune responses during hvKp infection.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was reviewed and approved by Institutional Research and Ethics Committee at Keimyung University.
Author contributions
JKK and H-JJ performed experiments and wrote the manuscript. MH and JYL collected clinical strains. J-HP provided Caspase-1-/- mice. S-IS supported analytical tools and reagents. W-KB and HAK designed and supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2020R1F1A1070470).
Acknowledgments
We would like to thank Hye Kyung Cho for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1207121/full#supplementary-material
References
1. Magill SS, O’Leary E, Janelle SJ, Thompson DL, Dumyati G, Nadle J, et al. Changes in prevalence of health care-associated infections in U.S. hospitals. N Engl J Med (2018) 379(18):1732–44. doi: 10.1056/NEJMoa1801550
2. WHO. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. (Geneva: World Health Organization) (2017).
3. Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev (2019) 32(3):e00001-19. doi: 10.1128/CMR.00001-19
4. Zhang Y, Zhao C, Wang Q, Wang X, Chen H, Li H, et al. High prevalence of hypervirulent Klebsiella pneumoniae infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob Agents Chemother (2016) 60(10):6115–20. doi: 10.1128/AAC.01127-16
5. Fursova AD, Fursov MV, Astashkin EI, Novikova TS, Fedyukina GN, Kislichkina AA, et al. Early response of antimicrobial resistance and virulence genes expression in classical, hypervirulent, and hybrid hvKp-MDR Klebsiella pneumoniae on antimicrobial stress. Antibiotics (Basel) (2021) 11(1):7. doi: 10.3390/antibiotics11010007
6. Wang L, Shen D, Wu H, Ma Y. Resistance of hypervirulent Klebsiella pneumoniae to both intracellular and extracellular killing of neutrophils. PloS One (2017) 12(3):e0173638. doi: 10.1371/journal.pone.0173638
7. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
8. Liang F, Zhang F, Zhang L, Wei W. The advances in pyroptosis initiated by inflammasome in inflammatory and immune diseases. Inflammation Res (2020) 69(2):159–66. doi: 10.1007/s00011-020-01315-3
9. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6(1):128. doi: 10.1038/s41392-021-00507-5
10. Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol (2021) 74(1):156–67. doi: 10.1016/j.jhep.2020.07.041
11. Wang Q, Wu J, Zeng Y, Chen K, Wang C, Yang S, et al. Pyroptosis: a pro-inflammatory type of cell death in cardiovascular disease. Clin Chim Acta (2020) 510:62–72. doi: 10.1016/j.cca.2020.06.044
12. Hu Y, Wang B, Li S, Yang S. Pyroptosis, and its role in central nervous system disease. J Mol Biol (2022) 434(4):167379. doi: 10.1016/j.jmb.2021.167379
13. Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys (2019) 670:32–42. doi: 10.1016/j.abb.2019.02.015
14. Chevriaux A, Pilot T, Derangere V, Simonin H, Martine P, Chalmin F, et al. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front Cell Dev Biol (2020) 8:167. doi: 10.3389/fcell.2020.00167
15. Shridas P, De Beer MC, Webb NR. High-density lipoprotein inhibits serum amyloid a-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J Biol Chem (2018) 293(34):13257–69. doi: 10.1074/jbc.RA118.002428
16. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
17. Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis (2019) 10(10):778. doi: 10.1038/s41419-019-2021-3
18. Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by Mycobacterium tuberculosis in infected macrophages. Front Immunol (2018) 9:1427. doi: 10.3389/fimmu.2018.01427
19. Wang Y, Jia L, Shen J, Wang Y, Fu Z, Su SA, et al. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PloS Pathog (2018) 14(1):e1006872. doi: 10.1371/journal.ppat.1006872
20. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (1995) 267(5206):2000–3. doi: 10.1126/science.7535475
21. Li W, Sun G, Yu Y, Li N, Chen M, Jin R, et al. Increasing occurrence of antimicrobial-resistant hypervirulent (hypermucoviscous) Klebsiella pneumoniae isolates in China. Clin Infect Dis (2014) 58(2):225–32. doi: 10.1093/cid/cit675
22. Compain F, Babosan A, Brisse S, Genel N, Audo J, Ailloud F, et al. Multiplex PCR for detection of seven virulence factors and K1/K2 capsular serotypes of Klebsiella pneumoniae. J Clin Microbiol (2014) 52(12):4377–80. doi: 10.1128/JCM.02316-14
23. Codo AC, Saraiva AC, Dos Santos LL, Visconde MF, Gales AC, Zamboni DS, et al. Inhibition of inflammasome activation by a clinical strain of Klebsiella pneumoniae impairs efferocytosis and leads to bacterial dissemination. Cell Death Dis (2018) 9(12):1182. doi: 10.1038/s41419-018-1214-5
24. Ruhl S, Broz P. The gasdermin-D pore: executor of pyroptotic cell death. Oncotarget (2016) 7(36):57481–2. doi: 10.18632/oncotarget.11421
25. Hua KF, Yang FL, Chiu HW, Chou JC, Dong WC, Lin CN, et al. Capsular polysaccharide is involved in NLRP3 inflammasome activation by Klebsiella pneumoniae serotype K1. Infect Immun (2015) 83(9):3396–409. doi: 10.1128/IAI.00125-15
26. Beckwith KS, Beckwith MS, Ullmann S, Saetra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun (2020) 11(1):2270. doi: 10.1038/s41467-020-16143-6
27. He S, Li L, Chen H, Hu X, Wang W, Zhang H, et al. PRRSV infection induces gasdermin D-driven pyroptosis of porcine alveolar macrophages through NLRP3 inflammasome activation. J Virol (2022) 96(14):e0212721. doi: 10.1128/jvi.02127-21
28. Seok JK, Kang HC, Cho YY, Lee HS, Lee JY. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch Pharm Res (2021) 44(1):16–35. doi: 10.1007/s12272-021-01307-9
29. Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta (2015) 1853(2):396–408. doi: 10.1016/j.bbamcr.2014.11.012
30. Liu YC, Cheng DL, Lin CL. Klebsiella pneumoniae liver abscess associated with septic endophthalmitis. Arch Intern Med (1986) 146(10):1913–6. doi: 10.1001/archinte.1986.00360220057011
31. Fang CT, Chuang YP, Shun CT, Chang SC, Wang JT. A novel virulence gene in Klebsiella pneumoniae strains causing primary liver abscess and septic metastatic complications. J Exp Med (2004) 199(5):697–705. doi: 10.1084/jem.20030857
32. Turton JF, Englender H, Gabriel SN, Turton SE, Kaufmann ME, Pitt TL. Genetically similar isolates of Klebsiella pneumoniae serotype K1 causing liver abscesses in three continents. J Med Microbiol (2007) 56(Pt 5):593–7. doi: 10.1099/jmm.0.46964-0
33. Gutierrez-Gutierrez B, Salamanca E, de Cueto M, Hsueh PR, Viale P, Pano-Pardo JR, et al. Effect of appropriate combination therapy on mortality of patients with bloodstream infections due to carbapenemase-producing enterobacteriaceae (INCREMENT): a retrospective cohort study. Lancet Infect Dis (2017) 17(7):726–34. doi: 10.1016/S1473-3099(17)30228-1
34. Fang CT, Lai SY, Yi WC, Hsueh PR, Liu KL, Chang SC. Klebsiella pneumoniae genotype K1: an emerging pathogen that causes septic ocular or central nervous system complications from pyogenic liver abscess. Clin Infect Dis (2007) 45(3):284–93. doi: 10.1086/519262
35. Zhang R, Lin D, Chan EW, Gu D, Chen GX, Chen S. Emergence of carbapenem-resistant serotype K1 hypervirulent Klebsiella pneumoniae strains in China. Antimicrob Agents Chemother (2016) 60(1):709–11. doi: 10.1128/AAC.02173-15
36. Fu L, Tang L, Wang S, Liu Q, Liu Y, Zhang Z, et al. Co-Location of the blaKPC-2, blaCTX-M-65, rmtB and virulence relevant factors in an IncFII plasmid from a hypermucoviscous Klebsiella pneumoniae isolate. Microb Pathog (2018) 124:301–4. doi: 10.1016/j.micpath.2018.08.055
37. Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature (2022) 602(7897):496–502. doi: 10.1038/s41586-021-04384-4
38. Crowley SM, Han X, Allaire JM, Stahl M, Rauch I, Knodler LA, et al. Intestinal restriction of Salmonella typhimurium requires caspase-1 and caspase-11 epithelial intrinsic inflammasomes. PloS Pathog (2020) 16(4):e1008498. doi: 10.1371/journal.ppat.1008498
39. Pudla M, Sanongkiet S, Ekchariyawat P, Luangjindarat C, Ponpuak M, Utaisincharoen P. TLR9 negatively regulates intracellular bacterial killing by pyroptosis in Burkholderia pseudomallei-infected mouse macrophage cell line (Raw264.7). Microbiol Spectr (2022) 10(5):e0348822. doi: 10.1128/spectrum.03488-22
40. You HS, Lee SH, Kang SS, Hyun SH. OmpA of Klebsiella pneumoniae ATCC 13883 induces pyroptosis in HEp-2 cells, leading to cell-cycle arrest and apoptosis. Microbes Infect (2020) 22(9):432–40. doi: 10.1016/j.micinf.2020.06.002
41. Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, et al. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol (2017) 307(8):490–6. doi: 10.1016/j.ijmm.2017.09.012
42. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213(10):2113–28. doi: 10.1084/jem.20151613
43. Brokatzky D, Mostowy S. Pyroptosis in host defence against bacterial infection. Dis Model Mech (2022) 15(7):dmm049414. doi: 10.1242/dmm.049414
44. Li Z, Liu W, Fu J, Cheng S, Xu Y, Wang Z, et al. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature (2021) 599(7884):290–5. doi: 10.1038/s41586-021-04020-1
45. Yang PY, Chen WX, Chang FY, Chen HW, Lin CH, Hsu YC, et al. HepG2 cells infected with Klebsiella pneumoniae show DNA laddering at apoptotic and necrotic stages. Apoptosis (2012) 17(2):154–63. doi: 10.1007/s10495-011-0666-1
46. Kobayashi SD, Porter AR, Dorward DW, Brinkworth AJ, Chen L, Kreiswirth BN, et al. Phagocytosis and killing of carbapenem-resistant ST258 Klebsiella pneumoniae by human neutrophils. J Infect Dis (2016) 213(10):1615–22. doi: 10.1093/infdis/jiw001
47. Jondle CN, Gupta K, Mishra BB, Sharma J. Klebsiella pneumoniae infection of murine neutrophils impairs their efferocytic clearance by modulating cell death machinery. PloS Pathog (2018) 14(10):e1007338. doi: 10.1371/journal.ppat.1007338
48. Morinaga Y, Yanagihara K, Nakamura S, Hasegawa H, Seki M, Izumikawa K, et al. Legionella pneumophila induces cathepsin B-dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir Res (2010) 11(1):158. doi: 10.1186/1465-9921-11-158
49. Lei L, Zhang X, Yang R, Jing H, Yuan Y, Chen Z, et al. Host immune response to clinical hypervirulent Klebsiella pneumoniae pulmonary infections via transcriptome analysis. J Immunol Res (2022) 2022:5336931. doi: 10.1155/2022/5336931
50. Zhao Y, Olonisakin TF, Xiong Z, Hulver M, Sayeed S, Yu MT, et al. Thrombospondin-1 restrains neutrophil granule serine protease function and regulates the innate immune response during Klebsiella pneumoniae infection. Mucosal Immunol (2015) 8(4):896–905. doi: 10.1038/mi.2014.120
51. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol (2009) 183(3):2008–15. doi: 10.4049/jimmunol.0900138
52. Wu KM, Li LH, Yan JJ, Tsao N, Liao TL, Tsai HC, et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol (2009) 191(14):4492–501. doi: 10.1128/JB.00315-09
Keywords: hypervirulent, Klebsiella pneumoniae, macrophages, pyroptosis, cathepsin B
Citation: Kim JK, Jung H-J, Hyun M, Lee JY, Park JH, Suh S-I, Baek W-K and Kim HA (2023) Resistance of hypervirulent Klebsiella pneumoniae to cathepsin B-mediated pyroptosis in murine macrophages. Front. Immunol. 14:1207121. doi: 10.3389/fimmu.2023.1207121
Received: 17 April 2023; Accepted: 31 May 2023;
Published: 29 June 2023.
Edited by:
Subash Babu, International Centers for Excellence in Research (ICER), IndiaReviewed by:
Arnaud John Kombe Kombe, Agricultural Research Service (USDA), United StatesVinícius S. Alves, Federal University of Rio de Janeiro, Brazil
Copyright © 2023 Kim, Jung, Hyun, Lee, Park, Suh, Baek and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyun ah Kim, aHl1bmFoMTExOEBkc21jLm9yLmty; Won-Ki Baek, d29ua2lAZHNtYy5vci5rcg==
†These authors have contributed equally to this work and share first authorship